effects of candesartan cilexetil in patients with systemic hypertension
TRANSCRIPT

Effects of Candesartan Cilexetil inPatients With Systemic Hypertension
Max Reif, MD, William B. White, MD, Timothy C. Fagan, MD, Suzanne Oparil, MD,Terry L. Flanagan, MPH, Dianne T. Edwards, BA, Daniel J. Cushing, PhD, and
Eric L. Michelson, MD, for the Candesartan Cilexetil Study Investigators*
The objectives of this double-blind, multicenter, random-ized, parallel-arm, placebo-controlled study were toevaluate the dose-related efficacy, tolerability, andsafety of candesartan cilexetil, a potent, AT1 selective,long-acting angiotensin II receptor blocker, in 365 adultpatients with systemic hypertension and mean sittingdiastolic blood pressure (BP) of 95 to 114 mm Hg.Patients received either placebo or candesartan cilexetil2, 4, 8, 16, or 32 mg once daily for 8 weeks. All dosesof candesartan cilexetil reduced trough (24 hours aftertreatment) sitting diastolic and systolic BP significantlycompared with placebo (p <0.005). A significant (p<0.0001) dose response was evident, with greater de-creases in BP at higher doses. Mean changes in BP were210.7/27.8 mm Hg and 212.6/210.2 mm Hg in the16- and 32-mg groups, respectively, versus 20.3/22.6
mm Hg in the placebo group. The 16- and 32-mg doseswere consistently significantly superior to placebo inantihypertensive effect with regard to all BP measure-ments, including peak (6 hours after treatment), trough,sitting, and standing measurements of diastolic and sys-tolic BP. Responder rates (trough sitting diastolic BP <90or >10 mm Hg BP decrease) were 54% and 64% for the16- and 32-mg groups, respectively. Tolerability andsafety profiles were similar to placebo at all doses. Inconclusion, candesartan cilexetil administered oncedaily effectively reduces BP in a dose-related mannerwhile maintaining safety and tolerability; doses of 16and 32 mg are most effective for treatment of hyperten-sion. Q1998 by Excerpta Medica, Inc.
(Am J Cardiol 1998;82:961–965)
The development of angiotensin receptor blockers isa major advance for the treatment of hypertension
and potentially for other cardiovascular disorders.1–3
These agents block the action of angiotensin II di-rectly at the AT1 receptor. Candesartan is a potent,highly selective, insurmountable AT1 receptor antag-onist that is devoid of agonist activity.4,5 Candesartanis administered as candesartan cilexetil, a prodrug(inactive) that is rapidly and completely hydrolyzed tocandesartan, the active drug, during absorption fromthe gastrointestinal tract.5,6 The antihypertensive ef-fect of candesartan cilexetil has been documented indoses up to 16 mg/day in European studies.7 Theobjective of this 8-week, randomized, double-blind,placebo-controlled, parallel-group study was to eval-uate the dose-related efficacy, tolerability, and safetyof candesartan cilexetil administered in doses rangingfrom 2 to 32 mg once daily in a diverse population ofpatients with systemic hypertension in the UnitedStates.
METHODSStudy patients: Eligible patients were men and
women without child-bearing potential, aged 18 to 80years, with essential hypertension, defined as an un-treated mean sitting diastolic blood pressure (BP) of95 to 114 mm Hg on 2 sequential clinic visits duringthe placebo run-in period. Patients with systolic BP$210 mm Hg or with secondary hypertension wereexcluded from the study. Also excluded were patientswith major systemic disorders, including clinicallysignificant cardiovascular, cerebrovascular, renal, he-patic, pulmonary, or hematologic disease, those onconcomitant medication use that might confoundstudy participation, or patients with a known hyper-sensitivity reaction to angiotensin II receptor blockers.
The study protocol was approved by the institu-tional review board at each site, and all patients pro-vided written informed consent.
Study design: This multicenter study, conductedfrom December 1995 to October 1996 in the UnitedStates, comprised a 4- to 5-week single-blind placeborun-in period followed by an 8-week double-blindtreatment period. After the screening visit, at whichtime all current antihypertensive agents were discon-tinued, patients were evaluated at 4 weekly visitsduring the placebo run-in period, at 2-week intervalsduring double-blind treatment, and then at a follow-upvisit 2 weeks after discontinuing treatment. Patientswere randomized to 1 of 6 treatment groups: placeboor candesartan cilexetil 2, 4, 8, 16, or 32 mg. Patientswere instructed to take their medication once daily, inthe morning.
