idiopathic inflammatory myositis
TRANSCRIPT

Idiopathic Inflammatory MyositisDR. AMAR MBBS MD

IntroductionThe inflammatory myopathies represent the largest group of acquired and potentially treatable causes of skeletal muscle weakness.
They are classified into three major groups:
Polymyositis (PM)
Dermatomyositis(DM), and
Inclusion body myositis (IBM)

If the inflammatory changes are restricted clinically to the striated muscles, the disease is called polymyositis (PM)
If in addition, the skin is involved, it is called dermatomyositis (DM),
Although the two diseases are now understood to be immunopathologically distinct.

HistoryBoth diseases have been known since the nineteenth century.
Polymyositis was first described by Wagner in 1863 and 1887.
DM was established as an entity by Unverricht in a series of articles written from 1887 to 1891.

Heinrich Unverricht"...it seems to me that the skin appearance plays such an important role in the disease picture that the designation Polymyositis is not completely accurate. In our case, the partnership of the skin and muscle disease allows us to use the elocution Dermatomyositis...“ (translation).[

EpidemiologyThe prevalence of inflammatory myopathies is estimated at 1 in 1,00,000.
PM as a stand-alone entity is a rare disease.
DM affects both children and adults and women more often than men
IBM is three times more frequent in men than in women, more common in whites than blacks, and is more likely to affect persons age > 50.

There is no internationally accepted classification system for inflammatory myopathies.
For discussion purpose, IM’s can be classified as
i. Polymyositis
ii. Dermatomyositis
iii. Inclusion body myositis
iv. Autoimmune necrotizing myopathies
v. Myositis associated with collagen vascular disorder
vi. Myositis associated with malignancy

Polymyositis (PM)Onset – Insidious.
Progression – Usually over a period of several weeks or months.
Age at onset – 30-60 years of age with a small peak at 15 years of age.
Sex – Female predominate in all age groups.

Inclusion body myositis (IBM)In patients ≥50 years of age, IBM is the most common of the inflammatory myopathies.
It is often misdiagnosed as PM and is suspected only later when a patient with presumed PM does not respond to therapy.

Aetiology & PathogenesisAn autoimmune etiology of the inflammatory myopathies is indirectly supported by
Association with other autoimmune or connective tissue diseases
Presence of various autoantibodies
Association with specific major histocompatibility complex (MHC) genes
Demonstration of T cell–mediated myocytotoxicity or complement-mediated microangiopathy
Response to immunotherapy.

Autoantibodies and ImmunogeneticsVarious autoantibodies against nuclear antigens (antinuclear antibodies) and cytoplasmic antigens are found in up to 30% of patients with inflammatory myopathies.
The antibodies to cytoplasmic antigens are directed against ribonucleoproteins involved in
i. protein synthesis (antisynthetases) (or)
ii. translational transport (anti-signal-recognition particles).

/
Anti Jo-1
The antibody directed against the histidyl-transfer RNA synthetase, called anti-Jo-1, accounts for 75% of all the
antisynthetases.
The clinical disorders associated with these
antibodies usually combine myositis with
(1) interstitial lung disease (80%)
(2) arthritis,
(3) Raynaud syndrome, and
(4) thickening of the skin of the hands ("mechanic’s
hands").

Immunopathologic MechanismsDermatomyositis – Humoral Immune mechanisms.
Polymyositis & IB Myositis – T-Cell mediated immune mechanisms.

PM & IBM - PathogenesisCD8 T cells, along with macrophages, initially surround and eventually invade and destroy healthy, nonnecrotic muscle fibers that aberrantly express class I MHC molecules.
The CD8/MHC-I complex is characteristic of PM and IBM and its detection can aid in confirming the histologic diagnosis of PM.
The cytotoxic CD8 T cells contain perforin and granzyme granules directed toward the surface of the muscle fibers and capable of inducing myonecrosis.

Non-immune factors in IBMIn addition to the autoimmune component, there is also a degenerative process.

Association with viral infectionsViruses including coxsackie viruses, influenza, paramyxoviruses, mumps, cytomegalovirus, and Epstein-Barr virus, have been indirectly associated with myositis but studies have repeatedly failed to confirm this association.

Role of RetrovirusesIndividuals infected with HIV or with human T cell lymphotropic virus 1 (HTLV-1) develop PM or IBM.
The inflammatory myopathy may occur as the initial manifestation of a retroviral infection, or myositis may develop later in the disease course.
Retroviral antigens have been detected only in occasional endomysial macrophages and not within the muscle fibers themselves, suggesting that persistent infection and viral replication within the muscle does not occur.

AZT induced myopathyRetroviral myopathy should be distinguished from a toxic myopathy related to long-term therapy with AZT, characterized by fatigue, myalgia, mild muscle weakness, and mild elevation of creatine kinase (CK).
AZT-induced myopathy, which generally improves when the drug is discontinued, is a mitochondrial disorder characterized histologically by “ragged-red” fibers.

Clinical FeaturesUsual mode of onset – Painless weakness of proximal limb muscles, especially of hips and thighs and to a lesser extent the shoulder, girdle and neck muscles.
The posterior and anterior neck muscles (the head may loll) may be involved.
The pharyngeal, striated esophageal, and laryngeal muscles (dysphagia and dysphonia) may be involved as well.

Other musclesIn restricted forms of the disease, only the neck and the paraspinal muscles may be implicated (camptocormia).
Distal muscles (forearm, hand, leg & foot) – Spared in 75% of cases.
Facial, Tongue and Jaw muscles – rarely affected.
Respiratory muscles slightly weakened but rarely to the extent that causes dyspnea.

Muscles are non-tender, reflexes are usually reduced.
As the weeks and months pass, the weakness and muscle atrophy progress unless treatment is initiated.
Without physical therapy, fibrous contracture of muscles eventually develops.
Some elderly individuals with a particularly chronic form of the disease may present with severe atrophy and fibrosis of muscles.

