gmp compliance for quality control laboratory...
TRANSCRIPT

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsIntroduction
Page Intro-1
©2014 西山経営研究所
GMP Compliance forQuality Control
Laboratory Operations
©2007 PharmaNet, Inc.
品質管理試験室作業のGMP遵守
©2007 PharmaNet, Inc.

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsIntroduction
Page Intro-2
©2014 西山経営研究所
Laboratory GMPs
Course Objectives
Review cGMP Requirements for LaboratoryOperations
Review Current cGMP Compliance Issues
Practical Application of cGMPs withExamples and Recommendations
試験室 GMP
コースの目的
試験室作業に関するcGMP要件のレビュー
cGMP遵守に関する現在の問題のレビュー
cGMPの実践的な適用およびその事例と提案

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsIntroduction
Page Intro-3
©2014 西山経営研究所
Laboratory GMPs
Topics
Walk-through Inspection Coverage
General Laboratory Controls
Laboratory Documentation practices
Validation of Analytical Methods
Laboratory OOS: Investigation and Retesting
試験室 GMP
話題
ウォークスルー査察の対象範囲
一般的な試験室管理
試験室の文書化の実践
分析法のバリデーション
試験室OOS:調査と再試験

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsWalk-through Inspection of the Laboratory
Page 2-1
©2014 西山経営研究所
Walk-throughInspection
of the Laboratory
試験室のウォークスルー査察

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsWalk-through Inspection of the Laboratory
Page 2-8
©2014 西山経営研究所
Laboratory Walk-through Inspection
Storage Refrigerators
Location of Temperature Sensor
Procedure and Frequency for TemperatureRecordings
Review and Filing of TemperatureRecordings
SOP With Temperature MonitoringFrequency and Limits
Storage of “Foreign” Materials
試験室のウォークスルー査察
冷蔵庫保管
温度センサーの場所
温度記録の手順と頻度
温度記録のレビューとファイリング
温度のモニタリングの頻度と温度の限度を示したSOP
「関係ない」物質の保管

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsWalk-through Inspection of the Laboratory
Page 2-9
©2014 西山経営研究所
Laboratory Walk-through Inspection
Storage Refrigerators
Adequate Sealing and Protection ofContainers
To minimize evaporation
To avoid cross-contamination
Solutions and Other Materials Have Properand Sufficient Labeling to Avoid Mix-ups
試験室のウォークスルー査察
冷蔵庫保管
容器の適切なシールと保護
蒸発を最小限にするため
交叉汚染を防ぐため
混同防止のための溶液と他の薬品の適切かつ十分な標識

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsWalk-through Inspection of the Laboratory
Page 2-10
©2014 西山経営研究所
Laboratory Walk-through Inspection
Storage Refrigerators
Status of Old Solutions; Still Appropriate forUse?
Standard or Stock Solutions With LongExpiration Dates (e,g., beyond 3 months)Might be Questioned
試験室のウォークスルー査察
冷蔵庫保管
古い溶液の状態:まだ使用に適しているか?
有効期限の長い(たとえば、3ヶ月超)標準溶液または保存溶液は問題があるかもしれない

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
GMP Compliance for Quality Control Laboratory OperationsWalk-through Inspection of the Laboratory
Page 2-11
©2014 西山経営研究所
Laboratory Walk-through Inspection
“Check for the reuse of stock solutionswithout assuring their stability. Stocksolutions are frequently stored in thelaboratory refrigerator. Examine thelaboratory refrigerators for these solutionsand when found check for appropriateidentification. Review records of standardsolution preparation to assure complete andaccurate documentation.”
FDA Laboratory Inspection Guide, 7/93
試験室のウォークスルー査察
「安定性を確認していない保存溶液の再使用をチェックしなさい。保存溶液は、試験室の冷蔵庫によく保管されている。冷蔵庫を調査して、これらの溶液があれば、識別が適切であることをチェックしなさい。完全で正確な文書化を確認するために、標準溶液の調製記録をレビューしなさい。」
FDA Laboratory Inspection Guide, 7/93

