flecainide prevents 2+ catecholaminergic polymorphic ... · ventricular tachycardia in mice and...
TRANSCRIPT

Flecainide preventscatecholaminergic polymorphicventricular tachycardia in miceand humansHiroshi Watanabe1,5,6, Nagesh Chopra1,6, Derek Laver2,6,Hyun Seok Hwang1, Sean S Davies1, Daniel E Roach3,Henry J Duff3, Dan M Roden1, Arthur A M Wilde4
& Bjorn C Knollmann1
Catecholaminergic polymorphic ventricular tachycardia (CPVT)
is a potentially lethal inherited arrhythmia syndrome in which
drug therapy is often ineffective. We discovered that flecainide
prevents arrhythmias in a mouse model of CPVT by inhibiting
cardiac ryanodine receptor–mediated Ca2+ release and thereby
directly targeting the underlying molecular defect. Flecainide
completely prevented CPVT in two human subjects who had
remained highly symptomatic on conventional drug therapy,
indicating that this currently available drug is a promising
mechanism-based therapy for CPVT.
CPVT is an inherited arrhythmia syndrome characterized by anormal baseline electrocardiogram (ECG), polymorphic ventriculartachycardia induced by adrenergic stress in the absence of structuralheart disease, and a high mortality rate in young individuals1.Treatment with b-adrenergic blockers reduces arrhythmia burdenand mortality but is not completely effective1, and implantablecardioverter defibrillators (ICDs) are used for the prevention ofsudden death. However, painful appropriate or inappropriatedefibrillation shocks can trigger further adrenergic stress andarrhythmias, and deaths have occurred despite appropriate ICDshocks2,3. In such instances, stellate ganglionectomy4 or evencardiac transplantation5 have been considered. Two CPVT dis-ease-related genes have been identified: RYR2, encoding the cardiacryanodine receptor Ca2+ release channel (RyR2), and CASQ2,encoding cardiac calsequestrin6,7. Mutations in these genes desta-bilize the RyR2 Ca2+ release complex8,9. In mice with CPVT-linkedmutations, catecholamines cause spontaneous sarcoplasmic reticu-lum Ca2+ release resulting in delayed after depolarizations (DADs),and they produce triggered activity in myocytes and polymorphicventricular tachycardia in vivo10,11. Here we identify a therapy thatdirectly targets the underlying arrhythmia mechanisms: we foundthat flecainide, an approved antiarrhythmic drug known to blocksodium channels, showed remarkable efficacy in suppressing
spontaneous sarcoplasmic reticulum Ca2+ release by inhibitingRyR2. Flecainide treatment completely prevented adrenergicstress–induced arrhythmias in a mouse model of CPVT and inhumans with CASQ2 or RYR2 mutations that are refractory tostandard drug treatment.
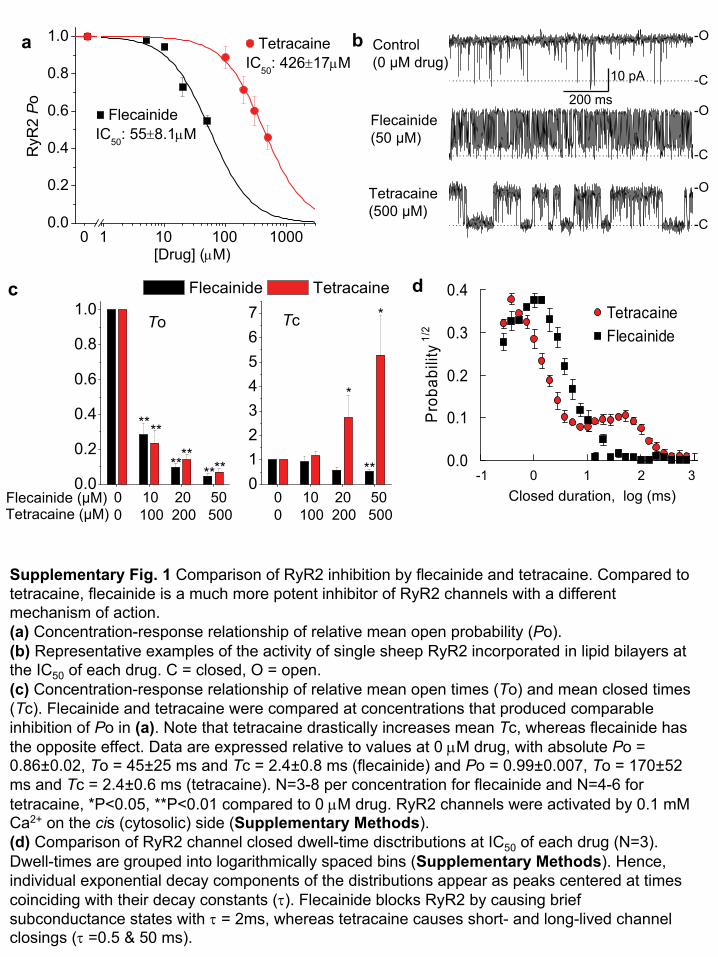
The local anesthetic tetracaine has been used to inhibit RyR2 andsuppress spontaneous sarcoplasmic reticulum Ca2+ release in iso-lated myocytes12. However, tetracaine causes a rebound increase insarcoplasmic reticulum Ca2+ release events during prolongedexposure13, effective inhibitory concentrations14 are too high forclinical use, and systemic administration is contraindicated inhumans. We searched among clinically available antiarrhythmicdrugs for a more useful RyR2 inhibitor and found that flecainideinhibited RyR2 more potently than tetracaine and by a differentmechanism. Whereas tetracaine caused long-lived channel closings,flecainide reduced the duration of channel openings and did notaffect closed channel duration (Supplementary Fig. 1 online).Flecainide’s inhibitory potency was higher when RyR2 was acti-vated by high luminal Ca2+ concentration mimicking spontaneoussarcoplasmic reticulum Ca2+ releases that trigger premature heartbeats (Fig. 1a,b; half-maximal inhibitory concentration ¼ 15 ±3 mM), compared to when RyR2 was activated by high cytosolicCa2+ concentration, such as would occur during a normal heartbeat (Supplementary Fig. 1a; half-maximal inhibitory concen-tration ¼ 55 ± 8 mM).
