
717
The mammalian heart can increase its pump work 3-fold, despite limited capacity to store chemical energy.
Changes in energy demand, therefore, require a rapid response from the mitochondria. The size of the total cardiac ATP pool changes little over a wide range of workloads, whereas ATP turnover varies substantially. At high workload, the total ATP pool can turn over in as little as 2 s.1,2 As a result, even modest changes in cardiac metabolism can have a significant impact on contractile function. Just as the heart has a large contractile reserve, the heart also has a large metabolic reserve to meet changes in energy demand. This review explores the function and dysfunction of the intracellular mechanisms that trans-duce the pathophysiological state of the heart to the mitochon-dria to provide this metabolic reserve.
Heart failure is a syndrome that is characterized by the heart’s inability to provide sufficient blood flow to the body. Changes in cardiac metabolism contribute to the organ’s im-paired contractile function.3 Here, we discuss 2 models in par-ticular, the hypertrophic heart and the diabetic heart, focusing on the changes in mitochondrial function and how these may contribute to heart failure. Although these 2 pathologies are vastly different in their clinical presentation, the hypertro-phic and diabetic hearts share signs of a return to the fetal
gene program and impaired metabolic reserve.4 We discuss potential defects in several parallel pathways that could lim-it the ability of the failing heart to meet energetic demand. Specifically, we examine the role of intracellular and mito-chondrial calcium in regulating mitochondrial metabolism, changes in mitochondrial transporter expression signaling between the mitochondria and the cytosol via intermediate exchange and redox regulation, the mitochondria as signaling organelles, and finally the role of the triacylglyceride pool in regulating mitochondrial function through fuel provision and lipid signaling.
Metabolic ReserveMetabolic reserve is defined as the available potential en-ergy that is demand accessible in response to an increase in cardiac work. In the hypertrophic heart, increased workload or β-adrenergic stimulation leads to a decrease in the ATP concentration and a reduction in intracellular buffer of ATP and the primary energy buffer in the heart–phosphocreatine (PCr).5 This, along with findings of restricted fuel utilization,6 suggests that the metabolic reserve of the hypertrophic heart is reduced, leading Ingwall7 and Neubauer8 to speculate initially that the hypertrophic heart is an engine out of fuel.
Review
© 2014 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/CIRCRESAHA.114.301863
Abstract: Metabolic signaling mechanisms are increasingly recognized to mediate the cellular response to alterations in workload demand, as a consequence of physiological and pathophysiological challenges. Thus, an understanding of the metabolic mechanisms coordinating activity in the cytosol with the energy-providing pathways in the mitochondrial matrix becomes critical for deepening our insights into the pathogenic changes that occur in the stressed cardiomyocyte. Processes that exchange both metabolic intermediates and cations between the cytosol and mitochondria enable transduction of dynamic changes in contractile state to the mitochondrial compartment of the cell. Disruption of such metabolic transduction pathways has severe consequences for the energetic support of contractile function in the heart and is implicated in the pathogenesis of heart failure. Deficiencies in metabolic reserve and impaired metabolic transduction in the cardiomyocyte can result from inherent deficiencies in metabolic phenotype or maladaptive changes in metabolic enzyme expression and regulation in the response to pathogenic stress. This review examines both current and emerging concepts of the functional linkage between the cytosol and the mitochondrial matrix with a specific focus on metabolic reserve and energetic efficiency. These principles of exchange and transport mechanisms across the mitochondrial membrane are reviewed for the failing heart from the perspectives of chronic pressure overload and diabetes mellitus. (Circ Res. 2014;114:717-729.)
Key Words: cytosol ■ diabetes mellitus ■ heart failure ■ metabolic pathways ■ metabolism ■ mobilization ■ mitochondria
Matrix RevisitedMechanisms Linking Energy Substrate Metabolism to the Function of the
Heart
Andrew N. Carley, Heinrich Taegtmeyer, E. Douglas Lewandowski
Original received September 28, 2013; revision received December 17, 2013; accepted December 17, 2013. In December 2013, the average time from submission to first decision for all original research papers submitted to Circulation Research was 11.66 days.
From the Center for Cardiovascular Research, University of Illinois at Chicago College of Medicine, Chicago IL (A.N.C., E.D.L.); and Department of Internal Medicine, Division of Cardiology, The University of Texas Medical School at Houston (H.T.).
Correspondence to E. Douglas Lewandowski, PhD, Center for Cardiovascular Research, UIC College of Medicine, MC-801, 909 S Wolcott Ave, Chicago, IL 60612. E-mail [email protected]
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

718 Circulation Research February 14, 2014
Metabolic reserve of the heart in diabetes mellitus is less understood and conceptually different from that of hypertro-phic heart. In diabetes mellitus, the heart resembles an engine oversupplied with fuel, yet no less restricted in metabolic signaling and the ability to recruit alternative mechanisms of ATP synthesis. Despite an oversupply of energy-rich long-chain fatty acids, the heart in diabetes mellitus is energetically compromised in a similar manner to the hypertrophic heart, that is, a decrease in the cellular energy charge, reflected by the relative contents of PCr and ATP.9 Despite this apparent oversupply, substrates are sequestered into storage pools and are uncoupled from mitochondrial metabolism during the later stages of disease progression into overt heart failure.10–13
Consideration of pathological energy starvation invokes the notion of metabolic reserve and efficiency, which becomes disrupted in the hypertrophic and diabetic heart. The efficien-cy of fuel pathways in the mitochondria exploits conservation of intermediates through metabolic cycles that transform a substrate with little or no change in the cycle’s constituents. Cycles have evolved because in a competitive environment the chances for survival are the greatest if resources are used in an optimal way.14 Steady-state concentrations of cycle metabo-lites depend on their rates of synthesis and degradation but not on the rate of cycle turnover.15–18 In the case of the citric acid cycle (CAC), substrates entering as citrate (as result of the condensation of acetyl CoA and oxaloacetate) do not cause a change in the CAC pool size because the 2 carbons of acetyl CoA are lost as CO
2 in the isocitrate and α-ketoglutarate de-
hydrogenase reactions, respectively. In contrast, the process of anaplerosis (eg, through the fixation of CO
2 by pyruvate
carboxylation) supplies compounds to the CAC through re-actions other than citrate synthase (Figure 1).19–22 These compounds replenish carbon intermediates lost as amino acids, citrate, or oxaloacetate.23 By maintaining CAC inter-mediate levels, anaplerosis facilitates energy transfer in the heart, an organ with high rates of energy turnover. Not sur-prisingly, changes in CAC flux antedate the functional de-cline in perfused hearts oxidizing acetoacetate as the sole
energy-providing substrate,24 and the collapse of metabolic cycles in the heart impairs metabolic flexibility and efficiency in the failing heart.25 Along with this influx of carbon into the cycle, efflux from the CAC intermediate pools also serves to maintain steady-state equilibrium during acute transitions in the individual, linked CAC reaction rates.17 Under pathologi-cal states that induce nonequilibrium conditions, metabolic reserve is challenged. For example, in myocardial ischemia, when glycolysis prevails during the loss of glucose and fatty acid oxidation because of the lack of oxygen, the collapse of cycles becomes detrimental to conserving pathways and thus metabolic efficiency.
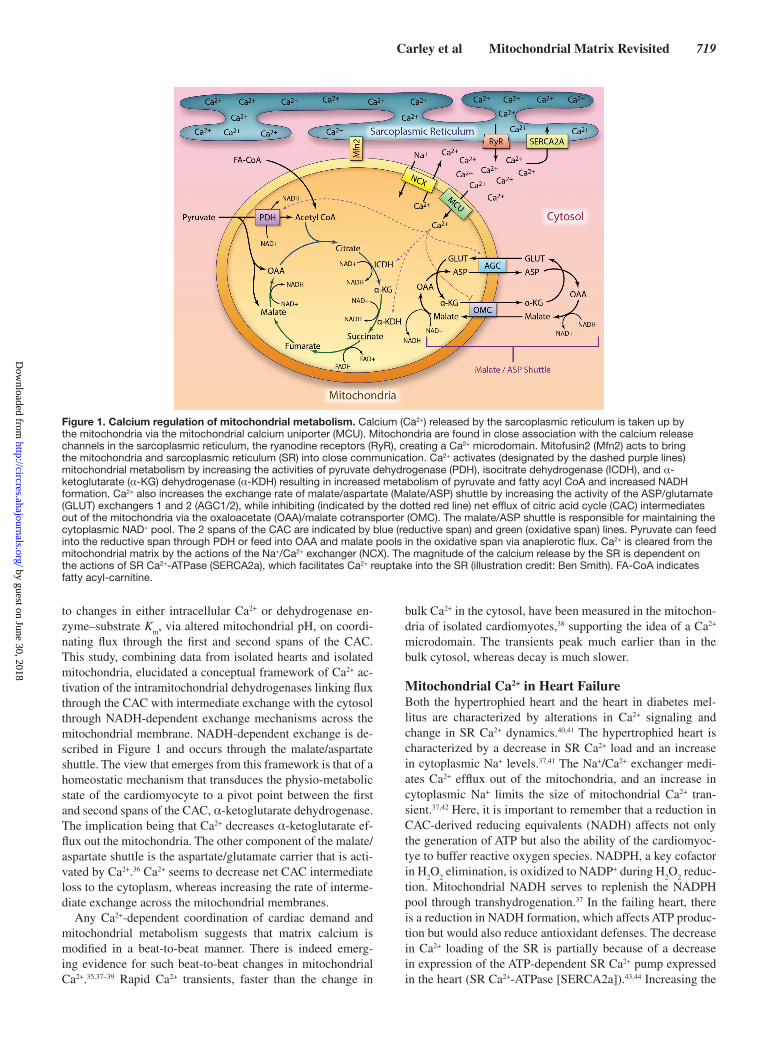
Ca2+ as a Metabolic RegulatorExtensive investigations have attempted to identify the key second messaging components that link mitochondrial respi-ration and cardiac demand. Although several candidates have been proposed, Ca2+ release by the sarcoplasmic reticulum (SR) now seems as a central player in modulating the meta-bolic rate of the heart in response to changes in energetic de-mand.26,27 Figure 1 depicts Ca2+ entry into mitochondria and the mechanisms, whereby Ca2+ modifies mitochondrial me-tabolism. Ca2+ enters the mitochondria through the recently identified mitochondrial calcium uniporter,28 a low affinity calcium transporter. Ca2+ entry through mitochondrial calcium uniporter is driven by the membrane potential across the inner mitochondrial membrane (IMM)29 and therefore dependent on electron transport chain (ETC) activity and mitochondrial me-tabolism. The regulation of mitochondrial calcium uniporter activity has yet to be fully elucidated, especially in heart fail-ure; however, mitochondrial calcium uptake 1 gates Ca2+ en-try through mitochondrial calcium uniporter, preventing Ca2+ overload and maintaining a wide dynamic range for Ca2+ entry into the mitochondria.30,31
Mitochondria are present in close proximity to the ryano-dine receptors of the SR, and large localized increases in Ca2+ in these microdomains facilitate sufficient mitochondrial Ca2+ entry.26 Mitofusin2 is integral in bringing the mitochondria and the SR into close communication.32 The loss of mitofusin2 se-verely limits mitochondrial Ca2+ uptake.32 These observations highlight the importance of viewing mitochondria not in isola-tion but within the context of the intact cell. The total flux of Ca2+ across the IMM remains unknown; however, a range of estimates in the literature has been reported, ranging from 10 nmol/L to 26% of total SR calcium load.33,34 Until this issue is resolved, modeling of mitochondrial dynamics remains difficult in isolation from appropriate calcium buffering concentrations.