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
Page 3-1
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Controls
©2014 西山経営研究所
Laboratory Controls
©2006 PharmaNet, Inc.
試験室管理
©2014 西山経営研究所

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
Page 3-6
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Controls
©2014 西山経営研究所
Laboratory Controls: General
Trending Programs
Environmental Monitoring
Water System Test Results
Deviations and Investigations
Written Trending Report
Calibration and Maintenance Problems
試験室管理: 一般
傾向分析プログラム
環境モニタリング
水システムの試験結果
逸脱と調査
傾向分析報告書
校正および保守の問題

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
Page 3-7
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Controls
©2014 西山経営研究所
FDA - 483
Laboratory Deviation Trending
Your firm has not been effective in identifying theissues and taking the necessary preventive actionsregarding the high percentage of Testing DeviationReports involving equipment malfunctions andlaboratory errors.
FDA - 483
試験室の逸脱の傾向分析
貴社は、装置の機能不良や試験室エラーを含む高比率の試験の逸脱報告に関して、問題の特定と必要な予防処置を実施しなかった。
©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
Page 3-69
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Controls
©2014 西山経営研究所
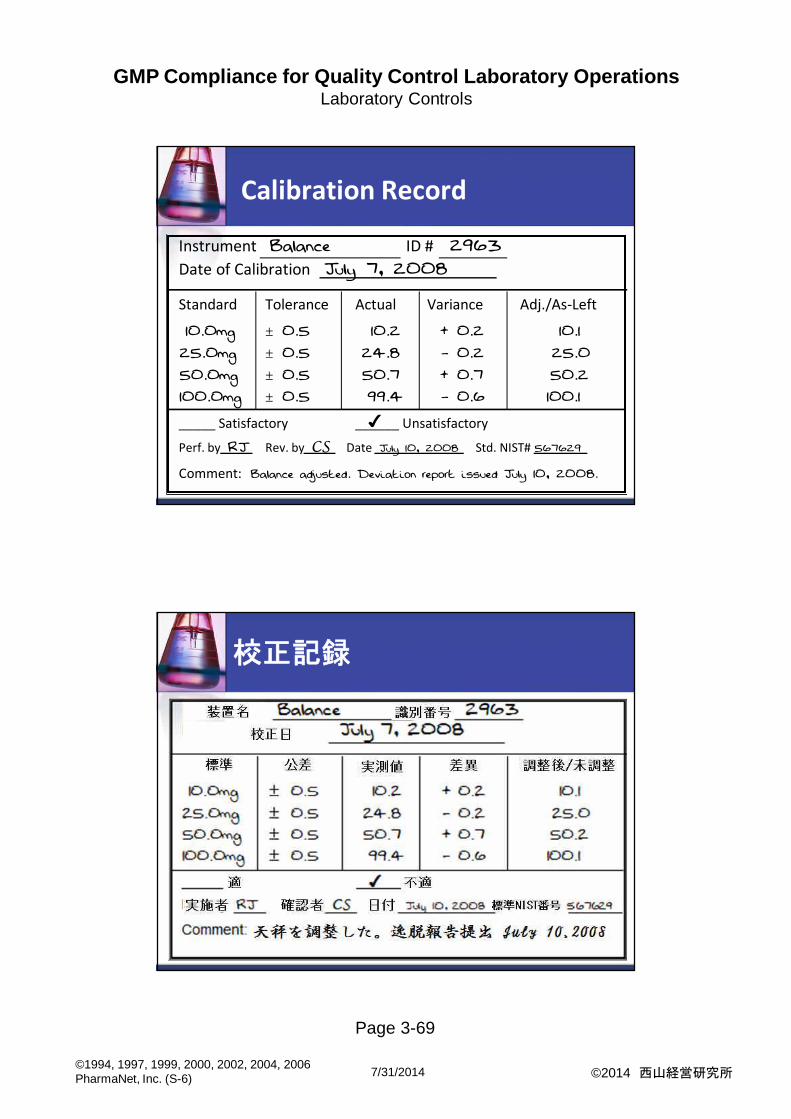
Calibration Record