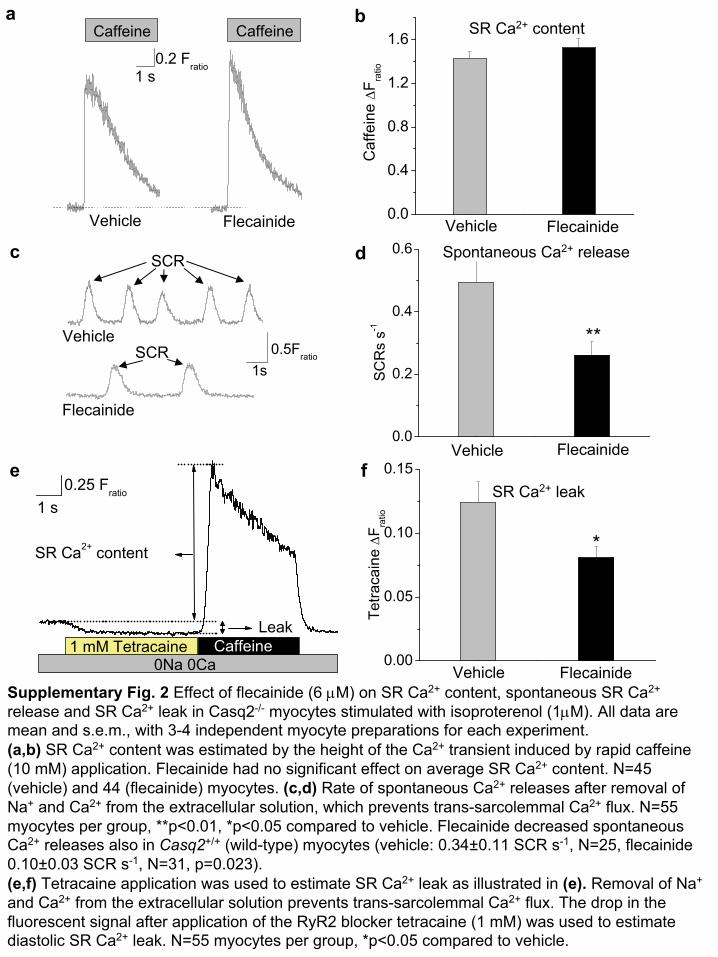
We next tested whether RyR2 block by flecainide translates into aninhibition of spontaneous sarcoplasmic reticulum Ca2+ release inventricular myocytes isolated from mice with gene-targeted deletionof Casq2 (Casq2–/– mice), a model of CASQ2-linked CPVT10. Uponcatecholaminergic challenge with isoproterenol, Casq2–/– myocytesexhibit frequent spontaneous sarcoplasmic reticulum Ca2+ releasesthat can trigger premature beats (Fig. 1c and ref. 10). Flecainidesignificantly suppressed the rate of spontaneous Ca2+ releases from thesarcoplasmic reticulum by 39% (Fig. 1d), even though Ca2+ content insarcoplasmic reticulum was not significantly changed (SupplementaryFig. 2a,b online). The reduction of spontaneous Ca2+ releases byflecainide remained significant even after Na+ and Ca2+ were removedfrom the extracellular bath solution (47% reduction, P ¼ 0.009,Supplementary Fig. 2c,d), indicating that inhibition of trans-sarco-lemmal Na+ or Ca2+ fluxes did not contribute to the reducedspontaneous Ca2+ releases; that is, indicating that flecainide doesnot act by blocking Na+ channels. Compared to vehicle, flecainidesignificantly decreased diastolic sarcoplasmic reticulum Ca2+ leak inisoproterenol-stimulated Casq2–/– myocytes (Supplementary Fig. 2e,f;P ¼ 0.02). Indeed, in contrast to tetracaine treatment13, flecainidetreatment did not result in a compensatory increase in sarcoplasmic
©20
09 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Received 22 October 2008; accepted 10 February 2009; published online 29 March 2009; doi:10.1038/nm.1942
1Departments of Medicine and Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee, USA. 2School of Biomedical Sciences, University ofNewcastle and Hunter Medical Research Institute, Callaghan, Australia. 3Libin Cardiovascular Institute, University of Calgary, Calgary, Alberta, Canada. 4Department ofCardiology, Academic Medical Center, University of Amsterdam, Amsterdam, The Netherlands. 5Current address: Division of Cardiology, Niigata University School ofMedical and Dental Sciences, Niigata, Japan. 6These authors contributed equally to this work. Correspondence should be addressed to B.C.K.([email protected]).
NATURE MEDICINE ADVANCE ONLINE PUBLICATION 1
BR I E F COMMUNICAT IONS
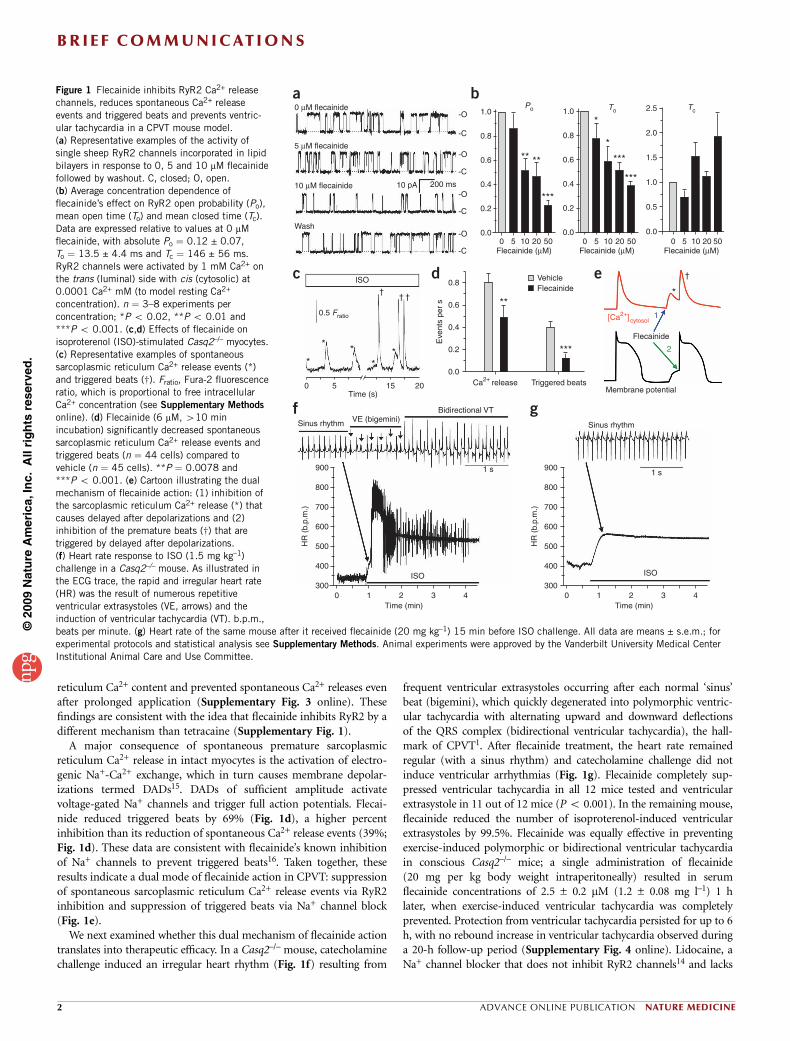
reticulum Ca2+ content and prevented spontaneous Ca2+ releases evenafter prolonged application (Supplementary Fig. 3 online). Thesefindings are consistent with the idea that flecainide inhibits RyR2 by adifferent mechanism than tetracaine (Supplementary Fig. 1).
A major consequence of spontaneous premature sarcoplasmicreticulum Ca2+ release in intact myocytes is the activation of electro-genic Na+-Ca2+ exchange, which in turn causes membrane depolar-izations termed DADs15. DADs of sufficient amplitude activatevoltage-gated Na+ channels and trigger full action potentials. Flecai-nide reduced triggered beats by 69% (Fig. 1d), a higher percentinhibition than its reduction of spontaneous Ca2+ release events (39%;Fig. 1d). These data are consistent with flecainide’s known inhibitionof Na+ channels to prevent triggered beats16. Taken together, theseresults indicate a dual mode of flecainide action in CPVT: suppressionof spontaneous sarcoplasmic reticulum Ca2+ release events via RyR2inhibition and suppression of triggered beats via Na+ channel block(Fig. 1e).
We next examined whether this dual mechanism of flecainide actiontranslates into therapeutic efficacy. In a Casq2–/– mouse, catecholaminechallenge induced an irregular heart rhythm (Fig. 1f) resulting from
frequent ventricular extrasystoles occurring after each normal ‘sinus’beat (bigemini), which quickly degenerated into polymorphic ventric-ular tachycardia with alternating upward and downward deflectionsof the QRS complex (bidirectional ventricular tachycardia), the hall-mark of CPVT1. After flecainide treatment, the heart rate remainedregular (with a sinus rhythm) and catecholamine challenge did notinduce ventricular arrhythmias (Fig. 1g). Flecainide completely sup-pressed ventricular tachycardia in all 12 mice tested and ventricularextrasystole in 11 out of 12 mice (Po 0.001). In the remaining mouse,flecainide reduced the number of isoproterenol-induced ventricularextrasystoles by 99.5%. Flecainide was equally effective in preventingexercise-induced polymorphic or bidirectional ventricular tachycardiain conscious Casq2–/– mice; a single administration of flecainide(20 mg per kg body weight intraperitoneally) resulted in serumflecainide concentrations of 2.5 ± 0.2 mM (1.2 ± 0.08 mg l–1) 1 hlater, when exercise-induced ventricular tachycardia was completelyprevented. Protection from ventricular tachycardia persisted for up to 6h, with no rebound increase in ventricular tachycardia observed duringa 20-h follow-up period (Supplementary Fig. 4 online). Lidocaine, aNa+ channel blocker that does not inhibit RyR2 channels14 and lacks
©20
09 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Figure 1 Flecainide inhibits RyR2 Ca2+ release
channels, reduces spontaneous Ca2+ release
events and triggered beats and prevents ventric-
ular tachycardia in a CPVT mouse model.