Ca2+ influences flux through competing pathways of mi-tochondrial metabolism, while also maintaining relative balance through the activation of calcium-dependent-dehy-drogenases17,35 (Figure 1); pyruvate dehydrogenase (PDH) catalyzes the conversion of pyruvate to acetyl CoA, and the rate-limiting enzymes of the CAC, isocitrate dehydrogenase, and α-ketoglutarate dehydrogenase. In addition, Ca2+ con-trols the efflux of CAC intermediates out of the mitochon-dria and shifts the efflux/oxidation ratio toward oxidation and continued formation of reducing equivalents.17 O’Donnell et al17 were able to quantify the influences of activating the NAD+-dependent, Ca2+-activated dehydrogenases in response
Nonstandard Abbreviations and Acronyms
ACC2 acetyl CoA carboxylase 2
CAC citric acid cycle
CAT carnitine acetyltransferase
CK creatine kinase
CPT1/2 carnitine palmitoyltransferase 1 and 2
ETC electron transport chain
HDAC histone deacetylase
IMM inner mitochondrial membrane
L-CPT1 liver CPT1
PCr phosphocreatine
PDH pyruvate dehydrogenase
PPARα peroxisome proliferator–activated receptor α
SERCA2a sarco/endoplasmic reticulum Ca2+-ATPase
Sirt1/3 sirtuin 1 and 3
SR sarcoplasmic reticulum
UPC3 uncoupling protein 3
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from
Carley et al Mitochondrial Matrix Revisited 719
to changes in either intracellular Ca2+ or dehydrogenase en-zyme–substrate K
m, via altered mitochondrial pH, on coordi-
nating flux through the first and second spans of the CAC. This study, combining data from isolated hearts and isolated mitochondria, elucidated a conceptual framework of Ca2+ ac-tivation of the intramitochondrial dehydrogenases linking flux through the CAC with intermediate exchange with the cytosol through NADH-dependent exchange mechanisms across the mitochondrial membrane. NADH-dependent exchange is de-scribed in Figure 1 and occurs through the malate/aspartate shuttle. The view that emerges from this framework is that of a homeostatic mechanism that transduces the physio-metabolic state of the cardiomyocyte to a pivot point between the first and second spans of the CAC, α-ketoglutarate dehydrogenase. The implication being that Ca2+ decreases α-ketoglutarate ef-flux out the mitochondria. The other component of the malate/aspartate shuttle is the aspartate/glutamate carrier that is acti-vated by Ca2+.36 Ca2+ seems to decrease net CAC intermediate loss to the cytoplasm, whereas increasing the rate of interme-diate exchange across the mitochondrial membranes.
Any Ca2+-dependent coordination of cardiac demand and mitochondrial metabolism suggests that matrix calcium is modified in a beat-to-beat manner. There is indeed emerg-ing evidence for such beat-to-beat changes in mitochondrial Ca2+.35,37–39 Rapid Ca2+ transients, faster than the change in
bulk Ca2+ in the cytosol, have been measured in the mitochon-dria of isolated cardiomyotes,38 supporting the idea of a Ca2+ microdomain. The transients peak much earlier than in the bulk cytosol, whereas decay is much slower.
Mitochondrial Ca2+ in Heart FailureBoth the hypertrophied heart and the heart in diabetes mel-litus are characterized by alterations in Ca2+ signaling and change in SR Ca2+ dynamics.40,41 The hypertrophied heart is characterized by a decrease in SR Ca2+ load and an increase in cytoplasmic Na+ levels.37,41 The Na+/Ca2+ exchanger medi-ates Ca2+ efflux out of the mitochondria, and an increase in cytoplasmic Na+ limits the size of mitochondrial Ca2+ tran-sient.37,42 Here, it is important to remember that a reduction in CAC-derived reducing equivalents (NADH) affects not only the generation of ATP but also the ability of the cardiomyoc-tye to buffer reactive oxygen species. NADPH, a key cofactor in H
2O
2 elimination, is oxidized to NADP+ during H
2O
2 reduc-
tion. Mitochondrial NADH serves to replenish the NADPH pool through transhydrogenation.37 In the failing heart, there is a reduction in NADH formation, which affects ATP produc-tion but would also reduce antioxidant defenses. The decrease in Ca2+ loading of the SR is partially because of a decrease in expression of the ATP-dependent SR Ca2+ pump expressed in the heart (SR Ca2+-ATPase [SERCA2a]).43,44 Increasing the
Figure 1. Calcium regulation of mitochondrial metabolism. Calcium (Ca2+) released by the sarcoplasmic reticulum is taken up by the mitochondria via the mitochondrial calcium uniporter (MCU). Mitochondria are found in close association with the calcium release channels in the sarcoplasmic reticulum, the ryanodine receptors (RyR), creating a Ca2+ microdomain. Mitofusin2 (Mfn2) acts to bring the mitochondria and sarcoplasmic reticulum (SR) into close communication. Ca2+ activates (designated by the dashed purple lines) mitochondrial metabolism by increasing the activities of pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (ICDH), and α-ketoglutarate (α-KG) dehydrogenase (α-KDH) resulting in increased metabolism of pyruvate and fatty acyl CoA and increased NADH formation. Ca2+ also increases the exchange rate of malate/aspartate (Malate/ASP) shuttle by increasing the activity of the ASP/glutamate (GLUT) exchangers 1 and 2 (AGC1/2), while inhibiting (indicated by the dotted red line) net efflux of citric acid cycle (CAC) intermediates out of the mitochondria via the oxaloacetate (OAA)/malate cotransporter (OMC). The malate/ASP shuttle is responsible for maintaining the cytoplasmic NAD+ pool. The 2 spans of the CAC are indicated by blue (reductive span) and green (oxidative span) lines. Pyruvate can feed into the reductive span through PDH or feed into OAA and malate pools in the oxidative span via anaplerotic flux. Ca2+ is cleared from the mitochondrial matrix by the actions of the Na+/Ca2+ exchanger (NCX). The magnitude of the calcium release by the SR is dependent on the actions of SR Ca2+-ATPase (SERCA2a), which facilitates Ca2+ reuptake into the SR (illustration credit: Ben Smith). FA-CoA indicates fatty acyl-carnitine.
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

720 Circulation Research February 14, 2014
expression of SERCA2a through gene therapy has shown the potential to mitigate some of the contractile dysfunction in the hypertrophied heart45,46; however, the underlying metabolic defects are not corrected and therefore the delivery of ATP for SERCA remains impaired, which has lessened the positive effects of increasing SERCA expression in some studies.47,48
In the diabetic heart there is an increase in diastolic [Ca2+] that is also partially mediated by a reduction in SERCA2a activity.40 There is also evidence that the SR calcium load is decreased.49 Overexpression of SERCA2a in the heart from diabetic animals is associated with improved contractile func-tion50; however, mitochondrial Ca2+ transients and their impact on cardiac metabolism have not yet been studied in the heart of diabetic individuals.
Energy TransferFigure 2 illustrates the energy transfer system within the car-diomyocyte. The diffusion of ATP and ADP is limited in the cell and therefore alternative mechanisms of transfer are re-quired. Much of this transfer is accomplished via the creatine kinase (CK) system.51 CK transfers the phosphoryl groups from ATP to PCr at a rate that is 10× faster than the rate of ATP synthesis by mitochondrial metabolism.52 Classically, this shuttle mechanism was thought to facilitate much of the energy transfer between the mitochondria and the sites of utilization. However, recent work has shown that a significant reduction in
the capacity of this system, either through loss of CK expres-sion or a decrease in available creatine, does not impair signifi-cantly cardiac function under basal conditions.53–55 In addition, under severe metabolic inhibition, direct ATP transfer seems to replace the CK shuttle as the primary energy transfer system.54 Just as microdomains exist between the tight junctional con-tacts of the mitochondria and the SR, interactions between the mitochondria and sights of ATP utilization (ie, MgATPases) regulate energy transfer. The CK null mouse undergoes mito-chondrial reorganization that seems to reduce the diffusion dis-tances between the mitochondria and the sites of utilization.51 Cytoskeletal mutations, which disrupt the structural organiza-tion of mitochondria within the cell and result in diffuse organi-zation of mitochondria within the heart, affect energy transfer and change in the apparent K
m of ADP for ATP synthesis.56,57
The latter point is of experimental significance and suggests, once again that isolated mitochondria behave much differently even than mitochondria in permeabilized cells. Not only does organelle localization minimize perfusion distances, but it also seems to affect enzyme activities/affinities significantly.56
Measurements of ATP turnover in vivo are technically challenging and therefore the relative importance of the dif-ferent phosphate transfer systems needs further investigation. Magnetic resonance spectroscopy–based methods have been used successfully to measure ATP flux via CK (ATP↔PCr) in vivo58,59; however, measuring ATP turnover or direct ATP
Figure 2. Phosphotransfer systems in the heart. ATP is formed within the mitochondria by ATP synthase, which uses the proton gradient across the inner mitochondrial membrane (IMM) to catalyze ATP formation from ADP and Pi (not depicted). The proton gradient is generated by actions of the electron transport chain (shown as complexes I–IV) after donation of an electron by NADH at complex I. Once formed ATP can cross the IMM via the adenine nucleotide transporter (ANT). ANT also brings ADP into the mitochondria from the cytoplasm. The energy contained within ATP is then transferred to the cytoplasmic phosphocreatine (PCr) pool through the actions of a mitochondrial isoform of creatine kinase (CK). PCr is less diffusion limited than ATP and is therefore an efficient means of transmitting energy to the peripheral sites of ATP utilization, depicted as sarcoplasmic reticulum (SR) Ca2+-ATPase (SERCA2a)–dependent calcium reuptake by the sarcoplasmic reticulum and myosin ATPase-dependent cross-bridge cycling within the sarcomere. PCr is converted back to free creatine (Cr) by cytoplasmic CK with the freed energy used for ATP formation and driving the peripheral ATPases. An alternative phosphotransfer pathway (depicted by a dashed line) is for ATP to directly travel from the mitochondria to the sites of utilization independent of the PCr:CK system. As ATP is diffusion limited, the direct transfer of ATP from the mitochondria to a site of utilization requires close localization between the mitochondria and the peripheral ATPase (illustration credit: Ben Smith).
