
E L S E V I E R Colloids and Surfaces
A: Physicochemical and Engineering Aspects 107 ( 1996} 245 262
COLLOIDS AND A SURFACES
Colloid release and transport processes in natural and model porous media
Sujoy B. Roy, David A. D z o m b a k *
Department o[ Civil and Environmental Engineering, Carnegie Mell(m University, Pittsburgh, PA 15213
Received 15 March 1995: accepted 21 July 1995
Abstract
Colloid release was observed from packed columns for two natural porous media (sands) and one model system (glass beads with deposited latex colloids). Colloid release was found to occur in all cases when the ionic strength was reduced in columns that were in equilibrium with Na ÷ ions. Most of the released colloids from the natural porous media were smaller than 1 ~tm in size, and comprised pure and impure forms of silica (60 70% by mass) and clay minerals (20-30% by mass). For greater reductions in ionic strength, the total mass of released colloids increased, although the shape of the effluent mass concentration profile did not change. Release rate coefficients were obtained by fitting a colloid transport model (an advection-dispersion transport model with source/sink terms for colloid release and deposition) to the column effluent data. To fit the data for different ionic strengths, the total available mass of releasable colloids had to be adjusted, and fitted release rate coefficients were not sensitive to the ionic strength. In contrast, calculations based on Derjaguin-Landau-Verwey Overbeek (DLVO) theory indicate a strong dependence of release rate constants on ionic strength for homogeneous colloids. This discrepancy can be attributed to charge and size heterogeneity in the colloids, and to our inability to determine accurately interparticle forces at small separations. The trend of greater mass release for greater reductions in ionic strength could be explained qualitatively by computing interparticle interactions with a constant-charge boundary condition (albeit with a charge density much lower than that experimentally determined) which showed a decreasing energy barrier for particle detachment with decreasing ionic slrength.
Keywords: Colloid release; Colloids; Glass beads; Latex colloids; Natural sands; Packed columns; Porous media
1. Introduction
Most na tura l po rous med ia such as soils and aquifer mater ia l s conta in some col lo idal part icles that are a t t ached to the surfaces of larger fixed part icles or are in a f locculated state. The mass fraction of col loid-s ize part icles (general ly defined as less than 2 gm in diameter) in a subsurface mineral gra in assemblage can vary over a wide range (i.e. from less than 1% to tens of per cent)
* C,~rrcsponding author.
0927-7757/96/$15.00 (9 1996 Elsevier Science B.V. All rights reserved SSI)I 0927-7757( 95)03367-X
and is typical ly higher for surface soils than for aquifer materials . Col lo ida l part icles in na tura l porous med ia are usual ly immobi le dur ing normal electrolyte and water flow condi t ions . However , when the e lec t ros ta t ic repuls ion between part icles is increased, most c o m m o n l y by lowering the ionic strength, they may disperse into the aqueous phase and be t r anspor t ed th rough the po rous medium. An under s t and ing of col loid release under changing ionic s t rength is of env i ronmenta l interest because this is one mechanism by which suspended colloi- dal par t ic les may be in t roduced in aquifers and

246 5:R Roy, D.A. Dzombak/Colloids SurJaces A: Physicochem. Eng. Aspects 107 (1996)245-262
potentially enhance the subsurface transport of contaminants sorbed on their surfaces [1,2].
The colloid release phenomenon has been studied in soil science and petroleum engineering where the primary concern is the decrease of permeability that results from clogging of pores from mobilized colloids in soils and oil-bearing rock formations [3-13]. More fundamental studies of colloid detachment (with a view to relating observed rates to interparticle forces) have been performed with model colloids and porous media [14-20] as well as flat plates [21-24].
The research presented here focused on measur- ing the rates of colloid release under changing ionic strengths from natural porous media of inter- est in contaminant transport, i.e. high permeability materials with low colloid contents. Release rates were also measured for a simpler system: latex colloids deposited in glass-bead-packed columns. The goals of this work were to gain insight into the influence of solution and surface chemistry on colloid release from porous media and to test the utility of available particle-particle interaction theories for predicting trends in colloid release.
2. Background
In this section we describe the theoretical frame- work for the detachment of flocculated colloids and their transport in a porous medium. We then summarize findings from previous experimental work on colloid release from porous media. The emphasis in this discussion is on mineral colloids (which may or may not contain organic coatings), rather than on dissolved organic carbon. Dissolved organic carbon exists as polymeric units and its interaction with larger mineral particles is sig- nificantly different from that of clays and oxidic colloidal particles. Studies on the transport of dissolved carbon have been described elsewhere (see, for example, Refs. [25,26]). Field evidence of colloid movement in porous media (e.g. data from several sites presented by Backhus et al. [27]) is also not included in this discussion. Field data are of limited use compared to laboratory systems in trying to understand basic processes responsible for colloid generation/release and transport.