(a) Representative examples of the activity of
single sheep RyR2 channels incorporated in lipid
bilayers in response to 0, 5 and 10 mM flecainide
followed by washout. C, closed; O, open.
(b) Average concentration dependence of
flecainide’s effect on RyR2 open probability (Po),
mean open time (To) and mean closed time (Tc).
Data are expressed relative to values at 0 mM
flecainide, with absolute Po ¼ 0.12 ± 0.07,
To ¼ 13.5 ± 4.4 ms and Tc ¼ 146 ± 56 ms.
RyR2 channels were activated by 1 mM Ca2+ onthe trans (luminal) side with cis (cytosolic) at
0.0001 Ca2+ mM (to model resting Ca2+
concentration). n ¼ 3–8 experiments per
concentration; *P o 0.02, **P o 0.01 and
***P o 0.001. (c,d) Effects of flecainide on
isoproterenol (ISO)-stimulated Casq2–/– myocytes.
(c) Representative examples of spontaneous
sarcoplasmic reticulum Ca2+ release events (*)
and triggered beats (w). Fratio, Fura-2 fluorescence
ratio, which is proportional to free intracellular
Ca2+ concentration (see Supplementary Methods
online). (d) Flecainide (6 mM, 410 min
incubation) significantly decreased spontaneous
sarcoplasmic reticulum Ca2+ release events and
triggered beats (n ¼ 44 cells) compared to
vehicle (n ¼ 45 cells). **P ¼ 0.0078 and
***P o 0.001. (e) Cartoon illustrating the dual
mechanism of flecainide action: (1) inhibition ofthe sarcoplasmic reticulum Ca2+ release (*) that
causes delayed after depolarizations and (2)
inhibition of the premature beats (w) that are
triggered by delayed after depolarizations.
(f) Heart rate response to ISO (1.5 mg kg–1)
challenge in a Casq2–/– mouse. As illustrated in
the ECG trace, the rapid and irregular heart rate
(HR) was the result of numerous repetitive
ventricular extrasystoles (VE, arrows) and the
induction of ventricular tachycardia (VT). b.p.m.,
beats per minute. (g) Heart rate of the same mouse after it received flecainide (20 mg kg–1) 15 min before ISO challenge. All data are means ± s.e.m.; for
experimental protocols and statistical analysis see Supplementary Methods. Animal experiments were approved by the Vanderbilt University Medical Center
Institutional Animal Care and Use Committee.
0 µM flecainide
5 µM flecainide
10 pA 200 ms
-O
-C
1.0
0.8
0.6
0 5Flecainide (µM)
Vehicle
Flecainide
1
2
Flecainide
Flecainide (µM) Flecainide (µM)10 20 50 0 5 10 20 50 0 5 10 20 50
0.4
0.2
0.0
1.0Po To Tc
0.8
0.6
0.4
0.2
0.0
2.5
2.0
1.5
1.0
0.5
0.0
-O
-C
-O
-C
-O
-C
10 µM flecainide
Wash
ISO
†
†
† †
0.5 Fratio
**
**
***
**
***
*
*
***
***
0.8
Eve
nts
per
s 0.6
0.4
0.2
0.0
0 5Time (s)
Ca2+ release
[Ca2+] cytosol
Triggered beatsMembrane potential
Sinus rhythm
1 s1 s
Bidirectional VT
Sinus rhythm
900
800
700
HR
(b.
p.m
.)
600
500
400
300
900
800
700
HR
(b.
p.m
.)600
500
400
3000 1 2
ISO
Time (min)3 4
VE (bigemini)
15 20
*
* **
*
*
0 1 2
ISO
Time (min)3 4
a
c
f g
d e
b
BR I E F COMMUNICAT IONS
2 ADVANCE ONLINE PUBLICATION NATURE MEDICINE
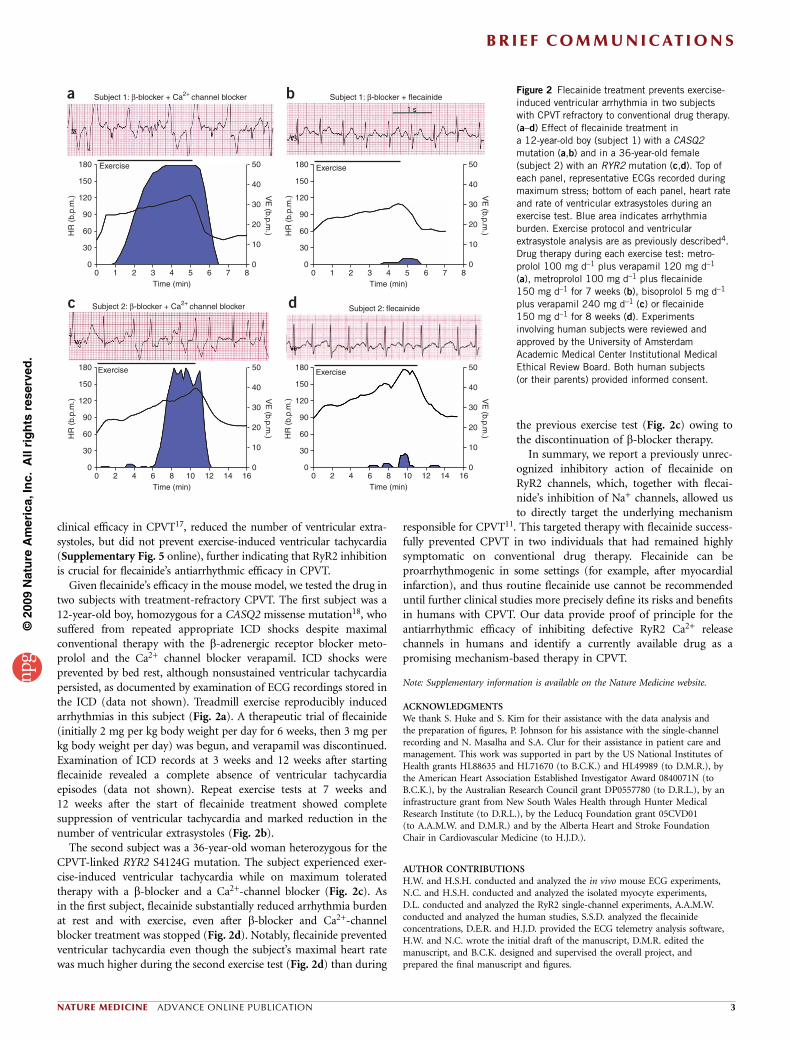
clinical efficacy in CPVT17, reduced the number of ventricular extra-systoles, but did not prevent exercise-induced ventricular tachycardia(Supplementary Fig. 5 online), further indicating that RyR2 inhibitionis crucial for flecainide’s antiarrhythmic efficacy in CPVT.
Given flecainide’s efficacy in the mouse model, we tested the drug intwo subjects with treatment-refractory CPVT. The first subject was a12-year-old boy, homozygous for a CASQ2 missense mutation18, whosuffered from repeated appropriate ICD shocks despite maximalconventional therapy with the b-adrenergic receptor blocker meto-prolol and the Ca2+ channel blocker verapamil. ICD shocks wereprevented by bed rest, although nonsustained ventricular tachycardiapersisted, as documented by examination of ECG recordings stored inthe ICD (data not shown). Treadmill exercise reproducibly inducedarrhythmias in this subject (Fig. 2a). A therapeutic trial of flecainide(initially 2 mg per kg body weight per day for 6 weeks, then 3 mg perkg body weight per day) was begun, and verapamil was discontinued.Examination of ICD records at 3 weeks and 12 weeks after startingflecainide revealed a complete absence of ventricular tachycardiaepisodes (data not shown). Repeat exercise tests at 7 weeks and12 weeks after the start of flecainide treatment showed completesuppression of ventricular tachycardia and marked reduction in thenumber of ventricular extrasystoles (Fig. 2b).