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

Carley et al Mitochondrial Matrix Revisited 721
transfer (ATP↔ADP+Pi) is problematic. Direct measurement of ATP turnover (ATP↔ADP+Pi) requires that the Pi peak can be visualized clearly on a magnetic resonance spectrum. The level of Pi in the heart is low and the peak attributed to Pi over-laps with the much larger peak for 2,3-disphosphoglycerate, a product glycolytic metabolism generated by erythrocytes. Recent work by Zhang and colleagues59,60 has pioneered an ap-proach to measure ATP turnover indirectly by measuring total ATP utilization and subtracting out the measurable component that can be attributed to ATP flux via CK. As will be discussed below, methodologies like these that try to quantify energy transfer/turnover in the intact heart are essential as static levels of metabolites have proven ineffective in adequately describ-ing the energetic status of the failing heart.58
Energy Transfer in Heart FailureThe complete notion of the failing heart as an engine out of fuel was challenged by the creatine-deficient mouse in which the PCr pool was almost completely abolished.53 This mouse model did not show differences in contractile function after myocardial infarction. This led the authors to question wheth-er the failing heart was indeed out of fuel. A follow-up study, however, showed that the mitochondrial organization was un-changed in this model55 and suggests that the tight coupling between the mitochondria and the sites of utilization was maintained, potentially allowing for direct exchange of ATP and ADP between the sites of utilization and the sites of gen-eration to compensate for the loss of CK shuttle.
In the hypertrophied heart, there is initially a decline in PCr pool before any changes in ATP.61,62 The decline in PCr is the result of a decrease in total creatine,63 with the decline in total creatine directly related to the severity of heart failure.64 As the heart moves closer to decompensation, both the PCr and ATP pools begin to decline.65 An important consideration is that even in late failure the concentration of ATP never falls below the K
m of ATP for the ATPase,63 further highlighting the need
for understanding ATP turnover (and not ATP content) as well as localized (rather than global) changes in metabolite concen-trations. Even indexing PCr:ATP can fail to uncover the degree of energetic deficit in the failing heart. Left ventricular hyper-trophy alone is associated with a reduction in the PCr:ATP; however, there is not a further decline in the PCr:ATP in pa-tients that have transitioned to chronic heart failure.58 There is, however, a significant decline in CK flux between left ven-tricular hypertrophy and chronic heart failure.58 Therefore, at a similar PCr:ATP, the delivery of energy via CK can be dramatically different. Interaction between mitochondria and myofibrils is reduced in heart failure, significantly reducing the capacity of direct ATP/ADP transfer between the mitochon-dria and the sites of utilization.66 Less is understood about the phosphotransfer system of the heart in diabetes mellitus, es-pecially in hearts from individuals who also present with obe-sity. The PCr:ATP is decreased in hearts from type 2 diabetic individuals,9 whereas other studies have suggested no change in PCr:ATP under basal conditions in type 1 diabetic hearts.67
Mitochondrial Metabolite TransportersKey metabolite transport proteins regulate substrate entry across outer and IMMs. We will now review the entry of fatty
acids and pyruvate into the mitochondria and how heart failure modifies these processes (illustrated in Figure 3).
Fatty AcidsLong-chain acyl coenzyme A synthetase isoform 1 catalyzes the synthesis of acyl coenzyme A from long-chain fatty ac-ids in the heart.68 Acyl CoA synthetase isoform 1 is expressed on the mitochondrial membrane.68 Hearts from mice lacking cardiac acyl CoA synthetase isoform 1 undergo hypertro-phy, most likely because of increased glucose 6-phosphate activation of the mammalian target of rapamycin.69 The rate-limiting step in fatty acid oxidation is the transport of fatty acids across the mitochondrial membrane.70 Carnitine palmi-toyltransferase 1 (CPT1), located on the outer mitochondrial membrane (Figure 3), controls the entry of fatty acids into the mitochondria by catalyzing acyl transfer from CoA to carni-tine. The heart expresses 2 isoforms of CPT1, muscle and, to a lesser extent, liver (l-CPT1). L-CPT1 is the primary isoform expressed in the neonatal heart when glucose metabolism pre-dominates, whereas muscle CPT1 dominates in adulthood.71 The activity of CPT1 is inhibited by the concentration of mal-onyl CoA in vitro; however, the in vivo relationship between the concentration of malonyl CoA and CPT1 activity is more complicated. The IC
50 for muscle CPT1 inhibition by malo-
nyl CoA is much lower than the intracellular concentration of
Figure 3. Protein transporters regulate metabolic substrate entry into the mitochondrial matrix and control the relative rates of oxidative metabolism. Fatty acyl CoA (FA-CoA) and pyruvate enter the mitochondria through protein transporters. FA-CoA is first converted to fatty acyl-carnitine (FA-carnitine) by carnitine palmitoyltransferase 1 (CPT1) located on the outer mitochondrial membrane (OMM). CPT1 activity is controlled by the concentration of malonyl CoA and therefore the activities of malonyl CoA decarboxylase (MCD) and acetyl CoA carboxylase 2 (ACC2) indirectly regulate CPT1 activity. FA-carnitine enters the mitochondria via carnitine-acylcarnitine translocase where it is then converted back to FA-CoA by CPT2 located on the inner mitochondrial membrane (IMM). FA-CoA is then able to undergo oxidation within the mitochondria to yield acetyl CoA for the citric acid cycle (CAC). Pyruvate enters the mitochondria via the newly discovered mitochondrial pyruvate carriers 1 and 2 (MPC1 and MPC2) protein complex. Pyruvate is then converted to acetyl CoA by the actions of pyruvate dehydrogenase (PDH). Acetyl CoA exerts product inhibition on the activity of PDH. CACT indicates carnitine-acylcarnitine translocase.
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

722 Circulation Research February 14, 2014
malonyl CoA,72,73 suggesting that CPT1 would be largely in-hibited under normal conditions. This is contrary to the known preference of the adult heart for fatty acid oxidation. A recent study by Smith et al73 in skeletal muscle suggests that the IC
50
for malonyl CoA inhibition of CPT1 is an order of magnitude greater in permeabilized muscle fibers compared with isolated mitochondria, and the IC
50 increases with increased palmitoyl
CoA concentration. This work suggests that high intracellular levels of fatty acyl CoA can compete with malonyl CoA to reduce CPT1 inhibition and may require us to re-evaluate how we interpret steady-state levels of malonyl CoA.
The concentration of malonyl CoA in the heart is regulated by 2 competing enzymes: acetyl CoA carboxylase 2 (ACC2) catalyzes the conversion of acetyl CoA to malonyl CoA, whereas malonyl CoA decarboxylase decarboxylates malo-nyl CoA to acetyl CoA.74,75 Mice deficient in ACC2 display a significant increase in fatty acid oxidation,76 whereas mice deficient in MCD show reduced rates of fatty acid oxidation and increased rates of glucose utilization.75
In failing hearts, the relationship between CPT1 activity and malonyl CoA is also becoming more complex than was previously appreciated. The ACC2-deficient mouse is protect-ed from some of the metabolic remodeling after aortic band-ing, and this correlates with a reduction in malonyl CoA levels and an increase in fatty acid oxidation.76 However, the expres-sion of l-CPT1 is significantly increased in the hypertrophied heart.77 L-CPT1 expression itself induces reduced fatty acid oxidation in whole hearts,78 independent of changes in malo-nyl CoA levels; a curious finding considering that l-CPT1 is much less sensitive to malonyl CoA inhibition.72 In the dia-betic heart, increased fatty acid oxidation is associated with elevated malonyl CoA.79 The elevated fatty acyl CoA content of diabetic hearts may lessen the impact of malonyl CoA on CPT1 activity.
The total CPT1 activity is decreased in the hypertrophied heart,77 which is in agreement with a decrease in the mitochon-drial metabolism of fatty acids. Studies in mice with either deficient muscle CPT1 to limit fatty acid oxidation80 (although with uncertain responses in the l-CPT1 isoform expression) or ACC2 knockout,76 to promote fatty acid oxidation, sug-gest that decrease in fatty acid oxidation in the hypertrophic heart is a maladaptive response. In diabetes mellitus, the heart shows no change in CPT1 activity at a time point where there is an observable increase in fatty acid oxidation,79 indicating that at least in the diabetic heart the delivery of substrates to the mitochondria, rather than the activity of the transport sys-tem across the mitochondria, is a more important determinant of fatty acid oxidation.
PyruvatePyruvate is the penultimate intermediate of the glycolytic pathway in the cytosol under aerobic conditions. The metabo-lism of pyruvate by the mitochondria is illustrated in Figure 3. Recent work has identified 2 proteins that are important com-ponents of the long sought after pyruvate transporter: mito-chondrial pyruvate carriers 1 and 2.81 Their expression was sufficient to promote pyruvate uptake in a model system, and mutations were identified in families displaying a significant impairment in pyruvate oxidation. How these proteins are
affected in heart failure is a point to consider and further in-vestigate. After transport into the mitochondria, pyruvate is converted to acetyl CoA by the PDH complex, feeding into the CAC. The CAC represents the convergence point for all oxidative metabolism, including glucose and fatty acids, with the acetyl CoA formed during β-oxidation also entering the CAC. Any distinction between acetyl CoA derived from these separate sources remains unknown, and none has been demonstrated.82,83
In both the hypertrophied heart and the heart in diabetes mellitus, the rate of pyruvate metabolism is reduced relative to the glycolytic rate.22,77,84 In both pathologies increasing flux through PDH has positive effects on reversing both the meta-bolic and contractile remodeling.22,85 Pyruvate provision itself has positive inotropic effects in the heart and improves cardiac function in patients with heart failure.86,87 Some of this effect can be traced to increased ATP generation through increased flux of pyruvate through the CAC, but an additional direct effect on the ryanodine receptors has also been suggested.88 Pyruvate seems to minimize increases in diastolic calcium by inhibiting SR Ca2+ release.
