guidelines on qualification - appendix 6...136 guideline on qualification – updated text proposed...
TRANSCRIPT

Working document QAS/16.673/Rev.2
September 2018
Draft document for comments
GUIDELINES ON QUALIFICATION - APPENDIX 6 1
VALIDATION 2
3
(September 2018) 4
DRAFT FOR COMMENTS 5
6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21
© World Health Organization 2018 22 23 All rights reserved. 24 25 This draft is intended for a restricted audience only, i.e. the individuals and organizations having received this draft. The draft 26 may not be reviewed, abstracted, quoted, reproduced, transmitted, distributed, translated or adapted, in part or in whole, in any 27 form or by any means outside these individuals and organizations (including the organizations' concerned staff and member 28 organizations) without the permission of the World Health Organization. The draft should not be displayed on any website. 29 30 Please send any request for permission to: 31 32 Dr Sabine Kopp, Group Lead, Medicines Quality Assurance, Technologies, Standards and Norms, Regulation of Medicines and 33 other Health Technologies, Department of Essential Medicines and Health Products, World Health Organization, CH-1211 34 Geneva 27, Switzerland, fax: (41-22) 791 4856; email: [email protected]. 35 36 The designations employed and the presentation of the material in this draft do not imply the expression of any opinion 37 whatsoever on the part of the World Health Organization concerning the legal status of any country, territory, city or area or of its 38 authorities, or concerning the delimitation of its frontiers or boundaries. Dotted lines on maps represent approximate border lines 39 for which there may not yet be full agreement. 40 41 The mention of specific companies or of certain manufacturers’ products does not imply that they are endorsed or recommended 42 by the World Health Organization in preference to others of a similar nature that are not mentioned. Errors and omissions 43 excepted, the names of proprietary products are distinguished by initial capital letters. 44 45 All reasonable precautions have been taken by the World Health Organization to verify the information contained in this draft. 46 However, the printed material is being distributed without warranty of any kind, either expressed or implied. The responsibility 47 for the interpretation and use of the material lies with the reader. In no event shall the World Health Organization be liable for 48 damages arising from its use. 49 50 This draft does not necessarily represent the decisions or the stated policy of the World Health Organization. 51
Please forward any comments you may have on the attached text to Dr Sabine Kopp, Group