Instrument Balance ID # 2963
Date of Calibration July 7, 2008
Standard Tolerance Actual Variance Adj./As-Left
10.0mg ± 0.5 10.2 + 0.2 10.1
25.0mg ± 0.5 24.8 - 0.2 25.0
50.0mg ± 0.5 50.7 + 0.7 50.2
100.0mg ± 0.5 99.4 - 0.6 100.1
_____ Satisfactory ______ Unsatisfactory
Perf. by RJ Rev. by CS Date July 10, 2008 Std. NIST# 567629
Comment: Balance adjusted. Deviation report issued July 10, 2008.
校正記録
©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
Page 3-78
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Controls
©2014 西山経営研究所
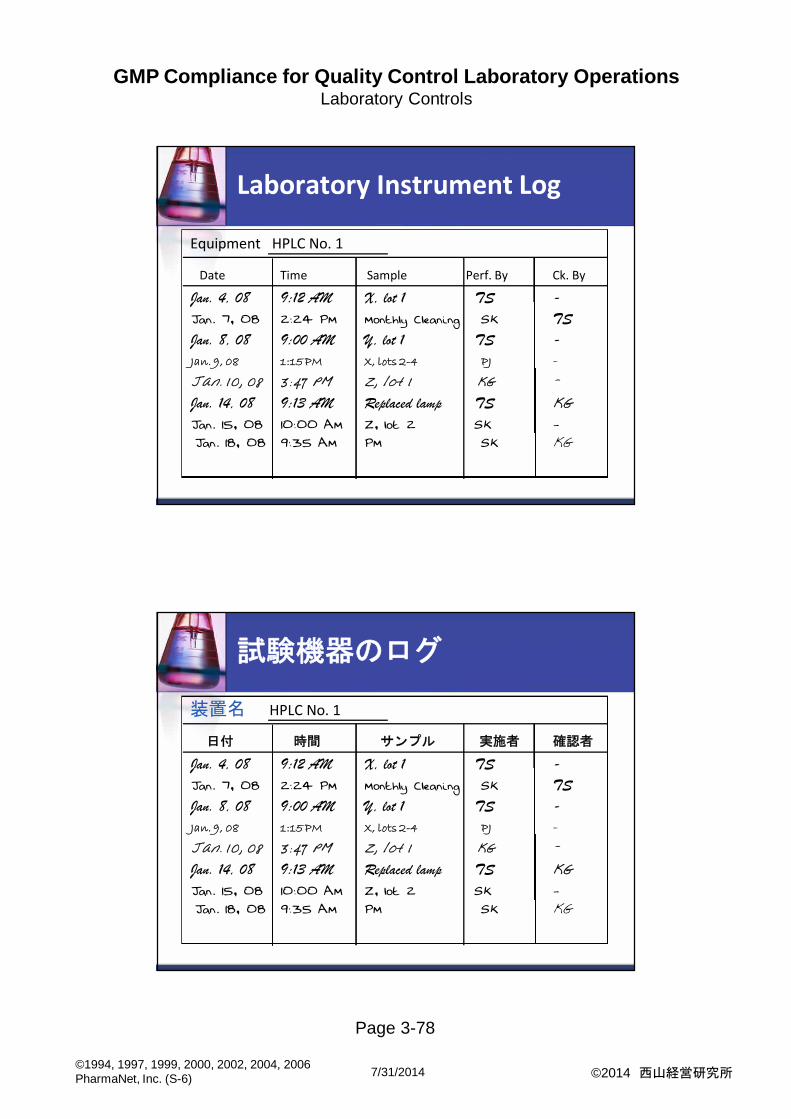
Laboratory Instrument Log
Equipment HPLC No. 1
Date Time Sample Perf. By Ck. By
Jan. 4, 08 9:12 AM X, lot 1 TS -
Jan. 7, 08 2:24 PM Monthly Cleaning SK TS
Jan. 8, 08 9:00 AM Y, lot 1 TS -
Jan. 9, 08 1:15 PM X, lots 2-4 PJ -
Jan. 10, 08 3:47 PM Z, lot 1 KG -
Jan. 14, 08 9:13 AM Replaced lamp TS KG
Jan. 15, 08 10:00 AM Z, lot 2 SK -
Jan. 18, 08 9:35 AM PM SK KG
試験機器のログ
装置名 HPLC No. 1
日付 時間 サンプル 実施者 確認者
Jan. 4, 08 9:12 AM X, lot 1 TS -
Jan. 7, 08 2:24 PM Monthly Cleaning SK TS
Jan. 8, 08 9:00 AM Y, lot 1 TS -
Jan. 9, 08 1:15 PM X, lots 2-4 PJ -
Jan. 10, 08 3:47 PM Z, lot 1 KG -
Jan. 14, 08 9:13 AM Replaced lamp TS KG
Jan. 15, 08 10:00 AM Z, lot 2 SK -
Jan. 18, 08 9:35 AM PM SK KG

