effect of sesquioxide coatings on surface charge of standard mineral and soil samples1
TRANSCRIPT

Effect of Sesquioxide Coatings on Surface Charge of Standard Mineral and Soil Samples1
W. H. HENDERSHOT AND L. M. LAVKULiCH2
ABSTRACTIn nature, Fe and Al hydrous oxides often form the interface be-
tween crystalline soil minerals and the soil solution. To understandbetter the effect of these coatings on surface charge properties, stan-dard minerals and cleaned C horizon samples were coated with eitherFe or Al hydrous oxide and the point(s) of zero salt effect (PZSE)were measured by potentiometric titration. Five standard mineralsamples (including illite, muscovite, kaolinite, microcline, and quartz)and three C horizon samples of mixed mineralogy were artificiallycoated with Fe and Al hydrous oxide material by precipitation ofFeClj or A1C13 solutions with the mineral and soil samples. Thesamples were then washed free of Cl~ and aged. Uncoated samplesand samples of pure sesquioxide material were prepared using sim-ilar procedures. Depending on the nature of the underlying surface,the addition of sesquioxide coatings to the samples changed thesurface charge properties in different ways. In every case the buff-ering capacity of the samples was increased by the addition of thesesquioxide coatings. The uncoated samples that had relatively highbuffering capacities had their surface charge properties changed moreby the Al- than the Fe-coating material. For example, the illite,muscovite, Memekay C horizon, and Marblehill C horizon all re-sponded to the Al-coating with a distinct shift of the PZSE to higherpH. The less reactive samples (kaolinite, microcline, quartz, and theCox Bay C horizon) responded little to the Al coatings but showeda strong response to the Fe coatings. The results indicate that in asystem containing both pH-dependent and permanent exchange sitesthere is no effective means of separating the two components. Belowthe point of zero net charge (PZNC) of the coating material, themeasured cation exchange capacity (CEC) will be decreased to apoint below the permanent CEC by the coatings, whereas at pHvalues above this point the measured CEC will be increased. ThePZSE of the pH-dependent charge material cannot be measuredseparately by potentiometric titration; this method measures thePZSE of the samples as a whole. At any pH the CEC measuredwill be the result of a complex interaction between the two types ofexchange sites.
Additional Index Words: point of zero salt effect, point of zero netcharge, zero point of charge, pH-dependent cation and anion ex-change.
Hendershot, W.H., and L.M. Lavkulich. 1983. Effect of sesquioxidecoatings on surface charge of standard mineral and soil samples.Soil Sci. Soc. Am. J. 47:1252-1260.
IN SOILS, MINERAL GRAINS are usually coated to agreater or lesser extent by the products of pedo-
genesis. Often these coatings consist of amorphousmaterials released from primary minerals by weath-ering or from the deposition of translocated material.In the presence of these coatings, the surface chargegenerated at the interface between the solid and liquidphases will depend not only on the charge of the min-eral grains themselves but also on the charge devel-oped by the amorphous coating materials. Thereforethe relationship between the charge developed by the
1 Contribution from the D6p. de geographic, Univ. de Montreal,C.P. 6128, Succursale A, Montreal, QueT H3C 3J7. Received 2 Feb.1983. Approved 27 July 1983.2 Chercheur adjoint, Dep. de geographic, Uiiiversite de Montreal,and Professor, Dep. of Soil Science, Unh. of British Columbia,respectively.
mineral grains, especially by clay minerals possessingpermanent negative charge, and the pH-dependentcharge developed largely by the amorphous coatingsdeserves further investigation.
Despite the large body of research findings to thecontrary, it is often stated that the pH-dependent orvariable charge in soils is due almost exclusively tothe presence of amorphous or crystalline metal oxidesand as a corollary that most temperate region soils,with the exception of the spodosols and andosols, havevirtually no pH-dependent charge. Early work on thissubject by Schofield (1949) and later work by Sumner(1963) led to the general conclusion that soils consistof two phases carrying different kinds of charge: a claymineral phase, with mainly permanent negative charge,and an iron and aluminum oxide phase, with pH-de-pendent charge. They recognized, however, that crys-talline minerals possess some pH-dependent charge inaddition to permanent negative charge and that theiron and aluminum oxide phase simply adds to thatpreexisting pH-dependent charge.
Admittedly, most of the pH-dependent exchange ca-pacity of soils does appear to be due to some metaloxide or hydroxide phase, and most of the permanentcharge is due to clay minerals, as indicated by thepresence of net negative charge even at low pH.
Early work on pure kaolinite indicated the impor-tance of both permanent and pH-dependent charge(Sumner, 1963). More recent work has shown that evenpure clay minerals such as illite or smectite possesspH-dependent charge when measured directly by cat-ion and anion adsorption at different pH values (Ren-gasamy and Oades, 1977; Hendershot, 1980).
Many methods of measuring the electric chargescarried by soil particles exist. One of these, potentio-metric titration, has been used in the study of surfacecharge chemistry of metal oxides and other com-pounds (Parks, 1967). By this method, a series of acid-base titration curves is plotted for a given material atdifferent electrolyte concentrations (pH vs. amount ofacid or base adsorbed); the point of intersection of thecurves (i.e., the pH at which salt concentration has noeffect on the amount of acid or base adsorbed) hasbeen termed the zero point of charge (ZPC) and re-named the point of zero salt effect (PZSE) by Parkeret al. (1979).