Lead, Medicines Quality Assurance, Technologies, Standards and Norms ([email protected])
with a copy to Mrs Xenia Finnerty ([email protected]) by 15 November 2018.
Medicines Quality Assurance working documents will only be sent out electronically and
will also be placed on the Medicines website for comments under “Current projects”. If
you have not already received our draft working documents, please send your email
address (to [email protected]) and we will add it to our electronic mailing list.
.
Working document QAS/16.673/Rev.2
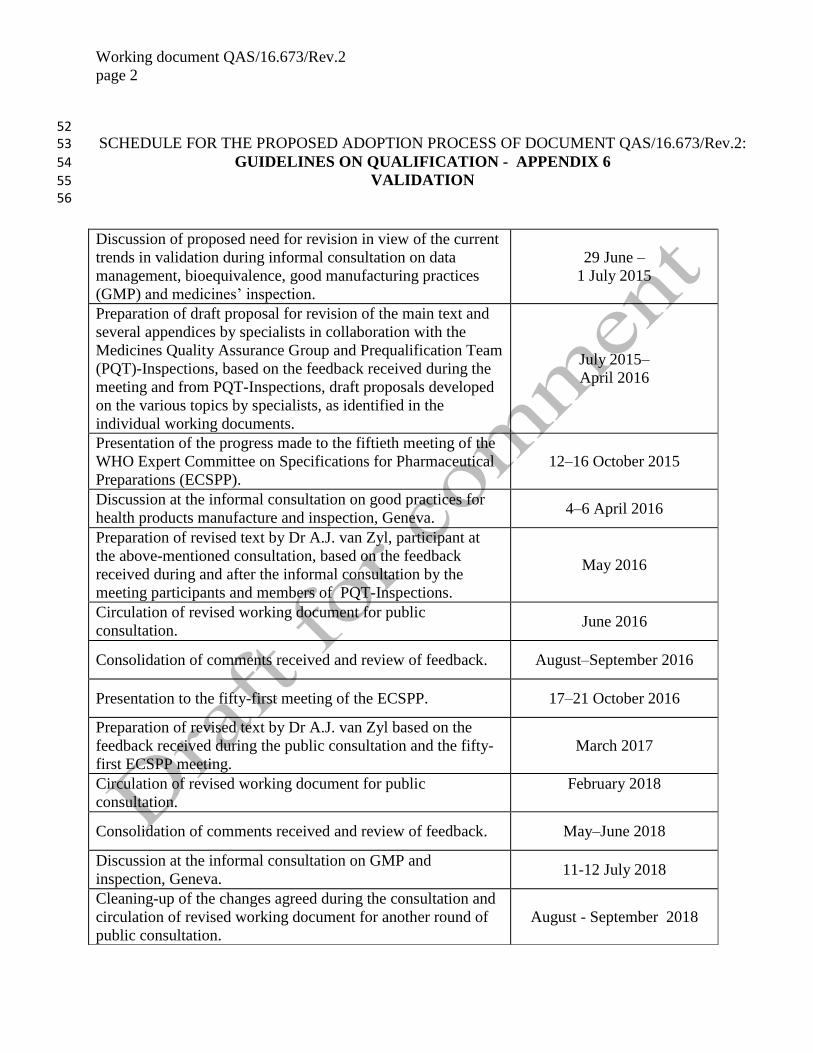
page 2 52 SCHEDULE FOR THE PROPOSED ADOPTION PROCESS OF DOCUMENT QAS/16.673/Rev.2: 53
GUIDELINES ON QUALIFICATION - APPENDIX 6 54
VALIDATION 55 56
Discussion of proposed need for revision in view of the current
trends in validation during informal consultation on data
management, bioequivalence, good manufacturing practices
(GMP) and medicines’ inspection.
29 June –
1 July 2015
Preparation of draft proposal for revision of the main text and
several appendices by specialists in collaboration with the
Medicines Quality Assurance Group and Prequalification Team
(PQT)-Inspections, based on the feedback received during the
meeting and from PQT-Inspections, draft proposals developed
on the various topics by specialists, as identified in the
individual working documents.
July 2015–
April 2016
Presentation of the progress made to the fiftieth meeting of the
WHO Expert Committee on Specifications for Pharmaceutical
Preparations (ECSPP).
12–16 October 2015
Discussion at the informal consultation on good practices for
health products manufacture and inspection, Geneva. 4–6 April 2016
Preparation of revised text by Dr A.J. van Zyl, participant at
the above-mentioned consultation, based on the feedback
received during and after the informal consultation by the
meeting participants and members of PQT-Inspections.
May 2016
Circulation of revised working document for public
consultation. June 2016
Consolidation of comments received and review of feedback. August–September 2016
Presentation to the fifty-first meeting of the ECSPP. 17–21 October 2016
Preparation of revised text by Dr A.J. van Zyl based on the
feedback received during the public consultation and the fifty-
first ECSPP meeting.
March 2017
Circulation of revised working document for public
consultation.
February 2018
Consolidation of comments received and review of feedback. May–June 2018
Discussion at the informal consultation on GMP and
inspection, Geneva. 11-12 July 2018
Cleaning-up of the changes agreed during the consultation and
circulation of revised working document for another round of
public consultation.
August - September 2018

