electronic supplementary information measurements 1h nmr
TRANSCRIPT

Electronic Supplementary Information
Measurements
1H NMR spectra were recorded at 300 MHz on a Mercury VX-300 spectrometer by using
tetramethylsilane (TMS) as the internal reference. 13C-NMR spectra were recorded at 75 MHz
with the solvent carbon signal as reference. Infrared spectra were recorded on a Nicolet Avator
360 FT-IR spectrometer. Size and distribution measurements of the micelles were performed on a
Nano-ZSZEN3600 (Malvern) instrument at room temperature. The critical micelle concentration
(CMC) was determined by using pyrene as a fluorescence probe. The concentration of polymer
was varied from approx. 1.0 × 10−3 to 0.5 mg/mL and the concentration of pyrene was fixed at 6 ×
10−7
The HABA/avidin assay was performed according to an early report described by Wooly.
M. The fluorescence spectra were recorded using an FLS920 fluorescence spectrometer with
an excitation wavelength of 337 nm. The emission fluorescence at 373 and 383 nm was monitored.
The CMC was estimated as the intersection when extrapolating the intensity ratio I373/I383 at low
and high concentration regions. Transmission electron microscopy images (TEM) were obtained
using a JEM-100CXⅡ transmission electron microscope. A drop of micelle solution (0.1 mg/mL)
was placed onto a copper grid with carbon film. The TEM images was observed at an acceleration
voltage of 80 keV.
[1] The
HABA solution was prepared by dissolving 24.2 mg of 4-hydroxyazobenzene-2-carboxylic acid
(0.1 mmol) in 10 mL of aqueous sodium hydroxide solution (10 mM). Then the HABA/Avidin
solution was made by dissolving 5.0 mg of avidin in 9.7 mL of 50 mM PBS with 50mM NaCl
(pH=7.2), followed by adding HABA solution (300 μL). This solution was stored up to one week
at 4ºC. The CPCB micelle stocks were prepared in different PBS solution (0.1 M, pH=6.8 or 7.4)
and kept in 37ºC for 4 h. The analyses were performed after mixing the HABA/Avidin solution
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

(300 μL) with the samples (33.3 μL). The final concentration of micelle was set at 0.1 mg/mL.
The UV spectra were recorded on a UV/Vis microplate spectrophotometer (Multiskan GO,
Thermo Fisher). The amount of biotin moieties available to avidin in the samples was calculated
by the formula: (ΔA500/34)×1000. (ΔA500
Cell internalization was observed on a confocal laser scanning microscopy (CLSM, Nikon C1-si).
MCF-7 cells were seeded in a 6-well plate and incubated at 37°C for 24 h. The mixed micelles
(CPCB/CPD/CPFITC= 0.1/0.1/0.01, CPC/CPD/CPFITC= 0.1/0.1/0.01) in fresh prepared DMEM
with 10 % fetal bovine serum (FBS) at different pH values were added to replace the medium.
After 4 h of incubation, the medium was removed and the cells were washed 3 times with PBS.
Then the cells were fixed with 4 wt % formaldehyde in PBS for 10 min at room temperature. A
drop of mounting media (10% PBS, 90% glycerol) were added to mount the cells. The
fluorescence of dox and fitc was examined under excitation at 488 nm.
means the differences of the UV absorbance at 500 nm
before and after the addition of the micelle samples).
The release profiles of DOX from nano-flowers were studied at 37°C under three different pH
conditions, i.e. (a) acetate buffer, pH 5.4; (b) phosphate buffer, pH 6.8; and (c) phosphate buffer,
pH 7.4. All the concentrations of the release media were 0.1 M. Every 1 mL of the mixed-micelles
solution was transferred to a dialysis tube (Mw 3500, cutoff). Then dialysis tube was immersed
into 20 mL of the corresponding buffer and the medium was shaken at 37°C. At desired time
intervals, 2 mL of release medium was replaced with an equal volume of fresh medium and then
dialyzed. The amount of released DOX was calculated by the results recorded on FLS920
fluorescence spectrometer. All the release experiments were conducted in triplicate. The results
presented are the average value ± standard deviation.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

The cytotoxicity assessment was carried out in MCF-7 cells by using the MTT assay.
100 μL of cell suspension (DMEM with 10% FBS) containing 6×103 MCF-7 cells
were seeded into each well of a 96-well plate and incubated at 37°C with 5% CO2
Cell viability (%) = (A
for
24 h. Then the cells were treated with mixed-micelles at various concentrations and
carried out a further incubation for 24 h. After that, all mediums were replaced with
MTT reagent (20 μL in PBS, 5 mg/mL) and incubated for another 4 h. The medium in
each well was carefully removed and replaced by 100 μL DMSO. When the purple
solution was homogeneous, the absorbance at 570 nm was recorded by a microplate
reader (Multiskan GO, Thermo Fisher). Cell viability was calculated by
treated/Acontrol
The data are shown as the average value ± standard deviation.
) ×100%
Materials: D-Biotin, 1,1'-carbonyldiimidazole (CDI), di-tert-butyl dicarbonate ((Boc)2O),
hydrazine hydrate,1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC·HCl),
sodium azide, 3-chloropropylamine hydrochloride, 1-octadecanol, 4-(dimethylamino)-pyridin
(DMAP), dicyclohexylcarbodiimide (DCC) and N-hydroxysuccinimide (NHS) were purchased
from Sinopharm (China) and used as received. N,N,N',N'',N''-pentamethyldiethylenetriamine
(PMDETA) , propargylamine, 4’-hydroxyazobenzene-2-carboxylic acid (HABA) and Fluorescein
isothiocyanate isomer I (FITC) were purchased from Alfa aesar and used as received. Doxorubicin
hydrochloride (Dox · HCl) was purchased from Zhejiang Hisun Pharmaceutical Co., Ltd. (China).
Trifluoroacetic acid, bromoacetyl bromide and 4-formylbenzoic acid were purchased from
Aladdin-reagent (China) and used as received. CuBr, Brij S 100 (average Mn ~4670) and
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

H-lys(Boc)-OH were purchased from Sigma-Aldrich and used as received. N,N-dimethyl
formamide (DMF) was dried over 4 Å molecular sieve and distilled over anhydrous MgSO4.
Dichloromethane (DCM) was distilled over CaH2
t-butyl carbazate 1
before used. Methanol was dried over 3 Å
molecular sieve and distilled by rectification. Bovine serum albumin (BSA), Dubelcco’s Modified
Eagle’s Medium (DMEM), penicillin–streptomycin, trypsin, and phosphate-buffered saline (PBS),
and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from
GIBCO Invitrogen Corporation.
Hydrazine hydrate (5.0 g, 0.1 mol) was dissolved in THF (30 mL) at 0ºC, and treated dropwise
with a solution of Boc2O (10.0 g, 45.8 mmol) in 10 mL of THF. Then the reaction was kept
stirring at room temperature over night. The solvent was removed and the residue was dissolved in
DCM, washed with distilled water and dried over anhydrous Na2SO4. The DCM was evaporated
and the residue was distilled under reduced pressure to obtain t-butyl carbazate as a white solid.
Yield: 4.5 g, (74.4 %). 1H NMR (300 MHz, CDCl3, δ ppm): 1.42 (s, 9H), 3.68 (s, 2H), 6.13 (s,
1H). 13C NMR (75 MHz, CDCl3
N-boc-2-bromoacetohydrazide 2
, δ ppm): 28.5, 77.2, 158.3.
t-Butyl carbazate (2.0 g, 15.2 mmol) and TEA (1.6 g, 15.8 mmol) were stirred in DCM (20 mL) at
0°C, and treated dropwise with a solution of bromoacetyl bromide (3.1 g, 15.4 mmol) in 1 mL of
DCM. The mixture was kept stirring at room temperature over night and filtrated, washed with
saturated NaHCO3 and brine, dried over anhydrous Na2SO4. The solvent was removed by rotary
evaporation and the residue was passed through a silica column (eluent, hexane/ethyl acetate =
1/1). N-boc-2-bromoacetohydrazide was obtained as colorless solid. Yield: 3.3 g (74.4 %). 1H
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

NMR (300MHz, CDCl3 δ ppm): 1.48 (s, 9H), 3.92(s, 2H), 7.14 (m, 1H), 8.92 (m, 1H). 13C NMR
(75 MHz, CDCl3
N-boc-2-azideacetohydrazide 3
, δ ppm): 26.9, 28.8, 83.1, 156.2, 166.7.
