histone deacetylases in kidney development: implications for disease and therapy
TRANSCRIPT

REVIEW
Histone deacetylases in kidney development: implicationsfor disease and therapy
Shaowei Chen & Samir S. El-Dahr
Received: 4 April 2012 /Revised: 15 May 2012 /Accepted: 17 May 2012 /Published online: 22 June 2012# IPNA 2012
Abstract Histone deacetylases (HDACs) are an evolution-arily conserved group of enzymes that regulate a broadrange of biological processes through removal of acetylgroups from histones as well as non-histone proteins. Recentstudies using a variety of pharmacological inhibitors andgenetic models of HDACs have revealed a central role ofHDACs in control of kidney development. These findingsprovide new insights into the epigenetic mechanisms under-lying congenital anomalies of the kidney and urinary tract(CAKUT) and implicate the potential of HDACs as thera-peutic targets in kidney diseases, such as cystic kidneydiseases and renal cell cancers. Determining the specificfunctions of individual HDAC members would be an im-portant task of future research.
Keywords Histone deacetylases . Histone acetylation .
Kidney development . CAKUT . Polycystic kidney disease
Introduction
Failure of normal kidney development results in a broadspectrum of congenital anomalies of the kidney and urinarytract (CAKUT) in children, ranging from mild unilateralobstructive uropathies to severe bilateral renal aplasia/dys-plasia. CAKUT occurs in one out of 500 newborns, constitut-ing approximately 20–30% of all identified birth defects.CAKUT has a major role in renal failure in children. Itaccounts for up to 30% of end-stage renal disease in children
less than 4 years of age (North American Pediatric RenalTrials and Collaborative Studies 2008 Annual report). Nota-bly, although some forms are familial and syndromic, mostcases of CAKUT are sporadic and nonsyndromic. Given thecomplexity of CAKUT, to date, only a few genetic mutationshave been identified for the syndromic forms of CAKUT,while the cellular and molecular basis of the more commonnonsyndromic forms of CAKUT are largely unknown [1].
Thanks to the advances in molecular biology and genetargeting technology, recent years have witnessed an emergingawareness of the critical role of epigenetic mechanisms inhealth and disease. During embryogenesis, epigenetic modifi-cations, such as DNA methylation, histone acetylation, histonephosphorylation and histone methylation, are set in the chro-matin of developmental regulators; this in turn, determines thegenome programming in a particular cell by modulating chro-matin structure and thus DNA accessibility to the transcription-al machinery. Disruptions of these epigenetic modificationsresulting from environmental exposures (e.g., diet, toxins,drugs, viral infections) can lead to dysregulation of gene func-tion, without altering the DNA sequence itself [2]. As epige-netic abnormalities depend on the interplay between genes andthe environment, they are often phenotypically variable, whichfits well with the broad phenotypic spectrum of CAKUT.Therefore, understanding the epigenetic basis of kidney devel-opment might provide new insights into the pathological mech-anisms of CAKUT and, hopefully, open new avenues fortreatment or prevention of CAKUT. This review focuses onhistone acetylation, a reversible epigenetic modification con-trolled by two opposing families of enzymes: histone acetyl-transferases (HATs) and histone deacetylases (HDACs). Wediscuss the developmental functions of HDACs in the kidneythat are revealed by a variety of pharmacological inhibitors andgenetic models of HDACs, and the potential of HDACs astherapeutic targets in kidney diseases.
S. Chen : S. S. El-Dahr (*)Department of Pediatrics, Section of Pediatric Nephrology,Tulane University Health Sciences Center,1430 Tulane Avenue, SL-37,New Orleans, LA 70112, USAe-mail: [email protected]
Pediatr Nephrol (2013) 28:689–698DOI 10.1007/s00467-012-2223-8

Brief overview of kidney morphogenesis
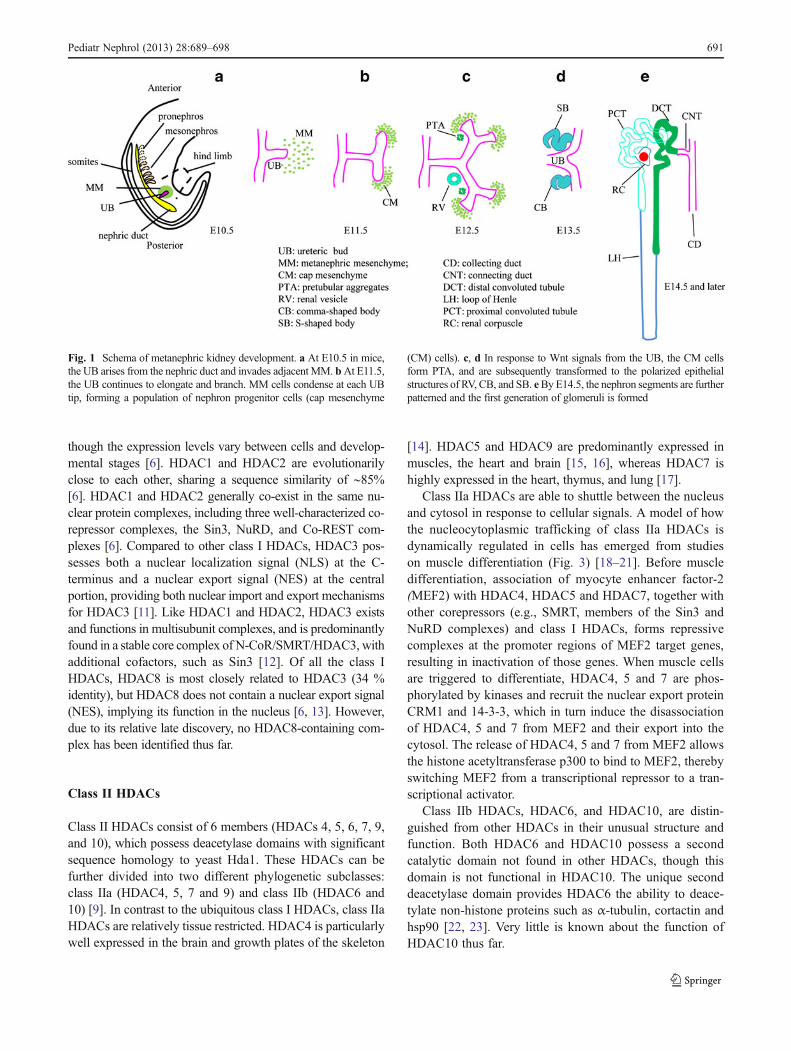
The kidney develops through reciprocal inductive interac-tions between the nephric duct (ND) and the surroundingmesenchyme (Fig. 1). At embryonic (E) day E22 in humansor E8 in mice, the ND arises in the intermediate mesodermbetween the anterior somites and lateral plate. The NDextends caudally and induces the adjacent mesenchyme tosequentially form three types of kidney: the pronephros (E8in mice), the mesonephros (E9 in mice), and the metaneph-ros (E10.5 in mice) [3, 4]. The pronephros and mesonephrosare transient structures in mammals but are absolutelyessential for the development of the later permanentmetanephros, as interruption of their development resultsin renal agenesis. The first step of metanephros forma-tion is the outgrowth of the ureteric bud (UB) from thecaudal part of the ND toward the mesenchyme at theprospective metanephros site (E10.5). Then, signalsfrom the metanephric mesenchyme (MM) continue toinduce the UB to elongate, enter the mesenchyme andbranch repeatedly by a process referred to as branchingmorphogenesis to form its daughter collecting ducts.The proximal part of the UB also forms the renalcalyces, pelvis and ureter. Simultaneously, in responseto the inductive signals from the UB, the MM cells areprevented from undergoing apoptosis, and some of theMM cells rapidly condense around the UB tips, forminga population of nephron progenitor cells (often referredto as cap mesenchyme (CM) cells). The nephron pro-genitor cells then undergo pretubular aggregation,mesenchymal-to-epithelial transition (MET), and subse-quently form the polarized structures of renal vesicle(RV), comma-shaped and S-shaped bodies (CB andSB), which in turn differentiate into the intricate struc-ture of the renal nephron, including the proximal con-voluted tubule, the loop of Henle, and the distalconvoluted tubule. Yet, not all the MM is fated tobecome epithelium, as some mesenchymal cells do notbecome aggregates and remain along the periphery asstromal mesenchyme (SM) (or interstitial mesenchyme)or potential mesenchymal stem cells. It has becomeclear that the SM also plays an active role in uretericbranching, nephron progenitor-cell commitment andnephron maturation, and contributes to vascularization.Iterations of dichotomous branching of the UB effec-tively double the number of branch tips or ampullaewith each branching event and are critical in determin-ing nephron number. It is estimated that, in humans, upto 20 generations of dichotomous UB branching are requiredto generate approximately 1 million nephrons normally pres-ent in the adult kidney. The sophisticated morphogeneticevents during renal organogenesis are tightly controlled bymultiple gene regulatory networks (reviewed in [3, 4]).