2.1. Basic theory
The interaction potential energy of two like- charged colloidal particles can be determined by summing the contribution of electrostatic repulsion and van der Waals attraction (i.e. the Derjaguin- Landau-Verwey-Overbeek (DLVO) theory), shown schematically in Fig. 1. For two particles that are flocculated at the primary minimum of potential energy, the DLVO theory predicts that the particles are at infinitesimal separation and that the attractive potential is infinite (the broken line in Fig. 1). In other words, two attached par- ticles would need an infinite amount of energy to detach. Experimentally, however, the detachment (or repeptization) of flocculated particles upon reduction in ionic strength has been observed in batch systems [28,29], often without any mechan- ical action. To account for the process of particle detachment, two simple modifications can be made to the DLVO theory. If the effect of short-range Born repulsion of electron clouds is included in the calculation of potential (see, for example, Ref. [30]), a finite energy minimum results (the solid line in Fig. 1). Similarly, if it is assumed that two surfaces cannot get any closer than the diame- ter of a hydrated counterion (a few AngstrOms), a finite minimum is obtained [31]. With either of these modifications to the DLVO theory, particles flocculated in the potential energy minimum can be shown to have a finite energy barrier for detach- ment. Frens and Overbeek [ 31 ] compared particle detachment to a chemical reaction, and stated that it would occur when the energy barrier for detach- ment was not very high (analogous to an activation energy), and when there was a net reduction in particle potential energy upon detachment. This approach was found adequate to explain qualita- tively data on the repeptization of flocculated colloids.
Theoretical expressions for computing rate con- stants for colloidal particle detachment over a potential energy barrier have also been derived [30,32-34]. Particle detachment over an energy barrier occurs by means of diffusion and can be described either by the Smoluchowski equation or by the Fokker-Planck equation when the particle motion changes rapidly over the time scales of the
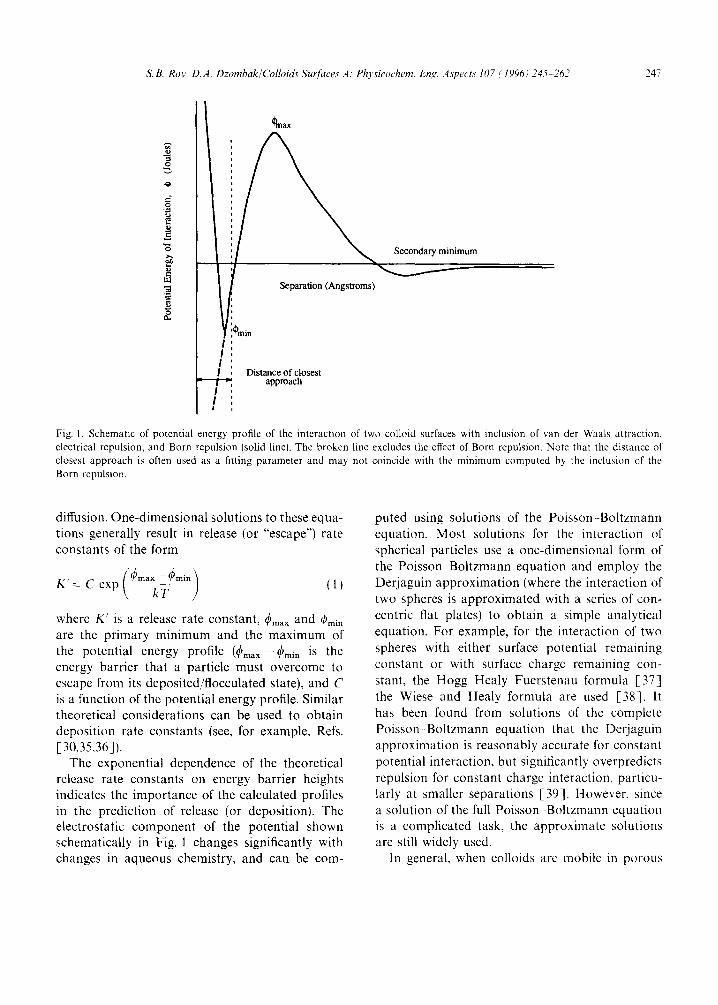
S.B. ROy, D, A. Dzombak/Colloids Surfaces A: Physicochem. Eng. As'pects 107 (1996) 245 262 247
-a
. o -
=g
oa =_
oa ,,=,
t'L
I , I : !
I•nlax
Secondary minimum
Separation (Angstroms)
Y i*mi. I'
[ i Distance of closest approach
Fig. 1. Schematic of potential energy profile of the interaction of two colloid surfaces with inclusion of van der Waals attraction, electrical repulsion, and Born repulsion (solid line). The broken line excludes the effect of Born repulsion. Note that the distance of closest approach is often used as a fitting parameter and may not coincide with the minimum computed by the inclusion of the Born repulsion.
diffusion. One-dimensional solutions to these equa- tions generally result in release (or "escape") rate constants of the form
(~max --_ ~min~ K ' = C e x p \ kT ] (1)
where K' is a release rate constant, q~ma~ and 4min are the primary minimum and the maximum of the potential energy profile (~bma x-~bmi n is the energy barrier that a particle must overcome to escape from its deposited/flocculated state), and C is a function of the potential energy profile. Similar theoretical considerations can be used to obtain deposition rate constants (see, for example, Refs. [30,35,36]).