The second subject was a 36-year-old woman heterozygous for theCPVT-linked RYR2 S4124G mutation. The subject experienced exer-cise-induced ventricular tachycardia while on maximum toleratedtherapy with a b-blocker and a Ca2+-channel blocker (Fig. 2c). Asin the first subject, flecainide substantially reduced arrhythmia burdenat rest and with exercise, even after b-blocker and Ca2+-channelblocker treatment was stopped (Fig. 2d). Notably, flecainide preventedventricular tachycardia even though the subject’s maximal heart ratewas much higher during the second exercise test (Fig. 2d) than during
the previous exercise test (Fig. 2c) owing tothe discontinuation of b-blocker therapy.
In summary, we report a previously unrec-ognized inhibitory action of flecainide onRyR2 channels, which, together with flecai-nide’s inhibition of Na+ channels, allowed usto directly target the underlying mechanism
responsible for CPVT11. This targeted therapy with flecainide success-fully prevented CPVT in two individuals that had remained highlysymptomatic on conventional drug therapy. Flecainide can beproarrhythmogenic in some settings (for example, after myocardialinfarction), and thus routine flecainide use cannot be recommendeduntil further clinical studies more precisely define its risks and benefitsin humans with CPVT. Our data provide proof of principle for theantiarrhythmic efficacy of inhibiting defective RyR2 Ca2+ releasechannels in humans and identify a currently available drug as apromising mechanism-based therapy in CPVT.
Note: Supplementary information is available on the Nature Medicine website.
ACKNOWLEDGMENTSWe thank S. Huke and S. Kim for their assistance with the data analysis andthe preparation of figures, P. Johnson for his assistance with the single-channelrecording and N. Masalha and S.A. Clur for their assistance in patient care andmanagement. This work was supported in part by the US National Institutes ofHealth grants HL88635 and HL71670 (to B.C.K.) and HL49989 (to D.M.R.), bythe American Heart Association Established Investigator Award 0840071N (toB.C.K.), by the Australian Research Council grant DP0557780 (to D.R.L.), by aninfrastructure grant from New South Wales Health through Hunter MedicalResearch Institute (to D.R.L.), by the Leducq Foundation grant 05CVD01(to A.A.M.W. and D.M.R.) and by the Alberta Heart and Stroke FoundationChair in Cardiovascular Medicine (to H.J.D.).
AUTHOR CONTRIBUTIONSH.W. and H.S.H. conducted and analyzed the in vivo mouse ECG experiments,N.C. and H.S.H. conducted and analyzed the isolated myocyte experiments,D.L. conducted and analyzed the RyR2 single-channel experiments, A.A.M.W.conducted and analyzed the human studies, S.S.D. analyzed the flecainideconcentrations, D.E.R. and H.J.D. provided the ECG telemetry analysis software,H.W. and N.C. wrote the initial draft of the manuscript, D.M.R. edited themanuscript, and B.C.K. designed and supervised the overall project, andprepared the final manuscript and figures.
©20
09 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
Subject 1: β-blocker + Ca2+ channel blocker
Exercise180
a b
c d
150
120
HR
(b.
p.m
.) VE
(b.p.m.)
90
60
30
0
50
40
30
20
10
00 1 2 3
Time (min)4 5 6 7 8
Exercise180
150
120
HR
(b.
p.m
.) VE
(b.p.m.)
90
60
30
0
50
40
30
20
10
00 1 2 3
Time (min)4 5 6 7 8
Exercise180
150
120
HR
(b.
p.m
.) VE
(b.p.m.)
90
60
30
0
50
40
30
20
10
00 2 4 6
Time (min)8 10 12 14 16
Exercise180
150
120
HR
(b.
p.m
.) VE
(b.p.m.)
90
60
30
0
50
40
30
20
10
00 2 4 6
Time (min)8 10 12 14 16
Subject 1: β-blocker + flecainide
Subject 2: β-blocker + Ca2+ channel blocker Subject 2: flecainide
Figure 2 Flecainide treatment prevents exercise-
induced ventricular arrhythmia in two subjects
with CPVT refractory to conventional drug therapy.
(a–d) Effect of flecainide treatment in
a 12-year-old boy (subject 1) with a CASQ2
mutation (a,b) and in a 36-year-old female
(subject 2) with an RYR2 mutation (c,d). Top of
each panel, representative ECGs recorded during
maximum stress; bottom of each panel, heart rate
and rate of ventricular extrasystoles during an
exercise test. Blue area indicates arrhythmia
burden. Exercise protocol and ventricular
extrasystole analysis are as previously described4.
Drug therapy during each exercise test: metro-
prolol 100 mg d–1 plus verapamil 120 mg d–1
(a), metroprolol 100 mg d–1 plus flecainide
150 mg d–1 for 7 weeks (b), bisoprolol 5 mg d–1
plus verapamil 240 mg d–1 (c) or flecainide
150 mg d–1 for 8 weeks (d). Experiments
involving human subjects were reviewed and
approved by the University of Amsterdam
Academic Medical Center Institutional Medical
Ethical Review Board. Both human subjects
(or their parents) provided informed consent.
BR I E F COMMUNICAT IONS
NATURE MEDICINE ADVANCE ONLINE PUBLICATION 3

Published online at http://www.nature.com/naturemedicine/
Reprints and permissions information is available online at http://npg.nature.com/
reprintsandpermissions/
1. Leenhardt, A. et al. Circulation 91, 1512–1519 (1995).2. Mohamed, U., Gollob, M.H., Gow, R.M. & Krahn, A.D. Heart Rhythm 3, 1486–1489
(2006).3. Pizzale, S., Gollob, M.H., Gow, R. & Birnie, D.H. J. Cardiovasc. Electrophysiol. 9,
1319–1321 (2008).4. Wilde, A.A. et al. N. Engl. J. Med. 358, 2024–2029 (2008).5. Robinson, M., Park, J. & Raff, G. Heart Rhythm 4 Suppl, S100 (2007).6. Priori, S.G. et al. Circulation 103, 196–200 (2001).
7. Lahat, H. et al. Am. J. Hum. Genet. 69, 1378–1384 (2001).8. Jiang, D. et al. Proc. Natl. Acad. Sci. USA 101, 13062–13067 (2004).9. di Barletta, M.R. et al. Circulation 114, 1012–1019 (2006).10. Knollmann, B.C. et al. J. Clin. Invest. 116, 2510–2520 (2006).11. Cerrone, M. et al. Circ. Res. 101, 1039–1048 (2007).12. Venetucci, L.A., Trafford, A.W., Diaz, M.E., O’Neill, S.C. & Eisner, D.A. Circ. Res. 98,
1299–1305 (2006).13. Gyorke, S., Lukyanenko, V. & Gyorke, I. J. Physiol. (Lond.) 500, 297–309 (1997).14. Shoshan-Barmatz, V. & Zchut, S. J. Membr. Biol. 133, 171–181 (1993).15. Marban, E., Robinson, S.W. & Wier, W.G. J. Clin. Invest. 78, 1185–1192 (1986).16. Rosen, M.R. & Danilo, P. Jr. Circ. Res. 46, 117–124 (1980).17. De Rosa, G. et al. Pediatr. Emerg. Care 20, 175–177 (2004).18. Postma, A.V. et al. Circ. Res. 91, e21–e26 (2002).
©20
09 N
atu
re A
mer
ica,
Inc.
All
rig
hts
res
erve
d.
