interaction of nitric oxide and endothelin-1 in ischemia/reperfusion injury of rat heart
TRANSCRIPT

J Mol Cell Cardiol 29, 2363–2374 (1997)
Interaction of Nitric Oxide andEndothelin-1 in Ischemia/reperfusionInjury of Rat HeartFriedrich BrunnerInstitut fur Pharmakologie und Toxikologie, Karl-Franzens-Universitat Graz, Universitatsplatz 2,A-8010 Graz, Austria
(Received 16 January 1997, accepted in revised form 2 May 1997)
F. B. Interaction of Nitric Oxide and Endothelin-1 in Ischemia/reperfusion Injury of Rat Heart. Journal ofMolecular and Cellular Cardiology (1997) 29, 2363–2374. Although several studies have demonstrated that nitricoxide appears to be cardioprotective and endothelin-1 (ET-1) deleterious in myocardial ischemia/reperfusioninjury, their interactions in the intact heart are unknown. Therefore, coronary effluent and interstitial fluid(“transudate”) levels of ET-1 and cyclic GMP, an indirect measure of nitric oxide production, were determinedsimultaneously in normoxic and reperfused hearts and compared with myocardial and coronary function. Rathearts were buffer-perfused at 9 ml/min/g heart wet weight for 45 min (baseline), followed either by another 45min of perfusion (normoxia), or 15 min of total global ischemia and 30 min reperfusion. Hearts received, from42–90 min, either vehicle, the inhibitor of nitric oxide formation NG-nitro-L-arginine (L-NNA; 200 lmol/l), thenitric oxide donor S-nitroso-N-acetyl-DL-penicillamine (SNAP; 200 lmol/l), or the ET receptor antagonist PD142893 (200 nmol/l). Both mediators were released preferentially into the vascular lumen which resulted insimilar luminal and interstitial concentrations of cyclic GMP, but three-fold higher levels of ET-1 in tissue becauseof the higher effluent than transudate flow rate. L-NNA increased the release of ET-1 and worsened coronaryfunction, whereas SNAP had opposite effects. On reperfusion, considerable functional impairment was observed,although levels of cyclic GMP both in the vascular and tissue compartment were not reduced, but even increased.Reperfusion functional impairment was aggravated after inhibiting the synthesis of nitric oxide, whereas SNAPrestored cardiac and coronary function close to pre-ischemic level. Deterioration of function corresponded withan increased level, and improvement with a decreased level of intersitial ET-1 at the onset of reperfusion. PD142893 was similarly cardioprotective as SNAP both in normoxia and reperfusion. These results suggest that inreperfusion, cardiac function is depressed, despite increased rather than decreased endogenous nitric oxideproduction, largely due to the prevalence of the deleterious effects of ET-1 which are overcome by antagonismof ET receptors or exogenous nitric oxide supplied by SNAP. 1997 Academic Press Limited
K W: Nitric oxide; Cyclic GMP; Endothelin-1; L-NNA; SNAP; Ischemia/reperfusion injury; Coronaryeffluent; Interstitial transudate; Rat heart.
oxide (Ignarro et al., 1987; Palmer et al., 1987). InIntroductionaddition to dilating vascular smooth muscle, nitricoxide resists the adhesion and aggregation of plate-The endothelium produces several vasodilator and
vasoconstrictor substances that regulate blood ves- lets and neutrophils, promotes the dissolution ofplatelet aggregates and preserves the integrity ofsel tone and exert a number of other cardiovascular
effects (Rubanyi, 1991). At present, no factor pro- vital organ functions such as myocardial mech-anical and electrical activity. Another importantduced by the endothelium appears to be more
important in the maintenance of vascular patency endothelial mediator is endothelin-1 (ET-1), whichappears to be a vasodilator at physiologically lowthan endothelium-derived relaxing factor (Furchgott
and Zawadzki, 1980), generally agreed to be nitric concentrations, and a potent vasoconstrictor in
Please address all correspondence to: F. Brunner at the above address.
0022–2828/97/092363+12 $25.00/0 mc970470 1997 Academic Press Limited

F. Brunner2364
several pathologies associated with a rise in ET- Materials and Methods1 plasma levels (Battistini et al., 1993; Luscher,1993). Perfusion procedure
Ischemia/reperfusion is associated with systolicand diastolic dysfunction and a decrease in endo- Sprague–Dawley rats weighing 260–320 g and fedthelium-dependent vasodilation. Recent studies a standard diet were anesthetized with diethyl ether,have implicated the ET and nitric oxide/cyclic GMP the hearts were excised, placed in ice-cold buffer,pathways in several aspects of ischemia/re- and rapidly mounted on the aortic cannula of aperfusion. Basal release of nitric oxide from rat Langendorff perfusion system. Hearts were perfusedhearts was diminished after ischemia and re- in the non-recirculating mode at a flow rate ofperfusion (Maulik et al., 1995), and the patho- 9 ml/min/g heart wet weight with a modified Krebs–physiological consequences due to impaired nitric Henseleit bicarbonate buffer with the followingoxide release were mitigated or abolished by pro- composition (mmol/l): NaCl 118, NaHCO3 25,viding exogenous nitric oxide donors (Fukuda et KH2PO4 1.2, KCl 4.8, MgSO4 1.2, CaCl2 1.25, gluc-al., 1995; Pabla et al., 1995). Specifically, these ose 11 (when measurements of cyclic GMP wereinterventions reduced myocardial infarct size in intended, 3-isobutyl-1-methylxanthine at 1 mmol/several models, both in vivo and in vitro. For ET-1, l final concentration was added to inhibit cyclica possible role in ischemic syndromes is suggested, nucleotide-dependent phosphodiesterases) usingbecause myocardial ischemia and reperfusion are the ISOHEART perfusion system (Hugo Sachs Elek-associated with increased cardiac release and el- tronik, March-Hugstetten, Germany). The ap-evated plasma levels of ET-1 in many experimental paratus was modified for perfusion of hearts in anmodels and cardiac patients (Brunner et al., 1992; inverted, upside-down position to allow the separateHasdai et al., 1994). However, no elevation of ET- collection of coronary effluent and interstitial tran-1 level was found in patients with either stable or sudate as described previously (Brunner, 1995b).unstable angina, and the effects of ET receptor Briefly, the original aortic cannula was replaced byantagonists on myocardial infarct size are con- a horizontal connecting piece to which the mobileflicting (Krause et al., 1994). aortic cannula was attached via an air-tight joint
