effect of chloride ions on the electrooxidation at low potentials of dissolved carbon monoxide on...
TRANSCRIPT

Journal of Electroanalytical Chemistry 511 (2001) 39–45
Effect of chloride ions on the electrooxidation at low potentials ofdissolved carbon monoxide on platinum
M.C. Perez, A. Rincon, C. Gutierrez *Instituto de Quımica Fısica ‘Rocasolano’, CSIC, C. Serrano 119, E-28006 Madrid, Spain
Received 29 January 2001; received in revised form 27 April 2001; accepted 13 May 2001
Abstract
We have studied by cyclic voltammetry and Fourier-transform infrared spectroscopy (FTIRS) the influence of chloride ions onthe adsorption and electrooxidation of CO on polycrystalline Pt in 0.5 M H2SO4 and in 0.1 M NaOH, using a low CO dosingpotential, 0.07 and 0.18 V versus RHE, respectively. As is well known, under these circumstances a subsequent stripping CV ofPt in CO-free solution shows a pre-peak and a main peak of chemisorbed CO, usually attributed to weakly and strongly adsorbedCO, respectively. It is also well known that in CO-saturated solutions, for low CO dosing potentials dissolved CO electrooxidationon Pt occurs already at 0.5 V, well below the usually reported potential of 0.85 V observed when CO is dosed at open circuit. Ithas been found here that in acidic, but not in alkaline medium, chloride ions inhibit the adsorption on Pt of the weaklychemisorbed CO far more strongly than that of the strongly chemisorbed CO, and also inhibit the electrooxidation of dissolvedCO on Pt at low potentials, in such a way that there exists a linear relationship between the peak current density of dissolved COelectrooxidation and the charge of the pre-peak in stripping CVs of chemisorbed CO. This correlation gives further confirmationto the hypothesis that electrooxidation of dissolved CO on Pt at low potentials occurs only on the Pt sites liberated from CO inthe pre-peak. Inhibition by chloride ions is due to their adsorption on Pt, since the peak current density of dissolved COelectrooxidation decreases linearly with the logarithm of the chloride concentration, and therefore with the adsorption of chlorideions, which increases linearly with the logarithm of the chloride concentration for [Cl−]�10−5 M. © 2001 Elsevier Science B.V.All rights reserved.
Keywords: Platinum; Carbon monoxide; Electrocatalytic oxidation; Chloride; Adsorption
www.elsevier.com/locate/jelechem
1. Introduction
In 1988 Kita et al. [1] found that if a Pt electrodeimmersed in 0.1 M HClO4 was held at 0.05 V versus thereversible hydrogen electrode (RHE) while CO wasbubbled in the electrolyte, the steady-state current ofdissolved CO electrooxidation at potentials lower than0.6 V was more than one order of magnitude higherthan that observed when CO was bubbled at opencircuit (the potentials are here always referred to theRHE). This quite unique influence of the CO dosingpotential (potential at which the electrode is held whilea reactant is dosed in the electrolyte cell) on the electro-
catalytic activity of Pt has been amply confirmed, bothfor polycrystalline [2,3] and single-crystal [4–7] Pt.
The low or high CO dosing potential also determinesthe presence or absence, respectively, of a pre-peak at0.46 V, preceding the main peak of chemisorbed CO, instripping CVs of chemisorbed CO in CO-free solution.The pre-peak appears in the stripping CV only if theCO dosing potential is lower than 0.36 V, which ledWieckowski et al. [4] and Kita et al. [5] to postulatethat electrooxidation of dissolved CO at 0.6 V occursonly on the small fraction of Pt atoms liberated fromCO in the aforementioned pre-peak at 0.46 V. Effec-tively, under widely differing experimental conditionsthe electrooxidation of dissolved CO at 0.60 V and thepresence of the small anodic peak at 0.46 V ofchemisorbed CO were always concomitant [8]. TheseCO-free Pt islands were found to be the active sites for
* Corresponding author. Tel.: +34-91-5619400, ext. 1327; fax:+34-91-5642431.
E-mail address: [email protected] (C. Gutierrez).