Methods have been developed to determine the cat-ion and anion exchange capacities (CEC and AEC)over a range of pH values that give pH-dependentCEC and AEC curves. Another ZPC that can be iden-tified using these methods is the point of zero net charge(PZNC), defined as the pH at which the capacity ofthe surface to adsorb the index cations and index an-ions is balanced (Schofield, 1949; van Raij and Peech,1972).
The PZNC is rarely equal to the PZSE since the twomethods measure the adsorption of different ions inarriving at the zero point. If there is any selective ad-sorption of either H or OH ions or of index ions suchas Ca2+ and Cl~, the PZSE and the PZNC will be
1252

HENDERSHOT & LAVKULICH: EFFECT OF SESQUIOXIDE COATINGS ON SURFACE 1253
displaced from one another. The difference may befurther exaggerated if dissolution of structural ions re-sults in solutions containing other ions that can act aspotential determining ions in the system. Accordingto Sposito (1981), the PZSE and PZNC will be equalonly when the amount of H+ adsorbed on exchangesites at a given pH (excluding H+ consumed in otherreactions) is equal to the amount of cation adsorbedand the amount of OH~ adsorbed is equal to theamount of anion adsorbed. This is, of course, a con-dition difficult to fulfill; Sposito (1981) suggests thatit will occur only when hydration energies for the twocompeting cations and the two competing anions arethe same.
For reasons that the PZSE is not comparable to thePZNC, Parker et al. (1979) have questioned the useof potentiometric titration and the resulting PZSE asa valid measure of the surface charge characteristics.Admittedly, the two values are seldom identical; how-ever, there is no theoretical basis for concluding thatone is correct and the other is not. Quite simply, theydo not measure the zero point of adsorption of thesame ions, but they are equally valid measurementsof surface charge.
In the present experiment we evaluate the effect ofsesquipxide coatings on the PZSE using the potenti-ometric titration method. The coatings on soil parti-cles are often composed of iron and aluminum ma-terial that tends to accumulate in B horizons; in naturethis material is not pure but contains varying amountsof silica, base elements, and organic material. For thepurposes of this experiment, the chemistry of the sys-tem was simplified by using standard mineral samplesand cleaned samples of C horizon material artificiallycoated with pure iron or aluminum hydrous oxides.
MATERIALS AND METHODSSample Preparation
Five standard mineral samples and three C horizon sam-ples (Table 1) were ground with an automatic mortar andpestle to pass through a 0.25-mm sieve; the C horizon sam-ples were extracted with three treatments of NaOCl to re-move organic matter (Lavkulich and Wiens, 1970); and alleight were washed six times with O.lAfHCl to remove easilysoluble Fe and Al surface coatings. The coated samples wereproduced by taking subsamples of this cleaned material andprecipitating Fe and Al hydrous oxide material with themas follows: 30-g samples were placed in a 250-mL poly-propylene beaker with 100 mL of Q.IM HC1, and enoughFeCl3 or A1C13 solution was added to produce approximately4% Fe(OH)3 or A1(OH)3 by weight. The samples were thentitrated slowly with \M NaOH to pH 7.0, and this pH was
Table 1—Identification and source/locationfor the eight samples.
Sample Identification Source/location
1234567
8
Illite(APIno.35)MuscoviteKaolinite (API no. 5)MicroclineQuartz (Rose)Memekay C horizon (Day et al., 1959)Marblehill C horizon (Luttmerding
and Sprout, 1967)Cox Bay, Site 1, C horizon
(Singleton, 1978)
Ward's ScientificWard's ScientificWard's ScientificWard's ScientificWard's ScientificCampbell River, B.C.
Agassiz, B.C.
Tofino, B.C.
maintained for 1 week by retitrating daily. The samples werethen placed in dialysis tubing, which was in turn suspendedin distilled water. The distilled water was changed twice daily,and the samples were allowed to remain until the exteriorwater was free of Cl~ (AgNO3 test). The samples were thenremoved from the dialysis tubing, returned to the beakers,and oven-dried at 40°C. The material was aged by alter-nately saturating the samples with distilled water and oven-drying them (40°C) an additional nine times. Uncoatedsamples consisting of the cleaned material were prepared inan identical manner without the addition of FeCl3 or A1C13.Pure samples of Fe and Al hydrous oxide materials wereprepared by similar procedures.
Characterization of SamplesPotentiometric titration curves of the samples were ob-
tained using the fast-adsorption method of Blok and de Bruyn(1970) with a soil-to-solution ratio of 1:20 (g:mL); the in-different electrolyte was NaCl in concentrations of 0.001A/,0.01M, and O.IM. The titrations with 0.1A/HC1 or NaOHwere carried out with aliquots ranging from 0.1 to 0.5 mL,such that approximately 10 additions would cover the de-sired range of pH values (3-11) in either direction from thezero point of titration (ZPT).
Selected samples were analysed for cation and anion ex-change capacities (CEC and AEC) with 0.05M CaCl2 solu-tions at pH 3, 5, and 7 by the method of Fey and Le Roux(1976). This procedure eliminates the washing step followingsaturation with the index ions; instead the occluded salt con-tent is determined by weighing, with the assumption thatthe concentration of Ca and Cl in the occluded solution isthe same as in the supernatant solution. The pH of the ex-change reaction was taken as being the pH of the clear su-pernatant solution following the last CaCl2 saturation andcentrifugation.
