hydrogen peroxide-assisted photocatalytic oxidation of phenolic compounds
TRANSCRIPT

Hydrogen peroxide-assisted photocatalytic oxidation of
phenolic compounds
M.A. Barakat a,*, J.M. Tseng b, C.P. Huang b
a Central Metallurgical R&D Institute, Helwan 11421,Cairo, Egyptb Department of Civil and Environmental Engineering, University of Delaware, DE 19716, USA
Received 17 September 2004; received in revised form 10 January 2005; accepted 14 January 2005
Available online 5 February 2005
Abstract
The effect of hydrogen peroxide (H2O2) on photocatalytic oxidation of phenol and monochlorophenols (CP) in aqueous suspensions of
commercial TiO2 rutile was investigated. Various concentrations of H2O2 were used without and with the presence of TiO2 under different
atmospheres, e.g., N2 or O2. Sources of hydroxyl radicals for photocatalytic processes are suggested through the surface hydroxyl group
reacting with hole, dissolved oxygen trapping an electron, and photolytic H2O2. The combination of TiO2 and H2O2 under UV illumination
can greatly enhance the degradation rates of the phenol and chlorophenols. The photocatalytic oxidation with the H2O2/UV/TiO2 system was
found to be much more effective than either UV/TiO2 or UV/H2O2 alone. The efficiency of the photocatalytic degradation of phenol was
improved from 30 to 97% due to the presence of H2O2. As the H2O2 concentration increases, more hydroxyl radicals are produced, and the
phenol oxidation rate increases. At high H2O2 concentration (�10�2 M), O2 or N2 atmospheres are not important factors for phenol oxidation
in the H2O2/UV/TiO2 system.
# 2005 Elsevier B.V. All rights reserved.
Keywords: H2O2/UV/TiO2 system; Photocatalytic degradation; H2O2 concentration
www.elsevier.com/locate/apcatb
Applied Catalysis B: Environmental 59 (2005) 99–104
1. Introduction
Phenol and phenolic substances are used as raw materials
in many petrochemical, chemical, and pharmaceutical
industries. Wastewater containing phenol has received
increased attention because of its toxicity and prevalence
in industrial processes [1–3]. In addition, phenol is
considered to be an intermediate product in the oxidation
of higher-molecular weight aromatic hydrocarbons [4].
Thus, it is usually taken as a model compound for advanced
wastewater treatment studies. Different methods were
reported for phenol degradation in aqueous solutions.
Catalytic wet oxidation is one of the conventional methods
which uses H2O2 as oxidative agent with homogenized
metal salts [5,6], or a combination of H2O2 with
heterogeneous catalysts such as Al–Fe pillared clay or iron
containing clays [7–9]. Advanced oxidation by using UV/
* Corresponding author. Tel.: +20 25010642; fax: +20 25010639.
E-mail address: [email protected] (M.A. Barakat).
0926-3373/$ – see front matter # 2005 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcatb.2005.01.004
H2O2 is another way to degrade phenolic compounds in
dilute solutions [10–15]. The UV/H2O2 processes result in
formation of OH radicals, which accelerate the oxidative
degradation of the phenolic compounds in water. In recent
years, photocatalytic degradation of phenolic compounds in
the presence of semiconductor powders such as titanium
dioxide (TiO2) has been considered as effective technology
[16–25]. TiO2 photocatalyst upon irradiation with UV light
produces electrons and positive holes. The holes are strong
oxidizing agents that can oxidize organic compounds such
as phenols to mineral acids, e.g., HCl and CO2. The main
disadvantage of using H2O2 is its high cost, whereas the low
oxidation rate is the major draw back of TiO2. The combined
use of H2O2 and TiO2 is a way of solving this problem.
Fewer studies have been devoted to the examination of
phenol and chlorophenols degradation in presence of H2O2
and TiO2 anatase [26–29].
In this study commercial TiO2 rutile was treated and
investigated as a photocatalyst. The effect of H2O2 on
improving the reactivity of the catalyst for phenol and

M.A. Barakat et al. / Applied Catalysis B: Environmental 59 (2005) 99–104100
monochlorophenols (CP) photocatalytic degradation was
studied. Two systems, H2O2 alone and H2O2 with TiO2,
under different atmospheres (O2 or N2) were investigated.