N-boc-2-bromoacetohydrazide (2.0 g, 7.9 mmol) and sodium azide (3.2g, 32 mmol) were
dissolved in 20 mL of anhydrous DMF and was stirred for 24h at 40°C. Then the solvent was
removed by rotary evaporation and the residue was dissolved in ether, washed with distilled water
for three times and dried over anhydrous Na2SO4. The solvent was removed and the product was
obtained as colorless solid. Yield: 1.6g (94.2%). 1H NMR (300MHz, CDCl3, δ ppm): 1.48 (s, 9H),
4.06(s, 2H), 6.97(m, 1H), 8.55 (m, 1H). 13C NMR (75 MHz, CDCl3
DOX-N
, δ ppm): 28.8, 51.9, 83.1,
156.1, 167.5.
3
N-boc-2-azideacetohydrazide (0.08g, 0.37 mmol) was stirred in a mixture solution of DCM and
TFA (1/1) for 30 min at room temperature. Then the solvent was removed by rotary evaporation
and the residue was co-evaporated three times with toluene to remove the excess TFA. The
obtained yellowish oil was dried over P
4
2O5 and NaOH under vacuum. The above yellowish oil
and Dox HCl (0.2g, 0.34 mmol) were dissolved in 15mL of anhydrous menthol, and treated with a
drop of TFA. After refluxed for 48h under dark, the mixture was cooled down. The solvent was
removed by rotary evaporation and the residue was precipitated in excess ethyl acetate. Precipitate
was collected by centrifugation and washed with ether. The crude product was obtained as a dark
red solid. DOX-N3
Aminopropylazide 5
was used in the next steps without further purification. Yeild: 97 mg (41.1%).
3-Chloropropylamine hydrochloride (10.0g, 78.0 mmol) and sodium azide (15.0g, 231.0 mmol)
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

and potassium iodide (1.3g, 7.8 mmol) were dissolved in 150 mL of distilled water and heated at
80°C for 18 h. The reaction solution was concentrated under vaccum and the pH was adjusted to
10 by adding aqueous NaOH solution (4M). The aqueous phase was extracted using diethyl ether,
and then the organic phase was dried over anhydrous Na2SO4, filtrated and concentrated. The
product was purified by distillation under vaccum and dried over molecular sieve. Yield: 5.3 g
(67.9 %).1H NMR (300 MHz, CDCl3, δ ppm): 1.14 (bs, 2H), 1.74 (m, 2H), 2.81 (t, 2H), 3.38 (t,
2H). 13C NMR (75 MHz, CDCl3
Octadecyl 4-formylbenzoate 6
, δ ppm): 32.1, 39.1, 49.1.
4-Formylbenzoic acid (3.0 g, 20.0 mmol), 1-octadecanol (5.4 g, 20.0 mmol), DMAP (0.24 g, 2.0
mmol) and DCC (4.1 g, 20.0 mmol) were dissolved in anhydrate ethyl ether (150 mL) and the
mixture was allowed to stir at room temperature for 24 h. Then the suspension was filtrated and
concentrated. The residue was purified by column chromatography (silica, EtOAc/petroleum ether
= 4:1 v/v). Octadecyl 4-formylbenzoate was obtained as a white solid. Yield: 5.8 g, (72.1 %). 1H
NMR (300MHz, CDCl3, δ ppm): 0.88 (t, 3H), 1.26 (m, 30H), 1.79 (m, 2H), 4.35 (t, 2H), 7.96 (d,
2H), 8.21 (d, 2H), 10.10 (s, 1H). 13C NMR (75 MHz, CDCl3
Octadecyl 4-((prop-2-ynylimino) methyl) benzoate 7
, δ ppm): 22.9, 26.3, 28.9, 29.5, 29.6,
29.9, 32.2, 66.0, 129.7, 130.4, 135.7, 139.3, 165.8, 191.8.
Octadecyl 4-formylbenzoate (2.0 g, 5.0 mmol) and propargylamine (0.28 g, 5.0 mmol) were
dissolved in anhydrous methanol (30 mL) and the solution was refluxed for 30 min. Then the hot
solution was cooled down to room temperature and recrystallization was carried out at 4ºC for a
further 10 h. The production was collected by filtration and then washed by methanol. Octadecyl
4-((prop-2-ynylimino) methyl) benzoate was obtained as a yellow solid. Yield: 1.8 g, (82.0 %). 1H
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

NMR (300MHz, CDCl3, δ ppm): 0.88 (t, 3H), 1.25 (m, 30H), 1.78 (m, 2H), 2.56 (s, 1H), 4.33 (t,
2H), 4.58 (s, 2H), 7.85 (d, 2H), 8.11 (d, 2H), 8.65 (s, 1H). 13C NMR (75 MHz, CDCl3
Octadecyl 4-((3-azidopropylimino) methyl) benzoate 8
, δ ppm):
22.9, 26.3, 28.9, 29.5, 29.6, 29.7, 29.8, 29.9, 32.2, 47.4, 65.6, 76.3, 78.7, 128.3, 130.0, 132.7,
139.8, 161.5, 166.3.
Octadecyl 4-formylbenzoate (2.0 g, 5.0 mmol) and aminopropylazide (0.5 g, 5mmol) were
dissolved in anhydrous methanol (30 mL) and the solution was refluxed for 30 min. Then the hot
solution was cooled down to room temperature and recrystallization was carried out at 4°C for a
further 10 h. The production was collected by filtration and then washed by methanol. Octadecyl
4-((3-azidopropylimino) methyl) benzoate was obtained as a white solid. Yield: 2.1 g (86.8 %). 1H
NMR (300MHz, CDCl3, δ ppm): 0.88 (t, 3H), 1.25 (m, 30H), 1.77 (m, 2H), 2.02 (m, 2H), 3.42 (t,
2H), 3.73 (t, 2H), 4.32 (t, 2H), 7.80 (d, 2H), 8.10 (d, 2H), 8.36 (s, 1H). 13C NMR (75 MHz, CDCl3
Biotin-NHS 9
,
δ ppm): 22.9, 26.3, 28.9, 29.5, 29.6, 29.7, 29.8, 29.9, 30.2, 32.2, 49.4, 58.5, 65.6, 128.2, 130.1,
132.7, 139.8, 161.2, 166.3.
Biotin (5 g, 20.5 mmol) was dissolved in 150 mL of DMF at 60°C, and then treated with DCC
(4.3 g, 20.8 mmol) and NHS (2.5 g, 21.7 mmol). The mixture was stirred at 60°C for 2 h and kept
stirring for 24 h at room temperature. The precipitate formed during reaction was filtrated and the
volume of solvent was concentrated to 30 mL by rotary evaporation, and then excess acetone was
added into the mixture until no more precipitate formed. The precipitate was collected and washed
with acetone. Biotin-NHS was purified by recrystallization in isopropanol. Yield: 4.3 g,
(61.5 %). 1H NMR (300MHz, DMSO-d6, δ ppm): 1.25~1.68 (m, 6H), 2.55 (d, 1H), 2.67 (t, 2H),
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

2.80~2.86 (m, 5H), 3.10 (t, 1H), 4.14 (m, 1H), 4.30 (m, 1H) 6.37 (d, 1H), 6.44 (d, 1H). 13C NMR
(75 MHz, DMSO-d6
Biotin-lys(Boc)-OH 10
, δ ppm): 24.7, 25.9, 28.0, 28.3, 30.4, 55.6, 59.86, 61.4, 163.1, 169.4, 170.7.
