improved pka calculations
DESCRIPTION
pkaTRANSCRIPT

Improved pKa calculations through flexibility basedsampling of a water-dominated interaction scheme
JIM WARWICKERDepartment of Biomolecular Sciences, University of Manchester Institute of Science and Technology (UMIST),Manchester M60 1QD, United Kingdom
(RECEIVED April 2, 2004; FINAL REVISION June 30, 2004; ACCEPTED July 6, 2004)
Abstract
Ionizable groups play critical roles in biological processes. Computation of pKas is complicated by modelapproximations and multiple conformations. Calculated and experimental pKas are compared for relativelyinflexible active-site side chains, to develop an empirical model for hydration entropy changes upon chargeburial. The modification is found to be generally small, but large for cysteine, consistent with small moleculeionization data and with partial charge distributions in ionized and neutral forms. The hydration modelpredicts significant entropic contributions for ionizable residue burial, demonstrated for components in thepyruvate dehydrogenase complex. Conformational relaxation in a pH-titration is estimated with a mean-fieldassessment of maximal side chain solvent accessibility. All ionizable residues interact within a low proteindielectric finite difference (FD) scheme, and more flexible groups also access water-mediated Debye-Hückel(DH) interactions. The DH method tends to match overall pH-dependent stability, while FD can be moreaccurate for active-site groups. Tolerance for side chain rotamer packing is varied, defining access to DHinteractions, and the best fit with experimental pKas obtained. The new (FD/DH) method provides a fastcomputational framework for making the distinction between buried and solvent-accessible groups that hasbeen qualitatively apparent from previous work, and pKa calculations are significantly improved for a mixedset of ionizable residues. Its effectiveness is also demonstrated with computation of the pH-dependence ofelectrostatic energy, recovering favorable contributions to folded state stability and, in relation to structuralgenomics, with substantial improvement (reduction of false positives) in active-site identification by elec-trostatic strain.
Keywords: protein electrostatics; pKas; ionization entropy; side-chain packing; active-site identification;structural genomics
Ionizable group interactions are important factors in variousbiological processes (Warshel 1981, 2003; Honig and Ni-cholls 1995; Warshel and Papazyan 1998; Simonson 2001),and are a focus of attempts to identify functional sites forstructural genomics (Elcock 2001; Ondrechen et al. 2001).Grid-based continuum electrostatics methods, such as FiniteDifference Poisson-Boltzmann (FDPB; Warwicker andWatson 1982; Klapper et al. 1986; Warwicker 1986) usinga low protein-relative dielectric (typically �p � 4; Gilson
and Honig 1986) have been applied to pKa calculations(Bashford and Karplus 1990). These can be useful whenapplied to regions with limited solvent accessibility (SA;Demchuk and Wade 1996; Warwicker 1998), but �p � 4calculations based on a single conformer have been largelyunreliable for overall pKa analysis. Generally �p � 20 per-forms better (Antosiewicz et al. 1994, 1996), presumablyaccounting to some degree for other factors (Schutz andWarshel 2001) such as conformational variation (Simonsonand Perahia 1995), proton/hydrogen-bond network relax-ation (Nielsen et al. 1999), or specific internal water binding(Fitch et al. 2002). Indeed, a Debye-Hückel (DH) modelwith water dielectric also gives reasonable agreement over-all for pKas, and for the pH dependence of folding energywhen combined with a simple model for ionizable group
Reprint requests to: Jim Warwicker, Department of Biomolecular Sci-ences, UMIST, P.O. Box 88, Manchester M60 1QD, UK; e-mail: [email protected]; fax: +44-(0)161-236-0409.
Article and publication are at http://www.proteinscience.org/cgi/doi/10.1110/ps.04785604.
Protein Science (2004), 13:2793–2805. Published by Cold Spring Harbor Laboratory Press. Copyright © 2004 The Protein Society 2793

interactions in the unfolded state (Warwicker 1999). ATanford-Kirkwood model gives reasonable estimates forcarboxylate pKas in ubiquitin (Sundd et al. 2002). As withFDPB/�p � 20, the DH and Tanford-Kirkwood models failto generate the large �pKas often associated with functionalgroups (Warwicker 1998).
The problem of accounting for large and small �pKas inone scheme is being addressed with inclusion of conforma-tional and proton configurational relaxation in �p � 4 cal-culations. Multiple conformers are generally sampled frommolecular dynamics simulations (You and Bashford 1995;Zhou and Vijayakumar 1997; van Vlijmen et al. 1998; Kou-manov et al. 2001; Gorfe et al. 2002), with some improve-ment in match to experiment and significant increase incomputational requirement. A key issue is to address thoseconformational adjustments of most relevance to a pH ti-tration. Multiconformation continuum electrostatics(MCCE) samples side-chain ionization and conformation(Alexov and Gunner 1997; Georgescu et al. 2002; Alexov2003), yielding a pKa root-mean-square (RMS) error of0.83. An approximation in this model is that all conformersare combined to produce a single dielectric boundary. Amethod that assigns higher or lower electrostatic screeningfunctions, (applied to Coulomb potentials), according to thehydrophobicity/hydrophilicity of residue microenviron-ments performs well, giving an RMS error of 0.5 for a largepKa set (Mehler and Guarnieri 1999).
The current work also pursues two interaction schemes,the DH model that is effective for flexible groups, andFDPB/�p � 4, which can give large �pKas of functionalinterest, with combination in a framework of conforma-tional relaxation. FD (for FDPB/�p � 4) interactions arealways sampled for the experimental conformer, and DHinteractions are selectively introduced as a mimic for relax-ation from this conformer. Two examples illustrate thisidea. First, a buried lysine, neutral at pH 7 due to dehydra-tion (lower residue in Fig. 1). The DH model omits the Born
term and underestimates such a �pKa, whereas FD with theBorn term provides the required destabilization. BecauseDH would give a higher statistical weight than FD, a com-bined scheme must exclude DH and reflect the restrictedconformation. Secondly (top of Fig. 1), in a relatively flex-ible salt bridge the Born contributions are balanced by fa-vorable charge–charge interactions. For a pH shift that neu-tralizes one of the partners, without conformational relax-ation FD will record a Born term for the remaining ionizedgroup that is not balanced by charge–charge interactions(other than potential hydrogen bonds). The ionized partneris likely to seek a more solvent accessible conformation,reducing the Born energy penalty. Interactions in this water-dominated situation could be estimated by DH modeling ofthe salt bridge, as an alternative to FD calculations on arange of generated conformers.
A mean-field algorithm is used to describe side-chainrotamer packing (Koehl and Delarue 1994; Cole and War-wicker 2002), and to define access to DH interactions,through assessment of maximal SA (SAmax) for each ion-izable group over rotamer variation. The method, termedFD/DH, reproduces �pKas over a wide range, and is com-pared with other techniques. The contribution of ionizablegroups to folded state stability is discussed, with FD/DHdelivering overall stabilization in contrast to much FD/�p � 4 single conformer work. The utility of a distinctionbetween flexible and buried groups is considered in thecontext of active-site finding for structural genomics, andwith regard to automation of active-site subset selection fordetailed electrostatic analysis (Nielsen and McCammon2003).
An earlier empirical analysis of hydration entropy andpKa calculations (Warwicker 1997) has been extended withstudy of relatively buried ionizable groups in the FD/�p � 4model, and comparison made to small molecule ionizationentropies. It is concluded that pKa adjustments due to hy-dration entropy are relatively small, except in the case ofcysteine. Changes in hydration entropy that can be impor-tant in binding energetics (Jung et al. 2002) are discussed inthe framework of the empirical analysis.
Results and Discussion
Estimation of hydration entropy changes uponionization and charge burial
A subset of ionizable groups with large �pKas and sur-rounding ionizations that can be reasonably assigned at a pHaround the pKa of interest are studied, partitioning theanalysis from the wider prediction of pH dependence (Table1). Figure 2 shows that these groups are relatively buried interms of subsequent FD/DH analysis (Fig. 3), and estimatesof changes in hydration entropy are made with the singleconformer FD/�p � 4 model. Polar hydrogen optimization
Figure 1. Schematic diagram of ionizable group relaxation with pH in twodifferent environments. An upper salt bridge can alter hydration upon pHtitration, while the lower basic group cannot (e.g., a lysine with a reducedpKa).
Warwicker
2794 Protein Science, vol. 13

is applied for the hydroxyl groups of serine and threonine.Tyrosine hydroxyls are included where the unionized formis used, which for entropic term modeling is in all calcula-tions other than those of Figure 2C. A previous study (War-wicker 1997) looked at carboxylate and thiolate groups in arange of SA environments, deriving differential hydrationnumbers upon ionization (Ns) of about 2 (carboxylate) and6 (thiolate). Current work extends the analysis for groupswith large �pKas, and includes polar hydrogen optimiza-tion.