0022-0728/01/$ - see front matter © 2001 Elsevier Science B.V. All rights reserved.PII: S 0 0 2 2 -0728 (01 )00543 -5

M.C. Perez et al. / Journal of Electroanalytical Chemistry 511 (2001) 39–4540
the electrooxidation in the lower potential range of H2
on Pt/Vulcan in the presence of 250 ppm CO [9].It is usually accepted that the pre-peak and the main
peak in stripping CVs correspond to weakly andstrongly chemisorbed CO, respectively. However, it hasbeen postulated [10] that, due to lateral repulsions, allthe CO molecules adsorbed on Pt(111) at dosing poten-tials lower than 0.25 V are weakly chemisorbed, but,once a fraction of them is electrooxidized in the pre-peak at 0.3–0.6 V, the remaining CO molecules relaxinto a strongly adsorbed state.
Chloride ions, the adsorption of which on Pt hasbeen much studied [11], could affect in different waysthe two types of chemisorbed CO that are electrooxi-dized in the pre-peak and in the main peak, respec-tively, of the stripping CV. Therefore, in this work wehave studied the influence of chloride ions on theCO-polycrystalline Pt system in 0.5 M H2SO4 and in0.1 M NaOH.
2. Experimental
A one-compartment Pyrex cell with a saturatedcalomel electrode (located in a long, narrow glass tubefilled with base electrolyte) as the reference and a Ptwire auxiliary electrode was used. The working elec-trode was a 15-mm Pt disk which was polished with 5,0.3 and 0.05 �m alumina in a Buehler Minimet polish-ing machine, sonicated in Milli-Q water, and activatedby potential cycling. All the reagents were of analyticalgrade. CO gas, 99.997% pure and contained in analuminum alloy cylinder in order to avoid the forma-tion of Fe(CO)5, was from Air Liquide. Milli-Q waterwas obtained from a Milli-RO+Milli-Q set-up fromMillipore, Bedford, MA. The potentiostat was a
�Autolab from EcoChemie B.V. Cyclic voltammograms(CVs) were recorded at 20 mV s−1 unless otherwisespecified.
CVs of Pt in CO-saturated solutions were carried outunder quiescent conditions after bubbling CO for 10min in the previously deaerated electrolyte, and withthe Pt electrode under potential control. Stripping CVsof chemisorbed CO were obtained after elimination ofthe CO in solution by N2 bubbling, also under potentialcontrol.
The FTIRS experiments were carried out in aPerkin–Elmer FTIR spectrophotometer, model 1725-X. A staircase potential program, in which the potentialwas increased in 0.1 V steps, was used for acquisition ofthe FTIR spectra. The reference was a spectrum at apotential positive enough for the Pt surface to beCO-free, and consequently chemisorbed CO gave riseto negative bands. The CO in solution was not elimi-nated after CO chemisorption, but it can be neglectedas compared with a CO monolayer.
Further details can be found in Ref. [12].
3. Results
3.1. Effect of chloride ions on the adsorption andelectrooxidation of CO on Pt in 0.5 M H2SO4
In Fig. 1 the marked effect of chloride ions on theCV of Pt in CO-saturated (while the electrode was heldat the initial potential of 0.07 V) 0.5 M H2SO4 can beclearly appreciated, even with a chloride concentrationas low as 10−4 M. This effect is twofold: chloride ionsdecrease the height of the peak of dissolved CO elec-trooxidation at 0.60 V, and shift positively the peak ofchemisorbed CO, to such an extent that in 0.5 MH2SO4 this peak appears at 0.78 V, but in 0.5 M HCl at1.21 V. The positive shift of the peak of chemisorbedCO was of 0.17 V already at the lowest chloride con-centration used, 10−4 M. The effect of HCl is essen-tially the same if it is added after the Pt electrode hasbeen equilibrated with CO, i.e. on an already formedCO monolayer.
