international journal of pharma and bio sciences issn 0975 ... · int j pharm bio sci 2013 july;...
TRANSCRIPT

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 788
Research Article Bioinformatics
International Journal of Pharma and Bio Sciences ISSN
0975-6299
IN-SILICO STUDY OF ACETYLCHOLINESTERASE INHIBITION BY
ORGANOPHOSPHATE PESTICIDES
MADHULIKA CHAUDHRY*, 1, 2, J. FEBIN PRABHU DASS2,
D.SELVAKUMAR1 AND DR. N.S.KUMAR1
1Defence Bio-Engineering and Electromedical Laboratory,
DRDO, CV Raman Nagar, Bangalore-5600093, Karnataka, India. 2
Bioinformatics Division, School of Biosciences and Technology,
Vellore Institute of Technology, Vellore-63014, Tamil Nadu, India.
ABSTRACT
Pesticides are widely used round the globe for a plethora of applications that vastly includes agricultural fields and commercial gardens. Despite their wide use that aid increase in crop yield, they have ill-effects on environment and non-target organisms. Organophosphates (OP) are an important class of pesticides, which cause irreversible inhibition of acetylcholinesterase (AChE). Irreversible inhibition of AChE may lead to severe pathological conditions such as muscular paralysis, bronchial constriction and even death by asphyxiation. An in-silico approach was employed to study the interaction of AChE with four commonly used OP pesticides namely, methyl parathion, dichlorvos, malathion, chlorpyrifos. AutoDock4.2 and iGEMDOCK and SwissDock are opensource docking tools which were used to form the AChE-OP complex. Further these complexes were explored on their protein-ligand binding affinity along with the mechanism of binding. Based on the results, It was found that chlorpyrifos has the best binding affinity with AChE as compared to the other pesticides. These observation would surely be helpful in development of precautionary antidotes for the people who have direct contact with these poisonous pesticides. In addition, this information may be utilized in developing non-toxic therapeutic leads from the best binding pesticides, for Alzheimer’s diseases, where AChE is considered as a potential drug target. KEYWORDS: Molecular Docking Analysis, Acetylcholinesterase (AChE), Organophosphates (OP), Protein-Ligand Interactions and Cholinesterase inhibition.
MADHULIKA CHAUDHRY
Defence Bio-Engineering and Electromedical Laboratory,
DRDO, CV Raman Nagar, Bangalore-5600093, Karnataka, India.

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 789
INTRODUCTION Insecticides and nerve agents based on organophosphate(OP) compounds are potent inhibitors of the serine hydrolase super family of enzymes, of which AChE (EC 3.1.1.7) is an important member1, 2. Organophosphate pesticides became more popular after the ban of dichlorodiphenyltrichloroethane (DDT) in 1970. They were also preferred because of their easy biodegradation compared to DDT, aldrin etc. However they have been found to have acute toxicity and also have poisoning effect on humans, apart from killing the non-target insects and plants. This poisoning has occurred due to the inhibition of AChE. The general structure of
OP is shown in Figure1. AChE is a serine protease, which terminates cholinergic transmission by causing the neurotransmitter acetylcholine to break down. Compounds which inhibit this important enzyme, have been used as chemical warfare agents suh as G-series and V-series agents acts on nerve cells . On the other hand they also sever as an important drug targets in treatments of diseases like Alzheimer’s, Lewy Body Dementia, Glaucoma, Myasthenia gravis by enhancing the neurotransmitter availability at the neuromuscular junction.
General structure of Organophosphates
Figure 1 The general structure of Organophosphates3 containing a central phosphorus atom with a double bond to sulfur or oxygen, R1 and R2 are either methyl or ethyl groups, and the leaving group is specific to the individual organophosphate (Schematic representation made using Marvin Sketch). The study of AChE inhibition by organophosphate pesticides is an importantaspect. Through which one can develop precautionary antidotes to the known binding sites. Moreover the study on the inhibition of AChE in various insect can be achived using the pesticides homologs depending on a unique binding residue in the insect AChE. Hence an improved target specific application of a pesticide which can only target the specific insect, and the off-target toxicity can be reduced. This kind of study has already been undertaken at experimental level4. Reducing off-target toxicity of pesticides has gained prime importance after cases of collective pesticide
poisoning due to the indirect peptide consumption through pesticide contaminated vegetables in China5,6, . Cases have been reported where detection of more than permitted levels of pesticide residues in fruits and their effect on vegetables7, wines , poultry birds8 and in some instances even in breast milk9. Such incidents enforce urgency to find less toxic alternative solutions to that of chemical pesticides. In the present study, a recently available structure of Human AChE - PDB ID: 4EY410 was used. This protein structure is considered to be much better than the earlier used structures of AChE from different species, Torpedo californica, Mus musculus. Human

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 790
AChE structure is advantageous due to its high quality structure (2.16Å) compared to its previous version, 1B41 with a resolution of 2.76Å. Besideit allows us to directly predict the interacting amino acids of the human protein. This computational modeling of protein-ligand prediction study has been carried out on four of the most widely used OP pesticides. The complexes of various pesticides with AChE as receptor have been studied using AutoDock 4.211, iGEMDOCK12, and SwissDock13, 14. The complexes have been analyzed with respect to binding affinity, stability, inhibition constants and interacting residues. Since very little work has been done in this field except for, for methyl parathion, this comparative study of the pesticides will go a long way in development of less toxic homologs of the least stable (and hence least harmful) pesticide. This work gives a brief description of the computational methodology undertaken by our research group.
