heme oxygenase inhibits human airway smooth muscle proliferation
TRANSCRIPT

1
Heme oxygenase inhibits human airway smooth muscle
proliferation via a bilirubin-dependent modulation
of ERK1/2 phosphorylation
by
Camille Taillé, Abdelhamid Almolki, Moussa Benhamed,
Christine Zedda, Jérôme Mégret,
Patrick Berger*, Guy Lesèche**, Elie Fadel ***, Tokio Yamaguchi ****,
Roger Marthan*, Michel Aubier and Jorge Boczkowski.
Institut National de la Santé et de la Recherche Médicale (INSERM), Unité 408,
Faculté de Médecine Xavier Bichat, Paris ; * INSERM, E9937, Université Victor Ségalen, Bordeaux 2;
** Service de Chirurgie Vasculaire et Thoracique, Hôpital Beaujon, Clichy ; *** Service de Chirurgie Thoracique, Centre Chirurgical Marie Lannelongue,
le Plessis Robinson , France ; **** Medical Research Institute, Tokyo Medical and Dental University, Tokyo, Japan.
Correspondance to : Jorge Boczkowski, MD, PhD. INSERM U408 Faculté de Médecine Xavier Bichat
16 rue Henri Huchard 75018 – Paris - France . tel : 33 1 44 85 62 51 Fax : 33 1 42 26 33 30 E-mail : [email protected]
Running title : Bilirubin and Human Airway Smooth Muscle proliferation.
Copyright 2003 by The American Society for Biochemistry and Molecular Biology, Inc.
JBC Papers in Press. Published on April 10, 2003 as Manuscript M300364200 by guest on February 24, 2018
http://ww
w.jbc.org/
Dow
nloaded from

2
Summary
The aim of this study was to investigate whether the heme oxygenase (HO) pathway could
modulate proliferation of airway smooth muscle (ASM), and the mechanism(s) involved in
this phenomenon. In cultured human ASM cells, 10% fetal calf serum (FCS) or 50 ng/ml
PDGF-AB induced cell proliferation, extra and intracellular ROS production and ERK 1/2
phosphorylation. Pharmacological HO-1 induction (by hemin 10 µM or cobalt-
protoporphyrin, CoPP, 20 µM), and HO inhibition (by tin-protoporphyrin, SnPP-IX, 25 µM or
by an antisense oligonucleotide), respectively reduced and enhanced significantly both cell
proliferation and ROS production. Nor the carbon monoxide (CO) scavenger myoglobin (5-
20µM) neither the guanylyl cyclase inhibitor 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one
(ODQ) could reverse ASM proliferation induced by SnPP, making unlikely a role of the CO-
cGMP pathway in HO-modulated proliferation. By contrast, bilirubin (1µM) and the
antioxidant N-acetyl-cysteine (1mM), significantly reduced mitogen-induced cell
proliferation, ROS production and ERK1/2 phosphorylation. Furthermore, both bilirubin and
N-acetyl-cysteine, and the ERK1/2 inhibitor PD 98059 significantly reversed the effects of
HO inhibition on ASM proliferation. These results could be relevant to ASM alterations
observed in asthma since activation of the HO pathway prevented the increase in bronchial
smooth muscle area induced by repeated ovalbumin challenge in immunized guinea pigs,
whereas inhibition of HO had the opposite effect. In conclusion, this study provides evidence
for an antiproliferative effect of the HO pathway in ASM in vitro and in vivo, through a
bilirubin-mediated redox modulation of phosphorylation of ERK1/2.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

3
Introduction
An increase in ASM1 mass is one of the features that characterise airway remodelling in
asthmatic patients [1]. Moreover, bronchial smooth muscle cells from asthmatic patients have
shown abnormal cellular proliferation in vitro [2]. Therefore, knowing the factors and the
mechanisms that modulate ASM proliferation can have important pathophysiological
implications. In the last years, various types of mitogens have been shown to induce human
ASM proliferation, including growth factors, contractile agonists or inflammatory mediators,
such as ROS [3]. Indeed, evidence is growing that in physiological conditions, ROS induce
signal transduction leading to gene transcription and cell growth [4]. As ASM can be
continuously exposed to large amounts of exogenous or endogenous ROS produced by
inhaled agents, inflammatory or ASM cells themselves [5], redox signalling might be of
particular importance in ASM proliferation. A mitogen-induced ROS production leading to
cell proliferation via activation of ERK1/2 has been described in ASM [6,7,8]. But, if the
oxidant signalling involved in ASM proliferation is well characterised, little is known about
involvement of antioxidant systems in the control of muscle proliferation.
Heme oxygenase, the enzyme responsible for heme degradation, is a powerful cytoprotective
antioxidant system [9,10]. Heme degradation produces CO and biliverdin, reduced into
bilirubin by the biliverdin reductase. In the airways, HO is expressed in the epithelium, the
smooth muscle, macrophages, parasympathic ganglia and endothelium [11,12], and is
involved in the protection against oxidative-mediated airway inflammation and
hyperreactivity [13,14]. Bilirubin is one of the most powerful antioxidant system in the
organism [15], mainly known for its cytoprotective effect in oxidative stress models [16].
1 Abbreviations used in the text are : ASM : airway smooth muscle; CO : carbon monoxide; CoPP : Cobalt-protoporphyrin; H2DCFH-DA : 2’-7’dichlorodihydrofluorescein diacetate; DMEM : Dulbecco’s modified Eagle’s medium; ERK 1/2 : extracellular signal-regulated kinases 1/2; FCS: fetal calf serum; HO : heme oxygenase; ODN : PDGF : platelet derived growth factor; PD 98059 : 2’-Amino-3’-methoxyflavone; PI : propidium iodide; ROS : reactive oxygen species; SM1 : smooth muscle myosin heavy chain isoform 1; SnPP IX : tin-protoporphyrin IX; SOD : superoxide dismutase.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

4
Moreover, we have recently shown that bilirubin could also modulate ROS production in
guinea pig tracheal smooth muscle in physiological conditions [14]. In this model, bilirubin
also modulated oxidant signaled phosphorylation of myosin light chain. This suggests that
HO, by the way of its antioxidant properties, could also play a role in the modulation of redox
signalling in ASM. Furthermore, HO has shown an antiproliferative effect in vascular smooth
muscle in vitro and in vivo [17,18]. However, no data is available in the current literature
investigating the effects of HO on ASM proliferation and the mechanism(s) involved in this
phenomenon.
Therefore, the aim of this study was to investigate whether the HO pathway could modulate
proliferation of human bronchial smooth muscle cells exposed to 2 mitogens, PDGF or FCS
[6], and the mechanism(s) involved in this phenomenon, with special attention to the role of
ROS signalling and ERK1/2 activation. We also investigated which of HO-end products, CO
and bilirubin, was responsible for the effect of HO. Finally, we evaluated the functional role
of the HO pathway on ASM in vivo in a model of airway remodelling secondary to multiple
ovalbumin challenge in immunized guinea pigs.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