The HDAC superfamily
Histone deacetylases (HDACs) are a large family of evolu-tionarily conserved enzymes which catalyze the removal ofacetyl groups from histone tails. Given that histone acetyla-tion is commonly associated with active transcription,HDACs were originally regarded as general transcriptionalco-repressors. However, later on, it has become clear thatHDACs regulate gene expression in a highly selective wayand exhibit both repressive and activating effects [5]. Due tolack of intrinsic DNA binding activities, HDACs do notoperate alone. Instead, HDACs are recruited to target genesvia association with transcriptional complexes (e.g., Sin3complex, NuRD complex, Co-REST complex, and SMRT/N-CoR complex) [6]. Thus, the specificity of HDACs ingene regulation depends on the partner proteins they asso-ciate with in different cell types, at specific develop-mental stages. It is likely that specific patterns ofhistone lysine acetylation may cooperate with otherforms of histone modifications (e.g., methylation) tocreate a “histone signature” for recruitment of additionaltranscriptional regulators, which in turn repress or acti-vate gene expression. Furthermore, while histones arethe primary substrates of HDACs, HDACs are alsofound to deacetylate many non-histone proteins, suchas p53, STAT3, Yin Yang transcription factor (YY1),GATA1, E2F1, tubulin and Hsp90. Acetylation of thoseproteins has been shown to affect multiple aspects oftheir functions, such as DNA binding affinity, transcrip-tional activity, protein stability, and protein–protein in-teraction [7, 8].
To date, 18mammalianHDACs have been identified. Basedon their sequence homology to yeast histone deacetylase genes,HDACs are divided into four classes: the class I RPD3-likeHDACs (1–3, and 8), the class II HDA1–like HDACs (4, 5, 6,7, 9, and 10), the class III SIR2-like HDACs (Sirtuins 1–7) andthe class IVHDAC 11- [6, 9, 10]. Class I, II and IVHDACs areclosely related to each other and belong to the classical familyof HDACs. These HDACs share a common catalytic domainstructure (∼390 amino acids) referred to as histone deacetylase-like protein (HDLP), which catalyzes the removal of acetylgroups from ε-N-acetyl lysine amino acids through a Zn2+-dependent charge relay system [6]. In comparison, the class IIIHDACs are homologous to yeast Sir2 and are NAD+dependent for their deacetylase activities [10]. In this review,we will focus on the classical family of HDACs, with anemphasis on their functions in kidney development.
Class I HDACs
Class I HDACs (HDAC1, 2, 3, and 8) share a high degree ofhomology with yeast Rpd3 and are expressed ubiquitously,
690 Pediatr Nephrol (2013) 28:689–698
though the expression levels vary between cells and develop-mental stages [6]. HDAC1 and HDAC2 are evolutionarilyclose to each other, sharing a sequence similarity of ∼85%[6]. HDAC1 and HDAC2 generally co-exist in the same nu-clear protein complexes, including three well-characterized co-repressor complexes, the Sin3, NuRD, and Co-REST com-plexes [6]. Compared to other class I HDACs, HDAC3 pos-sesses both a nuclear localization signal (NLS) at the C-terminus and a nuclear export signal (NES) at the centralportion, providing both nuclear import and export mechanismsfor HDAC3 [11]. Like HDAC1 and HDAC2, HDAC3 existsand functions in multisubunit complexes, and is predominantlyfound in a stable core complex of N-CoR/SMRT/HDAC3, withadditional cofactors, such as Sin3 [12]. Of all the class IHDACs, HDAC8 is most closely related to HDAC3 (34 %identity), but HDAC8 does not contain a nuclear export signal(NES), implying its function in the nucleus [6, 13]. However,due to its relative late discovery, no HDAC8-containing com-plex has been identified thus far.
Class II HDACs
Class II HDACs consist of 6 members (HDACs 4, 5, 6, 7, 9,and 10), which possess deacetylase domains with significantsequence homology to yeast Hda1. These HDACs can befurther divided into two different phylogenetic subclasses:class IIa (HDAC4, 5, 7 and 9) and class IIb (HDAC6 and10) [9]. In contrast to the ubiquitous class I HDACs, class IIaHDACs are relatively tissue restricted. HDAC4 is particularlywell expressed in the brain and growth plates of the skeleton
[14]. HDAC5 and HDAC9 are predominantly expressed inmuscles, the heart and brain [15, 16], whereas HDAC7 ishighly expressed in the heart, thymus, and lung [17].
Class IIa HDACs are able to shuttle between the nucleusand cytosol in response to cellular signals. A model of howthe nucleocytoplasmic trafficking of class IIa HDACs isdynamically regulated in cells has emerged from studieson muscle differentiation (Fig. 3) [18–21]. Before muscledifferentiation, association of myocyte enhancer factor-2(MEF2) with HDAC4, HDAC5 and HDAC7, together withother corepressors (e.g., SMRT, members of the Sin3 andNuRD complexes) and class I HDACs, forms repressivecomplexes at the promoter regions of MEF2 target genes,resulting in inactivation of those genes. When muscle cellsare triggered to differentiate, HDAC4, 5 and 7 are phos-phorylated by kinases and recruit the nuclear export proteinCRM1 and 14-3-3, which in turn induce the disassociationof HDAC4, 5 and 7 from MEF2 and their export into thecytosol. The release of HDAC4, 5 and 7 from MEF2 allowsthe histone acetyltransferase p300 to bind to MEF2, therebyswitching MEF2 from a transcriptional repressor to a tran-scriptional activator.