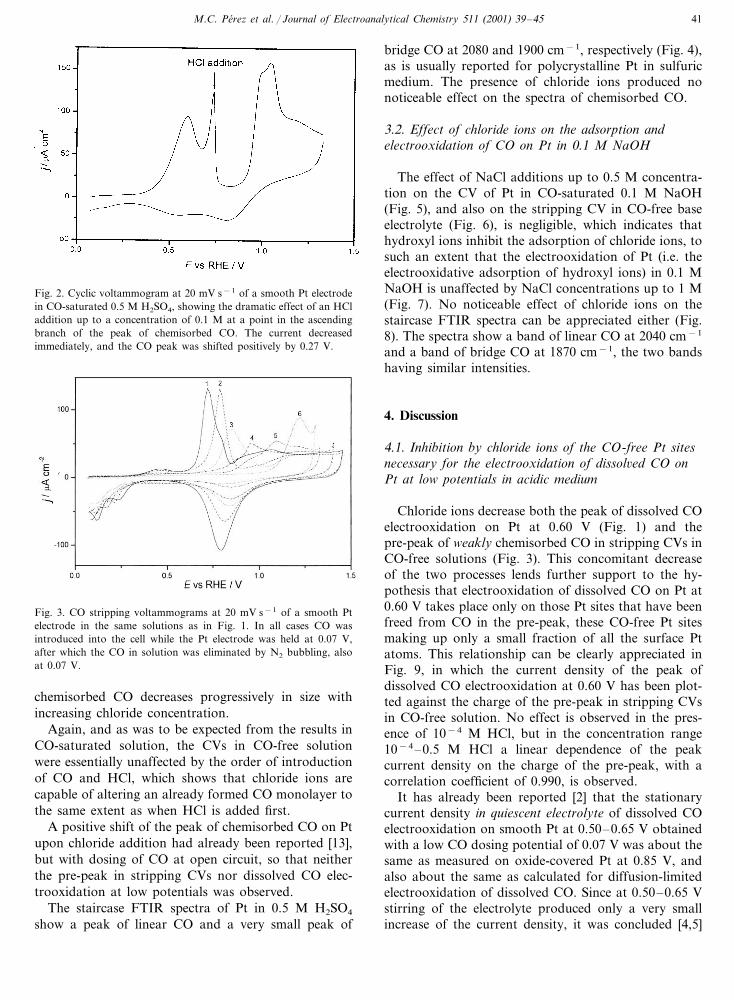
The effect of chloride on the electrooxidation ofchemisorbed CO was instantaneous within our timeresolution of a few seconds, so that if chloride wasadded up to a concentration of 0.1 M in the ascendingbranch of the peak of chemisorbed CO in a CV inCO-saturated 0.5 M H2SO4 the current decreased im-mediately, and the CO peak was shifted positively by0.27 V (Fig. 2).
As was to be expected, the effect of chloride ions isalso clearly apparent in the stripping CVs ofchemisorbed CO in CO-free solutions (Fig. 3). Theeffect is twofold: the main peak of chemisorbed CO isshifted positively, from 0.72 V in 0.5 M H2SO4 to 1.23V in 0.5 M HCl, and, furthermore, the pre-peak of
Fig. 1. Cyclic voltammograms at 20 mV s−1 of a smooth Pt electrodein the following CO-saturated solutions: 1; 0.5 M H2SO4. 2–5; 0.5 MH2SO4 with increasing chloride concentrations: 10−4, 10−3, 10−2
and 10−1 M HCl, respectively. 6; 0.5 M HCl. In all cases CO wasintroduced into the cell while the Pt electrode was held at 0.07 V.
M.C. Perez et al. / Journal of Electroanalytical Chemistry 511 (2001) 39–45 41
Fig. 2. Cyclic voltammogram at 20 mV s−1 of a smooth Pt electrodein CO-saturated 0.5 M H2SO4, showing the dramatic effect of an HCladdition up to a concentration of 0.1 M at a point in the ascendingbranch of the peak of chemisorbed CO. The current decreasedimmediately, and the CO peak was shifted positively by 0.27 V.