5
Experimental procedures
Reagents
[methyl-3H]- thymidine was purchased from NEN (Perkin-Elmer, Courtaboeuf, France) and
PDGF-AB was from R&D System (Abingdon, United-Kingdom). CoPP, hemin and SnPP IX
were from Porphyrin Products (London, England). Sense and antisense oligonucleotides were
supplied by Invitrogen (Cergy Pontoise, France), and transfected with the Superfect
Transfection Reagent (Qiagen SA, Courtaboeuf, France). H2DCFH-DA and PI were from
Molecular Probes (Eugene, OR). The selective MEK inhibitor PD 98059 was purchased from
Calbiochem (Merk Eurolab SA, Fontenay-sous-Bois, France). Anti phosphorylated p42/44
antibody was purchased from New England Biolabs (Ozyme, Saint-Quentin-en-Yvelines,
France), anti HO-1 antibodies were from StressGen (Tebu, Le-Perray-en-Yvelines, France),
anti SM1 antibody was from Seikagaku America (Palmouth, MA) and anti α-actin antibody
was from Sigma (Saint Quentin Fallavier, France). Except for the anti-phosphorylated p42/44
antibody, which was polyclonal, all of the antibodies used were monoclonal. Culture media,
supplements, and FCS were from Life Technologies SARL (Cergy Pontoise, France). Tissue
culture plastic ware was supplied by Costar Corp. (Cambridge, MA). Reagents for Western
blot were from BioRad Laboratories (Richmond, CA). Reagents for immunohistochemistry
were from Dako (Carpinteria, CA). Other reagents were from Sigma Chemical Co. (Saint
Quentin Fallavier, France).
Human bronchial smooth muscle isolation and cell culture
Primary cultures of human bronchial smooth muscle were established as described [19,20].
Briefly, human bronchi (5 to 15 mm internal diameter) were obtained from lobes resected
during thoracotomy for lung cancer in 11 different patients, and dissected from the
surrounding parenchyma. The epithelium was removed and bands of airway smooth muscle
were isolated by dissection under binocular microscope and cut into 1 mm square pieces.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

6
These muscular pieces, termed explants, were incubated in DMEM supplemented with 10%
heat inactivated FCS, antibiotics (streptomycin 10 mg/ml, penicillin G 10 000 UI/ml) and
amphotericin B (25µg/ml) in a humidified atmosphere of 5% CO2-95% air at 37°C, as
described [19]. On reaching confluence, cells were passed by lifting the cells with 0.05%
trypsin - 0.5mM EDTA. Cultures from passages 3 to 5 were used for experiments. At
confluence, cells exhibited the typical “hill and valley” aspect [20]. Cell characterization was
assessed by immunostaining using a monoclonal antibody against α-smooth muscle actin and
against the specific smooth muscle myosin heavy chain isoform 1 (SM1) [19, 22]. Greater
than 95% of cells from each patient used in the different experiments displayed positive
immunohistochemical staining for both antibodies.
The general experimental protocol was as follows : cells were seeded at an initial density
104/cm2 , grown to 70% - 80% confluence and serum deprived (1% FCS) for 24 hours. Then,
they were stimulated with 2 different mitogens : either 10% FCS or 50 ng/ml PDGF-AB.
Control non-stimulated cells were grown on 1% FCS. In some experiments, the cells were
incubated with different pharmacological agents and their respective vehicles for the indicated
time before the addition of the mitogen. Experiments were carried out in at least 3 different
cell lines, each one derived from a different individual.
HO expression and activity in ASM cells
HO-1 protein expression was measured by western blot and immunohistochemistry.
For western blot experiments, cells were cultured in 75 cm2 plates. Western blot was
performed as described previously [21,22]. The concentration of the anti HO-1 antibody was
1/1000. Detection was performed by a chemiluminescence substrate. Using the same blots,
the expression of the housekeeping protein β-actin was evaluated using a monoclonal anti β-
actin antibody. Optical densities were measured with a Perfect Image 2.01 image analysis
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

7
system (Iconix, Courtaboeuf, France). Results were expressed as the ratio of the expression of
HO-1 to that of β-actin.
For immunohistochemistry, cells were cultured in a Lab-Tek chamber slide (Nunc,
Naperville, Il). Immunohistochemistry was performed as described before [24], with a 1/1000
dilution of the anti HO-1 antibody. The specificity of the immunostaining was evaluated by
replacement of the primary antibody by a control isotype antibody at equivalent protein
concentration and by omitting the primary antibody.
HO activity was assessed by bilirubin production as already described [23], in cells grown on
75 cm2 plates. In addition, HO activity was evaluated in situ by immunohistochemistry using
the anti-bilirubin IX monoclonal antibody, 24G7 [25]. This monoclonal antibody specifically
recognizes the conjugated and unconjugated forms of bilirubin IX but not other isomers of
bilirubin [26]. Bilirubin IX is produced by the reduction of biliverdin IX, a product of the HO-
1 reaction. Thus, immunohistochemistry with the use of 24G7 allowed us to assess the HO-1-
specific heme degradation in situ in fixed cells. The antibody was used at 10 µg/ml
concentration. The specificity of the immunostaining was evaluated by replacement of the
primary antibody by a control isotype antibody at equivalent protein concentration and by
omitting the primary antibody.
[methyl-3H]-thymidine incorporation in ASM cells
Cells were cultured in 24-well plates and stimulated with mitogens for 24 h. For the final 18
hour incubation with the mitogen, 4µCi/ml of [methyl-3H]-thymidine was added to measure
DNA synthesis by scintillation counting. Results of individual treatments were obtained on
quadruplicate and were expressed as count per minute. Then the percent change from the
response of 1% FCS for each individual result was calculated. To minimize the influence of
variability between tissue donor, the value was calculated from the response of 1% FCS
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

8
treated cells from the same 24 well plate. In experiments with the MEK inhibitor PD 98056,
the control condition was FCS+DMSO, explaining the lower proliferative response. However,
we checked in preliminary experiments, that DMSO does not change the way of proliferative
response to mitogens but just decrease by about one third the absolute value of thymidine
incorporation.
Cellular toxicity and viability
Cellular toxicity and viability were assessed by 3 different methods : cell count and trypan
blue exclusion, LDH release in the medium, and incorporation of the fluorescent dye
propidium iodide (PI). For PI incorporation, cells seeded in 96-well plates were incubated
with 5 µM PI for 30 minutes and then washed in PBS before reading at 480-520 nm with a
multiwell fluorescence plate reader (Fluorostar BMG, Netherlands). Results are expressed in
fluorescent arbitrary units.
Measurement of ROS production by ASM cells
Intracellular ROS production : H2DCFH-DA oxidation.
Cells were cultured in 96-well plates as described previously. H2DCFH-DA (10µM final in
DMSO) was added one hour before stimulation. Immediately after addition of the mitogen,
fluorescence was measured every 5 minutes during a 45 minute period with a multiwell
fluorescence plate reader at 480-555 nm. Intracellular ROS (especially H2O2 or hydroxyl
radical) oxidize DCFH, yielding the fluorescent product DCF [24]. Results are expressed as
the ratio between fluorescence measured at 45 minutes and that measured at the first point
[27].
Extracellular superoxide anion release : cytochrome c reduction
Ferricytochrome c reduction was measured as previously described [24]. Briefly, cells were
cultured in 6-well plates. Before the addition of the mitogen, the medium was replaced with
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

9
HBSS without phenol red and incubated in 1 ml of the same buffer with and without 300
U/ml SOD. Subsequently, ferricytochrome c was added at a final concentration of 80µM to
the reaction buffer solution, followed by addition of the mitogens. After 1 h, the buffer was
removed and absorbance at 550 nm was measured immediately. Superoxide anion production
was calculated from the differences in the absorbances between samples with and without
SOD, using an extinction coefficient of 21.1 mM-1 cm–1 for reduced ferricytochrome c.
Measurement of ERK1/2 phosphorylation in ASM cells
Cells were cultured in 75 cm2 plates. In a first series of experiments, cells were stimulated for
different times with 50 ng/ml PDGF-AB or serum. Then the medium was removed, cells were
washed twice with cold PBS and removed in lysis buffer containing phosphatase inhibitors
(natrium fluoride 2.5 mM, β-glycerophosphate 1 mM, orthovanadate 1mM and para-
nitrophenol-phosphate 1mM). In a second series of experiments, cells were pre-treated with
different pharmacological agents before a 10 minutes-stimulation with PDGF or serum.
Western blot was performed as already described [14, 24]. The phosphorylated forms of the
enzyme were detected with a polyclonal antibody used at 1/1000 dilution. Optical density of
the band was measured as described (see below) and compared with �-actin expression.
Immunohistochemichal detection of HO-1 in human bronchial smooth muscle.
Detection of HO-1 protein in airway smooth muscle was examined in human bronchi.
Segments of 2 bronchi utilised for ASM cells isolation and culture were fixed in formol 10%
and embedded in paraffin. Immunohistochemistry was performed as previously published
[23,24].
HO-1 sense and antisense oligonucleotides (ODN) treatment.
The sense/antisense ODNs for HO-1 were directed against the flanking translation initiation
codon in the human HO-1 cDNA [28]. The antisense sequence was 5’-
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