©1994, 1997, 1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6)
Page 3-98
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Controls
©2014 西山経営研究所
FDA Draft Guidance (Contractor Quality Agreement)May 2013
Contract Laboratories are Contracted Facilities Subjectto CGMP Requirements “…the Contracted Facilities could be held responsible for
clear CGMP violations related to the laboratory activitiesthey conduct.”
“Additionally, the Owners could be responsible for CGMPviolations because, regardless of who tests the productsor the agreements in place regarding the manufacturingand testing of those products…”
“The Owners might further be cited for failure to followtheir own procedures for evaluating, qualifying, auditing,and monitoring contractors/suppliers.”
Lines 417, 449-453, 455-456
Contract Laboratories
FDAドラフトガイダンス(品質契約書)2013年5月
受託試験機関はCGMP要件の対象となる受託施設である 「...契約施設は、彼らが実施する試験室の活動に関連した明ら
かなCGMP違反に対する責任を負う可能性がある。」
「さらに、所有者は、誰が製品を試験したかに係わらず、またはそれらの製品の製造および試験に関する合意書の実施に係わらず、CGMP違反の責任を負う可能性がある...」
「所有者は、契約業者/供給業者の評価、適格性評価、監査およびモニタリングに関する彼らの手順を守らなかったことに対して、さらに指摘されるかもしれない。」
Lines 417, 449-453, 455-456
受託試験機関

©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
Page 5-1
GMP Compliance for Quality Control Laboratory OperationsLaboratory Documentation Practices
7/13/2014 ©2014 西山経営研究所
LABORATORY DOCUMENTATIONPRACTICES
©2007 PharmaNet, Inc.
試験室の文書化の実践

©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
Page 5-3
GMP Compliance for Quality Control Laboratory OperationsLaboratory Documentation Practices
7/13/2014 ©2014 西山経営研究所
LaboratoryDocumentation Practices
FDA Inspection Coverage
“Analyst’s Notebooks – Review SOP’s relating toanalysts’ notebooks, data recording, etc. Areview of the accuracy and completeness of thedata recorded in analysts’ notebooks orworksheets should be done and include reviewof raw data from associated chromatography,spectra, etc. and other sources generated bylaboratory instruments.”
FDA IOM, Subchapter 549, EIR
試験室の文書化の実践
FDA査察の対象
「分析者のノート-分析者のノート、データの記録等に関するSOPをレビューしなさい。
分析者のノートまたはワークシートに記録されたデータの正確性および完全性のレビューが実施されるべきで、関連したクロマトグラフィー、スペクトラ等から得た生データおよび試験機器から得たその他の情報のレビューも含めなさい。」
FDA IOM, Subchapter 549, EIR