Class IIb HDACs, HDAC6, and HDAC10, are distin-guished from other HDACs in their unusual structure andfunction. Both HDAC6 and HDAC10 possess a secondcatalytic domain not found in other HDACs, though thisdomain is not functional in HDAC10. The unique seconddeacetylase domain provides HDAC6 the ability to deace-tylate non-histone proteins such as α-tubulin, cortactin andhsp90 [22, 23]. Very little is known about the function ofHDAC10 thus far.
Fig. 1 Schema of metanephric kidney development. a At E10.5 in mice,the UB arises from the nephric duct and invades adjacent MM. bAt E11.5,the UB continues to elongate and branch. MM cells condense at each UBtip, forming a population of nephron progenitor cells (cap mesenchyme
(CM) cells). c, d In response to Wnt signals from the UB, the CM cellsform PTA, and are subsequently transformed to the polarized epithelialstructures of RV, CB, and SB. eByE14.5, the nephron segments are furtherpatterned and the first generation of glomeruli is formed
Pediatr Nephrol (2013) 28:689–698 691

Class IV HDACs
Class IV HDAC only has one member, HDAC11. HDAC11is enriched in the brain, heart, muscle, kidney, and testis, butits functions remain unknown [24, 25].
Developmental expression of HDACs in the kidney
To gain insights into potential functions of HDACs in renalorganogenesis, we examined their expression in the kidney.Based on the Gudmap database [26] and our data [27], themouse embryonic kidney expresses all 11 members of clas-sical HDACs. Among the nine HDAC genes (HDAC1–9)examined in our lab, HDAC 1–4, 7, and 9 are subject todevelopmental control and decline significantly during thematuration from embryonic to adult life. In comparison,HDAC5, 6, and 8 are constitutively expressed. In line withthese findings, immunofluorescence staining demonstratesthat expression of HDAC1, 2, and 3 is enriched in theundifferentiated MM, UB branches and interstitial stroma,but reduced with differentiation processes (Fig. 2 and [27]).Of note, HDAC3 is maintained at a high level in the podo-cytes, implicating its function in glomerular biology. Similarto other tissues, HDAC7, 8 and 9 are predominantlyexpressed in the renal microvasculature. Yet, interestingly,the global acetylation levels of histones H3 and H4 remainstable during kidney development. Maintenance of globalhistone acetylation levels suggests that the balance betweenHAT and HDAC activities is tightly regulated duringdevelopment.
Lessons from HDAC inhibitors
HDAC inhibitors (HDACi) are a group of compounds thatblock the activities of HDACs typically by binding to thezinc-containing catalytic domain of the HDACs. HDACican be grouped into 4 main structural classes, includinghydroxamate, aliphatic acids, cyclic peptide, and benza-mide. The hydroxamate class of HDACi, which includesTSA (trichostatin A), SAHA (suberoylanilide hydroxamicacid), and Scriptaid, are considered “pan-HDAC inhibitors”,as they generally affect all members of Class I and IIHDACs to similar degrees. In contrast, the other threeclasses of HDAC inhibitors show various selectivities to-ward class I HDACs. For example, aliphatic acid VPA(valproic acid) is modestly selective for class I HDACs;while benzamide HDAC inhibitor MS-275 almost exclu-sively inhibits HDAC 1, 2, and 3 [28, 29].
Utilizing HDACi, we examined the biological functionsof HDACs in ex vivo metanephric organ cultures, whichrecapitulate nephrogenesis and UB branching in vivo [27].
Long-term (>12-h) treatment with Scriptaid (2.0 μg/ml) inE13.5 kidneys impaired both nephrogenesis and UB branch-ing associated with remarkable cell cycle arrest and apopto-sis. When the kidneys were treated for a shorter-term (6-h),HDAC inhibition selectively dysregulated a small subset ofgenes, without effecting cell proliferation and cell survival.Genome-wide profiling revealed that ∼12% genes (4,800/41,000 transcripts) are significantly altered in response toHDACi (using a cutoff of 1.5-fold, p<0.05), with ∼5% ofgene transcripts upregulated (range 1.5 to 14-fold increase,n02,085) and 7% of gene transcripts down-regulated (range1.5- to 26-fold decrease, n02,865). The deregulated geneswere broadly distributed in five major pathways: cell cycle,Wnt/β-catenin, TGF- β /Smad, cancer and PI3K/AKT.CDK inhibitors/tumor suppressor genes, including p21,p15, p19, Bop1 and Htra1, were significantly up-regulated. And at the same time, many oncogenes, such asc-myc, N-myc, cyclin D1, cyclin J, cyclin B2 and thymidylatesynthase, were dramatically down-regulated. Therefore, theimbalance between growth promoters and suppressors col-lectively contribute to metanephric growth arrest induced byHDACi. Further analysis using the BINGO Network GeneOntology Tool revealed that expression of key developmen-tal renal regulators, including Osr1, Eya1, Pax2, Pax8,WT1, Gdnf, Emx2, Wnt9b, Wnt4, Sfrp1, Sfrp2, Lhx1 andFoxD1, is dependent on intact HDAC activity. Treatment ofcultured MK4 cells, an immortalized MM cell line, withScriptaid, largely reproduced the gene expression changesseen in the kidney explants, and chromatin immunoprecip-itation assays revealed that HDAC inhibition is associatedwith promoter/enhancer histone hyperacetylation of Pax2,Pax8, Gdnf, Sfrp1, and p21. Thus, HDACs most likelyregulate these MM genes via a direct effect on histoneacetylation. Furthermore, gene knockdown studies demon-strated that HDAC1 and 2 play a redundant role in regula-tion of Pax2, Pax8 and Sfrp1. This finding is consistent withour data gained in HDAC1 and 2 conditional mutant mice,which are discussed later in this review. Taken together,these data demonstrate that class I and II HDACs, particu-larly HDAC1 and 2, critically control a number of keydevelopmental renal regulators, and are essentially requiredfor cell proliferation, differentiation and survival in themetanephros.
Interestingly, HDACs appear to exert opposite func-tions on cell proliferation in the pronephros in zebrafish[30]. de Groh et al. reported that inhibition of class Iand II HDACs by TSA or PTBA stimulated renal pro-genitor cell proliferation in a retinoic acid (RA)-dependentmanner, resulting in expansion of renal progenitor cells. Theauthor proposed that HDACs probably are part of the RAtranscriptional repression complex. Therefore, inhibition ofHDAC activities releases the repressive effects of RA on genetranscription.