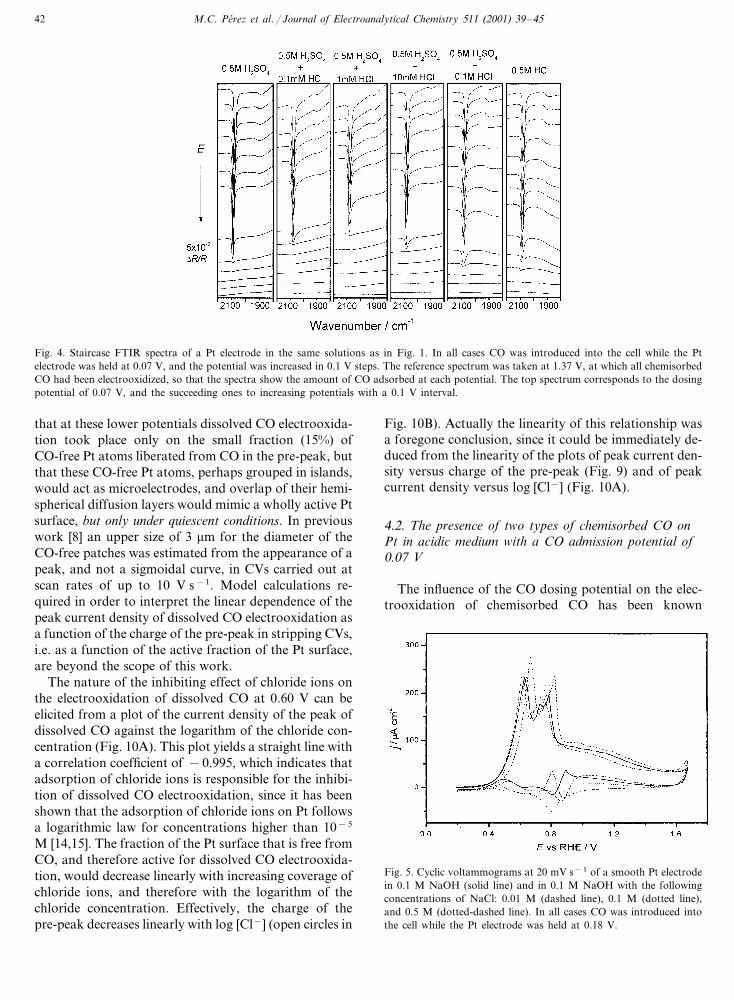
bridge CO at 2080 and 1900 cm−1, respectively (Fig. 4),as is usually reported for polycrystalline Pt in sulfuricmedium. The presence of chloride ions produced nonoticeable effect on the spectra of chemisorbed CO.
3.2. Effect of chloride ions on the adsorption andelectrooxidation of CO on Pt in 0.1 M NaOH
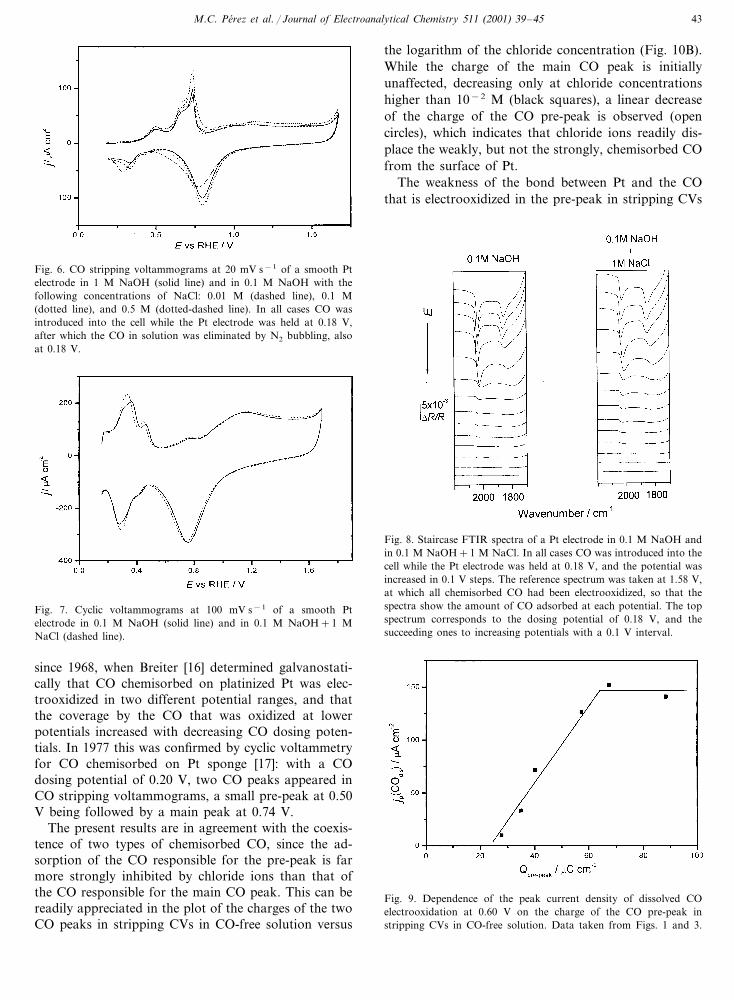
The effect of NaCl additions up to 0.5 M concentra-tion on the CV of Pt in CO-saturated 0.1 M NaOH(Fig. 5), and also on the stripping CV in CO-free baseelectrolyte (Fig. 6), is negligible, which indicates thathydroxyl ions inhibit the adsorption of chloride ions, tosuch an extent that the electrooxidation of Pt (i.e. theelectrooxidative adsorption of hydroxyl ions) in 0.1 MNaOH is unaffected by NaCl concentrations up to 1 M(Fig. 7). No noticeable effect of chloride ions on thestaircase FTIR spectra can be appreciated either (Fig.8). The spectra show a band of linear CO at 2040 cm−1
and a band of bridge CO at 1870 cm−1, the two bandshaving similar intensities.