Efficacy assessments: Patients reported for clinicvisits approximately 24 hours (“trough”) after the last
From the Hypertension Section, University of Cincinnati Medical Cen-ter, Cincinnati, Ohio; Section of Hypertension and Vascular Diseases,University of Connecticut Health Center, Farmington, Connecticut;Departments of Medicine and Pharmacology, University of Arizona,Tucson, Arizona; Department of Medicine, University of Alabama atBirmingham, Birmingham, Alabama; and Astra Merck Inc., Wayne,Pennsylvania. This study was supported by a grant from Astra MerckInc., Wayne, Pennsylvania. Manuscript received May 26, 1998;revised manuscript received and accepted July 27, 1998.
Address for reprints: Eric L. Michelson, MD, 725 ChesterbrookBlvd, D-3N, Wayne, Pennyslvania 19087.
*A list of investigators and their affiliations appears in the Appendix.
961©1998 by Excerpta Medica, Inc. 0002-9149/98/$19.00All rights reserved. PII S0002-9149(98)00627-4
dose of study medication and at the same time of daythroughout the study. At each visit, pulse rate wasrecorded after the patient had been sitting for 3 min-utes. Sitting BP was measured, using a mercurysphygmomanometer, from the right arm 3 times at1-minute intervals after the patient had been sitting forat least 5 minutes. The difference in 3 sitting diastolicBP measurements had to be#5 mm Hg. Standing BPmeasurements were then obtained 2 times after thepatient had been standing for at least 1 minute. Atweek 3 of the placebo run-in period and on day 1,week 2, and week 8 of double-blind treatment, pa-tients returned 6 hours after taking study medicationfor “peak” (drug effect) BP measurements. For thepatient to remain eligible for study participation, at theend of the week 4 visit of the run-in period, thepatient’s sitting diastolic BP had to be 95 to 114 mmHg, with #10 mm Hg difference from the sittingdiastolic BP at week 3.
Safety assessments: A medical history was recordedand a complete physical examination was performedat screening, with brief physical examinations at sub-sequent visits. Fasting laboratory samples were ob-tained at screening and at week 3 of the run-in period,at which time a chest x-ray and electrocardiogramwere done. Laboratory testing, a complete physicalexamination, and electrocardiogram were obtained atthe end of double-blind treatment or upon study dis-
continuation. Laboratory testing included a completeblood count, platelet count, blood chemistry, urinaly-sis, and human chorionic gonadotropin for women ofchild-bearing age. All laboratory tests were run at acentral laboratory (Corning SciCor Laboratories, Inc.,now known as COVANCE Central Laboratory Ser-vices, Indianapolis, Indiana). The occurrence of ad-verse events was recorded at each clinic visit, andassessment of causality to study drugs was made bythe investigators who remained blinded to treatmentallocation.
Patients could be discontinued during the double-blind treatment period if sitting diastolic BP was notcontrolled, namely, the diastolic BP was$105 mmHg after at least 4 weeks of double-blind treatment,.120 mm Hg at 1 visit, or$110 mm Hg at 2 sequen-tial visits.
The follow-up visit 2 weeks after study drug dis-continuation included a brief physical examination,trough BP measurements, and assessment of any ad-verse events occurring after withdrawal of study med-ication.