©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
Page 5-12
GMP Compliance for Quality Control Laboratory OperationsLaboratory Documentation Practices
7/13/2014 ©2014 西山経営研究所
LaboratoryDocumentation Practices
Electronic Recordsand
Raw Data
試験室の文書化の実践
電子記録と
生データ

©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
Page 5-13
GMP Compliance for Quality Control Laboratory OperationsLaboratory Documentation Practices
7/13/2014 ©2014 西山経営研究所
LaboratoryDocumentation Practices
Electronic Record Retention
“For High Performance Liquid Chromatography(HPLC) and Gas Chromatography (GC) systems (andother computerized systems involving user inputs,outputs, audit trails, etc.), the predicate rules, suchas 21 CFR 211.68 and 21 CFR 211.180(d), require theelectronic records themselves to be retained andmaintained in accordance with those regulations.”
FDA GMP Q&A; Updated August 4, 2004http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm
試験室の文書化の実践
電子記録の保管
「高速液体クロマトグラフィー(HPLC)とガスクロマトグラフィー(GC)システム(および、ユーザーの入力、
出力、監査証跡などを含むその他のコンピュータ化システム)については、21CFR211.68と21CFR211.180(d)などの関連規則が電子記録それ自
体を維持し、それらの規則に従って保持することを求めている。」
FDA GMP Q&A; Updated August 4, 2004http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm

©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
Page 5-14
GMP Compliance for Quality Control Laboratory OperationsLaboratory Documentation Practices
7/13/2014 ©2014 西山経営研究所
FDA Warning LetterElectronic Record Retention
“The printed paper copy of the chromatogram would not beconsidered a “true copy” of the entire electronic raw data used tocreate that chromatogram, as required by 21 CFR 211.180(d). Theprinted chromatogram would also not be considered an “exact andcomplete” copy of the electronic raw data used to create thechromatogram, as required by 21 CFR 211.68. The chromatogram doesnot generally include, for example, the injection sequence, instrumentmethod, integration method, or the audit trail, of which all were usedto create the chromatogram or are associated with its validity.Therefore, the printed chromatograms used in drug manufacturing andtesting do not satisfy the predicate rule requirements in 21 CFR Part211. The electronic records created by the computerized laboratorysystems must be maintained under these requirements.”
FDA GMP Q&A; Updated August 4, 2004http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm
FDA Warning Letter電子記録の保管
「クロマトグラムの印刷された紙のコピーは、21CFR211.180(d)で求め
るクロマトグラムを作成するために使用した電子生データ全体の『本当のコピー』とは見なせない。印刷されたクロマトグラムもまた、21CFR211.68で求めるクロマトグラムを作成するために使用した電子
生データの『正確で完全』なコピーをは見なせない。そのクロマトグラムは通常、たとえばインジェクションの順序、機器の方法、インテグレーションの方法、または監査証跡、その全てがクロマトグラムの作成に使われたか、その有効性と関わりがある。従って、医薬品の製造と試験に使用され印刷されたクロマトグラムは、21CFR211の関連規則
を満たしていない。コンピュータ化された試験システムにより作成された電子記録は、これらの要件の下で維持されなければならない。」
FDA GMP Q&A; Updated August 4, 2004http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm124787.htm

©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
Page 5-15
GMP Compliance for Quality Control Laboratory OperationsLaboratory Documentation Practices
7/13/2014 ©2014 西山経営研究所
FDA Warning LetterElectronic Record Retention
“You also informed our investigators that printedcopies of HPLC test results are treated as raw data.”
“Printed copies of HPLC test results from your firm’ssystems do not contain all of the analytical metadata(for example: instrument conditions, integrationparameters) that is considered part of the rawdata…This electronic HPLC data supports testing,disposition, and other significant quality controldecisions, and it is essential that you maintain thisinformation for each batch.”
FDA Warning Letter 320-12-08 (Gulf Pharm., Ind. (UAE)), Feb. 2012
FDA Warning Letter電子記録の保管
「あなたは私たちの査察官に、HPLC試験結果の印刷コピーは、生データとして扱われると伝えた。」
「貴社のシステムのHPLC試験結果の印刷コピーは、
生データの一部と見なされる分析のメタデータ(たとえば:機器の条件、インテグレーションパラメータ)の全てを含んでいない...このHPLCの電子データは、試
験、最終処分およびその他の重要な品質管理の意思決定をサポートしており、あなたが各バッチのこの情報を維持することは不可欠なことである。」
FDA Warning Letter 320-12-08 (Gulf Pharm., Ind. (UAE)), Feb. 2012

