leeuwenhoek the simple microscope microscope · leeuwenhoek microscope (circa late 1600s ) upright...
TRANSCRIPT

The simple microscope Leeuwenhoek Microscope
(circa late 1600s)

Upright microscope .
Inverted microscope
Transmitted and Fluorescence Illumination

The Objective
The Microscope’s Most Important Component
http://zeiss-campus.magnet.fsu.edu http://www.microscopyu.com/articles/optics/objectiveintro.html

The second most important component…
The Condenser

Condenser maximizes resolution
dmin = 1.22 λ / (NA objective +NA condenser)
Kohler Illumination: Condenser and objective focused at the same plane

Resolution versus Contrast
• d = 0.61λ/NA
• λ=wavelength; NA=Numerical Apeture

C ONTRAST
50 – 0 / 50 + 0 = 1
50 – 100 / 50 + 100 = -0.33
50 – 50 / 50 + 50 = 0
Background of BrightnessSpecimen of BrightnessBackground of Brightness-Specimen of Brightness
+
50 Units 0 Units 100 Units
50 Units 50 50


Electromagnetic Spectrum
Longer
Higher Resolution

Transmitted Light • Brightfield • Oblique
• Darkfield • Phase Contrast • Polarized Light • DIC (Differential Interference
Contrast)
Incident Light • Brightfield • Oblique
• Darkfield • Polarized Light • Fluorescence (Epi)
Illumination Techniques - Overview

DIC (Nomarski)
" High Contrast and high resolution
" Full Control of condenser aperture
" 3-D Image appearance
" Color DIC by adding a wave plate
" Selectable contrast / resolution via different DIC sliders
" Orientation-specific > orient fine details perpendicular to DIC prism

DIC (Differential Interference Contrast) after Nomarski
Observing local differences in phase retardation

9 Image
8 Tube lens 7 Analyzer (7a with Wave Plate) 6 Wollaston Prism behind objective 5 Objective
4 Specimen
3 Condenser 2 Wollaston Prism before condenser 1 Polarizer

Required Components for DIC:
• Nosepiece with DIC receptacles • Polarizer • Low Strain Condenser and Objective • DIC Prisms for Condenser (#I orII orIII) • Specific DIC Slider for each objective • Analyzer

Fluorescence
• Easy to set up > Objective = Condenser
• Highly specific technique, wide selection of markers
• Detection and Identification of Proteins, Bacteria, Viruses
• Basics for – Special Techniques eg. TIRF, FRET, FRAP etc. – 3-D imaging – Deconvolution – Structured Illumination – Confocal Techniques

Blue light absorbed
490nm 520nm
Green light emitted
Stokes Shift
Where does energy go?
Quantum Yield = light out/light in
Q ~ 0.8 fluorescein
~ 0.3 rhodamine

• Mercury (Hg)
• Xenon, Hg/Xe Combination
• Laser
• LED’s
• Tungsten Halogen
Light Sources

Epi - Fluorescence (Specimen containing green fluorescing Fluorochrome)
Dichromatic Mirror
Emission Filter
Excitation Filter
Observation port
FL
Light Source
Specimen containing green fluorescing Fluorochrome

How to improve Fluorescence Imaging in a major way:
• Optical Sectioning

Overview of Optical sectioning Methods
1. Confocal and Multi-photon Laser Scanning Microscopy
– Pinhole prevents out-of-focus light getting to the sensor(s) (PMT - Photomultiplier)
– Multi Photon does not require pinhole 2. Spinning disk systems
– A large number of pinholes (used for excitation and emission) is used to prevent out-of-focus light getting to the camera
– E.g. Perkin Elmer, Solamere 3. Deconvolution
– Point-Spread function (PSF) information is used to calculate light back to its origin
– Post processing of an image stack

Laser Scanning Confocal Microscopes (LSCM)
Zeiss LSM710 with Two-photon laser
Chameleon Ultra II Laser
Leica SP5 Spectral High Speed

Confocal Microscopy just a form of Fluorescence Microscopy
www.olympusfluoview.com

Optical Sectioning: Increased Contrast and Sharpness.
Examples: Zebrafish images, Inner ear
• Bit Depth • 8 bits = 256 • 12 ” = 4,096 • 16 ” = 65,536
• Maximize Histogram

3-D Reconstruction Zebrafish Cranial Ganglia
A P M L Neural Gata-2 Promoter GFP-Transgenic; Shuo Lin, UCLA

Spectral or Lambda Scanning
• Separate very similar colored fluorophores
– fluorescein and green fluorescent protein (GFP).
• Could be used to eliminate non-specific background fluorescence that has different emission spectra.
• Different technologies for spectrum detection
– Sequentially (Leica SP)
– Simultaneously (Zeiss QUASAR)