Statistical analysis: Efficacy analyses were per-formed using an intent-to-treat approach (including allpatients who took at least 1 dose of double-blind studymedication and had at least 1 subsequent BP measure-ment), with the last observation carried forward. Inaddition, a prespecified per protocol analysis was per-
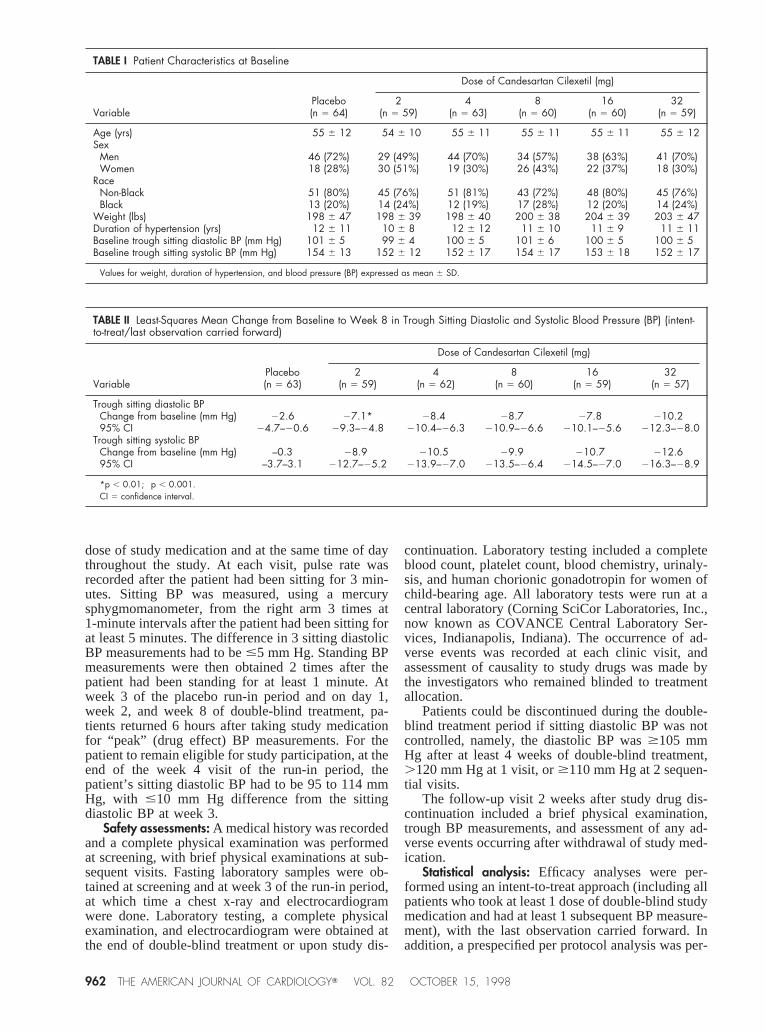
TABLE I Patient Characteristics at Baseline
VariablePlacebo(n 5 64)
Dose of Candesartan Cilexetil (mg)
2(n 5 59)
4(n 5 63)
8(n 5 60)
16(n 5 60)
32(n 5 59)
Age (yrs) 55 6 12 54 6 10 55 6 11 55 6 11 55 6 11 55 6 12Sex
Men 46 (72%) 29 (49%) 44 (70%) 34 (57%) 38 (63%) 41 (70%)Women 18 (28%) 30 (51%) 19 (30%) 26 (43%) 22 (37%) 18 (30%)
RaceNon-Black 51 (80%) 45 (76%) 51 (81%) 43 (72%) 48 (80%) 45 (76%)Black 13 (20%) 14 (24%) 12 (19%) 17 (28%) 12 (20%) 14 (24%)
Weight (lbs) 198 6 47 198 6 39 198 6 40 200 6 38 204 6 39 203 6 47Duration of hypertension (yrs) 12 6 11 10 6 8 12 6 12 11 6 10 11 6 9 11 6 11Baseline trough sitting diastolic BP (mm Hg) 101 6 5 99 6 4 100 6 5 101 6 6 100 6 5 100 6 5Baseline trough sitting systolic BP (mm Hg) 154 6 13 152 6 12 152 6 17 154 6 17 153 6 18 152 6 17
Values for weight, duration of hypertension, and blood pressure (BP) expressed as mean 6 SD.
TABLE II Least-Squares Mean Change from Baseline to Week 8 in Trough Sitting Diastolic and Systolic Blood Pressure (BP) (intent-to-treat/last observation carried forward)
VariablePlacebo(n 5 63)
Dose of Candesartan Cilexetil (mg)
2(n 5 59)
4(n 5 62)
8(n 5 60)
16(n 5 59)
32(n 5 57)
Trough sitting diastolic BPChange from baseline (mm Hg) 22.6 27.1* 28.4† 28.7† 27.8† 210.2†
95% CI 24.7–20.6 29.3–24.8 210.4–26.3 210.9–26.6 210.1–25.6 212.3–28.0Trough sitting systolic BP
Change from baseline (mm Hg) –0.3 28.9† 210.5† 29.9† 210.7† 212.6†
95% CI –3.7–3.1 212.7–25.2 213.9–27.0 213.5–26.4 214.5–27.0 216.3–28.9
*p , 0.01; †p , 0.001.CI 5 confidence interval.
