effect of co-inheritance of β-thalassemia and hemochromatosis mutations on iron overload
TRANSCRIPT

Hemoglobin, 36(1):85–92, (2012)Copyright © Informa Healthcare USA, Inc.ISSN: 0363-0269 print/1532-432X onlineDOI: 10.3109/03630269.2011.637148
SHORT COMMUNICATION
EFFECT OF CO-INHERITANCE OF β-THALASSEMIA AND
HEMOCHROMATOSIS MUTATIONS ON IRON OVERLOAD
Herminio López-Escribano,1 Joana F. Ferragut,2 Maria M. Parera,1
Pilar Guix,1 José A. Castro,2 M. Misericòrdia Ramon,2 and
Antònia Picornell2
1Servicio de Análisis Clínicos, Hospital Universitario Son Dureta, Palma de Mallorca, Illes Balears,España2Institut Universitari d’Investigació en Ciències de la Salut (IUNICS) i Laboratori de Genètica,Departament de Biologia, Universitat de les Illes Balears, Palma de Mallorca, Illes Balears, España
� Co-inheritance of mutations in the HFE gene underlying hereditary hemocromatosis (HH) mayplay a role in the variability of iron status in patients with β-thalassemia (β-thal) minor. Differentstudies have yielded conflicting results: some suggest iron overload might arise from the interactionof the β-thal trait with homozygosity or even heterozygosity for HFE mutations and others that itwas unrelated to the HFE genotype. Because of the high frequency of HFE mutations in the BalearicIslands, where the β-thal trait is also moderately common, it is of interest to evaluate the effect of theco-inheritance of mutations in both genes on the severity of iron loading. A retrospective analysis of142 individuals heterozygous for β-thal was performed to investigate the effect of HFE mutationson iron status of these patients. No significant differences were detected between β-thal carriers withand without HFE mutations. These results suggest that in the Balearic population the β-thal traitdoes not tend to be aggravated by the co-inheritance of HFE mutations.
Keywords β-Thalassemia (β-thal), Hereditary hemochromatosis (HH), HFE mutations,C282Y, H63D, Iron overload
Many diseases have been related to defects in iron metabolism leadingto complications of iron overload that is associated with different chronicdegenerative disorders. Thalassemia syndromes represent the main cause of
Received 21 March 2011; Accepted 11 June 2011.Address correspondence to Dr. Antònia Picornell, Laboratori de Genètica, Departament de Biolo-
gia, Universitat de les Illes Balears, Cra. Valldemossa, km 7.5, 07122-Palma de Mallorca, Illes Balears,España; Tel.: +34-971-172050; Fax: +34-971-173184; E-mail: [email protected]
85
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.

86 H. López-Escribano et al.
iron overload in Mediterranean countries. These patients are known to havepathologically increased iron stores due to greater iron absorption, regulartransfusions, anemia and increased erythropoiesis. However, only a minorityof individuals heterozygous for β-thalassemia (β-thal) develops iron over-load, despite excess iron absorption induced by ineffective erythropoiesis,indicating other hereditary and environmental variables are involved in ironloading (1,2). Among genetic factors, the most common cause of iron over-load is the presence of two main mutations (C282Y and H63D) in the HFEgene, mainly responsible for hereditary hemochromatosis (HH). Althoughthe role of the H63D mutation is not as clear as the role of the C282Y muta-tion, different studies claim H63D confers an increased risk of iron overloadand, therefore, genetic susceptibility to developing HH or aggravating otherdiseases (3–5). Therefore, the presence of mutations in the HFE gene mayadversely affect iron loading in β-thal carriers and explain the variability iniron overload observed in these patients.
The role of the HFE mutation on iron status in β-thal trait is controver-sial. The homozygous state for the C282Y mutation has been described asan aggravating factor (6,7); yet the consequences of co-inheritance of β-thaland H63D homozygosity have not been clearly elucidated, some authorsclaiming this genotype shows no effect on iron loading (8,9), while othersclaim it could induce iron overload in β-thal minor (6,10,11).
Because of the high prevalence of HFE mutations (especially H63D)in some Mediterranean countries, where the β-thal trait is also common,co-segregation of both inherited conditions in this region should not beconsidered exceptional. It is important for possible therapeutical implica-tions to investigate their possible synergistic effect on iron absorption. Thus,in this study we aimed to investigate the effect of co-inheritance of HFE andβ-thal in the Balearic Islands (where H63D carrier frequency is >31%) (12),to establish whether a fortuitous association is common and if it really affectsthe severity of iron overload.