692 Pediatr Nephrol (2013) 28:689–698

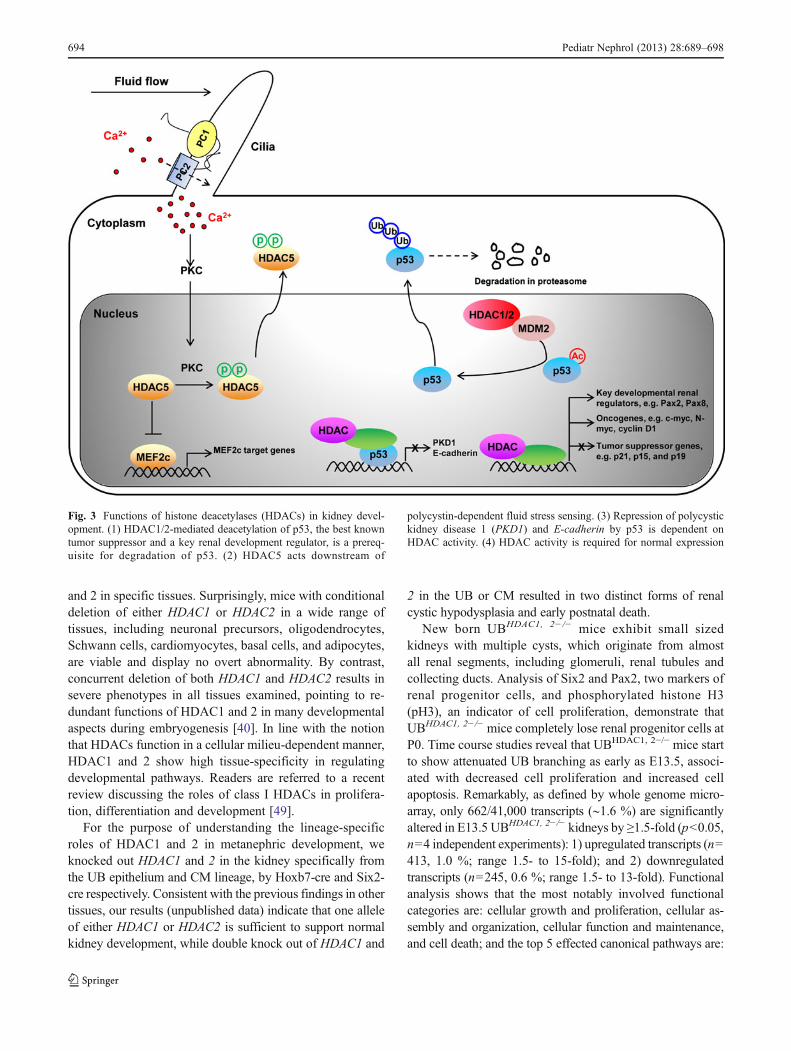
Studies in our laboratory also indicate that HDACs play acrucial role in gene–environment interactions during kidneydevelopment [31–33]. Mice lacking bradykinin B2 receptor(B2R) show no kidney abnormalities when receiving normalsodium intake. In contrast, B2R mutant mice exposed togestational salt stress exhibit renal dysgenesis and die short-ly after birth. The phenotype is caused by phosphorylationof p53 on Ser23, which stabilizes p53 by preventing theinteraction of p53 with Mdm2, an E3 ubiquitin ligase me-diating p53 degradation. Either genetic deletion of p53 orreplacement of Ser23 with Alanine restored normal renaldevelopment. Chromatin immunoprecipitation (ChIP) iden-tified E-cadherin as a direct target gene of p53, and therepression of E-cadherin by p53 depends on deacetylationof the E-cadherin promoter by HDACs. Treatment withTSA can reverse p53-mediated promoter deacetylation andrepression. Moreover, the same cooperation model of p53and HDACs in transcriptional repression is observed forPKD1 (polycystic kidney disease 1) [34].
Autosomal dominant polycystic kidney disease (ADPKD)affects nearly one in 1,000 Americans. It is characterized byprogressive cyst development and bilaterally enlarged kidneyswith multiple cysts. Most cases of ADPKD are caused bymutations in polycystic kidney disease 1 (PKD1) or polycystickidney disease 2 (PKD2) genes, which encode polycystin 1(PC1) and polycystin 2 (PC2), respectively [35]. There isevidence that PC1 and PC2 form a receptor-calcium channelcomplex at the surface of the primary cilium of renal epithelialcells. Within the complex, PC1 acts as a mechanosensor offluid flow, while PC2 is a Ca2+-translocating pore. The PC1/2
channel complex is required to induce a calcium influx inresponse to mechanical stimulation (shear stress) (Fig. 3) [36].Two recent studies showed therapeutic potentials of HDACinhibitors on ADPKD. Inhibition of class I and II HDACs byVPA and TSA was found to reduce cyst formation and slowthe decline of kidney function in PKD1 conditional knockoutmice and PKD2 null mice, respectively [37, 38]. The thera-peutic effects could be, at least partially, attributed to inhibi-tion of HDAC5, a class IIa HDAC member, which isdiscussed in detail later.
Lessons from mice genetic models
HDAC inhibitors provide valuable information of the de-velopmental functions of HDACs, but with limitation be-cause of their lack of specificity. In this respect, geneticdeletion or overexpression of individual HDACs in micehas revealed highly specific and redundant functions ofthese enzymes in development and disease (Table 1). Pres-ent evidence has indicated that HDAC1, 2 and 5 controlspecific developmental pathways in the kidney.
Redundant roles of HDAC1 and 2 in renal organogenesis
To circumvent the early lethality associated with global genedeletion of HDAC1 or HDAC2, conditional deletion strate-gies have been used to investigate the functions of HDAC1
Fig. 2 Immunofluorescencelocalization of histonedeacetylase 1 (HDAC1) andHDAC2 in newborn kidneys.Sections from newborn kidneysco-labeled with HDAC1 (a andc) or HDAC2 (b and d) (red),cytokeratin (CK, blue, a markerof UB branches), and lotustetragonolobus lectin aggluti-nin (LTL, green, a marker ofproximal tubules). UB uretericbud branch, RV renal vesicle,SB S-shaped body, NZnephrogenic zone, DZdifferentiation zone
Pediatr Nephrol (2013) 28:689–698 693
and 2 in specific tissues. Surprisingly, mice with conditionaldeletion of either HDAC1 or HDAC2 in a wide range oftissues, including neuronal precursors, oligodendrocytes,Schwann cells, cardiomyocytes, basal cells, and adipocytes,are viable and display no overt abnormality. By contrast,concurrent deletion of both HDAC1 and HDAC2 results insevere phenotypes in all tissues examined, pointing to re-dundant functions of HDAC1 and 2 in many developmentalaspects during embryogenesis [40]. In line with the notionthat HDACs function in a cellular milieu-dependent manner,HDAC1 and 2 show high tissue-specificity in regulatingdevelopmental pathways. Readers are referred to a recentreview discussing the roles of class I HDACs in prolifera-tion, differentiation and development [49].
For the purpose of understanding the lineage-specificroles of HDAC1 and 2 in metanephric development, weknocked out HDAC1 and 2 in the kidney specifically fromthe UB epithelium and CM lineage, by Hoxb7-cre and Six2-cre respectively. Consistent with the previous findings in othertissues, our results (unpublished data) indicate that one alleleof either HDAC1 or HDAC2 is sufficient to support normalkidney development, while double knock out of HDAC1 and
2 in the UB or CM resulted in two distinct forms of renalcystic hypodysplasia and early postnatal death.