962 THE AMERICAN JOURNAL OF CARDIOLOGYT VOL. 82 OCTOBER 15, 1998
formed, which included only patients with no majorprotocol violations to more accurately define the dose-response relation. The primary efficacy parameter wasthe change from baseline in trough sitting diastolic BPat week 8 of double-blind treatment. Analysis of co-variance, with baseline as the covariate and treatment,center, and treatment-by-center as fixed effects, wasused to compare treatment groups with respect tochanges from baseline in trough and peak, sitting andstanding BP. The arithmetic mean of 3 and 2 sequen-tial BP readings was used for analysis of sitting andstanding BP, respectively. Baseline BP was defined asthe mean reading at the last visit before the start ofdouble-blind treatment. Least-squares means andtreatment difference means were calculated along with95% confidence intervals. For non-normally distrib-uted variables, an analysis of variance was performedon the ranks of the observations using the Kruskal-Wallis test.
To assess dose response, 3 regression models werefit for the change from baseline in trough sitting dia-stolic and systolic BP at double-blind week 8 usingTukey’s method. These regressions were fit both withand without placebo as a 0 dose. The proportion ofresponders was calculated descriptively for each treat-ment group, as were placebo-corrected trough-to-peakratios for change in sitting diastolic BP. All patientsreceiving at least 1 dose of double-blind medicationwere included in the safety analyses. Adverse eventand laboratory data were descriptively comparedamong the 6 treatment groups.
With a sample size of 360 patients, the study had95% power to detect a difference of 5 mm Hg betweenany active treatment arm and placebo in change frombaseline to sitting diastolic BP, assuming a SD of 7.5mm Hg (2-tailed test,a 5 0.05).
RESULTSPatients: A total of 365 patients were randomized to
double-blind treatment at 29 sites, and 332 (91%)completed the study. The study population comprised64% male and 23% black patients with a collectivemean age of 55 years and mean sitting trough diastolicBP at baseline of 100 mm Hg. Patient characteristicsat baseline were similar among the 6 treatment groups(Table I). Reasons for study discontinuation were alsosimilar among the 6 groups. Overall, 9 patients (2.5%)withdrew because of lack of response and 9 (2.5%)because of an adverse event, whereas 8 patients(2.2%) withdrew consent and 7 (1.9%) discontinuedthe study for other reasons. All randomized patientswere evaluated for safety, whereas 5 of 365 random-ized patients were excluded from the intent-to-treatefficacy analyses because they lacked any post-treat-ment efficacy data. The per-protocol patient popula-tion numbered 312. Efficacy results reported here arefor the intent-to-treat population unless otherwisespecified.
Efficacy: All doses of candesartan cilexetil pro-duced significantly greater decreases in trough sittingdiastolic BP than placebo (p,0.005); changes frombaseline at week 8 are shown in Table II. Most of the
antihypertensive effects were seen within 2 weeks,although further BP reductions were seen thereafter(data not shown).