Table 1
Properties of the used titanium dioxide (rutile [17] and Degussa P25)
Property DuPont TiO2 Degussa P25 TiO2
Raw Treated
Structure Rutile Rutile Anatase:rutile (80:20)
pHzpc 9.3 9.0 6.6
Surface area (m2/g) 6.6 6.1 50 � 1.5
Band gab energy (eV) 3.2 3.2 3.2
Impurities (wt.%) 0.729 (Al) 0.708 (Al) 0.1
1.145 (Cl) 0.0 (Cl) 0.0
2. Methods and materials
2.1. Materials
Titanium dioxide (TiO2), commercial grade, was
provided by the DuPont Company (Wilmington, Delaware,
USA). Raw TiO2 powder was treated according to Tseng and
Huang [17] by rinsing with 1 M HClO4 solution followed by
washing with distilled water. Degussa P25 TiO2 was
provided by Degussa Company (Ridge-Field Park, NJ,
USA) and used without any pretreatment. Stock solutions of
both Dupont and Degussa TiO2 (10 g/L) were prepared,
shaken for 24 h and left for 3 days for hydration. The TiO2
suspension was shaken for 30 min every time before use.
Hydrogen peroxide was prepared from a 34% H2O2 stock
solution and diluted to the required concentration. All
phenols that used in this study were obtained from Aldrich
Chemical Co. (98–99%) and were used without further
purification.
The light sources used to illuminate TiO2 samples was a
1600 W medium pressure mercury vapor discharge lamp
(American Ultraviolet Co.). The spectral irradiance for the
UV lamp (260 W/m2) ranges from 228 to 420 nm at a
distance of 1 m from the light source according to the
information provided by the manufacturer. The light
intensity was recorded with a UV-radiometer (Model
365H, Spectronics Co.). A light intensity of 17.0 W/m2,
at a peak of 365 nm was provided at 1 m distance from test
tube reactors. The light wavelength (greater than 325 nm)
can completely pass through the Pyrex glass [17].
2.2. Methods
Pyrex glass tubes having dimensions of 20 mm internal
diameter and 13 cm long were used as reactors. All photo-
catalytic oxidation experiments were performed in the Pyrex
tubes containing 15 mL of 10�3 M phenol solution and
0.15 g of TiO2. A constant ionic strength (I = 5 � 10�2 M)
was obtained by addition of a given amount of NaClO4 to the
solutions. In order to study the effect of oxygen, different
atmospheres were created by introducing pure nitrogen or
oxygen into the reactors. Head space of each reactor also
was filled with the same gas as in solution. A pyrogallol
solution was used to remove oxygen prior to nitrogen
bubbling. The reactors were tightened with Teflon septa
airtight caps. The phenol solutions were aerated with
nitrogen or oxygen prior to experiments. Samples were
shaken over a reciprocal shaker (American Optical Co.) with
180 strokes per minute to insure complete mixing. The
temperature of the solutions was controlled and adjusted to
25 8C by a thermostat pump in conjunction with a cooler. At
the end of a given reaction time, samples were filtered using
0.45 mm microfilter (Gelman, Supor-450, 25 mm diameter).
2.3. Measurements
Dissolved oxygen was monitored by a dissolved oxygen
(DO) meter calibrated by the modified Winkler method [30].
Generally, 0.3 ppm oxygen was found in solution under
nitrogen bubbling. In the pure oxygen atmosphere 27.0 ppm
dissolved oxygen can be obtained. The residual concentra-
tion of phenol was measured with an UV–vis spectro-
photometer (Hitachi/Perkin-Elmer, Model 139) at a
wavelength of 271 nm. The optimal wavelength was
determined from the spectrum of phenol solutions at various
pHs and concentrations. Chloride was measured by using an
Orion Model 96-176 ion specific combination chloride
electrode.