4. Discussion
4.1. Inhibition by chloride ions of the CO-free Pt sitesnecessary for the electrooxidation of dissol�ed CO onPt at low potentials in acidic medium
Chloride ions decrease both the peak of dissolved COelectrooxidation on Pt at 0.60 V (Fig. 1) and thepre-peak of weakly chemisorbed CO in stripping CVs inCO-free solutions (Fig. 3). This concomitant decreaseof the two processes lends further support to the hy-pothesis that electrooxidation of dissolved CO on Pt at0.60 V takes place only on those Pt sites that have beenfreed from CO in the pre-peak, these CO-free Pt sitesmaking up only a small fraction of all the surface Ptatoms. This relationship can be clearly appreciated inFig. 9, in which the current density of the peak ofdissolved CO electrooxidation at 0.60 V has been plot-ted against the charge of the pre-peak in stripping CVsin CO-free solution. No effect is observed in the pres-ence of 10−4 M HCl, but in the concentration range10−4–0.5 M HCl a linear dependence of the peakcurrent density on the charge of the pre-peak, with acorrelation coefficient of 0.990, is observed.
It has already been reported [2] that the stationarycurrent density in quiescent electrolyte of dissolved COelectrooxidation on smooth Pt at 0.50–0.65 V obtainedwith a low CO dosing potential of 0.07 V was about thesame as measured on oxide-covered Pt at 0.85 V, andalso about the same as calculated for diffusion-limitedelectrooxidation of dissolved CO. Since at 0.50–0.65 Vstirring of the electrolyte produced only a very smallincrease of the current density, it was concluded [4,5]
Fig. 3. CO stripping voltammograms at 20 mV s−1 of a smooth Ptelectrode in the same solutions as in Fig. 1. In all cases CO wasintroduced into the cell while the Pt electrode was held at 0.07 V,after which the CO in solution was eliminated by N2 bubbling, alsoat 0.07 V.
chemisorbed CO decreases progressively in size withincreasing chloride concentration.
Again, and as was to be expected from the results inCO-saturated solution, the CVs in CO-free solutionwere essentially unaffected by the order of introductionof CO and HCl, which shows that chloride ions arecapable of altering an already formed CO monolayer tothe same extent as when HCl is added first.
A positive shift of the peak of chemisorbed CO on Ptupon chloride addition had already been reported [13],but with dosing of CO at open circuit, so that neitherthe pre-peak in stripping CVs nor dissolved CO elec-trooxidation at low potentials was observed.
The staircase FTIR spectra of Pt in 0.5 M H2SO4
show a peak of linear CO and a very small peak of
M.C. Perez et al. / Journal of Electroanalytical Chemistry 511 (2001) 39–4542
Fig. 4. Staircase FTIR spectra of a Pt electrode in the same solutions as in Fig. 1. In all cases CO was introduced into the cell while the Ptelectrode was held at 0.07 V, and the potential was increased in 0.1 V steps. The reference spectrum was taken at 1.37 V, at which all chemisorbedCO had been electrooxidized, so that the spectra show the amount of CO adsorbed at each potential. The top spectrum corresponds to the dosingpotential of 0.07 V, and the succeeding ones to increasing potentials with a 0.1 V interval.
that at these lower potentials dissolved CO electrooxida-tion took place only on the small fraction (15%) ofCO-free Pt atoms liberated from CO in the pre-peak, butthat these CO-free Pt atoms, perhaps grouped in islands,would act as microelectrodes, and overlap of their hemi-spherical diffusion layers would mimic a wholly active Ptsurface, but only under quiescent conditions. In previouswork [8] an upper size of 3 �m for the diameter of theCO-free patches was estimated from the appearance of apeak, and not a sigmoidal curve, in CVs carried out atscan rates of up to 10 V s−1. Model calculations re-quired in order to interpret the linear dependence of thepeak current density of dissolved CO electrooxidation asa function of the charge of the pre-peak in stripping CVs,i.e. as a function of the active fraction of the Pt surface,are beyond the scope of this work.