This study was performed on 142 anonymous, unrelated β-thal carriersfrom the Balearic Islands, detected during a pilot β-thal screening. Theywere diagnosed by hematological studies based on a full blood count andquantification of Hb A2 and Hb F by high performance liquid chromatog-raphy (HPLC), iron deficiency being excluded in all cases. An automatedfull blood count (ADVIA 120 Hematology System; Siemens HealthcareDiagnostics, Deerfield, IL, USA) was performed on all samples. Microcy-tosis indicator was the mean corpuscular volume (MCV) (<78 fL) andfor hypochromia, the mean corpuscular hemoglobin (Hb) (MCH) (<27pg). To discard microcytosis by ferropenic anemia, we evaluated the ironstatus in microcytosis samples in a Hitachi Modular 917 analyzer (RocheDiagnostic, Mannheim, Germany). The iron deficiency was defined as atransferrin saturation index (IST) <15% or ferritin levels <14 ng/mL. Hb
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.

HFE Mutations in β-Thalassemia Minor 87
A2 and Hb F levels were performed by HPLC (HA-8160, Menarini Diagnos-tic, Florence, Italy).The C282Y and H63D mutations in the HFE gene wereassessed on genomic DNA from peripheral leukocytes (QIAamp Blood kit;Qiagen, Crawley, West Sussex, UK), using the standard polymerase chainreaction-restriction fragment length polymorphism (PCR-RFLP) method.
The β-thal genotypes were characterized by the LightCycler PCRmethod. Amplification and melting analysis was carried out with the Light-Cycler instrument (Roche Diagnostic). This screening was performed forthe following mutations, β0 codon 39 (C>T), β0 codon 37 (G>A), β+ IVS-I-110 (G>A), β0 codon 6 (–A), β0 IVS-I-1 (G>A) and β+ IVS-I-6 (T>C),as these are the most frequent β-globin mutations in the Mediterraneanarea. In those individuals who did not carry any of the former mutations,the β-globin gene was sequenced with the primers described in Clark andThein (13) by using the Big Dye® Terminator Cycle Sequencing kit v.3.1 (Applied Biosystems, Foster City, CA, USA) and an ABI PRISM® 3100Genetic Analyzer (Applied Biosystems).
Twenty-eight of the 142 β-thal carriers (19.7%) presented with β+ muta-tions, mostly IVS-I-110 (14.8%) and IVS-I-6 (3.5%). Other β+ gene defectswere found in frequencies <1% [IVS-II-745 (C>G) and IVS-II-705 (T>G)].The β0 mutations were found in 80.3% of the β-thal minor patients. Themost common mutation, codon 39, accounted for 69.0% of the β0 alle-les. The IVS-I-1, codons 8/9 (+G), codon 6 and –32 (C>T) mutationsaccounted for 9.8%. The codon 44 (–C) and codon 37 (G>A) mutationswere only detected in one individual, respectively.
Of the β-thal carriers, 39% had mutations in the HFE gene: 49 H63Dheterozygotes, four H63D homozygotes, one subject was heterozygous forC282Y and one was a compound heterozygote for the C282Y/H63D muta-tions. The allelic frequencies of C282Y and H63D were 0.007 ± 0.005 and0.204 ± 0.024, respectively. As expected, no significant differences in allelicfrequencies were found between β-thal carriers and the general Balearicpopulation (12).
Concerning iron status, there is a statistically significant difference(p <0.01) between ferritin values detected in β-thal carriers and controls(Table 1, top), although a considerable inter patient variation was observed.In order to evaluate the influence of different β-globin mutations, the β-thal carriers were separated according to the type of mutation (β0 and β+)(Table 1, bottom). There was no significant difference in the iron profilebetween β0 and β+ minor thalassemic subjects, except in transferrin satura-tion in women; although, as expected, those with β0 had more iron loadingthan individuals with β+.