3. Results and discussion
The syntheses of the Dupont TiO2 catalyst were already
optimized and characterized in a previous study [17]. All
photo oxidation experiments were performed with the
pretreated Dupont TiO2, one run of photocatalytic degrada-
tion of phenol was carried out with Degussa P25 TiO2 for
comparison with the data of Dupont TiO2. The major
properties of the used TiO2, Dupont [17] and Degussa P25
(data supplied by the manufacturer, are summarized in
Table 1. The photo oxidation experiments for both phenol
and chlorophenols were conducted at optimized experi-
mental conditions that were reported in the previous works
[17,18]. The experimental conditions were: UV light
intensity of 17 W/m2, solution temperature of 25 8C,
solution pH of 7, and phenol or CP concentration of
10�3 M. The ionic strength of the solutions was adjusted by
adding 5 � 10�2 M NaClO4. At such experimental condi-
tions with dilute phenol concentration, it was very difficult
to identify intermediates of the degradation. The inter-
mediates were detected by GC/MS, and the data was
reported in a previous work [17]. Two intermediates,
hydroquinone and phenol dimmer were detected only at
higher phenol concentration, 5 � 10�2 M. Fig. 1 shows the
effect of H2O2 on the photolytic phenol oxidation under N2
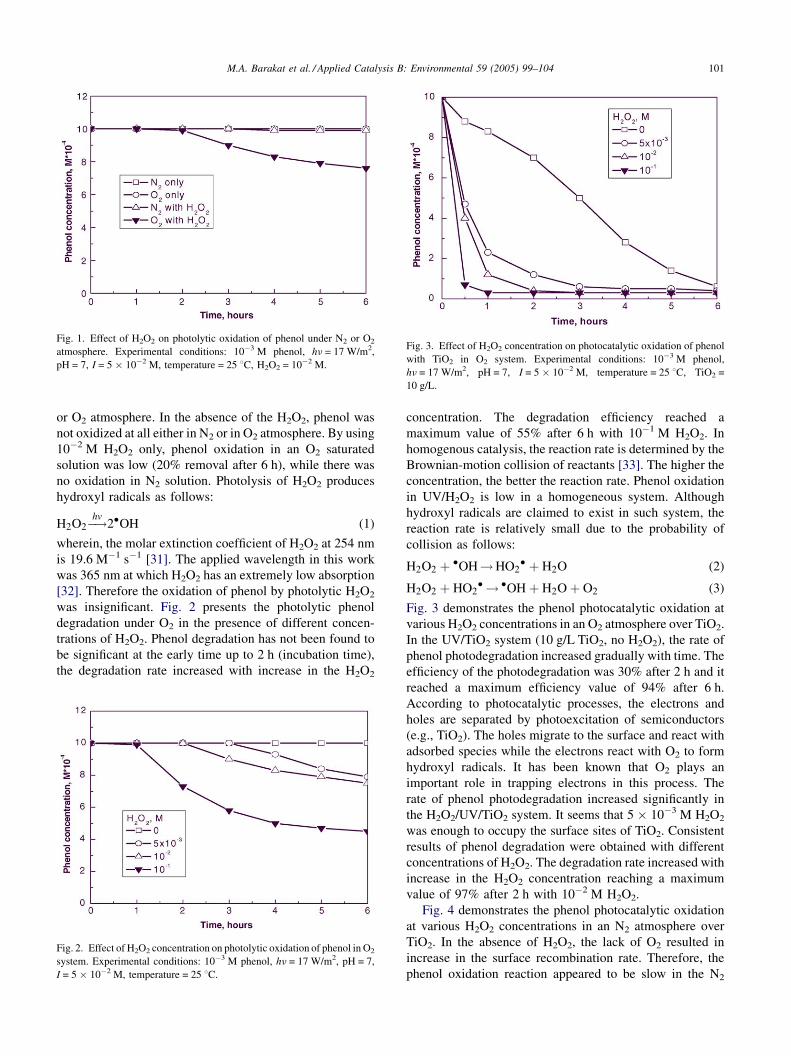
M.A. Barakat et al. / Applied Catalysis B: Environmental 59 (2005) 99–104 101
Fig. 1. Effect of H2O2 on photolytic oxidation of phenol under N2 or O2
atmosphere. Experimental conditions: 10�3 M phenol, hn = 17 W/m2,
pH = 7, I = 5 � 10�2 M, temperature = 25 8C, H2O2 = 10�2 M.
Fig. 3. Effect of H2O2 concentration on photocatalytic oxidation of phenol
with TiO2 in O2 system. Experimental conditions: 10�3 M phenol,
hn = 17 W/m2, pH = 7, I = 5 � 10�2 M, temperature = 25 8C, TiO2 =
10 g/L.
or O2 atmosphere. In the absence of the H2O2, phenol was
not oxidized at all either in N2 or in O2 atmosphere. By using
10�2 M H2O2 only, phenol oxidation in an O2 saturated
solution was low (20% removal after 6 h), while there was
no oxidation in N2 solution. Photolysis of H2O2 produces
hydroxyl radicals as follows:
H2O2�!hv
2�OH (1)
wherein, the molar extinction coefficient of H2O2 at 254 nm
is 19.6 M�1 s�1 [31]. The applied wavelength in this work
was 365 nm at which H2O2 has an extremely low absorption
[32]. Therefore the oxidation of phenol by photolytic H2O2
was insignificant. Fig. 2 presents the photolytic phenol
degradation under O2 in the presence of different concen-
trations of H2O2. Phenol degradation has not been found to
be significant at the early time up to 2 h (incubation time),
the degradation rate increased with increase in the H2O2
Fig. 2. Effect of H2O2 concentration on photolytic oxidation of phenol in O2
system. Experimental conditions: 10�3 M phenol, hn = 17 W/m2, pH = 7,
I = 5 � 10�2 M, temperature = 25 8C.
concentration. The degradation efficiency reached a
maximum value of 55% after 6 h with 10�1 M H2O2. In
homogenous catalysis, the reaction rate is determined by the
Brownian-motion collision of reactants [33]. The higher the
concentration, the better the reaction rate. Phenol oxidation
in UV/H2O2 is low in a homogeneous system. Although
hydroxyl radicals are claimed to exist in such system, the
reaction rate is relatively small due to the probability of
collision as follows:
H2O2 þ �OH!HO2� þ H2O (2)
H2O2 þ HO2� ! �OH þ H2O þ O2 (3)
Fig. 3 demonstrates the phenol photocatalytic oxidation at
various H2O2 concentrations in an O2 atmosphere over TiO2.