The exponential dependence of the theoretical release rate constants on energy barrier heights indicates the importance of the calculated profiles in the prediction of release (or deposition). The electrostatic component of the potential shown schematically in Fig. 1 changes significantly with changes in aqueous chemistry, and can be corn-
puted using solutions of the Poisson-Boltzmann equation. Most solutions for the interaction of spherical particles use a one-dimensional form of the Poisson Boltzmann equation and employ the Derjaguin approximation (where the interaction of two spheres is approximated with a series of con- centric fiat plates) to obtain a simple analytical equation. For example, for the interaction of two spheres with either surface potential remaining constant or with surface charge remaining con- stant, the H o g ~ H e a l y Fuerstenau formula [37] the Wiese and Healy formula are used [38]. It has been found from solutions of the complete Poisson-Boltzmann equation that the Derjaguin approximation is reasonably accurate for constant potential interaction, but significantly overpredicts repulsion for constant charge interaction, particu- larly at smaller separations [39]. However, since a solution of the full Poisson-Boltzmann equation is a complicated task, the approximate solutions are still widely used.
In general, when colloids are mobile in porous

248 S.B. Roy, D.A. Dzombak/Colloids Surfaces A." Physicochem. Eng. Aspects 107 (1996) 245-262
media, the release, deposition, and transport of colloidal particles occurs simultaneously. These processes can be described with a framework sim- ilar to that used for solute transport in porous media, i.e. with an advection-dispersion equation for transport and with first-order source/sink terms for colloid deposition and release, shown here for one-dimensional transport [40,41 ]
PC_ t~2Cp OCp 0 ~ = OD v -- q ~ X -- Kpd0Cp + KprCpaPb
(2)
Cpa p b ~ t - - = KpdOC p - KprCpaPb (3)
In these equations, Cp is the concentration of suspended colloids (M/L3), Cpa is the concentration of attached colloids (M/M), Kpd is the macroscopic deposition rate constant of colloids (l/T), Kpr is the macroscopic release rate constant of colloids (I/T), Op is the dispersion coefficient (LZ/T), Pb is the bulk density of the solids (M/L3), q is the Darcy velocity (L/T), and 0 is the water saturation. The macroscopic release constants can be esti- mated from theory or can be obtained from fitting experimental data. Eqs. (2) and (3) assume that there is no size exclusion of colloids in the pores of the medium and that the porosity does not change as a result of colloid deposition and release. In applying these equations to the results from the experiments described below, i.e. the release of previously deposited (or flocculated) colloids from columns, the appropriate initial and boundary conditions are
Cpa(X,t)=Cpa,O for t=0 (4)
Cp,(X,t)=Of for x = 0 (5)
aCva(x,t) ~?x =0 for x = L (6)
where Cpa,o is the mass concentration of releasable colloids (M/M), and L is the column length. In this work, the above equations were solved numeri- cally with an implicit finite difference technique.
2.2. Colloid release in porous media
Colloids in water-saturated porous media, either deposited on the surfaces of larger particles or flocculated as aggregates, can be dispersed by modifying the aqueous chemistry to increase double layer repulsion between colloidal surfaces. Experiments demonstrating colloid release have been performed for natural porous media (surface soils, aquifer materials, and rock cores) and for model porous media (e.g. spherical beads depos- ited with well-characterized colloids). The aqueous chemistry parameters most commonly modified to induce colloid release are the ionic strength, pH, and sodium adsorption ratio (SAR; given as Na+/(Ca2+ + Mg2*)°s). Key findings from previ- ous experimental studies are described below.
Experiments with packed soil columns have shown that colloids can be mobilized, and perme- ability reduced dramatically by the clogging of pores, when water with a high SAR is introduced into the columns [3,4]. Colloid release and perme- ability reduction is inhibited in the presence of organic matter, and oxides and hydroxides of aluminum and iron [6-8]. Permeability reduction is more significant at pH greater than 7 or 8 since many common soil minerals are negatively charged over this pH range E5,7]. Increasing colloid release at higher pH was also observed for an aquifer material containing iron oxides [9]. It was also found in the same study that colloid release upon ionic strength reduction was significantly increased when the iron oxides from the aquifer material had been removed by reductive dissolution. Trends in permeability reduction similar to those obtained for soils have also been observed for rock cores (i.e. increasing for higher pH and SAR, and for larger reductions in ionic strength) [10 13].
In a series of papers [14-18], Matijevi6 and co-workers described colloid release with model colloids and porous media (such as chromium hydroxide and ferric oxide colloids on glass and steel beads) and attempted to relate measured rates to interaction forces between colloids and porous medium (collector) surfaces. Colloid release was observed under a range of electrolyte conditions, pH, and flow rates. It was found that a significant fraction of the attached iron oxide and chromium

S.B. Roy, D.A. Dzombak/Colloids Surfaces A." Physicochem. Eng. Aspects 107 (1996) 245 262 249
hydroxide colloids could be detached from packed columns of spherical beads. The rates of release were not very sensitive to flow rate [14] and increased as the pH was increased [14,15]. The increased release with increasing pH can be explained by the greater negative charge that the oxide particles exhibit at higher pH. Greater release was observed at lower ionic strengths [ 17] although Kallay et al. [18] found that the release rate increased marginally as the ionic strength was increased. This was explained by the reduced height of the particle interaction potential energy barriers at higher ionic strength. However, this result con- tradicts experimental data on repeptization (see, for example, Ref. [29]), where greater dispersion is observed upon decrease of the ionic strength. Release data were interpreted with calculations of potential energy profiles at different conditions, and with a first-order model for release (without transport terms).