DermatomyositisMuscle weakness similar to that PM but the denominative feature is the rash.
Most often, the skin changes precede the muscle syndrome and take the form of a localized or diffuse erythema, maculopapular eruption, scaling eczematoiddermatitis, or exfoliative dermatitis.
Sometimes skin and muscle changes evolve together over a period of 3 weeks or even less.
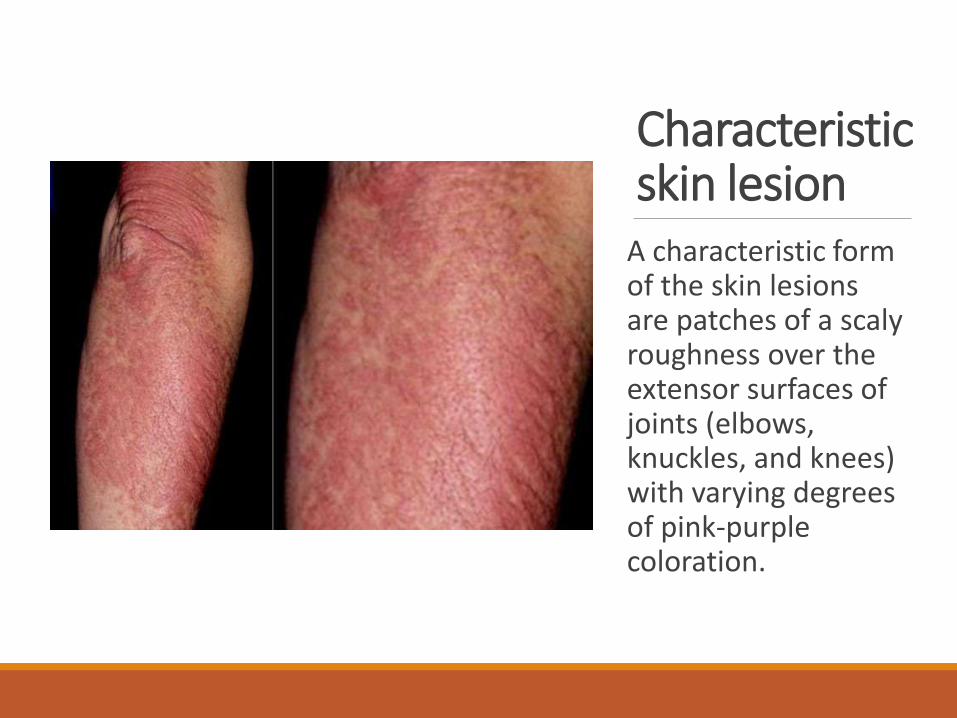
Characteristic skin lesion
A characteristic form of the skin lesions are patches of a scaly roughness over the extensor surfaces of joints (elbows, knuckles, and knees) with varying degrees of pink-purple coloration.