10
CGCCTTCATGGTGCC-3’, whereas the sense sequence was 5’-GGCACCATGAAGGCG-
3’. ODNs were phosphorothioated on the first 3 bases on the 3’ end. Cells were transfected
using the Superfect Transfection Reagent (Qiagen SA, Courtaboeuf, France) following
manufactor’s instructions. Briefly, cells were seeded in 24-well plates at a density of 20 000
cells /well 24 hours before transfection. The proportions used were 1µg DNA/ 5 µl
transfection reagent/ well. Cells were incubated for 3 hours with the ODNs; then the medium
was replaced with fresh medium containing 10% serum. Experiments for proliferation and
western blot were performed 48 hours after transfection.
Induction of airway remodelling in ovalbumin -sensitized and aerosol- challenged guinea pig
Pathogen-free male Hartley guinea pigs (250-300 g body wt; Charles River, France) were
housed in individual cages in climate-controlled animal quarters and were given water and
food ad libitum. As previously described [29], the animals were immunized with 0.5 ml of
0.9% wt/vol NaCl (saline) containing 100 mg ovalbumin (OVA), injected subcutaneously on
the neck, and another 0.5 ml intraperitoneally (i.p.) on day 1. On days 8, 9, 10, 13, 14 and 15,
the animals (called OO animals) were challenged in a 5 liter plastic chamber by a 10 minute
exposure to aerosolized OVA (0.1% OVA in 10 ml saline), using a Devilbiss nebulizer
(Sunrise, Devilbiss Medical, Nantes, France). The time of exposure was determined by the
appearance of respiratory distress signs (polypnea, bronchospasm, contraction of accessory
respiratory muscles and cyanose). Another group of animals were immunized to OVA as
described above and exposed to aerosolized saline (ON animals). This model is characterized
by bronchial hyperreactivity to histamine (measured by pulmonary inflation pressure) and an
increased number of polynuclear eosinophils in bronchoalveolar lavage in OO animals [29].
Both OO and ON animals were randomly divided into 2 groups. One group received the
inhibitor of HO activity SnPP IX, given i.p. at a dose of 50 µmol/kg on days 8, 10, 13 and 15,
6 hours before the challenge (OO-SnPP animals); a second group received received the HO-1
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

11
inductor, hemin, given i.p. at a dose of 50 mg/kg, on days 7, 10 and 13 (OO-Hemin animals).
In preliminary experiments we verified that SnPP IX and hemin's vehicle did not modify the
parameters measured in the study. We also verified that hemin and SnPP IX significantly
increased and decreased HO activity respectively, by measurement of bilirubin production in
lung microsomes, as previously described [23] (data not shown).
Animals were sacrificed 24 hours after the last challenge. They were anesthetized with
sodium pentobarbital (Nesdonal®, Specia-Rhone-Poulenc, Romainville, France), (50 mg/kg
of body weight i.p.), then the lung was inflated through a tracheal canula at 25 cm H2O with
10% formol and fixed in paraffine.
Evaluation of ASM area and HO-1 and bilirubin immunohistochemistry
Hematoxylin and eosin-stained tissues sections were examined microscopically and smooth
muscle area was measured with a microscope (Leitz, Germany) related to a camera (Olympus,
France) and a computerized image analysis system (AnalySIS, Soft Imaging System Münster,
Germany) as described by Palmans and coworkers [30]. Muscular area was determined by
delimitating the outer side of the basal membrane and the outer side of the muscular area. We
analyzed bronchi with similar diameter and with a ratio of minimal to maximal diameter of
more than 0.6. We analyzed 5 animals in each group and 3 to 4 different bronchi for each
animal.
HO-1 and bilirubin immunohistochemistry was performed as previously described [23].
Concentration of the anti-bilirubin antibody was 1µg/ml.
Statistical analysis
Values are given as mean ± S.E.M. Data were analysed by one way ANOVA; differences
between means were analysed with the Fisher's protected least-significant difference multiple
comparison test. Significance for all statistics was accepted at p<0.05.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

12
Results
HO-1 is expressed in human ASM
HO-1 was expressed in non stimulated ASM cells in culture (Figure 1, panel A). Incubation
with the mitogens FCS (10%) or PDGF (50 ng/ml) for 24 hours significantly and quite
similarly increased HO-1 expression and HO activity (p<0.05 as compared with 1% FCS;
Figure 1, panels A and B). CoPP (20 µM) could further potentiate HO activity induced by the
mitogens. As attempted, SnPP IX (25 µM) inhibited significantly HO activity (Figure 1,
panel B).
HO-1 expression and activity in human ASM cells in culture was further confirmed with
immunohistochemical analysis using anti HO-1 and anti-bilirubin antibodies respectively
(Figure1, panel C). No expression was observed with a control isotype antibody. HO-1 was
also expressed in ASM in human bronchi (Figure 1, panel D).
The HO pathway and bilirubin modulate human ASM cells proliferation
Mean thymidine incorporation in non-stimulated cells was 5448 ± 1228 cpm. ASM cell
proliferation was significantly enhanced by 10% FCS and PDGF, being 360% and 372%
higher respectively, than observed with 1% FCS (Figure 2, panel A and B, p<0.05
respectively). HO induction by CoPP significantly decreased the proliferative response to
FCS and PDGF. Indeed, in CoPP treated cells, proliferation induced by 10% FCS and PDGF
was only 135% and 155% higher respectively, than observed with 1% FCS (Figure 2, panels
A and B, p<0.05 respectively). Hemin (10µM), another HO inductor, has a similar effect on
ASM proliferation (data not shown). On the opposite, the HO inhibitor SnPP IX induced a
523% and 690% increase in proliferation induced by FCS and by PDGF (Figure 2, panels A
and B, p<0.05 respectively). For both mitogenic factors, SnPP IX reversed the
antiproliferative effect of CoPP, thus confirming a specific effect of HO activity. Transfection
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

13
with the HO-1 antisense effectively blocked HO-1 expression (Figure 3, panel A) and, like
pharmacological blockade, enhanced PDGF-induced proliferation (Figure 3, panel B).
No toxicity was observed with metalloporphyrins at the doses we used. Neither blue trypan
exclusion test nor LDH measurement or propidium iodide incorporation was modified (LDH
content between 6 ± 2 and 8 ± 4 UI/ml; propidium iodide incoporation between 32566 ± 1250
and 34559 ± 1452 arbitrary fluorescence units, NS, data not shown).
Having demonstrated that HO modulated ASM proliferation, we wondered which of the HO
end products, CO and bilirubin, was responsible for its antiproliferative effect. Concerning
CO, neither the guanylate cyclase inhibitor 1H-[1,2,4]oxadiazolo-[4,3-a]quinoxalin-1-one
ODQ (10 µM, given 1 hour before mitogenic stimulation) nor the CO scavenger myoglobin
(5-20 µM for 24 hours) could reverse inhibition of proliferation induced by SnPP IX, making
unlikely a role of the CO-cGMP pathway in HO-modulated proliferation (Figure 4, panels A
and B respectively).
By contrast, bilirubin (1 µM, 1 hour before stimulation) was able to significantly reduce FCS-
and PDGF-induced proliferation (Figure 2, panel C and D, p<0.05 vers 1% FCS).
Furthermore, bilirubin significantly reversed the mitogenic effect of SnPP IX : proliferation in
SnPP + bilirubin cultured cells was similar to that observed in cells cultured with the mitogen
alone (Figure 2, panels C and D). Considering the antioxidant properties of bilirubin, we
investigated if the antioxidant N-acetylcysteine (NAC) could mimic the effect of CoPP and
bilirubin. These experiments showed that pretreatment of cells with NAC (1 mM, 1 hour
before stimulation) mimicked the effect of CoPP and bilirubin on proliferation and reversed
the effect observed after treatment with SnPP IX (Figure 2, panels C and D).
The HO pathway modulates both intra and extracellular ROS production
Considering the inhibitory effect of bilirubin and NAC on cellular proliferation, we
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