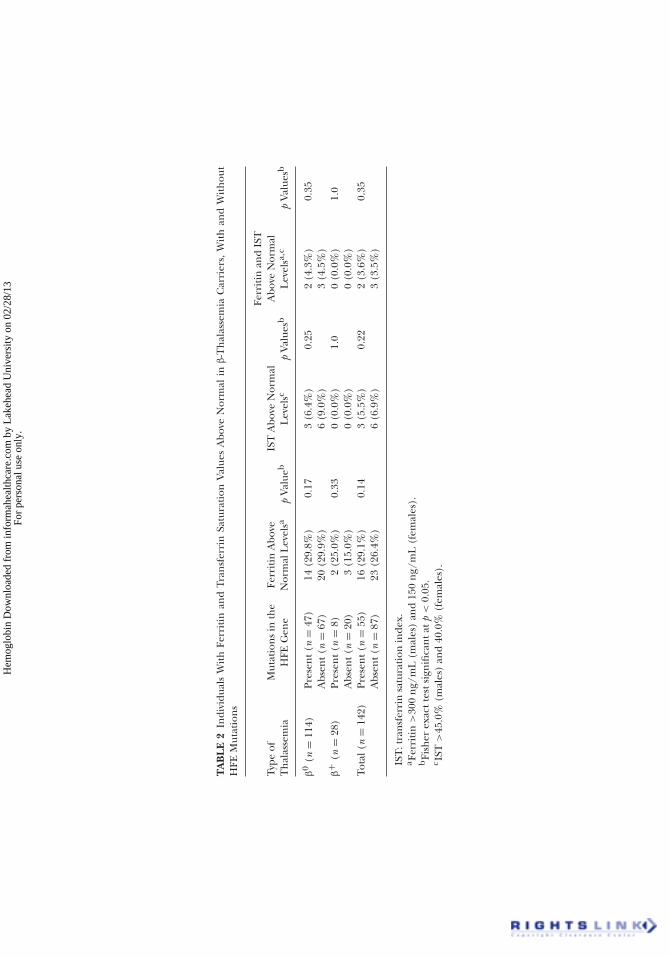
However, no statistical differences were detected between β-thal carriersubjects with and without HFE mutations (Figure 1). When data were ana-lyzed separately according to the type of mutation (β0 and β+) (Table 2),
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.

TA
BL
E1
Ferr
itin
and
Tran
sfer
rin
Satu
rati
onVa
lues
inth
eC
ontr
olG
roup
and
β-T
hal
asse
mia
Car
rier
s(t
op:t
otal
;bot
tom
:sep
arat
edac
cord
ing
toβ
0
and
β+
mut
atio
ns)
Ferr
itin
(ng/
mL
)Tr
ansf
erri
nSa
tura
tion
(%)
Gen
der
Gro
upn
Mea
n±
SDR
ange
pVa
luea
nM
ean
±SD
Ran
gep
Valu
ea
Mal
esC
ontr
ol18
616
8±
112
24–4
81<
0.01
939
25.7
±10
.110
.0–5
8.8
0.03
β-th
alca
rrie
rs57
273
±24
912
–138
357
29.9
±11
.115
.8–8
1.7
Fem
ales
Con
trol
222
82±
6116
–306
<0.
0171
424
.0±
8.9
10.0
–49.
10.
07β-th
alca
rrie
rs85
118
±13
48–
683
8525
.8±
9.4
15.6
–62.
2M
ales
β0
4927
7±
251
12–1
382
0.77
4930
.1±
11.9
15.8
–81.
70.
66β+
824
8±
248
82–8
258
28.3
±5.
123
.5–3
6.4
Fem
ales
β0
6512
9±
146
8–68
30.
1665
27.1
±10
.115
.6–6
2.2
0.02
β+
2080
±71
20–3
1320
21.6
±5.
416
.0–3
5.4
Ferr
itin
refe
ren
ceva
lues
:30
–300
ng/
ml
(mal
es)
and
15–1
50n
g/m
L(f
emal
es);
tran
sfer
rin
satu
rati
onre
fere
nce
valu
es:
15.0
–45.
0%(m
ales
)an
d15
.0–4
0.0%
fem
ales
;SD
:Sta
nda
rdde
viat
ion
.a S
tude
ntt
-test
orM
ann
-Wh
itn
eyte
stsi
gnifi
can
tatp
<0.
05.
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.