Lambda Stack
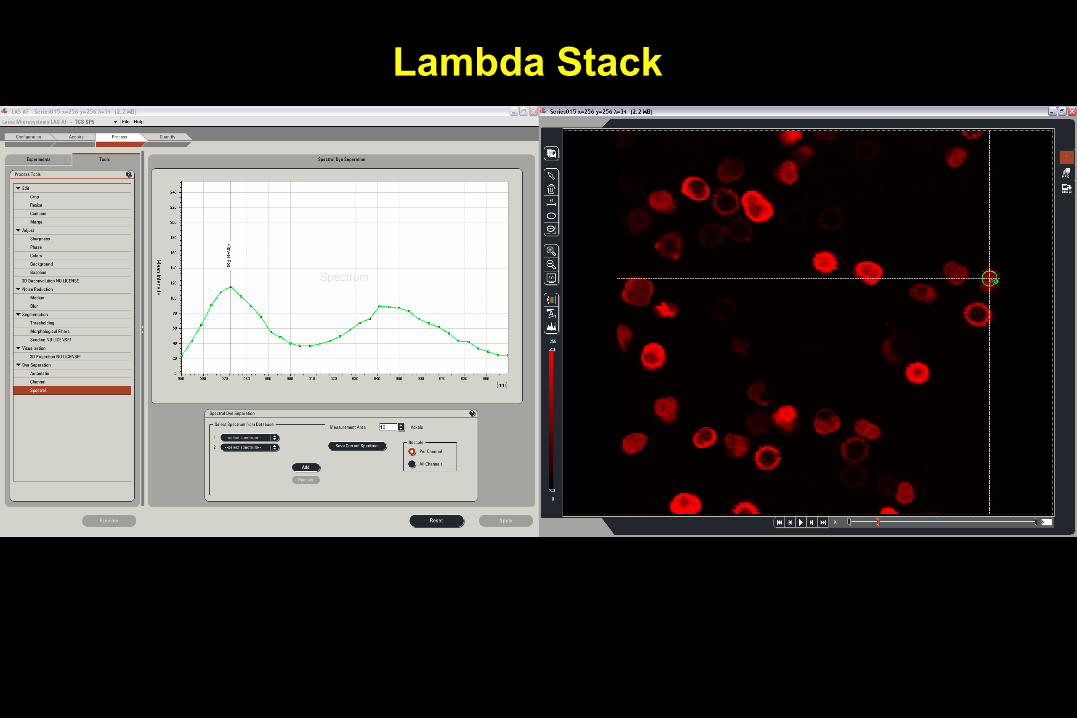
Lambda Stack

Lambda Stack

In vivo Hair Cell Dye, FM1-43 Spectra

High Speed Confocal Microscopy
1. Spinning disk systems
– A large number of pinholes with microlenses (used for excitation and emission) is used to prevent out-of-focus light getting to the camera
– E.g. Perkin Elmer, Solamere
2. Resonance Scanner (Leica, Nikon)
3. Double your scanning speed (Bidirectional)

http://zeiss-campus.magnet.fsu.edu/tutorials/spinningdisk/yokogawa/index.html

Confocal Speed - 90 fps
Crista Cilia Labeled in vivo with FM1-43
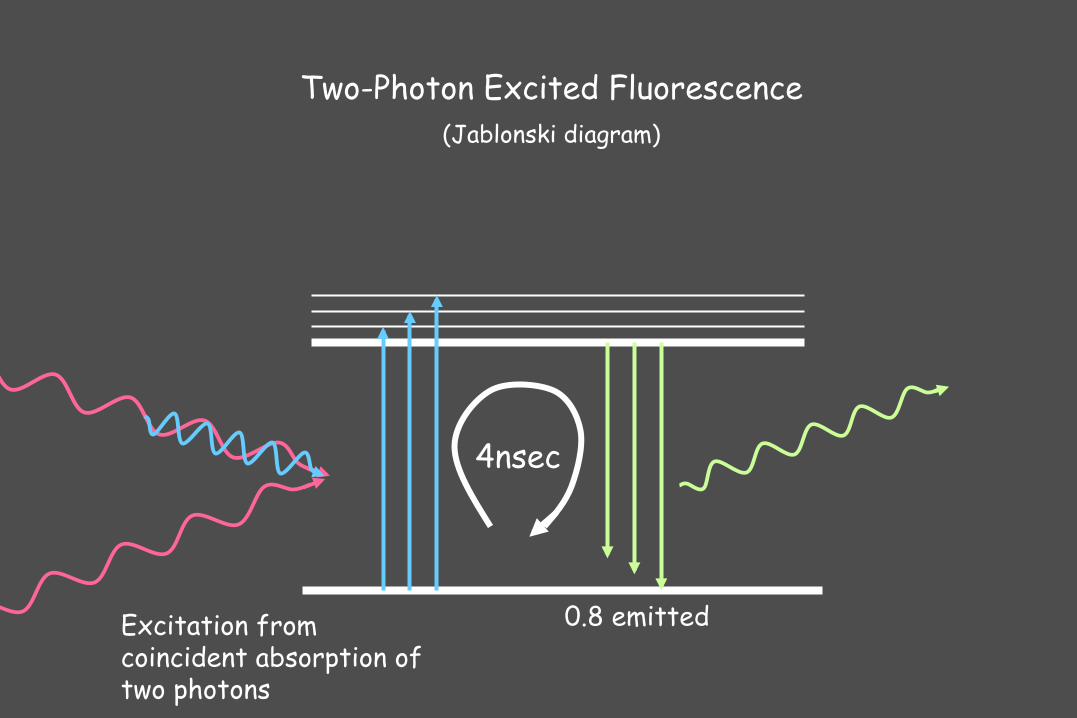
4nsec
Two-Photon Excited Fluorescence (Jablonski diagram)
0.8 emitted Excitation from coincident absorption of two photons