14
investigated the role of the HO pathway in intra and extracellular ROS production by
measurement of the oxidation of H2DCFH-DA and the reduction of cytochrome c
respectively.
Both FCS and PDGF increased significantly intra and extracellular ROS production. Indeed,
FCS and PDGF increased H2-DCFH-DA oxidation by 41 % and 38%, and cytochrome c
reduction by 108 % and 152% respectively, compared with 1% FCS (p<0.05 in each case,
Table 1). Treatment with the HO inductor CoPP prior to cell stimulation decreased mitogen-
induced H2DCFH-DA oxidation by 29 and 25% respectively and cytochrome c reduction by
68 and 48 % (p<0.05 for 10 % FCS and PDGF as compared to 1% FCS respectively, Table
1). The HO inhibitor SnPP IX increased ROS production by 53 and 58% for H2-DCFH-DA
oxidation and by 51 and 44.6% for cytochrome c reduction compared with stimulation with
mitogens alone (p<0.05 for 10 % FCS and PDGF as compared to 1% FCS respectively, Table
1). Bilirubin (1 µM) and N-acetylcysteine (1 mM) mimicked the effect observed with CoPP
and reversed the pro-oxidative effect of SnPP (p<0.05 respectively, Table 1).
The HO pathway modulates ERK 1/2 phosphorylation by a redox mechanism.
Considering that ROS modulate ERK 1/2 activation, and that these kinases are involved in
ASM cell proliferation, we investigated the role of the HO pathway in the modulation of ERK
1/2 phosphorylation.
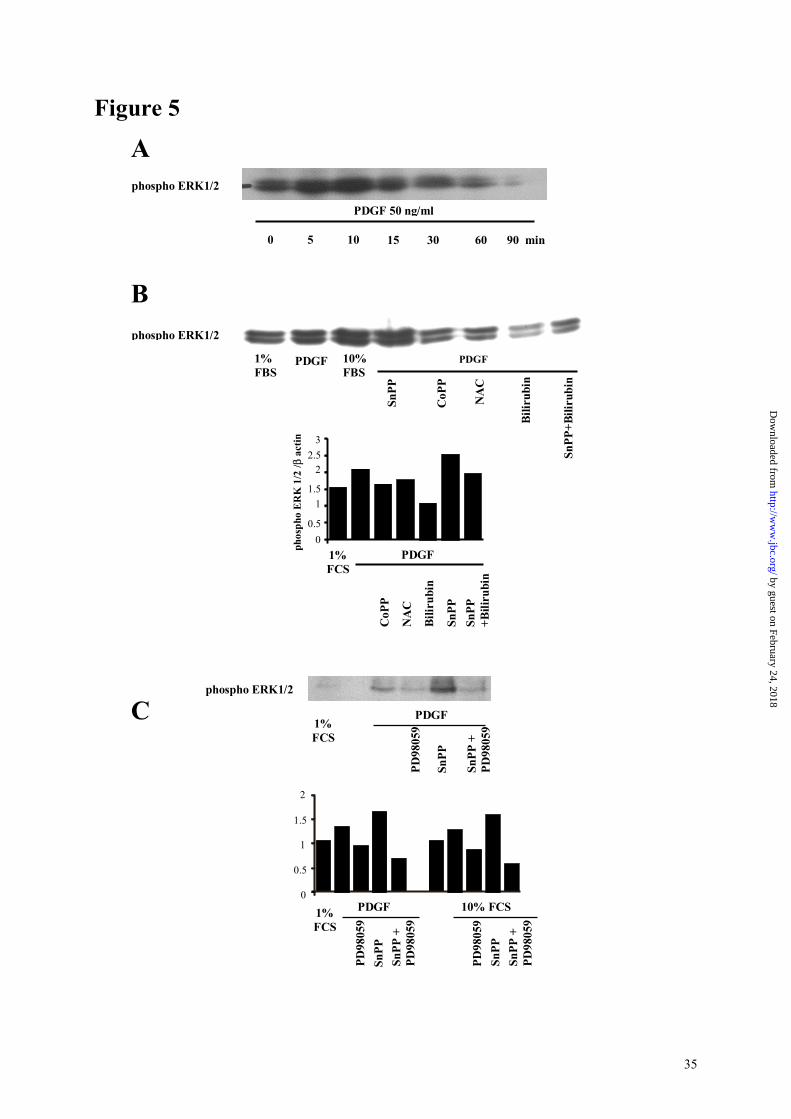
ASM cells exhibited a basal level of phosphorylated ERK 1/2 (Figure 5, panel A). PDGF
induced a time-dependent increase in ERK 1/2 phosphorylation in HASM cells, that peaked
after 10 minutes of stimulation (Figure 5, panel A). Pre-incubation with CoPP decreased
PDGF-induced ERK 1/2 phosphorylation at 10 minutes, while SnPP increased it (Figure 4,
panel B). Bilirubin (1 µM) strongly decreased SnPP-enhanced phosphorylation. A similar
effect was observed with N-acetylcysteine (1 mM), confirming the role of ROS in SnPP-
induced ERK 1/2 activation. Similar results were observed with serum-stimulated cells (data
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

15
not shown).
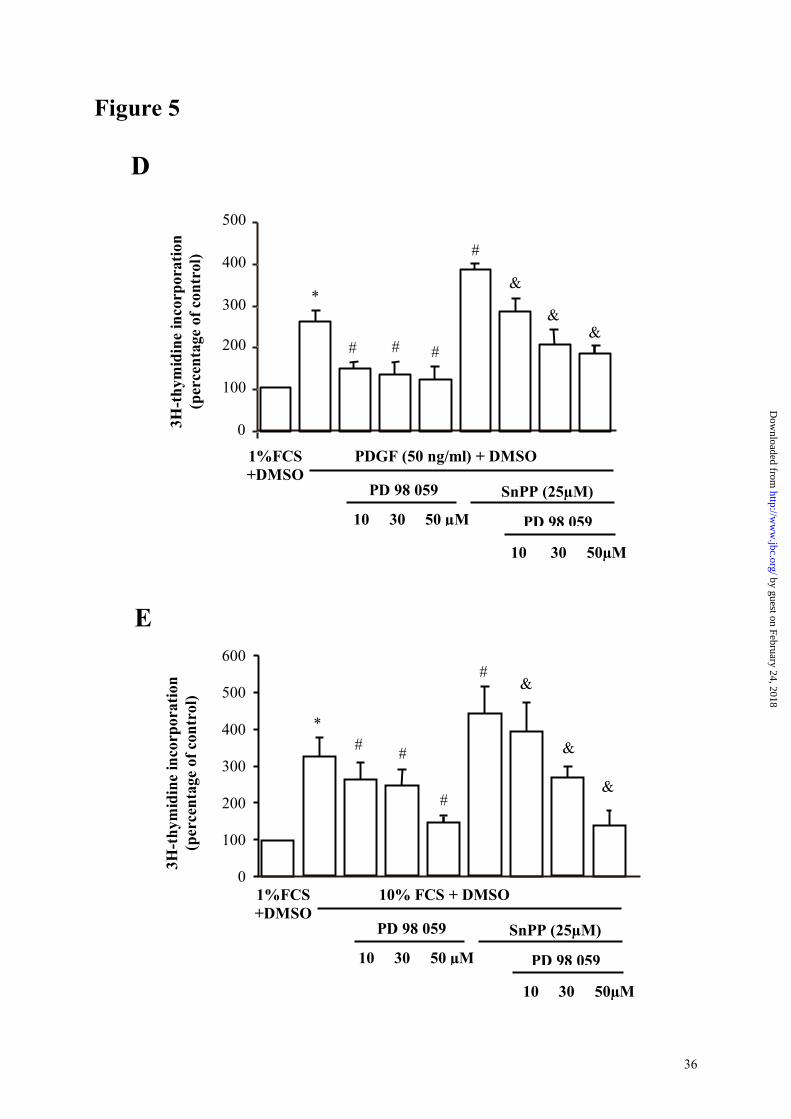
The MEK inhibitor PD98059 inhibited cell proliferation (Figure 5, panels D and E) and
blocked both FCS- and PDGF-induced ERK 1/2 phosphorylation at 10 minutes (Figure 5,
panel C), confirming involvement of the ERK 1/2 pathway in ASM cells growth. Moreover,
PD98059 inhibited the increase in cell proliferation induced by SnPP IX (Figure 5, panel E)
and reversed SnPP-enhanced ERK 1/2 phosphorylation, thus confirming that the effect of HO
on cell proliferation involves the MAP kinase pathway.
HO protects against the increase in ASM area in ovalbumin-sensitized and -challenged guinea
pig.
HO-1 expression in bronchial smooth muscle was increased in OO as compared to ON
animals, along with expression in epithelium and inflammatory cells (Figure 6, panel A).
Bilirubin, one of the end step product of heme catabolism, was found in a similar manner by
immunohistochemistry in bronchial smooth muscle, confirming the in vivo activity of
muscular HO (Figure 6, panel A).
Diameters of bronchi examined for measurement of smooth muscle area were not statistically
different, ranging from 375.78 ± 47.95 to 452.23 ± 66.72 µm within the various experimental
groups. Repeated allergen challenge in immunized animals induced a significant increase in
ASM area (88.40 ± 16.21 vs 119.32 ± 18.45 µm2, p<0.05 OO vs ON, Figure 6, panel B).
Treatment of OO animals with the HO inductor hemin inhibited the increase in bronchial
smooth muscle area and administration of the HO inhibitor SnPP produced the opposite
effect. Indeed, bronchial smooth muscle area was 90.25 ± 11.12µm2 for hemin treated- and
158.02 ± 18.23µm2 in SnPP-treated OO animals, p<0.05 vs OO animals, Figure 6, panel B).
In ON animals, the modulation of bronchial smooth mucle area induced by hemin and SnPP
was slight but non significant (data not shown).
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