©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
Page 5-16
GMP Compliance for Quality Control Laboratory OperationsLaboratory Documentation Practices
7/13/2014 ©2014 西山経営研究所
FDA Warning Letter
Electronic Record Retention
“…during the review of the chromatography data…ourinvestigator noticed that the raw data retained doesnot include the run sequence or the processingmethod used to perform the peak integrations…Moreover, the chromatography raw data does notinclude the processing method used to produce thefinal analytical results…therefore…it would not bepossible to detect any modification to the processingmethod.”
FDA Warning Letter WL:320-13-22; 30 Jul 2013
FDA Warning Letter
電子記録の保管
「...クロマトグラフィーのデータのレビュー中に...私た
ちの査察官は、保管された生データはピークのインテグレーションの実施に使われた分析順序または処理方法を含んでいないと伝えた...さらに、クロマトグラフ
ィーの生データは、最終分析結果を生成するすために使われた処理方法を含んでいない...従って...処理方法のいかなる修正も検出することができない。」
FDA Warning Letter WL:320-13-22; 30 Jul 2013

Page 6-1
©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsAnalytical Methods Validation
©2014 西山経営研究所
ANALYTICALMETHOD VALIDATION
Selected Compliance Issues:Verification for USP/Standard Methods
Validation for Method Transfer
Method Revalidation
Robustness Validation
Life Cycle Management
©2006 PharmaNet, Inc.
分析法のバリデーション
順守の問題の抜粋:USP/標準法のベリフィケーション
分析法の移転に関するバリデーション
分析法の再バリデーション
頑健性のバリデーション
ライフサイクル管理

Page 6-7
©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsAnalytical Methods Validation
©2014 西山経営研究所
Analytical Method Validation
Compendial Method Verification
USP <1226> Verification of CompendialProcedures
Verification not required for basiccompendial test procedures; e.g.:
Loss on drying
Residue on ignition
Wet chemical procedures (acid value)
Simple instrument methods (pH)
分析法のバリデーション
公定法のベリフィケーション
USP <1226> 公定法の手順のベリフィケーション
基本的な公定法の試験手順にベリフィケーションは必要ない;たとえば
乾燥減量
強熱残分
湿式の化学的手順(酸価)
単純な機器分析法(pH)

Page 6-8
©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsAnalytical Methods Validation
©2014 西山経営研究所
Analytical Method Validation
“Standard” Method Verification
“Verification of an analytical procedure is thedemonstration that a laboratory is capable ofreplicating with an acceptable level of performancea standard method. Verification under conditions ofuse is demonstrated by meeting system suitabilityspecifications established for the method, as well asa demonstration of accuracy and precision or othermethod parameters for the type of method.”
FDA Laboratory Procedure (LAB.5.4.5), Ver. 1.6, Rev. 1/21/12; Sec. 6.2(Current as of August 2014)
分析法のバリデーション
「標準」試験法のベリフィケーション
「分析手順のベリフィケーションは、試験室が標準法の遂行について許容可能なレベルで再現する能力があることを証明することである。使用条件下でのベリフィケーションは、真度および精度または試験法のタイプに対する他の試験法のパラメータの証明同様に、その試験法のために設定されたシステム適合性の規格を満たすことによって証明される。」
FDA Laboratory Procedure (LAB.5.4.5), Ver. 1.6, Rev. 1/21/12; Sec. 6.2(Current as of August 2014)