In the UV/TiO2 system (10 g/L TiO2, no H2O2), the rate of
phenol photodegradation increased gradually with time. The
efficiency of the photodegradation was 30% after 2 h and it
reached a maximum efficiency value of 94% after 6 h.
According to photocatalytic processes, the electrons and
holes are separated by photoexcitation of semiconductors
(e.g., TiO2). The holes migrate to the surface and react with
adsorbed species while the electrons react with O2 to form
hydroxyl radicals. It has been known that O2 plays an
important role in trapping electrons in this process. The
rate of phenol photodegradation increased significantly in
the H2O2/UV/TiO2 system. It seems that 5 � 10�3 M H2O2
was enough to occupy the surface sites of TiO2. Consistent
results of phenol degradation were obtained with different
concentrations of H2O2. The degradation rate increased with
increase in the H2O2 concentration reaching a maximum
value of 97% after 2 h with 10�2 M H2O2.
Fig. 4 demonstrates the phenol photocatalytic oxidation
at various H2O2 concentrations in an N2 atmosphere over
TiO2. In the absence of H2O2, the lack of O2 resulted in
increase in the surface recombination rate. Therefore, the
phenol oxidation reaction appeared to be slow in the N2
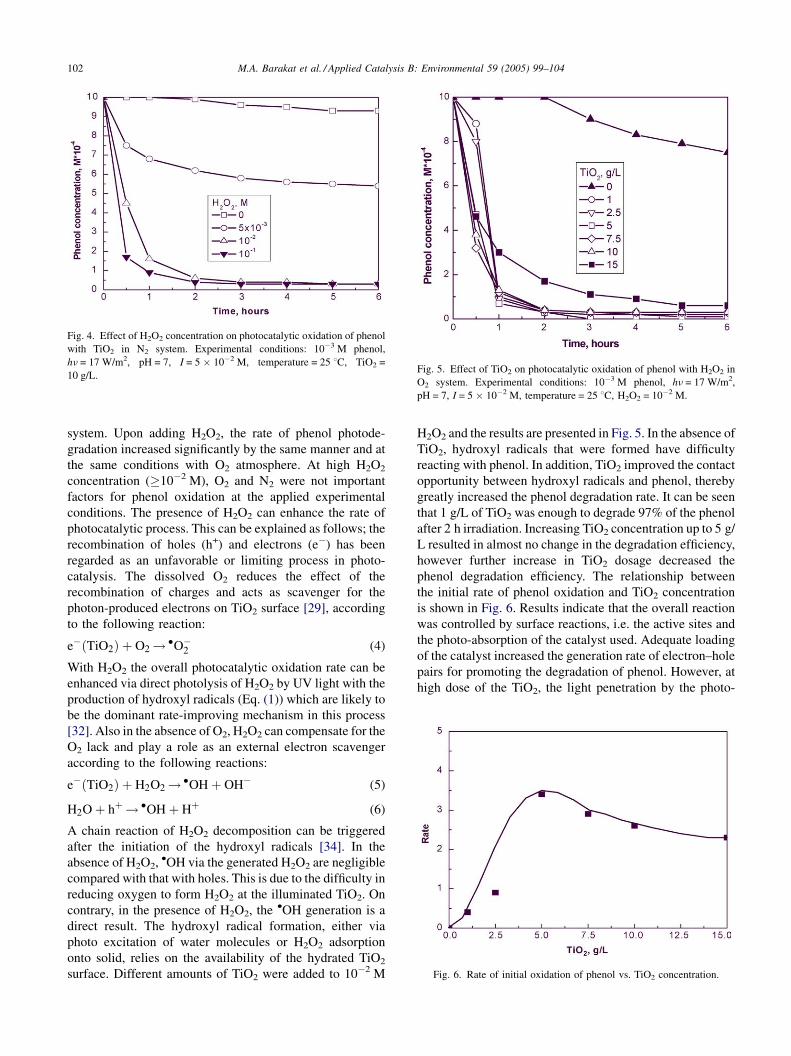
M.A. Barakat et al. / Applied Catalysis B: Environmental 59 (2005) 99–104102
Fig. 4. Effect of H2O2 concentration on photocatalytic oxidation of phenol
with TiO2 in N2 system. Experimental conditions: 10�3 M phenol,
hn = 17 W/m2, pH = 7, I = 5 � 10�2 M, temperature = 25 8C, TiO2 =
10 g/L.Fig. 5. Effect of TiO2 on photocatalytic oxidation of phenol with H2O2 in
O2 system. Experimental conditions: 10�3 M phenol, hn = 17 W/m2,
pH = 7, I = 5 � 10�2 M, temperature = 25 8C, H2O2 = 10�2 M.