BR I E F COMMUNICAT IONS
4 ADVANCE ONLINE PUBLICATION NATURE MEDICINE
0.0
0.1
0.2
0.3
0.4
Pro
babi
lity
1/2
TetracaineFlecainide
0 1 2 3Closed duration, log (ms)
-1
0.0
0.2
0.4
0.6
0.8
1.0 TetracaineIC50: 426±17μM
1000100101
RyR
2 P
o
[Drug] (μM)0
FlecainideIC50: 55±8.1μM
a
0 10 20 500 100 200 500
Flecainide (μM)Tetracaine (μM)
0 10 20 500 100 200 500
c
01234567
0.0
0.2
0.4
0.6
0.8
1.0 Flecainide Tetracaine
To Tc
****
**** **** **
*
*
Flecainide(50 μM)
Tetracaine(500 μM)
Control(0 μM drug)
200 ms
10 pA
b
-C
-O
-C
-O
-C
-O
d
Supplementary Fig. 1 Comparison of RyR2 inhibition by flecainide
and tetracaine. Compared to tetracaine, flecainide
is a much more potent inhibitor of RyR2 channels with a different mechanism of action. (a)
Concentration-response relationship of relative mean open probability (Po).(b)
Representative examples of the activity of single sheep RyR2 incorporated in lipid bilayers
at the IC50
of each drug. C = closed, O = open.(c) Concentration-response relationship of relative
mean open times (To) and mean closed times (Tc). Flecainide
and tetracaine
were compared at concentrations that produced comparable inhibition of Po in (a). Note that tetracaine
drastically increases mean Tc, whereas flecainide
has the opposite effect. Data are expressed relative to values at 0 μM
drug, with absolute Po = 0.86±0.02, To = 45±25 ms and Tc = 2.4±0.8 ms (flecainide) and Po = 0.99±0.007, To = 170±52 ms and Tc = 2.4±0.6 ms (tetracaine). N=3-8 per concentration for flecainide
and
N=4-6 for tetracaine, *P<0.05, **P<0.01 compared to 0 μM
drug. RyR2 channels were activated by 0.1 mM
Ca2+
on the cis
(cytosolic) side (Supplementary Methods). (d)
Comparison of RyR2 channel closed dwell-time disctributions
at IC50
of each drug (N=3). Dwell-times are grouped into logarithmically spaced bins (Supplementary Methods). Hence, individual exponential decay components of the distributions appear as peaks centered at times coinciding with their decay constants (τ). Flecainide
blocks RyR2 by causing brief subconductance
states with τ
= 2ms, whereas tetracaine
causes short-
and long-lived channel closings (τ
=0.5 & 50 ms).
0.00
0.05
0.10
0.15
*
Tetra
cain
e ΔF
ratio
Supplementary Fig. 2 Effect of flecainide
(6 μM) on SR Ca2+
content, spontaneous SR Ca2+
release and SR Ca2+
leak in Casq2-/-
myocytes stimulated with isoproterenol (1μM). All data are mean and s.e.m., with 3-4 independent myocyte
preparations for each experiment.(a,b)
SR Ca2+
content was estimated by the height of the Ca2+
transient induced by rapid caffeine (10 mM) application. Flecainide had no significant effect on average SR Ca2+
content. N=45 (vehicle) and 44 (flecainide) myocytes. (c,d)
Rate of spontaneous Ca2+
releases after removal of Na+
and Ca2+
from the extracellular solution, which prevents trans-sarcolemmal
Ca2+
flux. N=55 myocytes per group, **p<0.01, *p<0.05 compared to vehicle. Flecainide decreased spontaneous Ca2+
releases also in Casq2+/+
(wild-type) myocytes (vehicle: 0.34±0.11 SCR s-1, N=25, flecainide 0.10±0.03 SCR s-1, N=31, p=0.023).(e,f)
Tetracaine application was used to estimate SR Ca2+
leak as illustrated in (e).
Removal of Na+
and Ca2+
from the extracellular solution prevents trans-sarcolemmal
Ca2+
flux. The drop in the fluorescent signal after application of the RyR2 blocker tetracaine (1 mM) was used to estimate diastolic SR Ca2+
leak. N=55 myocytes per group, *p<0.05 compared to vehicle.
1 s0.2 Fratio
Caffeine Caffeine
0.0
0.4
0.8
1.2
1.6
Caf
fein
e ΔF
ratio
0.0
0.2
0.4
0.6
**
SC
Rs
s-11s
0.5Fratio
Vehicle
Vehicle Flecainide
Vehicle
Vehicle
Vehicle
SR Ca2+
content
Flecainide
Flecainide
Flecainide
Flecainide
SR Ca2+
leak
Spontaneous Ca2+
release
a
c
b
d
e f
SCR
SCR
1 s0.25 Fratio
Leak
SR Ca2+ content
1 mM Tetracaine Caffeine0Na 0Ca
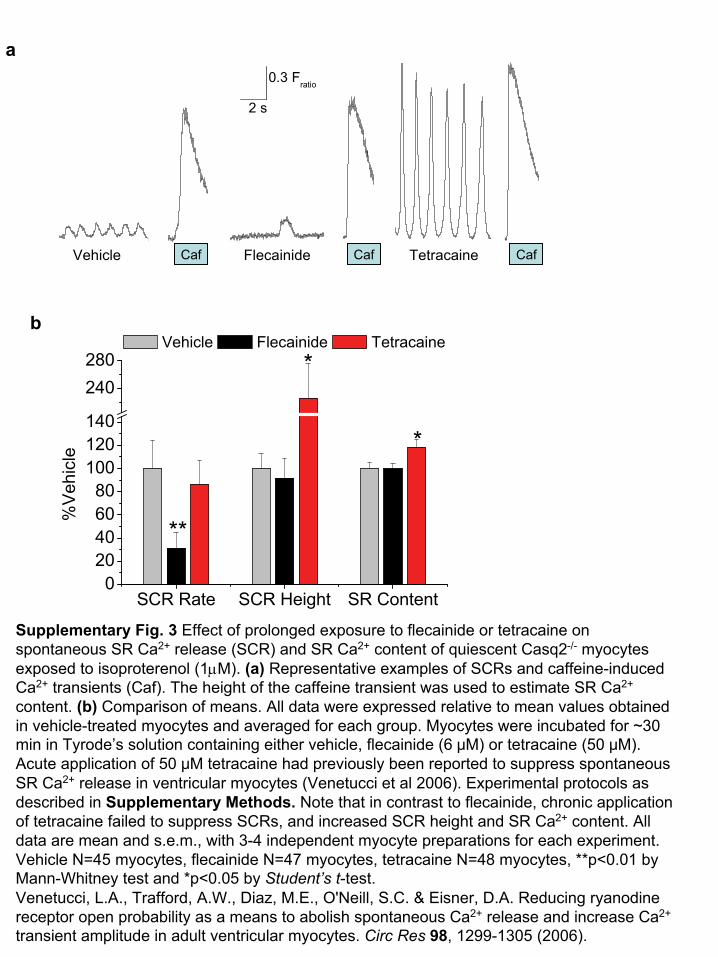
0.3 Fratio
2 s
SCR Rate SCR Height SR Content0
20406080
100120140
240280
%V
ehic
le
Vehicle Flecainide Tetracaine
**
*
*
Supplementary Fig. 3 Effect of prolonged exposure to flecainide or tetracaine on spontaneous SR Ca2+
release (SCR) and SR Ca2+
content of quiescent Casq2-/-
myocytes exposed to isoproterenol (1μM). (a)
Representative examples of SCRs
and caffeine-induced Ca2+
transients (Caf). The height of the caffeine transient was used to estimate SR Ca2+
content. (b)
Comparison of means. All data were expressed relative to mean values obtained in vehicle-treated myocytes and averaged for each group. Myocytes were incubated for ~30 min in Tyrode’s solution containing either vehicle, flecainide (6 μM) or tetracaine (50 μM). Acute application of 50 μM tetracaine had previously been reported to suppress spontaneous SR Ca2+
release in ventricular myocytes (Venetucci
et al 2006). Experimental protocols as described in Supplementary Methods. Note that in contrast to flecainide, chronic application of tetracaine failed to suppress SCRs, and increased SCR height and SR Ca2+
content. All data are mean and s.e.m., with 3-4 independent myocyte
preparations for each experiment.