Specific surface areas (SSA) were obtained from Ca-satu-rated samples by the ethylene-glycol-monoethyl-ether methodof Eltantawy and Arnold (1973).
Random powder mounts of the samples were analysed byx-ray diffraction to determine if there was crystallization ofthe Al and Fe coating materials. Oriented aggregate mountswere also analyzed according to the procedures of Whittig(1965).
RESULTS AND DISCUSSIONThe mineralogy of the < 2-^m fraction of the three
soil C horizons determined by x-ray diffraction anal-ysis is presented in Table 2. The mixed mineralogy ofthese samples, consisting of chlorite, illite (or a 1.0-nm mica), plagioclase, quartz, and amphibole, is rep-resentative of the mineralogy of the C horizons ofmany young, postglacial soils in western British Col-umbia, Canada.
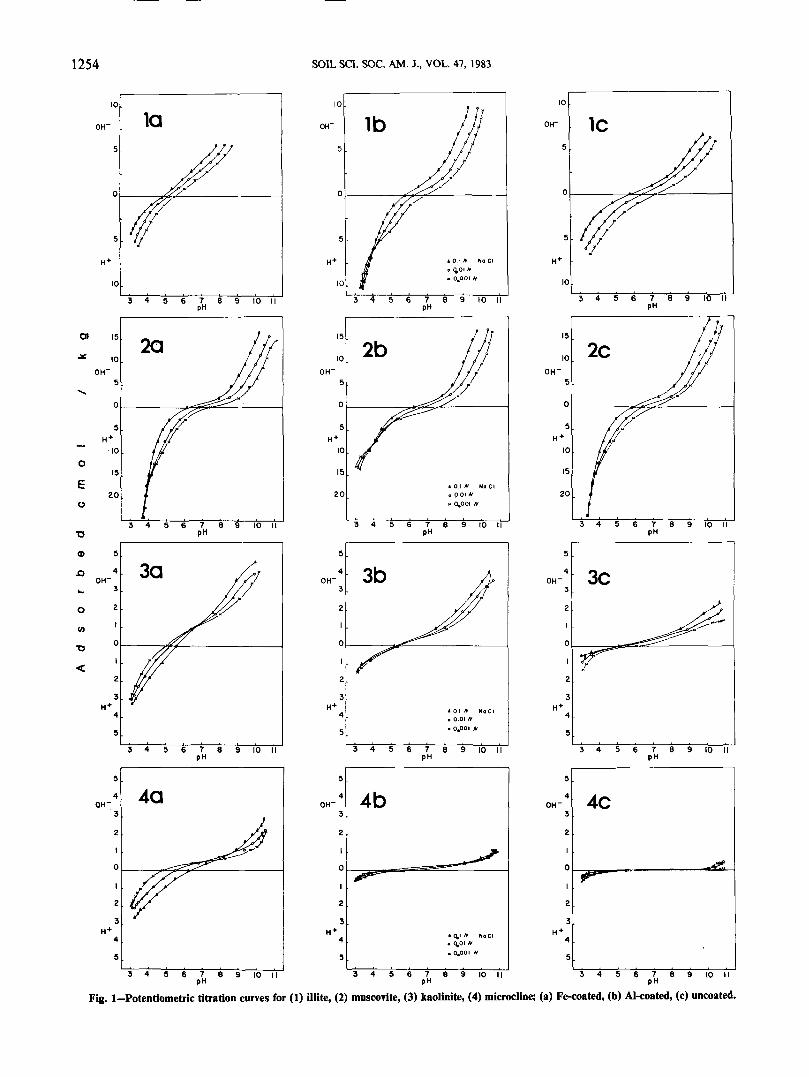
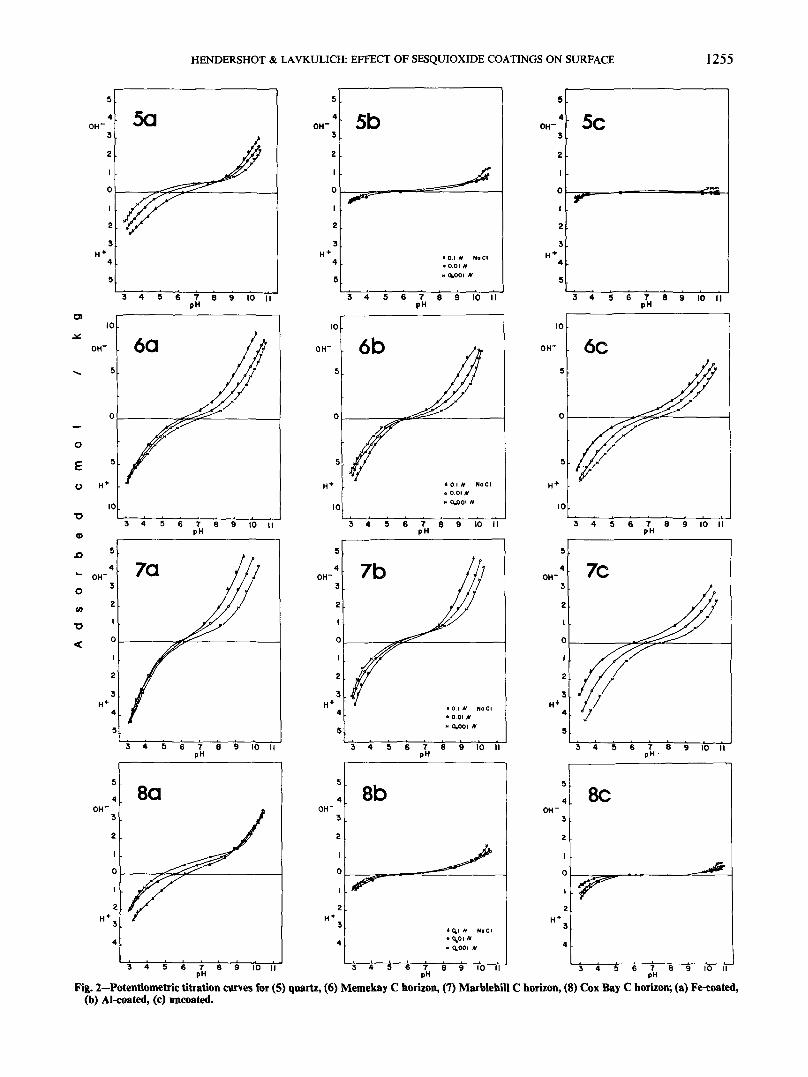
The potentiometric titration curves for the (i) Fe-coated samples, (ii) Al-coated samples, and (iii) un-coated samples are presented in Fig. 1 and 2. An ex-amination of the curves indicates that in some casesthe coatings have caused marked changes in the slopeand shape of the curves as well as changes in the pHat which the three curves intersect. This cross-over
Table 2—Dominant mineralogy of the < 2-̂ m fraction of theC horizon samples listed in order of abundance.