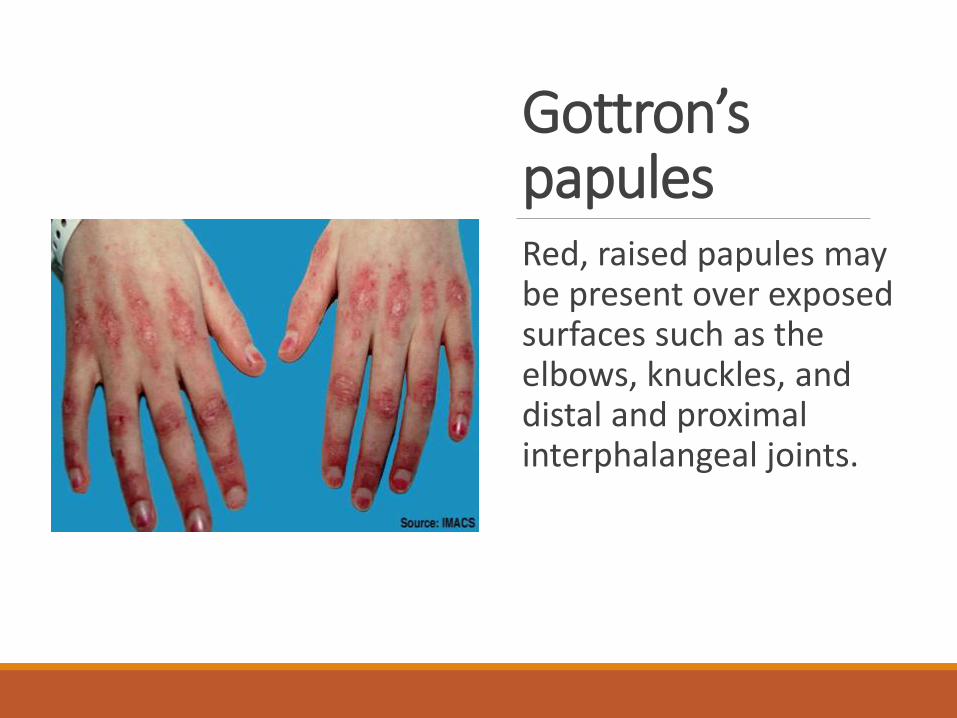
Gottron’spapulesRed, raised papules may be present over exposed surfaces such as the elbows, knuckles, and distal and proximal interphalangeal joints.
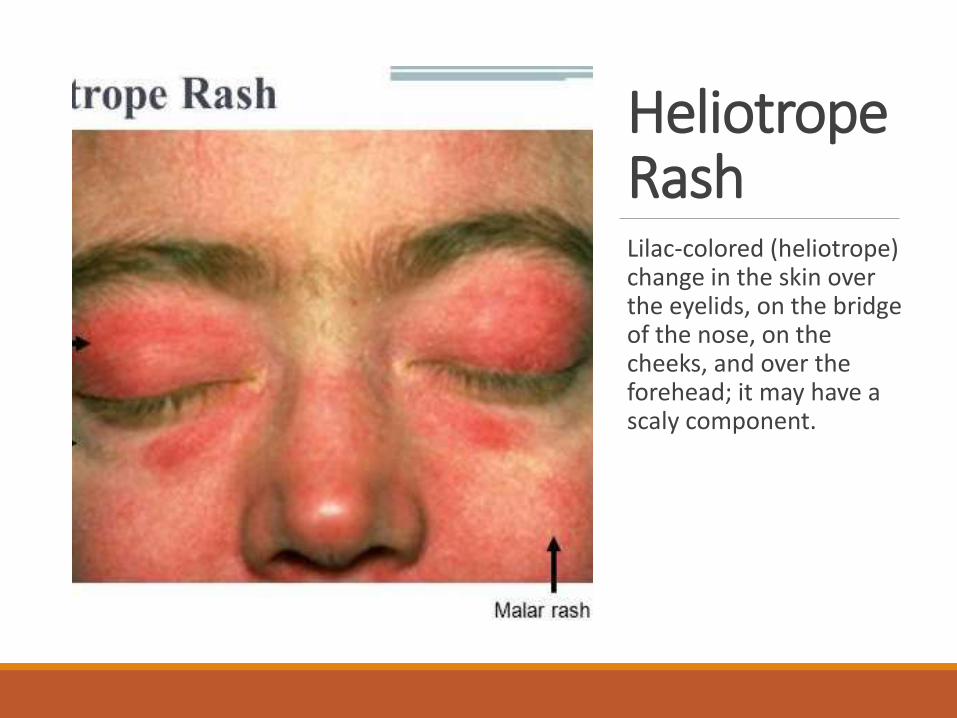
Heliotrope RashLilac-colored (heliotrope) change in the skin over the eyelids, on the bridge of the nose, on the cheeks, and over the forehead; it may have a scaly component.
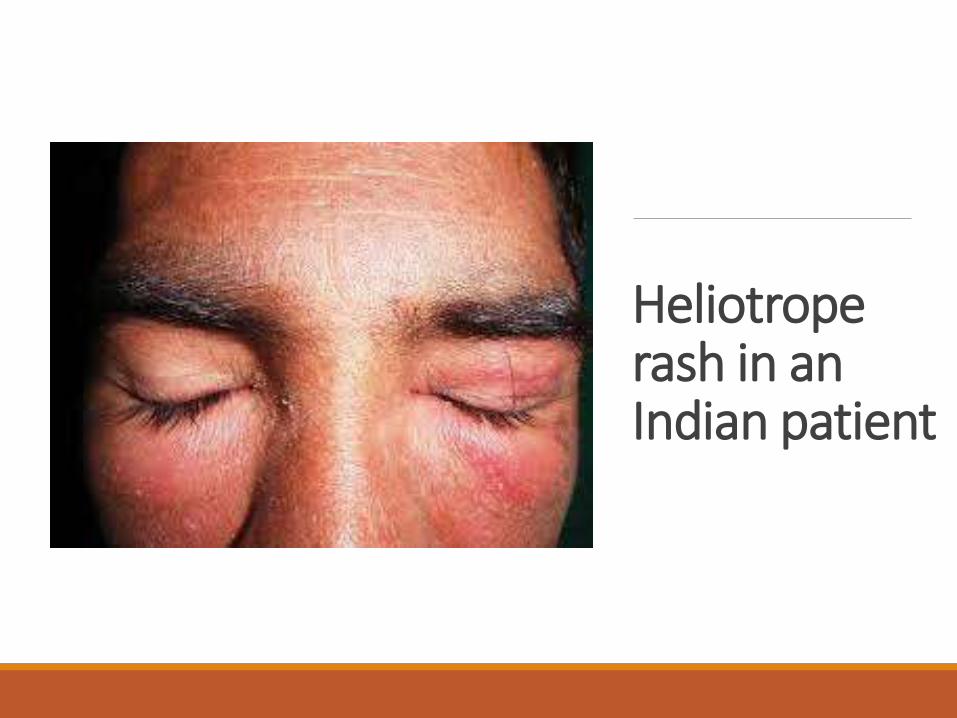
Heliotrope rash in an Indian patient
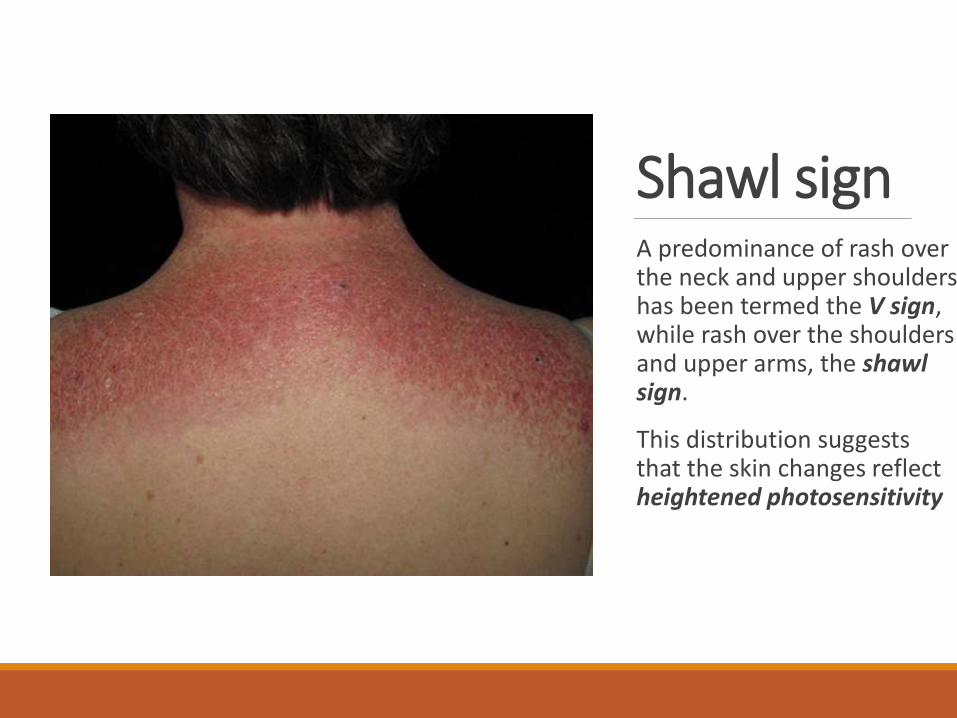