Page 6-9
©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsAnalytical Methods Validation
©2014 西山経営研究所
Analytical Method Validation
“Standard” Method Verification
“Method performance is accomplished byusing performance characteristics such as:”
Blanks in chemistry, or un-inoculated mediain microbiology, to assess contamination;
Laboratory control samples - spikedsamples for chemistry or positive culturecontrols for microbiology, to assessaccuracy;
FDA Laboratory Procedure (LAB.5.4.5), Ver. 1.6, Rev. 1/21/12; Sec. 6.2(Current as of August 2014)
分析法のバリデーション
「標準」試験法のベリフィケーション
「試験法の遂行は、以下の性能特性を用いることによって成し遂げられる:」
汚染を評価するための、化学的なブランク、または微生物学的に植え付けていない培地;
試験のコントロールサンプル-真度を評価するための、化学試験に対するスパイクサンプルまたは微生物試験に対する陽性コントロール;
FDA Laboratory Procedure (LAB.5.4.5), Ver. 1.6, Rev. 1/21/12; Sec. 6.2(Current as of August 2014)

Page 6-10
©1994, 1997, 1999, 2000, 2002, 2004, 2007PharmaNet, Inc. (S-6)
7/31/2014
GMP Compliance for Quality Control Laboratory OperationsAnalytical Methods Validation
©2014 西山経営研究所
Analytical Method Validation
“Standard” Method Verification
“Method performance is accomplished by usingperformance characteristics such as:”
Precision based on the analysis of duplicates;
Calibration check standards analyzedperiodically in the analytical batch forquantitative analyses; and
Monitoring quality control samples, usuallythrough the use of control charts.
FDA Laboratory Procedure (LAB.5.4.5), Ver. 1.6, Rev. 1/21/12; Sec. 6.2(Current as of August 2014)
分析法のバリデーション
「標準」試験法のベリフィケーション
「試験法の遂行は、以下の性能特性を用いることによって成し遂げられる:」
複製の分析に基づいた精度;
定量分析のための分析バッチで定期的に分析された標準品の校正チェック;そして
一般的に管理図の使用による、品質管理サンプルのモニタリング。
FDA Laboratory Procedure (LAB.5.4.5), Ver. 1.6, Rev. 1/21/12; Sec. 6.2(Current as of August 2014)

Page 7-1©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory Out-of-specification(OOS)
Investigation and Retesting
©2006 PharmaNet, Inc.
試験室の規格外 (OOS)調査と再試験

Page 7-54©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory OOSInvestigation & Retesting
Interpretation of Investigation Results
Quality Unit Responsible for Interpreting OOSInvestigation Results
Suspected Laboratory Error Required to Invalidate anOOS Test Result
“Invalidation of a discrete test result may be done onlyupon the observation and documentation of a testevent that can reasonably be determined to havecaused the OOS result.”
FDA OOS Guidance, 10/11/06; V-A
試験室OOS調査と再試験
調査結果の解釈
品質部門はOOS調査結果の解釈の責任を負う
はOOS試験結果を無効にするために疑わしい試験室エラーが要求される
「個別の試験結果の無効は、試験がOOS結果の原因となったことを合理的に決定できる試験の観察と記録によってのみ可能である。」
FDA OOS Guidance, 10/11/06; V-A

Page 7-55©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory OOSInvestigation & Retesting
OOS and Retesting
“The investigation should include an appropriate evaluationof events associated with the laboratory analysis thatscientifically supports any determination that the OOS resultis caused by laboratory error. Strong evidence and science-based justification is necessary for your quality unit to permitinvalidation of an initial OOS result.”
FY 2010 Warning Letter: Laboratorios L.O. Oftalmi, C.A. (Venezuela)
試験室OOS調査と再試験
OOSと再試験
「その調査は、OOS結果は試験室エラーに起因するという決定を、科学的にサポートする試験室の分析に関連した出来事の適切な評価を含むべきである。強い証拠および科学に基づいた正当化は、あなたの品質部門が初期のOOS結果の無効を許可するために必要である。」
FY 2010 Warning Letter: Laboratorios L.O. Oftalmi, C.A. (Venezuela)