system. Upon adding H2O2, the rate of phenol photode-
gradation increased significantly by the same manner and at
the same conditions with O2 atmosphere. At high H2O2
concentration (�10�2 M), O2 and N2 were not important
factors for phenol oxidation at the applied experimental
conditions. The presence of H2O2 can enhance the rate of
photocatalytic process. This can be explained as follows; the
recombination of holes (h+) and electrons (e�) has been
regarded as an unfavorable or limiting process in photo-
catalysis. The dissolved O2 reduces the effect of the
recombination of charges and acts as scavenger for the
photon-produced electrons on TiO2 surface [29], according
to the following reaction:
e�ðTiO2Þ þ O2 !�O�2 (4)
With H2O2 the overall photocatalytic oxidation rate can be
enhanced via direct photolysis of H2O2 by UV light with the
production of hydroxyl radicals (Eq. (1)) which are likely to
be the dominant rate-improving mechanism in this process
[32]. Also in the absence of O2, H2O2 can compensate for the
O2 lack and play a role as an external electron scavenger
according to the following reactions:
e�ðTiO2Þ þ H2O2 !�OH þ OH� (5)
H2O þ hþ!�OH þ Hþ (6)
Fig. 6. Rate of initial oxidation of phenol vs. TiO2 concentration.
A chain reaction of H2O2 decomposition can be triggered
after the initiation of the hydroxyl radicals [34]. In the
absence of H2O2, �OH via the generated H2O2 are negligible
compared with that with holes. This is due to the difficulty in
reducing oxygen to form H2O2 at the illuminated TiO2. On
contrary, in the presence of H2O2, the �OH generation is a
direct result. The hydroxyl radical formation, either via
photo excitation of water molecules or H2O2 adsorption
onto solid, relies on the availability of the hydrated TiO2
surface. Different amounts of TiO2 were added to 10�2 M
H2O2 and the results are presented in Fig. 5. In the absence of
TiO2, hydroxyl radicals that were formed have difficulty
reacting with phenol. In addition, TiO2 improved the contact
opportunity between hydroxyl radicals and phenol, thereby
greatly increased the phenol degradation rate. It can be seen
that 1 g/L of TiO2 was enough to degrade 97% of the phenol
after 2 h irradiation. Increasing TiO2 concentration up to 5 g/
L resulted in almost no change in the degradation efficiency,
however further increase in TiO2 dosage decreased the
phenol degradation efficiency. The relationship between
the initial rate of phenol oxidation and TiO2 concentration
is shown in Fig. 6. Results indicate that the overall reaction
was controlled by surface reactions, i.e. the active sites and
the photo-absorption of the catalyst used. Adequate loading
of the catalyst increased the generation rate of electron–hole
pairs for promoting the degradation of phenol. However, at
high dose of the TiO2, the light penetration by the photo-
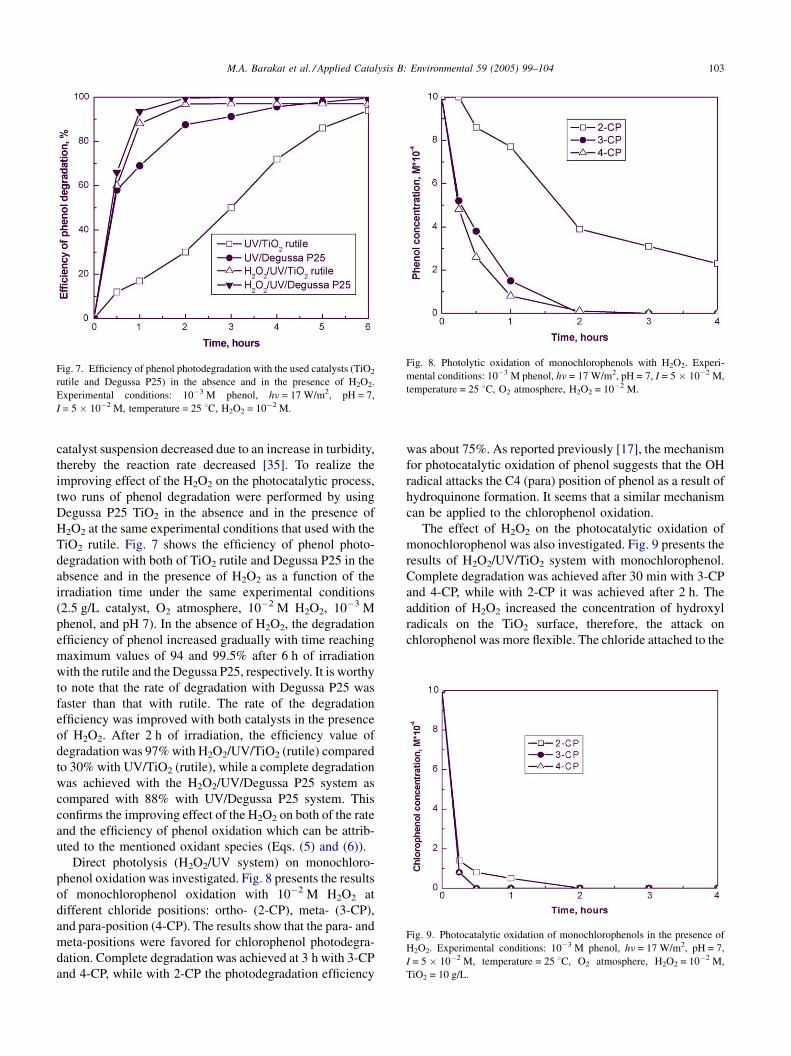
M.A. Barakat et al. / Applied Catalysis B: Environmental 59 (2005) 99–104 103
Fig. 7. Efficiency of phenol photodegradation with the used catalysts (TiO2
rutile and Degussa P25) in the absence and in the presence of H2O2.