To clarify the role of nitric oxide and ET-1 in made of Plexiglass. Hearts were mounted with thethe coronary vascular bed and myocardium, the aortic cannula pointing downward, a latex balloondirection of release and interstitial (tissue) levels of was mounted on a rigid bi-luminal catheter insertedthese mediators appear of interest. Presumably, the into the left ventricle, and the cannula togetherrelative rates of nitric oxide and ET-1 release to the with the heart was turned upward, the cathetervascular lumen and the cardiac interstitium are fixing the heart in an upright position. Interstitialregulated physiologic variables, and the resulting transudate produced by the ventricles and ap-local concentrations account for the effects of these pearing on their surface was collected under amediators on coronary tone, hemostasis, and myo- latex cap using slight suction and was sampledcardial function. In addition, there are important in exchangeable vials (Wienen and Kammermeier,interactions between these mediators in cultured 1988). The hearts were thermostated at 37°C (isch-cells, in particular the production of ET-1 is inhibited emia: 34–35). Cardiac parameters were monitoredby nitric oxide, apparently at the level of tran- continuously and included heart rate, coronaryscription (Kourembanas et al., 1993), and these perfusion pressure (CPP), left ventricular developedinteractions may be reinforced or antagonized by pressure (LVDP; difference between left ventricularchanges occurring as a result of ischemia (Brunner, peak systolic pressure and end-diastolic pressure),1995a). The purpose of this study was (1) to deter- and left ventricular end-diastolic pressure (LVEDP).mine the luminal and abluminal release of nitric The baseline pressure in the intraventricular bal-oxide and ET-1 under normoxic conditions and after loon was set at 0 mmHg.ischemia/reperfusion; and (2) to assess the effectsof a nitric oxide donor and a nitric oxide synthaseinhibitor on mediator levels and heart function bothin normoxia and reperfusion. Because nitric oxide Experimental protocol and sample collectionlevels were too low to allow direct detection witha nitric oxide-sensitive electrode (Schmidt et al., Hearts were perfused for 45 min at 9 ml/min/g heart
wet weight (baseline), followed by one of two pro-1994), nitric oxide generation was assessed in-directly by measuring the production of cyclic GMP tocols: (1) another 45 min normoxic perfusion; or (2)
15 min of global ischemia, and 30 min reperfusion at(Rapoport and Murad, 1983).

Nitric Oxide and Endothelin in Ischemia/reperfusion Injury 2365
normal flow (total duration in each case: 90 min). 142893 was a gift from Dr Annette Doherty, Parke-Davis Pharmaceutical Research, Ann Arbor, MI,Vehicle or drugs were added to the perfusion medium
from 42–90 min, i.e. in ischemia/reperfusion ex- USA; all other chemicals, including L-NNA hydro-chloride, were obtained from Sigma Chemicals Co.,periments hearts were exposed to drugs from 3 min
preischemia to end of reperfusion. In both protocols, Vienna, Austria. All solutions were made in distilledwater and stored as stock solutions at −18°C.hemodynamic measurements were taken at 45, 50,
60, 70, 80, and 90 min. In protocol 1, coronaryeffluents were collected in intervals of 5 min, i.e. from40–45 (baseline), 55–60, 70–75, and 85–90 min.
Statistical analysisInterstitial transudates were collected quantitativelyfrom 15–45 min (baseline), 45–60 min, 60–75, and
All values in the text and figures are presented as75–90 min. In Protocol 2, for determination of cyclicmeans±standard error of the mean (S.E.M.) of nGMP in coronary effluent, the intervals were (s) 0–5,independent hearts or determinations. Changes in5–10, 10–15, 15–20, 20–25, 25–30, 30–60, and 9hemodynamic parameters were subjected to a two-min 55 s–10 min, and 29 min 55 s–30 min [Fig.way analysis of variance (ANOVA) for repeated5(a)]; for determination of cyclic GMP in transudate,measurements to account for different treatmentsthe intervals were (min) 0–0.5, 0.5–1.25, 1.25–(control, ischemia, reperfusion) and factors (vehicle,2.25, 2.25–3.5, 3.5–5, 5–10, and 20–30 [Fig. 5(b)];drugs). When a significant overall effect was de-for determination of ET-1 in coronary effluent, thetected, the Scheffe test was used to compare singleintervals were (min) 0–3, 3–6, 6–20, and 20–30mean values. Data are presented as secretion rates(Fig. 6); and for determination of ET-1 in interstitial(amount of mediator/min) normalized to 1 g hearttransudate, they were 0–15 and 15–30 min of re-wet weight, and/or as concentrations (amount ofperfusion (Fig. 7). The materials were stored frozenmediator/ml). A P value <0.05 was considereduntil analysis. The drugs used were the nitric oxidesignificant. P values less than 0.01 were not in-donor S-nitroso-N-acetyl-DL-penicillamine (SNAP)dicated separately.(Kojda et al., 1996), the nitric oxide synthase in-
hibitor NG-nitro-L-arginine (L-NNA) (Mayer et al.,1993) and the mixed ETA/B receptor antagonist PD142893 (Cody et al., 1992). The concentrations of Resultsdrugs were chosen to achieve maximal effects; inparticular, a concentration of -NNA >60 times Transudate flowhigher than its IC50 value as determined with endo-thelial nitric oxide synthase (Mayer et al., 1993) was Under normoxic perfusion conditions, transudateused. flow was 50±1 ll/min, i.e. 0.5% of perfusate flow
(Fig. 1). The rate slowly increased with time up to1.3-fold within 90 min, reflecting gradual loss ofMeasurement of cyclic GMP and ET-1endothelial barrier function due to wash-out ofalbumin from the coronary vessels (Mann, 1981).Cyclic GMP levels in venous effluents and tran-During reperfusion (i.e. after no-flow ischemia),sudates were determined by radioimmunoassaytransudate flow was increased above the normoxic(RIA) essentially, as described previously (Schmidtcontrol rate (2.2-fold).et al., 1989). Standard curves were established in
Krebs–Henseleit buffer (pH 8.0) containing 1 mmol/l3-isobutyl-1-methylxanthine to match the con-centration present in effluent and transudate
Normoxic perfusionsamples. ET-1 was concentrated by solid-phase ex-traction followed by RIA using a commercial anti-
To evaluate the validity of measurements of cyclicbody specific for ET-1 (RAS 6901, Peninsula,
GMP instead of nitric oxide, we first determined theBelmont, CA, USA), as described previously (Brunner,
cyclic GMP response to acetylcholine, an established1995b).