Shawl signA predominance of rash over the neck and upper shoulders has been termed the V sign, while rash over the shoulders and upper arms, the shawl sign.
This distribution suggests that the skin changes reflect heightened photosensitivity
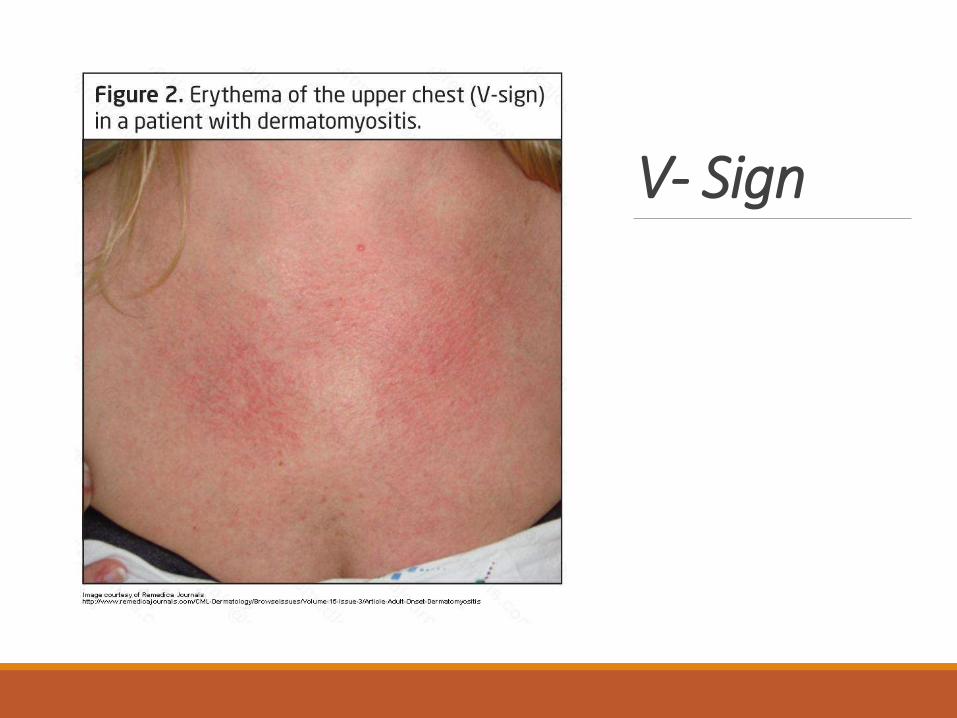
V- Sign
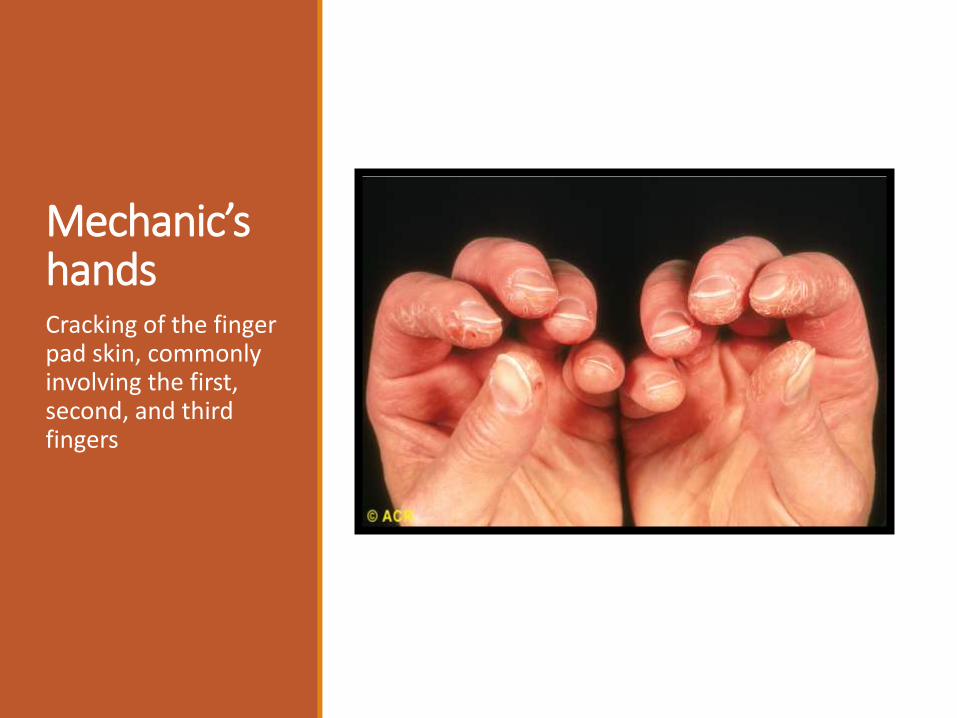
Mechanic’s handsCracking of the finger pad skin, commonly involving the first, second, and third fingers
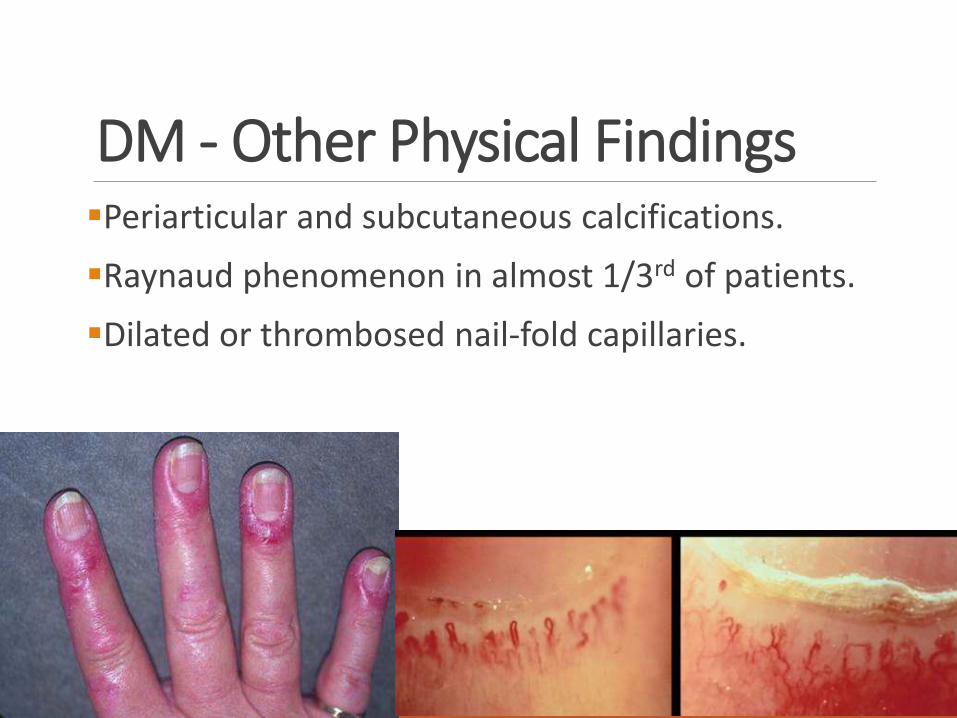
DM - Other Physical FindingsPeriarticular and subcutaneous calcifications.
Raynaud phenomenon in almost 1/3rd of patients.
Dilated or thrombosed nail-fold capillaries.