Sample Identification Mineralogy
6 Memekay Chlorite, quartz, plagioclase, illite, amphibole7 Marblehill Quartz, plagioclase, illite, chlorite, amphibole8 Cox Bay Quartz, plagioclase, amphibole, chlorite, mica
1254 SOIL SCI. SOC. AM. J., VOL. 47, 1983
10
OH- la
?y
3 4 5 6 7 8 9 10pH
OH
O
Eo
T>
a>
o<n
•o
2d
3 4 5 6 7 8 9 10 I IpH
3 4 5 6 7 8 9 10 I IpH
3 4 5 6 7 8 9 10
OH" ib
* O.I N No Clo 401 *- 0.001 »
3 4 5 6 7 8 9 10pH
15
10-
5
0
5H +
10
15
20
2b
i O.I N Na Clo 0.01 H- 0.001 N
3 4 5 6 7 8 9 10PH
3b
"O. I # NoCID 0.01 A/. 0.001 »
3 4 5 6 7 8 9 10pH
OH- 4b
• O,' * NoCIo q,OI N„ 0*001 N
OH~ 1C
3 4 5 6 7 8 9 10pH
OH-
2C
3 4 5 6 7 8 9 10pH
3C
3 4 5 6 7 8 9 10pH
OH- 4C
3 4 5 6 7 8 9 10 I IpH
3 4 5 6 7 8 9 10 I I
Fig. 1—Potentiometric titration curves for (1) illite, (2) muscovite, (3) kaolinite, (4) microcline; (a) Fe-coated, (b) Al-coated, (c) uncoated.
HENDERSHOT & LAVKULICH: EFFECT OF SESQUIOXIDE COATINGS ON SURFACE 1255
OH'80
3 4 5 6 7 8 9 10 I IpH
10
5b
» O.I (V NoCI« 0.01 N» 0.001 #
3 4 5 6 7 8 9 10 I IPH
H +
6b
» O.I # NaC(o 0.01 *»OJ»OI *
3 4 5 6 7 8 9 10 I Ip H
OH
7b
* O.t N N o C I• 0.01 N
» 0*OOI N
34 5 6 7 8 9 10 II
8b
* 0.1 # N o C I« Q.OI H> Q.OO i n
3 4 5 6 7 8 9 10pH
5
4OH-
3
Z
1
0
1
z3
4
5
5c
r—
3 4 5 6 7 8 9 1 0 1 1pH
OH" 6C
3 4 5 6 7 8 9 10 I IpH
7C
3 4 5 6 7 8 9 10pH •
8C
3 4 5 6 7 8 9 10 I IpH
Fig. 2—Potentiometric titiation curves for (5) quartz, (6) Memekay C horizon, (7) Marblehill C horizon, (8) Cox Bay C horizon; (a) Fe-coated,(b) Al-coated, (c) uncoated.