H-lys(Boc)-OH (1.5 g, 6.1 mmol)was suspended in 100 mL of DMF, and treated with biotin-NHS
(2.0 g, 5.8 mmol) and TEA (2.4 g, 11.8 mmol). The mixture was stirred at room temperature until
most of solid were dissolved (this process needed 2 days at least) and another TEA (2.4 g, 11.8
mmol) was added. After the solution sirred for 48 h at room temperature, the small amount of
insoluble substances were filtrated and the solvent was removed by rotary evaporation. The
residue was dissolved in n-butanol and then washed three times with 0.5 M KHSO4, three times
with distilled water, once with brine and dried over hydrous Na2SO4. The biotin-lys(Boc)-OH was
dried over P2O5 and NaOH under vacuum and was obtained as a yellowish solid. Yield: 2.1 g
(76.7 %). 1H NMR (300MHz, DMSO-d6, δ ppm): 1.18~1.72 (m, 21H), 2.11 (t, 2H), 2.56 (d, 1H),
2.81~2.86 (m, 3H), 3.09 (t, 1H), 4.11~4.14 (m, 2H), 4.30 (m, 1H), 6.35 (d, 1H), 6.41 (d, 1H), 6.74
(t, 1H), 7.87~8.00 (m, 1H). 13C NMR (75 MHz, DMSO-d6
Biotin-lys(Boc)-N
, δ ppm): 23.3, 25.7, 28.7, 29.6, 31.2,
33.8, 35.2, 36.2, 52.2, 55.9, 59.7, 61.5, 77.8, 156.0, 163.2, 172.7, 174.3.
3
Biotin-lys(Boc)-OH (2g, 4.2 mmol) and EDC·HCl (0.81g, 4.2 mmol) were dissolved in 100 mL of
DMF and then aminopropylazide (0.5g, 5.0 mmol) was added. After the solution was stirred for
24h at room temperature, the solvent was removed by rotary evaporation. The residue was
dissolved in n-butanol and washed once with 0.5 M KHSO
11
4, three times with distilled water, once
with brine, dried over hydrous Na2SO4 and filtrated. The filtrate was concentrated and added into
excess ethyl ether for precipitation. The precipitate was collected and purified with column
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

chromatography (silica, DCM/MeOH=10:1 v/v). Biotin-lys(Boc)-N3 was obtained as a yellowish
solid. Yield: 1.9g (81.6 %). 1H NMR (300MHz, DMSO-d6, δ ppm): 1.18~1.72 (m, 23H), 2.11 (t,
2H), 2.56 (d, 1H), 2.78~2.87 (m, 3H), 3.09 (m, 3H), 3.32~3.37 (m, 2H), 4.14 (m, 2H), 4.30 (m,
1H), 6.39 (d, 1H), 6.47 (d, 1H), 6.73 (t, 1H), 7.87~7.98 (m, 2H). 13C NMR (75 MHz, DMSO-d6
CDI-activated PEG-b-C18 12
, δ
ppm): 23.3, 25.6, 28.4, 28.5, 28.7, 29.6, 32.2, 35.3, 36.2, 48.7, 52.9, 55.9, 59.7, 61.4, 77.7, 156.0,
163.2, 172.4, 172.5.
Brij (100) (6.1g, 1.3 mmol) was dissolved in DCM (60 mL) and CDI (2.07 g, 12.8 mmol) was
added, and then the solution was allowed to stir for 24h at room temperature. The reaction mixture
was concentrated to a small volume and poured into excess anhydrous diethyl ether. The white
precipitate was collected by centrifugation and washed with diethyl ether and dried under vacuum.
The product with trace imidazole was directly used in the next step without further purification.
Yield5.4 g, (87.2%). 1H NMR (300MHz, CDCl3,
Biotin-lys(PEG-C18)-N
δ ppm): 0.88 (t, 3H), 1.25 (m, 30H), 1.57 (m,
2H), 3.4~3.9 (m, ~400H), 4.56 (t, 2H), 7.08 (d, 1H), 7.46 (d, 1H), 8.16 (s, 1H).
3
Biotin-lys(Boc)-N
13
3 (0.55g, 1.0 mmol) was stirred in a mixture solution of DCM and TFA (1/9) for
2 h at room temperature. The solvent was then removed by rotary evaporation and the residue was
added into excess ethyl ether. The precipitate was collected and dried over P2O5 and NaOH under
vaccum for two days. The de-protected Biotin-lys(Boc)-N3 and PEG-b-C18-CDI (1.0g, 0.2 mmol)
were dissolved in hydrous DMF, and treated with TEA (0.21g, 2.1 mmol) to neutralize the trace
mount of TFA and catalyze the reaction. The mixture was stirred for 72 h at room temperature and
dialyzed against with DMSO for 72h (1L×6) and then dialyzed extensively against with DI water.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012

Biotin-lys(PEG-C18)-N3 was obtained by freeze-drying. Yield: 0.72g (69.9 %). 1H NMR
(300MHz, CDCl3
Alkyne-PEG-b-C18 14
, δ ppm): 0.88 (t, 3H), 1.25 (m, 34H), 1.57 (m, 12H), 3.4~3.9 (m, ~400H),
4.00~4.30 (m, 5H), 6.39 (d, 1H), 6.47 (d, 1H).
CDI-activated PEG-b-C18 (2g, 0.42 mmol), propargylamine (0.23g 4.2mmol) and triethylamine
(0.61mL, 4.2mmol) were dissolved in DCM (15mL) and the solution was allowed to stir for 48 h
at room temperature. The procedure of purification was same as that of compound 12. Yield: 1.6g
(80.2 %). 1H NMR (300MHz, CDCl3
Click chemistry
, δ ppm): 0.88 (t, 3H), 1.25 (m, 30H), 1.57 (m, 2H), 2.25 (s,
1h), 3.4~3.9 (m, ~400H), 4.15~4.31 (m, 4H).
C18-PEG-(biotin)-C18, C18-PEG-C18 and C18-PEG-DOX were synthesized in a similar
procedure. Typically, C18-PEG-(biotin)-N3 and octadecyl 4-((prop-2-ynylimino)methyl)benzoate
were dissolved in 2mL of DMF. After three freeze-pump-thaw cycles, PMDETA and Cu(Ⅰ)Br were
added and the solution was stirred under Ar atmosphere at 35°C for 24 h. After reaction, the
mixture was diluted with THF and passing through a short alumina column to eliminate the copper.
The product was obtained by three times of precipitation in excess ice-cooled ethyl ether and dried
under vacuum.
C18-PEG-DOX was synthesized in a similar procedure but purified by dialysis (DMSO for 72h
(1L×6), and then DI water for 24h (1L×4)).