Ryan and Gschwend [19] observed colloid release in hematite-quartz systems. Release was generally greater at lower ionic strength, and at higher flow rates. Data were explained by con- sidering that particles were released only when the potential energy was repulsive (i.e. there was no energy barrier for attached particles to overcome), and that the release rate was limited by diffusion over the fluid boundary layer. Colloid release was described as a first-order process without transport terms. In obtaining a fitted value of the release rate constant from experimental data, an additional parameter that was changed was the total mass of particles that could be released. Thus the effect of ionic strength could not be captured with a modi- fied release rate alone; a different initial concen- tration of attached particles was also needed. McDowell-Boyer [20] found that latex colloids deposited on a quartz sand could be released upon reduction of the ionic strength, and that greater total mass release of colloids occurred at higher flow velocities.
Although there are numerous studies of colloid release from natural porous media, most have emphasized the permeability changes rather than colloid transport (with the exception of Ref. [9]). Experiments of colloid release where colloid trans- port is significant have typically been performed
for model systems. Mechanistic interpretation of data has also been limited to these model systems (again, with the exception of Ref. [9]). Our objec- tives in this work were to help to elucidate the processes responsible for colloid release from natu- ral porous media of interest in contaminant trans- port, i.e. high permeability materials with low fines contents; to estimate release rates with the frame- work described by Eqs. (2) and (3); and to assess the utility of particle-particle interaction theories in predicting trends in colloid release in the context of Eq. (1).
3. Experimental
3.1. Materials
The primary natural material used was a sand belonging to the Lincoln series obtained from near the surface in Pontotoc County, Oklahoma (here- after referred to as Lincoln sand). A second sand material was obtained from 35 40 ft below the surface from a coastal aquifer near the Otis Air Base, Cape Cod, Massachusetts (hereafter referred to as Otis sand). The Lincoln sand was used as received for the experiments. The Otis sand was first sieved through a 1 mm sieve to remove larger, gravel-sized particles to improve reproducibility in the experiments. The ds0 (i.e. the grain diameter below which 50% of the total sand mass is con- tained) obtained by dry sieving for the unmodified Lincoln sand was 250 lam and for the sub-l-mm Otis sand was 550 p.m. Both sands were provided as disturbed samples.
For the model experiments, glass beads (0.40 0.52 mm diameter, Thomas Scientific, Swedesboro. NJ) were employed as the porous medium. The beads were first washed with metha- nol to remove organic impurities, then washed repeatedly with deionized (DI) water to remove all traces of methanol, and soaked in chromic acid for several minutes. The chromic acid was washed out with DI water, and the beads were dried in an oven at 100'C before use. The model colloids used were 0.468 lam polystyrene latex spheres (Interracial Dynamics Corporation, Portland~ ORI. The latex spheres were obtained suspended in

250 S.B. Roy, D.A. Dzombak/Colloids Surfaces A: Physicochem. Eng. Aspects 107 (1996) 245-262
deionized water and were used without further purification.
3.2. Methods
The experimental apparatus consisted of a 10 cm long, 2.2 cm inner diameter stainless steel column for the sand experiments (Alltech, Deerfield, IL). A 10 cm long, 2.5 cm inner diameter glass column was used for the experiments with glass beads (Ace Glass, Vineland, NJ). Both columns had an 80 p.m stainless steel wire mesh on both ends. Columns were packed for each release experiment in steps by adding a small amount of sand or glass beads and tapping the side of the column at each step. The columns were placed vertically and water was passed through each column in upflow mode with a high-performance liquid chromatography (HPLC) pump. The effluent from the column was collected in approximately 1 pore volume increments with a fraction collector (Scanivalve, San Diego, CA). The flow rate was maintained at 21 ml h 1 for the experiments with the sand col- umns (corresponding to an average pore-water velocity of 14.7 cm h -1) and at 158 ml h -1 for the glass-bead columns (corresponding to an average pore-water velocity of 85 cm h 1). The flow rate had an accuracy of _+ 5%. The flow rate accuracy improved as flow rates were increased. For the model system, with two pumps running in parallel, a variation in the flow rates would affect the influent concentrations of colloids. Higher flow rates were therefore used for greater stability and accuracy. However, because we were comparing the effects of different ionic strength changes in different systems, the fact the flow rates were not identical does not affect the calculations. Back- pressure in the columns was monitored with a 0-100 inch water column pressure gauge (Cole Parmer, Niles, IL). Pressure drops in the columns were low (a few pounds per square inch), and no pulsing in pressure was observed. The porosity of the sand columns was between 0.35 and 0.39 and close to 0.38 for the glass-bead-packed columns (measured by the mass of water required for saturation).