Vehicle N=45 myocytes, flecainide N=47 myocytes, tetracaine N=48 myocytes, **p<0.01 by Mann-Whitney test and *p<0.05 by Student’s
t-test.Venetucci, L.A., Trafford, A.W., Diaz, M.E., O'Neill, S.C. & Eisner, D.A. Reducing ryanodine receptor open probability as a means to abolish spontaneous Ca2+
release and increase Ca2+
transient amplitude in adult ventricular myocytes. Circ Res
98, 1299-1305 (2006).
a
b
Caf Caf CafFlecainide TetracaineVehicle
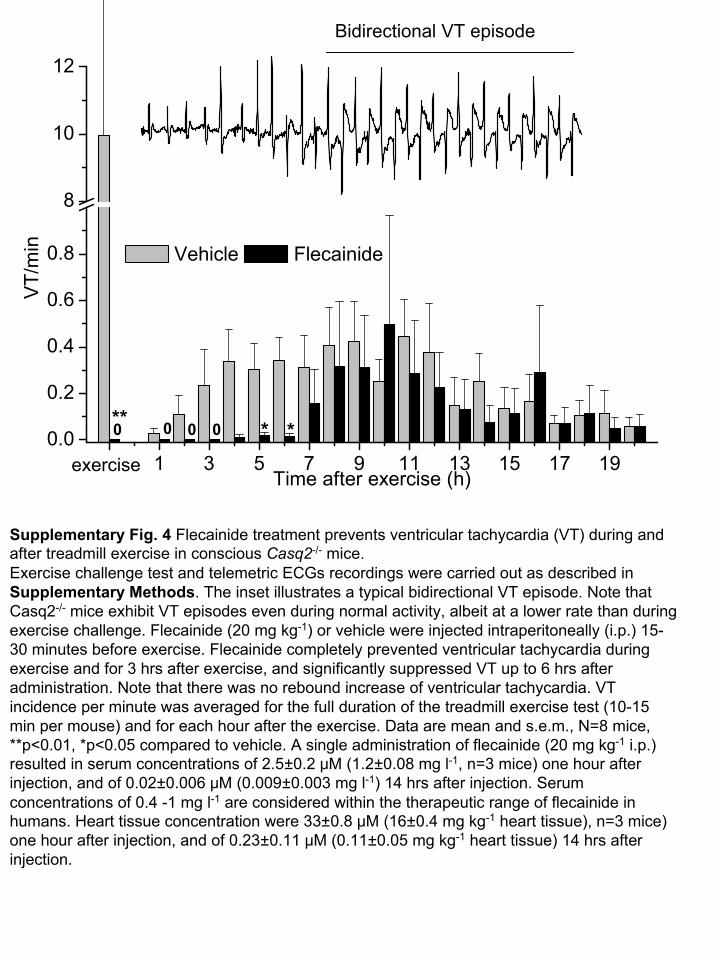
-1 1 3 5 7 9 11 13 15 17 190.0
0.2
0.4
0.6
0.8
8
10
12
VT/
min
Time after exercise (h)
Vehicle Flecainide
Supplementary Fig. 4 Flecainide treatment prevents ventricular tachycardia (VT) during and after treadmill exercise in conscious Casq2-/-
mice. Exercise challenge test and telemetric ECGs recordings were carried out as described in Supplementary Methods. The inset illustrates a typical bidirectional VT episode. Note
that Casq2-/-
mice exhibit VT episodes even during normal activity, albeit at
a lower rate than during exercise challenge. Flecainide
(20 mg kg-1) or vehicle were injected intraperitoneally
(i.p.) 15-
30 minutes before exercise. Flecainide
completely prevented ventricular tachycardia during exercise and for 3 hrs after exercise, and significantly suppressed VT up to 6 hrs after administration. Note that there was no rebound increase of ventricular tachycardia. VT incidence per minute was averaged for the full duration of the treadmill exercise test (10-15 min per mouse) and for each hour after the exercise. Data are mean and s.e.m., N=8 mice, **p<0.01, *p<0.05 compared to vehicle. A single administration of flecainide
(20 mg kg-1
i.p.) resulted in serum concentrations of 2.5±0.2 μM (1.2±0.08 mg l-1, n=3 mice) one hour after injection, and of 0.02±0.006 μM
(0.009±0.003 mg l-1) 14 hrs after injection. Serum concentrations of 0.4 -1 mg l-1
are considered within the therapeutic range of flecainide
in humans. Heart tissue concentration were 33±0.8 μM (16±0.4 mg kg-1
heart tissue), n=3 mice) one hour after injection, and of 0.23±0.11 μM (0.11±0.05 mg kg-1
heart tissue) 14 hrs after injection.
Bidirectional VT episode
exercise
0 0 0** * *0
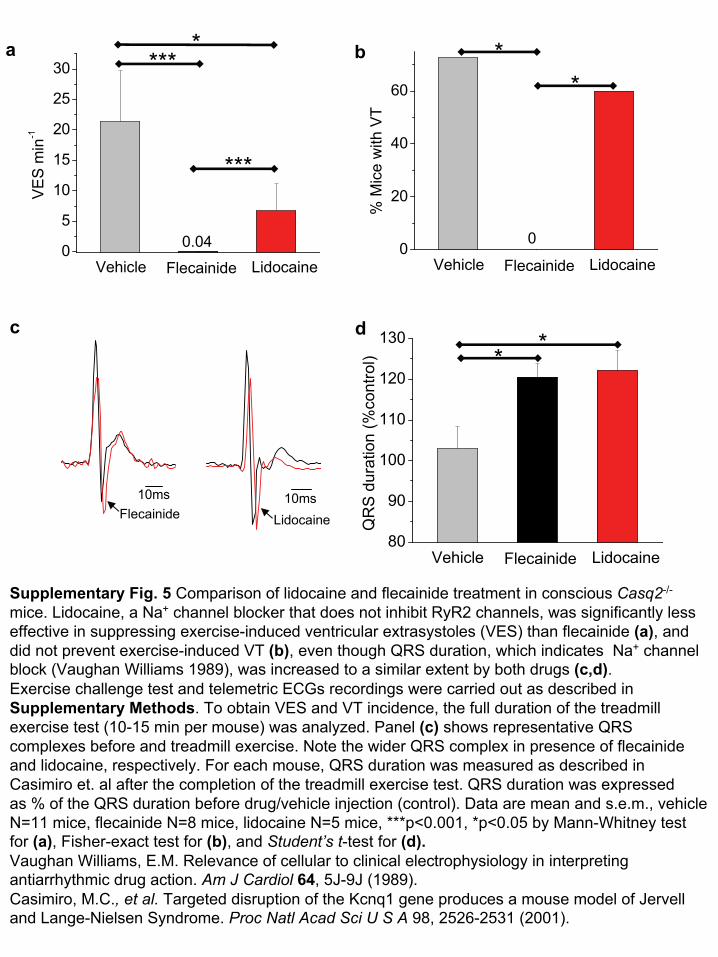
Supplementary Fig. 5 Comparison of lidocaine
and flecainide treatment in conscious Casq2-/-
mice. Lidocaine, a Na+
channel blocker that does not inhibit RyR2 channels, was significantly less effective in suppressing exercise-induced ventricular extrasystoles
(VES) than flecainide (a), and did not prevent exercise-induced VT (b), even though QRS duration, which indicates Na+
channel block (Vaughan Williams 1989), was increased to a similar extent
by both drugs (c,d). Exercise challenge test and telemetric ECGs recordings were carried out as described in Supplementary Methods. To obtain VES and VT incidence, the full duration of the treadmill exercise test (10-15 min per mouse) was analyzed. Panel (c)
shows representative QRS complexes before and treadmill exercise. Note the wider QRS complex in presence of flecainide and lidocaine, respectively. For each mouse, QRS duration was measured as described in Casimiro
et. al after the completion of the treadmill exercise test. QRS
duration was expressed as % of the QRS duration before drug/vehicle injection (control). Data are mean and s.e.m., vehicle N=11 mice, flecainide N=8 mice, lidocaine
N=5 mice, ***p<0.001, *p<0.05 by Mann-Whitney test for (a), Fisher-exact test for (b), and Student’s
t-test for (d).Vaughan Williams, E.M. Relevance of cellular to clinical electrophysiology in interpreting antiarrhythmic
drug action. Am J Cardiol
64, 5J-9J (1989). Casimiro, M.C., et al.
Targeted disruption of the Kcnq1 gene produces a mouse model of
Jervell
and Lange-Nielsen Syndrome. Proc Natl
Acad
Sci
U S A
98, 2526-2531 (2001).