Hydration entropy and pKas: Asparticand glutamic acids, cysteine, and tyrosine
Bacillus circulans xylanase E172 has an elevated pKa (6.7;Joshi et al. 1997) that, along with E78 (pKa � 4.6), definesthe pH optimum for hydrolysis. Calculated electrostatic in-teractions are shown for E172 in Figure 2A. Ionization as-signment for other groups was made with reference to mea-sured pKas (Joshi et al. 1997), and with model compoundpKas at neutral pH otherwise. H149 has a measured pKa of<2.3 and H156 of 6.7. The distance between carboxylate(172) and imidazole (156) groups is about 23 Å, and theprotonation state of H156 makes no significant difference tothe calculations. Modeled hydrogen bonds between E172and N35, Y80 are shown in Figure 2A. Discrepancy be-tween the sum of calculated �pKa contributions and themeasured �pKa, with a burial fraction (Vf) of 0.57, gives
Ns � 0.7. A second calculation, with partially chargedforms for ionized and neutral E172 and torsioning of theY80 hydroxyl to point away from neutral E172, givesNs = 1.9. Without Ns modification, partial charge analysisgives �pKa � 3.7 versus 2.8 for the net charge model and2.3 experimentally. Both calculations qualitatively capturethe pKa shift.
Hen egg white lysozyme is a well-known model for elec-trostatic calculations, particularly E35 (pKa � 6.1; Kura-mitsu and Hamaguchi 1980). Figure 2A shows the restrictedenvironment of E35 and the elements that give Ns � 1.5,again with qualitative agreement to the experiment withoutNs modification (Warwicker 1997). If discrepancies in thecarboxylate group calculations of Figure 2A are approxi-mated as hydration entropy within the FD framework, thena related single Ns value is relatively small and positive(Table 2; Warwicker 1997).
In contrast, cysteine residues with large �pKas require ahigh value of Ns to match experiment (Table 2; Warwicker1997). Figure 2B shows the active site around papain C25and H159, with a pKa of 3.3 for C25 (Noble et al. 2000).Oxygen atoms were removed from oxidized C25 in thecrystal structure. Calculations were made with H159 proto-nated and carboxylates deprotonated. Of the two closestacidic groups, D158 has a pKa around 2.8 (Noble et al.2000), and the mutation E50A in caricain has only a smallaffect on the pH dependence of activity (Ikeuchi et al.1998).
Table 1. Proteins and groups used for pKa calculations
PDB ID FD/DH gps Ns group Protein Source
4pti 11 — trypsin inhibitor bovine pancreas3icb 10 — calbindin bovine intestine1b0d 18 E35 lysozyme hen egg-white1pga 13 — protein G Streptococcus3rn3 15 — ribonuclease A bovine pancreas2rn2 22 — ribonuclease H Escherichia coli1a2p 10 — barnase Bacillus amyloliquefaciens1ppf 11 — ovomucoid inhibitor 3rd domain turkey1xnb 1 E172 xylanase Bacillus circulans9pap 1 C25 papain papaya1a21 1 C30 DsbA (reduced) Escherichia coli
1ado 1 K229fructose 1,6-bisphosphatealdolase rabbit muscle
1axt 1 K93 (H) immunoglobulin/aldolase mouse1gsd 1 Y9 glutathione S-transferase A1-1 human1nai 1 Y149 UDP-galactose 4-epimerase Escherichia coli2trx — C32 thioredoxin Escherichia coli1p2p — H48 phospholipase A2 porcine pancreas4cha — H57 �-chymotrypsin bovine1163 — H31 lysozyme (C54T, C97A mutant) T4 phage
FD/DH gps gives the number of ionisable groups used in FD/DH calculation. Ns group specifies thoseused in Ns derivation. Monomers were used in all cases (selecting the first subunit from crystal coordi-nates where required). For thioredoxin, a model of the reduced D26N mutant was derived from theoxidised wild-type structure. Details (e.g. inclusion of ligands) are given in the text.
Side-chain packing and ionizable group energetics
www.proteinscience.org 2795
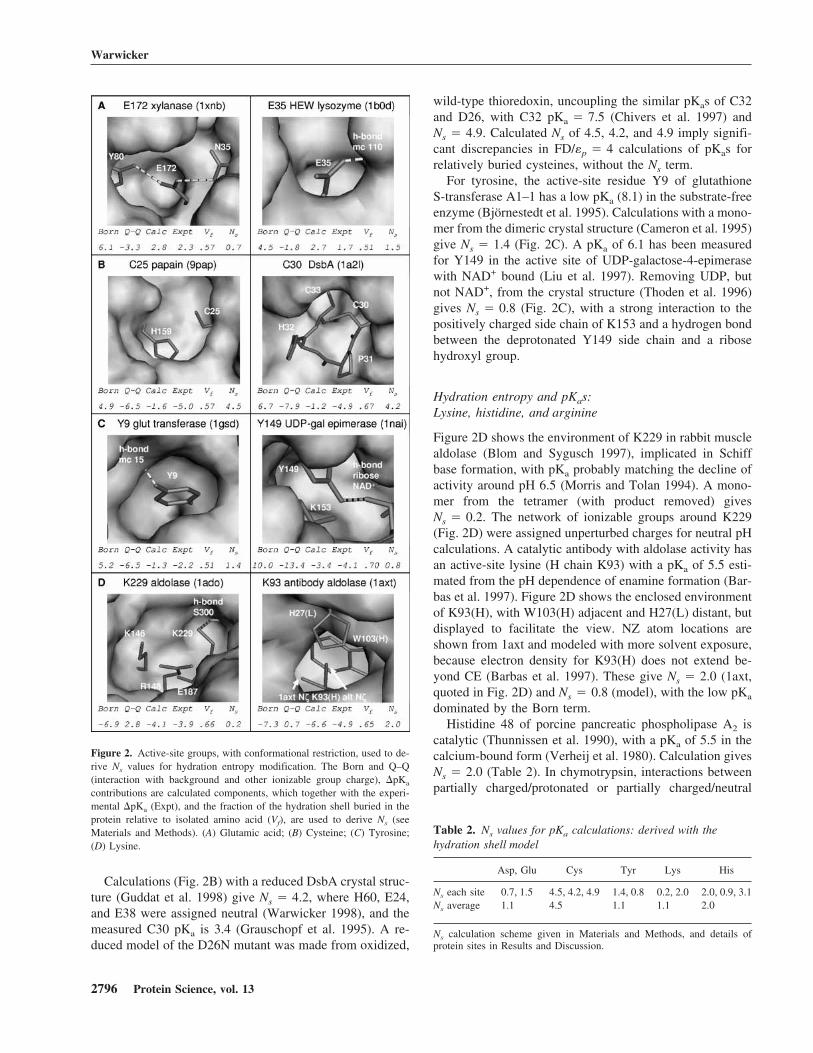
Calculations (Fig. 2B) with a reduced DsbA crystal struc-ture (Guddat et al. 1998) give Ns � 4.2, where H60, E24,and E38 were assigned neutral (Warwicker 1998), and themeasured C30 pKa is 3.4 (Grauschopf et al. 1995). A re-duced model of the D26N mutant was made from oxidized,
wild-type thioredoxin, uncoupling the similar pKas of C32and D26, with C32 pKa � 7.5 (Chivers et al. 1997) andNs � 4.9. Calculated Ns of 4.5, 4.2, and 4.9 imply signifi-cant discrepancies in FD/�p � 4 calculations of pKas forrelatively buried cysteines, without the Ns term.
For tyrosine, the active-site residue Y9 of glutathioneS-transferase A1–1 has a low pKa (8.1) in the substrate-freeenzyme (Björnestedt et al. 1995). Calculations with a mono-mer from the dimeric crystal structure (Cameron et al. 1995)give Ns � 1.4 (Fig. 2C). A pKa of 6.1 has been measuredfor Y149 in the active site of UDP-galactose-4-epimerasewith NAD+ bound (Liu et al. 1997). Removing UDP, butnot NAD+, from the crystal structure (Thoden et al. 1996)gives Ns � 0.8 (Fig. 2C), with a strong interaction to thepositively charged side chain of K153 and a hydrogen bondbetween the deprotonated Y149 side chain and a ribosehydroxyl group.