New born UBHDAC1, 2−/− mice exhibit small sizedkidneys with multiple cysts, which originate from almostall renal segments, including glomeruli, renal tubules andcollecting ducts. Analysis of Six2 and Pax2, two markers ofrenal progenitor cells, and phosphorylated histone H3(pH3), an indicator of cell proliferation, demonstrate thatUBHDAC1, 2−/− mice completely lose renal progenitor cells atP0. Time course studies reveal that UBHDAC1, 2−/− mice startto show attenuated UB branching as early as E13.5, associ-ated with decreased cell proliferation and increased cellapoptosis. Remarkably, as defined by whole genome micro-array, only 662/41,000 transcripts (∼1.6 %) are significantlyaltered in E13.5 UBHDAC1, 2−/− kidneys by ≥1.5-fold (p<0.05,n04 independent experiments): 1) upregulated transcripts (n0413, 1.0 %; range 1.5- to 15-fold); and 2) downregulatedtranscripts (n0245, 0.6 %; range 1.5- to 13-fold). Functionalanalysis shows that the most notably involved functionalcategories are: cellular growth and proliferation, cellular as-sembly and organization, cellular function and maintenance,and cell death; and the top 5 effected canonical pathways are:
Fig. 3 Functions of histone deacetylases (HDACs) in kidney devel-opment. (1) HDAC1/2-mediated deacetylation of p53, the best knowntumor suppressor and a key renal development regulator, is a prereq-uisite for degradation of p53. (2) HDAC5 acts downstream of
polycystin-dependent fluid stress sensing. (3) Repression of polycystickidney disease 1 (PKD1) and E-cadherin by p53 is dependent onHDAC activity. (4) HDAC activity is required for normal expression
694 Pediatr Nephrol (2013) 28:689–698
sonic hedgehog signaling, basal cell carcinoma signaling,IGF-1 signaling, amyloid processing, and ascorbate and alda-rate metabolism.
Newborn CMHDAC1, 2−/− mice also display bilateral renalcystic dysplasia. But the majority of renal cysts originatefrom proximal tubules, while collecting ducts do not con-tribute to the cysts. Interestingly, renal progenitor cells arestill found in the CM at P0, though at lower cell number andwith much lower expression of Pax2 and Six2. In agreementwith this observation, examination of pH3 and caspase 3indicates that loss of HDAC1 and 2 in the CM cells onlyblocks cell proliferation but has no effects on cell apoptosis.A careful histological examination revealed that nephronformation is impaired in the CMHDAC1, 2−/− mice. Renalvesicles are still formed, though no well-formed comma-shaped bodies or s-shaped bodies were detected.
Our data reveal that p53, the best known tumor suppres-sor and a key renal development regulator, is a non-histonetarget of HDAC1 and 2 in the UB and CM. A robustincrease of acetylated p53 and total p53 was detected inthe HDAC1 and 2 deficient UB and CM cells. This findingis consistent with two recent studies showing that HDAC1and 2 mediate p53 deacetylation in mice epidermal progen-itor cells and oocytes [50, 51]. Moreover, it has been shownthat the HDAC-mediated deacetylation of p53 is requiredfor degradation of p53 in 293 T cells [52]. GOF (gain offunction) experiments in our lab also demonstrate that ex-cessive p53 in the UB or MM causes bilateral renal hypo-dysplasia [53].
Taken together, our results demonstrate that HDAC1 and2 selectively regulate key kidney developmental pathways,including sonic hedgehog pathway, Pax2-Six2-GDNF-cRetpathway and p53 pathway.
HDAC5
A recent study revealed that HDAC5 and MEF2c aredownstream targets of polycystin-dependent fluid stresssensing in renal epithelial cells [38]. Fluid flow stimula-tion in polarized mouse embryonic kidney (MEK) cells isfound to induce calcium influx into the cells. Increasedintracellular calcium activates protein kinase C (PKC),which in turn leads to phosphorylation of HDAC5, disso-ciation of HDAC5 from MEF2c and nuclear export ofHDAC5. Thus, fluid stress releases MEF2c from therepressive influence of HDAC5, switching MEF2 from atranscriptional repressor to a transcriptional activator. In-triguingly, deletion of the PKD1−/− gene eliminates cal-cium influx and its subsequent events, suggesting thatdefective responses to fluid stress by HDAC5 and MEF2cprobably contribute to ADPKD. Supporting this idea,conditional knockout of MEF2c in renal tubules and glo-meruli results in renal tubule dilation and epithelial cystsin adult mice by 5 months of age, while HDAC5 hetero-zygosity alleviates cyst formation in PKD2−/− mouseembryos.
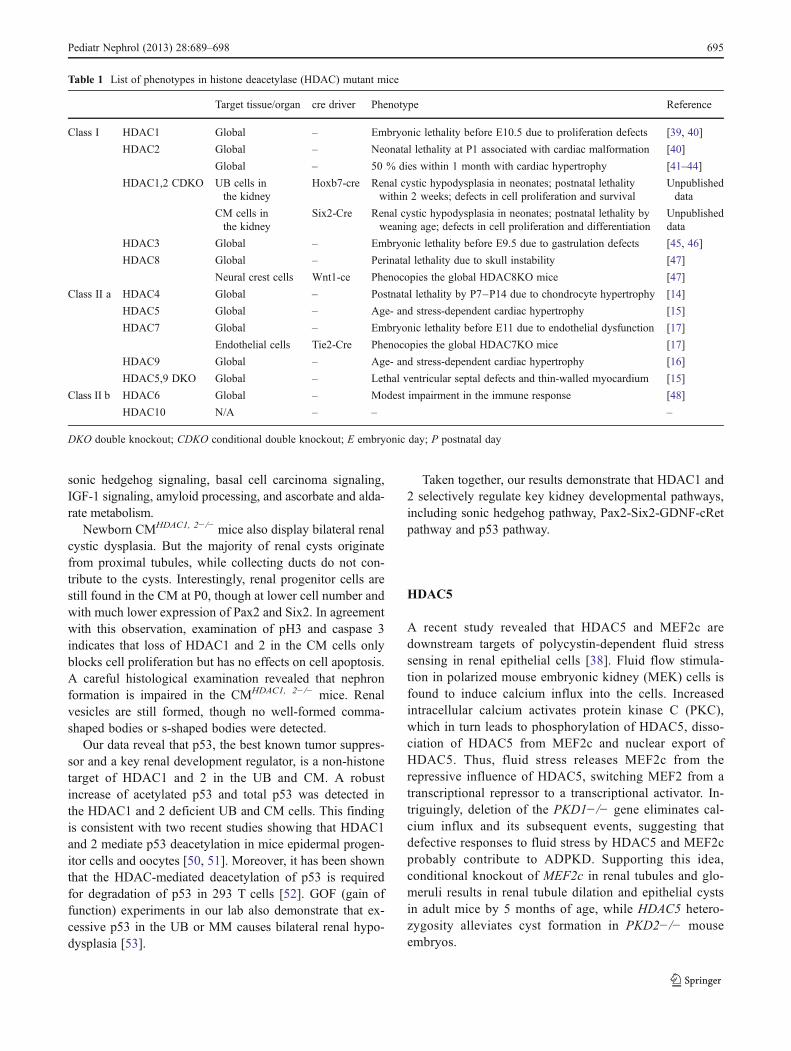
Table 1 List of phenotypes in histone deacetylase (HDAC) mutant mice
Target tissue/organ cre driver Phenotype Reference
Class I HDAC1 Global – Embryonic lethality before E10.5 due to proliferation defects [39, 40]
HDAC2 Global – Neonatal lethality at P1 associated with cardiac malformation [40]
Global – 50 % dies within 1 month with cardiac hypertrophy [41–44]
HDAC1,2 CDKO UB cells inthe kidney
Hoxb7-cre Renal cystic hypodysplasia in neonates; postnatal lethalitywithin 2 weeks; defects in cell proliferation and survival
Unpublisheddata