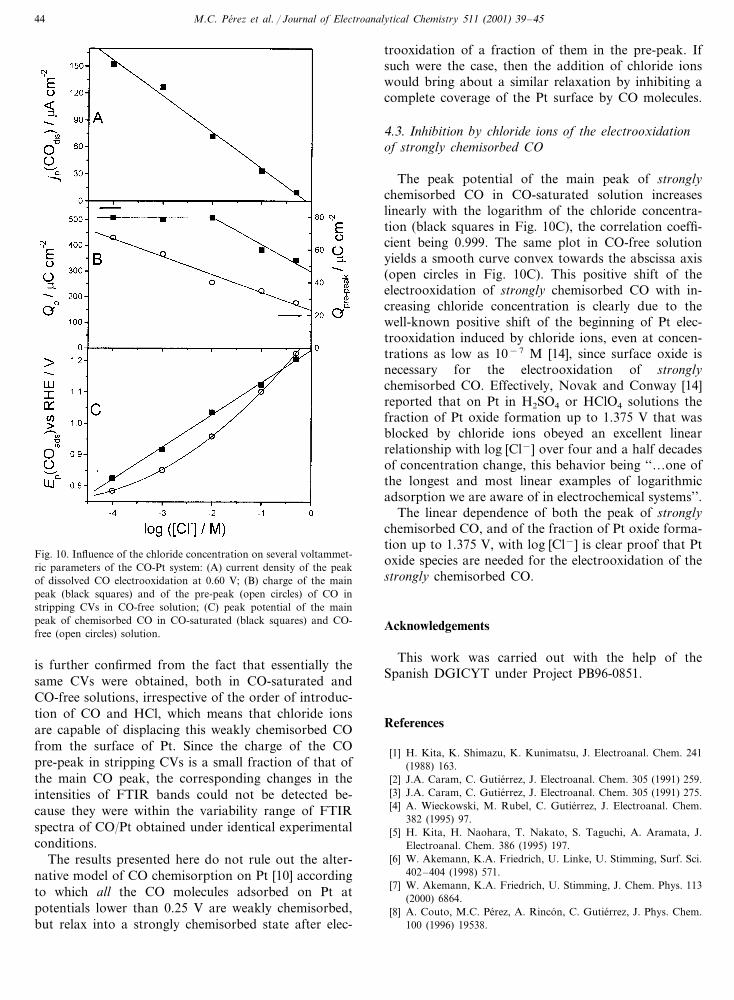
The nature of the inhibiting effect of chloride ions onthe electrooxidation of dissolved CO at 0.60 V can beelicited from a plot of the current density of the peak ofdissolved CO against the logarithm of the chloride con-centration (Fig. 10A). This plot yields a straight line witha correlation coefficient of −0.995, which indicates thatadsorption of chloride ions is responsible for the inhibi-tion of dissolved CO electrooxidation, since it has beenshown that the adsorption of chloride ions on Pt followsa logarithmic law for concentrations higher than 10−5
M [14,15]. The fraction of the Pt surface that is free fromCO, and therefore active for dissolved CO electrooxida-tion, would decrease linearly with increasing coverage ofchloride ions, and therefore with the logarithm of thechloride concentration. Effectively, the charge of thepre-peak decreases linearly with log [Cl−] (open circles in
Fig. 10B). Actually the linearity of this relationship wasa foregone conclusion, since it could be immediately de-duced from the linearity of the plots of peak current den-sity versus charge of the pre-peak (Fig. 9) and of peakcurrent density versus log [Cl−] (Fig. 10A).