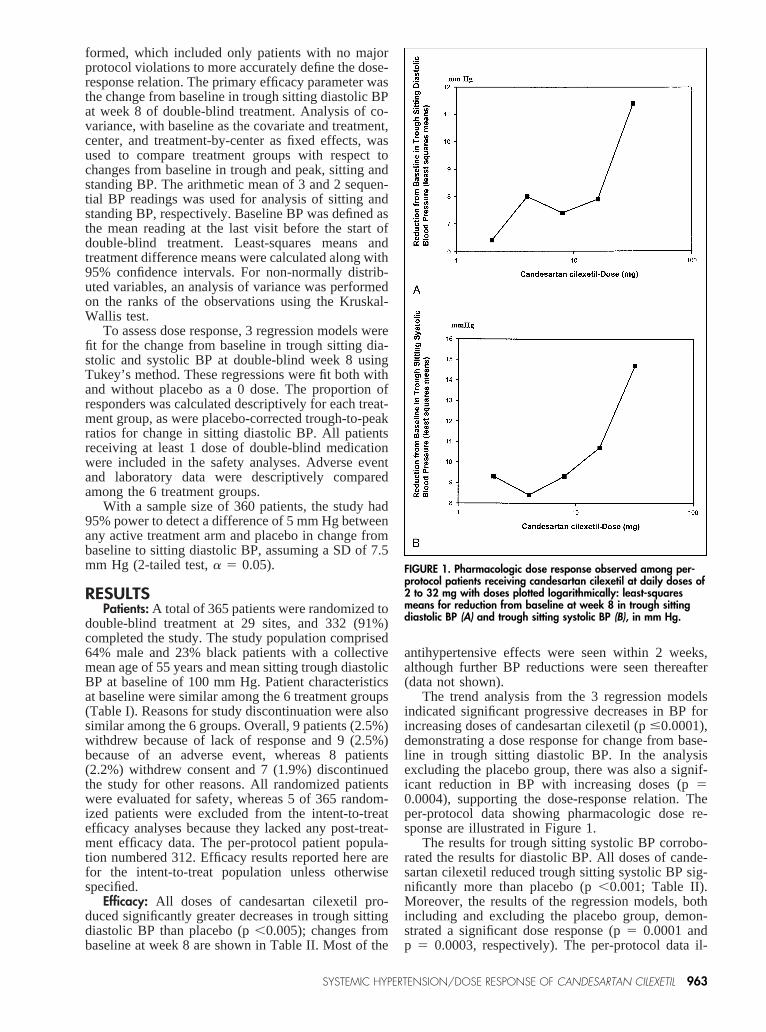
The trend analysis from the 3 regression modelsindicated significant progressive decreases in BP forincreasing doses of candesartan cilexetil (p#0.0001),demonstrating a dose response for change from base-line in trough sitting diastolic BP. In the analysisexcluding the placebo group, there was also a signif-icant reduction in BP with increasing doses (p50.0004), supporting the dose-response relation. Theper-protocol data showing pharmacologic dose re-sponse are illustrated in Figure 1.
The results for trough sitting systolic BP corrobo-rated the results for diastolic BP. All doses of cande-sartan cilexetil reduced trough sitting systolic BP sig-nificantly more than placebo (p,0.001; Table II).Moreover, the results of the regression models, bothincluding and excluding the placebo group, demon-strated a significant dose response (p5 0.0001 andp 5 0.0003, respectively). The per-protocol data il-
FIGURE 1. Pharmacologic dose response observed among per-protocol patients receiving candesartan cilexetil at daily doses of2 to 32 mg with doses plotted logarithmically: least-squaresmeans for reduction from baseline at week 8 in trough sittingdiastolic BP (A) and trough sitting systolic BP (B), in mm Hg.
SYSTEMIC HYPERTENSION/DOSE RESPONSE OF CANDESARTAN CILEXETIL 963
lustrating this dose response are displayed in Figure 1.In addition, the BP lowering effects of candesartancilexetil were consistent across demographic groups,including age, race, and sex.
Examination of all other BP parameters, includingpeak sitting diastolic, peak sitting systolic, trough andpeak standing diastolic, and trough and peak standingsystolic, indicated that, although doses of 2, 4, and 8mg provided reductions in most BP parameters, the16- and 32-mg doses of candesartan cilexetil oncedaily were consistently statistically superior to pla-cebo (all p,0.01).
The placebo-corrected trough-to-peak ratios for sit-ting diastolic BP reductions were$1 for all doses ofcandesartan cilexetil, indicating a consistent BP re-ducing effect over a 24-hour period. The percentage ofresponders, defined as patients who completed thestudy and had a sitting diastolic BP of,90 mm Hg ora reduction from baseline of$10 mm Hg at week 8,ranged from 20% in the placebo group to 54% in the16-mg group, and 64% in the 32-mg group.