CM cells inthe kidney
Six2-Cre Renal cystic hypodysplasia in neonates; postnatal lethality byweaning age; defects in cell proliferation and differentiation
Unpublisheddata
HDAC3 Global – Embryonic lethality before E9.5 due to gastrulation defects [45, 46]
HDAC8 Global – Perinatal lethality due to skull instability [47]
Neural crest cells Wnt1-ce Phenocopies the global HDAC8KO mice [47]
Class II a HDAC4 Global – Postnatal lethality by P7–P14 due to chondrocyte hypertrophy [14]
HDAC5 Global – Age- and stress-dependent cardiac hypertrophy [15]
HDAC7 Global – Embryonic lethality before E11 due to endothelial dysfunction [17]
Endothelial cells Tie2-Cre Phenocopies the global HDAC7KO mice [17]
HDAC9 Global – Age- and stress-dependent cardiac hypertrophy [16]
HDAC5,9 DKO Global – Lethal ventricular septal defects and thin-walled myocardium [15]
Class II b HDAC6 Global – Modest impairment in the immune response [48]
HDAC10 N/A – – –
DKO double knockout; CDKO conditional double knockout; E embryonic day; P postnatal day
Pediatr Nephrol (2013) 28:689–698 695

Clinical implications and issues for the future
In recent years, there has been a dramatic expansion ofclinical investigation on HDACi. Besides the current clini-cal utility of SAHA as an anticancer agent for cutaneous T-cell lymphoma, a number of HDACi belonging to differentclasses are being tested in clinical/preclinical trials for awide variety of diseases, including renal cell carcinoma(RCC) [54]. As in many other types of solid tumors, mono-therapy of RCC with single HDACi fails to show significantantitumor activity, though combination therapy using sever-al HDACi with other antitumor drugs (e.g., Entinostat with13-cis-retinoic acid or Interleukin 2, Vorinostat with VEGFinhibitor bevacizumab, and LDH589 with mTOR Inhibitor)has already demonstrated therapeutic benefits in RCC. Theexact mechanisms responsible for the anti-tumor activitiesof HDACi are still elusive, though multiple lines of evi-dence indicate the potential roles of HDACi in inducing cellcycle arrest and differentiation, promoting cell apoptosis andROS (reactive oxygen species)-induced cell death, andinhibiting metastasis and angiogenesis by altering the acety-lation status of histones as well as non-histone proteins. Ourstudy demonstrates that administration of HDACi in embry-onic kidneys results in many comparable changes in cellbiology and gene expression to studies using HDACi intumors. Therefore, understanding the genetic pathways con-trolled by HDACs in kidney development would help us togain insights into the mechanisms whereby HDACi exert theiranti-tumor effects. For example, a number of tumor suppres-sor genes and oncogenes are dysregulated following HDACitreatment in embryonic kidneys. These genes should be care-fully examined in tumor studies using HDACi.
More importantly, in addition to cancer, administration ofHDACi at low concentrations has been used to treat a broadrange of diseases that are not related to cancer, such as HIVinfection, asthma, cardiac hypertrophy, and hepatitis[55–59]. The observation of therapeutic effects of HDACiin PKD1 and PKD2 mutant mice raises the possibility thatHDACi could be explored as candidate drugs for the treat-ment of ADPKD. In particular, genetic evidence shows thatHDAC5 phosphorylation is a key step in response to fluidstress in renal epithelial cells, though the functions of otherHDAC members require further investigation.
Although pan-HDAC inhibitors are well tolerated in themajority of patients, they display many side effects, includ-ing gastrointestinal symptoms, bone marrow suppression,cardiac toxicity, thrombocytopenia, neutropenia and fatigue[60, 61]. Given the specificity and redundancy of HDACsevidenced by the unique phenotypes resulting from geneticdeletion of individual HDACs, a promising strategy is to usemore selective HDACi or isotype-specific HDACi to pre-cisely target the disease relevant HDAC(s) while reducingor eliminating the side effects [62]. So far, phase I trials of
class I selective HDACi (e.g., Depsipeptide and MS-275)show similar toxicity as pan-HDACi, suggesting that class IHDAC inhibition, particularly HDAC1 and 2, is largelyresponsible for the observed side effects [63, 64]. Evaluationof class II selective HDACi has just started. ACY-1215, aselective HDAC6 inhibitor, has recently entered Phase I/IIclinical trials in North America [65]. In its preclinical study,ACY-1215, in combination with bortezomib, significantlyrepressed tumor growth and enhanced animal survival intwo different mouse models for multiple myeloma. As dis-cussed above, global deletion of HDAC6 leads to no obvi-ous phenotype in mice. So, it is tempting to speculate thatACY-1215 will be likely devoid of the side effects commonto pan-HDACi. Given the potential usage of HDACi inkidney diseases, such as ADPKD and kidney cancers, animportant task in the future will be deciphering the role ofindividual HDACs in renal physiology and disease, to allowdevelopment of isoform-specific HDACi for optimizedtherapy.