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
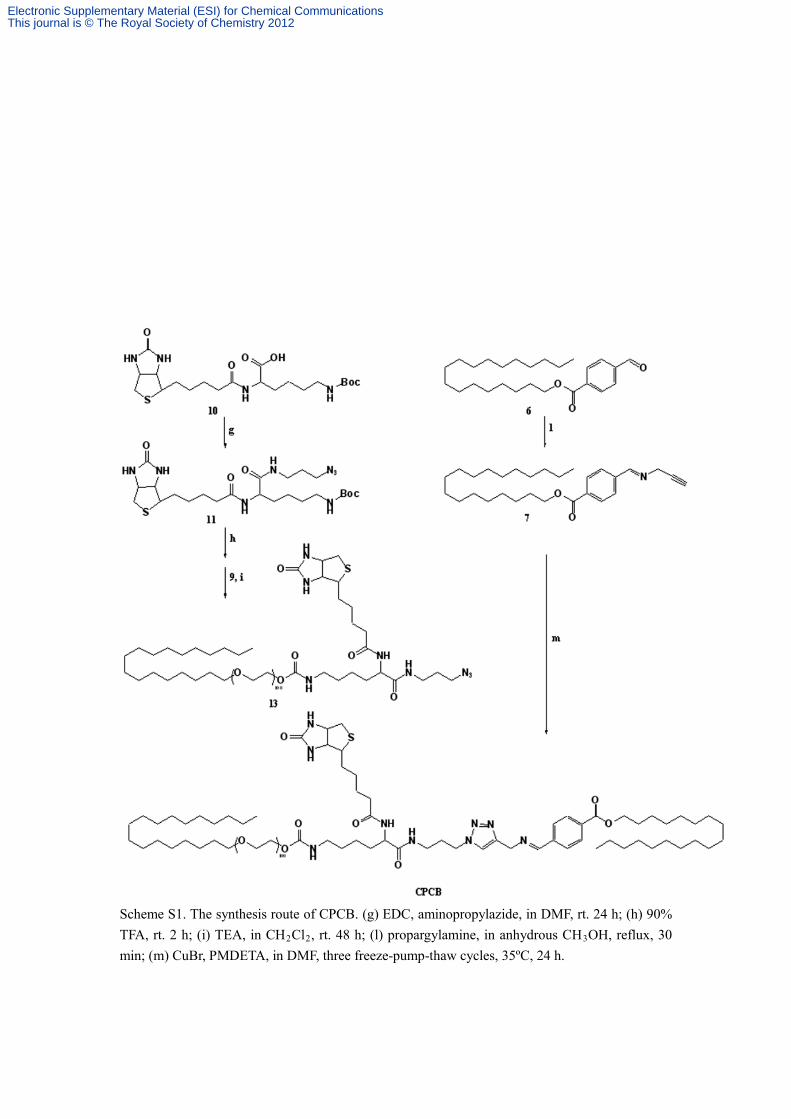
Scheme S1. The synthesis route of CPCB. (g) EDC, aminopropylazide, in DMF, rt. 24 h; (h) 90% TFA, rt. 2 h; (i) TEA, in CH2Cl2, rt. 48 h; (l) propargylamine, in anhydrous CH3OH, reflux, 30 min; (m) CuBr, PMDETA, in DMF, three freeze-pump-thaw cycles, 35ºC, 24 h.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
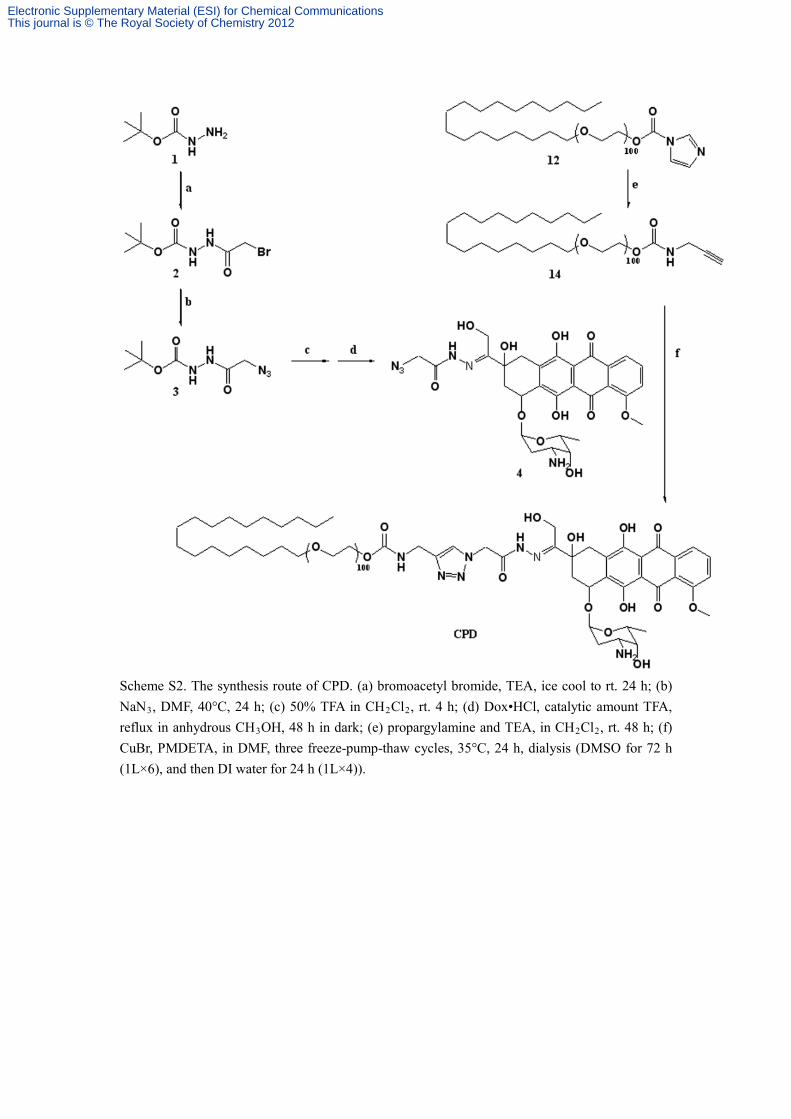
Scheme S2. The synthesis route of CPD. (a) bromoacetyl bromide, TEA, ice cool to rt. 24 h; (b) NaN3, DMF, 