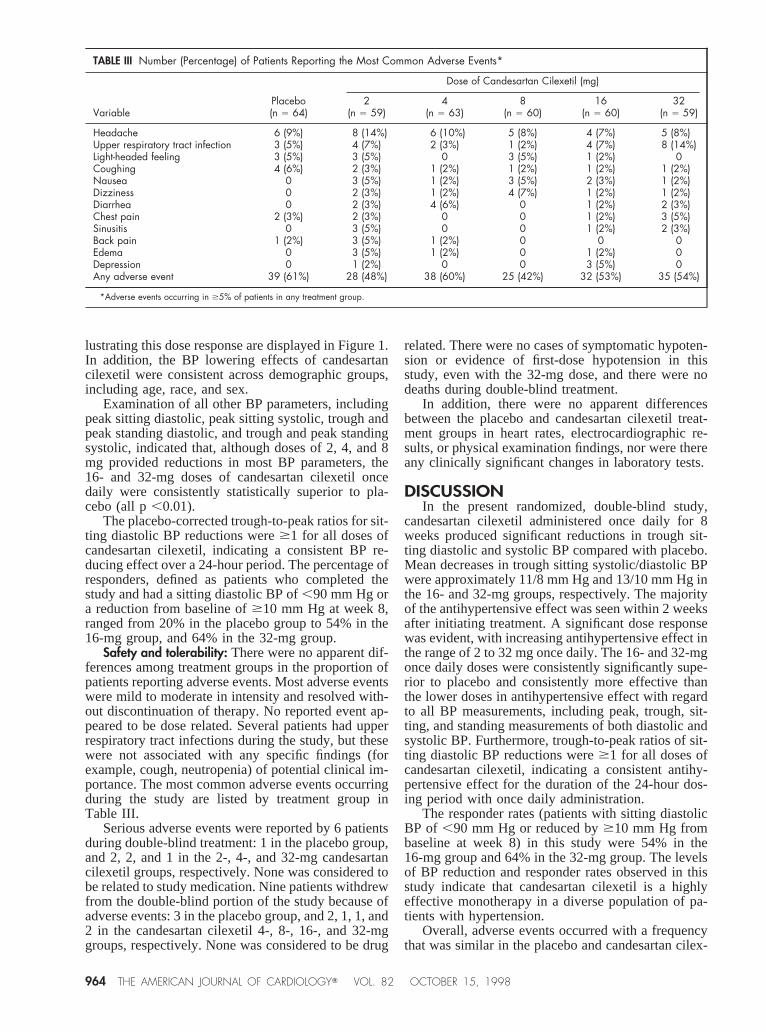
Safety and tolerability: There were no apparent dif-ferences among treatment groups in the proportion ofpatients reporting adverse events. Most adverse eventswere mild to moderate in intensity and resolved with-out discontinuation of therapy. No reported event ap-peared to be dose related. Several patients had upperrespiratory tract infections during the study, but thesewere not associated with any specific findings (forexample, cough, neutropenia) of potential clinical im-portance. The most common adverse events occurringduring the study are listed by treatment group inTable III.
Serious adverse events were reported by 6 patientsduring double-blind treatment: 1 in the placebo group,and 2, 2, and 1 in the 2-, 4-, and 32-mg candesartancilexetil groups, respectively. None was considered tobe related to study medication. Nine patients withdrewfrom the double-blind portion of the study because ofadverse events: 3 in the placebo group, and 2, 1, 1, and2 in the candesartan cilexetil 4-, 8-, 16-, and 32-mggroups, respectively. None was considered to be drug
related. There were no cases of symptomatic hypoten-sion or evidence of first-dose hypotension in thisstudy, even with the 32-mg dose, and there were nodeaths during double-blind treatment.
In addition, there were no apparent differencesbetween the placebo and candesartan cilexetil treat-ment groups in heart rates, electrocardiographic re-sults, or physical examination findings, nor were thereany clinically significant changes in laboratory tests.