The effluent was analyzed for mass colloid concentration and pH. Colloid mass concentra-
tion was measured using a turbidimeter (Hach Chemical Company, Ames, Iowa) or a light- absorbance spectrophotometer (Milton Roy, Rochester, NY). Both techniques provided linear calibrations for colloid mass concentration for Lincoln sand colloids (over the range 0-100 mg 1-1) and latex colloids (over the range 0-35 mg 1-1).
Experiments with the Lincoln and Otis sand were performed by first obtaining stable flow for several pore volumes through the columns with a solution of 0.01 M CaCI2. This step was imple- mented to displace common exchangeable ions that might have been present in the sands (e.g. K +, Mg 2 + ), and to result in the flocculation of colloidal material in the primary minimum. In the next stage of the experiment, the Ca 2 + ions were exchanged for Na + ions using a solution of 0.1 M NaC1. (Both these influent solutions were open to the atmosphere, with a pH of approximately 5.8 due to dissolved carbon dioxide.) Finally, the concen- tration of Na + was reduced with a step change using solutions of either NaC1 or NaHCO3. Colloid release generally began 1 pore volume after the introduction of Na + ions at low concentration. The pH of the reduced ionic strength solutions was approximately 9.2 (when obtained by dissolu- tion of NaHCO3) or 4.0 (by dissolution of NaC1 and with the addition of HC1). The exchange of Ca 2+ ions for Na + ions using 0.1 M NaC1 was a key step, because minimal colloid release was observed when a low concentration NaC1 solution was injected directly into a column with sand in the Ca 2+ form. Experiments were generally per- formed over 10-50 pore volumes depending on the electrolyte concentration used. The pH in all cases was measured off-line with a pH meter (Fisher Scientific, Pittsburgh, PA).
Experiments with the glass bead-packed col- umns included an additional deposition step at the start. A suspension of the latex colloids in DI water and NaC1 at 0.2 M (open to the atmosphere, pH approximately 5.8) was injected into the column for several pore volumes. Flows of the two liquids were mixed with a T-fitting located before the inlet to the column to minimize prior flocculation. The colloid suspension influent flow was then stopped

S.B. Roy, D.A. Dzombak/Colloids Sur]aces A: Physicochem. Eng. Aspects 107 (1996)245 262 251
and only 0.2 M NaC1 was passed through the column. No colloid release occurred at this stage.
To initiate colloid release in the glass-bead col- umns, the influent was changed to a low concen- tration of NaC1 (pH approximately 5.8). The effluent was monitored for latex particle mass concentrations as in the column experiments with the sands above.
For some effluent samples from the sand col- umns, as well as for the size fractions of particles obtained by sieving sands through a 75 lain sieve, particle size and composition were determined with a scanning electron microscope equipped with an energy-dispersive X-ray (EDX) probe and auto- mated particle counting and sizing capability (RJ Lee Group, Monroeville, PA). Column efflu- ents were diluted in DI water, sonicated for 10 s and filtered through 0.2 I~m polycarbonate mem- brane filters. The sub-75-gm fraction was sus- pended in DI water and similarly filtered. The particles trapped on the membrane were coated with carbon for scanning electron microscope (SEM) analysis. The particle density on the filters was kept sufficiently low to prevent particles from touching or overlapping. A few microliters of effluent provided sufficient particle density on the filters. The automated particle scanning instrument function provided size (i.e. average diameter) and mass distributions of the particles and a breakdown of the particle elemental composition from an analysis of 600 randomly selected particles. The mass distribution was estimated from the cross- sectional area, the oxide density, and by assuming that the particle depth was equal to the narrowest measured dimension. This technique is described in greater detail elsewhere [-42,43].
The magnitude of the negative surface charge of the Lincoln sand colloids and the latex colloids was estimated over a range of pH by sorption of 45Ca. Colloids from column experiments were washed with NaC1 at 0.1 M, followed by repeated washing and centrifuging with DI water to remove exchangeable ions. Forty milliliters of this suspen- sion were equilibrated in polypropylene vials with NaOH or HCI, and radiolabeled CaCI2 solution added to obtain a concentration of 0.0002 M. The suspensions were equilibrated in an end-over-end rotator for 24 h. Subsequently, the suspensions
were centrifuged, and the supernatant was filtered through a 0.22 ~tm PTFE filter. The aqueous activ- ity of 4SCa in the filtrate was measured using an Opti-Fluor scintillation cocktail (Packard Instrument Company, Downers Grove, IL) with a liquid scintillation counter (Beckman Instruments, Fullerton, CA). Three replicate samples were counted for each vial. The surface charge was estimated from the number of equivalents of Ca 2 ~ ions sorbed.
The surface area of the Lincoln sand colloids, and of whole Lincoln sand (before and after a release experiment) was determined by the single- point BET method with the Quantasorb sorption apparatus (Quantachrome Corp,, Syosset, NYI. Colloids from column effluents were washed and centrifuged las above) and dried at 8 0 C for BET analysis.