HFE Mutations in β-Thalassemia Minor 89
N= 34 23
N HFE HFE mutations
800-
700-
600-
500-
400-
300-
200-
100-
0-
P = 0.86
32
N HFE HFE mutations
400-
300-
200-
100-
0-
P= 0.34
a)
23
N HFE HFE mutations
60-
40-
20-
0-
P = 0.32
b)
N = 53
N = 34 N = 53 32
N HFE HFE mutations
P = 0.0760-
40-
20-
0-
FIGURE 1 (a) Ferritin (ng/mL) and (b) IST (%) in β-thal carriers without (N) and with HFE mutations(white: males; grey: females).
the frequency of individuals with high ferritin and/or IST values was verysimilar between those with or without HFE gene mutations. Therefore, theinter patient variation in iron loading in our thalassemia minor patientsdoes not seem to depend on the type of inherited mutation in the β-globingene or the presence of mutations on the HFE gene.
Taking into account that most (49 out of 55) patients were heterozygousfor the H63D mutation, the results suggest iron overload in β-thal carriersis not accentuated by H63D heterozygosity; even the H63D homozygotesdid not have elevated ferritin and/or IST levels. Only the single compoundheterozygote (H63D/C282Y) individual had an unusual iron overload. Alarge number of subjects carrying different HFE mutated genotypes wouldbe necessary to conclude their effect on iron status.
Our results are not in agreement with those obtained in some stud-ies (2,14,15), suggesting iron overload in β-thal trait tends to be aggra-vated by co-inheritance of the H63D mutation, even when present in the
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.
TA
BL
E2
Indi
vidu
als
Wit
hFe
rrit
inan
dTr
ansf
erri
nSa
tura
tion
Valu
esA
bove
Nor
mal
inβ-T
hal
asse
mia
Car
rier
s,W
ith
and
Wit
hou
tH
FEM
utat
ion
s
Type
ofT
hal
asse
mia
Mut
atio
ns
inth
eH
FEG
ene
Ferr
itin
Abo
veN
orm
alL
evel
sap
Valu
ebIS
TA
bove
Nor
mal
Lev
elsc
pVa
lues
b
Ferr
itin
and
IST
Abo
veN
orm
alL
evel
sa,c
pVa
lues
b
β0
(n=
114)
Pres
ent(
n=
47)
14(2
9.8%
)0.
173
(6.4
%)
0.25
2(4
.3%
)0.
35A
bsen
t(n
=67
)20
(29.
9%)
6(9
.0%
)3
(4.5
%)
β+
(n=
28)
Pres
ent(
n=
8)2
(25.
0%)
0.33
0(0
.0%
)1.
00
(0.0
%)
1.0
Abs
ent(
n=
20)
3(1
5.0%
)0
(0.0
%)
0(0
.0%
)To
tal(
n=
142)
Pres
ent(
n=
55)
16(2
9.1%
)0.
143
(5.5
%)
0.22
2(3
.6%
)0.
35A
bsen
t(n
=87
)23
(26.
4%)
6(6
.9%
)3
(3.5
%)
IST
:tra
nsf
erri
nsa
tura
tion
inde
x.a F
erri
tin
>30
0n
g/m
L(m
ales
)an
d15
0n
g/m
L(f
emal
es).
bFi
sher
exac
ttes
tsig
nifi
can
tatp
<0.
05.
c IST
>45
.0%
(mal
es)
and
40.0
%(f
emal
es).
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.

HFE Mutations in β-Thalassemia Minor 91
heterozygous state; but are in agreement with those found in Thai andIranian populations (8,16) for β-thal minor and even β-thal intermedia inGreeks (17). An earlier study from Italy (7) also concluded that their find-ings do not support the hypothesis that the association of the β-thal traitwith a single C282Y or H63D allele might lead to iron overload. Diversityof results are probably due to the fact that iron status depends on com-plex interactions and is influenced by factors like sex, age, other mutationsin the HFE or other genes related to iron metabolism and environmentalpopulational variables such as diet or alcohol intake.
In conclusion, in the Balearic population we found no significantchange in ferritin or transferrin saturation levels in β-thal minor patientswith HFE mutations compared to patients without them. Therefore, system-atic screening of HFE mutations in β-thal carriers does not seem warrantedunless they develop evidence of iron overload. Large studies would beneeded to further assess the possible effect of less frequent and morecommitted HFE genotypes than H63D heterozygosity.
ACKNOWLEDGMENTS
This study was partially supported by grants PRDIB-2006-687872 andPROGECIB-10A from the Direcció General de R+D+I (Comunitat Autònoma deles Illes Balears), Palma, Balearic Islands, España.