Hydration entropy and pKas:Lysine, histidine, and arginine
Figure 2D shows the environment of K229 in rabbit musclealdolase (Blom and Sygusch 1997), implicated in Schiffbase formation, with pKa probably matching the decline ofactivity around pH 6.5 (Morris and Tolan 1994). A mono-mer from the tetramer (with product removed) givesNs � 0.2. The network of ionizable groups around K229(Fig. 2D) were assigned unperturbed charges for neutral pHcalculations. A catalytic antibody with aldolase activity hasan active-site lysine (H chain K93) with a pKa of 5.5 esti-mated from the pH dependence of enamine formation (Bar-bas et al. 1997). Figure 2D shows the enclosed environmentof K93(H), with W103(H) adjacent and H27(L) distant, butdisplayed to facilitate the view. NZ atom locations areshown from 1axt and modeled with more solvent exposure,because electron density for K93(H) does not extend be-yond CE (Barbas et al. 1997). These give Ns � 2.0 (1axt,quoted in Fig. 2D) and Ns � 0.8 (model), with the low pKa
dominated by the Born term.Histidine 48 of porcine pancreatic phospholipase A2 is
catalytic (Thunnissen et al. 1990), with a pKa of 5.5 in thecalcium-bound form (Verheij et al. 1980). Calculation givesNs � 2.0 (Table 2). In chymotrypsin, interactions betweenpartially charged/protonated or partially charged/neutral
Figure 2. Active-site groups, with conformational restriction, used to de-rive Ns values for hydration entropy modification. The Born and Q–Q(interaction with background and other ionizable group charge), �pKa
contributions are calculated components, which together with the experi-mental �pKa (Expt), and the fraction of the hydration shell buried in theprotein relative to isolated amino acid (Vf), are used to derive Ns (seeMaterials and Methods). (A) Glutamic acid; (B) Cysteine; (C) Tyrosine;(D) Lysine.
Table 2. Ns values for pKa calculations: derived with thehydration shell model
Asp, Glu Cys Tyr Lys His
Ns each site 0.7, 1.5 4.5, 4.2, 4.9 1.4, 0.8 0.2, 2.0 2.0, 0.9, 3.1Ns average 1.1 4.5 1.1 1.1 2.0
Ns calculation scheme given in Materials and Methods, and details ofprotein sites in Results and Discussion.
Warwicker
2796 Protein Science, vol. 13

forms of H57, and other catalytic residues D102 and S195have been evaluated, with S195 hydroxyl directed towardneutral H57 and away from protonated H57. Comparisonwith the measured pKa of 6.8 (Fersht and Renard 1974)gives Ns � 0.9 (Table 2). For T4 lysozyme, the noncatalyticresidue H31 (pKa � 9.1) is significantly buried in a saltbridge with D70 (Anderson et al. 1990), giving Ns � 3.1(Table 2). This Ns-based study of buried side chains withknown pKas has excluded histidine residues with compli-cating factors such as strongly coupled titrations and ligandinvolvement. Variability in Ns values for histidine (prior toaveraging) probably reflects relatively mixed environmentsand the omission of local relaxations (Edgcomb and Mur-phy 2002).
There is some evidence for reduced arginine pKas (Le-houx and Mitra 1999; Morillas et al. 1999), but not a clearexample coupled to an atomic structure. Arginine side chainand N-terminal groups are assigned (Ns) as the lysine side-chain average, and C-terminal groups follow the average ofthe carboxylate containing side chains.
Comparison with ionization datafor amino acids and analogs
The empirical modification for hydration entropy is gener-ally small in �pKa terms compared with the Born andcharge–charge contributions, with the exception of cysteine(Fig. 2; Table 2). The sense of the modification is in linewith water molecule release upon ionizable group burialbeing greater (albeit slightly in most cases) for ionized ver-sus unionized forms. Table 3 shows the enthalpies and en-tropies of ionization for amino acid side chains and molecu-lar analogs (Izatt and Christensen 1976). Hydration entro-pies make the major contributions to ionization entropies(Alberty 1983), which show a wide variation for acidic and
basic side chains in amino acids, partially due to overlap-ping proton binding equilibria. In contrast, the analog datashow similar ionization �S within acidic and within basicgroups, distinguishing between dissociation from one neu-tral to two charged species (acidic, larger �S), and from onecharged to one charged and one neutral species (basic,smaller �S). Differencing between average �S values foreach grouping (−103.5 J/deg/mole for AH ⇒ A− + H+ and−17.0 J/deg/mole for BH+ ⇒ B + H+), cancelling the protonterm, gives �S � −86.5 J/deg/mole for AH ⇒ A− summedwith B ⇒ BH+. Approximating equal �S for each of theseunionized to ionized form transitions, �S � −43.3 J/deg/mole is obtained, equivalent to Ns � 1.7 (300 K) in thehydration shell model and consistent overall with the pKa
calculations (other than cysteine). This favorable compari-son supports use of the empirical model.
A relatively small hydration entropy modification forpKas is expected where charged and neutral species differlittle in water ordering for a first hydration shell, due to thepropensity of both forms to accommodate multiple hydro-gen bonds. This is consistent with high Ns for cysteine,which is relatively nonpolar in the neutral form. Table 4compares �S values measured for small carboxylic acidsand thiols (Irving et al. 1964). Consistently more negative�S for thiol group dissociation compared to carboxylic ac-ids qualitatively matches the results of the hydration shellmodel, supporting a significant role for hydration entropy incysteine pKa calculation.
Entropic contributions to interactions withinthe pyruvate dehydrogenase complex
The interaction between dihydrolipoyl dehydrogenase (E3)dimer and the peripheral subunit-binding domain (PSBD) ofdihydrolipoyl acyltransferase (E2) in the pyruvate dehydro-genase complex of Bacillus stearothermophilus has beensubject to thermodynamic investigation (Jung et al. 2002).The binding (�Go � −52.7 kJ/mole) is entropically driven(−T�So � −61.9 kJ/mole), and alanine mutations demon-strate that one side chain (R135) makes a major entropiccontribution (Table 5; Jung et al. 2002), leading the authorsto conclude that water liberation upon charge network for-mation in the complex is important.
Table 3. Amino acid side chain and analog ionization data
Amino acid/analog pKa �H (kJ/mole) �S (J/deg/mole)
Aspartic acid 3.87 4.0 −60.6Glutamic acid 4.27 1.6 −76.5Cysteine 8.39 36.0 −39.7Tyrosine 10.05 25.1 −108.7Arginine 12.48 51.8 −64.8Lysine 10.53 48.5 −38.9Histidine 6.00 29.9 13.0Propanoic acid 4.87 −0.6 −95.3Phenol 9.98 23.6 −111.6phenyl-arginine 12.40 50.0 −20.9Monomethylamine 10.63 54.7 −19.7Imidazole 6.99 36.7 −10.5
Measurements at 25°C and ionic strength between 0 and 0.15 M for theproton dissociations, AH <�> A− + H+ (acid) and BH+ <�> B + H+
(base) (Izatt and Christensen 1976). Only side-chain ionization pKa shownfor amino acids.
Table 4. Comparison of ionization �S values for carboxylatesand thiolates
Compound �S (X�COOH) �S (X�SH) ��S (COOH-SH)
CH3CH2X −95.3 −112.9 17.6(CH3)2CHX −107.0 −132.5 25.5(CH3)3CX −106.6 −140.0 33.4
Data from Irving et al. (1964), at 25°C, with �S units of J/deg/mole.
Side-chain packing and ionizable group energetics
www.proteinscience.org 2797

Whereas hydration modeling for pKa calculations in-volves differences for ionized/unionized forms and forcharge burial, hydration modeling of complexation needs toconsider primarily charge burial (although pKa changesupon burial could play a secondary role). Making the ap-proximation that unionized cysteine has a weakly boundhydration shell, then pKa modification for this case is usedto estimate the change in hydration entropy �(T�S)QWAT
for burial of a full ionizable group hydration shell in the E3dimer:PSBD interface. The fractions of hydration shell oc-cluded upon complexation are summed and multiplied bythe 36 kJ/mole of a full shell. Mutant complexes were mod-elled with arginine side chain reduction to alanine (Fig. 4).Component parts (E3 dimer and PSBD) were modeled with-out conformational relaxation.
Estimates of changes upon complexation for side chainrotameric entropy and water structure associated with bur-ied nonpolar area are added (Table 5). Side-chain rotamerentropies derived from a mean-field calculation (Cole andWarwicker 2002). Nonpolar surface burial is converted to afree energy estimate (assumed due to water ordering) by theempirical factor 0.1 kJ/mole/Å2. The entropic term fromionizable group burial is the most significant in the wild-type to mutant differences. Summed values are qualitativelyconsistent with experiment for the two mutants (Table 5),demonstrating potential for the hydration model in bindingstudies.