Page 7-56©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory OOSInvestigation & Retesting
Interpretation of Investigation Results
Investigation Cannot Confirm OOS and/or Identify aCause for OOS
OOS result given full consideration in batchdisposition decision
FDA OOS Guidance, 10/11/06; V-A
試験室OOS調査と再試験
調査結果の解釈
調査がOOSの確認および/またはOOSの原因を特定できない
バッチの最終処分の決定において、OOS結果が全て考慮される
FDA OOS Guidance, 10/11/06; V-A

Page 7-57©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory OOSInvestigation & Retesting
Interpretation of Investigation Results
QA Decision to Release Batch In Spite of an OOS ResultThat Has Not Been Invalidated
FDA Example
Specification: 90.0 – 110.0%
Initial OOS: 89.5%
Retest results: 99.0, 98.9, 99.0, 99.1, 98.8, 99.1,and 99.0 %
FDA OOS Guidance, 10/11/06; V-A
試験室OOS調査と再試験
調査結果の解釈
無効にされていないOOS結果にもかかわらず、バッチを出荷判定するQAの意思決定
FDA の事例
規格:90.0 – 110.0%
初回の OOS: 89.5%
再試験結果:99.0, 98.9, 99.0, 99.1, 98.8, 99.1,および 99.0 %
FDA OOS Guidance, 10/11/06; V-A

Page 7-58©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory OOSInvestigation & Retesting
Interpretation of Investigation Results
QA Should Evaluate Following to Conclude That theOOS Result Does Not Reflect Actual Batch Quality
1. The retest results must not support the initial OOStest result
FDA OOS Guidance, 10/11/06; IV-C-1-b
試験室OOS調査と再試験
調査結果の解釈
OOS結果は実際のバッチの品質を反映していないことを結論づけるために、QAは以下のことを評価すべきである
1. 再試験結果は、初回のOOS試験結果をサポートしてはならない
FDA OOS Guidance, 10/11/06; IV-C-1-b

Page 7-59©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory OOSInvestigation & Retesting
Interpretation of Investigation Results
QA Should Evaluate Following to Conclude That theOOS Result Does Not Reflect Actual Batch Quality
2. Satisfactory retest results should be within thevariability of the test method
3. Related test results (e.g., content uniformity,dissolution, in-process assay) are consistent withpassing retest results
FDA OOS Guidance, 10/11/06; V-A
試験室OOS調査と再試験
調査結果の解釈
OOS結果は実際のバッチの品質を反映していないことを結論づけるために、QAは以下のことを評価すべきである
2. 満足な再試験結果が試験法のばらつきの範囲内に収まっている
3. 関連する試験結果(たとえば、含量均一性、溶出試験、工程内分析)が、合格した再試験の結果と一貫性がある
FDA OOS Guidance, 10/11/06; V-A

Page 7-60©1994, 1997,1999, 2000, 2002, 2004, 2006PharmaNet, Inc. (S-6) 7/31/2014
GMP Compliance for Quality Control Laboratory OperationsLaboratory Out-of-Specification Investigation and Retesting
©2014 西山経営研究所
Laboratory OOSInvestigation & Retesting
Interpretation of Investigation Results
QA Should Evaluate Following to Conclude That TheOOS Result Does Not Reflect Actual Batch Quality
4. Investigation of Production department revealed norelated process deviations
5. Process and product history indicates a robustprocess
FDA OOS Guidance, 10/11/06; V-A
試験室OOS調査と再試験
調査結果の解釈
OOS結果は実際のバッチの品質を反映していないことを結論づけるために、QAは以下のことを評価すべきである
4. 工程の逸脱に関係しないことを明らかにした製造部門の調査
5. 工程および製品の履歴が頑健な工程を示す
FDA OOS Guidance, 10/11/06; V-A