4.2. The presence of two types of chemisorbed CO onPt in acidic medium with a CO admission potential of0.07 V
The influence of the CO dosing potential on the elec-trooxidation of chemisorbed CO has been known
Fig. 5. Cyclic voltammograms at 20 mV s−1 of a smooth Pt electrodein 0.1 M NaOH (solid line) and in 0.1 M NaOH with the followingconcentrations of NaCl: 0.01 M (dashed line), 0.1 M (dotted line),and 0.5 M (dotted-dashed line). In all cases CO was introduced intothe cell while the Pt electrode was held at 0.18 V.
M.C. Perez et al. / Journal of Electroanalytical Chemistry 511 (2001) 39–45 43
Fig. 6. CO stripping voltammograms at 20 mV s−1 of a smooth Ptelectrode in 1 M NaOH (solid line) and in 0.1 M NaOH with thefollowing concentrations of NaCl: 0.01 M (dashed line), 0.1 M(dotted line), and 0.5 M (dotted-dashed line). In all cases CO wasintroduced into the cell while the Pt electrode was held at 0.18 V,after which the CO in solution was eliminated by N2 bubbling, alsoat 0.18 V.
the logarithm of the chloride concentration (Fig. 10B).While the charge of the main CO peak is initiallyunaffected, decreasing only at chloride concentrationshigher than 10−2 M (black squares), a linear decreaseof the charge of the CO pre-peak is observed (opencircles), which indicates that chloride ions readily dis-place the weakly, but not the strongly, chemisorbed COfrom the surface of Pt.
The weakness of the bond between Pt and the COthat is electrooxidized in the pre-peak in stripping CVs
Fig. 8. Staircase FTIR spectra of a Pt electrode in 0.1 M NaOH andin 0.1 M NaOH+1 M NaCl. In all cases CO was introduced into thecell while the Pt electrode was held at 0.18 V, and the potential wasincreased in 0.1 V steps. The reference spectrum was taken at 1.58 V,at which all chemisorbed CO had been electrooxidized, so that thespectra show the amount of CO adsorbed at each potential. The topspectrum corresponds to the dosing potential of 0.18 V, and thesucceeding ones to increasing potentials with a 0.1 V interval.
Fig. 7. Cyclic voltammograms at 100 mV s−1 of a smooth Ptelectrode in 0.1 M NaOH (solid line) and in 0.1 M NaOH+1 MNaCl (dashed line).
Fig. 9. Dependence of the peak current density of dissolved COelectrooxidation at 0.60 V on the charge of the CO pre-peak instripping CVs in CO-free solution. Data taken from Figs. 1 and 3.
since 1968, when Breiter [16] determined galvanostati-cally that CO chemisorbed on platinized Pt was elec-trooxidized in two different potential ranges, and thatthe coverage by the CO that was oxidized at lowerpotentials increased with decreasing CO dosing poten-tials. In 1977 this was confirmed by cyclic voltammetryfor CO chemisorbed on Pt sponge [17]: with a COdosing potential of 0.20 V, two CO peaks appeared inCO stripping voltammograms, a small pre-peak at 0.50V being followed by a main peak at 0.74 V.
The present results are in agreement with the coexis-tence of two types of chemisorbed CO, since the ad-sorption of the CO responsible for the pre-peak is farmore strongly inhibited by chloride ions than that ofthe CO responsible for the main CO peak. This can bereadily appreciated in the plot of the charges of the twoCO peaks in stripping CVs in CO-free solution versus
M.C. Perez et al. / Journal of Electroanalytical Chemistry 511 (2001) 39–4544
Fig. 10. Influence of the chloride concentration on several voltammet-ric parameters of the CO-Pt system: (A) current density of the peakof dissolved CO electrooxidation at 0.60 V; (B) charge of the mainpeak (black squares) and of the pre-peak (open circles) of CO instripping CVs in CO-free solution; (C) peak potential of the mainpeak of chemisorbed CO in CO-saturated (black squares) and CO-free (open circles) solution.
trooxidation of a fraction of them in the pre-peak. Ifsuch were the case, then the addition of chloride ionswould bring about a similar relaxation by inhibiting acomplete coverage of the Pt surface by CO molecules.