Extramuscular Manifestations of DM/PMSystemic symptoms, such as fever, malaise, weight loss, arthralgia, and Raynaud’s phenomenon, especially when inflammatory myopathy is associated with a connective tissue disorder.
Joint contractures, mostly in DM and especially in children. (related to skin thickening)
Dysphagia and gastrointestinal symptoms, due to involvement of oropharyngeal striated muscles and upper esophagus, especially in DM and IBM.

Cardiac disturbances, including atrioventricular conduction defects, tachyarrhythmias, dilated cardiomyopathy, a low ejection fraction, and congestive heart failure, which may rarely occur either from the disease itself or from hypertension associated with longterm use of glucocorticoids.

Pulmonary dysfunction, due to weakness of the thoracic muscles, interstitial lung disease, or drug-induced pneumonitis (e.g., from methotrexate), which may cause dyspnea, nonproductive cough, and aspiration pneumonia.
Interstitial lung disease may precede myopathy or occur early in the disease and develops in up to 10% of patients with PM or DM, most of whom have antibodies to t-RNA Synthetases.

Subcutaneous calcifications, in DM, sometimes extruding on the skin and causing ulcerations and infections.
Arthralgias, synovitis, or deforming arthropathy with subluxation in the interphalangeal joints, which can occur in some patients with DM and PM who have Jo-1 antibodies

Malignancy associated with DM/PMMany studies have been done to prove the association between Malignancies and Myostitis especially PM & DM.
The association ranging between 29-66% according to various studies.
Commonest malignancies are Lung & Colon Cancer in males and Breast & Ovarian Cancer in females.

Systemic autoimmune diseases with PM/DMIn both PM and DM, the inflammatory changes are often not confined to muscle but are associated with systemic autoimmune diseases such as rheumatoid arthritis, scleroderma, lupus erythematosus, or combinations thereof (mixed connective tissue disease).

Overlap syndromeIn overlap syndromes that incorporate autoimmune disease and myositis, there is usually greater muscular weakness and atrophy than can be accounted for by the muscle changes alone.
Arthritis or periarticular inflammation may limit motion because of pain, result in disuse atrophy, and also at times cause a vasculitic polyneuropathy, the interpretation of diminished strength in these autoimmune diseases is not simple.

Malaise, aches, and pains points to a systemic disease.
Sometimes the diagnosis of myositis must depend on muscle biopsy, EMG findings, and measurements of muscle enzymes in the serum.

Inclusion Body MyositisIn patients ≥50 years of age, IBM is the most common of the inflammatory myopathies.
Weakness and atrophy of the distal muscles, especially foot extensors and deep finger flexors, occur in almost all cases of IBM and may be a clue to early diagnosis.
Most common presentation - weakness in the small muscles of the hands, especially finger flexors, and complain of inability to hold objects such as golf clubs or perform tasks such as turning keys or tying knots.
Recurrent Falls – Early Quadriceps weakness.

IBM – Continued Occasionally, the weakness and accompanying atrophy can be asymmetric and selectively involve the quadriceps, iliopsoas, triceps, biceps, and finger flexors, resembling a lower motor neuron disease.
Dysphagia is common (60% of patients).
Sensory examination is normal.
Disease progression is slow but steady.

In at least 20% of cases, IBM is associated with systemic autoimmune or connective tissue diseases.
Familial aggregation of typical IBM may occur; such cases have been designated as familial inflammatory IBM.

Lab Diagnosis Creatine Kinase
Tends to be higher in Polymyositis than in Dermatomyositis because of the widespread single fiber necrosis in Polymyositis.
In DM, if there are infarcts, CK will be raised moderately as well.
Usually elevated up to 5- 50 times ULN.
Elevated up to 10 times in IBM.

RA and ANA are positive in fewer than half of cases.
Other antibodies can be found on occasion that are directed against constituents of a nucleolar protein complex (PM-Scl) and ribonucleoproteins (Ro /SS-A and La/SS-B).

20 percent of patients with PM and DM have antibodies against various cellular components of muscle, in particular, antibodies directed against cytoplasmic transfer ribonucleic acid (tRNA) synthetases (anti-Jol), or against the tRNA itself.
These are fairly specific to PM and, less frequently to DM, they are found when the myositis is coupled with an expanded illness that involves other organs or connective tissues.

Myoglobinuria can be detected in majority of patients with any form of myositis, particularly a necrotizing form.

EMG A typical Myopathic pattern is found on EMG - many abnormally brief action potentials of low voltage in addition to numerous fibrillation potentials, trains of positive sharp waves, occasional polyphasic units, and myotonic activity-all but the brief potentials possibly reflecting irritability of the muscle membranes.
The EMG is also helpful in choosing a muscle for biopsy sampling.
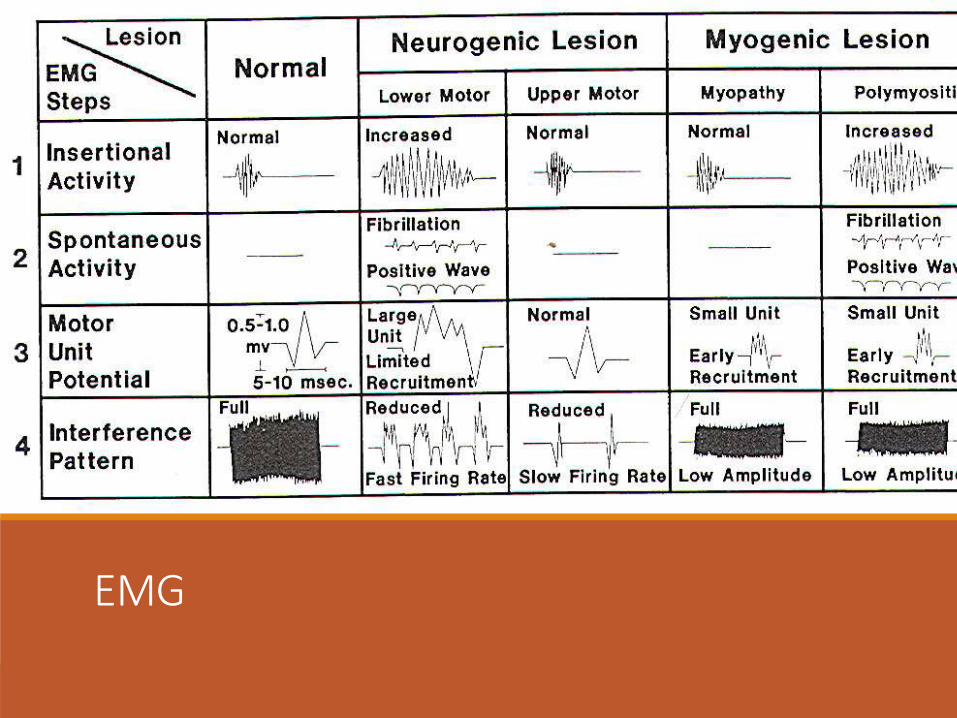
EMG