In diabetes mellitus, the heart exhibits an increased reliance on fatty acid metabolism, and upregulation of fatty acid utili-zation limits the metabolism of pyruvate. This occurs, in part, because of the fatty acid–induced expression of PDH kinase through increased peroxisome proliferator–activated receptor (PPAR)α activation.89–92 In the hypertrophied heart, the oxida-tion of pyruvate, as a glycolytic product, is mismatched to the elevated rate of glycolysis, which could potentially lead to a buildup of glycolytic intermediates (including lactate) in the cytoplasmic compartment. This mismatch of glycolysis and glucose oxidation is accounted for by elevated pyruvate car-boxylation to form malate.50,69 The hypertrophic heart works to maintain CAC flux by upregulating anaplerosis (described in Metabolic Reserve and Figure 1). In the hypertrophied heart, the elevated anaplerotic flux occurs via carboxylation of pyru-vate through increased expression of the NADPH-dependent, cytosolic malic enzyme-1.22 The consequences of shifting 1 pyruvate molecule from oxidation to anaplerosis reduces ATP generation from 14 to 3 ATP. Anaplerotic entry into the second span of the CAC bypasses several of the NADH-generating re-actions. In addition, the carboxylation of pyruvate to form ma-late via malic enzyme-1 consumes and depletes NADPH. The reduction in NADPH may represent one of the mechanisms responsible for the reduced triacylglyceride content within the hypertrophic myocardium.22,93
Uncoupling Protein 3Uncoupling protein 3 (UCP3) was originally proposed to act as an uncoupler of mitochondrial metabolism by dissipating some of the proton gradient across the IMM and preventing reactive oxygen species formation.94,95 However, more recent evidence suggests that UCP3 acts as a fatty acid ion carrier, providing an overflow pathway when fatty acids accumulate within the mitochondria and buffering the mitochondrial concentration of CoA.96,97 More significantly, our under-standing as to whether UCP3 expression is a positive adap-tive response to help maintain fatty acid oxidation within the mitochondria or maladaptive is also changing. The induction
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

Carley et al Mitochondrial Matrix Revisited 723
of UCP3 during ischemia in insulin-resistant rat hearts im-proves recovery and increases fatty acid oxidation while also increasing cardiac efficiency.97 The latter point argues against UCP3 inducing uncoupling of ETC flux from ATP synthesis. In addition, ischemia results in a greater decrease in myocardial ATP levels in the UCP3 null mouse.98 This further supports the emerging realization that UCP3 expres-sion is positive in the heart and may not be related to reactive oxygen species production as previously envisioned. Thus, revisiting some of the past observations concerning heart failure and UCP3 expression may be necessary.99,100
Coordinating Metabolic Compartments via Intermediate Exchange Across the
Mitochondrial MembraneAcetyl CoAThe concentrations of acetyl CoA, in both mitochondria and cytoplasm, have a significant impact on metabolic rates and mitochondrial fuel selection. In addition, as discussed later, nuclear acetyl CoA levels can exert significant control over gene expression through post-translational modifica-tion of proteins. As mentioned in Mitochondrial Metabolite Transporters, the cytoplasmic pool of acetyl CoA is thought to originate from exchange of the CAC intermediate ci-trate.83,101,102 Figure 4 illustrates the sources of cytoplasmic acetyl CoA. The primary mechanism whereby intramitochon-drial acetyl CoA levels are buffered occurs through the actions of carnitine acetyltransferase (CAT), which controls the mito-chondrial acetyl CoA concentration by shunting acetyl CoA to acetyl carntine.103,104 CAT helps to maintain the mitochondrial CoA pool and reduces product inhibition by mitochondrial acetyl CoA accumulation (Figure 4). In addition, as will be discussed in Mitochondrial Contribution to Post-Translational
Modification of Proteins, preventing the accumulation of acetyl CoA could have beneficial effects in heart failure as it would lead to reduced acetylation of the mitochondrial pro-teome. Acetyl carnitine, unlike acetyl CoA, can be exported out of the mitochondria and either re-enter the mitochondrial acetyl CoA pool or be exported out of the cell.104 Acetyl carni-tine does not seem to contribute to the cytoplasmic acetyl CoA pool nor to participate in malonyl CoA formation and CPT1 inhibition.104 After adrenergic challenge, pyruvate incorpora-tion into acetyl carnitine is reduced, as acetyl CoA is fully metabolized in response to increased ATP demand.103 The ace-tyl carnitine pool represents a previously unappreciated meta-bolic reserve within the heart.
A muscle-specific CAT null mouse was created recently, and the results suggest an alternative mechanism describing the metabolic inflexibility in heart failure.104 With this mech-anism, contrasting regulation via the Randle cycle,105,106 the ability of pyruvate to inhibit fatty acid oxidation was reduced by CAT loss, and the muscle-specific loss of CAT leads to glucose intolerance. Re-expressing CAT restored the capac-ity of pyruvate to inhibit fatty acid oxidation; however, this inhibition was independent of changes in CPT1 activity. CAT maintains metabolic flexibility by allowing for uninterrupted CAC flux and metabolite clearance irrespective of ETC activ-ity through direct competition of glucose and fatty acid–de-rived acetyl CoA for entry into the CAC. Carnitine deficiency has been documented in heart failure107,108 and pressure over-load hypertrophy specifically.109 Supplementation with carni-tine has improved cardiac function,110 although a metabolic reason for this effect is just now being elucidated. Initially, the carnitine deficit was thought to be limiting for fatty acyl CoA entry into the mitochondria, but now such deficiency is known to limit the buffering capacity for acetyl CoA levels within the mitochondria.104
Figure 4. Intermediate exchange across the mitochondrial membrane buffers the cytoplasmic and mitochondrial acetyl CoA concentrations. The concentration of acetyl CoA formed during both pyruvate and fatty acyl CoA (FA-CoA) oxidation is buffered within the mitochondrial matrix by carnitine acetyltransferase (CAT). CAT converts acetyl CoA to acetyl carnitine. Acetyl carnitine can then be exported out of the mitochondria by carnitine-acylcarnitine translocase and potentially be cleared from the cell into the circulation or re-enter the mitochondria to undergo oxidative metabolism. Cytoplasmic acetyl CoA does not originate from mitochondrial acetyl CoA but rather from citric acid cycle (CAC) intermediate citrate, which is converted to acetyl CoA via ATP citrate lyase (ACL). Partial oxidation of FA-CoA in the peroxisome has also been suggested to contribute to the cytoplasmic acetyl CoA pool. The activities of malonyl CoA decarboxylase (MCD) and acetyl CoA carboxylase 2 (ACC2) regulate the ratio of cytoplasmic acetyl CoA to malonyl CoA. CACT indicates carnitine-acylcarnitine translocase; IMI, inner mitochondrial membrane; and OMM, outer mitochondrial membrane.
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

724 Circulation Research February 14, 2014
NAD+/NADH+H+
The NAD+/NADH ratio can influence cardiac metabolism at several levels.111 Flux through glycolysis is regulated by the NAD+/NADH ratio via the activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and lactate dehydro-genase. NAD+ is reduced to NADH by GAPDH, and NADH must be continually reoxidized to NAD+ in the mitochondria to maintain glycolytic flux. The IMM is impermeable to pyri-dine nucleotides and therefore alternative mechanisms have been devised to transfer reducing equivalents between the cytoplasmic and mitochondrial environments.112 The prima-ry mechanism in the heart is the malate–aspartate shuttle113 discussed in Ca2+ as a Metabolic Regulator (Figure 1). The challenge of pressure overload leads to an initial increase in NADH shuttling, which returns to baseline levels after the de-velopment of compensated hypertrophy and elevates again on decompensation of the hypertrophied myocardium.114 The rate of NADH shuttling is proportional to the coupling of glycoly-sis and glucose oxidation, with glycolysis increasing rapidly with the induction of hypertrophy with proportionally, less of an increase in glucose oxidation at pyruvate dehydrogenase, in part, because of elevated malic enzyme activity.77,114
The ratio of NAD+/NADH influences intramitochondrial substrate metabolism by regulating flux through the PDH com-plex (glucose) and CAC flux through the regulation of dehydro-genases within the CAC (glucose and fatty acids). In addition, the NAD+/NADH ratio may also influence ETC activity. There are also nonmetabolic effects of the NAD+/NADH ratio. SR calcium load is reduced when the cytoplasmic concentration of NADH is increased, and this seems to be mediated with a reduction in SERCA2a activity.115 NAD+ levels are reduced in failing heart possibly because of the downregulation of nicotin-amide phosphoribosyltransferase,116 the rate-limiting enzyme in NAD+ salvage pathway. Nicotinamide phosphoribosyltrans-ferase expression is decreased by pressure overload and nico-tinamide phosphoribosyltransferase overexpression improves metabolic recovery after myocardial infarction.116 Recent data highlight a potential role for changing NAD+ levels to affect post-translational modifications in the failing heart.117,118
Mitochondrial Contribution to Post-Translational Modification of Proteins
Lysine acetylation is now recognized as a significant modi-fier of nonhistone proteins within the nucleus, the myofila-ment and mitochondria that rival protein phosphorylation in terms of the number of targets modified and the significance to cellular function.119 The level of lysine acetylation is a bal-ance between the rate of acetylation by histone acetyl trans-ferases and deacetylation by histone deacetylases (HDACs) named after their initial identification in histone modification. Recent in vitro studies have also suggested that nonenzymat-ic processes may contribute to lysine acetylation, especially within mitochondria.120 Lysine acetylation regulates gene transcription targets and non-nuclear protein targets that are responsible for cardiomyocyte growth and cardiac remodel-ing, cardiac metabolism, reactive oxygen species buffering, and cardiac contractility.119 In a recent undertaking Foster et al119 tried to characterize the total cardiac acetyl-lysine
proteome in guinea pig hearts. The majority of acetylated proteins were identified in the mitochondria, with the major-ity of nonmitochondrial acetylation sites in the sarcomere and cytoskeletal proteins. The significance of these modifications on protein function still requires classification. However, pro-tein acetylation highlights the significant impact that mito-chondrial metabolism holds on the cardiac proteome through the production of acetyl CoA.