References
1. Schedl A (2007) Renal abnormalities and their developmentalorigin. Nat Rev Genet 8:791–802
2. Kiefer JC (2007) Epigenetics in development. Dev Dyn 236:1144–1156
3. Reidy KJ, Rosenblum ND (2009) Cell and molecular biology ofkidney development. Semin Nephrol 29:321–337
4. Dressler GR (2009) Advances in early kidney specification, devel-opment and patterning. Development 136:3863–3874
5. Smith CL (2008) A shifting paradigm: histone deacetylases andtranscriptional activation. BioEssays 30:15–24
6. De Ruijter AJM, Van Gennip AH, Caron HN, Kemp S, VanKuilenburg ABP (2003) Histone deacetylases (HDACs): charac-terization of the classical HDAC family. Biochem J 370:737–749
7. Glozak MA, Sengupta N, Zhang XH, Seto E (2005) Acetylationand deacetylation of non-histone proteins. Gene 363:15–23
8. Spange S, Wagner T, Heinzel T, Kramer OH (2009) Acetylation ofnon-histone proteins modulates cellular signalling at multiple lev-els. J Biochem Cell Biol 41:185–198
9. Yang XJ, Gregoire S (2005) Class II histone deacetylases: fromsequence to function, regulation, and clinical implication. Mol CellBiol 25:2873–2884
10. Sauve AA, Wolberger C, Schramm VL, Boeke JD (2006) Thebiochemistry of sirtuins. Annu Rev Biochem 75:435–465
11. Yang WM, Tsai SC, Wen YD, Fejer G, Seto E (2002) Functionaldomains of histone deacetylase-3. J Biol Chem 277:9447–9454
12. Karagianni P, Wong J (2007) HDAC3: taking the SMRT-N-CoRrect road to repression. Oncogene 26:5439–5449
13. Haberland M, Montgomery RL, Olson EN (2009) The many rolesof histone deacetylases in development and physiology: implica-tions for disease and therapy. Nat Rev Genet 10:32–42
14. Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E,McAnally J, Pomajzl C, Shelton JM, Richardson JA, Karsenty G,Olson EN (2004) Histone deacetylase 4 controls chondrocytehypertrophy during skeletogenesis. Cell 119:555–566
15. Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, OlsonEN (2004) Histone deacetylases 5 and 9 govern responsiveness of
696 Pediatr Nephrol (2013) 28:689–698

the heart to a subset of stress signals and play redundant roles inheart development. Mol Cell Biol 24:8467–8476
16. Zhang CL, McKinsey TA, Chang SR, Antos CL, Hill JA, OlsonEN (2002) Class II histone deacetylases act as signal-responsiverepressors of cardiac hypertrophy. Cell 110:479–488
17. Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN (2006)Histone deacetylase 7 maintains vascular integrity by repressingmatrix metalloproteinase 10. Cell 126:321–334
18. Choi SJ, Park SY, Han TH (2001) 14-3-3tau associates with andactivates the MEF2D transcription factor during muscle cell dif-ferentiation. Nucleic Acids Res 29:2836–2842
19. Dressel U, Bailey PJ, Wang SC, Downes M, Evans RM, MuscatGE (2001) A dynamic role for HDAC7 in MEF2-mediated muscledifferentiation. J Biol Chem 276:17007–17013
20. Kim MS, Fielitz J, McAnally J, Shelton JM, Lemon DD, McKinseyTA, Richardson JA, Bassel-Duby R, Olson EN (2008) Protein kinaseD1 stimulates MEF2 activity in skeletal muscle and enhances muscleperformance. Mol Cell Biol 28:3600–3609
21. McKinsey TA, Zhang CL, Lu J, Olson EN (2000) Signal-dependent nuclear export of a histone deacetylase regulates muscledifferentiation. Nature 408:106–111
22. Grozinger CM, Hassig CA, Schreiber SL (1999) Three proteinsdefine a class of human histone deacetylases related to yeastHda1p. Proc Natl Acad Sci USA 96:4868–4873
23. Verdel A, Khochbin S (1999) Identification of a new family ofhigher eukaryotic histone deacetylases. Coordinate expression ofdifferentiation-dependent chromatin modifiers. J Biol Chem274:2440–2445
24. Gao L, Cueto MA, Asselbergs F, Atadja P (2002) Cloning andfunctional characterization of HDAC11, a novel member of thehuman histone deacetylase family. J Biol Chem 277:25748–25755
25. Liu H, Hu Q, Kaufman A, D’Ercole AJ, Ye P (2008) Develop-mental expression of histone deacetylase 11 in the murine brain. JNeurosci Res 86:537–543
26. McMahon AP, Aronow BJ, Davidson DR, Davies JA, Gaido KW,Grimmond S, Lessard JL, Little MH, Potter SS, Wilder EL, ZhangP, Project G (2008) GUDMAP: the Genitourinary DevelopmentalMolecular Anatomy Project. J Am Soc Nephrol 19:667–671
27. Chen S, Bellew C, Yao X, Stefkova J, Dipp S, Saifudeen Z,Bachvarov D, El-Dahr SS (2011) Histone deacetylase (HDAC)activity is critical for embryonic kidney gene expression, growth,and differentiation. J Biol Chem 286:32775–32789
28. Ma X, Ezzeldin HH, Diasio RB (2009) Histone deacetylase inhib-itors: current status and overview of recent clinical trials. Drugs69:1911–1934
29. Dokmanovic M, Clarke C, Marks PA (2007) Histone deacetylaseinhibitors: overview and perspectives. Mol Cancer Res 5:981–989
30. de Groh ED, Swanhart LM, Cosentino CC, Jackson RL, Dai W,Kitchens CA, Day BW, Smithgall TE, Hukriede NA (2010) Inhi-bition of histone deacetylase expands the renal progenitor cellpopulation. J Am Soc Nephrol 21:794–802
31. El-Dahr SS, Harrison-Bernard LM, Dipp S, Yosipiv IV, Meleg-Smith S (2000) Bradykinin B2 null mice are prone to renal dys-plasia: gene–environment interactions in kidney development.Physiol Genomics 3:121–131
32. Fan H, Harrell JR, Dipp S, Saifudeen Z, El-Dahr SS (2005) A novelpathological role of p53 in kidney development revealed by gene–environment interactions. Am J Physiol Renal Physiol 288:F98–F107
33. El-Dahr SS, Aboudehen K, Dipp S (2008) Bradykinin B2 receptornull mice harboring a Ser23-to-Ala substitution in the p53 gene areprotected from renal dysgenesis. Am J Physiol Renal Physiol 295:F1404–F1413
34. Van Bodegom D, Saifudeen Z, Dipp S, Puri S, Magenheimer BS,Calvet JP, El-Dahr SS (2006) The polycystic kidney disease-1 geneis a target for p53-mediated transcriptional repression. J Biol Chem281:31234–31244
35. Ong AC, Harris PC (2005) Molecular pathogenesis of ADPKD:the polycystin complex gets complex. Kidney Int 67:1234–1247
36. Yoder BK (2007) Role of primary cilia in the pathogenesis ofpolycystic kidney disease. J Am Soc Nephrol 18:1381–1388
37. Cao Y, Semanchik N, Lee SH, Somlo S, Barbano PE, Coifman R,Sun Z (2009) Chemical modifier screen identifies HDAC inhib-itors as suppressors of PKD models. Proc Natl Acad Sci USA106:21819–21824
38. Xia S, Li X, Johnson T, Seidel C, Wallace DP, Li R (2010)Polycystin-dependent fluid flow sensing targets histone deacety-lase 5 to prevent the development of renal cysts. Development137:1075–1084