Experimental conditions: 10�3 M phenol, hn = 17 W/m2, pH = 7,
I = 5 � 10�2 M, temperature = 25 8C, H2O2 = 10�2 M.
Fig. 8. Photolytic oxidation of monochlorophenols with H2O2. Experi-
mental conditions: 10�3 M phenol, hn = 17 W/m2, pH = 7, I = 5 � 10�2 M,
temperature = 25 8C, O2 atmosphere, H2O2 = 10�2 M.
Fig. 9. Photocatalytic oxidation of monochlorophenols in the presence of
H2O2. Experimental conditions: 10�3 M phenol, hn = 17 W/m2, pH = 7,
I = 5 � 10�2 M, temperature = 25 8C, O2 atmosphere, H2O2 = 10�2 M,
TiO2 = 10 g/L.
catalyst suspension decreased due to an increase in turbidity,
thereby the reaction rate decreased [35]. To realize the
improving effect of the H2O2 on the photocatalytic process,
two runs of phenol degradation were performed by using
Degussa P25 TiO2 in the absence and in the presence of
H2O2 at the same experimental conditions that used with the
TiO2 rutile. Fig. 7 shows the efficiency of phenol photo-
degradation with both of TiO2 rutile and Degussa P25 in the
absence and in the presence of H2O2 as a function of the
irradiation time under the same experimental conditions
(2.5 g/L catalyst, O2 atmosphere, 10�2 M H2O2, 10�3 M
phenol, and pH 7). In the absence of H2O2, the degradation
efficiency of phenol increased gradually with time reaching
maximum values of 94 and 99.5% after 6 h of irradiation
with the rutile and the Degussa P25, respectively. It is worthy
to note that the rate of degradation with Degussa P25 was
faster than that with rutile. The rate of the degradation
efficiency was improved with both catalysts in the presence
of H2O2. After 2 h of irradiation, the efficiency value of
degradation was 97% with H2O2/UV/TiO2 (rutile) compared
to 30% with UV/TiO2 (rutile), while a complete degradation
was achieved with the H2O2/UV/Degussa P25 system as
compared with 88% with UV/Degussa P25 system. This
confirms the improving effect of the H2O2 on both of the rate
and the efficiency of phenol oxidation which can be attrib-
uted to the mentioned oxidant species (Eqs. (5) and (6)).
Direct photolysis (H2O2/UV system) on monochloro-
phenol oxidation was investigated. Fig. 8 presents the results
of monochlorophenol oxidation with 10�2 M H2O2 at
different chloride positions: ortho- (2-CP), meta- (3-CP),
and para-position (4-CP). The results show that the para- and
meta-positions were favored for chlorophenol photodegra-
dation. Complete degradation was achieved at 3 h with 3-CP
and 4-CP, while with 2-CP the photodegradation efficiency
was about 75%. As reported previously [17], the mechanism
for photocatalytic oxidation of phenol suggests that the OH
radical attacks the C4 (para) position of phenol as a result of
hydroquinone formation. It seems that a similar mechanism
can be applied to the chlorophenol oxidation.
The effect of H2O2 on the photocatalytic oxidation of
monochlorophenol was also investigated. Fig. 9 presents the
results of H2O2/UV/TiO2 system with monochlorophenol.