16
Discussion
The main results of this study indicate that HO acts in an autocrine negative feedback manner
to limit ROS-dependent phosphorylation of the ERK 1/2 MAP kinase and the ensuing
proliferation induced by serum and PDGF in human ASM cells. These effects are secondary
to a reduction in ROS production by the action of the HO end-product bilirubin. These data
provide the first evidence that HO takes part in the control of ASM proliferation by
modulating ROS signalling, via the effect of bilirubin. Furthermore, these results could be
relevant to the increase in ASM mass observed in asthma. Indeed, using a model of airway
remodelling secondary to multiple ovalbumin challenges in immunized guinea pigs, we found
that activation of the HO pathway prevents the increase in bronchial smooth muscle area,
whereas inhibition of HO has the opposite effect.
An antiproliferative effect of the HO pathway in smooth muscle was first described in rat
vascular smooth muscle cells in vitro [17,31], then confirmed in vivo in animal models of
vascular remodeling after hypoxia or balloon-induced wall injury [32,33]. Indeed, to our
knowledge, all data concerning HO and smooth muscle proliferation were obtained in
vascular smooth muscle cells, either from rat or guinea pig. This antiproliferative effect is cell
type-dependant, since opposite effects of HO on cell cycle have been described in endothelial
and in smooth muscle cells [34] : HO inhibition decreases S and G2/M phases in endothelial
cells and increases it in smooth muscle cells, while HO induction exhibits opposite effects. To
our knowledge, no information is available concerning the effect of HO on the proliferation of
ASM. Our results confirm and expand data obtained on vascular smooth muscle. Indeed,
induction of HO-1 protein expression and HO activity may represent a mechanism by which
mitogens such as serum or PDGF regulate ASM growth, as described in vascular smooth
muscle [35]. This hypothesis is supported by our data showing that serum or PDGF-
stimulated DNA synthesis was augmented in the presence of the HO inhibitor SnPP and
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

17
reduced in the presence of the HO inductors hemin or CoPP, indicating a growth inhibitory
effect of HO on human ASM. It must be noted, however, that definitive conclusions from
drug-based experiments should not be drawn without verifying the biological activity of the
drug in a particular experimental setting. In the present study, we used 2 different HO-1
inductors (hemin and CoPP) and we verified that both inductors and the HO inhibitor SnPP
IX effectively modulated HO-1 protein expression and HO activity in ASM cells and lung
microsomes. Moreover, the effect of CoPP was inhibited by the HO inhibitor SnPP IX, thus
confirming the specificity of these compounds. The similar inhibitor effects of the two
different HO-1 inductors on ASM proliferation further supports this interpretation. To finish
with, ASM transfection with an HO-1 antisense oligonucleotide induced a similar effect than
the HO inhibitor SnPP IX on cell proliferation. Furthermore, we also found HO-1 protein
expression in both human and guinea pig airway smooth muscle in situ, thus ensuing the in
vivo relevance of the cellular data.
In vascular smooth muscle, the antiproliferative effect of HO has been mainly related to
carbon monoxide production, since hemoglobin, a scavenger for CO, was able to reverse the
effect of HO induction [17,31]. The antiproliferative effect of CO has been suggested to be
secondary to soluble guanylyl cyclase activation, because inhibition of this enzyme or of its
end-product, cGMP, can restore DNA synthesis in vascular smooth muscle cells transfected
with HO-1 cDNA [18]. Exogenous administered CO has also an antiproliferative effect on
vascular as well as airway smooth muscle. Indeed, exogenous CO at low doses (between 100
and 250 ppm, that is considered to be comparable to the gaseous production by the enzyme
itself) arrests rat vascular smooth muscle cells at the G1/S transition of the cell cycle [17] and
human ASM cells at the G0/G1 phase [36]. However, in ASM, exogenous CO acts
independently from a guanylyl cyclase/cGMP pathway [36]. This result is in line with our
data showing that the soluble guanylyl cyclase inhibitor ODQ did not impair the anti-
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

18
proliferative effect of HO activation. However, in the present study, a predominant role of the
endogenous CO-HO pathway in the control of ASM cell proliferation is unlikely since
application of different concentrations (5 to 20 µM) of the CO scavenger myoglobin did not
modify the antiproliferative effect of HO activation. Differences in cell type (airway versus
vascular), species (human versus rat) and exogenous versus endogenous HO-produced CO,
may explain these discrepancies.
In contrast with CO, we found that bilirubin exerted a clear antiproliferative effect on human
ASM cells. This result agrees with data published in osteoblasts, neural and hepatoma cells
[36]. The concentration of bilirubin is a very important point to consider because of the
potential cellular toxicity of high concentrations of bilirubin [16]. Indeed, a recent study by
Liu and associates [38] shows that biliverdin and bilirubin can induce apoptosis in rat vascular
smooth muscle cells, as done by HO induction, while CO and iron failed to induce such
phenomenon. In this study, bilirubin required concentrations as high as 500 µM to induce
apoptosis, while no effect was observed at concentrations similar to the one we used in the
present study (5 µM). Although both cell type and experimental conditions were different,
this can explain why we did not observe any cell toxicity and death in our model. In the
present study, the simultaneous decrease in both ASM oxidants production and cell
proliferation observed with 1 µM of bilirubin, and the fact that this last effect was mimicked
by the antioxidant NAC, strongly suggests a major role of the antioxidant properties of
bilirubin in the decreased proliferation. Furthermore, bilirubin was able to reverse the pro-
oxidant and pro-proliferative effect of HO inhibition, stressing therefore its involvement in the
effects of HO. This conclusion is further supported by the in situ immunohistochemical
detection of bilirubin in ASM cells, demonstrating that this molecule was well synthesized in
living cells during the different experimental conditions. The present results emphasize the
concept that the antioxidant properties of the HO-bilirubin pathway are not only related to its
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