Working document QAS/16.673/Rev.2
page 3
57 58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
Consolidation of comments received and review of feedback. October-November 2018
Presentation to the fifty-third meeting of ECSPP. 22–26 October 2018
Any other follow-up action as required. …

Working document QAS/16.673/Rev.2
page 4
BACKGROUND INFORMATION 92
93
The need for revision of the published Supplementary guidelines on good manufacturing 94
practices: validation (World Health Organization (WHO) Technical Report Series, No. 937, 95
2006, Annex 4) was identified by the Prequalification of Medicines Programme and a draft 96
document was circulated for comments in early 2013. The focus of the revision was the 97
Appendix on non-sterile process validation (Appendix 7), which had been revised and was 98
adopted by the WHO Expert Committee on Specifications for Pharmaceutical Preparations 99
(ECSPP) at its forty-ninth meeting in October 2014. 100
101
The main text was sent out for consultation as a working document QAS/15.639 entitled 102
Guidelines on validation which constitute the general principles of the new guidance on 103
validation. 104
105
The draft on the specific topics, the appendices to this main text, will follow. One of them, the 106
Validation on qualification of systems, utilities and equipment, newly entitled Guidelines on 107
qualification, constitutes this working document. 108
109
The following is an overview on the appendices that are intended to complement the general text 110
on validation: 111
112
Appendix 1 113
Validation of heating, ventilation and air-conditioning systems (HVAC) 114
will be replaced by cross-reference to the WHO Guidelines on GMP for HVAC 115
systems for considerations in qualification of HVAC systems 116
(– Annex 8 in TRS 1010, 2018) 117
118
Appendix 2 119
Validation of water systems for pharmaceutical use 120

Working document QAS/16.673/Rev.2
page 5
will be replaced by cross-reference to WHO Guidelines on water for pharmaceutical 121
use for consideration in qualification of water purification systems 122
123
Appendix 3 124
Cleaning validation – consensus to retain 125
126
Appendix 4 127
Analytical method validation 128
will be replaced by update – working document QAS/16.671 129
130
Appendix 5 131
Validation of computerized systems 132
will be replaced by update – working document QAS/16.667 133
134
Appendix 6 135
Guideline on Qualification – updated text proposed in this working document 136
(new title) 137
138
Appendix 7 139
Non-sterile process validation – update already published as Annex 3, WHO Technical 140
Report Series, No. 992, 2015 141
142
Brief background on the changes in this document 143
144
There was some confusion regarding the title. It is therefore suggested to change the title to 145
GUIDELINES ON QUALIFICATION. In this way, the general principles in qualification are 146
addressed which can be applied for systems, equipment, and so on. 147
148
Based on the comments, the general chapters on objective and scope were written to make it 149
clear that the guidelines address principles of qualification that can be applied, as appropriate, to 150

Working document QAS/16.673/Rev.2
page 6
premises, systems, utilities and equipment and to include the application of risk management 151
principles. 152
153
Moreover, duplication was removed, logical flow of concepts addressed and aligned with 154
international texts and the comments. The V Model has been removed based on the feedback 155
received. In the former published text on qualification, protocol formats were included. These 156
protocol formats were extracted from training materials and were intended to serve as examples. 157
In view of the feedback that seemingly manufacturers took them as absolute examples to be 158
used, these examples have been removed in the current version. 159
160 161
162

Working document QAS/16.673/Rev.2
page 7
APPENDIX 6 163
GUIDELINES ON QUALIFICATION 164
165
1. Principle 166
2. Scope 167
3. Glossary 168
4. General 169
5. User requirement specifications 170
6. Design qualification 171
7. Factory acceptance test and site acceptance test 172
8. Installation qualification 173
9. Operational qualification 174
10. Performance qualification 175
11. Periodic review and requalification 176
177
1. PRINCIPLE 178
179
1.1 In principle, premises, systems, utilities and equipment should be appropriately designed, 180
installed, qualified, operated, cleaned and maintained to suit their intended purpose. 181
182
1.2 Quality management systems should be in place to ensure that these remain in a qualified 183
state throughout their life cycle. 184
185
1.3 Products should be manufactured on qualified equipment. 186
187
1.4 Manufacturers who may use an alternative verification framework to achieve qualification 188
should ensure the qualification expectations within this guide are satisfied. 189
190
191