Histone Acetyl Transferases and Heart FailureThe transcriptional co-activator CREB-binding protein and p300 display histone acetylation activity and have been shown to be integral in the development of cardiac hypertrophy in response to chronic phenylephrine treatment or after myo-cardial infarction.121,122 Overexpression of CREB-binding protein or p300 induces cell remodeling and hypertrophy and these changes are dependent on acetylase activity.121 Both CREB-binding protein and p300 are extramitochondrial his-tone acetyl transferases. GCN5L1 was identified recently as an acetyltransferase, capable of localizing to the mitochondria in various cells lines, that may mediate acetylation of the mi-tochondrial proteome.123 A role for GCNL51 in cardiac mito-chondria still needs to be investigated. Others have suggested that a significant portion of mitochondrial acetylation could be nonenzymatic.120 Incubating isolated heart or liver mitochon-drial proteins in a model environment meant to recapitulate the environment of the mitochondrial matrix induces extensive protein acetylation in the presence of acetyl CoA. The level of acetylation was unaffected by previously denaturing the mito-chondrial proteins or increasing the concentration of CoA, both of which should reduce the activity acetyltransferases. Under diseased states in which acetyl CoA accumulates within mito-chondria, nonenzymatic acetylation could become significant.
HDACs and Heart FailureA great deal more research has been done on the impact of HDACs on cardiac function and heart failure. The HDACs are a varied class of enzymes that display both detrimental and positive effects on cardiac function.124 The sirtuins, specifi-cally sirtuin1 (Sirt1) and sirtuin3 (Sirt3), are class III HDACs that are generally thought to be cardiac protective and exert significant control over cardiac metabolism.118,124,125 Sirt1 is thought to localize to the nucleus and controls gene transcrip-tion through both histone and nonhistone deacetylation. Sirt3 is found in the mitochondria and regulates cardiac metabo-lism and mitochondrial fuel selection. The activity of Sirt3 is increased by caloric restriction, and the deacetylation of mitochondrial proteins is a key event of the cardio-protective effects of caloric restriction.126 The class III HDACs coordi-nate metabolic rates with protein function as their activities are dependent on NAD+ and their actions are opposed by the production of acetyl CoA. In contrast, the class I and class II HDACs are zinc-dependent and therefore represent less of an integrative control system between cardiac metabolic rates and protein function.127
In heart failure there is a decrease in NAD+ and increased mitochondrial protein acetylation, whereas NAD+ supplemen-tation reduces the level of acetylation and prevents some of the hypertrophic remodeling.117,128 Inhibiting complex I of
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

Carley et al Mitochondrial Matrix Revisited 725
the ETC in mouse hearts through genetic knockdown of one of the subunits of complex I reduced NADH oxidation and the NAD+/NADH ratio and increased mitochondrial protein acetylation and sensitivity to hypertrophic remodeling after pressure overload.129 Therefore, not only is the absolute level of NAD+ important in regulating Sirt3 activity, so too is the ra-tio of NAD+/NADH. Surprisingly, even stimuli that normally induce physiological hypertrophy lead to pathological remod-eling when protein acetylation is increased by inhibiting com-plex I.129 This suggests that one of the factors that differentiate pathological from physiological hypertrophy could be the ac-tivity of Sirt3. This is in agreement with a previous study that demonstrated that physiological hypertrophy leads to a larger increase in total Sirt3 expression than does pressure overload or chronic isoproterenol infusion.130
A model emerges of acetylation acting as a break when rates of acetyl CoA formation exceed oxidative metabolism, whereas Sirt3 acts in opposition to maintain CAC flux as ace-tyl CoA increases. Just as in any biological system, a balance between such opposing forces is most likely required to pre-vent metabolic dysfunction.
Metabolic Signals as Atrophic and Hypertrophic Remodelers
Thus far, we have discussed adaptive responses in mitochon-drial metabolism and how they may contribute to the ob-served limitation in metabolic reserve in the failing heart. What is also possible is that some of the metabolic changes that occur in the development of heart failure could mediate the hypertrophic remodeling. Although we have discussed the heart in diabetes mellitus as being pathogenically dif-ferent pathology from the hypertrophied heart, there is an
emerging realization that diabetes mellitus and obesity can result in hypertrophic remodeling even in normotensive pa-tients or animal models of diabetes mellitus.131,132 Because the diabetic heart is characterized by an oversupply of nu-trients, a concept that emerged was the induction of hyper-trophy was because of an increase in metabolic load.133 In animal models of diabetes mellitus, the use of thiazolidin-ediones and other nonthiazolidinediones results in improved circulating substrate milieu and a reversal of both cardiac metabolism and contractile function.10,11
In models of hypertrophy, changing mitochondrial metabo-lism has led to reduced left ventricular remodeling and hyper-trophy, suggesting that mitochondrial activity can also exert some control over hypertrophy and atrophy. As an example, the knockdown of ACC2 prevents much of the hypertrophic remodeling in response to pressure overload by preventing the classic substrate switching observed in the hypertrophic heart.76 As we have already discussed, the sirtuins may play a role in this capacity. An additional candidate for integrat-ing the metabolic and growth signals in response to mito-chondrial metabolism is the nutrient sensor AMP kinase.134,135 Signaling through AMP kinase has been shown to regulate protein degradation through E3 ligases.134 Pressure-overload hypertrophy and diabetes mellitus induce increased flux of glucose through the hexosamine biosynthesis pathways, gen-erating O-linked N-acetylglucosamine. Inhibiting O-linked N-acetylglucosamine accumulation reduces hypertrophic remodeling and heart failure.136,137 Although studies such as these are in their infancy,138 changes in mitochondrial metab-olism observed in the failing heart may not be simply mal-adaptive responses that are induced by structural changes, but rather mediate the structural changes.139
Figure 5. Turnover of long-chain fatty acids (LCFAs) within the myocardial triacylglyceride (TAG) pool and their importance in myocardial signaling. LCFAs, found within the circulation either complexed to albumin or esterified into triacylglyceride rich lipoproteins (TG-lipoprotein), enter the cell via a protein-mediated mechanism across the sarcolemma that is sensitive to CD36 expression. The insulin sensitive fatty acid transport protein 1 (FATP1) and the cardiac-specific FATP6 have also been implicated in LCFA uptake by the heart; however, at present only a direct effect of CD36 expression on TAG dynamics has been demonstrated.140 Lipoprotein lipase (LPL) is required to liberate LCFA from their esterified form in TG-lipoprotein. After entry into the myocyte LCFA can either enter the mitochondria for oxidation or cycle through the TAG pool (TAG turnover). TAG turnover is defined by the diacylglycerol acyltransferase 1 (DGAT1) dependent on rate (esterification) and the adipose triglyceride lipase (ATGL) dependent on off rate (lipolysis). After LCFA cycling through the TAG pool, LCFA can then be transported into the mitochondria for oxidation or used to initiate gene transcription via peroxisome proliferator–activated receptor α (PPARα). LCFA must first be cycled through the TAG pool to efficiently activate gene transcription.
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

726 Circulation Research February 14, 2014
Triglycerides as a Source of Signaling Molecules
The myocardial triacylglyceride pool was initially viewed as an inert storage depot; however, recent work has identified the tria-cylglyceride pool as a dynamic organelle within the heart that provides key signaling molecules for the regulation of myocar-dial metabolism140,141 while buffering the intracellular availabil-ity of fatty acids for oxidation.142 Figure 5 illustrates the key role played by the triacylglyceride pool in regulating PPARα activ-ity. PPARα controls the expression of many of the regulatory enzymes responsible for fatty acid oxidation in the heart,143 with increased PPARα expression coinciding with increased fatty acid utilization.90,144 It was shown recently that fatty acids lib-erated from the triacylglyceride pool by the actions of adipose triglyceride lipase are required to activate PPARα signaling.141 Cardiac-specific loss of adipose triglyceride lipase expression prevents PPARα activation in the heart and suggests that fatty acids must first be esterified into the triacylglyceride pool to sig-nal through the PPARα as fatty acid uptake was not impaired in this model. Following up on this work, we have determined that the uptake of fatty acids into the cell and the turnover of fatty ac-ids within the triacylglyceride pool are reciprocally regulated.140 This ensures that the uptake of fatty acids and triacylglyceride turnover are proportional so that rates of fatty acid uptake and mitochondrial metabolism are appropriately matched.
In heart failure, there is dysregulation in triacylglyceride turnover. In the hypertrophic heart we have shown that triacyl-glyceride turnover is decreased, and this decrease is a key lim-itation in substrate provision to the mitochondria.93 A decrease in myocardial adipose triglyceride lipase activity exacerbates the hypertrophic response to pressure overload, whereas adi-pose triglyceride lipase overexpression is protective.145,146 In opposition to the hypertrophic heart, the diabetic heart shows increased triacylglyceride turnover.147 This may explain some of the upregulation of PPARα activity and downstream targets observed in this model. What is striking, however, is that these 2 different models both show increased ceramide content, in-dicating a mismatch between fatty acid uptake, triacylglycer-ide turnover, PPARα activation.
ConclusionsThe past 2 decades have witnessed dramatic changes in the per-ception of cardiac metabolism. Here, we have drawn attention to a new understanding of mitochondrial physiology beyond the synthesis of ATP, to the importance of metabolic signals in the adaptive and maladaptive responses of the heart to a variety of environmental changes, to the importance of the turnover of endogenous substrates (eg, triacylglycerides), and to the im-portance of moiety conserved cycles for the efficient transfer of energy in the context of metabolic flexibility. Our review is far from a complete presentation of cardiac metabolism. Yet, we hope it offers new impulses for further investigations.
DisclosuresNone.
References 1. Balaban RS. Cardiac energy metabolism homeostasis: role of cytosolic
calcium. J Mol Cell Cardiol. 2002;34:1259–1271.
2. Mootha VK, Arai AE, Balaban RS. Maximum oxidative phosphorylation capacity of the mammalian heart. Am J Physiol. 1997;272:H769–H775.
3. Aubert G, Vega RB, Kelly DP. Perturbations in the gene regulatory path-ways controlling mitochondrial energy production in the failing heart. Biochim Biophys Acta. 2013;1833:840–847.
4. Kassiotis C, Rajabi M, Taegtmeyer H. Metabolic reserve of the heart: the forgotten link between contraction and coronary flow. Prog Cardiovasc Dis. 2008;51:74–88.
5. Bache RJ, Zhang J, Murakami Y, Zhang Y, Cho YK, Merkle H, Gong G, From AH, Ugurbil K. Myocardial oxygenation at high workstates in hearts with left ventricular hypertrophy. Cardiovasc Res. 1999;42:616–626.
6. Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interac-tions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–616.
7. Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81:412–419.
8. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. 2007;356:1140–1151.
9. Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, Radda GK, Neubauer S, Clarke K. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. 2003;107:3040–3046.
10. Golfman LS, Wilson CR, Sharma S, Burgmaier M, Young ME, Guthrie PH, Van Arsdall M, Adrogue JV, Brown KK, Taegtmeyer H. Activation of PPARgamma enhances myocardial glucose oxidation and improves contractile function in isolated working hearts of ZDF rats. Am J Physiol Endocrinol Metab. 2005;289:E328–E336.
11. Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000;97:1784–1789.
12. Razeghi P, Young ME, Cockrill TC, Frazier OH, Taegtmeyer H. Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation. 2002;106:407–411.
13. Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H. Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes. 2002;51:2587–2595.
14. Baldwin JE, Krebs H. The evolution of metabolic cycles. Nature. 1981;291:381–382.
15. Balaban RS, Kantor HL, Katz LA, Briggs RW. Relation between work and phosphate metabolite in the in vivo paced mammalian heart. Science. 1986;232:1121–1123.
16. Cooney GJ, Taegtmeyer H, Newsholme EA. Tricarboxylic acid cycle flux and enzyme activities in the isolated working rat heart. Biochem J. 1981;200:701–703.
17. O’Donnell JM, Doumen C, LaNoue KF, White LT, Yu X, Alpert NM, Lewandowski ED. Dehydrogenase regulation of metabolite oxidation and ef-flux from mitochondria in intact hearts. Am J Physiol. 1998;274:H467–H476.
18. Taegtmeyer H. Tracing cardiac metabolism in vivo: one substrate at a time. J Nucl Med. 2010;51 Suppl 1:80S–87S.
19. Russell RR 3rd, Taegtmeyer H. Pyruvate carboxylation prevents the de-cline in contractile function of rat hearts oxidizing acetoacetate. Am J Physiol. 1991;261:H1756–H1762.
20. Gibala MJ, Young ME, Taegtmeyer H. Anaplerosis of the citric acid cy-cle: role in energy metabolism of heart and skeletal muscle. Acta Physiol Scand. 2000;168:657–665.
21. Panchal AR, Comte B, Huang H, Kerwin T, Darvish A, des Rosiers C, Brunengraber H, Stanley WC. Partitioning of pyruvate between oxida-tion and anaplerosis in swine hearts. Am J Physiol Heart Circ Physiol. 2000;279:H2390–H2398.
22. Pound KM, Sorokina N, Ballal K, Berkich DA, Fasano M, Lanoue KF, Taegtmeyer H, O’Donnell JM, Lewandowski ED. Substrate-enzyme com-petition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res. 2009;104:805–812.
23. Vincent G, Comte B, Poirier M, Rosiers CD. Citrate release by perfused rat hearts: a window on mitochondrial cataplerosis. Am J Physiol Endocrinol Metab. 2000;278:E846–E856.
24. Russell RR 3rd, Taegtmeyer H. Changes in citric acid cycle flux and anaplerosis antedate the functional decline in isolated rat hearts utilizing acetoacetate. J Clin Invest. 1991;87:384–390.
25. Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and dis-eased heart. Ann N Y Acad Sci. 2004;1015:202–213.
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

Carley et al Mitochondrial Matrix Revisited 727
26. Dorn GW 2nd, Maack C. SR and mitochondria: calcium cross-talk be-tween kissing cousins. J Mol Cell Cardiol. 2013;55:42–49.
27. Liu T, Brown DA, O’Rourke B. Role of mitochondrial dysfunction in car-diac glycoside toxicity. J Mol Cell Cardiol. 2010;49:728–736.
28. Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential com-ponent of the mitochondrial calcium uniporter. Nature. 2011;476:341–345.
29. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uni-porter is a highly selective ion channel. Nature. 2004;427:360–364.
30. Mallilankaraman K, Cárdenas C, Doonan PJ, et al. MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabo-lism. Nat Cell Biol. 2012;14:1336–1343.
31. Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151:630–644.
32. Chen Y, Csordás G, Jowdy C, Schneider TG, Csordás N, Wang W, Liu Y, Kohlhaas M, Meiser M, Bergem S, Nerbonne JM, Dorn GW 2nd, Maack C. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ Res. 2012;111:863–875.
33. Andrienko TN, Picht E, Bers DM. Mitochondrial free calcium regulation during sarcoplasmic reticulum calcium release in rat cardiac myocytes. J Mol Cell Cardiol. 2009;46:1027–1036.
34. Pacher P, Csordás P, Schneider T, Hajnóczky G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochon-dria. J Physiol. 2000;529 Pt 3:553–564.
35. Maack C, O’Rourke B. Excitation-contraction coupling and mitochondrial energetics. Basic Res Cardiol. 2007;102:369–392.
36. Contreras L, Gomez-Puertas P, Iijima M, Kobayashi K, Saheki T, Satrústegui J. Ca2+ Activation kinetics of the two aspartate-glutamate mi-tochondrial carriers, aralar and citrin: role in the heart malate-aspartate NADH shuttle. J Biol Chem. 2007;282:7098–7106.
37. Kohlhaas M, Liu T, Knopp A, Zeller T, Ong MF, Böhm M, O’Rourke B, Maack C. Elevated cytosolic Na+ increases mitochondrial forma-tion of reactive oxygen species in failing cardiac myocytes. Circulation. 2010;121:1606–1613.
38. Liu T, O’Rourke B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ Res. 2008;103:279–288.
39. Bell CJ, Bright NA, Rutter GA, Griffiths EJ. ATP regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J Biol Chem. 2006;281:28058–28067.
40. Belke DD, Dillmann WH. Altered cardiac calcium handling in diabetes. Curr Hypertens Rep. 2004;6:424–429.
41. Nickel A, Löffler J, Maack C. Myocardial energetics in heart failure. Basic Res Cardiol. 2013;108:358.
42. Bay J, Kohlhaas M, Maack C. Intracellular Na+ and cardiac metabolism. J Mol Cell Cardiol. 2013;61:20–27.
43. Piacentino V 3rd, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658.
44. Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium con-tent is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584.
45. Ito K, Yan X, Feng X, Manning WJ, Dillmann WH, Lorell BH. Transgenic expression of sarcoplasmic reticulum Ca(2+) atpase modifies the transi-tion from hypertrophy to early heart failure. Circ Res. 2001;89:422–429.
46. Miyamoto MI, del Monte F, Schmidt U, DiSalvo TS, Kang ZB, Matsui T, Guerrero JL, Gwathmey JK, Rosenzweig A, Hajjar RJ. Adenoviral gene transfer of SERCA2a improves left-ventricular function in aortic-banded rats in transition to heart failure. Proc Natl Acad Sci USA. 2000;97:793–798.
47. O’Donnell JM, Fields A, Xu X, Chowdhury SA, Geenen DL, Bi J. Limited functional and metabolic improvements in hypertrophic and healthy rat heart overexpressing the skeletal muscle isoform of SERCA1 by adenoviral gene transfer in vivo. Am J Physiol Heart Circ Physiol. 2008;295:H2483–H2494.
48. Pinz I, Tian R, Belke D, Swanson E, Dillmann W, Ingwall JS. Compromised myocardial energetics in hypertrophied mouse hearts di-minish the beneficial effect of overexpressing SERCA2a. J Biol Chem. 2011;286:10163–10168.
49. Lagadic-Gossmann D, Buckler KJ, Le Prigent K, Feuvray D. Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. Am J Physiol. 1996;270:H1529–H1537.
50. Trost SU, Belke DD, Bluhm WF, Meyer M, Swanson E, Dillmann WH. Overexpression of the sarcoplasmic reticulum Ca(2+)-ATPase im-proves myocardial contractility in diabetic cardiomyopathy. Diabetes. 2002;51:1166–1171.
51. Kaasik A, Veksler V, Boehm E, Novotova M, Minajeva A, Ventura-Clapier R. Energetic crosstalk between organelles: architectural integration of en-ergy production and utilization. Circ Res. 2001;89:153–159.
52. Bittl JA, Ingwall JS. Reaction rates of creatine kinase and ATP synthesis in the isolated rat heart. A 31P NMR magnetization transfer study. J Biol Chem. 1985;260:3512–3517.
53. Lygate CA, Aksentijevic D, Dawson D, ten Hove M, Phillips D, de Bono JP, Medway DJ, Sebag-Montefiore L, Hunyor I, Channon KM, Clarke K, Zervou S, Watkins H, Balaban RS, Neubauer S. Living without creatine: unchanged exercise capacity and response to chronic myocardial infarc-tion in creatine-deficient mice. Circ Res. 2013;112:945–955.
54. Vendelin M, Hoerter JA, Mateo P, Soboll S, Gillet B, Mazet JL. Modulation of energy transfer pathways between mitochondria and myofibrils by changes in performance of perfused heart. J Biol Chem. 2010;285:37240–37250.
55. Branovets J, Sepp M, Kotlyarova S, Jepihhina N, Sokolova N, Aksentijevic D, Lygate CA, Neubauer S, Vendelin M, Birkedal R. Unchanged mito-chondrial organization and compartmentation of high-energy phosphates in creatine-deficient GAMT-/- mouse hearts. Am J Physiol Heart Circ Physiol. 2013;305:H506–H520.
56. Saks V, Guzun R, Timohhina N, Tepp K, Varikmaa M, Monge C, Beraud N, Kaambre T, Kuznetsov A, Kadaja L, Eimre M, Seppet E. Structure-function relationships in feedback regulation of energy fluxes in vivo in health and disease: mitochondrial interactosome. Biochim Biophys Acta. 2010;1797:678–697.
57. Wilding JR, Joubert F, de Araujo C, Fortin D, Novotova M, Veksler V, Ventura-Clapier R. Altered energy transfer from mitochondria to sarco-plasmic reticulum after cytoarchitectural perturbations in mice hearts. J Physiol. 2006;575:191–200.
58. Smith CS, Bottomley PA, Schulman SP, Gerstenblith G, Weiss RG. Altered creatine kinase adenosine triphosphate kinetics in failing hyper-trophied human myocardium. Circulation. 2006;114:1151–1158.
59. Xiong Q, Ye L, Zhang P, Lepley M, Tian J, Li J, Zhang L, Swingen C, Vaughan JT, Kaufman DS, Zhang J. Functional consequences of human induced pluripotent stem cell therapy: myocardial ATP turnover rate in the in vivo swine heart with postinfarction remodeling. Circulation. 2013;127:997–1008.