MATERIALS AND METHODS 1. Materials Structures of all the compounds in this study have been obtained from various online
databases and many online servers. Open-source software have been used in this study. A brief overview has been given in the subsequent sections. 1.1 Structure retrieval The structure of the acetylcholine receptor (AChE) has been downloaded from Protein Data Bank (PDB) 15, PDB ID: 4EY4. A very brief summary of the 4EY4 entry has been given in Table 1. The structure has been visualized in Accerlys Discovery Studio Visualizer 3.516 in its single chain form complexed with the ligands (labeled and shown in Figure 2). The four pesticides, methyl parathion (MP), dichlorvos (DDVP), malathion (ML) and chlorpyrifos (CLP) chosen for this study are already well characterized in terms of their structures. The 2D structures, along with their common and IUPAC names, are shown in Figure 3. For an accurate docking analysis, precise 3D coordinates of the ligands are required. Here, the SMILES notation for each of the four pesticides was retrieved from PUBCHEM17, 18, 19, 20, database and their 3D coordinates were generated using CORINA online server21, 22. These were used as input to all the softwares for our computational study.
Table 1
Summary of human acetylcholinesterase (4EY4) entry.
Chains Method External Ligands Natural Ligands Mol. Wt. (Da) Resolu--tion (Å) A(542 aa) B(542 aa) aa- amino acids
X-ray Crystallography
1.EDO (ethylene diol) 2.PE8(3,6,9,12,15,18,21-Heptaoxa tricosane-1,2,3-Diol)
1. NAG (N-acetyl-D-glucosamine) 2. FUC (Alpha-L-Fucose) 3. Nitrate ion
67,796 2.16
Structure of AChE
Figure 2 Single chain of AChE in complex with ligands.

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 791
1.2 Tools and Softwares Protein and Lingand energy minimization was performed using YASARA (Yet Another Scientific Artificial Reality Application) 23. Q-site Finder24, 25,
26 was used for active site prediction in AChE. AutoDock, iGEMDOCK and SwissDock were used for protein ligand docking. Accerlys Discovery Studio Visualizer 3.5, Jmol applet, Chimera25, Marvin Beans27 and SwissPDBViewer28 were used for visualization of macro and small molecules. 2. Methodology 2.1 Energy Minimization Energy minimization is considered as one of the important step toward docking as it optimize the molecules towards stable state.Energy of a molecule is inversely proportional to its stability. Hence to mimic the in-vivo environmental stability of AChE molecule, energy minimization was carried out using YASARA server.
Moreover, energy minimization also removes the bad contacts in the protein. 2.2 Active Site Prediction Apart from the detailed literature review about the catalytic gorge of AChE29, it’s always better to rely on prediction software because every compound will have a different site of most stable binding. Hence the best binding sites were predicted using Q-site Finder Server at Leeds University website. The server gives the 10 best binding site locations along with their respective site volume and the involved residues. It uses the interaction energy between the protein and a simple van der Waals probe to locate energetically favorable binding sites. Energetically favorable probe sites are clustered according to their spatial proximity and clusters are then ranked according to the sum of interaction energies for sites within each cluster30.
The pesticides under study
Figure 3 2D structures and IUPAC names of the pesticides used for the study.

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 792
2.3 Molecular Docking Molecular Docking was done using three tools, to arrive at a consensus solution. AutoDock 4.2 The 3D purified (no external ligands) and minimized structure of AChE was opened and given as input to the docking software. From the AChE structure, solvent was removed, polar hydrogen atoms were added and merged with non-polar, and all missing residues were repaired. After this Kollman Charges were added, and excess charges were equally dispersed over all the residues of AChE. Active torsions were defined for each of the pesticide structures and Gasteiger charges added. The Map types were manually defined. The number of points in grid in each dimension was defined as 60, 60 and 60 and default grid spacing was maintained. The center of each grid was defined with 10 active site locations obtained from Q-Site Finder. Hence for each of the 4 pesticides, 10 dockings were done. The best binding energy helped to find the best active site for each of the pesticide. The Grid log file was generated using Autogrid program. The docking simulation was carried out using Lamarckian Genetic Algorithm, with 20 GA runs, population size set at 150 and 25000000 energy evaluations. All this was performed using AutoDock Tools as a part of MGL Tools31 with graphical user interface for easy visualization of AutoDock results.
iGEMDOCK The full structure of AChE was uploaded in the “Prepare Binding Site” and the “By current file” option was maintained because the whole surface will be checked for binding, instead of specifying a single cavity. The ligand files were uploaded in “.pdb” format. The GA parameters were set to: Default setting – Accurate Docking (very slow) in which population is set to 800, generations to 80 and number of solutions to 10. The default settings were maintained in the “scoring function”. SwissDock A single chain of AChE was uploaded, along with a Tripos mol2 format (with explicit Hydrogens intact) of the ligands. The region of interest coordinates were left blank to ensure blind docking approach. Accurate docking was carried out, allowing flexibility within 5Å of the ligand atoms in the reference binding mode.