Working document QAS/16.673/Rev.2
page 8
2. SCOPE 192
193
2.1 These guidelines describe the general approach to qualification, for example, premises, 194
systems, computerized system, utilities and equipment. 195
196
2.2 The principles in these guidelines may also be applied to the qualification of instruments, 197
analytical instruments and testing devices, where appropriate. 198
199
2.3 These may include and are not limited to: certain rooms; water purification systems; 200
cleaning systems; heating, ventilation and air conditioning systems; compressed air systems; gas 201
systems; steam systems; as well as production equipment and analytical instruments. 202
203
2.4 Separate guidelines in this series address other principles in validation such as process 204
validation and cleaning validation (see references at the end of this document). 205
2.5 The principle can be used when de-commissioning equipment to show that it remains fit 206
for its purpose throughout the life cycle. 207
208
3. GLOSSARY 209
210
computerized system. A computerized system collectively controls the performance 211
and execution of one or more automated processes and/or functions. It includes computer 212
hardware, software, peripheral devices, networks and documentation, for example, manuals and 213
standard operating procedures (SOPs), as well as personnel interacting with hardware and 214
software. 215
216
design qualification. Documented evidence that, for example, the premises, supporting 217
systems, utilities and equipment have been designed for their intended purposes and in 218
accordance with the requirements of good manufacturing practices (GMP). 219
220

Working document QAS/16.673/Rev.2
page 9
factory acceptance test. A test conducted, usually at the vendor’s premises, to verify 221
that the system, equipment or utility, as assembled or partially assembled, meets approved 222
specifications. (new) 223
224
installation qualification. The performance of tests to ensure that the installations (such 225
as machines, measuring devices, utilities and manufacturing areas) used in a manufacturing 226
process are appropriately selected and correctly installed and operate in accordance with 227
established specifications. 228
229
operational qualification. Documented verification that the system or subsystem 230
performs as intended over all anticipated operating ranges. 231
232
performance qualification. Documented verification that the equipment or system 233
operates consistently and gives reproducibility within defined specifications and parameters for 234
prolonged periods. (In the context of systems, the term “process validation” may also be used.) 235
236
site acceptance test. A test conducted at the site of use to verify that the system, 237
equipment or utility, as assembled or partially assembled meets approved specifications. (new) 238
239
system. A regulated pattern of interacting activities and techniques that are united to 240
form an organized whole. 241
242
user requirement specifications. An authorized document that defines the requirements 243
for use of the system, equipment or utility in its intended production environment. (amended) 244
245
utility. A system consisting of one or more components to form a structure designed to 246
collectively operate, function or perform and provide a service such as electricity, water, 247
ventilation or other. (new) 248
249

Working document QAS/16.673/Rev.2
page 10
4. GENERAL 250
251
Note: The remainder of the text in these guidelines will refer to utilities and equipment as 252
examples, even though the principles may be applicable to others such as premises and systems. 253
254
4.1 The validation master plan, or other relevant document, should specify the policy, 255
organization, planning, scope and stages applied in qualification on site, and should cover, for 256
example, production, quality control and engineering. 257
258
4.2 Quality risk management principles should be applied in qualification. These include: 259
260
A clear understanding of the system, and the role it plays in 261
establishing/protecting the process and quality and all of the potential ways (risks) the 262
process or quality could be impacted by failures, events, errors, or time/use-based factors 263
(deterioration, out of tolerance instruments, wear and tear, and so on); 264
Defining all of the Design, Procedural and/or Quality System Controls required to 265
protect against these potential risks. These controls either mitigate/reduce the risks 266
and/or detect the impact to quality or process – should the risk occur. (To ensure the 267
“failure” does not impact final product quality); 268
Compiling evidence during the design, engineering, commissioning and 269
qualification to demonstrate that all of these required “controls” have been properly 270
implemented and verified. (Including “function” where applicable – such as alarms on 271
operating parameters); 272
Appropriate control and oversight of change once the controls have been verified. 273
274
4.3 The scope and extent of qualification and requalification should be determined based on 275
the principles of impact assessment and risk management principles. 276
277
4.4 Qualification should be executed by trained personnel. Training records should be 278
maintained. 279