19
ROS scavenging properties [39], but also to the modulation of ROS production. Since recent
studies demonstrated that a NAD(P)H oxidase like system is the main source of ROS in
animal and human ASM [24,40], inhibition of NAD(P)H oxidase by the HO-bilirubin
pathway could explain its inhibitory effect on ROS production. This hypothesis is supported
by previous data obtained in vitro, in acellular preparations, showing that bilirubin can inhibit
reconstituted NAD(P)H oxidase [41]. Further studies are needed to verify if bilirubin
modulates NAD(P)H oxidase in ASM and the mechanism(s) involved in this effect.
Involvement of the ERK 1/2 MAP kinase in PDGF- and serum-induced ASM proliferation
and the role of ROS in the kinase cascade activation has been extensively described in smooth
muscle cells [6,7,40,42-44]. In our model, the critical role of ERK was confirmed by the
inhibitory effect of the MEK inhibitor PD98059 on muscle proliferation. The present results
show that HO is an important pathway to control activation of the ROS-sensitive ERK 1/2
pathway in ASM. Indeed, ERK activation was blocked by induction of HO-1 by CoPP, by the
antioxidant NAC and by bilirubin, thus confirming the sensibility of the ERK pathway to
oxygen species in human ASM cells. Moreover, the MEK inhibitor PD98059 significantly
reduced the pro-proliferative effect of HO inhibition, thus confirming that the MEK-ERK1/2
pathway is a major target of the modulatory effect of HO on human ASM proliferation.
However, whether or not the HO-bilirubin pathway can modulate other ROS-dependant
signaling pathway, such as the JAK-STAT pathway [45,46], remains to be investigated.
Furthermore, if bilirubin antioxidant properties appear to be an important mechanism
explaining the decrease in ASM proliferation, other mechanisms cannot be excluded. Indeed,
bilirubin actually inhibits protein phosphorylation, probably by interacting with different
domains of the kinase [47]. The relative importance of these pathways in modulating ASM
proliferation, as well as the role of other HO-end products, such as iron and ferritin, warrants
further investigations.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

20
The antiproliferative effect of HO on ASM might be protective in vivo under conditions
leading to bronchial smooth muscle proliferation. Indeed, using a model of ovalbumin-
immunized and multiple aerosol challenged guinea-pigs characterized by airway
inflammation [29] and an increase in ASM muscle area, we found that bronchial muscle mass
was augmented in the presence of the HO inhibitor SnPP IX and reduced in the presence of
the HO inductor hemin. In these animals, HO-1 protein was induced in bronchial smooth
muscle, probably representing a mechanism by which HO regulates ASM growth. In this
model, we cannot exclude however that HO may have a local anti-inflammatory effect that, in
turn, could participate to the modulation of ASM growth, as observed in animals treated with
anti-inflammatory drugs such as cysteinyl leukotriene or endothelin receptor antagonist
[48,49]. However, immunohistochemical detection of bilirubin in bronchial muscle suggests
an autocrine effect of the HO-bilirubin pathway in vivo in ovalbumin-challenged animals.
In conclusion, this study provides evidence that HO is involved in the control of ASM cells
proliferation through a bilirubin-mediated redox modulation of phosphorylation of ERK1/2.
These data are relevant in terms of in vivo protection against some features of airway
remodelling, such as increased ASM area. Collectively, these results, and previous data
showing that the HO-bilirubin pathway modulated negatively ASM contractility [14], suggest
that induction of the HO pathway could be beneficial in asthma or other respiratory diseases
leading to airway remodelling and hyperreactivity.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

21
Acknowledgements
We are in debt to Drs Roberta Foresti and Roberto Motterlini (Harrow, Middlesex, United
Kingdom.) and Pr Bruno Crestani (Paris, France) for their helpful and encouraging comments.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

22
References 1. Dunnill MS, Massarella GR, Anderson JA. (1969) Thorax 24,176-179. 2. Johnson PRA, Roth M, Tamm M, Hughes JM, Ge Q, Burgess JK, King G, Black JL.
(2001) Am J Respir Crit Care Med. 164,474-477. 3. Ammit AJ, Panettieri RA. (2001). J Appl Physiol. 91,1431-1437.
4. Shackelford RE, Kaufmann WK, Paules RS. Oxidative stress and cell cycle checkpoint function. Free Rad Biol Med. 2000; 28,1387-1404.
5. Henricks PAJ, Nijkamp FP. Reactive oxygen species as mediators in asthma. Pulm Pharmacol Therap. 2001;14,409-421.
6. Brar SS, Kennedy TP, Whorton AR, Murphy TM, Chitano P, Hoidal JR. (1999) J Biol Chem. 274,20017-20026.
7. Page K, Li J, Hodge JA, Liu PT, Vanden Hoeck TL, Becker LB, Pestell RG, Rosner MR, Hershenson MB. (1999) J Biol Chem. 274,22065-22071.
8. Abe MK, Chao TS, Solway J, Rosner MR, Hershenson MB. (1994) Am J Respir Cell Mol Biol.11,577-585.
9. Choi AMK, Alam J. (1996) Am J Respir Cell Mol Biol. 15,9-19. 10. Maines MD. (1997) Annu Rev Pharmacol Toxicol. 37,517-554.
11. Donnelly LE, Barnes PJ. (2001) Am J Respir Cell Mol Biol. 24,295-303. 12. Lim S, Groneberg D, Fischer A, Oates T, Caramori G, Mattos W, Adcock I, Barnes
PJ, Chung KF. (2000) Am J Respir Crit Care Med. 162,1912-1918. 13. Jia YX, Sekizawa K, Okinaga S, Lie R, Sasaki H. (1999) Int Arch Allergy Immunol.
120,141-145. 14. Samb A, Taillé C, Almolki A, Mégret J, Staddon JM, Aubier M, Boczkowski J.
(2002) Am J Physiol Lung Cell Mol Physiol. 283,L596-L603. 15. Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. (1997) Science
235,1043-1046. 16. Doré S, Takahashi M, Ferris CD, Hester LD, Guastella D, Snyder SH. (1999) Proc
Natl Acad Sci USA. 96,2445-1450.
17. Peyton KJ, Reyna SV, Chapman GB, Ensenat D, Liu XM, Wang H, Schafer AI, Durante W. (2002) Blood. 99,4443-4448.
18. Duckers HJ, Boehm M, True AL, Yet SF, San H, Park JL, Webb RC, Lee ME, Nabel GJ, Nabel EG. (2001) Nat Med. 7,693-698.
19. Johnson PR, Armour CL, Carey D, Black JL. (1995) Am J Physiol 269,L514-519.
20. Berger P, Perng DW, Thabrew H, Compton SJ, Cairns JA, McEuen AR, Marthan R, Tunon de Lara JM, Walls AF. (2001) J Appl Physiol. 91,1372-1379.
21. Chamley-Campbell J, Campbell GR, Ross R. (1979) Physiol Rev. 59,1-61. 22. Wong JZ, Woodcock-Mitchell J, Mitchell J, Rippetoe P, White S, Absher M, Baldor
L, Evans J, McHugh KM, Low RB. (1998) Am J Physiol. 274,L786-792. 23. Taillé C, Foresti R, Lanone S, Zedda C, Green C, Aubier M, Motterlini R,
Boczkowski J. (2001) Am J Respir Crit Care Med. 163,753-761.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