40°C, 24 h; (c) 50% TFA in CH2Cl2, rt. 4 h; (d) Dox•HCl, catalytic amount TFA, reflux in anhydrous CH3OH, 48 h in dark; (e) propargylamine and TEA, in CH2Cl2
, rt. 48 h; (f) CuBr, PMDETA, in DMF, three freeze-pump-thaw cycles, 35°C, 24 h, dialysis (DMSO for 72 h (1L×6), and then DI water for 24 h (1L×4)).
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
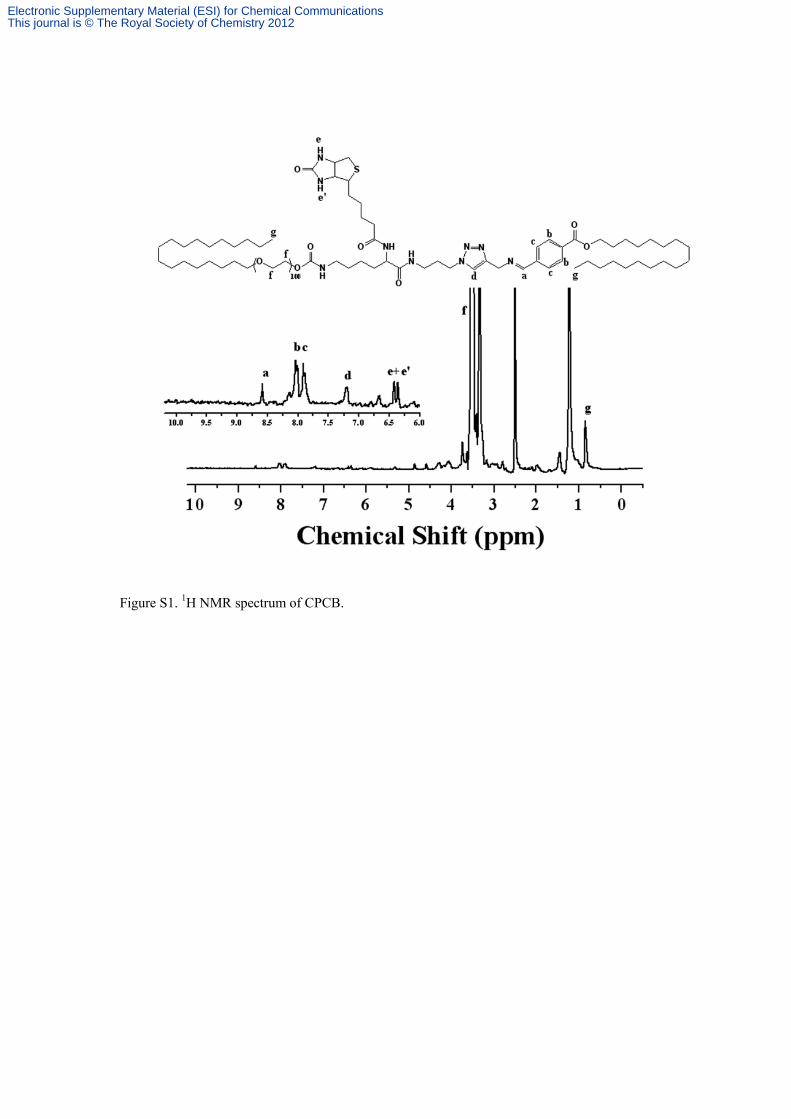
Figure S1. 1
H NMR spectrum of CPCB.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
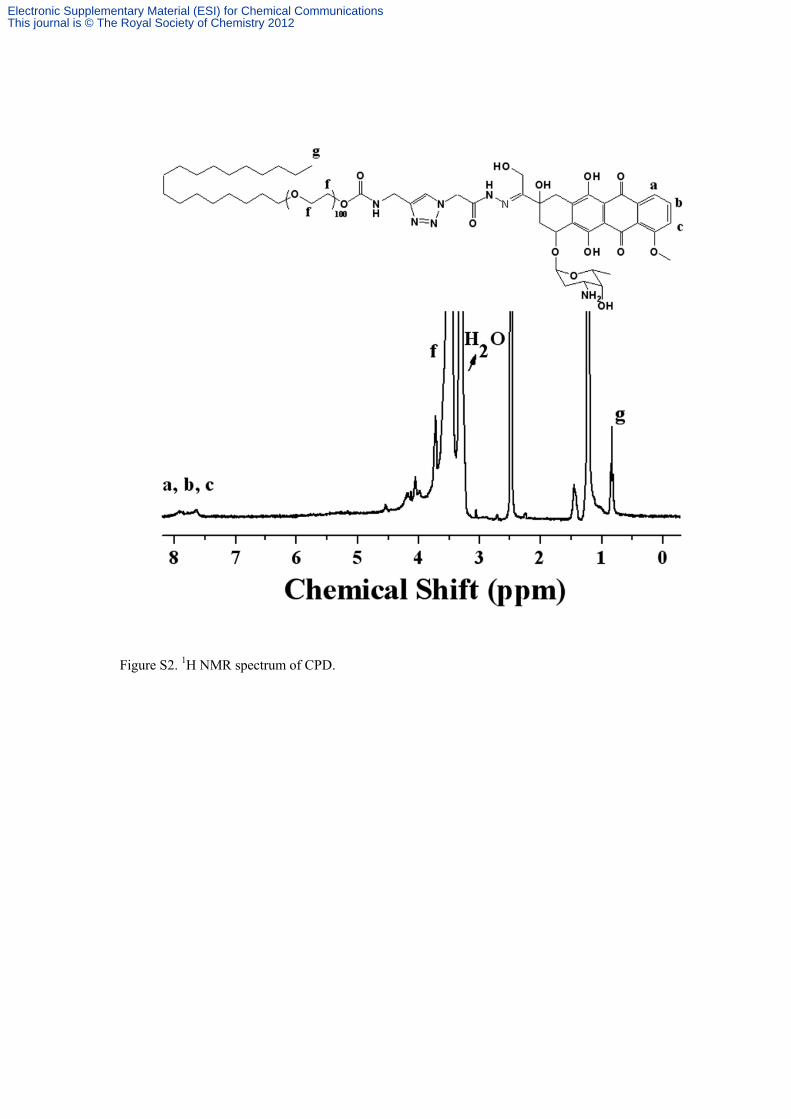
Figure S2. 1