0
5
10
15
20
25
30
0.04LidocaineFlecainide
VE
S m
in-1
Vehicle0
20
40
60
LidocaineFlecainide
% M
ice
with
VT
Vehicle
0
80
90
100
110
120
130
LidocaineFlecainide
QR
S d
urat
ion
(%co
ntro
l)
Vehicle
***
***
* **
* *
a
c
b
d
10ms 10msFlecainide Lidocaine

1
Flecainide Prevents Catecholaminergic Polymorphic Ventricular Tachycardia in Mice and Humans Hiroshi Watanabe, MD, PhD*; Nagesh Chopra, MD*; Derek Laver, PhD*; Hyun Seok Hwang, PhD; Sean S. Davies, PhD; Daniel E. Roach, PhD, Henry J. Duff, MD, Dan M Roden, MD; Arthur A.M. Wilde, MD, PhD; Björn C. Knollmann, MD, PhD Supplementary Methods Human Studies Both CPVT patients described in our report were refractory to standard drug therapy and awaiting surgical stellate ganglionectomy, an investigational procedure currently being evaluated for effectiveness in CPVT. Given this therapeutic dilemma, the treating physician (AAMW) elected a therapeutic trial of flecainide, which is an approved anti-arrhythmic drug. Given the lack of clinical data in CPVT, the standard dose for treating atrial fibrillation was used. The patient or the parents provided informed consent, and the study was reviewed and approved by the institutional medical ethical review board of the Academic Medical Center, University of Amsterdam, Amsterdam, Netherlands. Treadmill exercise testing and ICD interrogations were performed as part of routine clinical care. Arrhythmia burden during exercise testing was quantified as previously described.1 Animal studies All studies were approved by the institutional animal care and use committees at Vanderbilt University, and performed in accordance with NIH guidelines. Adult Casq2-/- mice (3-5 months old) were used for all experiments2. Casq2-/- mice consistently develop VT during exercise or after catecholamine challenge. Single RyR2 Ca2+ release channel measurements SR vesicles containing ryanodine receptor Ca2+ release channels were obtained from sheep hearts and were reconstituted into artificial lipid bilayers as previously described 3. Lipid bilayers were formed from phosphatidylethanolamine and phosphatidylcholine (8:2 wt/wt, Avanti Polar Lipids, Alabaster, AL) in n-decane, (50 mg ml-1, ICN Biomedicals) across an aperture of 150–250 µm diameter in a Delrin cup which separated two baths (cis and trans). During SR-vesicle incorporation, the cis (cytoplasmic) bath contained (in mM) 250 Cs+ (230 CsCH3O3S, 20 CsCl), 1.0 CaCl2 and 500 mannitol, while the trans (luminal) solution contained 50 Cs+ (30 CsCH3O3S, 20 CsCl2) and 1.0 CaCl2. After detection of channels in the bilayer the compositions of the cis and trans solutions were changed as follows. The [CsCH3O3S] in the trans solution was increased to 230 mM (i.e. establishing 250 mM Cs+ in both cis and trans baths) by means of aliquot addition of 4 M stock. The cis solution was exchanged for one containing 250 mM Cs+, 2 mM ATP, 0.1 � M free Ca2+ and the desired flecainide or tetracaine concentration. Rapid solution exchange was performed using a tube (300 � m ID) located within 100 � m of the bilayer. Flow rates of ~1 � l per second produced total solution exchange at the bilayer in <1 s.4 Free [Ca2+] of 0.1 � M was achieved by a combination of 4.5 mM BAPTA and 1 mM CaCl2. Mannitol was obtained from Ajax chemicals and CaCl2 from BDH Chemicals. Solutions were pH-buffered with 10 mM N-tris [Hydroxymethyl]methyl-2-aminoethanesulfonic acid (TES, ICN Biomedicals) and solutions were titrated to pH 7.4 using CsOH (optical grade, ICN Biomedicals) and were redox buffered with 5 mM glutathione (ICN Biomedicals). Flecainide (acetate salt), tetracaine (hydrochloride) and caesium salts were obtained from Sigma Corporation. Bilayer potential was controlled using an Axopatch 200B amplifier (Axon Instruments). Electrical potentials are expressed using standard physiological convention (i.e., cytoplasmic side relative to the luminal side at virtual ground). Single channel recordings were obtained using bilayer potential difference of +40 mV. The current signal was digitized at 5 kHz and low-pass filtered at 1 kHz with a Gaussian digital filter. Open probability (Po) as well as open and closed durations was measured by the 50% threshold detection method. Analysis was carried out using Channel2 software (P.W. Gage and M. Smith, Australian National University, Canberra). Dwell-time distributions were compiled from
Nature Medicine: doi:10.1038/nm.1942

2
single channel records and displayed using log-binned histograms with 7 bins per decade.5 This method transforms individual exponential decay components to peaked distributions which are centered on time values that correspond to their decay constants. Treadmill exercise testing in mice Exercise testing in conscious mice was carried out as previously described.2 Briefly, mice were initially anesthetized (pentobarbital, 70 mg g-1) and an electrocardiogram (ECG) transmitter (Data Sciences International, St. Paul, MN) was implanted into the abdominal cavity with subcutaneous electrodes in lead 2 configuration. Animals were allowed to recover for at least 72 h after surgery before participating in the treadmill exercise studies. Flecainide (20 mg kg-1), lidocaine (20 mg kg-1) or vehicle (70%ethanol) was injected intraperitoneally 15-30 minutes before exercise. Mice were placed individually into a special chamber of the motorized rodent treadmill (Exer-6M, Columbus Instruments, Columbus, OH) and exercised until they exhibited signs of exhaustion, as described previously.2 Exhaustion was defined as the mouse spending more than 50% of the time or more than 15 s consecutively on the shock grid. For each mouse, treadmill testing was performed two times (drug and placebo) at an interval of >72 h. High-quality ECGs were recorded from 15 min before exercise until 20 h after exercise. An analysis program (Dataquest A.R.T. version 2.3, Data Sciences International) was used to review the ECG records during the exercise test and quantify ventricular extrasystoles (VEs) and runs of ventricular tachycardia (>3 consecutive ventricular beats). To analyze arrhythmia incidence during the 20 h recording period, we utilized the multi-purpose MultiJobECG analysis and visualization system ([email protected]), which applies multi-scale, hierarchical, contextual signal processing (CSP) to high volumes of raw ECG data.6 Using the MultiJobECG's episode learning library, we defined different spectral classes of ECG episodes (sinus rhythm, VEs, run of ventricular tachycardia); these episodes were then automatically located and displayed on the user-interface for confirmation by an experienced reviewer. Once confirmed, ventricular arrhythmias were categorized, counted and quantified as incidence rate per minute. ECG analysis was performed blinded to the treatment group. ECG recordings in anesthetized, isoproterenol- treated mice The effects of flecainide on isoproterenol-induced ventricular arrhythmia were studied in anesthetized Casq2-/- mice as previously described.2 Briefly, mice were anesthetized with isoflurane vapor titrated to maintain the lightest anesthesia as possible. On average, 1.0% vol/vol isoflurane vapor was sufficient to maintain adequate anesthesia. Loss of toe pinch reflex and respiration rate were used to monitor levels of anesthesia. Average respiration rate was not different between placebo and flecainide administration. Baseline ECG was recorded for 5 minutes, followed by an additional 20 minutes after administration of isoproterenol (1.5 mg kg-1 i.p.). Flecainide (20 mg kg-1 i.p.) or vehicle (70% ethanol) was administered 15 minutes before administration of isoproterenol. Isoproterenol challenge was performed two times in the same mouse at an interval of >72 h, with half the animals receiving first placebo, then flecainide, and the other half receiving first flecainide, then placebo. ECG analysis was performed by an experienced cardiologist blinded to the treatment group. Myocyte isolation and Ca2+ indicator loading Single ventricular myocytes were isolated by a modified collagenase/protease method as previously described.7 Ventricular myocytes were incubated with 2 µM fura-2 acetoxymethyl ester (Fura-2 AM) for 8 minutes at room temperature to load the indicator into the cytosol. Myocytes were washed two times for 10 minutes with 1.2 mM Ca2+ Tyrode’s solution containing 250 µM probenecid to retain the indicator in the cytosol. An additional 30 minutes were allowed for de-esterfication of the indicator before imaging the cells. All the experiments were performed in 2 mM Ca2+ Tyrode’s solution containing (in mM): NaCl 134, KCl 5.4, MgCl2 1, glucose 10, and HEPES 10, pH adjusted to 7.4 with NaOH. All chemicals were obtained from Sigma (St. Louis, MO) unless otherwise specified. Ca2+ fluorescence measurements Ca2+ transients were measured using a dual beam excitation fluorescence photometry set-up (IonOptix, Milton, MA) as described previously.2 Briefly, fura-2 loaded ventricular myocytes were incubated with 6 µM flecainide or vehicle (70% ethanol) for 10 minutes. In an additional set of experiments,