MRIMRI can refine the distribution of all lesions and aid in targeting the muscle biopsy.
May show abnormalities in Tl, T2 and STIR signal intensity define regions of increased water content and inflammation and spectroscopic studies demonstrate regional deficits in energy production.
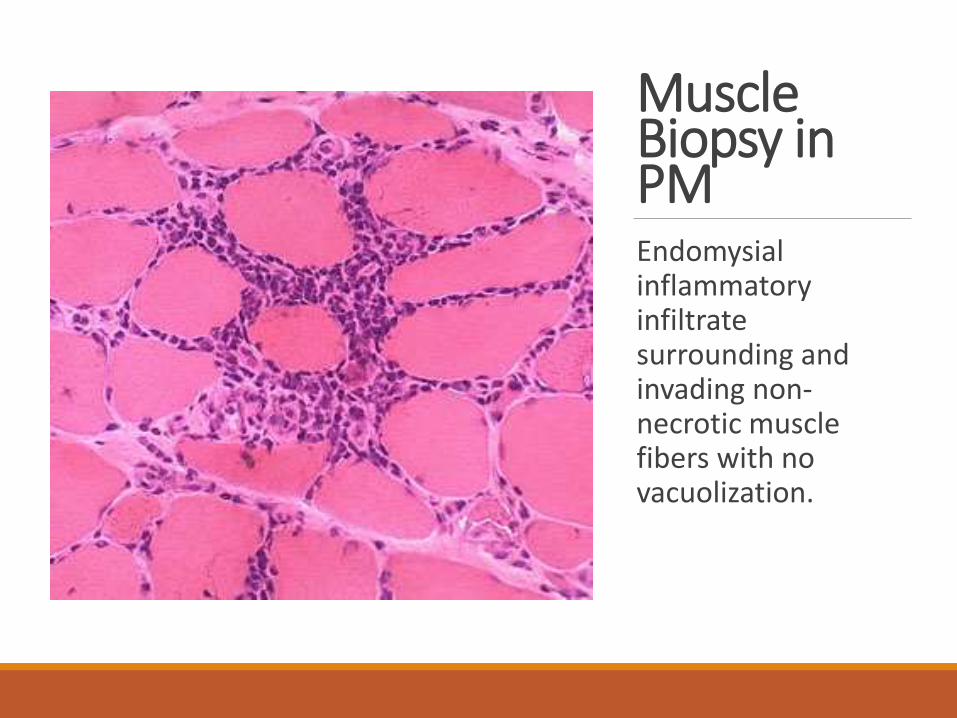
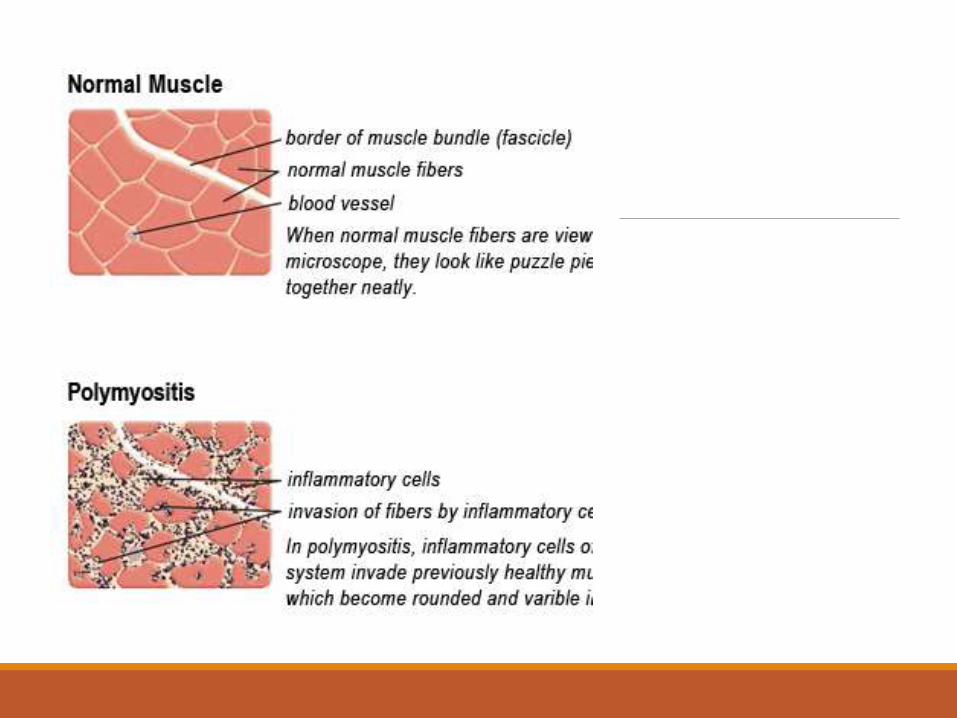