Two-Photon microscopy
Optical sectioning by non-linear absorbance --> broad excitation maxima

0
0.1
0.2
0.3
0.4
0.5
450 500 550 600nanometers
norm
aliz
ed in
tens
ityYFPCFPDilGFPEtBrRFP
TPLSM excitation at 900nm excites multiple dyes and GFP variants
Two-photon microscopy is somewhat color-blind

Two Photon Microscopy
• No need for pinhole
• No bleaching beyond focal plane
• Potentially more sensitive
• IR goes deeper into tissue
• Laser $$$
• Samples with melanin
• Samples with multiple fluorescent labels
Advantages Disadvantages

Super-Resolution Confocal Imaging: Below the wavelength of light
• STED: STimulated Emission Depletion (Deterministic)
• PALM: PhotoActivated Localization Microscopy (Stochastic)
• STORM: STochastic Optical Reconstruction Microscopy
http://zeiss-campus.magnet.fsu.edu
“True” Super-resolution
“Functional” Super-resolution

STED: STimulated Emission Depletion
http://zeiss-campus.magnet.fsu.edu
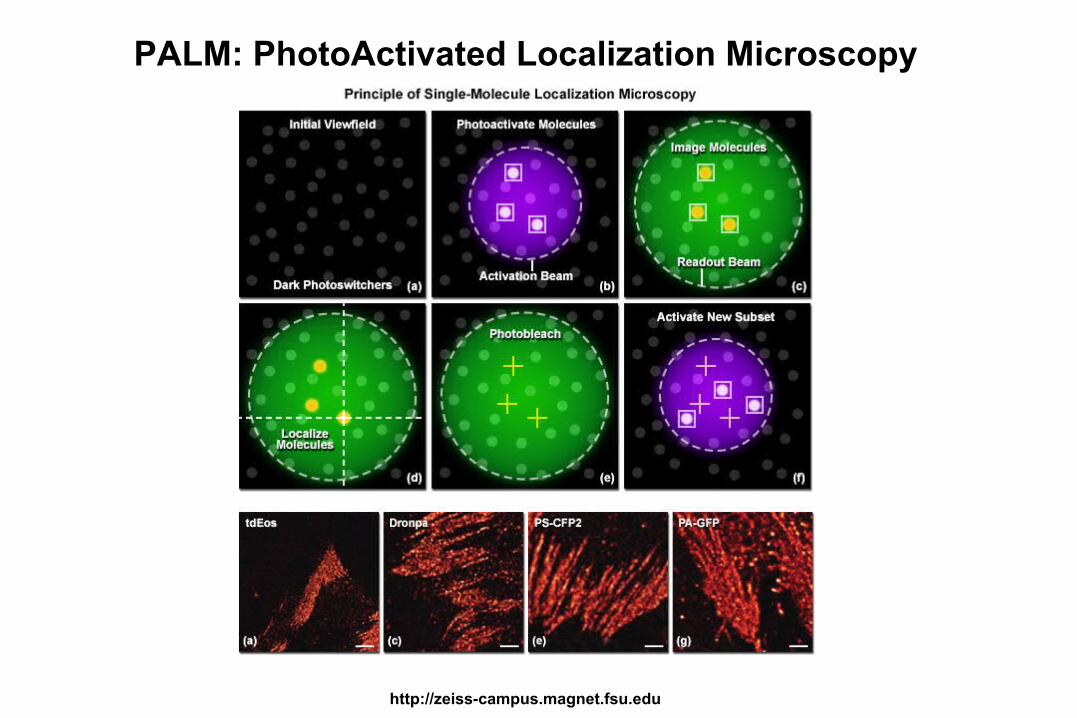
PALM: PhotoActivated Localization Microscopy
http://zeiss-campus.magnet.fsu.edu

Microscopy Resources on the Web
• http://www.olympusmicro.com
– Olympus
• http://www.microscopyu.com
– Nikon
• http://zeiss-campus.magnet.fsu.edu
– Zeiss

Acknowledgements
Shuo Lin, UCLA Caryl Forristall, University of Redlands Rudi Rottenfusser, Carl Zeiss Carlos Alonso, Leica Supported by NIH and NIDCD
Olivier Bricaud Aldo Castillo Aicha Castillo Frank Stellabotte Kalpana Desai
Bill Dempsey Periklis Pantazis
• Caltech Scott Fraser
Sung-Hee Kil Erik Waldman
Le Trinh

Field aperture
Condenser aperture

Kohler Step 1: Close field aperture Move condenser up-down to focus image of the field aperture

Kohler Step 2: Center image of field aperture Move condenser adjustment
centered

Kohler Illumination gives best resolution
Set Condenser aperture so NAcondenser = 0.9 x NAobjective
Open field aperture to fill view