1256 SOIL SCI. SOC. AM. J., VOL. 47, 1983
10 II
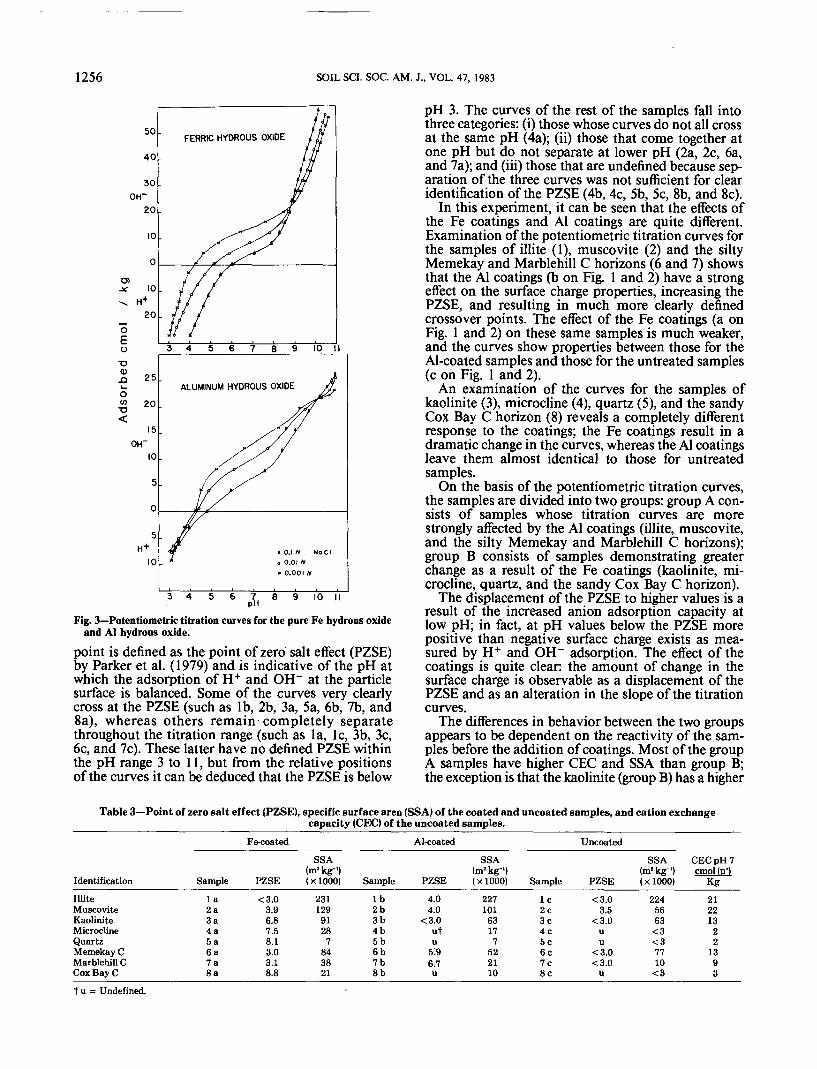
Fig. 3—Potentiometric titration curves for the pure Fe hydrous oxideand AI hydrous oxide.
point is denned as the point of zero salt effect (PZSE)by Parker et al. (1979) and is indicative of the pH atwhich the adsorption of H+ and OH~ at the particlesurface is balanced. Some of the curves very clearlycross at the PZSE (such as Ib, 2b, 3a, 5a, 6b, 7b, and8a), whereas others remain completely separatethroughout the titration range (such as la, Ic, 3b, 3c,6c, and 7c). These latter have no denned PZSE withinthe pH range 3 to 11, but from the relative positionsof the curves it can be deduced that the PZSE is below
pH 3. The curves of the rest of the samples fall intothree categories: (i) those whose curves do not all crossat the same pH (4a); (ii) those that come together atone pH but do not separate at lower pH (2a, 2c, 6a,and 7a); and (iii) those that are undefined because sep-aration of the three curves was not sufficient for clearidentification of the PZSE (4b, 4c, 5b, 5c, 8b, and 8c).
In this experiment, it can be seen that the effects ofthe Fe coatings and Al coatings are quite different.Examination of the potentiometric titration curves forthe samples of illite (1), muscovite (2) and the siltyMemekay and Marblehill C horizons (6 and 7) showsthat the Al coatings (b on Fig. 1 and 2) have a strongeffect on the surface charge properties, increasing thePZSE, and resulting in much more clearly dennedcrossover points. The effect of the Fe coatings (a onFig. 1 and 2) on these same samples is much weaker,and the curves show properties between those for theAl-coated samples and those for the untreated samples(c on Fig. 1 and 2).
An examination of the curves for the samples ofkaolinite (3), microcline (4), quartz (5), and the sandyCox Bay C horizon (8) reveals a completely differentresponse to the coatings; the Fe coatings result in adramatic change in the curves, whereas the Al coatingsleave them almost identical to those for untreatedsamples.
On the basis of the potentiometric titration curves,the samples are divided into two groups: group A con-sists of samples whose titration curves are morestrongly affected by the Al coatings (illite, muscovite,and the silty Memekay and Marblehill C horizons);group B consists of samples demonstrating greaterchange as a result of the Fe coatings (kaolinite, mi-crocline, quartz, and the sandy Cox Bay C horizon).
The displacement of the PZSE to higher values is aresult of the increased anion adsorption capacity atlow pH; in fact, at pH values below the PZSE morepositive than negative surface charge exists as mea-sured by H+ and OH~ adsorption. The effect of thecoatings is quite clear: the amount of change in thesurface charge is observable as a displacement of thePZSE and as an alteration in the slope of the titrationcurves.
The differences in behavior between the two groupsappears to be dependent on the reactivity of the sam-ples before the addition of coatings. Most of the groupA samples have higher CEC and SSA than group B;the exception is that the kaolinite (group B) has a higher
Table 3—Point of zero salt effect (PZSE), specific surface area (SSA) of the coated and uncoated samples, and cation exchangecapacity (CEC) of the uncoated samples.