L-arginine/nitric oxide-dependent agonist. Cumu-lative application of 3–300 nmol/l through a side-line just above the heart resulted in coronaryDrugsdilatation as evident from a 5–40% decrease in CPPas well as a concentration-dependent increase inODQ (code 0880) and SNAP (code 0598) were
obtained from Tocris Cookson, Bristol, UK; PD cyclic GMP in the coronary effluent (maximum,

F. Brunner2366
100
160
Time (min)
Tran
suda
te f
low
(l/m
in)
80
140
120
100
60
50 60 70 80 9040
No-
flow
isch
emia
Figure 1 Interstitial transudate flow rates in isolatedhearts under normoxic perfusion (control, open symbols)and after 15 min global no-flow ischemia (reperfusion,closed symbols). Coronary flow was 9 ml/min. Tran-sudates were collected as described in Materials andMethods. Values are mean±S.E.M. from five hearts each.The increase in transudate flow in reperfusion is due topartial loss of the endothelial barrier function as a resultof ischemia.
2.5-fold; n=3, data not shown). (In these ex-periments, CPP had previously been raised to100 mmHg by increasing coronary flow.) Therefore,the cyclic nucleotide appears to be a reliable indexof nitric oxide production in this model.
The release of cyclic GMP from rat hearts perfusedwith oxygenated buffer is shown in Figure 2. Thebasal rate of cyclic GMP release was5.7±0.68 pmol/min into the coronary lumen and0.03±0.003 pmol/min into interstitial transudate[ratio: 183; Fig. 2(a)]. Addition of L-NNA
30
0Vehicle
Sec
reti
on r
ate
in e
fflu
ent
(pm
ol/m
in)
L-NNA SNAP
15
25
5
20
10
0.3
0
0.05
0.25
0.2
Sec
reti
on r
ate
in t
ran
suda
te (
pmol
/min
)
0.15
0.1
*†
*
*†*†
(a)
3
0Vehicle
Con
cen
trat
ion
(n
mol
/l)
L-NNA SNAP
1.5
2.5
0.5
2
1
**
**
(b)
(200 lmol/l) reduced cyclic GMP release into ef-fluent and transudate by>one third, and addition Figure 2 Secretion of cyclic GMP into coronary effluentof SNAP (200 lmol/l) increased it 4.2-fold (effluent) and interstitial transudate (a) (note dissimilar ordinates)
and resultant concentrations (b) in normoxic-perfusedand 4.4-fold (transudate). As shown in Figure 2(b),hearts in the presence of vehicle, L-NNA (200 lmol/l) orthese different release rates resulted in similar cyclicSNAP (200 lmol/l). Effluent samples (Φ) were takenGMP concentrations in both fluids because of the between 85 and 90 min, and transudate samples (Ε)
>180-fold higher effluent than transudate flow. between 75 and 90 min perfusion. Columns areThe inhibitory effect of L-NNA and the stimulatory mean±S.E.M. from four hearts each. †P<0.05 effluent v
transudate; ∗P<0.05 L-NNA or SNAP v vehicle.effect of SNAP were clearly reflected in the respective

Nitric Oxide and Endothelin in Ischemia/reperfusion Injury 2367
cyclic GMP concentrations in effluent and tran-sudate.
The release of ET-1 under normoxic conditionsis shown in Figure 3. The basal rate of release was0.26±0.01 pg/min into the coronary lumen and0.005±0.001 pg/min into interstitial transudate[ratio: 52; Fig. 3(a)]. The nitric oxide synthaseinhibitor L-NNA increased ET-1 release 3.4-fold (ef-fluent) and 10.2-fold (transudate), whereas SNAPreduced it to 0.17±0.01 (effluent) and0.002±0.001 (transudate) pg/min (65 and 40%,respectively). The corresponding ET-1 con-centrations [Fig. 3(b)] were higher in transudatethan effluent (vehicle, 3.2-fold; L-NNA, 7.9-fold;SNAP, 2.3-fold).
The heart was hemodynamically stable through-out the experiment (Table 1 and Fig. 4). The heartrate was >300 beats/min under all experimentalconditions, LVDP was 90±2 mmHg, CPP was54±2 mmHg, and LVEDP remained at baseline(0 mmHg; not shown). L-NNA reduced LVDP by26%, and increased CPP 2.7-fold after 90 minperfusion; in the presence of SNAP or the ET re-ceptor antagonist PD 142893, all parameters weresimilar to vehicle values.