RESULTS 1. Energy Minimization The result of the YASARA server was viewed using YASARA View. After the minimization step (which uses AMBER force field) the energy of 4EY4 structure reduced from -26419.06 Kcal/mol (-110540.6 kJ/mol) to -139143.88 Kcal/mol (-582192.0 kJ/mol), where the stability increased almost 5 fold. The minimized structure is given inFigure 4

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 793
Minimized Energy scene in YASARA View
Figure 4: 3D structure of AChE before and after Energy minimization.
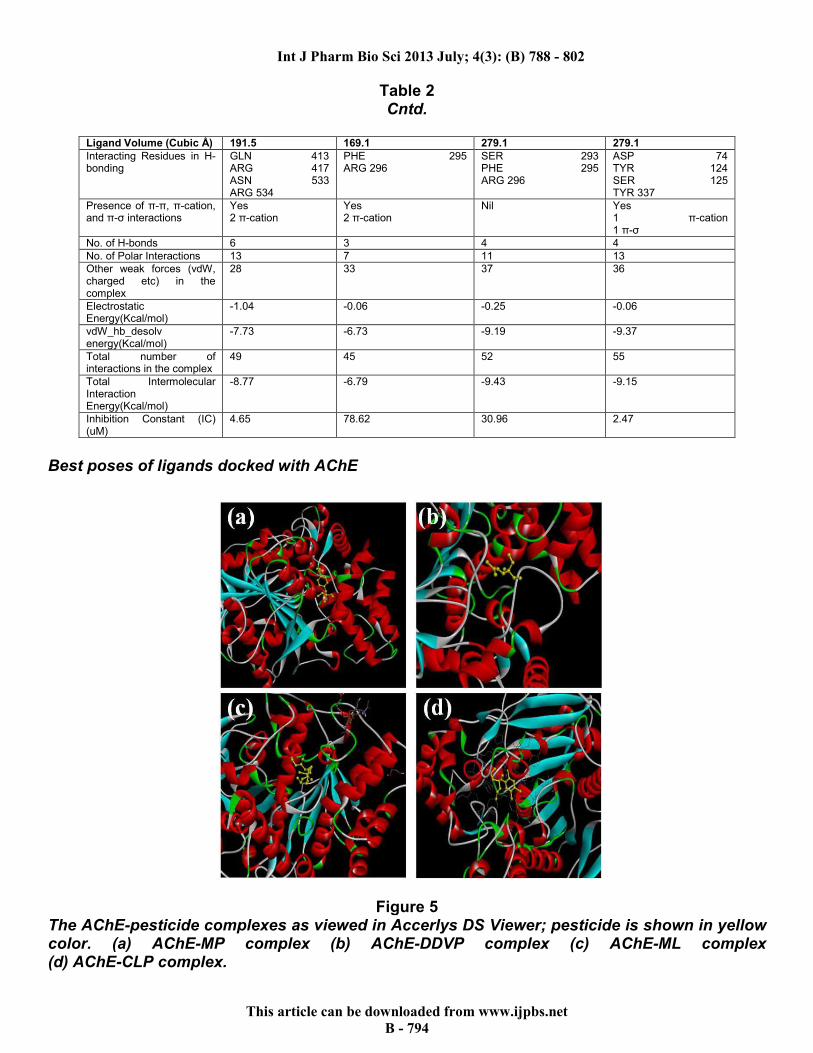
2. Molecular Docking Analysis AutoDock 4.2 The various complexes were visualized and the best run and pose of each compound was decided. The results are summarized in Table 2.The best predicted binding free energy of AChE with MP, DDVP, ML and CLP were, -7.28Kcal/mol, -5.60Kcal/mol, -6.15Kcal/mol and -
7.65Kcal/mol respectively, and the respective bound complexes are given in Figure 5. They bind to different active sites as found by the Q-site Finder, which is shown in Figure 6. Since CLP has the best (least) estimated free energy of binding, and hence owing to its stability, it may be the fastest in stopping cholinergic transmission.
Table 2
Summary of docking results generated using AutoDock 4.2
AChE-MP AChE-DDVP AChE-ML AChE-CLP Free Energy of Binding(Kcal/mol)
-7.28 -5.60 -6.15 -7.65
Active Site Location (-1.5, -40.5,12.5) (-16.5,-43.0,25) (-11.5,-44.5,31) (11,-57.5,-26) Site Volume (Cubic Å)
265 287 442 761
Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 794
Table 2 Cntd.
Ligand Volume (Cubic Å) 191.5 169.1 279.1 279.1 Interacting Residues in H-bonding
GLN 413 ARG 417 ASN 533 ARG 534
PHE 295 ARG 296
SER 293 PHE 295 ARG 296
ASP 74 TYR 124 SER 125 TYR 337
Presence of π-π, π-cation, and π-σ interactions
Yes 2 π-cation
Yes 2 π-cation
Nil
Yes 1 π-cation 1 π-σ
No. of H-bonds 6 3 4 4 No. of Polar Interactions 13 7 11 13 Other weak forces (vdW, charged etc) in the complex
28 33 37 36
Electrostatic Energy(Kcal/mol)
-1.04 -0.06 -0.25 -0.06
vdW_hb_desolv energy(Kcal/mol)
-7.73 -6.73 -9.19 -9.37
Total number of interactions in the complex
49 45 52 55
Total Intermolecular Interaction Energy(Kcal/mol)
-8.77 -6.79 -9.43 -9.15
Inhibition Constant (IC) (uM)
4.65 78.62 30.96 2.47
Best poses of ligands docked with AChE
Figure 5 The AChE-pesticide complexes as viewed in Accerlys DS Viewer; pesticide is shown in yellow color. (a) AChE-MP complex (b) AChE-DDVP complex (c) AChE-ML complex (d) AChE-CLP complex.
Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 795
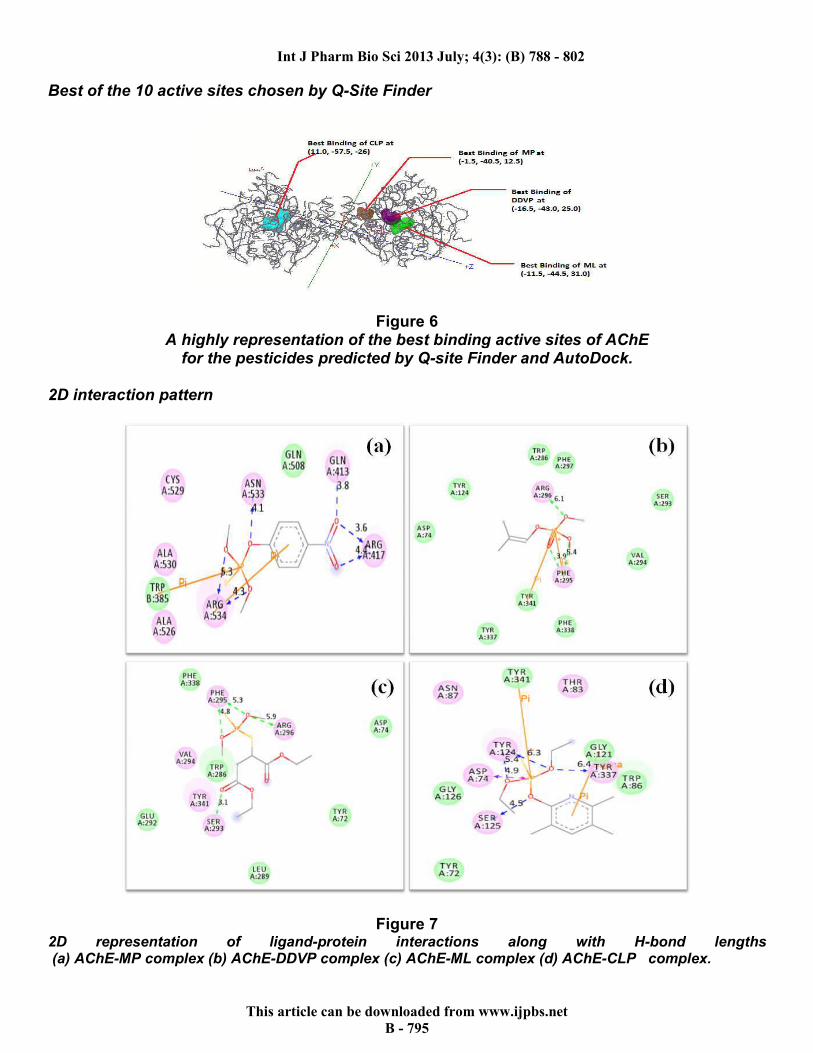
Best of the 10 active sites chosen by Q-Site Finder
Figure 6 A highly representation of the best binding active sites of AChE
for the pesticides predicted by Q-site Finder and AutoDock. 2D interaction pattern
Figure 7 2D representation of ligand-protein interactions along with H-bond lengths (a) AChE-MP complex (b) AChE-DDVP complex (c) AChE-ML complex (d) AChE-CLP complex.
Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 796
Figure 7 throws light on the various interactions of the ligand with its receptor, along with bond lengths. The figure helps to easily visualize what is happening at molecular level. iGEMDOCK The four pesticides were docked into AChE with the help of the accurate docking function. Table 3 gives a summary of docking results.
Table 3 Docking results for the OP pesticides, obtained using iGEMDOCK.