Muscle Biopsy in PMEndomysialinflammatory infiltrate surrounding and invading non-necrotic muscle fibers with no vacuolization.
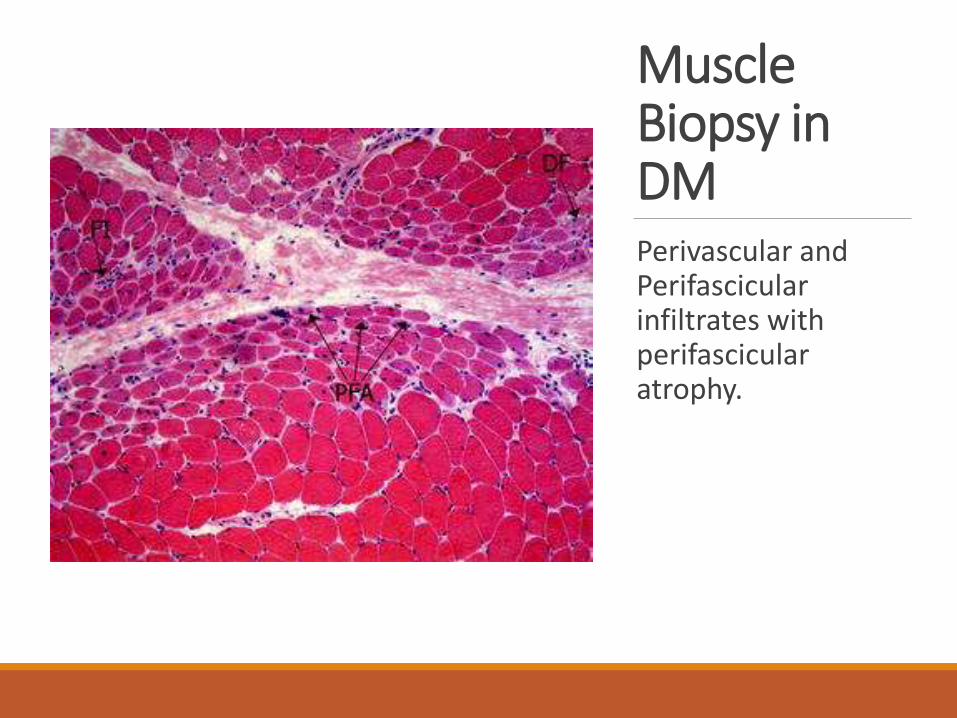
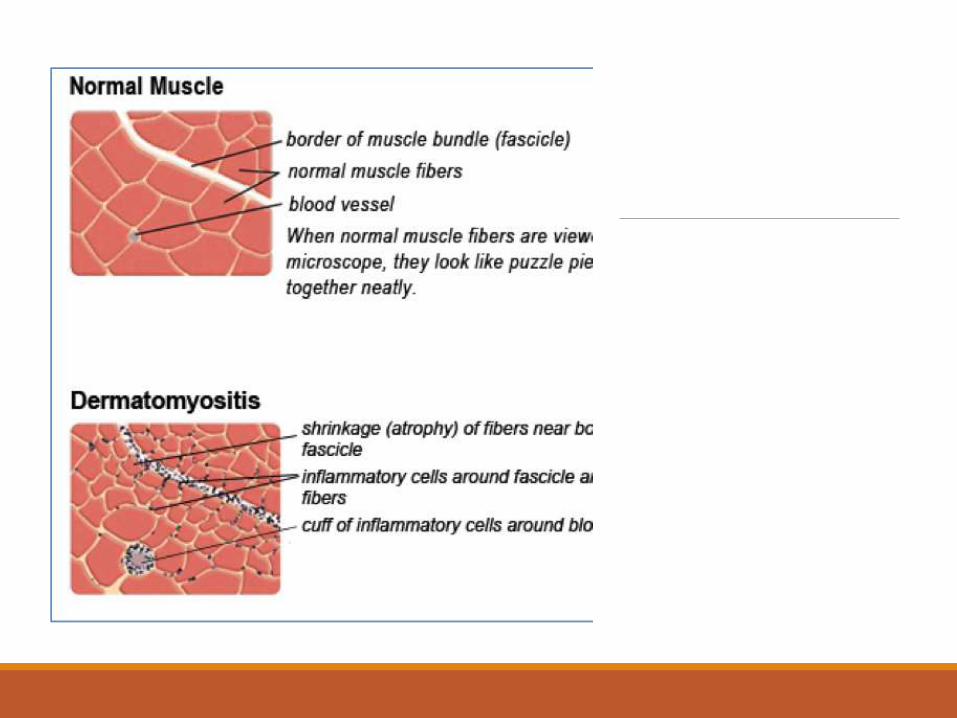
Muscle Biopsy in DMPerivascular and Perifascicular infiltrates with perifascicular atrophy.
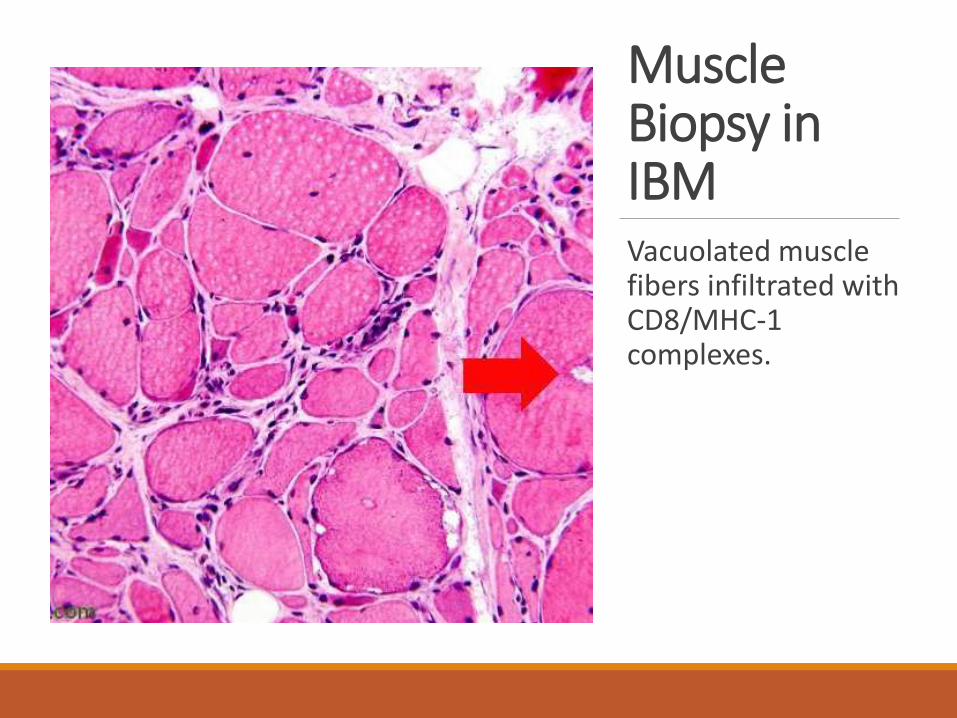
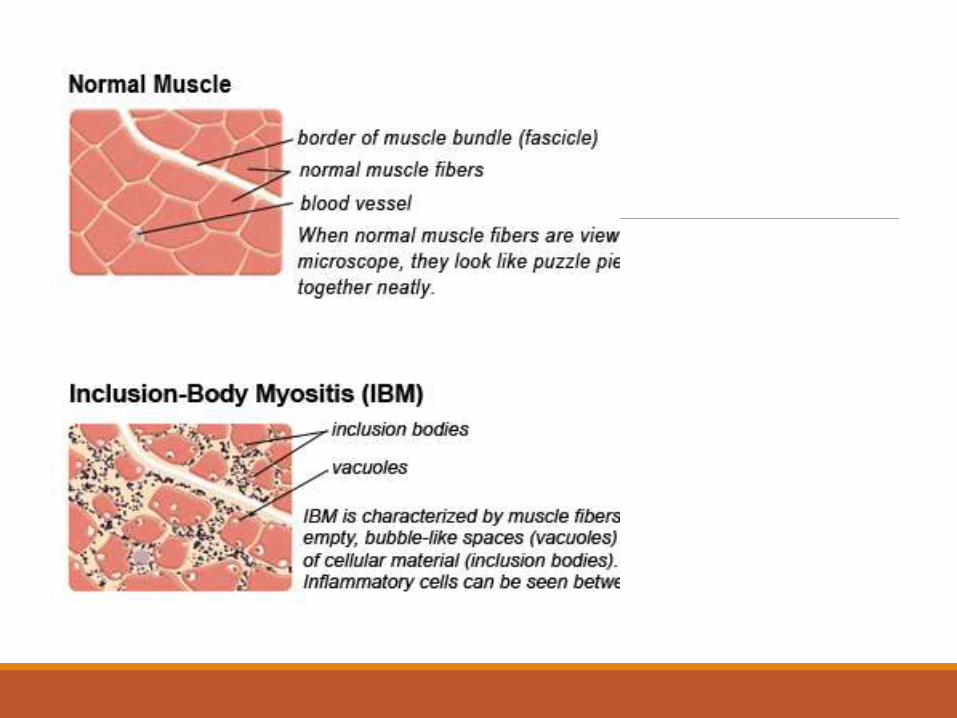
Muscle Biopsy in IBMVacuolated muscle fibers infiltrated with CD8/MHC-1 complexes.