Combination of FDPB and DH models:The FD/DH interaction scheme
The empirical modification of the FD/�p � 4 single con-former pKa calculations for hydration entropy is applied tothe FD component of the FD/DH scheme. Figure 3 sche-matizes side-chain flexibility in different environments, andthe derivation of SAmax and access to DH interactions. AVdW clash tolerance (VdWtol) is included in mean-fieldcalculations that determine allowed rotamers for SA analy-sis. This parameter is varied to approximate conformational
relaxation not explicitly included (nonlibrary rotamer pack-ing and main-chain movement), and the best value deter-mined with respect to experimental pKas (Fig. 5B). VdWtol
Table 5. Binding energies for PSBD mutants relative to wild type, in complex with E3dimer, and estimated entropic contributions
Experiment (kJ/mole) Calculation (kJ/mole)
��G0 ��H0 �(T�S0) �(T�S)QWAT �(T�S)SC �(T�S)NP �{�(T�S)}
R135A −11.7 +20.1 +31.8 +20.8 −1.0 −0.3 +19.5R139A −10.5 −10.5 0.0 +12.1 −3.5 −6.0 +2.6
Experimental data from Jung et al. (2002), differenced for wild type-mutant values. Calculations differ-enced for complexation, T�S � TScomplex − [TSE3dimer + TSPSBD], followed by wild type to mutantdifferencing giving �(T�S). Charge hydration entropy �(T�S)QWAT derived from fraction of first hy-dration shell that is buried at interface for ionizable groups, multiplied by the cysteine shell value frompKa fitting of 36 kJ/mole. Side chain entropy �(T�S)SC taken from mean-field calculations (Koehl andDelarue 1994; Cole and Warwicker 2002). Entropy changes associated with nonpolar surface burial�(T�S)NP were approximated with the multiplicative factor 0.1 kJ/mole/Å2.
Figure 3. Schematic diagram for determination of access to DH interac-tions. (A) Solvent probing of a side chain gives two SA arcs according towhether probed in the context of the drawn rotamer set (SAconfig) or fixedatoms only (SAfixed-atoms). (B) A different rotamer set gives higher solventaccessibility, such that [SAmax/SAfixed-atoms] � 0.75 and access to DHinteractions is allowed. (C) In this case, the rotamer set of B is not possible,SA remains small, and DH interactions are disallowed.
Warwicker
2798 Protein Science, vol. 13

of around 0.8 Å is typically required to give a packingsolution for united atom VdW radii, so that VdWtol > 0.8 Årepresents additional flexibility that will lead to greaterSAmax and successive entry of more ionizable groups to theDH calculation regime.
Variation of RMS pKa errors with VdWtol
Averages of fractional SAmax, over ionizable groups in rep-resentative proteins 1b0d and 1ado, show that most groupsattain close to full SA by VdWtol � 0.8 Å, illustrative ofgeneral surface location (Fig. 5A). Specific groups (E35 of1b0d and K229 of 1ado shown) retain SAmax < 0.75 (Figs.3, 5A) at VdWtol � 1.4 Å. A fraction of 0.75 describes apivot point largely separating relatively buried, active-sitegroups from surface, flexible side chains.
Figure 5B shows that for the 110 groups, excluding mostof the large active-site �pKas, FD performs poorly and DHwell in comparison to the null hypothesis. For the 117 set,the large �pKas impair DH performance, but have relativelylittle impact overall on FD, which remains dominated byinaccuracy within the 110 set. A broad region of optimal fitto experiment is found as VdWtol is varied for FD/DH.Increasing VdWtol adds flexibility that reduces the errone-ously large �pKas for surface groups in the 110 set (sche-matically at the top of Fig. 1), while too much flexibility(beyond VdWtol � 1.4 Å) allows buried group relaxation(bottom of Fig. 1) to an extent that is inconsistent withexperiment. From the optimal region, VdWtol � 1.4 Å istaken for FD/DH pKa calculations.
Comparison of pKa calculation methods
Table 6 gives overall RMS pKa errors for various methods.FD/�p � 20 (Antosiewicz et al. 1994) is similar to DH,consistent with a water-like environment (Warwicker1999). Similarly, FD/�p � 4 with SA derived from VdWradii rather than a probe reentrant surface (Dong and Zhou
2002), has a large degree of internal water, and is compa-rable overall to DH. FD/DH performs moderately betterwith hydration entropy modification than without, with thelargest difference for the active-site cysteine pKas of papainand DsbA (Table 7). Overall results are improved somewhatwith the simple polar hydrogen optimization algorithm,consistent with previous observations (Nielsen et al. 1999).Differences associated with application of either the hydra-tion entropy or polar hydrogen placement schemes are gen-erally smaller than those from FD/DH introduction relativeto FD (Table 6). The MCCE method gives an RMS pKa
error for the 110 groups of 0.92 (Georgescu et al. 2002),compared with 0.58 for DH and 0.79 for FD/DH, while thesingle conformer FDPB method used by these authors gives2.05, close to the current FD value of 2.10.
Scatter plots for �pKas and pKas (Fig. 6) show that FDhas a bias toward overestimation of pKa shifts, while for DHan underestimation of �pKas is evident. These trends are
Figure 5. VdWtol variation, SAmax, and RMS pKa errors. (A) Represen-tative averages over a protein and active-site groups are shown for SAmax
vs. VdWtol. Intersection of the SAmax � 0.75 and VdWtol � 1.4 linesgives FD/DH parameterization. (B) RMS pKa errors are shown for FD/DHcalculations for each of the 110 and 117 group sets (with FD only and DHonly at each extreme), and horizantal lines (“null”) give RMS errors formodel compound pKas.
Figure 4. PSBD (blue backbone):E3 dimer (green and orange surfaces)interface, showing mutated charge network.
Side-chain packing and ionizable group energetics
www.proteinscience.org 2799

corrected to a large extent in the FD/DH model, althoughseveral groups remain significantly in error.
Remaining discrepancies with the FD/DH method
Several of the groups used in the Ns analysis have FD�pKa > 1, despite derivation of Ns values from experimentalpKas (Table 7). Discrepancy arises upon moving to multipleionizable groups from a fixed charge environment, and fromuniform Ns for each class of ionizable group. The DH modelgives low RMS pKa errors for some proteins, but also somelarge values for active-site pKas. FD/DH largely matchesthe low RMS errors of DH within the 110 set, illustratingDH dominance of statistical weightings (Table 7). Ribo-nucleases 3rn3 and 2rn2 are the biggest exceptions.
For 3rn3, groups with �pKa to experiment >1.5 are H48and H119. There are alternate locations for H119 (“A” wasused), and a sulphate ion that may give overestimation ofstabilizing interactions (FD/DH pKa � 8.3 and measuredpKa � 6.1). H48 of 3rn3 has an FD/DH pKa of 4.6 and ameasured pKa of 6.3. H48 is relatively buried and excludedfrom the DH scheme. FD interactions are dominated by anion pair with D14 that varies between ribonuclease A struc-tures (Georgescu et al. 2002).
Calculations for 2rn2 were made without magnesium tomatch experiment. FD/DH discrepancies with experiment>1.5 pKa units are E48 and H114. For E48 (1.7 calculatedand 4.4 measured), DH is incorporated at VdWtol � 1.4 Åbut not at VdWtol � 1.2 Å. However, the calculated pKa
remains the same, because interactions are dominated byhydrogen bonding to background charges in the FD scheme.It is possible that unfavorable interactions with the D10,D70, and D134 charge cluster surrounding E48 have beenunderestimated. These groups are DH accessible, so thatrelatively weak DH interactions dominate. For H114 anFD/DH pKa at VdWtol � 1.4 Å of 1.0 compares with ameasured pKa of 5.0. This residue is relatively buried, withan enforced FD-only scheme. The key (FD) interaction is ahydrogen bond to the peptide NH of C63, so that H114protonation must involve some change in structure.
Of the seven additional proteins with individual groups,1xnb/E172 and 9pap/C25 give the worst results for FD/DH(Table 7). For 1xnb, this discrepancy (calculatedpKa � 4.4, measured � 6.7) is a failure of the FD/DHscheme, because FD alone gives a large positive �pKa forE172 with or without hydration entropy modification.Again, more detailed FD analysis of coupled clusters mayimprove the FD/DH model, a conclusion supported forxylanase by pKa calculations using subsets of ionizablegroups (Nielsen and McCammon 2003).