Ischemia/reperfusion
Release of cyclic GMP and ET-1 was also measuredafter a period of 15 min total global ischemia. Thecyclic GMP concentrations resulting from releaseinto coronary effluent and interstitial transudateover 30 min reperfusion are shown in Figures 5(a)and 5(b). The cyclic nucleotide concentration waselevated 11.1-fold in the first effluent sample col-lected after initiation of reperfusion (vehicle; P<0.05v baseline), and this increase was unaffected by200 lmol/l L-NNA (8.0-fold; P>0.05 v vehicle), butpotentiated by SNAP (52-fold) and almost com-pletely suppressed by the specific inhibitor of solubleguanylyl cyclase, (1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one [ODQ] 20 lmol/l) (Garthwaite etal., 1995). After addition of SNAP, the cyclic GMPconcentration remained elevated throughout re-
1.2
0Vehicle
Sec
reti
on r
ate
in e
fflu
ent
(pg/
min
)
L-NNA SNAP
0.6
1.0
0.2
0.8
0.4
0.12
0
0.02
0.1
0.08
Sec
reti
on r
ate
in t
ran
suda
te (
pg/m
in)
0.06
0.04
*†
*
*†
*
†
(a)
1.2
0Vehicle
Con
cen
trat
ion
(pg
/ml)
L-NNA SNAP
0.6
1.0
0.2
0.8
0.4
**
*
*
(b)
†
†
†
perfusion (P<0.05 v baseline). The effect of theFigure 3 Secretion of ET-1 into coronary effluent anddrugs on cyclic GMP levels in transudate wereinterstitial transudate (a) (note dissimilar ordinates) andsimilar, albeit in part more protracted [Fig. 5(b)].resultant concentrations (b) in normoxic-perfused hearts
Cyclic GMP levels were never lower than baseline. in the presence of vehicle, L-NNA (200 lmol/l) or SNAPThe effect of L-NNA and SNAP on the release of (200 lmol/l). Effluent samples (Φ) were taken between
85 and 90 min, and transudate samples (Ε) betweenET-1 in ischemia/reperfusion is shown in Figures 675 and 90 min perfusion. Columns are mean±S.E.M. fromand 7. As expected, control ET-1 secretion wassix hearts each. †P<0.05 effluent v transudate; ∗P<0.05increased several-fold on reperfusion compared toL-NNA or SNAP v vehicle.
basal rate (0.72±0.03 (effluent) and

F. Brunner2368
Table 1 Heart rate values for individual treatment groups during normoxic perfusion
Treatment Time after mounting of heart (min)
45 50 60 70 80 90
Vehicle 311±6 312±6 310±7 312±8 311±9 309±9L-NNA 304±10 300±10 304±5 307±12 300±8 295±11SNAP 303±5 302±5 301±5 305±4 306±5 306±5PD 142893 308±12 305±9 305±7 300±8 302±10 298±10
Values represent means±S.E.M. of five hearts and are presented as beats/min. There were no significant differences betweenbaseline (45 min) and later time points for any of the treatments nor between treatment groups and vehicle for matched timepoints.
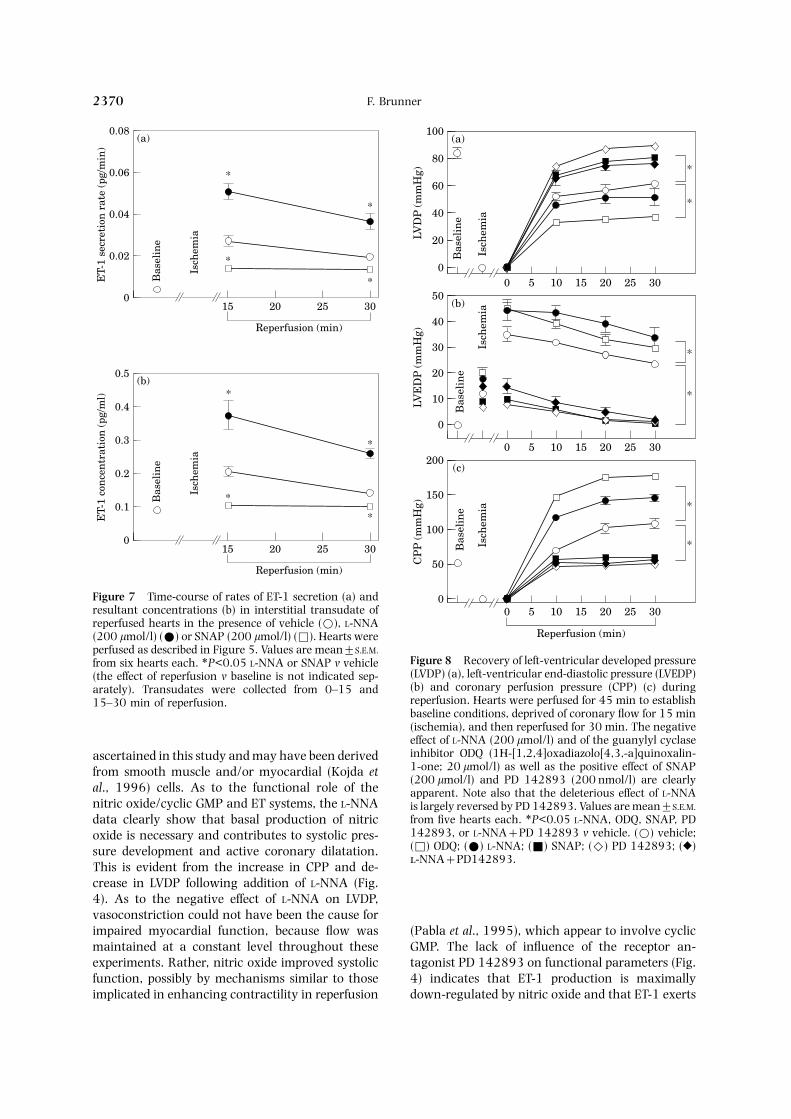
was secreted into the lumen than the interstitium[ratios: 26 (control), 24 (L-NNA), and 39 (SNAP)].The corresponding calculated ET-1 concentrations[Figs 6(b) and 7(b)], however, were 2–3-fold higherin transudate than effluent under all conditions. Atthe end of reperfusion, ET-1 levels remained abovebaseline in the vehicle and L-NNA groups, butreturned to baseline in hearts treated with SNAP.
The hemodynamic parameters (Fig. 8) showedthe expected changes in ischemia, i.e. cessation ofleft-ventricular contractile activity [Fig. 8(a)], anincrease in LVEDP [Fig. 8(b)], and a collapse ofCPP [Fig. 8(c)]; heart beat slowly ceased. Afterreperfusion for 30 min, heart rate returned to pre-ischemic level in all experimental groups (Table 2),LVDP remained depressed to 74% of pre-ischemicvalue, LVEDP was elevated (24±1 mmHg), andCPP was increased 2.1-fold (P<0.05 in each case).In the presence of L-NNA or ODQ, all indices ofcardiac and vascular function deteriorated, whereasSNAP and the ET receptor antagonist PD 142893considerably improved (LVDP), or even normalized(CPP and LVEDP) them compared to vehicle. Im-portantly, the deleterious effect of L-NNA was ef-fectively antagonized in the presence of the ETreceptor antagonist.