Differential DiagnosisSubacute - Denervating conditions such as SMA or ALS.
Acute – Acute neuropathy such as GBS, Transverse myelitis, or a neurotropic viral infection such as poliomyelitis or west nile virus.
Myofascitis
Necrotizing autoimmune myositis.
Drug induced myopathies – D-Penicillamine, procainamide and statins. AZT causes a mitochondrial myopathy.

ManagementThe goal of therapy is to improve muscle strength, thereby improving function in activities of daily living, and ameliorate the extramuscularmanifestations (rash, dysphagia, dyspnea, fever).
The level of CK and degree of leukocyte infiltration often reduces on treatment with glucocorticoids.
But CK levels should never be the target of therapy.

Glucocorticoids Oral prednisone is the initial treatment of choice, the effectiveness and side effects of this therapy determine the future need for stronger immunosuppressive drugs.
Initial dose – 1mg/kg/day
After 3-4 weeks – Dose tapering to be started
Over 10 weeks – dose reduced to 1mg/kg alternate day
Every 3-4 weeks – Dosage reduced by 5-10 mg (until lowest possible dose that controls the disease is achieved)

Efficacy of GlucocorticoidsDetermined by an objective increase in muscle strength and activities of daily living.
If prednisolone provides no objective benefit after 3 months of therapy, it is advisable to taper and stop the drug so that the next in-line immunosuppressive therapy can be started.
Almost all patients with DM/PM respond to steroids to some degree or for some period of time.

In acute and particularly severe cases, high dose Methylprednisolone (1gm infused over 2 hours daily for 5 days) is used.

Steroid myopathyThe long-term use of prednisone may cause increased weakness associated with a normal or unchanged CK level, this effect is referred to as steroid myopathy.
In a patient who previously responded to high doses of prednisone, the development of new weakness may be related to steroid myopathy or to disease activity that either will respond to a higher dose of glucocorticoids or has become glucocorticoid-resistant.
In uncertain cases, the prednisone dosage can be steadily increased or decreased as desired: the cause of the weakness is usually evident in 2–8 weeks.

Other immunosuppressive drugsApproximately 75% of the patients ultimately require additional treatment during the course of the illness.
Indications –
i. patient fails to respond adequately to glucocorticoids after a 3-month trial
ii. the patient becomes glucocorticoid-resistant
iii. glucocorticoid-related side effects appear
iv. attempts to lower the prednisone dose repeatedly result in a new relapse
v. rapidly progressive disease with evolving severe weakness and respiratory failure develops.

Other immunosuppressive drugsAzathioprine – Dose up to 3mg/kg/day is used. As effective as other drugs when used on a long term basis.
Methotrexate - Faster onset of action than azathioprine. It is given orally starting at 7.5 mg weekly for the first 3 weeks (2.5 mg every 12 h for 3 doses), with gradual dose escalation by 2.5 mg per week to a total of 25 mg weekly.
A rare side effect is methotrexate pneumonitis, which can be difficult to distinguish from the interstitial lung disease of the primary myopathy associated with Jo-1 antibodies

Mycophenolate Mofetil – Also has a faster onset of action than azathioprine. 2.5-3gm/day in 2 divided doses. It is well tolerated for long term therapy.
Rituximab – Monoclonal anti-CD20 antibody (rituximab) has been shown in a small uncontrolled series to benefit patients with DM and PM at a dose of intravenously 750 mg/m2, repeated in 2 weeks and sometimes required every 6 to 18 months.

Cyclophosphamide (0.5–1 g/m2 IV monthly for 6 months) has limited success and significant toxicity.
Tacrolimus (formerly known as Fk506) has been effective in some difficult cases of PM especially with interstitial lung disease.

ImmunomodulationIn a controlled trial of patients with refractory DM, IVIg improved not only strength and rash but also the underlying immunopathology.
The benefit is often short-lived (≤8 weeks), and repeated infusions every 6–8 weeks are generally required to maintain improvement.
A dose of 2 g/kg divided over 2–5 days per course is recommended.
Uncontrolled observations suggest that IVIg may also be beneficial for patients with PM.
Neither plasmapheresis nor leukapheresis appears to be effective in PM and DM.

Sequential empirical approach to the treatment of PM and DM
High-dose prednisone
01
Azathioprine, Mycophenolate orMethotrexate for steroid-sparing effect
02
IVIg
03
A trial, with one of the following agents, chosen according to age, degree of disability, tolerance, experience with the drug, and general health: rituximab, cyclosporine, cyclophosphamide or tacrolimus.
04

Treatment strategyA combination of prednisone in low dosage and one of these immunosuppressant drugs.
This approach is generally necessary when myocarditis or interstitial pneumonitis is coupled with DM.

Patients with Interstitial Lung Disease may benefit from aggressive treatment with Cyclophosphamide (or) Tacrolimus.

Poor Response to therapy A patient with presumed PM who has not responded to any form of immunotherapy most likely has IBM or another disease, usually a metabolic myopathy, a muscular dystrophy, a drug-induced myopathy, or an endocrinopathy.
In these cases, a repeat muscle biopsy and a renewed search for another cause of the myopathy is indicated.

Calcinosis - ManagementCalcinosis, a manifestation of DM, is difficult to treat however, new calcium deposits may be prevented if the primary disease responds to the available therapies.
Bisphosphonates, aluminium hydroxide, probenecid, colchicine, low doses of warfarin, calcium blockers, and surgical excision have all been tried without success.

Inclusion Body Myositis -ManagementGenerally resistant to immunosuppressive therapies.
Prednisolone together with azathioprine or methotrexate is often tried for a few months in newly diagnosed patients.
IVIg in IBM with minimal benefit in up to 30% of patients was found in one study.
A 2- to 3-month trial with IVIg may be reasonable for selected patients with IBM who experience rapid progression of muscle weakness or choking episodes due to worsening dysphagia.

PrognosisThe 5-year survival rate for treated patients with PM and DM is ~95%, and the 10-year survival rate is 84%.
Death is usually due to pulmonary cardiac, or other systemic complications.
The prognosis is worse for patients who are severely affected at presentation, when initial treatment is delayed, and in cases with severe dysphagia or respiratory difficulties.

DM responds more favorably to therapy than PM and thus has a better prognosis. Most patients improve with therapy, and many make a full functional recovery, which is often sustained with maintenance therapy.
Up to 30% may be left with some residual muscle weakness. Relapses may occur at any time.

IBM has the least favorable prognosis of the inflammatory myopathies.
Most patients will require the use of an assistive device such as a cane, walker, or wheelchair within 5–10 years of onset.
In general, the older the age of onset in IBM, the more rapidly progressive is the course.
Thank you