60. Xiong Q, Du F, Zhu X, Zhang P, Suntharalingam P, Ippolito J, Kamdar FD, Chen W, Zhang J. ATP production rate via creatine kinase or ATP synthase in vivo: a novel superfast magnetization saturation transfer method. Circ Res. 2011;108:653–663.
61. Shen W, Spindler M, Higgins MA, Jin N, Gill RM, Bloem LJ, Ryan TP, Ingwall JS. The fall in creatine levels and creatine kinase isozyme changes in the failing heart are reversible: complex post-transcriptional regulation of the components of the CK system. J Mol Cell Cardiol. 2005;39:537–544.
62. Shen W, Asai K, Uechi M, Mathier MA, Shannon RP, Vatner SF, Ingwall JS. Progressive loss of myocardial ATP due to a loss of total purines dur-ing the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation. 1999;100:2113–2118.
63. Ingwall JS, Weiss RG. Is the failing heart energy starved? On using chemi-cal energy to support cardiac function. Circ Res. 2004;95:135–145.
64. Nakae I, Mitsunami K, Omura T, Yabe T, Tsutamoto T, Matsuo S, Takahashi M, Morikawa S, Inubushi T, Nakamura Y, Kinoshita M, Horie M. Proton magnetic resonance spectroscopy can detect creatine depletion associated with the progression of heart failure in cardiomyopathy. J Am Coll Cardiol. 2003;42:1587–1593.
65. Beer M, Seyfarth T, Sandstede J, Landschütz W, Lipke C, Köstler H, von Kienlin M, Harre K, Hahn D, Neubauer S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnet-ic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–1274.
66. Joubert F, Wilding JR, Fortin D, Domergue-Dupont V, Novotova M, Ventura-Clapier R, Veksler V. Local energetic regulation of sarcoplas-mic and myosin ATPase is differently impaired in rats with heart failure. J Physiol. 2008;586:5181–5192.
67. Spindler M, Saupe KW, Tian R, Ahmed S, Matlib MA, Ingwall JS. Altered creatine kinase enzyme kinetics in diabetic cardiomyopathy. A(31)P NMR
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

728 Circulation Research February 14, 2014
magnetization transfer study of the intact beating rat heart. J Mol Cell Cardiol. 1999;31:2175–2189.
68. Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman GI, Willis MS, Coleman RA. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy. Mol Cell Biol. 2011;31:1252–1262.
69. Sen S, Kundu BK, Wu HC, et al. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2013;2:e004796.
70. Eaton S. Control of mitochondrial beta-oxidation flux. Prog Lipid Res. 2002;41:197–239.
71. Onay-Besikci A, Campbell FM, Hopkins TA, Dyck JR, Lopaschuk GD, Onay Besikci A. Relative importance of malonyl CoA and carnitine in maturation of fatty acid oxidation in newborn rabbit heart. Am J Physiol Heart Circ Physiol. 2003;284:H283–H289.
72. McGarry JD, Mills SE, Long CS, Foster DW. Observations on the af-finity for carnitine, and malonyl-CoA sensitivity, of carnitine palmi-toyltransferase I in animal and human tissues. Demonstration of the presence of malonyl-CoA in non-hepatic tissues of the rat. Biochem J. 1983;214:21–28.
73. Smith BK, Perry CG, Koves TR, Wright DC, Smith JC, Neufer PD, Muoio DM, Holloway GP. Identification of a novel malonyl-CoA IC(50) for CPT-I: implications for predicting in vivo fatty acid oxidation rates. Biochem J. 2012;448:13–20.
74. Saddik M, Gamble J, Witters LA, Lopaschuk GD. Acetyl-CoA car-boxylase regulation of fatty acid oxidation in the heart. J Biol Chem. 1993;268:25836–25845.
75. Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, Kawase Y, Jishage K, Lopaschuk GD. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation. 2006;114:1721–1728.
76. Kolwicz SC Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111:728–738.
77. Sorokina N, O’Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K, Taegtmeyer H, Buttrick PM, Lewandowski ED. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes in-efficiency in energy metabolism in hypertrophied hearts. Circulation. 2007;115:2033–2041.
78. Lewandowski ED, Fischer SK, Fasano M, Banke NH, Walker LA, Huqi A, Wang X, Lopaschuk GD, O’Donnell JM. Acute liver carnitine palmitoyl-transferase I overexpression recapitulates reduced palmitate oxidation of cardiac hypertrophy. Circ Res. 2013;112:57–65.
79. Carley AN, Atkinson LL, Bonen A, Harper ME, Kunnathu S, Lopaschuk GD, Severson DL. Mechanisms responsible for enhanced fatty acid utili-zation by perfused hearts from type 2 diabetic db/db mice. Arch Physiol Biochem. 2007;113:65–75.
80. He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, Ding Y, Prasain J, Wood PA, Yang Q. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation. 2012;126:1705–1716.
81. Bricker DK, Taylor EB, Schell JC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. 2012;337:96–100.
82. Reszko AE, Kasumov T, David F, Jobbins KA, Thomas KR, Hoppel CL, Brunengraber H, Des Rosiers C. Peroxisomal fatty acid oxidation is a sub-stantial source of the acetyl moiety of malonyl-CoA in rat heart. J Biol Chem. 2004;279:19574–19579.
83. Poirier M, Vincent G, Reszko AE, Bouchard B, Kelleher JK, Brunengraber H, Des Rosiers C. Probing the link between citrate and malonyl-CoA in perfused rat hearts. Am J Physiol Heart Circ Physiol. 2002;283:H1379–H1386.
84. Belke DD, Larsen TS, Lopaschuk GD, Severson DL. Glucose and fat-ty acid metabolism in the isolated working mouse heart. Am J Physiol. 1999;277:R1210–R1217.
85. How OJ, Larsen TS, Hafstad AD, Khalid A, Myhre ES, Murray AJ, Boardman NT, Cole M, Clarke K, Severson DL, Aasum E. Rosiglitazone treatment improves cardiac efficiency in hearts from diabetic mice. Arch Physiol Biochem. 2007;113:211–220.
86. Hermann HP, Zeitz O, Keweloh B, Hasenfuss G, Janssen PM. Pyruvate po-tentiates inotropic effects of isoproterenol and Ca(2+) in rabbit cardiac mus-cle preparations. Am J Physiol Heart Circ Physiol. 2000;279:H702–H708.
87. Hasenfuss G, Maier LS, Hermann HP, Lüers C, Hünlich M, Zeitz O, Janssen PM, Pieske B. Influence of pyruvate on contractile performance and Ca(2+) cycling in isolated failing human myocardium. Circulation. 2002;105:194–199.
88. Zima AV, Kockskämper J, Mejia-Alvarez R, Blatter LA. Pyruvate modulates cardiac sarcoplasmic reticulum Ca2+ release in rats via mitochondria-dependent and -independent mechanisms. J Physiol. 2003;550:765–783.
89. Wu P, Sato J, Zhao Y, Jaskiewicz J, Popov KM, Harris RA. Starvation and diabetes increase the amount of pyruvate dehydrogenase kinase iso-enzyme 4 in rat heart. Biochem J. 1998;329(Pt 1):197–201.
90. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP. The cardiac pheno-type induced by PPARalpha overexpression mimics that caused by dia-betes mellitus. J Clin Invest. 2002;109:121–130.
91. Houten SM, Chegary M, Te Brinke H, Wijnen WJ, Glatz JF, Luiken JJ, Wijburg FA, Wanders RJ. Pyruvate dehydrogenase kinase 4 expression is synergistically induced by AMP-activated protein kinase and fatty acids. Cell Mol Life Sci. 2009;66:1283–1294.
92. Taegtmeyer H, McNulty P, Young ME. Adaptation and maladapta-tion of the heart in diabetes: Part I: general concepts. Circulation. 2002;105:1727–1733.
93. O’Donnell JM, Fields AD, Sorokina N, Lewandowski ED. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol. 2008;44:315–322.
94. Murray AJ, Cole MA, Lygate CA, Carr CA, Stuckey DJ, Little SE, Neubauer S, Clarke K. Increased mitochondrial uncoupling proteins, respiratory uncoupling and decreased efficiency in the chronically in-farcted rat heart. J Mol Cell Cardiol. 2008;44:694–700.
95. Murray AJ, Panagia M, Hauton D, Gibbons GF, Clarke K. Plasma free fatty acids and peroxisome proliferator-activated receptor al-pha in the control of myocardial uncoupling protein levels. Diabetes. 2005;54:3496–3502.
96. Himms-Hagen J, Harper ME. Physiological role of UCP3 may be export of fatty acids from mitochondria when fatty acid oxidation predominates: an hypothesis. Exp Biol Med (Maywood). 2001;226:78–84.
97. Harmancey R, Vasquez HG, Guthrie PH, Taegtmeyer H. Decreased long-chain fatty acid oxidation impairs postischemic recovery of the insulin-resistant rat heart. FASEB J. 2013;27:3966–3978.
98. Ozcan C, Palmeri M, Horvath TL, Russell KS, Russell RR 3rd. Role of uncoupling protein 3 in ischemia-reperfusion injury, arrhythmias, and preconditioning. Am J Physiol Heart Circ Physiol. 2013;304: H1192–H1200.
99. Murray AJ, Anderson RE, Watson GC, Radda GK, Clarke K. Uncoupling proteins in human heart. Lancet. 2004;364:1786–1788.
100. Opie LH. The metabolic vicious cycle in heart failure. Lancet. 2004;364:1733–1734.
101. Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080.
102. Randle PJ. Regulation of glycolysis and pyruvate oxidation in cardiac muscle. Circ Res. 1976;38:I8–15.
103. Schroeder MA, Atherton HJ, Dodd MS, Lee P, Cochlin LE, Radda GK, Clarke K, Tyler DJ. The cycling of acetyl-coenzyme A through acetylcar-nitine buffers cardiac substrate supply: a hyperpolarized 13C magnetic resonance study. Circ Cardiovasc Imaging. 2012;5:201–209.
104. Muoio DM, Noland RC, Kovalik JP, et al. Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell Metab. 2012;15:764–777.
105. Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic distur-bances of diabetes mellitus. Lancet. 1963;1:785–789.
106. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578–E591.
107. Paulson DJ. Carnitine deficiency-induced cardiomyopathy. Mol Cell Biochem. 1998;180:33–41.
108. Waber LJ, Valle D, Neill C, DiMauro S, Shug A. Carnitine deficiency presenting as familial cardiomyopathy: a treatable defect in carnitine transport. J Pediatr. 1982;101:700–705.