90
110
5040
Time (min)
LVD
P (
mm
Hg)
50
90
80
70
60
45 60 70 80
100
(a)
Bas
elin
e
90
160
Time (min)
CP
P (
mm
Hg)
50
80
60
45 60 70 80
140
(b)
Bas
elin
e
120
40
** * *
**
*
100
DiscussionFigure 4 Left-ventricular developed pressure (LVDP) (a)and coronary perfusion pressure (CPP) (b) in normoxic- This work is the first report of simultaneous meas-perfused hearts in the presence of vehicle (Β), L-NNA urements of vascular and interstitial cyclic GMP(200 lmol/l) (Χ), SNAP (200 lmol/l) (Ε), or PD 142893
and ET-1 levels. The results show that (1) in nor-(200 nmol/l) (Η). Values are mean±S.E.M. from fivemoxia and reperfusion, both mediators were re-hearts each. ∗P<0.05 L-NNA v vehicle (the differences
for SNAP and PD 142893 were not significant). leased preferentially to the vascular lumen whichresulted in similar luminal and interstitial cyclicGMP concentrations, whereas the ET-1 con-centration was higher in the interstitium; (2) func-0.028±0.003 (transudate) pg/min (2.7- and 5.6-
fold, respectively) [Figs 6(a) and 7(a), respectively]. tional impairment was aggravated after inhibitingthe synthesis of nitric oxide or cyclic GMP, whereasL-NNA further increased and SNAP decreased ET-
1 secretion in both fluids. In all cases, more ET-1 SNAP restored cardiac and coronary function; (3)

Nitric Oxide and Endothelin in Ischemia/reperfusion Injury 2369
30
1.4
0
Reperfusion (min)
ET-
1 se
cret
ion
rat
e (p
g/m
in)
0.2
0 5
0.8
(a)
Bas
elin
e
Isch
emia
25
30
0.14
0
Reperfusion (min)
ET-
1 co
nce
ntr
atio
n (
pg/m
l)
0.04
0.02
0
0.10
(b)
Bas
elin
e
Isch
emia
1.2
1.0
0.6
0.4
0.12
0.08
0.06
10 15 20
5 2510 15 20
**
*
*
*
*
*
*
**
*
*
*
*
*
*
30
30
0
Reperfusion (min)
Cyc
lic
GM
P c
once
ntr
atio
n (
nm
ol/l)
15
5
0 0.5 1
25
(a)B
asel
ine
Isch
emia
10
30
30
0
Reperfusion (min)
Cyc
lic
GM
P c
once
ntr
atio
n (
nm
ol/l)
15
5
0 2 4
25
(b)
Bas
elin
e
Isch
emia
10
Figure 6 Time-course of rates of ET-1 secretion (a) andFigure 5 Cyclic GMP concentration in coronary effluentresultant ET-1 concentrations (b) in coronary effluent of(a) and interstitial transudate (b) during reperfusion.reperfused hearts in the presence of vehicle (Β), L-NNAHearts were perfused for 45 min to establish baseline(200 lmol/l) (Χ) or SNAP (200 lmol/l) (Φ). Hearts wereconditions, deprived of coronary flow for 15 min (isch-perfused as described in Figure 5. Values are mean±S.E.M.emia), and then reperfused for 30 min. The effects offrom five hearts each. ∗P<0.05 L-NNA or SNAP v vehiclevehicle (Β), L-NNA (200 lmol/l) (Χ), SNAP (200 lmol/(the effect of reperfusion v baseline is not indicated sep-l) (Ε) and the guanylyl cyclase inhibitor ODQ (1H-[1,arately).2,4]oxadiazolo[4,3,-a]quinoxalin-1-one; 20 lmol/l) (Φ)
are shown. Values are mean±S.E.M. from five hearts each.Reperfusion resulted in a significant increase in cyclic
Direction of release and function of basal cyclic GMPGMP concentration (vehicle). This increase was po-tentiated by SNAP and almost completely abolished by and ET-1ODQ, whereas L-NNA was without effect. Statistical sym-bols are omitted for clarity (see text). We have previously shown that ET-1 levels are
higher in interstitium than coronary fluid in per-fused rat hearts (Brunner, 1995b). Using this model,it was shown here that basal release of cyclic GMPwas higher into the venous effluent than interstitialtransudate, but resulted in identical concentrationsin both fluids due to the much lower transudateon reperfusion, deterioration of function cor-
responded with an increased level, and im- than effluent flow rate. Because the hearts wereperfused with a physiological buffer without plasmaprovement with a decreased level of tissue ET-1,
and an ET receptor antagonist largely restored components, on which luminally released nitricoxide might have acted, the observed stability ofnormal cardiac function; and (4) nitric oxide pro-
duction was not impaired in ischemia, as indicated myocardial and coronary function (Fig. 4) waslikely due to abluminally released nitric oxide. Theby increased, rather than decreased levels of cyclic
GMP in reperfusion. cellular source of interstitial cyclic GMP was not
F. Brunner2370
30
0.08
0
Reperfusion (min)
ET-
1 se
cret
ion
rat
e (p
g/m
in)
15
(a)
Bas
elin
e
Isch
emia
25
0.06
0.04
0.02
*
20
*
*
*
30
0.5
0
Reperfusion (min)
ET-
1 co
nce
ntr
atio
n (
pg/m
l)
15
(b)
Bas
elin
e
Isch
emia
25
0.4
0.3
0.2
*
20
*
*
*0.1
Figure 7 Time-course of rates of ET-1 secretion (a) andresultant concentrations (b) in interstitial transudate ofreperfused hearts in the presence of vehicle (Β), L-NNA(200 lmol/l) (Χ) or SNAP (200 lmol/l) (Φ). Hearts were
30
100
0
LVD
P (
mm
Hg)
0
(a)
Isch
emia
25
60
20
20
*80
40
155 10
Bas
elin
e30
50
0LV
ED
P (
mm
Hg)
0
(b)
Isch
emia
25
30
20
20
10
40
155 10B
asel
ine
30
200
0
Reperfusion (min)
CP
P (
mm
Hg)
0
(c)Is
chem
ia
25
100
50
20
150
155 10
Bas
elin
e
*
*
*
*
*
perfused as described in Figure 5. Values are mean±S.E.M.Figure 8 Recovery of left-ventricular developed pressurefrom six hearts each. ∗P<0.05 L-NNA or SNAP v vehicle(LVDP) (a), left-ventricular end-diastolic pressure (LVEDP)(the effect of reperfusion v baseline is not indicated sep-(b) and coronary perfusion pressure (CPP) (c) duringarately). Transudates were collected from 0–15 andreperfusion. Hearts were perfused for 45 min to establish15–30 min of reperfusion.baseline conditions, deprived of coronary flow for 15 min(ischemia), and then reperfused for 30 min. The negativeeffect of L-NNA (200 lmol/l) and of the guanylyl cyclaseinhibitor ODQ (1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-ascertained in this study and may have been derived1-one; 20 lmol/l) as well as the positive effect of SNAPfrom smooth muscle and/or myocardial (Kojda et(200 lmol/l) and PD 142893 (200 nmol/l) are clearly
al., 1996) cells. As to the functional role of the apparent. Note also that the deleterious effect of L-NNAnitric oxide/cyclic GMP and ET systems, the L-NNA is largely reversed by PD 142893. Values are mean±S.E.M.