AChE-MP AChE-DDVP AChE-ML AChE-CLP Fitness(Kcal/mol) -87.68 -66.08 -80.34 -84.31 van der Waal’s Energy (kcal/mol)
-59.49 -49.83 -73.25 -71.02
H-bonding Energy(Kcal/mol)
-28.48 -16.26 -7.09 -13.29
Electrostatic Energy(Kcal/mol)
0.291 0 0 0
Interacting Amino Acids GLN 413 ARG 417 TYR 503
GLY 122 SER 203 HIS 447
TYR 124 TYR 337
TYR 124 TYR 337
It can be deduced that according to iGEMDOCK, MP is the best binding pesticide because of its best fitness (or total energy) of -87.68 Kcal/mol. Using iGEMDOCK post-screening analysis tools, all the docked poses were clustered based on the default “identity consensus residues” parameters where pharmacological energies of Electrostatics, H-bonding and van der Waal’s were set at -2.5, -2.5 and -4.0 respectively. Their Z-scores were set at 1.645, 1.645 and 1.645
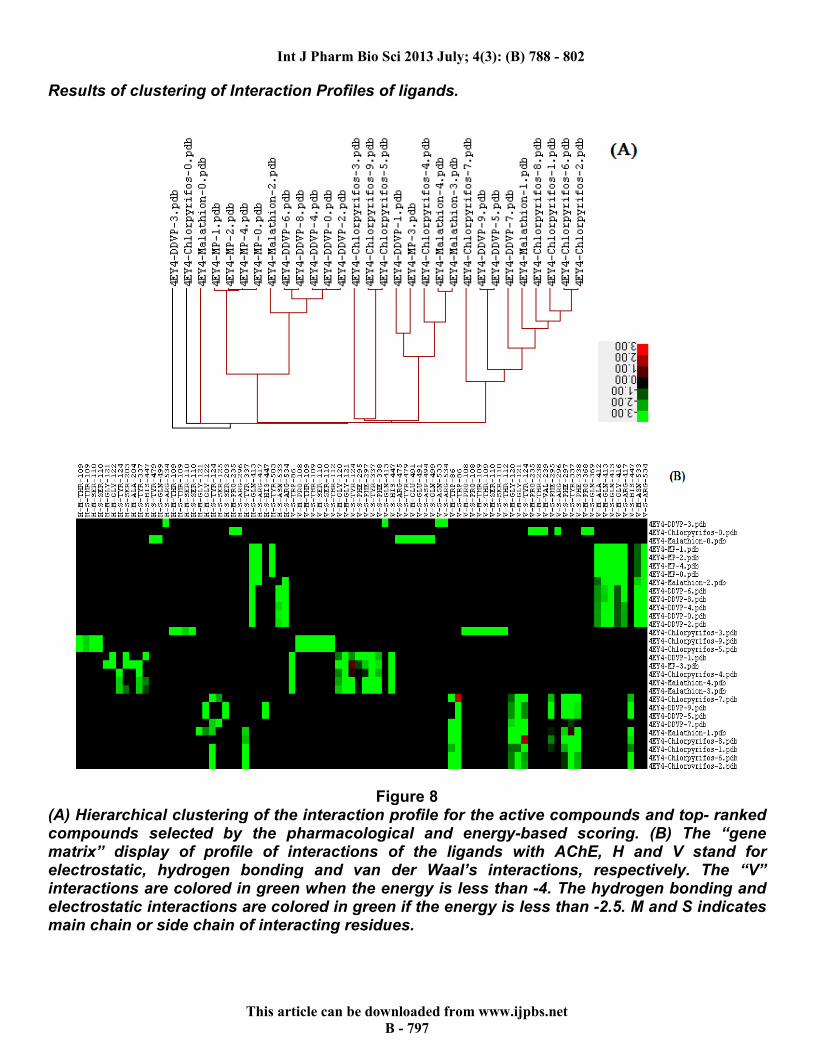
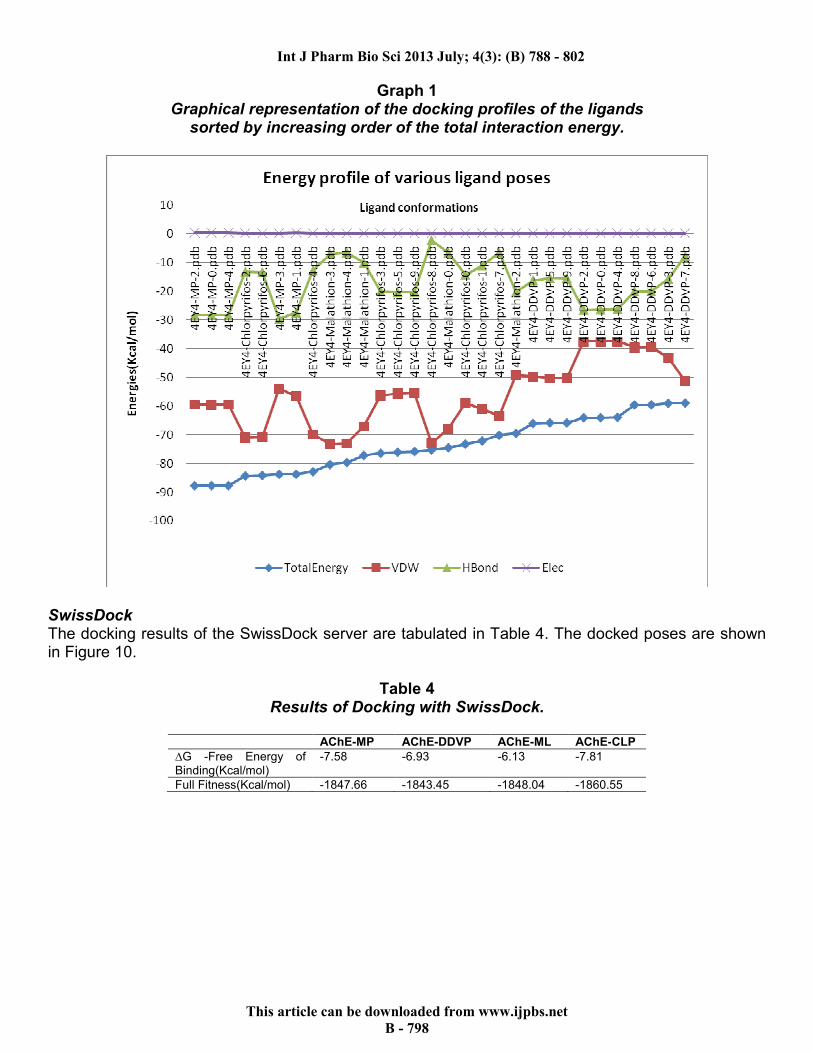
respectively. Using the clustering options, hierarchal clustering was done on basis of protein-ligand interactions and heat maps. The 30 poses (10 each of CLP and DDVP, and 5 each of MP and ML) were selected with all their interacting residues to generate a data tree and a “gene matrix” which are shown in Figure 8. The summary of their various energies is shown in Graph 1.
Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 797
Results of clustering of Interaction Profiles of ligands.
Figure 8 (A) Hierarchical clustering of the interaction profile for the active compounds and top- ranked compounds selected by the pharmacological and energy-based scoring. (B) The “gene matrix” display of profile of interactions of the ligands with AChE, H and V stand for electrostatic, hydrogen bonding and van der Waal’s interactions, respectively. The “V” interactions are colored in green when the energy is less than -4. The hydrogen bonding and electrostatic interactions are colored in green if the energy is less than -2.5. M and S indicates main chain or side chain of interacting residues.
Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 798
Graph 1 Graphical representation of the docking profiles of the ligands
sorted by increasing order of the total interaction energy.
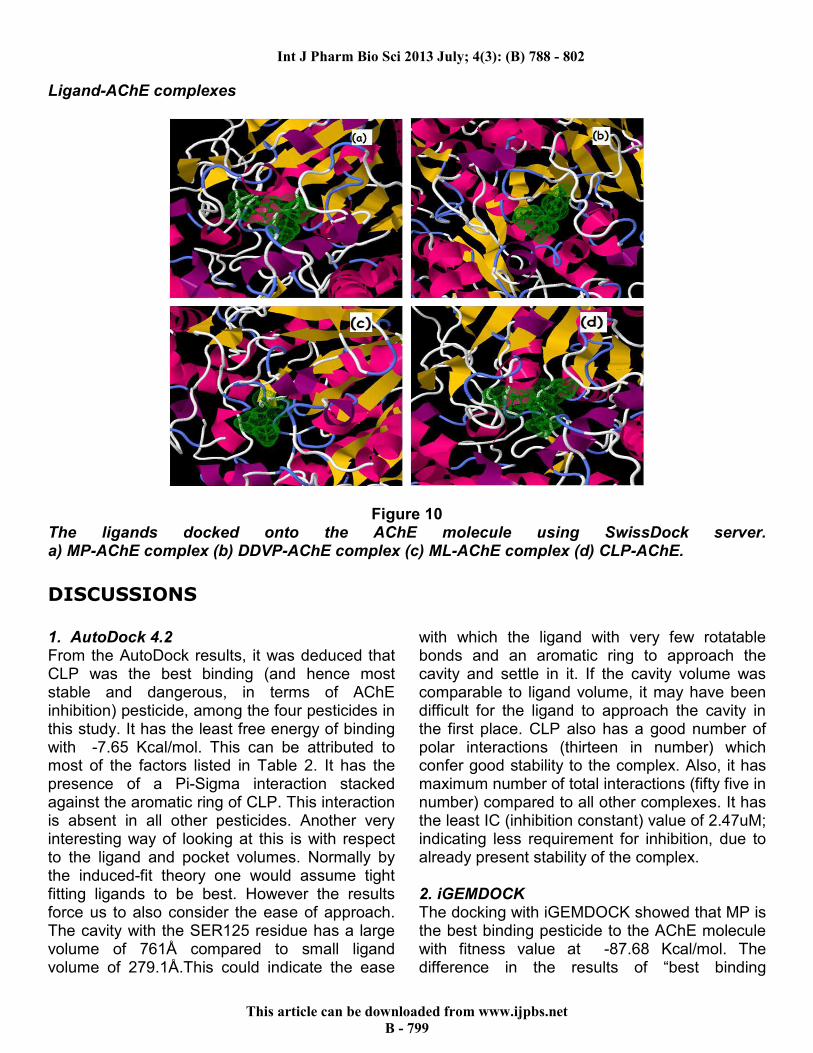
SwissDock The docking results of the SwissDock server are tabulated in Table 4. The docked poses are shown in Figure 10.
Table 4 Results of Docking with SwissDock.
AChE-MP AChE-DDVP AChE-ML AChE-CLP ∆G -Free Energy of Binding(Kcal/mol)
-7.58 -6.93 -6.13 -7.81
Full Fitness(Kcal/mol) -1847.66 -1843.45 -1848.04 -1860.55
Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 799
Ligand-AChE complexes
Figure 10 The ligands docked onto the AChE molecule using SwissDock server. a) MP-AChE complex (b) DDVP-AChE complex (c) ML-AChE complex (d) CLP-AChE.