Electrophoretic mobil)ties and distributions of the electrophoretic mobility of Lincoln sand col- loids were measured over a range of ionic strengths at pH 9 + 0.1 using an automated electrokinetics analyzer (System 3000, Pen Kern Corporation, Bedford Hills, NY). Colloid suspensions were equilibrated for 24 h with NaOH prior to analysis to allow chemical equilibrium (a 0.01 M solution of NaOH was added dropwise until the pH was very close to 9). The potential dissolution of minerals over this time was not studied directly. However, of the minerals present, silica would be expected to dissolve to an upper limit of approx- imately 10 3 M [44]. At pH 9, of the dissolved species, undissociated silicic acid is dominant, and will not affect the ionic strength of the solutions [44]. Mean zeta potentials were calculated from the mean mobility using Smoluchowski's equation for electrophoresis [45].
The zeta potential, surface charge, and surface area were not estimated for the Otis sand colloids due to the small masses of colloids released.
4. Results
4.1. Characterization of~ne f)'action
For the sands used in this study, the mass fraction of particles passing through a 75 ~m sieve
252 S.B. Roy, D.A. Dzombak/Colloids Surfaces A: Physicochem. Eng. Aspects 107 (1996) 245 262
was fairly small and was dependent on the mode of sieving (wet or dry). Wet sieving of whole sands with DI water resulted in approximately twice the mass fraction of particles being passed through the 75 jam sieve than dry sieving. Wet sieving was used to estimate the sub-75-~tm mass fraction because dry sieving would not account for fines that were adhered to larger particles or existed as aggregates. For Lincoln sand, the sub-75-gm mass fraction was 8.6% (compared with 3.9% for dry sieving), and for Otis sand the sub-75-~tm mass fraction was 1% (compared with 0.5% for dry sieving).
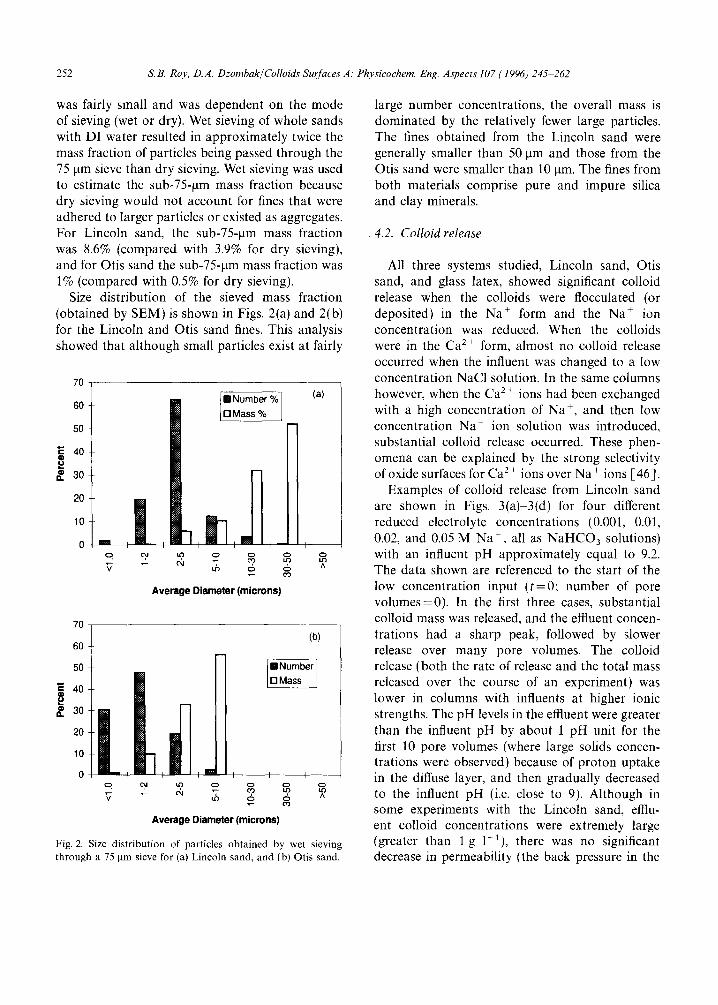
Size distribution of the sieved mass fraction (obtained by SEM) is shown in Figs. 2(a) and 2(b) for the Lincoln and Otis sand fines, This analysis showed that although small particles exist at fairly
40
~. 30
7O
60~ ~ {"Number %] (a)
5O
2O
10
0
Average Diameter (microns)
70
60
50
40
~. 30
20
10
0
(b)
b p i , i m I ~ I I I
~ ~ ~ ~ A
Average Diameter (microns)
Fig. 2. Size distribution of particles obtained by wet sieving through a 75 ~tm sieve for (a) Lincoln sand, and (b) Otis sand.
large number concentrations, the overall mass is dominated by the relatively fewer large particles. The fines obtained from the Lincoln sand were generally smaller than 50 pm and those from the Otis sand were smaller than 10 ~tm. The fines from both materials comprise pure and impure silica and clay minerals.
• 4.2. Co l lo id re lease
All three systems studied, Lincoln sand, Otis sand, and glass latex, showed significant colloid release when the colloids were flocculated (or deposited) in the Na + form and the Na + ion concentration was reduced. When the colloids were in the Ca 2+ form, almost no colloid release occurred when the influent was changed to a low concentration NaC1 solution. In the same columns however, when the Ca 2+ ions had been exchanged with a high concentration of Na +, and then low concentration Na + ion solution was introduced, substantial colloid release occurred. These phen- omena can be explained by the strong selectivity of oxide surfaces for Ca 2+ ions over Na + ions [46].