from five hearts each. ∗P<0.05 L-NNA, ODQ, SNAP, PDdata clearly show that basal production of nitric142893, or L-NNA+PD 142893 v vehicle. (Β) vehicle;oxide is necessary and contributes to systolic pres-(Φ) ODQ; (Χ) L-NNA; (Ε) SNAP; (Η) PD 142893; (Ο)sure development and active coronary dilatation.-NNA+PD142893.
This is evident from the increase in CPP and de-crease in LVDP following addition of L-NNA (Fig.4). As to the negative effect of L-NNA on LVDP,vasoconstriction could not have been the cause forimpaired myocardial function, because flow was (Pabla et al., 1995), which appear to involve cyclic
GMP. The lack of influence of the receptor an-maintained at a constant level throughout theseexperiments. Rather, nitric oxide improved systolic tagonist PD 142893 on functional parameters (Fig.
4) indicates that ET-1 production is maximallyfunction, possibly by mechanisms similar to thoseimplicated in enhancing contractility in reperfusion down-regulated by nitric oxide and that ET-1 exerts

Nitric Oxide and Endothelin in Ischemia/reperfusion Injury 2371
Table 2 Heart rate values for individual treatment groups during reperfusion
Treatment Baseline Reperfusion (min)(45 min)
65 70 80 90
Vehicle 310±7 300±8 300±9 302±9 307±9ODQ 295±12 290±14 294±7 295±8 295±8L-NNA 311±8 281±19 291±12 301±11 305±9SNAP 310±11 296±12 293±14 305±12 307±9PD 142893 310±13 298±15 307±12 310±9 310±6
Values represent means±S.E.M. of five hearts and are presented as beats/min. There were no significantdifferences between baseline (45 min) and any time point after initiation of reperfusion (at 60 min). Duringischaemia (45–60 min), the hearts did not beat.
no appreciable deleterious effect at normoxic con- or unaffected (Richard et al., 1994) rates of nitricditions. However, this was clearly the case after oxide release consequent to ischemia. Yet, cardiacischemia, as discussed below. function clearly deteriorated in the presence of L-
The continuous basal release of nitric oxide, pre- NNA, and SNAP reversed all aspects of functionalviously shown for a number of different vascular impairment. It can be excluded that the transientlypreparations, represented a sizable portion of the elevated level of cyclic GMP is due to activationtotal nitric oxide-releasing capacity in our model, of particulate guanylyl cyclase, e.g. by atrialsince a maximal concentration of SNAP increased natriuretic peptide (Lochner et al., 1992), becausecyclic GMP release only>four-fold (compare vehicle post-ischemic cyclic GMP release was almostand SNAP effects in Fig. 2). Shear stress appears to completely suppressed by ODQ, which does notbe a major stimulus of basal nitric oxide release inhibit particulate guanylyl cyclase (Garthwaite et(Busse et al., 1993), and it is conceivable that in this al., 1995). A speculative interpretation, presentlymodel of crystalloid high-flow perfusion, basal release being investigated, is that ischemia causes a defectof nitric oxide might be augmented compared to the further down the effector cascade, which can bein vivo situation. In addition, the half-life of nitric overcome by a substantial increase in cyclic GMPoxide might be prolonged in the effluent in our such as generated by SNAP. Alternatively ormodel due to the absence of oxyhemoglobin, which additionally, the cyclic GMP released in reperfusioneffectively binds and destroys luminally released nitric may not be representative for the functionallyoxide in vivo (Noack et al., 1992), so that the inter- active pool, e.g. it might have accumulated institial nitric oxide concentration in vivo might in fact ischemia and then been washed out on resumptionbe higher than the luminal level. of coronary flow. Another possibility is that the
steps leading to activation of guanylyl cyclase bynitric oxide are more complex than presentlyknown, possibly involving storage forms of nitricRole of cyclic GMP in ischemia/reperfusion injuryoxide, as previously reported for vascular smoothmuscle tissue (Venturini et al., 1993). This spec-The present results are of relevance to theulation might explain why L-NNA had little effectimpairment of endothelium-dependent vascularon cyclic GMP release. The considerable functionalrelaxation consistently observed after experimentalimpairment in the presence of L-NNA would thenischemia. They clearly show that cyclic GMPbe due to continued inhibition of nitric oxide/release is not attenuated on reperfusion, nor iscyclic GMP production, which is, however, notthe direction of release changed; rather it is evenreflected in a reduced rate of cyclic GMP effluxtransiently elevated, both into the vascular lumenfrom the heart. Clearly, the small effect of L-and toward the interstitium, albeit only for severalNNA on cyclic GMP production was not due tomin. Thereafter, cyclic GMP release returnedinsufficient time of contact with nitric oxideto the pre-ischemic level, and remained theresynthase, because we have recently shown thatthroughout reperfusion, whereas cardiac and cor->2 min suffice for complete inhibition of theonary function remained depressed much longer.enzyme by L-NNA in rat coronary arteries (PfeifferThese results contradict previous observations of
reduced (Fukuda et al., 1995; Maulik et al., 1995) et al., 1996).