Working document QAS/16.673/Rev.2
page 11
4.5 Where appropriate, new premises, systems, utilities and equipment should be subjected to 280
all stages of qualification. This includes the preparation of user requirement specifications 281
(URS), design qualification (DQ), installation qualification (IQ), operational qualification (OQ) 282
and performance qualification (PQ). 283
284
4.6 Justification should be provided where it is decided that not all stages of qualification are 285
required. 286
287
4.7 Qualification should be done in accordance with predetermined and approved 288
qualification protocols. The protocol should specify the prerequisites and test details, including 289
acceptance criteria. 290
291
4.8 The results of the qualification should be recorded and reflected in qualification reports. 292
293
4. 9 A qualification report prepared at the completion of each protocol or stage of 294
qualification (Installation/Operational/Performance) should include, or reference as appropriate, 295
the following: 296
297
test results, including supporting calculations, documentation and raw/original 298
data, 299
test failures, 300
protocol departures, 301
recommendations and justification for issue resolution, and 302
conclusions. 303
304
4.10 There should be a logical sequence for executing qualification, such as premises (rooms), 305
then utilities and equipment. 306
307
4.11 Normally, qualification stages should be sequential. (For example, operational 308
qualification should follow after the successful completion of installation qualification.) In some 309

Working document QAS/16.673/Rev.2
page 12
cases, different stages of qualification may be executed concurrently. This should be justified 310
and documented in the validation master plan (or qualification protocol). 311
312
4.12 Equipment should be released for routine use only once there is documented evidence 313
that the qualification has been successful. 314
315
4.13 Certain stages of the qualification may be done by a supplier or a third party, subject to 316
the conditions and responsibilities as defined in a written agreement between the parties. The 317
contract giver remains responsible to ensure that the qualification is done in accordance with the 318
principles of GMP. 319
320
4.14 The relevant documentation associated with qualification, including SOPs, specifications 321
and acceptance criteria, certificates and manuals, should be available. 322
323
4.15 Utilities and equipment should be maintained in a qualified state and should be 324
periodically reviewed for the need for requalification. Requalification should be considered 325
when changes are made. 326
327
5. USER REQUIREMENT SPECIFICATIONS 328
329
5.1 URS should be prepared for, but not limited to, utilities and equipment, as appropriate. 330
331
5.2 URS should be used at later stages in qualification to verify that the purchased and 332
supplied utility or equipment is in accordance with the user’s needs. 333
334
6. DESIGN QUALIFICATION 335
336
6.1 DQ should demonstrate that the system, as designed, is appropriate for its intended use as 337
defined in the URS. 338
339

Working document QAS/16.673/Rev.2
page 13
6.2 A suitable supplier should be selected and approved for the relevant utility or equipment. 340
341
7. FACTORY ACCEPTANCE TEST AND SITE ACCEPTANCE TEST 342
343
7.1 Where a utility or equipment is assembled, or partially assembled at a site other than that 344
of the purchaser or end-user, testing and verification may be done, based on quality risk 345
management principles, to ensure that it is appropriate and ready for dispatch. 346
347
7.2 The checks and tests during factory acceptance test (FAT) should be recorded. 348
349
7.3 The acceptability of the assembly and overall status of the utility or equipment should be 350
described in a conclusion of the report for the FAT, prior to shipment. 351
352
7.4 Tests, based on quality risk management principles, may be performed to verify the 353
acceptability of the utility or equipment when it is received at the end-user. This is a site 354
acceptance test (SAT). 355
356
7.5 The results of the tests should be evaluated and the outcome of the acceptability of the 357
utility or equipment should be recorded in the conclusion section of the report for the SAT. 358
359
8. INSTALLATION QUALIFICATION 360
361
8.1 Utilities and equipment should be correctly installed, in an appropriate location. 362
363
8.2 There should be documented evidence of the installation. This should be in accordance 364
with the IQ protocol which contains all the relevant details. 365
366
8.3 IQ should include identification and installation verification of relevant components 367
identified, e.g. services, controls and gauges. 368
369