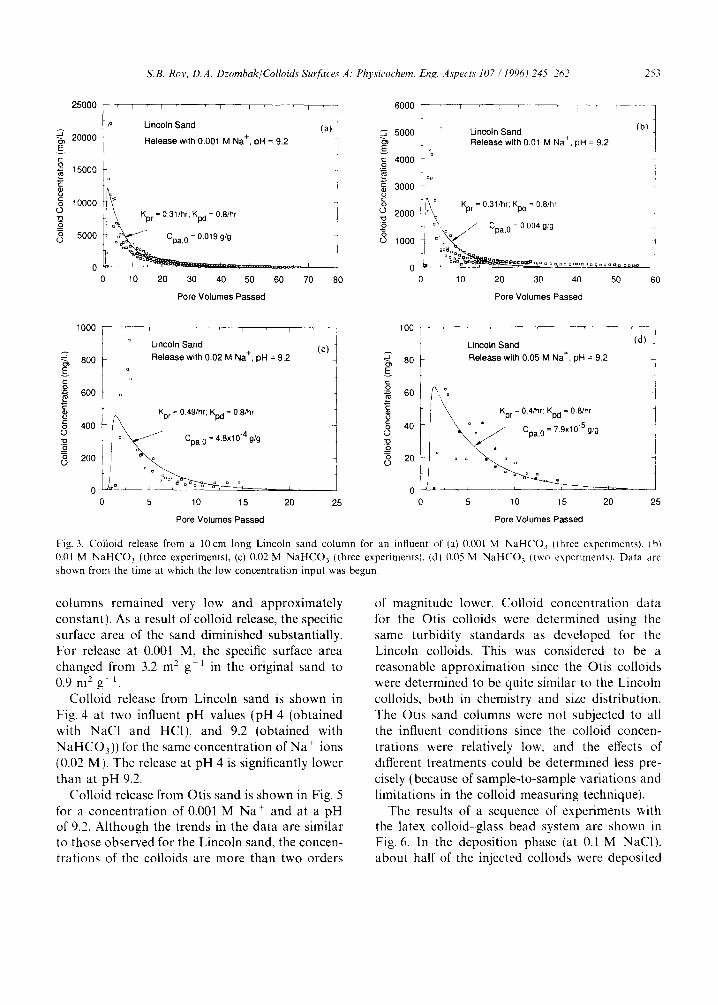
Examples of colloid release from Lincoln sand are shown in Figs. 3(a)-3(d) for four different reduced electrolyte concentrations (0.001, 0.01, 0.02, and 0.05 M Na +, all as NaHCO3 solutions) with an influent pH approximately equal to 9.2. The data shown are referenced to the start of the low concentration input ( t=0 ; number of pore volumes=0). In the first three cases, substantial colloid mass was released, and the effluent concen- trations had a sharp peak, followed by slower release over many pore volumes. The colloid release (both the rate of release and the total mass released over the course of an experiment) was lower in columns with influents at higher ionic strengths. The pH levels in the effluent were greater than the influent pH by about 1 pH unit for the first 10 pore volumes (where large solids concen- trations were observed) because of proton uptake in the diffuse layer, and then gradually decreased to the influent pH (i.e. close to 9). Although in some experiments with the Lincoln sand, efflu- ent colloid concentrations were extremely large (greater than 1 g 1-1), there was no significant decrease in permeability (the back pressure in the
S.B. Ro_v, D.A. Dzombak/Colloids Surfaces A: Physicochem. Eng. Aspects 107 (1996) 245 262 253
25000
20000 E g • ~ 15000
10000 o
5000
i ~ i i i [ i i i i Lincoln Sand (a)
R e l e a s e with 0.001 M Na +, pH = 9.2
Kpr = 031/hr; Kpd = 0.8/hr 1 = ; ; 1 0 0 , 0 °co
~ . . . . J ......... :~,,h.oo~m~ I
10 20 30 40 50 60 70 80
Pore Volumes P a s s e d
6000 ~ 1 ~ [ , - r ~ ~ - " I - - 1
~ 5 0 0 0 ~ Lincoln Sand i b ) ] Release with 0.01 M Na +, pH = 9.2
v = 4000 .o
3000
2000 Kpr = 0.31/hr; Kpd = 0.8/hr
=o [ e ~ J Cpa,0 = 0.004 g/g
10 20 30 40 50 60
Pore Volumes Passed
1000
8oo
g 600
400 0 "o
8 200
0
i , [ i
Lincoln Sand (c) R e l e a s e with 0.02 M Na +, pH = 9.2
5 10 15
Pore Volumes Passed
100
~ 80
g 60
i .0 L)
-6 20 O
i 0 20 25 0
i ; i r |
Lincoln Sand (d) +
R e l e a s e with 0.05 M Na , pH = 9.2
~ Kpr = 0.4/hr; KIX I = 0.8/hr
5 10 15 20 25
Pore Volumes P a s s e d
Fig. 3. Colloid release from a 10cm long Lincoln sand column for an influent of (a) 0.001 M NaHCO3 lthree experiments), (bl 0.01 M N a H C O 3 (three experiments), (c) 0.02 M NaHCO3 (three experiments), (d} 0.05 M N a H C O 3 (two experiments). Data are shown from the time at which the low concentration input was begun.
columns remained very low and approximately constant). As a result of colloid release, the specific surface area of the sand diminished substantially. For release at 0.001 M, the specific surface area changed from 3.2 m 2 g - t in the original sand to 0.9 m 2 g-1.