39. Lagger G, O’Carroll D, Rembold M, Khier H, Tischler J, WeitzerG, Schuettengruber B, Hauser C, Brunmeir R, Jenuwein T, SeiserC (2002) Essential function of histone deacetylase 1 in prolifera-tion control and CDK inhibitor repression. EMBO J 21:2672–2681
40. Montgomery RL, Davis CA, Potthoff MJ, Haberland M, Fielitz J,Qi X, Hill JA, Richardson JA, Olson EN (2007) Histone deacety-lases 1 and 2 redundantly regulate cardiac morphogenesis, growth,and contractility. Genes Dev 21:1790–1802
41. Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M, GaisbergerM, Hartl A, Epstein MM, Matthias P, Seiser C, Ellmeier W (2010)Conditional deletion of histone deacetylase 1 in T cells leads toenhanced airway inflammation and increased Th2 cytokine pro-duction. J Immunol 185:3489–3497
42. Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W, Wang T, Floss T,Goettlicher M, Noppinger PR, Wurst W, Ferrari VA, Abrams CS,Gruber PJ, Epstein JA (2007) Hdac2 regulates the cardiac hyper-trophic response by modulating Gsk3 beta activity. Nat Med13:324–331
43. Zimmermann S, Kiefer F, Prudenziati M, Spiller C, Hansen J,Floss T, Wurst W, Minucci S, Gottlicher M (2007) Reduced bodysize and decreased intestinal tumor rates in HDAC2-mutant mice.Cancer Res 67:9047–9054
44. Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N,Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE,DePinho RA, Jaenisch R, Tsai LH (2009) HDAC2 negativelyregulates memory formation and synaptic plasticity. Nature459:55–60
45. Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S,Humphries KM, Richardson JA, Bassel-Duby R, Olson EN (2008)Maintenance of cardiac energy metabolism by histone deacetylase3 in mice. J Clin Invest 118:3588–3597
46. Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, WilsonAJ, Zheng S, Yenamandra A, Locke K, Yuan JL, Bonine-SummersAR, Wells CE, Kaiser JF, Washington MK, Zhao Z, Wagner FF,Sun ZW, Xia F, Holson EB, Khabele D, Hiebert SW (2010) Hdac3is essential for the maintenance of chromatin structure and genomestability. Cancer Cell 18:436–447
47. Haberland M, Mokalled MH, Montgomery RL, Olson EN (2009)Epigenetic control of skull morphogenesis by histone deacetylase8. Genes Dev 23:1625–1630
48. Zhang Y, Kwon S, Yamaguchi T, Cubizolles F, Rousseaux S,Kneissel M, Cao C, Li N, Cheng HL, Chua K, Lombard D,Mizeracki A, Matthias G, Alt FW, Khochbin S, Matthias P(2008) Mice lacking histone deacetylase 6 have hyperacetylatedtubulin but are viable and develop normally. Mol Cell Biol28:1688–1701
49. Reichert N, Choukrallah MA, Matthias P (2012) Multiple roles ofclass I HDACs in proliferation, differentiation, and development.Cell Mol Life Sci. doi:10.1007/s00018-012-0921-9
50. LeBoeuf M, Terrell A, Trivedi S, Sinha S, Epstein JA, Olson EN,Morrisey EE, Millar SE (2010) Hdac1 and Hdac2 act redundantlyto control p63 and p53 functions in epidermal progenitor cells. DevCell 19:807–818
Pediatr Nephrol (2013) 28:689–698 697

51. Ma P, Pan H, Montgomery RL, Olson EN, Schultz RM (2012)Compensatory functions of histone deacetylase 1 (HDAC1) andHDAC2 regulate transcription and apoptosis during mouse oocytedevelopment. Proc Natl Acad Sci USA 109:E481–E489
52. Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, AppellaE, Yao TP (2002) MDM2-HDAC1-mediated deacetylation of p53is required for its degradation. EMBO J 21:6236–6245
53. Hilliard S, Aboudehen K, Yao X, El-Dahr SS (2011) Tight regu-lation of p53 activity by Mdm2 is required for ureteric bud growthand branching. Dev Biol 353:354–366
54. Hudes GR, Carducci MA, Choueiri TK, Esper P, Jonasch E,Kumar R, Margolin KA, Michaelson MD, Motzer RJ, Pili R,Roethke S, Srinivas S (2011) NCCN Task Force report: optimizingtreatment of advanced renal cell carcinoma with molecular tar-geted therapy. J Nat Compr Cancer Netw 9(Suppl 1):S1–S29
55. Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, ModenaD, Moras ML, Pozzi P, Reznikov LL, Siegmund B, Fantuzzi G,Dinarello CA, Mascagni P (2005) The histone deacetylase inhib-itor ITF2357 reduces production of pro-inflammatory cytokines invitro and systemic inflammation in vivo. Mol Med 11:1–15
56. Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, WiegandA, Landay AL, Coombs RW, Richman DD, Mellors JW, CoffinJM, Bosch RJ, Margolis DM (2005) Depletion of latent HIV-1infection in vivo: a proof-of-concept study. Lancet 366:549–555
57. Choi JH, Oh SW, Kang MS, Kwon HJ, Oh GT, Kim DY (2005)Trichostatin A attenuates airway inflammation in mouse asthmamodel. Clin Exp Allergy 35:89–96
58. Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, OlsonEN, Hill JA (2006) Suppression of class I and II histonedeacetylases blunts pressure-overload cardiac hypertrophy. Circula-tion 113:2579–2588
59. Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, ZhangCL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN (2003) Dose-dependent blockade to cardiomyocyte hypertrophy by histonedeacetylase inhibitors. J Biol Chem 278:28930–28937
60. Walkinshaw DR, Yang XJ (2008) Histone deacetylase inhibitors asnovel anticancer therapeutics. Curr Oncol 15:237–243
61. Walkinshaw DR, Tahmasebi S, Bertos NR, Yang XJ (2008) His-tone deacetylases as transducers and targets of nuclear signaling. JCell Biochem 104:1541–1552
62. Karagiannis TC, El-Osta A (2007) Will broad-spectrum histonedeacetylase inhibitors be superseded by more specific compounds?Leukemia 21:61–65
63. Sandor V, Bakke S, Robey RW, Kang MH, Blagosklonny MV,Bender J, Brooks R, Piekarz RL, Tucker E, Figg WD, Chan KK,Goldspiel B, Fojo AT, Balcerzak SP, Bates SE (2002) Phase I trialof the histone deacetylase inhibitor, depsipeptide (FR901228, NSC630176), in patients with refractory neoplasms. Clin Cancer Res8:718–728
64. Ryan QC, Headlee D, Acharya M, Sparreboom A, Trepel JB, Ye J,Figg WD, Hwang K, Chung EJ, Murgo A, Melillo G, Elsayed Y,Monga M, Kalnitskiy M, Zwiebel J, Sausville EA (2005) Phase Iand pharmacokinetic study of MS-275, a histone deacetylase in-hibitor, in patients with advanced and refractory solid tumors orlymphoma. J Clin Oncol 23:3912–3922
65. Santo L, Hideshima T, Kung AL, Tseng JC, Tamang D, Yang M,Jarpe M, van Duzer JH, Mazitschek R, Ogier WC, Cirstea D,Rodig S, Eda H, Scullen T, Canavese M, Bradner J, AndersonKC, Jones SS, Raje N (2012) Preclinical activity, pharmacody-namic, and pharmacokinetic properties of a selective HDAC6inhibitor, ACY-1215, in combination with bortezomib in multiplemyeloma. Blood 119:2579–2589
698 Pediatr Nephrol (2013) 28:689–698