Complete degradation was achieved after 30 min with 3-CP
and 4-CP, while with 2-CP it was achieved after 2 h. The
addition of H2O2 increased the concentration of hydroxyl
radicals on the TiO2 surface, therefore, the attack on
chlorophenol was more flexible. The chloride attached to the
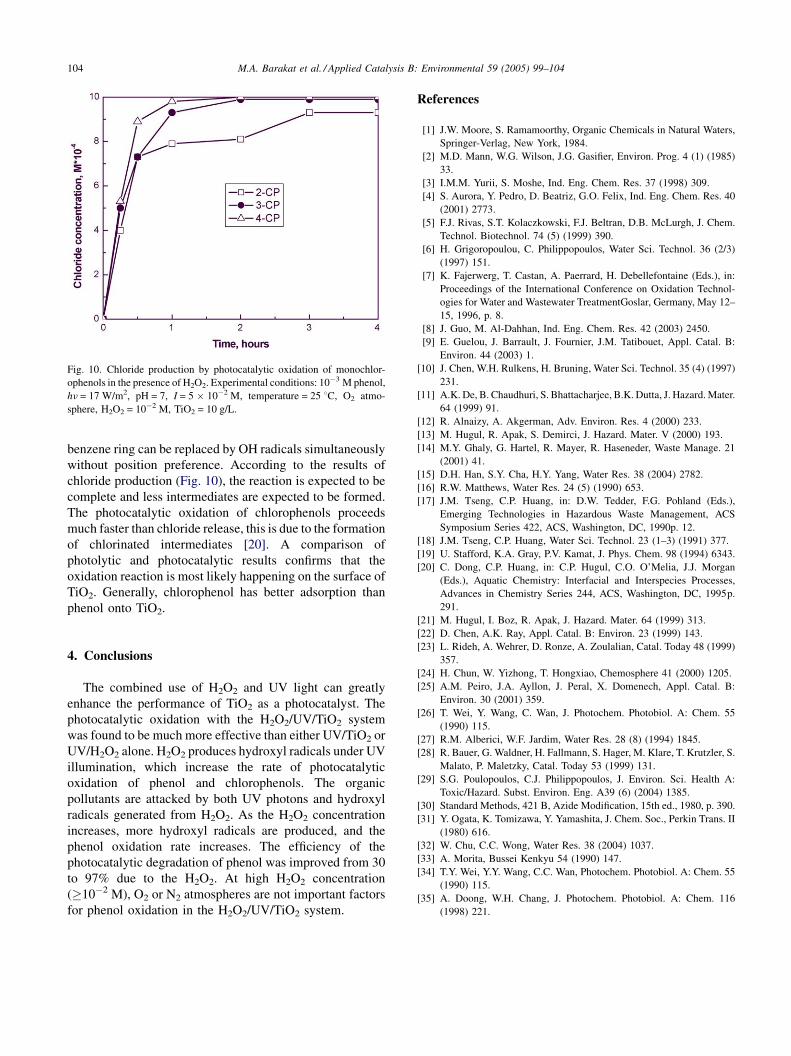
M.A. Barakat et al. / Applied Catalysis B: Environmental 59 (2005) 99–104104
Fig. 10. Chloride production by photocatalytic oxidation of monochlor-
ophenols in the presence of H2O2. Experimental conditions: 10�3 M phenol,
hn = 17 W/m2, pH = 7, I = 5 � 10�2 M, temperature = 25 8C, O2 atmo-
sphere, H2O2 = 10�2 M, TiO2 = 10 g/L.
benzene ring can be replaced by OH radicals simultaneously
without position preference. According to the results of
chloride production (Fig. 10), the reaction is expected to be
complete and less intermediates are expected to be formed.
The photocatalytic oxidation of chlorophenols proceeds
much faster than chloride release, this is due to the formation
of chlorinated intermediates [20]. A comparison of
photolytic and photocatalytic results confirms that the
oxidation reaction is most likely happening on the surface of
TiO2. Generally, chlorophenol has better adsorption than
phenol onto TiO2.
4. Conclusions
The combined use of H2O2 and UV light can greatly
enhance the performance of TiO2 as a photocatalyst. The
photocatalytic oxidation with the H2O2/UV/TiO2 system
was found to be much more effective than either UV/TiO2 or
UV/H2O2 alone. H2O2 produces hydroxyl radicals under UV
illumination, which increase the rate of photocatalytic
oxidation of phenol and chlorophenols. The organic
pollutants are attacked by both UV photons and hydroxyl
radicals generated from H2O2. As the H2O2 concentration
increases, more hydroxyl radicals are produced, and the
phenol oxidation rate increases. The efficiency of the
photocatalytic degradation of phenol was improved from 30
to 97% due to the H2O2. At high H2O2 concentration
(�10�2 M), O2 or N2 atmospheres are not important factors
for phenol oxidation in the H2O2/UV/TiO2 system.
References
[1] J.W. Moore, S. Ramamoorthy, Organic Chemicals in Natural Waters,
Springer-Verlag, New York, 1984.