H NMR spectrum of CPD.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
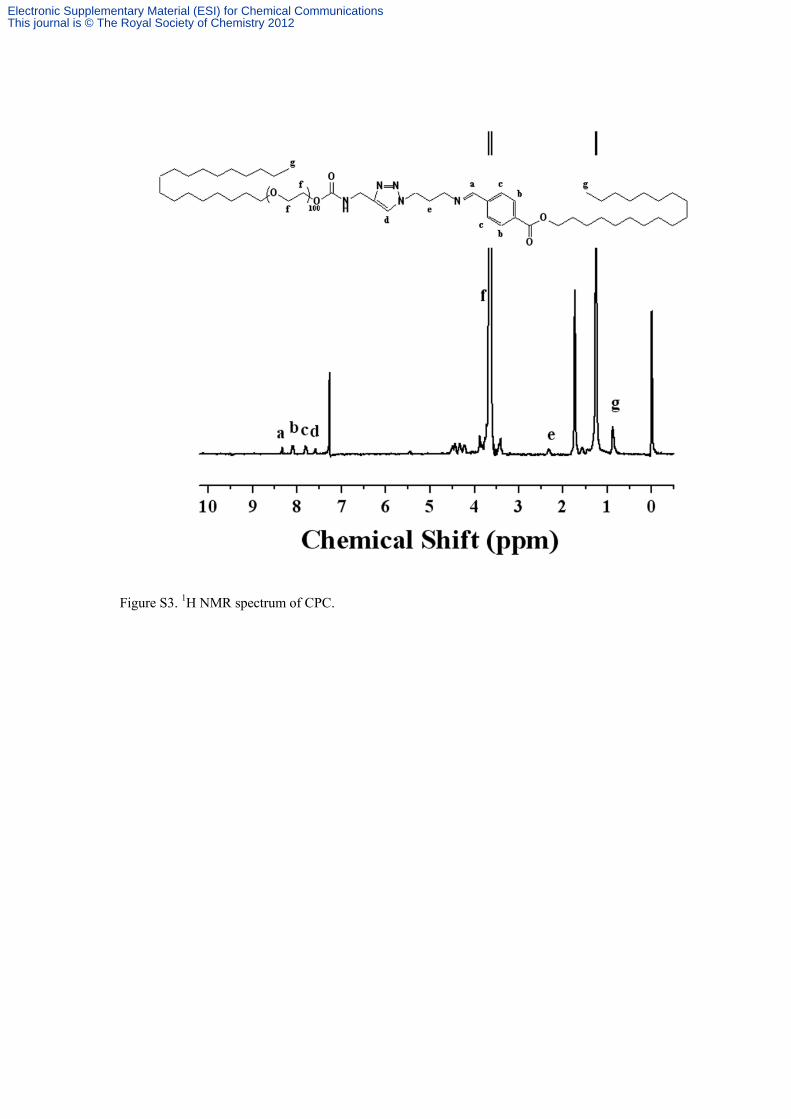
Figure S3. 1
H NMR spectrum of CPC.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
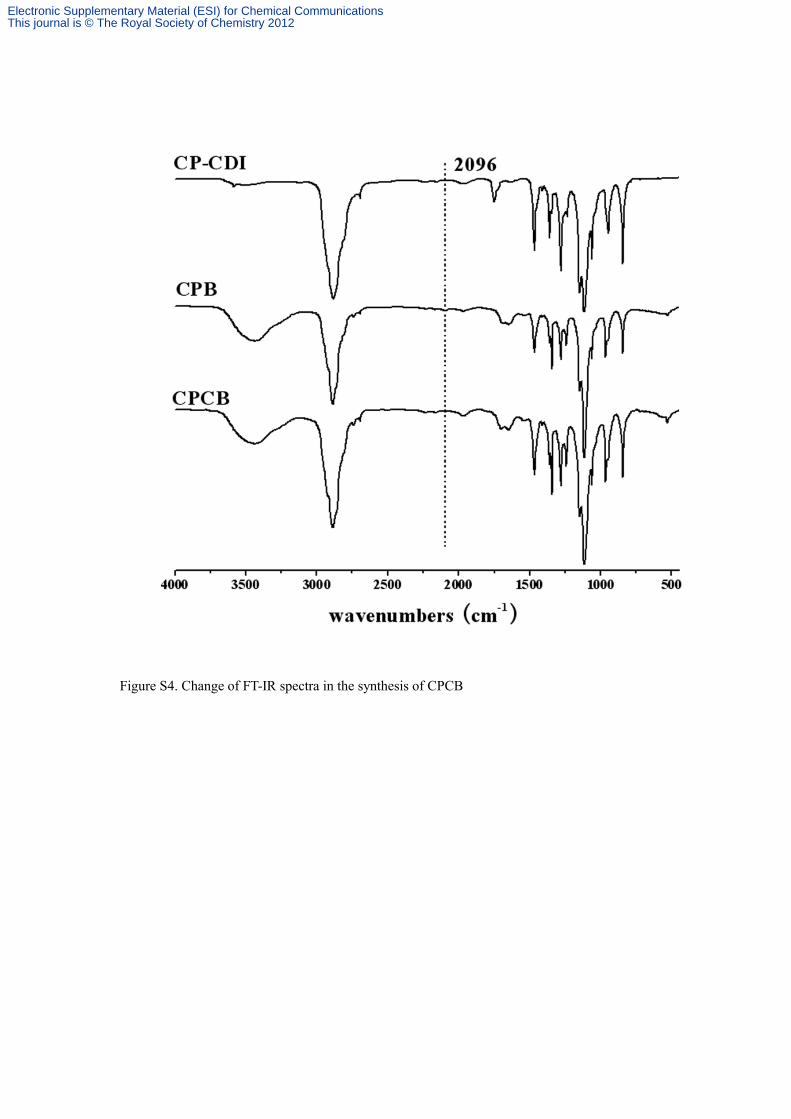
Figure S4. Change of FT-IR spectra in the synthesis of CPCB
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
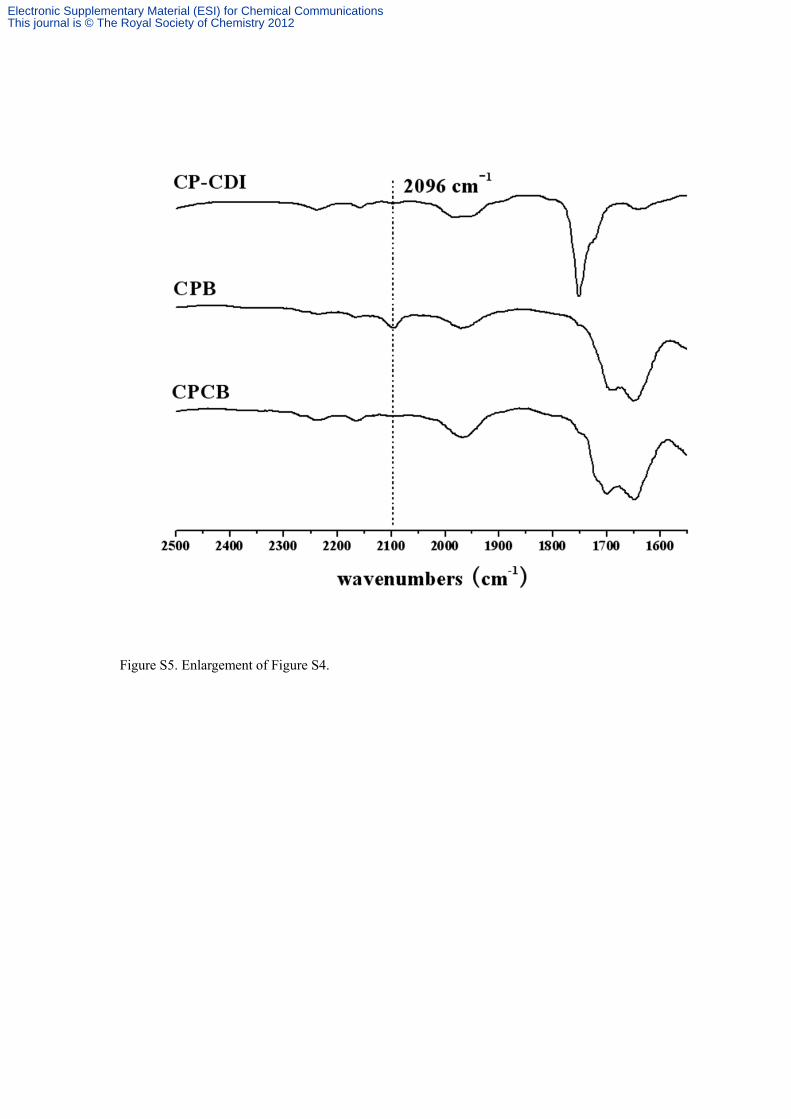
Figure S5. Enlargement of Figure S4.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
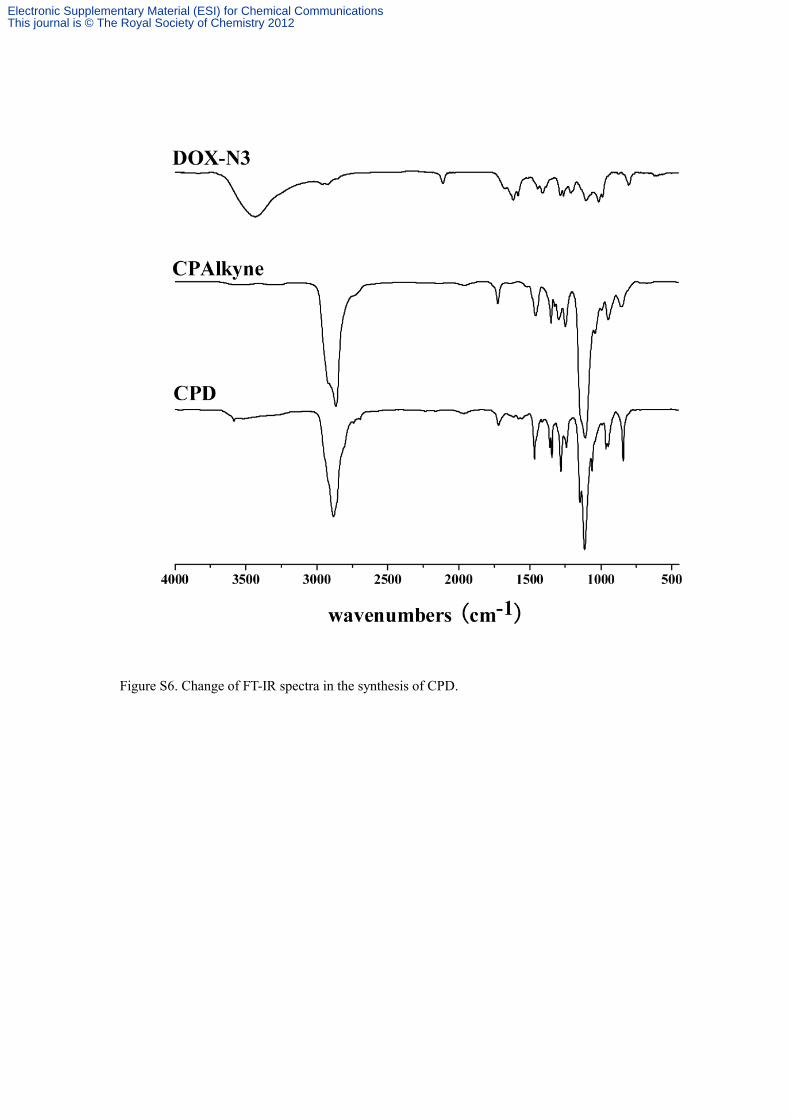