DISCUSSIONS
1. AutoDock 4.2 From the AutoDock results, it was deduced that CLP was the best binding (and hence most stable and dangerous, in terms of AChE inhibition) pesticide, among the four pesticides in this study. It has the least free energy of binding with -7.65 Kcal/mol. This can be attributed to most of the factors listed in Table 2. It has the presence of a Pi-Sigma interaction stacked against the aromatic ring of CLP. This interaction is absent in all other pesticides. Another very interesting way of looking at this is with respect to the ligand and pocket volumes. Normally by the induced-fit theory one would assume tight fitting ligands to be best. However the results force us to also consider the ease of approach. The cavity with the SER125 residue has a large volume of 761Å compared to small ligand volume of 279.1Å.This could indicate the ease
with which the ligand with very few rotatable bonds and an aromatic ring to approach the cavity and settle in it. If the cavity volume was comparable to ligand volume, it may have been difficult for the ligand to approach the cavity in the first place. CLP also has a good number of polar interactions (thirteen in number) which confer good stability to the complex. Also, it has maximum number of total interactions (fifty five in number) compared to all other complexes. It has the least IC (inhibition constant) value of 2.47uM; indicating less requirement for inhibition, due to already present stability of the complex. 2. iGEMDOCK The docking with iGEMDOCK showed that MP is the best binding pesticide to the AChE molecule with fitness value at -87.68 Kcal/mol. The difference in the results of “best binding

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 800
compound” of AutoDock and iGEMDOCK may be due to difference in exhaustiveness of search, difference in algorithm etc. There have been studies where AutoDock is known to outperform iGEMDOCK in accuracy, however with regard to this study; such solid conclusions cannot be made until rigorous further studies are done. 3. SwissDock Docking with SwissDock help to conclude that CLP with free energy of binding of -7.81 Kcal/mol, has the best binding and stability with AChE. This result is corroborated with the results of AutoDock 4.2 and helps us reach a consensus decision about CLP at the end.
CONCLUSION The present computational study helped to conclude that out of the most commonly used OP pesticides, chlorpyrifos is the best binder to acetylcholinesterase, making it the best inhibitor. This can be supported by the fact that out of the three docking softwares used, chlorpyrifos has been reported best binder by two of them. Also with the closeness of the values of free energies of binding reported by the three softwares for methyl parathion and chlorpyrifos, it can be concluded that methyl parathion and chlorpyrifos have comparable stabilities when in complex with AChE. From the view of danger, being the most stable complex, chlorpyrifos will hamper cholinergic transmission fastest and cause variety of symptoms like nausea, breathlessness and paralysis. The acute toxic effect of chlorpyrifos on aquatic life32,33 and rodents34 has been studied experimentally. The study in this
paper can help in finding compounds which go and bind at the best identified active sites, and are not toxic so that they can be used as precautionary antidotes for people who are exposed to large amounts of pesticides everyday as a part of their occupation. From the drug discovery point of view, if one can generate less toxic homologs (therapeutic leads) of chlorpyrifos using the Qualitative Structure Activity Relationship (QSAR) principle, it can be used as potential AChE inhibitor drugs for treatment of Dementia, Alzheimer’s etc. This study also highlights the difference obtained in results using different softwares, attributed to difference of algorithm, search parameters etc. It forces us to realize the importance of carrying out multiple dockings to arrive at results which are of sensitive nature because results of a single software cannot be relied upon completely. One must either carry out multiple dockings using software repeatedly, or refine results by using software which are known to have better accuracy.
ACKNOWLEDGEMENT
The authors acknowledge Dr. V C Padaki, Director and Mrs. S N Vijayalakshmi, Scientist and in- charge of HRD from DEBEL, Bangalore for their support. The authors also thank Management, VIT University for their cooperation. Conflict of Interest The authors declare no conflict of interest.
REFERENCES
1. Epstein T.M., Samanta U., Kirby S.D.,
Cerasoli D.M., Bahnson, B.J., Crystal structures of brain group-VIII phospholipase A2 in non-aged complexes with the organophosphorus nerve agents soman and sarin, Biochemistry, 48: 3425-3435(2009) .
2. Kavrut M., Development of acetylcholinesterase biosensor for the detection of pesticides. M.S Thesis, Middle-East Technical University, Turkey, September 2010.
3. National Pesticide Information Center. Medical Case Profiles- Biomarkers of exposure: Organophosphates. Available

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 801
online: http://npic.orst.edu/mcapro/opbiomarkers.html (accessed on 1 April 2013).
4. Pang Y.P., Brimijoin S., Ragsdale, D.W., Zhu K.Y., Suranyi R., Novel and viable acetylcholinesterase target site for developing effective and environmentally safe insectides, Current Drug Targets, 13: 471-482, (2012).
5. Wu J., Luan T., Lan C., Hung T.W., Chan G.Y., Removal of residual pesticides on vegetables using ozonated water, Practical Preventive Medicine, 10: 766-767,(2003).
6. Jiang G.H., Huo F., Li J., Wang Y.G., Cao H.L., Studies on use and residue level of pesticides in fruit and vegetable in Tianjin Area and its control measures, Zhonghua Yu Fang Yi Xhe Za Zhi , 37: 351-354, (2003).
7. Nasrabadi M., Ghayal N., Dhumal K.N., Effect of Chlorpyrifos and Malathion on Antioxidant Enzymes in Tomato and Brinjal, International Journal of Pharma and Bio Sciences, 2(2):202-209, (2011).