F. Brunner2372
Role of ET-1 in ischemia/reperfusion injury in this study raises the cytosolic free Ca2+ con-centration in endothelial cells in situ has not been
Several recent studies indicate that the endogenous determined.nitric oxide/cyclic GMP system serves as a func-tionally important modulator of the vasoconstrictorand cardiac actions of ET-1. In canine and humanarteries and veins, the contraction to ET-1 can be
Inference to in vivo mediator levelsrapidly and effectively reversed by nitric oxide(Miller et al., 1989; Luscher et al., 1990), and
It is of interest to compare the coronary effluentinhibition of endothelium-derived relaxing factor
and interstitial transudate levels of cyclic GMP and(nitric oxide) with NG-monomethyl-L-arginine
ET-1 with the likely levels encountered in vivo. Anmarkedly enhanced the vasoconstriction to ET-1 in
approximate answer can be obtained by comparinganesthetized dogs (Lerman et al., 1992), indicating
the respective flow rates. With a cardiac blood flowa functional antagonism between these two me-
of 6.6±2.2 ml/g heart in conscious rats aged 12diators. In the present study, ET-1 release from
months (Tuma et al., 1985), plasma flow throughreperfused hearts was increased several-fold, and
the entire heart (average weight: >1 g) isthis increase was suppressed by SNAP and po- >2.5–4 ml/min, i.e. >2.5- to four-times less thantentiated by L-NNA. Inspection of Figures 6, 7,
perfusate flow in the present in vitro model underand 8 clearly shows that the degree of reperfusion
normoxic conditions. Therefore, coronary arterialmyocardial and vascular dysfunction corresponds
ET-1 concentrations may be several-fold higher inwith ET-1 release at the onset of reperfusion. A
vivo than measured here. To our knowledge, lymphtemporal correlation between ET-1 levels in effluent
flow rates in rats have not been determined, so thator transudate and the recovery of myocardial and
the likely in vivo levels are not known. In analogycoronary function during reperfusion cannot be
to the lymph flow rate in the dog (10–75 [mean:expected, because of the very slow rate of dis-
29±15)] ll/min/100 g heart weight (Nordbeck,sociation of ET-1 bound to ET receptors (dissociation
1978), which is >100 times less than transudatehalf life: 77 h for rat heart) (Waggoner et al., 1992).
flow in the present hearts at baseline, in vivo inter-Therefore, the levels of ET-1 observed in early re-
stitial ET-1 concentrations of cyclic GMP and ET-1perfusion determine the ultimate degree of func-
might be >100-fold higher than shown here, i.e.tional recovery, irrespective of the duration of
in the nanomolar (cyclic GMP) and low pg/ml (ET-reperfusion. Accordingly, SNAP entirely restored
1) range.both cardiac function and ET-1 release to baselinelevel, whereas the deterioration of function in thepresence of L-NNA coincided with a significantincrease in interstitial ET-1 levels, strongly im-plicating ET-1 as an important contributing factor Conclusions and implicationsin the reperfusion dysfunction observed. This wassubstantiated by use of the ET receptor antagonist, The present study shows that cardiac interstitial
levels of important mediators of myocardial andwhich largely abolished functional depression, in-cluding the additional impairments caused by L- vascular function can reliably be determined at
baseline (normoxia) and after ischemia/reperfusionNNA (see effects of L-NNA in the presence of PD142893 in Fig. 8), thus restoring CPP and LVEDP in this buffer-perfused rat heart model. Cardiac
interstitial ET-1 levels were elevated in reperfusion,to pre-ischemic, and LVDP close to pre-ischemiclevel. Collectively, these data indicate that endo- this increase closely corresponded with impaired
recovery of myocardial and vascular function, andgenous ET-1 causes myocardial and vascular dys-function in the setting of ischemia/reperfusion, and an ET receptor antagonist largely restored normal
myocardial and coronary function. Cardiac ET-1that the beneficial effects of SNAP were, in alllikelihood, mainly due to the reduction of ET-1 production is also stimulated in patients undergoing
reperfusion procedures, and may be related to poorrelease. Examining the cellular mechanism of isch-emia-induced ET-1 release in rat hearts was beyond prognosis after myocardial infarction (Omland et
al., 1994). It is suggested that the beneficial effectsthe scope of our present objectives. In culturedendothelial cells, we have recently shown that Ca2+ of experimental donors of nitric oxide such as SNAP
and clinically used organic nitrates may be mediatedstimulates, and nitric oxide inhibits, the synthesisand release of ET-1 (Brunner et al., 1995), but in part by inhibition of the deleterious actions of
endogenous ET-1.whether a period of experimental ischemia as used

Nitric Oxide and Endothelin in Ischemia/reperfusion Injury 2373
K SM, L JJJ, S II, W RF, 1994.AcknowledgementsIntravenous administration of the endothelin-1 an-tagonist BQ-123 does not ameliorate myocardial isch-The author thanks Mr G. Wolkart for expert tech- aemic injury following acute coronary artery occlusion
nical assistance and Dr B. Mayer for critical reading in the dog. Cardiovasc Res 28: 1672–1678.of the manuscript. This work was supported by L A, S EK, H FLJ, B JCJ,
1992. Inhibition of endothelium-derived relaxing fac-the Austrian Research Council (Osterreichischertor enhances endothelin-mediated vasoconstriction.Forschungs-Fonds), Projects 11040 and 10655.Circulation 85: 1894–1898.
L A, G S, M R, 1992. Massive atrialnatriuretic peptide (ANP) release in ischemia re-References perfusion. Cardiovasc Drugs Ther 6: 447–449.