Figure S6. Change of FT-IR spectra in the synthesis of CPD.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
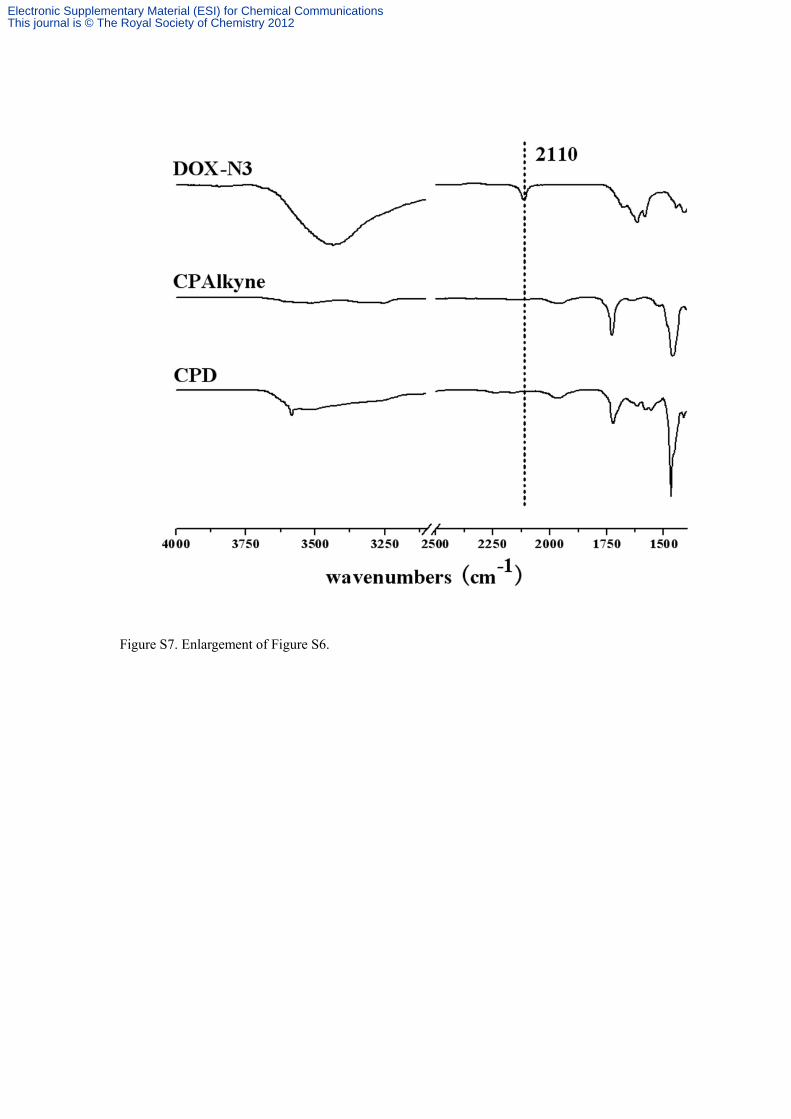
Figure S7. Enlargement of Figure S6.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
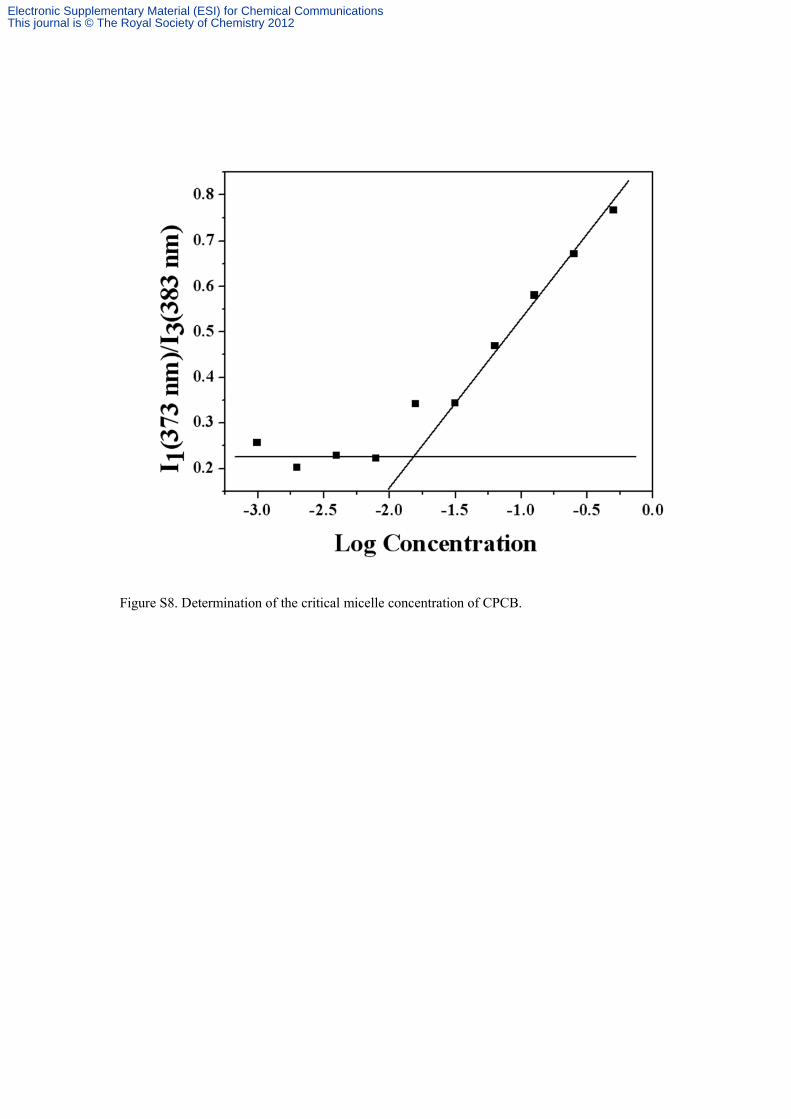
Figure S8. Determination of the critical micelle concentration of CPCB.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
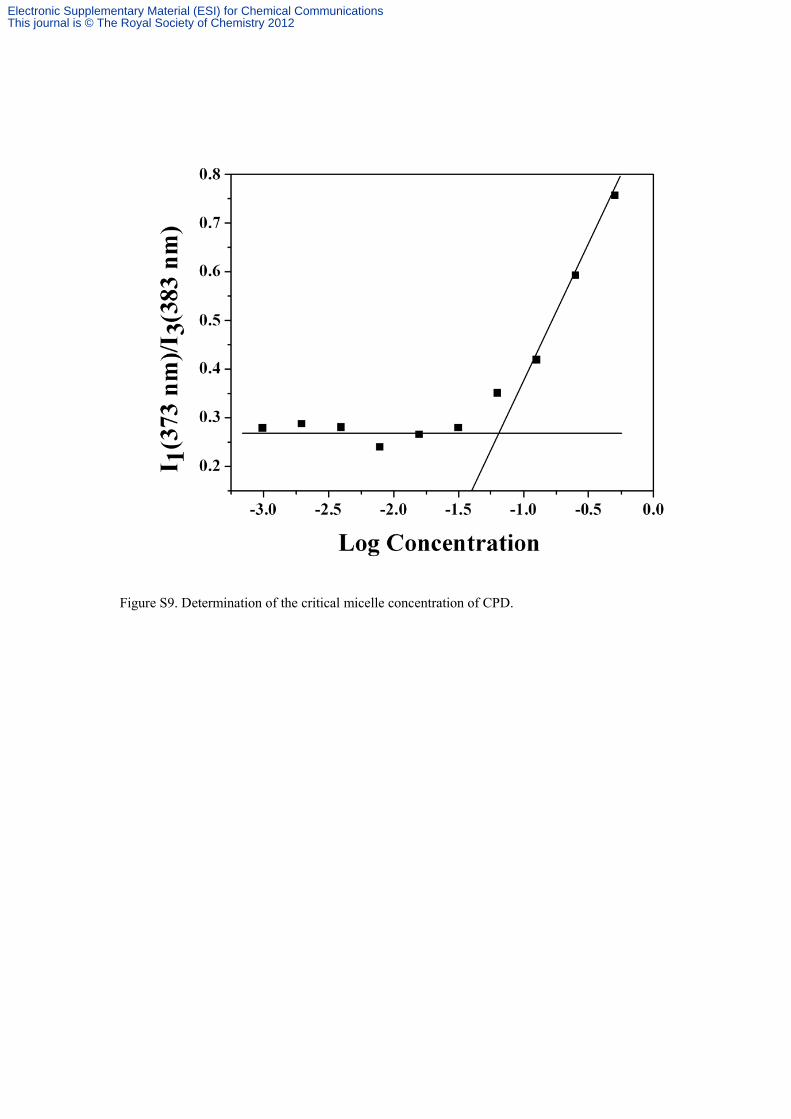
Figure S9. Determination of the critical micelle concentration of CPD.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