Identification
IlliteMuscoviteKaoliniteMicroclineQuartzMemekay CMarblehill CCox Bay C
Sample
la2a3a4a5a6a7a8a
Fe-coated
PZSE
<3.03.96.87.58.13.03.18.8
SSA(m'kg-'l(x lOOO)
2311299128
7843821
Sample
I b2b3b4b5b6b7b8b
Al-coated
PZSE
4.04.0
<3.0utu
5:96.7u
SSA(m'kg-1)( x 1000)
22710163177
522110
Sample
Ic2c3c4c5c6c7c8c
Uncoated
PZSE
<3.03.5
<3.0uu
<3.0<3.0
u
SSA(m'kg-')( x 1000)
2245663<3<37710<3
CECpH7cmol (p'l
Kg
21221322
1393
t u = Undefined.
HENDERSHOT & LAVKULICH: EFFECT OF SESQUIOXIDE COATINGS ON SURFACE 1257
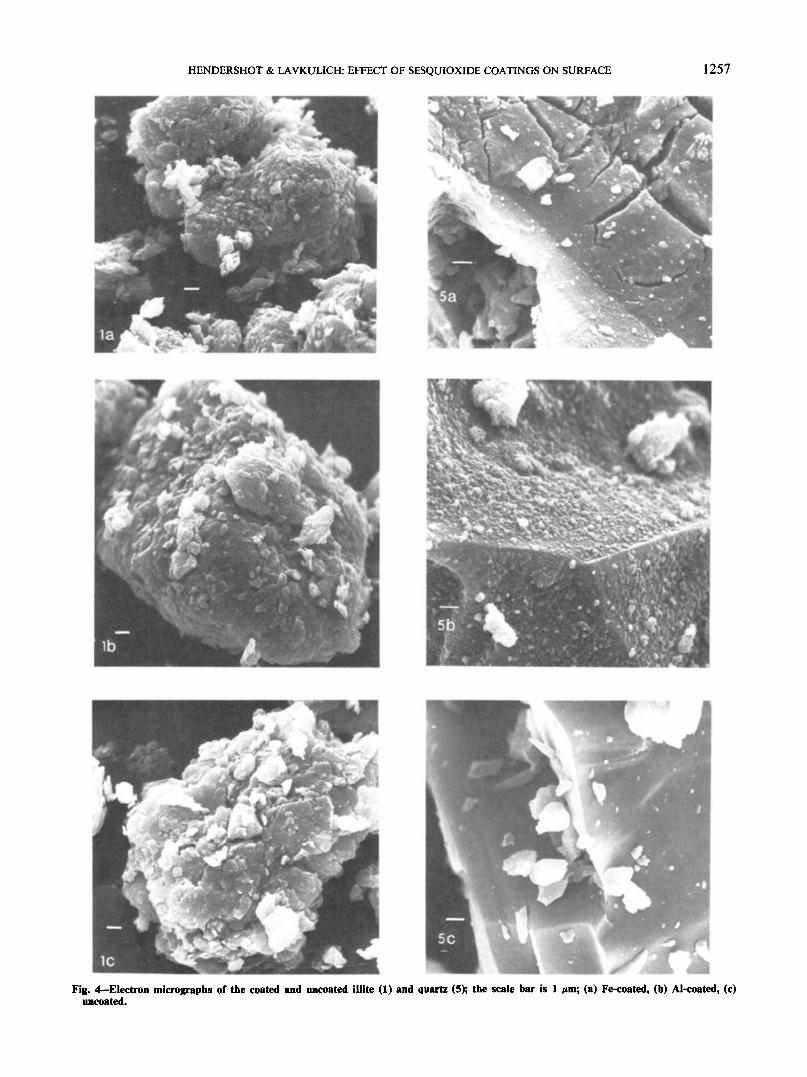
Fig. 4—Electron micrographs of the coated and uncoated illite (1) and quartz (5); the scale bar is 1 Mm; (a) Fe-coated, (b) Al-coated, (c)uncoated.

1258 SOIL SCI. SOC. AM. J., VOL. 47, 1983
CEC and SSA than the Marblehill C horizon (groupA) (Table 3). This is most clearly evidenced by thebuffering capacity of the uncoated samples; as shownby the slope of the titration curves on Fig. 1 and 2,group A is better buffered than group B.
An examination of SSA of the samples before andafter the addition of the hydrous oxides provides apartial explanation of the behavior of the differentsamples. Generally the addition of the coating mate-rial increases the SSA, although one sample, Al-coatedMemekay C, is actually lower than the uncoated sam-ple. Some of the samples show a dramatic increase inSSA as a result of the coatings, for example, the mus-covite and microcline coated with Fe or Al and theFe-coated kaolinite, Marblehill C, and Cox Bay C. Inevery case the Fe-coated samples have the same orhigher SSA values than the Al-coated samples. Thesedifferences, resulting from the interaction of the coat-ing material and the mineral grains, cannot be ex-plained satisfactorily by referring to the two groupsdenned earlier. The results suggest that there is a com-plex interaction that produces coatings with widelydifferent surface areas depending on the chemistry ofthe underlying minerals.
The potentiometric titration curves of the pure Feand Al hydrous oxide materials are presented in Fig.3. The Fe hydrous oxide, with a very clearly dennedPZSE of 8.8, has smooth, well-defined curves. In con-trast, the Al hydrous oxide with a higher PZSE of 10.2produces curves that are very irregular, although re-producible. It is also clear from the slope of the titra-tion curves that on a weight basis the pure Al materialis much less reactive than the pure Fe material. Spe-cific surface area measurements help to explain thislarge difference in reactivity: the Fe hydrous oxide hasan SSA of 531 000 m2/kg, whereas the Al hydrous ox-ide has a much lower SSA of only 49 000 m2/kg.