8. Bisht S.P., Kumar R., Praveen B., Rajakumar V., Mishra S.R., Effect of Dichlorvos on Serum Enzymes of Poultry Birds, International Journal of Pharma and Bio Sciences, 3(1):553-557, (2012).
9. Ennaceur S., Gandoura N., Driss M.R., Organochloride pesticide residues in Human milk of mothers living in Northern Tunisia, Bulletin of Environmental Contamination and Toxicology, 78: 325-329, (2007).
10. Cheung J., Rudolph M.J., Burshteyn F., Cassidy M.S., Gary E.N., Love J., Franklin M.C., Height J.J., Structures of human acetylcholinesterase in complex with pharmacologically important ligands, Journal of Medicinal Chemistry, 55: 10282-10286, (2012).
11. Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J., AutoDock 4 and AutoDockTools4: automated docking with selected receptor flexibility, Journal of Computational Chemistry, 16: 2785-2791, (2009).
12. Hsu K.C., Chen Y.F., Lin S.R., Yang J.M., iGEMDOCK: a graphical environment of enhancing GEMDOCK using pharmacological interactions and post-screening analysis, BMC Bioinformatics, 12: S33, (2011).
13. Grosdidier A., Zoete V., Michielin O., Fast docking using the CHARMM force field with EADock D S S, Journal of Computational Chemistry, PMID: 21541944, (2011).
14. Grosdidier A., Zoete V., Michielin O., SwissDock, a protein-small molecule docking web server based on EADock D S S, Nucleic Acids Research, 39(Web server issue):W270-W277, (2011).
15. Research Collaboratory for Structural Bioinformatics online database- Protein Data Bank. Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 1 April 2013).
16. Accelrys Software Inc., Discovery Studio Modeling Environment, Release 3.5, San Diego: Accelrys Software Inc., 2007. Available online: http://accelrys.com/products/discovery-studio/visualization-download.php (Accessed 15 March 2013).
17. National Center for Biotechnology Information. PubChem Compound Database- CID=4130, Available online: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=4130 (accessed 26 February 2013).
18. National Center for Biotechnology Information. PubChem Compound Database- CID=3039, Available online: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=3039 (accessed 26 February 2013).
19. National Center for Biotechnology Information. PubChem Compound Database- CID=4004, Available online: http://pubchem.ncbi.nlm.nih.gov/summary/summary.cgi?cid=4004 (accessed 26 February 2013).
20. National Center for Biotechnology Information. PubChem Compound Database- CID=2730, Available online: http://pubchem.ncbi.nlm.nih.gov/summary/s

Int J Pharm Bio Sci 2013 July; 4(3): (B) 788 - 802
This article can be downloaded from www.ijpbs.net
B - 802
ummary.cgi?cid=2730 (accessed 26 February 2013).
21. Sadowski J., Gasteiger J., Klebe G., Comparison of automatic three-dimensional model builders using 639 X-Ray structures, Journal of chemical information and computer sciences, 34: 1000-1008, (1994).
22. Molecular Networks. CORINA online Demo. Available online: http://www.molecular-networks.com/online_demos/corina_demo (accessed on 27 February 2013).
23. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K., Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling: Four approaches that performed well in CASP8 , Proteins, 77(9): 114-122, (2009).
24. Laurie A.T., Jackson R.M., Q-SiteFinder: an energy-based method for prediction of protein-ligand binding sites, Bioinformatics, 21: 1908-1916, (2005).
25. Burgoyne N.J., Jackson R.M., Predicting protein interaction sites: binding hot-spots in protein-protein and protein-ligand interfaces, Bioinformatics, 22: 1335-1342, (2006).
26. University of Leeds. Faculty of Biological Sciences, Q-SiteFinder. Available online: http://www.modelling.leeds.ac.uk/qsitefinder/ (accessed on 1 March 2013).
27. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E., UCSF Chimera- A Visulialzation system for exploratory research and analysis, Journal of Computational Chemistry, 25: 1605-1612, (2004).
28. Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin 5.8.3, 2012, ChemAxon (http://www.chemaxon.com ).
29. Guex N., Peitsch M.C, SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling, Electrophoresis, 18: 2714-2723, (1997).
30. Bartolucci C., Haller L.A., Jordis U., Fels G., Lamba D., Probing Torpedo californica acetylcholinesterase catalytic gorge with two-novel bis-functional galanthamine derivatives, Journal of Medicinal Chemistry, 53(2): 745-751, (2010).
31. Sanner M.F., Python: A programming language for software integration and development, Journal of Molecular Graphics and Modeling, 17: 57-61, (1999).
32. Malla F.A., Assessment of Total Protein Concentration in Liver of Fresh Water Fish, Channa Punctuates(Bloch.) with special reference to an organophosphate insecticide, Chlorpyrifos, International Journal of Pharma and Bio Sciences, 3(2):567-571, (2012).
33. Malla F.A., Changes in oxygen carrying capacity of blood in fish, Channa Punctatus(Bloch.) exposed to chlorpyrifos, International Journal of Pharma and Bio Sciences, 3(2):423-425, (2012).
34. Savithri Y., Sekhar P.R., Doss P.J., Changes in hematological profiles of albino rats under chlorpyrifos toxicity, International Journal of Pharma and Biosciences, 1(3), (2010).