L TF, 1993. Do we need endothelin antagonists?B B, D’O-J P, S P, 1993. En- Cardiovasc Res 27: 2089–2093.
dothelins: circulating plasma levels and presence in L TF, Y Z, T M, S L, S P,other biologic fluids. Lab Invest 68: 600–628. B C, S R, T M, B FR,
B F, 1995a. Dependence of endothelin-1 secretion 1990. Interaction between endothelin-1 and endo-on Ca2+. Biochem Pharmacol 49: 1785–1791. thelium-derived relaxing factor in human arteries and
B F, 1995b. Tissue endothelin-1 levels in perfused veins. Circ Res 66: 1088–1094.rat heart following stimulation with agonists and in M GE, 1981. Alterations in myocardial capillaryischaemia and reperfusion. J Mol Cell Cardiol 27: 1953– permeability by albumin in the isolated, perfused rabbit1963. heart. J Physiol 319: 311–323.
B F, T EF, O LH, 1992. Endothelin release M N, E DT, W M, E RM,during ischaemia and reperfusion of isolated perfused M G, C GA, D DK, 1995. Nitric oxiderat hearts. J Mol Cell Cardiol 24: 1291–1305. signaling in ischemic heart. Cardiovasc Res 30: 593–
B F, S H, K WR, 1995. Novel gua- 601.nylyl cyclase inhibitor, ODQ reveals role of nitric oxide, M B, S M, K P, S K, 1993. Re-but not of cyclic GMP in endothelin-1 secretion. FEBS versible inactivation of endothelial nitric oxide syn-Lett 376: 262–266. thase by NG-nitro-L-arginine. FEBS Lett 333: 203–206.
B R, M A, F I, H M, 1993. Mech- M VM, K K, B JCJ, V PM,anisms of nitric oxide release from the vascular endo- 1989. Differential sensitivity to endothelin in caninethelium. Circulation 87 (Suppl. V): V-18–V-25. arteries and veins. Am J Physiol 257: H1127–H1131.
C WL, D AM, H JX, DP PL, R N E, K D, K G, 1992. SpectrophotometricST, H GA, M TC, P RL, D determination of nitric oxide using hemoglobin. Neuro-DT, H SJ, LD D, H KE, F MA, protocols 1: 133–139.R EE, 1992. Design of a functional hexapeptide
N H, 1978. Lymphphysiologie des Herzens. Stutt-antagonist of endothelin. J Med Chem 35: 3301–3303.gart: Georg Thieme Verlag, 1–169.F H, S Y, K K, T K, S
O T, L RT, A A, A T, DY, M H, 1995. Supplement of nitric oxide at-K, 1994. Plasma endothelin determination as a prog-tenuates neutrophil-mediated reperfusion injury. Cir-nostic indicator of 1-year mortality after acute myo-culation 92 (Suppl. II): II-413–II-416.cardial infarction. Circulation 89: 1573–1579.F RF, Z JV, 1980. The obligatory role
P R, B AJ, F DM, S DB, L DJ,of endothelial cells in the relaxation of arterial smooth1995. Intracoronary nitric oxide improves post-muscle by acetylcholine. Nature 288: 373–376.ischemic coronary blood flow and myocardial con-G J, S E, B CL, N EB,tractile function. Am J Physiol 269: H1113–H1121.S K, M B, 1995. Potent and selective
P RMJ, F AG, M S, 1987. Nitricinhibition of nitric oxide-sensitive guanylyl cyclaseoxide release accounts for the biological activity ofby 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Molendothelium-derived relaxing factor. Nature 327: 524–Pharmacol 48: 184–188.526.H D, K R, B A, 1994. Endothelin
P S, L E, S K, B F, M B,and myocardial ischemia. Cardiovasc Drugs Ther 8:1996. Inhibition of nitric oxide synthase by NG-nitro-589–599.L-arginine methyl ester (L-NAME): requirement for bio-I LJ, B GM, W KS, B RE, 1987. Endo-activation to the free acid, NG-nitro-L-arginine. Br Jthelium-derived relaxing factor produced and releasedPharmacol 118: 1433–1440.from artery and vein is nitric oxide. Proc Natl Acad Sci
R RM, M F, 1983. Endothelium-dependentUSA 84: 9265–9269.and nitrovasodilator-induced relaxation of vascularK G, K K, N P, S KD, P HM,smooth muscle: role of cyclic GMP. J Cycl Nucl ProtN E, 1996. Low increase in cGMP induced byPhosphor Res 9: 281–296.organic nitrates and nitrovasodilators improves con-
R V, K N, T C, T C, 1994. Isch-tractile response of rat ventricular myocytes. Circ Resemic preconditioning protects against coronary endo-78: 91–101.thelial dysfunction induced by ischemia andK S, MQ LP, L GK, F DV,reperfusion. Circulation 89: 1254–1261.1993. Nitric oxide regulates the expression of vaso-
R GM, 1991. Endothelium-derived relaxing andconstrictors and growth factors by vascular endo-contracting factors. J Cell Biochem 46: 27–36.thelium under both normoxia and hypoxia. J Clin Invest
92: 99–104. S K, M B, K WR, 1989. Effect of

F. Brunner2374
calcium on endothelium-derived relaxing factor forma- cular smooth muscle contains a depletable store ofa vasodilator which is light-activated and restoredtion and cGMP levels in endothelial cells. Eur J Phar-
macol 170: 157–166. by donors of nitric oxide. J Pharmacol Exp Ther 266:1497–1500.S K, K P, M B, 1994. Reaction of per-
oxynitrite with oxyhaemoglobin: interference with W WG, G SL, R VA, 1992. Kineticanalyses demonstrate that the equilibrium assumptionphotometrical determination of nitric oxide. Biochem J
301: 645–647. does not apply to [125I]Endothelin-1 binding data. LifeSci 51: 1869–1876.T WF, I GL, V US, H LA, 1985.
Age-related changes in regional blood flow in the rat. W W, K H, 1988. Intra- and extra-cellular markers in interstitial transudate of perfusedAm J Physiol 249: H485–H491.
V CM, P RMJ, M S, 1993. Vas- rat hearts. Am J Physiol 254: H785–H794.