DISCUSSIONIn the present randomized, double-blind study,
candesartan cilexetil administered once daily for 8weeks produced significant reductions in trough sit-ting diastolic and systolic BP compared with placebo.Mean decreases in trough sitting systolic/diastolic BPwere approximately 11/8 mm Hg and 13/10 mm Hg inthe 16- and 32-mg groups, respectively. The majorityof the antihypertensive effect was seen within 2 weeksafter initiating treatment. A significant dose responsewas evident, with increasing antihypertensive effect inthe range of 2 to 32 mg once daily. The 16- and 32-mgonce daily doses were consistently significantly supe-rior to placebo and consistently more effective thanthe lower doses in antihypertensive effect with regardto all BP measurements, including peak, trough, sit-ting, and standing measurements of both diastolic andsystolic BP. Furthermore, trough-to-peak ratios of sit-ting diastolic BP reductions were$1 for all doses ofcandesartan cilexetil, indicating a consistent antihy-pertensive effect for the duration of the 24-hour dos-ing period with once daily administration.
The responder rates (patients with sitting diastolicBP of ,90 mm Hg or reduced by$10 mm Hg frombaseline at week 8) in this study were 54% in the16-mg group and 64% in the 32-mg group. The levelsof BP reduction and responder rates observed in thisstudy indicate that candesartan cilexetil is a highlyeffective monotherapy in a diverse population of pa-tients with hypertension.
Overall, adverse events occurred with a frequencythat was similar in the placebo and candesartan cilex-
TABLE III Number (Percentage) of Patients Reporting the Most Common Adverse Events*
VariablePlacebo(n 5 64)
Dose of Candesartan Cilexetil (mg)
2(n 5 59)
4(n 5 63)
8(n 5 60)
16(n 5 60)
32(n 5 59)
Headache 6 (9%) 8 (14%) 6 (10%) 5 (8%) 4 (7%) 5 (8%)Upper respiratory tract infection 3 (5%) 4 (7%) 2 (3%) 1 (2%) 4 (7%) 8 (14%)Light-headed feeling 3 (5%) 3 (5%) 0 3 (5%) 1 (2%) 0Coughing 4 (6%) 2 (3%) 1 (2%) 1 (2%) 1 (2%) 1 (2%)Nausea 0 3 (5%) 1 (2%) 3 (5%) 2 (3%) 1 (2%)Dizziness 0 2 (3%) 1 (2%) 4 (7%) 1 (2%) 1 (2%)Diarrhea 0 2 (3%) 4 (6%) 0 1 (2%) 2 (3%)Chest pain 2 (3%) 2 (3%) 0 0 1 (2%) 3 (5%)Sinusitis 0 3 (5%) 0 0 1 (2%) 2 (3%)Back pain 1 (2%) 3 (5%) 1 (2%) 0 0 0Edema 0 3 (5%) 1 (2%) 0 1 (2%) 0Depression 0 1 (2%) 0 0 3 (5%) 0Any adverse event 39 (61%) 28 (48%) 38 (60%) 25 (42%) 32 (53%) 35 (54%)
*Adverse events occurring in $5% of patients in any treatment group.
964 THE AMERICAN JOURNAL OF CARDIOLOGYT VOL. 82 OCTOBER 15, 1998

etil treatment groups. Treatment discontinuation rateswere low and similar in candesartan cilexetil- andplacebo-treated patients. Physical, laboratory, andelectrocardiographic findings were also similar in bothgroups.
In conclusion, candesartan cilexetil is an effectiveonce daily antihypertensive agent with dose-relatedefficacy and a tolerability and safety profile similar toplacebo.
Acknowledgment: We gratefully acknowledge theassistance of J. Denise Hardison, MS, with statisticalanalyses, James Hoffman, MD, Joyce Weiner, PhD,MBA, and Deborah Brangman, MBA, with manu-script preparation, and the diligent efforts of the studycoordinators at the investigative sites.