Declaration of Interest: The authors report no conflicts of interest. The authorsalone are responsible for the content and writing of this article.
REFERENCES
1. Kontoghiorghes GJ, Spyrou A, Kolnagou A. Iron chelation therapy in hereditary hemochromato-sis and thalassemia intermedia: regulatory and non-regulatory mechanisms of increased ironabsorption. Hemoglobin. 2010;34(3):251–264.
2. Ruiz-Argüelles GJ, Garcés-Eisele J, Reyes-Nunez V, et al . Heterozygosity for the H63D mutation inthe hereditary hemochromatosis (HFE) gene may lead into severe iron overload in β-thalassemiaminor: observations in a thalassemic kindred. Rev Invest Clin. 2001;53(2):117–120.
3. Barton JC, Bertoli LF, Acton RD. HFE C282Y and H63D in adults with malignancies in a communitymedical oncology practice. BMC Cancer. 2004;4(1):6.
4. Matas M, Guix P, Castro JA, et al . Prevalence of HFE C282Y and H63D in Jewish populations andclinical implications of H63D homozygosity. Clin Genet. 2006;69(2):155–162.
5. Bukvic N, Sportelli F, Sessa F, et al . Coexistence of β-thalassemia and hereditary hemochromatosisin homozygosity: a possible synergic effect? Hemoglobin. 2009; 33(2):155–157.
6. Fairbanks VF, Thibodeau SN, Granfortuna JM, Lubin IM, Silverman LM, Rohlfs EM. Iron over-loading in β-thalassemia trait and spherocytosis: analysis of C282Y and H63D mutations. Blood.1997;190(Suppl. 1):Abstract 2740.
7. Piperno A, Mariani R, Arosio C, et al . Haemochromatosis in patients with β-thalassaemia trait. Br JHaematol. 2000;111(3):908–914.
8. Jazayeri M, Bakayev V, Adibi P, et al . Frequency of HFE gene mutations in Iranian β-thalassaemiaminor patients. Eur J Haematol. 2003;71(6):408–411.
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.

92 H. López-Escribano et al.
9. Garewal G, Das R, Ahluwalia J, Marwaha RK. Prevalence of the H63D mutation of the HFE innorth India: its presence does not cause iron overload in β thalassemia trait. Eur J Haematol.2005;74(4):333–336.
10. Melis MA, Cau M, Deidda F, Barella S, Cao A, Galanello R. H63D mutation in the HFE geneincreases iron overload in β-thalassemia carriers. Haematologica. 2002;87(3):242–245.
11. Miniero R, Tardivo I, Roetto A, De Gobbi M. Heterozygous β-thalassemia and homozygous H63Dhemochromatosis in a child: an 18-year follow-up. Pediatr Hematol Oncol. 2005;22(2):163–166.
12. Guix P, Picornell A, Parera M, et al . Distribution of HFE C282Y and H63D mutations in the BalearicIslands (NE Spain). Clin Genet. 2002;61(1):43–48.
13. Clark BE, Thein SL. Molecular diagnosis of haemoglobin disorders. Clin Lab Haematol.2004;26(3):159–176.
14. Agarwal S, Tewari D, Arya V, et al . Status of HFE mutation in thalassemia syndromes in north India.Ann Hematol 2007;86(7):483–485.
15. Martins R, Picanço I, Fonseca A, et al . The role of HFE mutations on iron metabolism inβ-thalassemia carriers. J Hum Genet. 2004;49(12):651–655.
16. Yamsri S, Sanchaisuriya K, Fucharoen S, et al . H63D Mutation of the hemochromatosis gene andserum ferritin levels in Thai thalassemia carriers. Acta Haematol. 2007;118(2):99–105.
17. Politou M, Kalotychou V, Pissia M, Rombos Y, Sakellaropoulus N, Papanikolaou G. The impact ofthe mutations of the HFE gene and of the SLC11A3 gene on iron overload in Greek thalassemiaintermedia and βS/βThal anemia patients. Haematologica. 2004;89(4):490–492.
Hem
oglo
bin
Dow
nloa
ded
from
info
rmah
ealth
care
.com
by
Lak
ehea
d U
nive
rsity
on
02/2
8/13
For
pers
onal
use
onl
y.