The FD/DH pKa for C25 in papain (9pap) is 5.9, com-pared with 3.3 by experiment, and in contrast the FD pKa isa good match. Both C25 and H159 are accessible to the DHscheme at VdWtol � 1.4 Å. Because the FD result corre-sponds to significant stabilization of C25, it might be ex-pected to figure more strongly in FD/DH calculation atVdWtol � 1.4 Å. Of the four possible samplings of C25 andH159 net charged forms, only one (both FD) will give a
Table 6. RMS pKa errors compared across calculations
NullFD
�p � 20FD
VdW SA DH FD/DHFD/DH no
�(T�S)QWAT
FD/DH noHmove FD
110 gps 0.77 0.78 1.06 0.58 0.79 1.02 1.11 2.10117 gps 1.24 1.25 1.46 1.18 0.86 1.25 1.16 2.06
Sets of 110 and 117 groups explained in Materials and Methods. All calculations include polar hydrogenoptimization except for the column labeled “no Hmove”. FD/DH schemes include hydration modificationfor the FD component except for the “no �(T�S)QWAT” column. Of the FD calculations, only the FDcolumn uses this modification. It is not appropriate for either �p � 20 or VdW solvent accessibilityschemes, which are closer to DH in terms of water domination. FD/DH calculations were made withVdWtol � 1.4 Å. Null refers to model compound pKas in place of calculated values. RMS pKa error �sqrt {[�(pKa
expl − pKacalc)2]/N}, where the sum runs over the N ionizable groups with measured pKas.
Table 7. RMS pKa errors compared for DH, FD/DH, andFD methods
PDB ngps DH FD/DHFD/DH no
�(T�S)QWAT FD
4pti 11 0.29 0.35 0.35 1.903icb 10 0.40 0.37 0.39 0.971b0d 18 0.74 0.47 0.61 1.491pga 13 0.46 0.80 0.47 1.683rn3 15 0.45 0.87 2.02 2.432rn2 22 0.59 1.17 1.20 1.971a2p 10 0.58 0.76 0.59 3.221ppf 11 0.81 0.77 0.74 2.761xnb 1 2.48 2.35 2.34 0.629pap 1 4.85 2.64 4.95 0.651a21 1 4.86 0.71 5.35 1.141ado 1 4.84 0.84 0.33 1.441axt 1 6.04 1.06 1.85 1.551gsd 1 1.59 1.36 1.69 0.221nai 1 3.34 0.23 1.9 0.64
All FD component calculations are single conformer �p � 4, and use the�(T�S)QWAT modification unless stated. Polar hydrogen optimization isapplied in all cases. FD/DH calculations made with VdWtol � 1.4 Å.
Warwicker
2800 Protein Science, vol. 13

large favorable interaction. Alteration from FD to FD/DHmay therefore result from relative weighting toward weak(DH) interactions. Active-site residue Y9 of 1gsd is allowedaccess to the DH scheme at VdWtol � 1.4 Å, (FD/DHpKa � 9.5 and measured 8.1), but not at VdWtol � 1.3 Å(calculated pKa � 8.7). It is tightly coupled to R20, whichalso has a DH accessibility transition over this VdWtol
range.
Ionizable group electrostatic energy contributions
A move away from neutral pH generally destabilizes thefolded state (Fink et al. 1994), consistent with a stabilizingnetwork of positive and negative charges (Wada and Naka-mura 1981), and with destabilization by charge reversal ofsurface amino groups (Hollecker and Creighton 1982).Computational methods with FD/�p � 20 or DH interac-tions accommodate such pH dependence, albeit with adjust-ment for pH-dependent effects in unfolded states (Schaeferet al. 1997; Warwicker 1999).
However, use of single conformer FD/�p � 4 has givenrise to questions of salt-bridge stability (Hendsch and Tidor1994; Dong and Zhou 2002), as is shown with the mostlyunfavorable contributions of ionizable group interactions tostability at pH 7 (FD column of Table 8). In contrast to theDH column, FD calculations suggest that most of thesefolded proteins would be more stable with global replace-ment of ionizable groups, at variance with the prior discus-sion. The FD/DH method recovers favorable contributionsto neutral pH stability. In many cases FD/DH matches DHclosely, with DH interactions dominating FD overall. Theparticularly favorable values in Table 8 for 3icb/calbindin
with FD and FD/DH calculation result from the strong bind-ing of calcium ions by acidic side chains.
Improved active-site prediction
Figure 7 demonstrates the utility of a method that focuses onlarge �pKas in a background of smaller �pKas. Four rep-resentative examples are shown, with ionizable groups dis-allowed from DH interactions (VdWtol � 1.4 Å), furtherrestricted to those with calculated pKas between 3 and 10.This latter feature excludes side chains such as tyrosine orhistidine that are largely buried but remain neutral over a pHrange around physiological, and buried groups in particu-larly stable charge networks. In Figure 7, just active-siteresidues remain, including D31 of 1nai that lies on the op-posite end of the NAD binding groove to Y149. The largenumber of tyrosine side chains that are relatively buried(fractional SAmax < 0.75 at VdWtol � 1.4 Å), but withpKa > 10, show the potential for FD/DH to improve active-site identification methods (Elcock 2001; Ondrechen et al.2001) that tend to give such groups as false positives. Inaddition, FD/DH allows detailed investigation of active-siteelectrostatics, coupled to existing biochemical data or on thebasis of structural genomics predictions.
Conclusions
Extension of the hydration entropy modification for pKa
calculations (Warwicker 1997) with a set of active-site�pKas (Fig. 2; Table 2) gives results that are consistent withsmall molecule ionization data (Tables 3,4). For cysteine,the modification can significantly influence calculated
Figure 6. Scatter plots (calculation vs. experiment) for �pKas and pKas with FD, DH, and FD/DH calculations (117 group set).
Side-chain packing and ionizable group energetics
www.proteinscience.org 2801

pKas. In general, the empirical modification makes rela-tively small changes (Tables 6,7), agreeing with the fruitfulapplication of FD methods to active-site pKas in previouswork. For some binding processes, entropy gain upon de-hydration makes a significant contribution (Fig. 4; Jung etal. 2002), and the empirical hydration model could proveuseful (Table 5).
The effectiveness of FD/DH calculations in combiningFD/�p � 4 and DH computation is illustrated in Figure 5B.The simple DH model is relatively good for the mostlysmall �pKas of the 110 groups set (Table 6), with an RMSpKa error of 0.58, matching the performance of more com-plex methods (Mehler and Guarnieri 1999; Georgescu et al.2002). However, DH performance is eroded when larger�pKas are included (117 groups). The FD/�p � 4 single-conformer method fails to consistently predict �pKas in the110 set. As the system is relaxed in the FD/DH model, withselective access to DH interactions mimicking side-chainand possibly main-chain readjustment on a limited scale, anoptimal relaxation is evident before key groups are errone-ously solvent-exposed. Large-scale conformational changecannot be reliably predicted, and is not the subject of thiswork. A VdWtol parameter controls conformational relax-ation through mean-field rotamer packing and estimationof SAmax for each ionizable group (Figs. 3, 5A). AtVdWtol � 1.4 Å, FD/DH gives an RMS pKa error of 0.86for the 117 groups, compared with null, DH and FD valuesof 1.24, 1.18, and 2.06, respectively. The tendencies of FDto overestimate small �pKas and DH to underestimate large�pKas are clear in Figure 6. Factors that could further im-prove the FD/DH scheme include modeling of larger scaleflexibility and pH-dependent ion binding, and detailed
water structure (Koumanov et al. 2002). Tightly coupledclusters, with groups that are borderline for access to the DHscheme at VdWtol � 1.4 Å, may benefit from FD analysisof individual rotamer combinations. This refers to relativelyfew groups; the majority would be included in FD/DHsampling.
Generally, the FD/DH model is improved by hydrationentropy modification and polar hydrogen optimization, butnot to the extent of FD/DH improvement over FD (Tables6,7). Two areas further demonstrate potential. First (Table8), FD/DH recovers overall stabilizing contributions forionizable group interactions at pH 7, in contrast to the de-stabilization of the FD/�p � 4 single-conformer model.Second, Figure 7 demonstrates active-site identification,which could be used in a structural genomics context orprovide subsets of residues to seed active-site–centeredelectrostatics calculations (Nielsen and McCammon 2003).The computational speed of the FD/DH method, coupled toaccuracy over large and small pKa deviations, make it suit-able for large-scale database analysis.