23
24. Thabut G, El-Benna J, Samb A, Corda S, Megret J, Lesèche G, Vicaut E, Aubier M, Boczkowski J. (2002) J Biol Chem. 277,22814-22821.
25. Nakayama M, Takahashi K, Komaru T, Fukuchi M, Shioiri H, Sato K, Kitamuro T, Shirato K, Yamaguchi T, Suematsu M, Shibahara S. (2001) Arterioscler Thromb Vasc Biol. 21,1373-1377.
26. Ozawa N, Goda N, Makino N, Yamaguchi T, Yoshimura Y, Suematsu M. (2002). J Clin Invest. 109,457-467.
27. Wang H, Joseph JA. (1999) Free Rad Biol Med. 27,612-616. 28. Wagener FA, Da Silva JL, Farley T, De Witte T, Kappas A, Abraham NG. (1999). J
Pharmacol Exp Ther 291,416-423. 29. Samb A, Pretolani M, Dinh-Xuan AT, Ouksel H, Callebert J, Lisdero C, Aubier M,
Boczkowski J. (2001) Am J Respir Crit Care Med. 164,149-154. 30. Palmans E, Kips JC, Pauwels RA. (2000) Am J Respir Crit Care Med. 161,627-635.
31. Morita T, Mitsialis SA, Koike H, Liu Y, Kourembanas S. (1997) J Biol Chem. 272, 32804-32809.
32. Christou H, Morita T, Hsieh CM, Koike H, Arkonac B, Perrella MA, Kourembanas S. (2000) Circ Res. 86,1224-1229.
33. Aizawa T, Ishizaka N, Taguchi JI, Kimura S, Kurokawa K, Ohno M. (1999) Biochem Biophys Res Com. 261,302-307.
34. Li Volti G, Wang J, Traganos F, Kappas A, Abraham NG. (2002) Biochem Biophys Res Com. 296,1077-1082
35. Durante W, Peyton KJ, Schafer AI. (1999) Arterioscler Thromb Vasc Biol. 19,2666-2672.
36. Song R, Mahidhara RS, Liu F, Ning W, Otterbein LE, Choi AMK. (2002) Am J Respir Cell Mol Biol. 27,603-610.
37. Janes CH, Dickson ER, Okazaki R, Bonde S, McDonagh AF, Riggs BL. (1995) J Clin Invest. 95,2581-2586.
38. Liu X, Chapman GB, Wang H, Durante W. (2002) Circulation. 105,79-84. 39. Minetti M, Mallozzi C, Di Stasi AM, Pietraforte D. (1998) Arch Biochem Biophys.
352,165-174. 40. Brar SS, Kennedy TP, Sturrock AB, Huecksteadt TP, Quinn MT, Murphy TM,
Chitano P, Hoidal JR. (2002) Am J Physiol Lung Cell Mol Physiol. 282, L782-L795.
41. Kwak JY, Takeshige K, Cheung BS, Minakami S. (1991) Biochem Biophys Acta. 1076,369-373.
42. Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. (1995) Science. 270,296-299.
43. Lee JH, Johnson PRA, Roth M, Hunt NH, Black JL. (2001) Am J Physiol Lung Cell Mol Physiol. 280,L1019-L1029.
44. Ravenhall C, Guida E, Harris T, Koutsoubos V, Stewart A. (2000) Br J Pharmacol. 131,17-28.
45. Simon A, Rai U, Fanburg B, Cochran B. (1998) Am J Physiol Cell Physiol. 275, C1640-1652.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

24
46. Simon Ar, Takahashi S, Severgnini M, Fanburg BL, Cochran BH. (2002) Am J Physiol Lung Cell Mol Physiol. 282,L1296-L1304.
47. Hansen TW, Mathiesen SB, Walaas SI. (1996) Pediatric Res. 39,1072-1078.
48. Henderson WR, Tang L, Chu S, Tsao S, Chiang G, Jones F, Jonas M, Pae C, Wang H, Chi EY. (2002) Am J Respir Crit Care Med. 165,108-116.
49. Salmon M, Liu Y, Mak JCW, Rousell J, Huang TJ, Hisada T, Nicklin PL, Chung KF. (2000) Am J Respir Cell Mol Biol. 23,618-625.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

25
Table 1. Cellular ROS production.
H2DCFHDA oxidation (T45/T0 ratio)
SOD-inhibitable
extracellular production of ROS
(nmol of superoxide anion/ 106 cells/hour)
1% FCS
1.09 ± 0.02
4.96 ± 0.82
10 % FCS 1.55 ± 0.05 * 10.01 ± 1.52 *
+ CoPP (20µM) 1.09 ± 0.09 # 3.03 ± 1.02 # + bilirubin (1µM) 1.08 ± 0.07 # 2.25 ± 0.65 # + NAC (1mM) 1.06 ± 0.02 # 4.30 ± 0.41 # + SnPP (25 µM) 2.37 ± 0.22 # 15.10 ± 1.10 # + CoPP (20µM)+ SnPP (25µM) 1.57 ± 0.42 & 9.51 ± 0.61 & + SnPP (25µM)+ Bilirubin (1µM) 1.30 ± 0.07 & 5.25 ± 1.35 & + SnPP (25µM)+ NAC (1mM) 1.01 ± 0.08 & 5.01 ± 0.55 & PDGF (50 ng/ml) 1.52 ± 0.04
* 12.10 ± 1.81
*
+ CoPP (20µM) 1.14 ± 0.02 # 6.25 ± 0.51 # + bilirubin (1µM) 1.13 ± 0.09 # 4.85 ± 0.41 # + NAC (1mM) 0.99 ± 0.19 # 5.81 ± 1.45 # + SnPP (25 µM) 2.41 ± 0.21 # 17.51 ± 0.72 # + CoPP (20µM)+ SnPP (25µM) 1.98 ± 0.15 & 9.65 ± 0.71 & + SnPP (25µM)+ Bilirubin (1µM) 1.23 ± 0.07 & 3.51 ± 0.51 & + SnPP (25µM)+ NAC (1mM)
1.17 ± 0.05 & 8.15 ± 2.61 &
Intra- and extracelular ROS production was measured by H2DCFHDA oxidation and SOD-
inhibitable reduction of cytochrome c respectivelly, as described in the Experimental
Procedures section. Results are expressed as mean ± SEM. n=6-12 for each condition. *:
P<0.05 vs 1% FCS ; # : P<0.05 vs 10% FCS or PDGF ; &: P<0.05 vs SnPP+10% FCS or
SnPP+PDGF
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

26
Figure legends
Figure 1
HO-1 expression in human airway smooth muscle cells.
Panel A : Western blot analysis of HO-1 expression. Whole-cell proteins were extracted from
human ASM cells after a 24-hour serum deprivation followed by a stimulation with 10% FCS,
50 ng /ml PDGF or 20µM CoPP for 24 hours. The histogram represents the ratio between
HO-1 optical density and that of β-actin for the above typical experiment. Panel B : HO
activity was assessed by measuring bilirubin production by human ASM cells with a
spectrophotometer according to the method described in the “Experimental procedures”
section. n=4-6 for each condition. Bars are mean ± SE. * P<0.05 vs 1% FCS; # P<0.05 vs
mitogen alone. Panel C : immunohistochemical analysis of HO-1 and bilirubin expression in
ASM cells. Cells were cultured in a chamber slide, fixed with acetone and stained by
immunoreaction with monoclonal antibodies. No staining was observed with isotype
antibodies. Panel D : immunohistochemical analysis of HO-1 expression in airway smooth
muscle in human bronchi. Staining was observed in airway smooth muscle (*) and epithelium
(arrow). No staining was observed with the isotype antibody.
Figure 2
Role of the HO pathway on human ASM cells proliferation. Cells were stimulated for 24
hours with 10% FCS (panel A and C) or PDGF (panel B and D). 3H-thymidine incorporation
(4µCi/ml) was measured by scintillation counting. Each condition was done on quadruplicate.
Three different muscular explants were tested for each experiment. Values are mean ± SE.
*:P<0.05 vs 1% FCS, #: P<0.05 vs mitogen alone, &: P<0.05 vs mitogen+SnPP.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