Scanning electron micrographs were taken of se-lected samples from groups A and B (Fig. 4). The un-coated sample of quartz has a very clean, fresh surfaceand a clearly visible fracture pattern caused by grind-ing the sample during initial preparation. When coatedwith Fe, the quartz displays a distinct layer of Fe ma-terial with the smooth underlying quartz surface vis-ible through cracks in the coating. Although the Al-coated quartz also has distinct coatings, they containwhat appear to be microcrystals, suggesting the pres-ence of gibbsite. By contrast, examination of the coat-ings on the illite aggregates does not reveal the pres-ence of microcrystals, and, in fact, the coatings areevident only as a slight rounding of the edges of theillite flakes and by the more compact structure of theaggregates.
X-ray diffraction analysis confirms that gibbsite ispresent in the pure Al hydrous oxide sample, but be-cause of the low content (4%) of this material in thecoated samples, the presence of gibbsite could not beconfirmed. In addition, x-ray diffraction provided noevidence of crystallinity of the Fe hydrous oxide inthe pure sample or in the Fe-coated samples.
It is concluded from the titration curves, the SEMphotographs, and the x-ray diffraction data that in thecase of the group B samples, the Al hydrous oxideformed microcrystalline gibbsite. This material has less
capacity to develop pH-dependent charge than theamorphous Fe material coating these same samples.For the samples in group A, since the Al material re-mained in the amorphous state, it was more reactivethan the Fe-coating material and therefore had a morepronounced effect on the surface charge properties.
It has been suggested that potentiometric titrationmeasures the PZSE of the pH-dependent charge phaserather than the PZSE of the sample as a whole (Ueharaand Gillman, 1980; Gillman and Uehara, 1980; Tes-sens and Zauyah, 1982). If this hypothesis were cor-rect, all of the Fe-coated samples should have had aPZSE near 8.8, and all of the Al-coated samples a PZSEnear 10.2, which is not the case. In the present exper-iment, particularly in those cases where the materialhad high reactivity, the coatings were the source of thegreater part of the pH-dependent charge. It thereforeappears that there is no basis, either experimental ortheoretical, for the assumption that the PZSE, as de-termined by potentiometric titration, is a measure ofanything other than the properties of the system as awhole (Parks, 1967; Sposito, 1981).
The variation in surface charge as a function of pHcan also be measured by the adsorption of anions andcations at different pH values. In this experiment, CECand AEC were measured by the adsorption of Ca2+
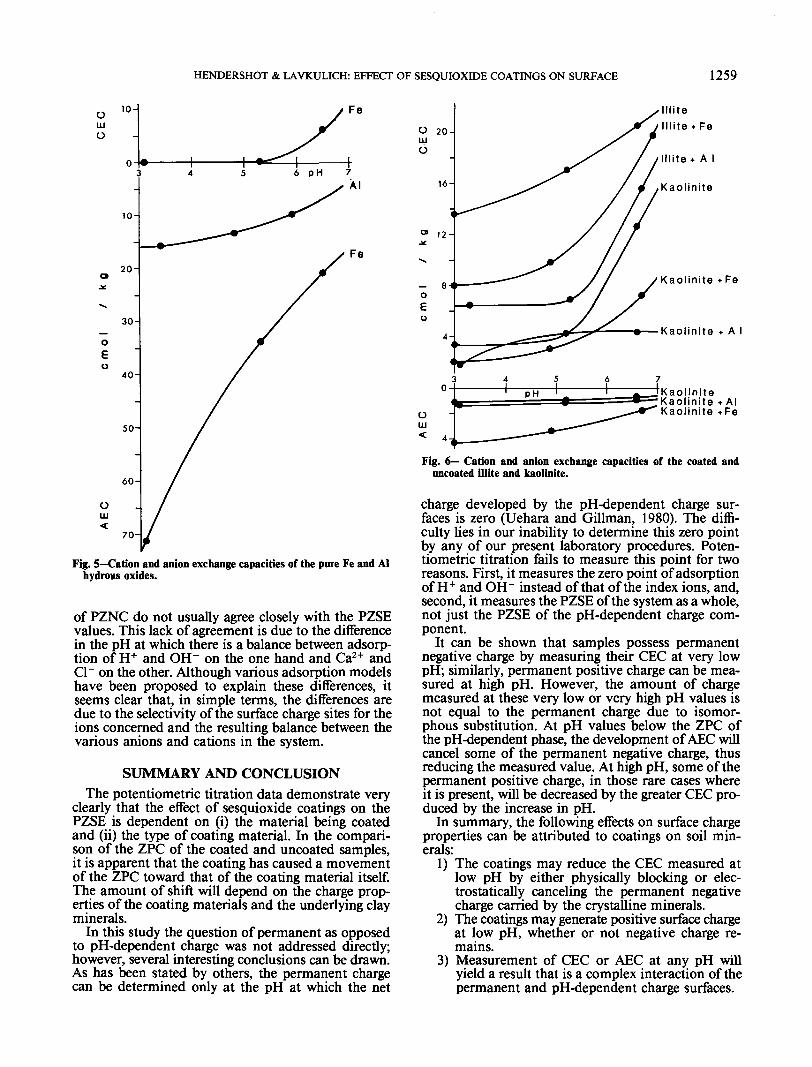
and Cl~ at pH values of approximately 3, 5, and 7.The pH-dependent CEC and AEC curves for the pureFe and Al hydrous oxides are presented in Fig. 5. Itcan be seen that reducing the pH of the solution usedto saturate the exchange complex causes an increasein the positive surface charge or AEC, and in keepingwith the results of the potentiometric titrations, thepure Fe material is more reactive than the pure Almaterial. The results also indicate that whereas the Fesample begins to develop CEC above pH 5, the Alsample possesses only AEC, even at a pH near 6. Inthis case the PZNC, defined as the pH at which theadsorption of the index anion and cation is balanced,is not obtained, but would be above pH 7 for both theAl and Fe hydrous oxides.