Materials and methods
Coordinates and pKas
Proteins and coordinate sets (Berman et al. 2000) are given inTable 1. A previous study of computational methods (Georgescu etal. 2002) has collated pKa data for the first eight proteins in Table1, covering 126 ionizable groups, although the authors report that
Table 8. Predicted ionizable group array energies at pH7 (kJ/mole)
FD DH FD/DH
4pti 24.1 −5.8 −5.23icb −368.9 −57.4 −423.31b0d 5.3 −26.6 −35.31pga −11.3 −23.5 −43.13rn3 26.3 −21.7 −22.02rn2 67.4 −29.7 −30.51a2p −21.3 −38.9 −69.41ppf 0.1 −8.3 −17.51xnb 53.6 −29.1 −22.49pap 73.3 −44.2 −24.91a21 51.8 −39.4 −42.91ado 90.4 −101.5 −92.91axt 84.4 −101.2 −106.31gsd 135.6 −72.8 −65.31nai 81.2 −81.6 −73.4
Calcium ions were included in all 3icb calculations leading to significantstabilization. Ionizable group energy calculations given in Materials andMethods.
Figure 7. Active-site identification using FD/DH. Four proteins (greenbackbones) used in pKa calculations are shown, with ionizable groupsassessed as disallowed from DH interactions in orange, and a subset withcalculated pKas between 3 and 10 shown in purple and labeled.
Warwicker
2802 Protein Science, vol. 13

some of these ionizations may overlap either a limit of the mea-sured pH range and/or a protein unfolding transition. Further in-vestigation of the literature cited by Georgescu et al. (2002), withregard to these criteria, lead to the exclusion of the followinggroups in the current study: D54, E73, D93, D101 of 1a2p; Y23,Y35, and K41 of 4pti; D66 of 1b0d; D7, D27, Y31, and C-t of1ppf; K13 of 1pga; D14 of 3rn3; D102 and D148 of 2rn2. These16 exclusions reduce 126 groups for the eight proteins studied byGeorgescu et al. (2002) to 110, which also excludes the C-t of1pga.
This set of 110 groups was supplemented by a further sevenactive-site groups, with large �pKas for FD/DH analysis, formingthe “117” set. Groups and coordinates used for development of theempirical hydration model (Warwicker 1997), including theseseven active-site residues, are listed in Table 1. In addition, coor-dinate set 1ebd was used to study interactions within the pyruvatedehydrogenase complex.
Electrostatics calculations (FD and DH)
DH (Warwicker 1999) and FD (�p � 4, single conformer) (War-wicker 1998) calculations followed previous work, except for hy-dration entropy and polar hydrogen optimization modifications(next section). Model compound pKas used and charge assignmentfor ionizable group atoms were Arg 12.0 0.5/NH1 0.5/NH2; Lys10.4 1.0/NZ; His 6.3 0.5/ND1 0.5/NE2; Asp 4.0–0.5/OD1–0.5/OD2; Glu 4.4–0.5/OE1–0.5/OE2; Cys 8.3–1.0/SG (where not di-sulfide bonded); Tyr 10.2–1.0/OH; N-t 7.5 1.0/N; C-t 3.8–0.5/O–0.5/OXT. Cysteine side chains were excluded from pKa calcula-tions, except for papain C25, DsbA C30 and C33, and thioredoxinC32 and C35. Partial charges were assigned from the GROMOSlibrary (van Gunsteren and Berendsen 1987). Where specified,�p � 20 was used in place of �p � 4 for the FD model. A waterrelative dielectric of 78.4 was used throughout. An alternative to asolvent-probed (probe radius � 1.4 Å) reentrant surface wastested, a VdW sphere derived surface giving greater water pen-etration into the protein. All DH and FD computations were madewith a linear ion response at 0.15 Molar.
The energy of a protonation microstate relative to the fullyunprotonated state for M titratable groups is (modified from Bash-ford and Karplus 1990):
Gs = �m=1,M
xm�2.303�kBT(pH-pKm,model�
+ �m=1,M
�xm+qm0� (�GBorn + �G(T�S)QWAT + �Gback�
+ 0.5 �m=1,M
�n=1,M,n�m
�xm+qm0��xn+qn
0��Gmn
where xm,xn are members of vectors (lengths M) taking the values0 (unprotonated) and 1 (protonated), qm
o,qno are the charges of the
unprotonated sites m,n, and pKm,model is the model compound pKa
for site m. Interactions of group m with the protein dielectric andnonionizable (background) charge environments are differenced tocalculations for each ionizable group extracted from the protein,but remaining on the same FD grid (�GBorn, �Gback), while �Gmn
gives the interaction between ionizable groups. The term�G(T�S)QWAT arises from the hydration entropy model discussedin the next section. This definition of Gs applies to either FD or DHcalculations, but in the DH case �GBorn and �G(T�S)QWAT termsare zero. In addition �G(T�S)QWAT is set to zero for �p � 20calculations. Average fractional protonation for site m is de-rived as:
�xm = �states,s
xmexp(−GskBT�� �states,s
exp(−GskBT�
where a full evaluation of the partition function (Z) is possible forsmall numbers of groups using the reduced sites method (Bashfordand Karplus 1990) at pH extremes, or alternatively monte carlosampling of lowest energy states is used (Beroza et al. 1991). Thecalculated pKa of site m is that pH for which ⟨xm⟩ � 0.5.
Electrostatic energy contributions from ionizable groups werecalculated by summing increments over pH according to �(�G)/�pH � 2.303RT�Q, where �G, �Q differences are relative to pHtitration of the same set of ionizable groups with model compoundpKas (Antosiewicz et al. 1994). These sums were extended to anextreme pH at which a full evaluation of ionization states waspossible, G � −RTlnZ, again with differencing to a calculation forthe same groups titrating with model compound pKas.
Hydration entropy modificationand polar hydrogen placement
An empirical estimate of the contribution that water orderingmakes to the change in ionization energy upon transfer into proteinis written as �G(T�S)QWAT � Vf·Es (Warwicker 1997), where Vf isthe fractional change in first hydration shell volume, and Es is afree energy associated with water ordering for the complete shell.Vf is calculated from FD grids for a group in the protein relative tothat whole amino acid extracted from the protein. It is convenientto consider Es in terms of a notional number of water molecules(Warwicker 1997) in the first hydration shell (Ns), using 25 J/K/mole entropy cost of immobilization of a single water molecule or7.5 kJ/mole at 300 K. In pKa calculations, Es and Ns correspond todifferences between water structure in ionized and unionizedforms. Averages of Ns are taken over groups within a class, andindividual Ns values derived with charge environments fixed ac-cording to expected ionization at physiological pH (see Resultsand Discussion). Thus, Ns derivation was separated from globalpKa computation in the FD/DH method through the use of separatecalculations with fixed ionization on all groups but one. The Ns
parameter gives the empirically derived difference in hydrationnumbers between ionized and unionized forms, for a completehydration shell. It is uniform for a class of ionizable group, buteach individual modification is proportional to burial (Vf).
Optimization of polar hydrogen locations can improve pKa cal-culations (Nielsen et al. 1999; Koumanov et al. 2003). A simpleprocedure was implemented which optimizes polar hydrogen po-sitions for the OH groups of Ser and Thr side chains (and Tyr ifpresent in its neutral form). Torsions are sampled, with 12° reso-lution, against coulombic interactions from charges other thanthose belonging to the set of OH groups being optimized. Ionizablegroups are specified in their ionized forms, to focus pKa calcula-tions on the net difference upon titration.
Mean-field packing algorithm and accessto the DH interaction scheme
A mean-field algorithm (Koehl and Delarue 1994), adjusted withhard sphere clashes replacing Lennard-Jones interactions (Coleand Warwicker 2002), establishes a set of allowed rotamers froma standard library (Tuffery et al. 1997) as well as those in theexperimental structure. The conformational matrix (CM) or weightfor rotamer k of side chain i (of N side chains), depends on clasheswith fixed atoms and the rotamers of other side chains j:
Side-chain packing and ionizable group energetics
www.proteinscience.org 2803

CM (i,k� = �1, no clash with fixed atoms0, clash with fixed atoms �
� �j=1,j�i
N
�1=1
Kj �1, no clash with �j,1�0, clash with �j,1� �CM�j,1�
Weights for rotamers l � 1 to Kj of side chain j are summed,and the multiplication of these side-chain and fixed-atom weightsrequires that each be nonzero for a packing solution for rotamer kof residue i. CM values are iterated from an initial assignment ofequal weights. These mean-field methods were used previously toassess the entropy associated with side-chain rotamer distributions,but are used here simply to estimate possible rotamers for a givenstructure and packing tolerance (VdWtol).
Hydrogen atoms are absent in the packing calculations, whichuse united atom VdW radii, so that a VdWtol of about 0.8 Å istypically required to repack rotamers of the experimental structure,due particularly to side-chain/main-chain clashes. Above this valueclashes may partially account for conformational relaxation, suchas variation from library side-chain rotamers or small main-chainmovements.