4.3. Inhibition by chloride ions of the electrooxidationof strongly chemisorbed CO
The peak potential of the main peak of stronglychemisorbed CO in CO-saturated solution increaseslinearly with the logarithm of the chloride concentra-tion (black squares in Fig. 10C), the correlation coeffi-cient being 0.999. The same plot in CO-free solutionyields a smooth curve convex towards the abscissa axis(open circles in Fig. 10C). This positive shift of theelectrooxidation of strongly chemisorbed CO with in-creasing chloride concentration is clearly due to thewell-known positive shift of the beginning of Pt elec-trooxidation induced by chloride ions, even at concen-trations as low as 10−7 M [14], since surface oxide isnecessary for the electrooxidation of stronglychemisorbed CO. Effectively, Novak and Conway [14]reported that on Pt in H2SO4 or HClO4 solutions thefraction of Pt oxide formation up to 1.375 V that wasblocked by chloride ions obeyed an excellent linearrelationship with log [Cl−] over four and a half decadesof concentration change, this behavior being ‘‘…one ofthe longest and most linear examples of logarithmicadsorption we are aware of in electrochemical systems’’.
The linear dependence of both the peak of stronglychemisorbed CO, and of the fraction of Pt oxide forma-tion up to 1.375 V, with log [Cl−] is clear proof that Ptoxide species are needed for the electrooxidation of thestrongly chemisorbed CO.
Acknowledgements
This work was carried out with the help of theSpanish DGICYT under Project PB96-0851.
References
[1] H. Kita, K. Shimazu, K. Kunimatsu, J. Electroanal. Chem. 241(1988) 163.
[2] J.A. Caram, C. Gutierrez, J. Electroanal. Chem. 305 (1991) 259.[3] J.A. Caram, C. Gutierrez, J. Electroanal. Chem. 305 (1991) 275.[4] A. Wieckowski, M. Rubel, C. Gutierrez, J. Electroanal. Chem.
382 (1995) 97.[5] H. Kita, H. Naohara, T. Nakato, S. Taguchi, A. Aramata, J.
Electroanal. Chem. 386 (1995) 197.[6] W. Akemann, K.A. Friedrich, U. Linke, U. Stimming, Surf. Sci.
402–404 (1998) 571.[7] W. Akemann, K.A. Friedrich, U. Stimming, J. Chem. Phys. 113
(2000) 6864.[8] A. Couto, M.C. Perez, A. Rincon, C. Gutierrez, J. Phys. Chem.
100 (1996) 19538.
is further confirmed from the fact that essentially thesame CVs were obtained, both in CO-saturated andCO-free solutions, irrespective of the order of introduc-tion of CO and HCl, which means that chloride ionsare capable of displacing this weakly chemisorbed COfrom the surface of Pt. Since the charge of the COpre-peak in stripping CVs is a small fraction of that ofthe main CO peak, the corresponding changes in theintensities of FTIR bands could not be detected be-cause they were within the variability range of FTIRspectra of CO/Pt obtained under identical experimentalconditions.
The results presented here do not rule out the alter-native model of CO chemisorption on Pt [10] accordingto which all the CO molecules adsorbed on Pt atpotentials lower than 0.25 V are weakly chemisorbed,but relax into a strongly chemisorbed state after elec-

M.C. Perez et al. / Journal of Electroanalytical Chemistry 511 (2001) 39–45 45
[9] T.J. Schmidt, H.A. Gasteiger, R.J. Behm, J. Electrochem. Soc.146 (1999) 1296.
[10] N.M. Markovic, B.N. Grgur, C.A. Lucas, P.N. Ross, J. Phys.Chem. B 103 (1999) 487.
[11] N. Li, J. Lipkowski, J. Electroanal. Chem. 491 (2000)95.
[12] A. Couto, A. Rincon, Ma C. Perez, C. Gutierrez, Electrochim.Acta 46 (2001) 1285.
[13] A.M. de Becdelievre, J. de Becdelievre, J. Clavilier, J. Elec-troanal. Chem. 294 (1990) 97.
[14] D.M. Novak, B.E. Conway, J. Chem. Soc. Faraday Trans. I 77(1981) 2341.
[15] J.O’M. Bockris, M. Gamboa-Aldeco, M. Szklarczyk, J. Elec-troanal. Chem. 339 (1992) 355.
[16] M.W. Breiter, J. Phys. Chem. 72 (1968) 1305.[17] L. Grambow, S. Bruckenstein, Electrochim. Acta 2 (1977) 377.
.