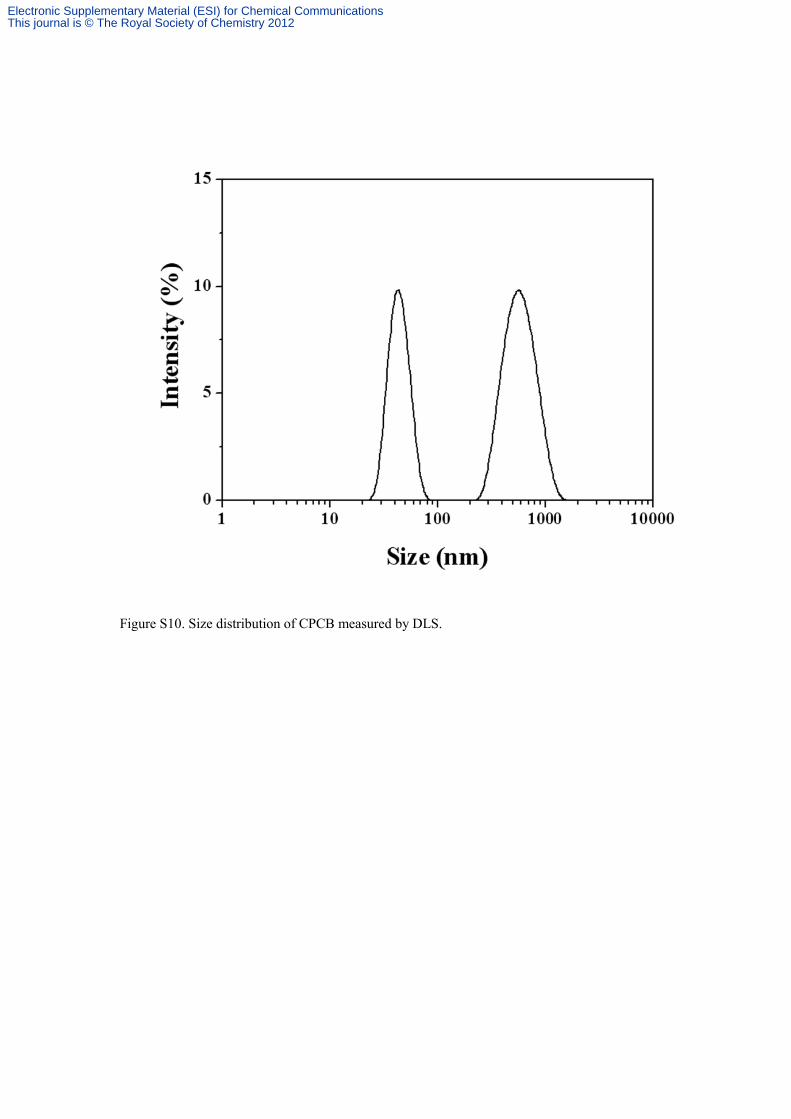
Figure S10. Size distribution of CPCB measured by DLS.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
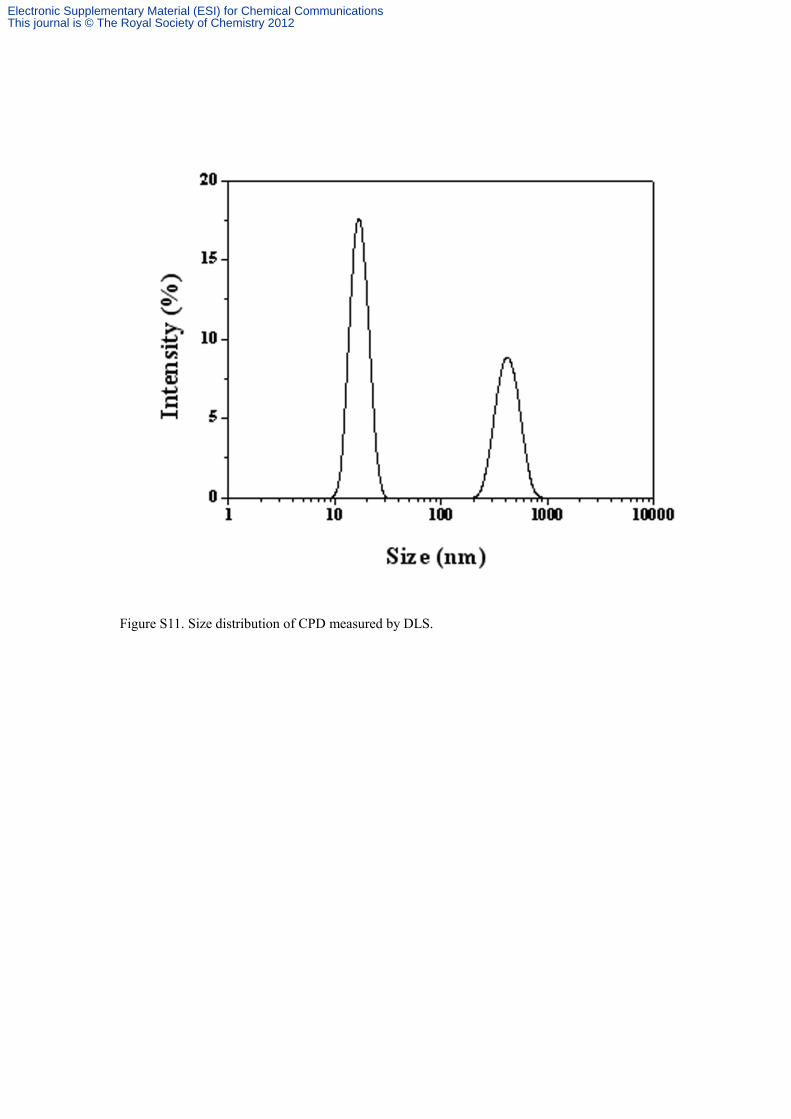
Figure S11. Size distribution of CPD measured by DLS.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012
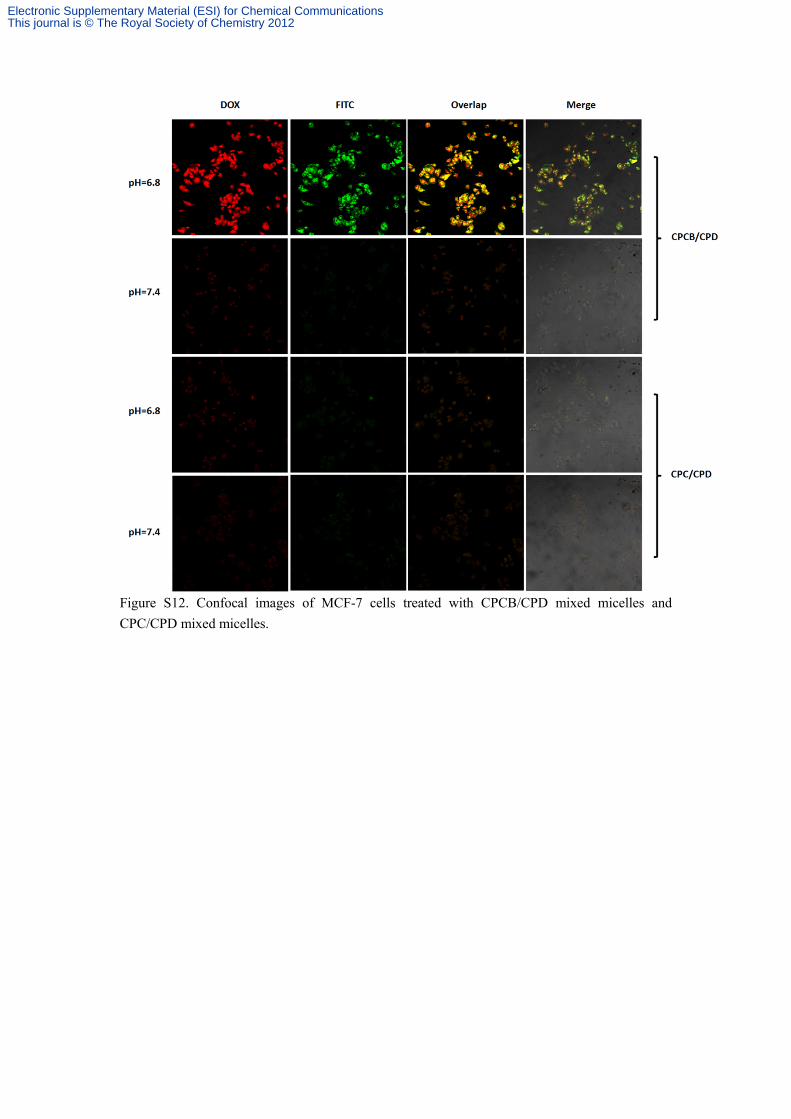
Figure S12. Confocal images of MCF-7 cells treated with CPCB/CPD mixed micelles and CPC/CPD mixed micelles.
Electronic Supplementary Material (ESI) for Chemical CommunicationsThis journal is © The Royal Society of Chemistry 2012