[2] M.D. Mann, W.G. Wilson, J.G. Gasifier, Environ. Prog. 4 (1) (1985)
33.
[3] I.M.M. Yurii, S. Moshe, Ind. Eng. Chem. Res. 37 (1998) 309.
[4] S. Aurora, Y. Pedro, D. Beatriz, G.O. Felix, Ind. Eng. Chem. Res. 40
(2001) 2773.
[5] F.J. Rivas, S.T. Kolaczkowski, F.J. Beltran, D.B. McLurgh, J. Chem.
Technol. Biotechnol. 74 (5) (1999) 390.
[6] H. Grigoropoulou, C. Philippopoulos, Water Sci. Technol. 36 (2/3)
(1997) 151.
[7] K. Fajerwerg, T. Castan, A. Paerrard, H. Debellefontaine (Eds.), in:
Proceedings of the International Conference on Oxidation Technol-
ogies for Water and Wastewater TreatmentGoslar, Germany, May 12–
15, 1996, p. 8.
[8] J. Guo, M. Al-Dahhan, Ind. Eng. Chem. Res. 42 (2003) 2450.
[9] E. Guelou, J. Barrault, J. Fournier, J.M. Tatibouet, Appl. Catal. B:
Environ. 44 (2003) 1.
[10] J. Chen, W.H. Rulkens, H. Bruning, Water Sci. Technol. 35 (4) (1997)
231.
[11] A.K. De, B. Chaudhuri, S. Bhattacharjee, B.K. Dutta, J. Hazard. Mater.
64 (1999) 91.
[12] R. Alnaizy, A. Akgerman, Adv. Environ. Res. 4 (2000) 233.
[13] M. Hugul, R. Apak, S. Demirci, J. Hazard. Mater. V (2000) 193.
[14] M.Y. Ghaly, G. Hartel, R. Mayer, R. Haseneder, Waste Manage. 21
(2001) 41.
[15] D.H. Han, S.Y. Cha, H.Y. Yang, Water Res. 38 (2004) 2782.
[16] R.W. Matthews, Water Res. 24 (5) (1990) 653.
[17] J.M. Tseng, C.P. Huang, in: D.W. Tedder, F.G. Pohland (Eds.),
Emerging Technologies in Hazardous Waste Management, ACS
Symposium Series 422, ACS, Washington, DC, 1990p. 12.
[18] J.M. Tseng, C.P. Huang, Water Sci. Technol. 23 (1–3) (1991) 377.
[19] U. Stafford, K.A. Gray, P.V. Kamat, J. Phys. Chem. 98 (1994) 6343.
[20] C. Dong, C.P. Huang, in: C.P. Hugul, C.O. O’Melia, J.J. Morgan
(Eds.), Aquatic Chemistry: Interfacial and Interspecies Processes,
Advances in Chemistry Series 244, ACS, Washington, DC, 1995p.
291.
[21] M. Hugul, I. Boz, R. Apak, J. Hazard. Mater. 64 (1999) 313.
[22] D. Chen, A.K. Ray, Appl. Catal. B: Environ. 23 (1999) 143.
[23] L. Rideh, A. Wehrer, D. Ronze, A. Zoulalian, Catal. Today 48 (1999)
357.
[24] H. Chun, W. Yizhong, T. Hongxiao, Chemosphere 41 (2000) 1205.
[25] A.M. Peiro, J.A. Ayllon, J. Peral, X. Domenech, Appl. Catal. B:
Environ. 30 (2001) 359.
[26] T. Wei, Y. Wang, C. Wan, J. Photochem. Photobiol. A: Chem. 55
(1990) 115.
[27] R.M. Alberici, W.F. Jardim, Water Res. 28 (8) (1994) 1845.
[28] R. Bauer, G. Waldner, H. Fallmann, S. Hager, M. Klare, T. Krutzler, S.
Malato, P. Maletzky, Catal. Today 53 (1999) 131.
[29] S.G. Poulopoulos, C.J. Philippopoulos, J. Environ. Sci. Health A:
Toxic/Hazard. Subst. Environ. Eng. A39 (6) (2004) 1385.
[30] Standard Methods, 421 B, Azide Modification, 15th ed., 1980, p. 390.
[31] Y. Ogata, K. Tomizawa, Y. Yamashita, J. Chem. Soc., Perkin Trans. II
(1980) 616.
[32] W. Chu, C.C. Wong, Water Res. 38 (2004) 1037.
[33] A. Morita, Bussei Kenkyu 54 (1990) 147.
[34] T.Y. Wei, Y.Y. Wang, C.C. Wan, Photochem. Photobiol. A: Chem. 55
(1990) 115.
[35] A. Doong, W.H. Chang, J. Photochem. Photobiol. A: Chem. 116
(1998) 221.