An estimate of maximal SA (SAmax) is obtained for each ion-izable group. For each neighboring side chain, the (allowed) rota-mer that provides the most SA for the charged atom of the ioniz-able group under study is used to mark SA information on aspherical polar grid around that atom. This grid is scanned aftercomplete neighbor analysis for the overall SAmax. MaximalSA estimates for an ionizable group with more than one chargedatom in our model are averaged. The likelihood that small con-formational adjustment could lead to substantial solvent expo-sure of each group is assessed with SAmax (Fig. 3). If SAmax/SAfixed-atoms � 0.75, where SAfixed-atoms is calculated in the con-text of fixed atoms only (i.e., excluding atoms that move in therotamer library), then that group is allowed to sample DH (water-dominated) interactions.
Combined FD/DH calculations
In the FD/DH combination, each site m in the equation for mi-crostate energy Gs is sampled as protonated or unprotonated ineach of FD and DH (if allowed) schemes, with the componentenergies of Gs assigned appropriately. Thus, a standard singleconformer pKa calculation with 2M states for M groups, ap-proaches 4M states in FD/DH, because most sites are relativelyflexible and have access to the DH scheme. Note that for any groupm sampled as DH, all mn (and nm) interactions are DH whethergroup n is sampled as FD or DH, that is, the water-dominatedscheme persists for any interaction involving a group sampled in apresumed water-rich environment. Ionizable group array energiesand pKas in FD/DH calculations are derived as for FD or DHalone. No attempt is made to assign a density of conformationalstates, other than DH access or not, so that electrostatic energydetermines the relative weightings of FD and DH. A moderateenergy (favorable or unfavorable) from DH interaction would out-weigh a highly unfavorable FD interaction, while a large and fa-vorable FD term will dominate DH.
Acknowledgments
The European Union is thanked for funding during the course ofthis work.
The publication costs of this article were defrayed in part by
payment of page charges. This article must therefore be herebymarked “advertisement” in accordance with 18 USC section 1734solely to indicate this fact.
References
Alberty, R.A. 1983. Physical chemistry, 6th ed. John Wiley & Sons, New York.Alexov, E. 2003. Role of the protein side-chain fluctuations on the strength of
pair-wise electrostatic interactions: Comparing experimental with computedpKas. Proteins 50: 94–103.
Alexov, E. and Gunner, M.R. 1997. Incorporating protein conformational flex-ibility into the calculation of pH-dependent protein properties. Biophys. J.74: 2075–2093.
Anderson, D.E., Becktel, W.J., and Dahlquist, F.W. 1990. pH-induced denatur-ation of proteins: A single salt bridge contributes 3–5 kcal/mol to the freeenergy of folding of T4 lysozyme. Biochemistry 29: 2403–2408.
Antosiewicz, J., McCammon, J.A., and Gilson, M.K. 1994. Prediction of pH-dependent properties in proteins. J. Mol. Biol. 238: 415–436.
———. 1996. The determinants of pKas in proteins. Biochemistry 35: 7819–7833.
Barbas III, C.F., Heine, A., Zhong, G., Hoffmann, T., Gramatikova, S.,Björnestedt, R., List, B., Anderson, J., Stura, E.A., Wilson, I.A., et al. 1997.Immune versus natural selection: Antibody aldolases with enzymic rates butbroader scope. Science 278: 2085–2092.
Bashford, D. and Karplus, M. 1990. pKa’s of ionizable groups in proteins:Atomic detail from a continuum electrostatic model. Biochemistry 29:10219–10225.
Berman, H.M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T.N., Weissig, H.,Shindyalov, I.N., and Bourne, P.E. 2000. The Protein Data Bank. NucleicAcids Res. 28: 235–242.
Beroza, P., Fredkin, D.R., Okamura, M.Y., and Feher, G. 1991. Protonation ofinteracting residues in a protein by a Monte Carlo method: Application tolysozyme and the photosynthetic reaction center of Rhodobacter sphaeroi-des. Proc. Natl. Acad. Sci. 88: 5804–5808.
Björnestedt, R., Stenberg, G., Widersten, M., Board, P.G., Sinning, I., Jones,T.A., and Mannervik, B. 1995. Functional significance of arginine 15 in theactive site of human class � glutathione transferase A1–1. J. Mol. Biol. 247:765–773.
Blom, N. and Sygusch, J. 1997. Product binding and role of the C-terminalregion in class I D-fructose 1,6-bisphosphate aldolase. Nat. Struct. Biol. 4:36–39.
Cameron, A.D., Sinning, I., L’Hermite, G., Olin, B., Board, P.G., Mannervik,B., and Jones, T.A. 1995. Structural analysis of human �-class glutathionetransferase A1–1 in the apo-form and in complexes with ethacrynic acid andits glutathione conjugate. Structure 3: 717–727.
Chivers, P.T., Prehoda, K.E., Volkman, B.F., Kim, B.M., Markley, J.L., andRaines, R.T. 1997. Microscopic pKa values of Escherichia coli thioredoxin.Biochemistry 36: 14985–14991.
Cole, C. and Warwicker, J. 2002. Side-chain conformational entropy at protein–protein interfaces. Protein Sci. 11: 2860–2870.
Demchuk, E. and Wade, R.C. 1996. Improving the continuum dielectric ap-proach to calculating pKas of ionizable groups in proteins. J. Phys. Chem.100: 17373–17387.
Dong, F. and Zhou, H.X. 2002. Electrostatic contributions to T4 lysozymestability: Solvent-exposed charges versus semi-buried salt bridges. Biophys.J. 83: 1341–1347.
Edgcomb, S.P. and Murphy, K.P. 2002. Variability in the pKa of histidineside-chains correlates with burial within proteins. Proteins 49: 1–6.
Elcock, A.H. 2001. Prediction of functionally important residues based solelyon the computed energetics of protein structure. J. Mol. Biol. 312: 885–896.
Fersht, A.R. and Renard, M. 1974. pH-dependence of chymotrypsin catalysis.Appendix: Substrate binding to dimeric � chymotrypsin studied by x-raydiffraction and equilibrium method. Biochemistry 13: 1416–1426.
Fink, A.L., Calciano, L.J., Goto, Y., Kurotsu, T., and Palleros, D.R. 1994.Classification of acid denaturation of proteins: Intermediates and unfoldedstates. Biochemistry 33: 12505–12511.
Fitch, C.A., Karp, D.A., Lee, K.K., Stites, W.E., Lattman, E.E., and Garcia-Moreno, E.B. 2002. Experimental pKa values of buried residues: Analysiswith continuum methods and role of water penetration. Biophys. J. 82:3289–3304.
Georgescu, R.E., Alexov, E., and Gunner, M.R. 2002. Combining conforma-tional flexibility and continuum electrostatics for calculating pKas in pro-teins. Biophys. J. 83: 1731–1748.
Warwicker
2804 Protein Science, vol. 13

Gilson, M.K. and Honig, B.H. 1986. The dielectric constant of a folded protein.Biopolymers 25: 2097–2119.
Gorfe, A.A., Ferrara, P., Caflisch, A., Marti, D.N., Bosshard, H.R., and Jelesa-rov, I. 2002. Calculation of protein ionization equilibria with conformationalsampling: pKas of a model leucine zipper, GCN4 and barnase. Proteins 46:41–60.
Grauschopf, U., Winther, J.R., Korber, P., Zander, T., Dallinger, P., andBardwell, J.C. 1995. Why is DsbA such an oxidizing catalyst? Cell 83:947–955.
Guddat, L.W., Bardwell, J.C., and Martin, J.L. 1998. Crystal structures of re-duced and oxidized DsbA: Investigation of domain motion and thiolatestabilization. Structure 6: 757–767.
Hendsch, Z.S. and Tidor, B. 1994. Do salt-bridges stabilize proteins? A con-tinuum electrostatic analysis. Protein Sci. 3: 211–226.
Hollecker, M. and Creighton, T.E. 1982. Effect on protein stability of reversingthe charge on amino groups. Biochim. Biophys. Acta 701: 395–404.
Honig, B. and Nicholls, A. 1995. Classical electrostatics in biology and chem-istry. Science 268: 1144–1149.
Ikeuchi, Y., Katerelos, N.A., and Goodenough, P.W. 1998. The enhancing of acysteine protease activity at acidic pH by protein engineering, the role ofglutamic 50 in the enzyme mechanism of caricain. FEBS Lett. 437: 91–96.
Irving, R.J., Nelander, L., and Wadso, I. 1964. Thermodynamics of the ioniza-tion of some thiols in aqueous solution. Acta. Chem. Scand. 18: 769–787.
Izatt, R.M. and Christensen, J.J. 1976. Heats of proton ionization, pK, andrelated thermodynamic quantities. In Handbook of biochemistry and mo-lecular biology: Physical and chemical data (ed. G.D. Fasman), 3rd ed.,Vol. I, pp. 151–269. CRC Press, Cleveland, OH.