Nature Medicine: doi:10.1038/nm.1942

3
myocytes were incubated for 30 min with flecainide (6 µM), tetracaine (50 µM) or vehicle. After the cells attached to the bottom of the laminin coated chamber, they were superfused with Tyrode’s solution containing 2 mM Ca2+, 6 µM flecainide and 1 µM isoproterenol. SR Ca2+ content was estimated by measuring the amplitude of a Ca2+ transient induced by rapid application of 10 mM caffeine.8 All experiments were conducted at room temperature (∼23 oC). The emission at excitation wavelengths of 360 and 380 nm (background subtracted) was used to monitor the fluorescence signals of Ca2+-bound and Ca2+-free Fura-2. Intracellular Ca2+ concentration [Ca2+]i is proportional to the fluorescence ratio at 360 nm and 380 nm excitation.9 Fura-2 AM compartmentalizes into intracellular organelles,10 and therefore calculating [Ca2+]i from Fura-2 fluorescence ratios may not be accurate in intact cells. Thus, [Ca2+]i measurements are reported as fluorescence ratios (Fratio). Data were analyzed using a specialized analysis software package (IonWizardTM , IonOptix). Analysis of spontaneous SR Ca2+ releases and triggered beats A spontaneous Ca2+ release event was defined as a spontaneous increase of 0.07 Fratio (two times the average background noise) or more from the diastolic Fratio as previously described.11 Spontaneous Ca2+ releases and triggered beats were recorded while myocytes were exposed to 2 mM Ca2+ and1 µM isoproterenol Tyrode’s solution additionally containing 6 µM flecainide, 50 µM tetracaine, or 70% ethanol respectively. To eliminate the effect of trans-sarcolemmal Ca2+ or Na+ fluxes,11 spontaneous Ca2+ releases were recorded in a subset of myocytes bathed for 30s in 0 mM Na+ and Ca2+ Tyrode’s solution containing either 6 µM flecainide or vehicle and 1 µM isoproterenol. The 0 mM Na+ and Ca2+ Tyrode’s solution contained (in mM): LiCl 134, KCl 5.4, MgCl2 1, EGTA 1, glucose 10, and HEPES 10, pH adjusted to 7.4 with LiOH. Measurement of diastolic SR Ca2+ leak Leak was measured as previously described.2,11 Briefly, quiescent Fura-2 loaded cardiac myocytes were bathed in 0 mM Na+ and Ca2+ Tyrode’s solution containing 6 µM flecainide (or vehicle) and 1 µM isoproterenol. After spontaneous Ca2+ release frequency reached a steady-state, the external solution was quickly changed to 0 mM Na+ and Ca2+ Tyrode’s solution containing 1 mM tetracaine. Tetracaine blocks RyR2 channels and as a result Ca2+ shifts from the cytosol into the SR. The tetracaine-induced decline in diastolic Fura-2 fluorescence ratio was used as an estimate of SR Ca2+ leak,2,11 which is insensitive to changes in SR Ca2+ uptake.12 The amplitude of the caffeine-induced Ca2+ transient was used as an estimate of total [Ca2+]i, which included the Ca2+ leak. Analysis of [flecainide] in mouse plasma and heart tissue The concentration of flecainide in plasma and whole heart tissue was measured by reversed phase high performance liquid chromatography with positive electrospray ionization mass spectrometric detection HPLC-MS as previously described13 with slight modifications. Briefly, 50 � l of 20 � M verapamil was added to the samples as an internal standard, the tissue homogenized in phosphate buffered saline, protein precipitated with 2 volumes acetonitrile:methanol (85:15), and acidified supernatant loaded on Waters Oasis MCX cartridges. After washing with acidic phosphate buffer and 60% methanol, the drugs were eluted with acetonitrile containing 0.1% triethylamine and analyzed by MS using selective reaction monitoring of the m/z 415 ◊ 301 transition for flecainide and the m/z 455 ◊ 165 transition for verapamil. The HPLC gradient went from 95% Solvent A (water with 0.1% acetic acid to 90%) initially to 90% Solvent B (acetonitrile) over 5 minutes using a C18 column (Magic Bullet 3A, Michrom BioResources, Auburn, CA). Under these conditions flecainide eluted at 2.7 min and verapamil at 3.0 min. Tissue concentration was calculated as the ratio of peak heights for the sample relative to flecainide calibration standards and normalized to volume of plasma or tissue (1 mg heart tissue assumed to be 1 � l.) Statistical analysis Unless otherwise stated, data are presented as mean ± s.e.m.. All statistical tests were two-tailed and conducted with SPSS, version 12.0 or 16.0 (SPSS Inc, Chicago, IL). For normally distributed data, differences between groups were assessed using a one-way analysis of variance (ANOVA). If
Nature Medicine: doi:10.1038/nm.1942

4
statistically significant differences were found, individual groups were compared with Student's t-test. For non-normally distributed data such as VT incidence rates, Wilcoxon signed ranks test (for paired comparisons) or Mann-Whitney test (for unpaired comparisons) were used. The level of significance is stated in the text. References 1. Wilde, A.A., et al. Left cardiac sympathetic denervation for catecholaminergic polymorphic
ventricular tachycardia. N Engl J Med 358, 2024-2029 (2008). 2. Knollmann, B.C., et al. Casq2 deletion causes sarcoplasmic reticulum volume increase,
premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 116, 2510-2520 (2006).
3. Laver, D.R., et al. Cytoplasmic Ca2+ inhibits the ryanodine receptor from cardiac muscle. J Membr Biol 147, 7-22 (1995).
4. Laver, D.R. & Curtis, B.A. Response of ryanodine receptor channels to Ca2+ steps produced by rapid solution exchange. Biophys J 71, 732-741 (1996).
5. Sigworth, F.J. & Sine, S.M. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J 52, 1047-1054 (1987).
6. Guo, J., et al. Decrease in density of INa is in the common final pathway to heart block in murine hearts overexpressing calcineurin. Am J Physiol Heart Circ Physiol 291, H2669-2679 (2006).
7. Knollmann, B.C., et al. Familial hypertrophic cardiomyopathy-linked mutant troponin T causes stress-induced ventricular tachycardia and Ca2+-dependent action potential remodeling. Circ Res 92, 428-436 (2003).
8. Bers, D.M. Sarcoplasmic reticulum. . in In Excitation and Contraction Coupling and Cardiac Contractile Force 161-202 (Kluwer Academic Publishers; 2001, Dordrecht, The Netherlands/Boston, Massachusetts, USA/London, United Kingdom ).
9. Grynkiewicz, G., Poenie, M. & Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260, 3440-3450 (1985).
10. Williford, D.J., Sharma, V.K., Korth, M. & Sheu, S.S. Spatial heterogeneity of intracellular Ca2+ concentration in nonbeating guinea pig ventricular myocytes. Circ Res 66, 241-248 (1990).
11. Chopra, N., Laver, D., Davies, S.S. & Knollmann, B.C. Amitriptyline Activates Cardiac Ryanodine Channels And Causes Spontaneous Sarcoplasmic Reticulum Calcium Release. Mol Pharmacol (2008).
12. Curran, J., Hinton, M.J., Rios, E., Bers, D.M. & Shannon, T.R. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res 100, 391-398 (2007).
13. Kristoffersen, L., et al. Simultaneous determination of 6 beta-blockers, 3 calcium-channel antagonists, 4 angiotensin-II antagonists and 1 antiarrhythmic drug in post-mortem whole blood by automated solid phase extraction and liquid chromatography mass spectrometry. Method development and robustness testing by experimental design. J Chromatogr B Analyt Technol Biomed Life Sci 850, 147-160 (2007).
Nature Medicine: doi:10.1038/nm.1942