The pH-dependent CEC and AEC of the coated anduncoated illite and kaolinite were also measured (Fig.6). Both of these samples show significant amounts ofpH-dependent negative charge in the uncoated statein keeping with the results of previous research (Bow-den et al., 1977; Rengasamy and Oades, 1977; Hen-dershot, 1980). The PZNC values for both uncoatedsamples are < 3, as are the PZSE values. In all cases,the addition of coatings has decreased the CEC at pHvalues below 7, and in the case of illite, the coatingshave resulted in steeper slopes to CEC curves abovepH 5. Coatings produce a different result for kaolinite,in that the slope of the CEC curves is decreased andthere is an increase in AEC. Even with the coatings(even at pH 3), the illite samples have no measurableAEC. Rengasamy and Oades (1977) had similar re-sults for Fe- and Al-coated clay minerals, although intheir experiment, at higher Fe and Al contents moreAEC was developed at low pH.
In the present experiment the PZNC values are <3 for all of the samples except the Fe-coated kaolinite,which had a PZNC of 5.0 compared with its PZSEvalue of 6.8. As has been mentioned earlier, the values
HENDERSHOT & LAVKULICH: EFFECT OF SESQUIOXIDE COATINGS ON SURFACE 1259
oLUO
10-
10-
20-
30-
oEo
Oai
40-
50-
60-
70-
Fig. 5—Cation and anion exchange capacities of the pure Fe and AIhydrous oxides.
of PZNC do not usually agree closely with the PZSEvalues. This lack of agreement is due to the differencein the pH at which there is a balance between adsorp-tion of H+ and OH~ on the one hand and Ca2+ andCl~ on the other. Although various adsorption modelshave been proposed to explain these differences, itseems clear that, in simple terms, the differences aredue to the selectivity of the surface charge sites for theions concerned and the resulting balance between thevarious anions and cations in the system.
SUMMARY AND CONCLUSIONThe potentiometric titration data demonstrate very
clearly that the effect of sesquioxide coatings on thePZSE is dependent on (i) the material being coatedand (ii) the type of coating material. In the compari-son of the ZPC of the coated and uncoated samples,it is apparent that the coating has caused a movementof the ZPC toward that of the coating material itself.The amount of shift will depend on the charge prop-erties of the coating materials and the underlying clayminerals.
In this study the question of permanent as opposedto pH-dependent charge was not addressed directly;however, several interesting conclusions can be drawn.As has been stated by others, the permanent chargecan be determined only at the pH at which the net
Kaol ini te + Fe
Kaolinite + A I
Kaol ini teKaol in i te * AIKaolinite »Fe
Fig. 6— Cation and anion exchange capacities of the coated anduncoated illite and kaolinite.
charge developed by the pH-dependent charge sur-faces is zero (Uehara and Gillman, 1980). The diffi-culty lies in our inability to determine this zero pointby any of pur present laboratory procedures. Poten-tiometric titration fails to measure this point for tworeasons. First, it measures the zero point of adsorptionof H+ and OH~ instead of that of the index ions, and,second, it measures the PZSE of the system as a whole,not just the PZSE of the pH-dependent charge com-ponent.
It can be shown that samples possess permanentnegative charge by measuring their CEC at very lowpH; similarly, permanent positive charge can be mea-sured at high pH. However, the amount of chargemeasured at these very low or very high pH values isnot equal to the permanent charge due to isomor-phous substitution. At pH values below the ZPC ofthe pH-dependent phase, the development of AEC willcancel some of the permanent negative charge, thusreducing the measured value. At high pH, some of thepermanent positive charge, in those rare cases whereit is present, will be decreased by the greater CEC pro-duced by the increase in pH.
In summary, the following effects on surface chargeproperties can be attributed to coatings on soil min-erals:
1) The coatings may reduce the CEC measured atlow pH by either physically blocking or elec-trostatically canceling the permanent negativecharge carried by the crystalline minerals.
2) The coatings may generate positive surface chargeat low pH, whether or not negative charge re-mains.
3) Measurement of CEC or AEC at any pH willyield a result that is a complex interaction of thepermanent and pH-dependent charge surfaces.

1260 SOIL SCI. SOC. AM. J., VOL. 47, 1983
4) Since even standard clay minerals possess bothpH-dependent and permanent charge, it is im-possible to assign the permanent charge to theclay minerals and the pH-dependent charge to acrystalline or amorphous metal oxide material,whether this last acts as a coating or as a separatephase.
5) Sesquioxide coatings add to the pH-dependentcharge of soil samples, resulting in a shift of theZPC of the sample as a whole toward the ZPCof the coating material.