Working document QAS/16.673/Rev.2
page 14
8.4 Identified measuring, control and indicating devices, should be calibrated on site unless 370
otherwise appropriately justified. The calibration should be traceable to national or international 371
standards. Traceable certificates should be available. 372
373
8.5 Deviations and non-conformances, including those from URS, DQ and acceptance 374
criteria specified and observed during installation, should be recorded, investigated, and 375
corrected or justified. 376
377
8.6 Normally, the outcome of the IQ should be recorded in the conclusion of the report, 378
before OQ is started. 379
380
9. OPERATIONAL QUALIFICATION 381
382
9.1 Requirements and procedures for operation (or use), calibration, maintenance and 383
cleaning should normally be prepared normally before OQ and approved prior to PQ. 384
385
9.2 Utilities and equipment should operate correctly and their operation should be verified in 386
accordance with an OQ protocol. OQ normally follows IQ but, depending on the complexity of 387
utility or equipment, it may be performed as a combined installation/operation qualification 388
(IOQ). This should be justified and documented in the validation master plan (or qualification 389
protocol). 390
391
9.3 OQ should include, but is not limited to, the following: 392
393
tests that have been developed from the knowledge of processes, systems and 394
equipment to ensure the utility or equipment is operating as designed; and 395
396
tests to confirm upper and lower operating limits, and/or “worst case” conditions. 397
398

Working document QAS/16.673/Rev.2
page 15
9.4 Training of operators for the utilities and equipment should be provided and training 399
records maintained. 400
401
9.5 Calibration, cleaning, maintenance, training and related tests and results should be 402
verified to be acceptable. 403
404
9.6 Deviations and non-conformances observed should be recorded, investigated and 405
corrected or justified. 406
407
9.7 The results for the verification of operation should be documented in the OQ report. 408
409
The outcome of the OQ should be recorded in the conclusion of the report, normally before PQ is 410
started. 411
412
10. PERFORMANCE QUALIFICATION 413
414
10.1 PQ should normally follow the successful completion of IQ and OQ. In some cases, it 415
may be appropriate to perform PQ in conjunction with OQ or process validation. This should be 416
justified and documented in the validation master plan (or qualification protocol). 417
418
10.2 PQ should include, but is not limited to, the following: 419
420
tests using production materials, qualified substitutes or simulated products 421
proven to have equivalent behaviour under normal operating conditions with worst case 422
scenario and batch sizes where appropriate; and 423
tests should cover the intended operating range. 424
425
10.3 Utilities and equipment should consistently perform in accordance with their design 426
specifications and URS. The performance should be verified in accordance with a PQ protocol. 427
428

Working document QAS/16.673/Rev.2
page 16
10.4 There should be records (for example, a PQ report) for the PQ to indicate the satisfactory 429
performance over a predefined period of time. Manufacturers should justify the period over 430
which PQ is done. 431
432
11. PERIODIC REVIEW AND REQUALIFICATION 433
434
11.1 Utilities and equipment should be maintained in a qualified state through the life cycle of 435
the utility or equipment. 436
437
11.2 Utilities and equipment should be reviewed periodically to confirm that they remain in a 438
qualified state or to determine the need for requalification. 439
440
11.3 Where the need for requalification is identified, this should be performed. 441
442
11.4 Risk management principles should be applied in the review and requalification and the 443
possible impact of small changes over a period of time should further be considered (such as, 444
through change control). 445
446
11.5 Risk management principles may include factors such as calibration, verification, 447
maintenance data and other information. 448
449
11.6 The qualification status and requalification due dates should be documented, for example, 450
in a qualification matrix, schedule or plan. 451
452
11.7 In case a utility or equipment in use is identified, where it had not been subjected to 453
qualification, a qualification protocol should be prepared where elements of URS, design 454
specifications, operation and performance are verified for acceptability. The outcome of this 455
qualification should be recorded in a report. 456
457
458

Working document QAS/16.673/Rev.2
page 17
Reference documents for additional reading 459
460
[Note from the Secretariat: The references below will be updated upon finalization of the related 461
texts.] 462
463
See WHO TRS 970, 2012, Annex 2, for aspects to be considered for inclusion in qualification of 464
water purification systems. 465
466
See WHO TRS 1010, 2018, Annex 8, for aspects to be considered for inclusion in qualification 467
of heating, ventilation and air-conditioning (HVAC) systems. 468
469
See WHO TRS XXX for aspects to be considered for inclusion in qualification and validation of 470
computerized systems (QAS working document QAS/16.667). 471
472
See WHO TRS 992, 2015, Annex 3, for aspects to be considered in process validation. 473
474
See WHO TRS XXX for aspects to be considered in analytical method validation (QAS working 475
document QAS/16.671) 476
477
*** 478