27
Figure 3
Effect of cell transfection with HO sense and antisense oligonucleotides (ODNs).
Panel A : effect of HO-1 ODNs on PDGF-induced HO-1 expression. Experimental procedure
is the same than described in Figure 1.
Panel B : Thymidine incorporation in ODN transfected cells. Cells were stimulated for 24
hours with PDGF. 3H-thymidine incorporation (4µCi/ml) was measured by scintillation
counting. Each condition was done on quadruplicate. Values are mean ± SE. *:P<0.05 vs 1%
FCS, #: P<0.05 vs mitogen alone.
Figure 4
Role of the guanylyl cyclase-cGMP pathway.
Panel A : thymidine incorporation by human ASM cells stimulated for 24 hours with serum or
PDGF plus DMSO (that is used as the solvent for ODQ). Cells were treated with the HO
inductor CoPP (20µM) and by the guanylate cyclase inhibitor 1H-(1,2,4)oxadiazolo(4,3-
a)quinoxalin-1-one (ODQ, 10µM).
Panel B : role of myoglobin, a scavenger for CO, on cell proliferation. For panel A and B,
p<0.05 in all conditions. n=12 for each condition. Bars are mean ± SE. *:P<0.05 vs 1% FCS;
#: P<0.05 vs mitogen.
Figure 5
Role of HO on phosphorylation of the ERK1/2 MAPkinase.
Panel A : time course of phosphorylation of ERK1/2 MAPKinase induced by 50 ng/ml
PDGF, assessed by western blot. Panel B : typical western blot analysis of ERK1/2
phosphorylation after a 10 minute stimulation with 50 ng/ml PDGF-AB. Cells were pre
treated with the different reagents for one hour before stimulation. Results were similar with
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

28
cells from three different muscular explants. Graph represents optical density of the bands of
one typical western blot, compared with expression of β-actin. Panel C : effects of the MEK
inhibitor PD 98059 on ERK phosphorylation induced by a 10 minutes stimulation with serum
and PDGF. The histogram represents the ratio between phosphorylated ERK1/2 optical
density and that of β-actin for a typical experiment.
Panels D and E : assessment of the role of PD 98059 on cell proliferation by incorporation of
3H-thymidine after a 24 hour stimulation with 10% serum (panel D) and PDGF (panel E).
Results are expressed as mean±SE of three different experiments with cells from three
different explants. *:P<0.05 vs 1%FCS+DMSO; #:P<0.05 vs mitogen + DMSO; &:P<0.05 vs
mitogen+DMSO+SnPP.
Figure 6
Role of the HO pathway on airway smooth muscle increase in allergic airway inflammation
Panel A : HO-1 and bilirubin were expressed in ASM (*) in OO guinea pigs. Expression was
also observed in inflammatory cells and epithelium (arrows). No staining was observed with
isotype antibodies.
Panel B : Airway smooth muscle area measurement. Animals were either sensitized with
ovalbumin and challenged with NaCl (ON animals) or sensitized and challenged with
ovalbumin (OO animals), chronically treated or not with the HO inductor hemin or the HO
inhibitor SnPP as described in “Experimental Procedures” section. Results represent the mean
± SE of 3 to 4 bronchi in 5 different animals in each group. *: P<0.05 vs ON; #: P<0.05 vs
OO.
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

29
Figure 1
CoPP
A32k
1% FCS 10% FCS PDGF
0
0.5
1
1.5
2
2.5
HO
-1/β
act
in
(arb
itrar
y un
its)
PDGF10% FCS 1% FCS CoPP
PDGF (50 ng/ml)10% FCS
* *
#
&
#
&
0
100
200
300
400
500
600
HO
act
ivity
(p
mol
bili
rubi
n/m
g pr
otei
n/60
min
)
CoP
P (2
0µM
)
CoP
P (2
0µM
)
SnPP
(25
µM)
SnPP
(25
µM)
B by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

30
Figure 1
D
C
HO-1
Bilirubin
IsotypeAntibody
HO-1
Isotype
*
* *
*
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

31
Figure 2
1% FCS PDGF 50 ng/ml
CoP
P 20
µM
SnPP
25µ
M
CoP
P 20
µM
+ Sn
PP 2
5µM
B
*
#
#
&
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
0
200
400
600
800
1% FCS PDGF 50 ng/ml C
oPP
20µM
SnPP
25µ
M
CoP
P 20
µM
+ Sn
PP 2
5µM
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
0
200
400
600
300
500
100
*
#
#
&
A
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

32
Figure 2
NA
C 1
mM
*# #
#
& &
0
100
200
300
400
500
SnPP 25µM 1 % FCS
10% FCS B
iliru
bin
1µM
NA
C 1
mM
Bili
rubi
n 1µ
M
C
1 % FCS
PDGF (50 ng/ml)
Bili
rubi
n 1µ
M
NA
C 1
mM
SnPP 25µM
Bili
rubi
n 1µ
M
NA
C 1
mM
D
*
# #
#
&&
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
ofco
ntro
l)
0
100
200
300
400
500
600
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

33
Figure 3
HO-1
β-Actin
Positive control (spleen)
Non transfected
cells
Transfection reagent alone
HO-1 HO-1 antisense
A
0
100
200
300
400
500
600
700
1%FCS PDGF (50 ng/ml)
HO
-1 a
ntis
ense
HO
-1 se
nse
Tra
nsfe
ctio
n re
agen
t al
one
*#
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
B
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

34
Figure 4
10% FCS PDGF (50 ng/ml)
CoP
P (2
0µM
)
OD
Q (1
0µM
)
C
oPP
(20µ
M)
+ O
DQ
(10µ
M)
CoP
P (2
0µM
)
OD
Q (1
0µM
)
C
oPP
(20µ
M)
+ O
DQ
(10µ
M)
A
1% FCS
*
*
# #
0
100
200
300
400
500
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
0
100
200
300
400
500
B
CoPP (20µM) Myoglobin
5 10 20µM
CoPP (20µM) Myoglobin
5 10 20µM
10% FCS PDGF (50 ng/ml) 1% FCS
**
# ## #
## #
#
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from
35
Figure 5
PDGF 50 ng/ml
0 5 10 15 30 60 90 min
Aphospho ERK1/2
0
0.5
1
1.5
2 2.5
3
phos
pho
ER
K 1
/2 /β
act
in
Bilir
ubin
SnPP
+B
iliru
bin
PDGF
CoP
P
NA
C
SnPP
1%
FCS
Bphospho ERK1/2
PDGF 1% FBS
10% FBS
SnPP
CoP
P
NA
C
Bilir
ubin
SnPP
+Bili
rubi
n
PDGF
0
0.5
1
1.5
2
Cphospho ERK1/2
1%FCS
PDGF
PD98
059
SnPP
+
PD98
059
SnPP
1% FCS
PDGF 10% FCS
PD98
059
SnPP
+
PD98
059
SnPP
PD98
059
SnPP
+
PD98
059
SnPP
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from
36
Figure 5
E
D
0
100
200
300
400
500
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
PD 98 059
PDGF (50 ng/ml) + DMSO
SnPP (25µM)
10 30 50µM
PD 98 059
10 30 50 µM
1%FCS +DMSO
# # #
#
&
& &
*
# #
#
#&
&
&
*
0
100
200
300
400
500
600
PD 98 059
10% FCS + DMSO
SnPP (25µM)
10 30 50µM
PD 98 059
10 30 50 µM
1%FCS +DMSO
3H-t
hym
idin
e in
corp
orat
ion
(per
cent
age
of c
ontr
ol)
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

37
Figure 6
A
B
Bilirubin
HO-1
OO ON Isotype
0
50
100
150
200
Mus
cula
r ar
ea
(µm
2 )
ON OO+ hemin + SnPP
OO OO
* ##
*
*
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from

Michel Aubier and Jorge BoczkowskiMegret, Patrick Berger, Guy Leseche, Elie Fadel, Tokio Yamaguchi, Roger Marthan, Camille Taille, Abdelhamid Almolki, Moussa Benhamed, Christine Zedda, Jerome
bilirubin-dependent modulationof ERK1/2 phosphorylationHeme oxygenase inhibits human airway smooth muscle proliferation via a
published online April 10, 2003J. Biol. Chem.
10.1074/jbc.M300364200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on February 24, 2018http://w
ww
.jbc.org/D
ownloaded from