109. Reibel DK, Uboh CE, Kent RL. Altered coenzyme A and carnitine metabolism in pressure-overload hypertrophied hearts. Am J Physiol. 1983;244:H839–H843.
110. Schönekess BO, Allard MF, Lopaschuk GD. Propionyl L-carnitine im-provement of hypertrophied heart function is accompanied by an in-crease in carbohydrate oxidation. Circ Res. 1995;77:726–734.
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

Carley et al Mitochondrial Matrix Revisited 729
111. Ussher JR, Jaswal JS, Lopaschuk GD. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ Res. 2012;111:628–641.
112. Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di Lisa F. Transient and long-lasting openings of the mitochondrial perme-ability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys J. 1999;76:725–734.
113. Griffin JL, O’Donnell JM, White LT, Hajjar RJ, Lewandowski ED. Postnatal expression and activity of the mitochondrial 2-oxoglutarate-malate carrier in intact hearts. Am J Physiol Cell Physiol. 2000;279:C1704–C1709.
114. Lewandowski ED, O’donnell JM, Scholz TD, Sorokina N, Buttrick PM. Recruitment of NADH shuttling in pressure-overloaded and hypertro-phic rat hearts. Am J Physiol Cell Physiol. 2007;292:C1880–C1886.
115. Zima AV, Copello JA, Blatter LA. Effects of cytosolic NADH/NAD(+) levels on sarcoplasmic reticulum Ca(2+) release in permeabilized rat ventricular myocytes. J Physiol. 2004;555:727–741.
116. Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phos-phoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–491.
117. Pillai JB, Isbatan A, Imai S, Gupta MP. Poly(ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2alpha deacetylase activ-ity. J Biol Chem. 2005;280:43121–43130.
118. Sack MN. The role of SIRT3 in mitochondrial homeostasis and cardiac adaptation to hypertrophy and aging. J Mol Cell Cardiol. 2012;52:520–525.
119. Foster DB, Liu T, Rucker J, O’Meally RN, Devine LR, Cole RN, O’Rourke B. The cardiac acetyl-lysine proteome. PLoS One. 2013;8:e67513.
120. Wagner GR, Payne RM. Widespread and enzyme-independent Nε-acetylation and Nε-succinylation of proteins in the chemical condi-tions of the mitochondrial matrix. J Biol Chem. 2013;288:29036–29045.
121. Gusterson RJ, Jazrawi E, Adcock IM, Latchman DS. The transcriptional co-activators CREB-binding protein (CBP) and p300 play a critical role in cardiac hypertrophy that is dependent on their histone acetyltransfer-ase activity. J Biol Chem. 2003;278:6838–6847.
122. Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, Komeda M, Fujita M, Shimatsu A, Kita T, Hasegawa K. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest. 2008;118:868–878.
123. Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular com-ponent of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J. 2012;443:655–661.
124. Xie M, Hill JA. HDAC-dependent ventricular remodeling. Trends Cardiovasc Med. 2013;23:229–235.
125. Tanno M, Kuno A, Horio Y, Miura T. Emerging beneficial roles of sirtu-ins in heart failure. Basic Res Cardiol. 2012;107:273.
126. Sack MN, Finkel T. Mitochondrial metabolism, sirtuins, and aging. Cold Spring Harb Perspect Biol. 2012;4:.
127. Codd R, Braich N, Liu J, Soe CZ, Pakchung AA. Zn(II)-dependent his-tone deacetylase inhibitors: suberoylanilide hydroxamic acid and tricho-statin A. Int J Biochem Cell Biol. 2009;41:736–739.
128. Pillai VB, Sundaresan NR, Kim G, Gupta M, Rajamohan SB, Pillai JB, Samant S, Ravindra PV, Isbatan A, Gupta MP. Exogenous NAD blocks cardi-ac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J Biol Chem. 2010;285:3133–3144.
129. Karamanlidis G, Lee CF, Garcia-Menendez L, Kolwicz SC Jr, Suthammarak W, Gong G, Sedensky MM, Morgan PG, Wang W, Tian R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013;18:239–250.
130. Sundaresan NR, Gupta M, Kim G, Rajamohan SB, Isbatan A, Gupta MP. Sirt3 blocks the cardiac hypertrophic response by augmenting
Foxo3a-dependent antioxidant defense mechanisms in mice. J Clin Invest. 2009;119:2758–2771.
131. Rodrigues SL, Angelo LC, Pereira AC, Krieger JE, Mill JG. Determinants of left ventricular mass and presence of metabolic risk factors in normo-tensive individuals. Int J Cardiol. 2009;135:323–330.
132. Barouch LA, Berkowitz DE, Harrison RW, O’Donnell CP, Hare JM. Disruption of leptin signaling contributes to cardiac hypertrophy inde-pendently of body weight in mice. Circulation. 2003;108:754–759.
133. Algahim MF, Lux TR, Leichman JG, Boyer AF, Miller CC 3rd, Laing ST, Wilson EB, Scarborough T, Yu S, Snyder B, Wolin-Riklin C, Kyle UG, Taegtmeyer H. Progressive regression of left ventricular hypertrophy two years after bariatric surgery. Am J Med. 2010;123:549–555.
134. Baskin KK, Taegtmeyer H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ Res. 2011;109:1153–1161.
135. Baskin KK, Taegtmeyer H. An expanded role for AMP-activated protein kinase: regulator of myocardial protein degradation. Trends Cardiovasc Med. 2011;21:124–127.
136. Darley-Usmar VM, Ball LE, Chatham JC. Protein O-linked β-N-acetylglucosamine: a novel effector of cardiomyocyte metabolism and function. J Mol Cell Cardiol. 2012;52:538–549.
137. Chatham JC, Marchase RB. Protein O-GlcNAcylation: A critical regulator of the cellular response to stress. Curr Signal Transduct Ther. 2010;5:49–59.
138. Young ME, Yan J, Razeghi P, Cooksey RC, Guthrie PH, Stepkowski SM, McClain DA, Tian R, Taegtmeyer H. Proposed regulation of gene expres-sion by glucose in rodent heart. Gene Regul Syst Bio. 2007;1:251–262.
139. Chatham JC, Young ME. Metabolic remodeling in the hypertrophic heart: fuel for thought. Circ Res. 2012;111:666–668.
140. Carley AN, Bi J, Wang X, Banke NH, Dyck JR, O’Donnell JM, Lewandowski ED. Multiphasic triacylglycerol dynamics in the intact heart during acute in vivo overexpression of CD36. J Lipid Res. 2013;54:97–106.
141. Haemmerle G, Moustafa T, Woelkart G, et al. Atgl-mediated fat catabo-lism regulates cardiac mitochondrial function via ppar-alpha and pgc-1. Nat Med. 2011;17:1076–1085
142. Banke NH, Wende AR, Leone TC, O’Donnell JM, Abel ED, Kelly DP, Lewandowski ED. Preferential oxidation of triacylglyceride-derived fat-ty acids in heart is augmented by the nuclear receptor PPARalpha. Circ Res. 2010;107:233–241.
143. Gilde AJ, van der Lee KA, Willemsen PH, Chinetti G, van der Leij FR, van der Vusse GJ, Staels B, van Bilsen M. Peroxisome proliferator-activated receptor (PPAR) alpha and PPARbeta/delta, but not PPARgamma, modu-late the expression of genes involved in cardiac lipid metabolism. Circ Res. 2003;92:518–524.
144. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology. 2005;146:5341–5349.
145. Kienesberger PC, Pulinilkunnil T, Sung MM, Nagendran J, Haemmerle G, Kershaw EE, Young ME, Light PE, Oudit GY, Zechner R, Dyck JR. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dys-function. Mol Cell Biol. 2012;32:740–750.
146. Kienesberger PC, Pulinilkunnil T, Nagendran J, Young ME, Bogner-Strauss JG, Hackl H, Khadour R, Heydari E, Haemmerle G, Zechner R, Kershaw EE, Dyck JR. Early structural and metabolic cardiac remodelling in response to inducible adipose triglyceride lipase ablation. Cardiovasc Res. 2013;99:442–451.
147. O’Donnell JM, Zampino M, Alpert NM, Fasano MJ, Geenen DL, Lewandowski ED. Accelerated triacylglycerol turnover kinetics in hearts of diabetic rats include evidence for compartmented lipid storage. Am J Physiol Endocrinol Metab. 2006;290:E448–E455.
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

Andrew N. Carley, Heinrich Taegtmeyer and E. Douglas Lewandowskithe Heart
Matrix Revisited: Mechanisms Linking Energy Substrate Metabolism to the Function of
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2014 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/CIRCRESAHA.114.3018632014;114:717-729Circ Res.
http://circres.ahajournals.org/content/114/4/717World Wide Web at:
The online version of this article, along with updated information and services, is located on the
/content/114/8/e34.full.pdfAn erratum has been published regarding this article. Please see the attached page for:
http://circres.ahajournals.org/content/suppl/2014/03/13/CIRCRESAHA.114.301863.DC1Data Supplement (unedited) at:
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on June 30, 2018http://circres.ahajournals.org/
Dow
nloaded from

e34
Correction
In the Circulation Research article by Carley et al (Matrix Revisited: Mechanisms Linking Energy Substrate Metabolism to the Function of the Heart. Circ Res. 2014;114:717–729. DOI: 10.1161/CIRCRESAHA.114.301863), a correction was needed.
The correction is to an incorrect word that appears on page 718, left column, 2nd paragraph, line 13: instead of “α-ketoglutarate”, the correct term should be “oxaloacetate”.
The error has been corrected in the online version of the article, which is available at http://circres.ahajournals.org/content/114/4/717.
(Circ Res. 2014;114:e34.)© 2014 American Heart Association, Inc.
Circulation Research is available at http://circres.ahajournals.org DOI: 10.1161/RES.0000000000000019

In the Circulation Research article by Carley et al (Matrix Revisited: Mechanisms Linking Energy Substrate Metabolism to the Function of the Heart. Circ Res. 2014;114:717-729. DOI: 10.1161/CIRCRESAHA.114.301863), a correction was needed. The correction is to an incorrect word that appears on page 718, left column, 2nd paragraph, line 13: instead of “α-ketoglutarate”, the correct term should be “oxaloacetate”. The error has been corrected in the online version of the article, which is available at http://circres.ahajournals.org/content/114/4/717.