APPENDIXCandesartan Cilexetil Study Investigators: Richard Albery, MD,
Lovelace Scientific Resources, Phoenix, AZ; John Bagdade, MD, Peace HealthMedical Group/Oregon Research Group, Eugene, OR; Grace Bialy, MD,UMDNJ-Robert Wood Johnson Medical School, New Brunswick, NJ; PaulBresnan, MD, Watson Clinic, Lakeland, FL; Albert Carr, MD, SoutheasternClinical Research and Management, Inc., Augusta, GA; Youssef Chami, MD,Westside VAMC, Chicago, IL; Arlene Chapman, MD, Colorado PreventionCenter, Denver, CO; William Cushman, MD, Memphis VAMC, Memphis, TN;Timothy C. Fagan, MD, University of Arizona, Tucson, AZ; Robert Fiddes, MD,Southern California Research Institute, Whittier, CA; Cynthia Gaboury, MD,Clinical Research Group of Oregon, Portland, OR; Larry Gilderman, DO, Uni-versity Clinical Research Associates, Inc., Pembroke Pines, FL; Ronald Gove,MD, Jersey Research Foundation, Inc., Pleasantville, NJ; Isaac Hammond, MD,PhD, Tulane University Medical Center, New Orleans, LA/American ResearchAssociates, Rockville, MD; Robert Holloway, MD, InSite Clinical Trials, At-lanta, GA; Joseph Izzo, MD, Millard Fillmore Hospital, Buffalo, NY; KirkJacobson, MD, Peace Health Medical Group/Oregon Research Group, Eugene,
OR; Kenneth C. Lasseter, MD, Clinical Pharmacology Associates, Miami, FL;Alan Marcus, MD, Southern California Research Center/InSite Clinical Trials,Mission Viejo, CA; Howard B. Miller, MD, Advanced Research Management,L.P., Seattle, WA; Mark Okusa, MD, University of Virginia, Charlottesville, VA;Suzanne Oparil, MD, University of Alabama at Birmingham, Birmingham, AL;Max Reif, MD, University of Cincinnati Medical Center, Cincinnati, OH; JeffreyRosen, MD, Clinical Research of South Florida, Coral Gables, FL; JacksonRhudy, MD, Clinical Research Advantage, Salt Lake City, UT; ArvindkumarShah, MD, St. Alexis Medical Center/InSite Clinical Trials, Cleveland, OH; GezaSimon, MD, Minneapolis VAMC, Minneapolis, MN; Wayne Weart, PharmD,Medical University of South Carolina, Charleston, SC; William B White, MD,University of Connecticut Health Center, Farmington, CT; Paul Ziajka, MD,PhD, Florida Lipid Association, Orlando, FL.
1. Goodfriend TL, Elliott ME, Catt KJ. Angiotensin receptors and their antago-nists.N Engl J Med1996;334:1649–1654.2. Gavras I, Gavras H. Angiotensin II—possible adverse effects on arteries, heart,brain, and kidney: experimental, clinical, and epidemiological evidence. In:Robertson JIS, Nicholls MG, eds. The Renin-Angiotensin System. Vol. 1. Bio-chemistry/Physiology. London: Gower Medical Publishing, 1993:40.1–40.11.3. Liao Y, Husain A. The chymase-angiotensin system in humans: biochemistry,molecular biology and potential role in cardiovascular diseases.Can J Cardiol1995;11(suppl F):13F–19F.4. Shibouta Y, Inada Y, Ojima M, Wada T, Noda M, Sanada T, Kubo K, KoharaY, Naka T, Nishikawa K. Pharmacological profile of a highly potent and long-acting angiotensin II receptor antagonist, 2-ethoxy-1-[[29-(1H-tetrazol-5-yl)bi-phenyl-4-yl]methyl]-1H-benzimidazol e-7-carboxylic acid (CV-11974), and itsprodrug, (6)-1-(cyclohexyloxycarbonyloxy)-ethyl 2-ethoxy-1-[[29-(1H-tetrazol-5-yl)biphenyl-4-yl]methyl]-1H-benzimidazole-7-carboxylate (TCV-116).J Phar-macol Exp Ther1993;266:114–120.5. Nishikawa K, Naka T, Chatani F, Yoshimura Y. Candesartan cilexetil: areview of its preclinical pharmacology.J Human Hypertens1997;11(suppl 2):S9–S18.6. van Lier JJ, van Heiningen PNM, Sunzel M. Absorption, metabolism andexcretion of14C-candesartan and14C-candesartan cilexetil in healthy volunteers.J Human Hypertens1997;11(suppl 2):S27–S28.7. Elmfeldt D, George M, Hu¨bner R, Olofsson B. Candesartan cilexetil, a newgeneration angiotensin II antagonist, provides dose dependent antihypertensiveeffect.J Human Hypertens1997;11(suppl 2):S49–S53.
SYSTEMIC HYPERTENSION/DOSE RESPONSE OF CANDESARTAN CILEXETIL 965