Joshi, M.D., Hedberg, H., and McIntosh, L.P. 1997. Complete measurement ofthe pKa values of the carboxyl and imidazole groups in Bacillus circulansxylanase. Protein Sci. 6: 2667–2670.
Jung, H.-I., Cooper, A., and Perham, R.N. 2002. Identification of key aminoacid residues in the assembly of enzymes into the pyruvate dehydrogenasecomplex of Bacillus stearothermophilus: A kinetic and thermodynamicanalysis. Biochemistry 41: 10446–10453.
Klapper, I., Hagstrom, R., Fine, R., Sharp. K., and Honig, B. 1986. Focusing ofelectric fields in the active site of Cu-Zn superoxide dismutase: Effects ofionic strength and amino-acid modification. Proteins 1: 47–59.
Koehl, P. and Delarue, M. 1994. Application of a self-consistent mean fieldtheory to predict side-chains conformation and estimate their conforma-tional entropy. J. Mol. Biol. 239: 249–275.
Koumanov, A., Karshikoff, A., Friis, E.P., and Borchert, T.V. 2001. Confor-mational averaging in pK calculations: Improvement and limitations in pre-diction of ionization properties of proteins. J. Phys. Chem. B 105: 9339–9344.
Koumanov, A., Ruterjans, H., and Karshikoff, A. 2002. Continuum electrostaticanalysis of irregular ionization and proton allocation in proteins. Proteins46: 85–96.
Koumanov, A., Benach, J., Atrian, S., Gonzalez-Duarte, R., Karshikoff, A., andLadenstein, R. 2003. The catalytic mechanism of Drosophila alcohol de-hydrogenase: Evidence for a proton relay modulated by the coupled ion-ization of the active site Lysine/Tyrosine pair and a NAD+ ribose OHswitch. Proteins 51: 289–298.
Kuramitsu, S. and Hamaguchi, K. 1980. Analysis of the acid-base titration curveof hen lysozyme. J. Biochem. 87: 1215–1219.
Lehoux, I.E. and Mitra, B. 1999. (S)-Mandelate dehydrogenase from Pseudo-monas putida: Mechanistic studies with alternate substrates and pH andkinetic isotope effects. Biochemistry 38: 5836–5848.
Liu, Y., Thoden, J.B., Kim, J., Berger, E., Gulick, A.M., Ruzicka, F.J., Holden,H.M., and Frey, P.A. 1997. Mechanistic roles of tyrosine 149 and serine 124in UDP-galactose 4-epimerase from Escherichia coli. Biochemistry 36:10675–10684.
Mehler, E.L. and Guarnieri, F. 1999. A self-consistent, microenvironmentmodulated screened coulomb potential approximation to calculate pH-de-pendent electrostatic effects in proteins. Biophys. J. 75: 3–22.
Morillas, M., Goble, M.L., and Virden, R. 1999. The kinetics of acylation anddeacylation of penicillin acylase from Escherichia coli ATCC11105: Evi-dence for lowered pKa values of groups near the catalytic centre. Biochem.J. 338: 235–239.
Morris, A.J. and Tolan, D.R. 1994. Lysine-146 of rabbit muscle aldolase isessential for cleavage and condensation of the C3–C4 bond of fructose1,6-bis(phosphate). Biochemistry 33: 12291–12297.
Nielsen, J.E. and McCammon, J.A. 2003. Calculating pKa values in enzymeactive sites. Protein Sci. 12: 1894–1901.
Nielsen, J.E., Andersen, K.V., Honig, B., Hooft, R.W.W., Klebe, G., Vriend, G.,and Wade, R.C. 1999. Improving macromolecular electrostatics calcula-tions. Protein Eng. 12: 657–662.
Noble, M.A., Gul, S., Verma, C.S., and Brocklehurst, K. 2000. Ionization char-acteristics and chemical influences of aspartic acid residue 158 of papainand caricain determined by structure-related kinetic and computational tech-niques: Multiple electrostatic modulators of active-centre chemistry. Bio-chem. J. 351:723–733.
Ondrechen, M.J., Clifton, J.G., and Ringe, D. 2001. THEMATICS: A simplecomputational predictor of enzyme function from structure. Proc. Natl.Acad. Sci. 98: 12473–12478.
Schaefer, M., Sommer, M., and Karplus, M. 1997. pH-dependence of proteinstability: Absolute electrostatic free energy differences between conforma-tions. J. Phys. Chem. B 101: 1663–1683.
Schutz, C.N. and Warshel, A. 2001. What are the dielectric “constants” ofproteins and how to validate electrostatic models? Proteins 44: 400–417.
Simonson, T. 2001. Macromolecular electrostatics: Continuum models and theirgrowing pains. Curr. Opin. Struct. Biol. 11: 243–252.
Simonson, T. and Perahia, D. 1995. Internal and interfacial dielectric propertiesof cytochrome c from molecular dynamics in aqueous solution. Proc. Natl.Acad. Sci. 92: 1082–1086.
Sundd, M., Iverson, N., Ibarra-Molero, B., Sanchez-Ruiz, J.M., and Robertson,A.D. 2002. Electrostatic interactions in ubiquitin: Stabilization of carbox-ylates by lysine amino groups. Biochemistry 41: 7586–7596.
Thoden, J.B., Frey, P.A., and Holden, H.M. 1996. Crystal structures of theoxidized and reduced forms of UDP-galactose 4-epimerase isolated fromEscherichia coli. Biochemistry 35: 2557–2566.
Thunnissen, M.M.G.M., Eiso, A.B., Kalk, K.H., Drenth, J., Dijkstra, B.W.,Kuipers, O.P., Dijkman, R., de Haas, G.H., and Verheij, H.M. 1990. X-raystructure of phospholipase A2 complexed with a substrate-derived inhibitor.Nature 347: 689–691.
Tuffery, P., Etchebest, C., and Hazout, S. 1997. Prediction of protein side chainconformations: A study on the influence of backbone accuracy on confor-mation stability in the rotamer space. Protein Eng. 10: 361–372.
van Gunsteren, W.F. and Berendsen, H.J.C. 1987. GROMOS manual. Univer-sity of Groningen, The Netherlands.
van Vlijmen, H.W., Schaefer, M., and Karplus, M. 1998. Improving the accu-racy of protein pKa calculations: Conformational averaging versus the av-erage structure. Proteins 33: 145–158.
Verheij, H.M., Volwerk, J.J., Jansen, E.H.J.M., Puyk, W.C., Dijkstra, B.W.,Drenth, J., and de Haas, G.H. 1980. Methylation of histidine 48 in pancre-atic phospholipase A2. Role of histidine and calcium ion in the catalyticmechanism. Biochemistry 19: 743–750.
Wada, A. and Nakamura, H. 1981. Nature of the charge distribution in proteins.Nature 293: 757–758.
Warshel, A. 1981. Calculations of enzymatic reactions: Calculations of pKa,proton transfer reactions, and general acid catalysis reactions in enzymes.Biochemistry 20: 3167–3177.
———. 2003. Computer simulations of enzyme catalysis: Methods, progress,and insights. Annu. Rev. Biophys. Biomol. Struct. 32: 425–443.
Warshel, A. and Papazyan, A. 1998. Electrostatic effects in macromolecules:Fundamental concepts and practical modelling. Curr. Opin. Struct. Biol. 8:211–217.
Warwicker, J. 1986. Continuum dielectric modelling of the protein–solventsystem, and calculation of the long-range electrostatic field of the enzymephosphoglycerate mutase. J. Theor. Biol. 121: 199–210.
———. 1997. Improving pKa calculations with consideration of hydration en-tropy. Protein Eng. 10: 809–814.
———. 1998. Modeling charge interactions and redox properties in DsbA. J.Biol. Chem. 273: 2502–2504.
———. 1999. Simplified methods for pKa and acid pH-dependent stabilityestimation in proteins: Removing dielectric and counterion boundaries. Pro-tein Sci. 8: 418–425.
Warwicker, J. and Watson, H.C. 1982. Calculation of the electric potential in theactive site cleft due to �-helix dipoles. J. Mol. Biol. 157: 671–679.
You, T.J. and Bashford, D. 1995. Conformation and hydrogen ion titration ofproteins: A continuum model with conformational flexibility. Biophys. J.69:1721–1733.
Zhou, H.X. and Vijayakumar, M. 1997. Modeling of protein conformationalfluctuations in pKa predictions. J. Mol. Biol. 267: 1002–1011.
Side-chain packing and ionizable group energetics
www.proteinscience.org 2805