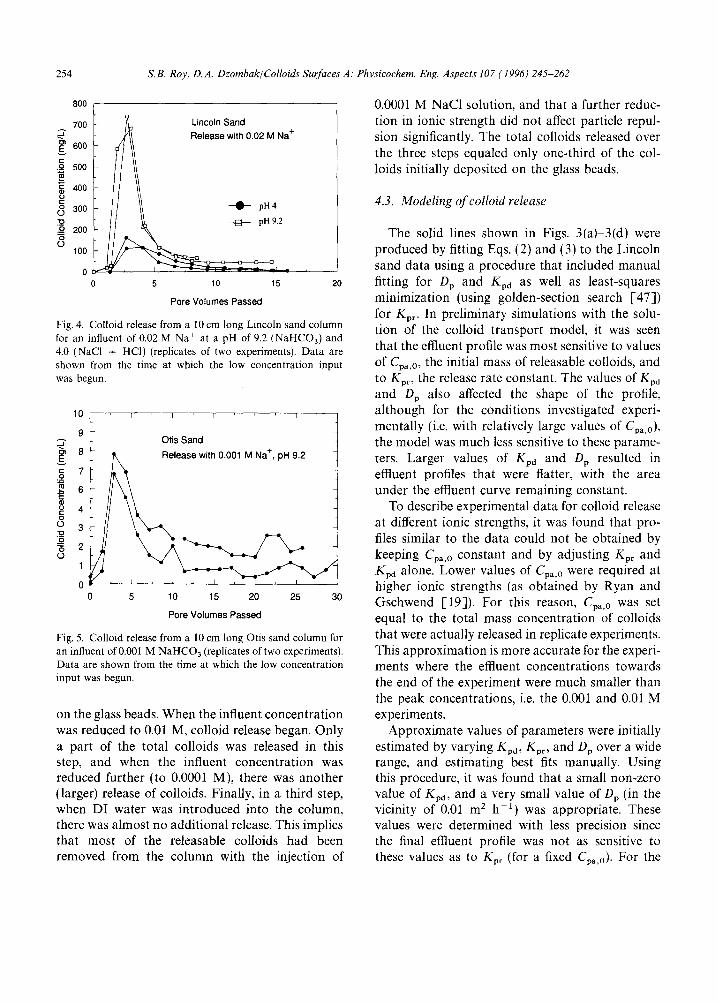
Colloid release from Lincoln sand is shown in Fig, 4 at two influent pH values (pH 4 (obtained with NaCI and HC1), and 9.2 (obtained with NaHCO~)) for the same concentration of Na + ions (0.02 M), The release at pH 4 is significantly lower than at pH 9.2.
Colloid release from Otis sand is shown in Fig. 5 for a concentration of 0.001 M Na ÷ and at a pH of 9.2. Although the trends in the data are similar to those observed for the Lincoln sand, the concen- trations of the colloids are more than two orders
of magnitude lower. Colloid concentration data for the Otis colloids were determined using the same turbidity standards as developed for the Lincoln colloids. This was considered to be a reasonable approximation since the Otis colloids were determined to be quite similar to the Lincoln colloids, both in chemistry and size distribution. The Otis sand columns were not subjected to all the influent conditions since the colloid concen- trations were relatively low, and the effects of different treatments could be determined less pre- cisely (because of sample-to-sample variations and limitations in the colloid measuring technique).
The results of a sequence of experiments with the latex colloid-glass bead system are shown in Fig. 6. In the deposition phase (at 0.1 M NaC1), about half of the injected colloids were deposited
254 S.B. Roy, D.A, Dzombak/Colloids Surfaces A." Physicochem. Eng. Aspects 107 (1996) 245-262
800
700
s00
400
8 300
_o 200 o o
100
0 2 0
o .. .a+
pH 4.2
5 10 15
Pore Volumes Passed
Fig. 4. Colloid release from a 10 cm long Lincoln sand column for an influent of 0.02 M Na + at a pH of 9.2 (NaHCO3) and 4.0 (NaCI + HCI) (replicates of two experiments). Data are shown from the time at which the low concentration input was begun.
10
9
8 g __. 7
~- 6
8 a o -6 2
1
0
I I I f 1
Otis Sand
I I I J I
5 10 15 20 25 30
Pore Volumes Passed
Fig. 5. Colloid release from a 10 cm long Otis sand column for an influent of 0.001 M NaHCO3 (replicates of two experiments). Data are shown from the time at which the low concentration input was begun.
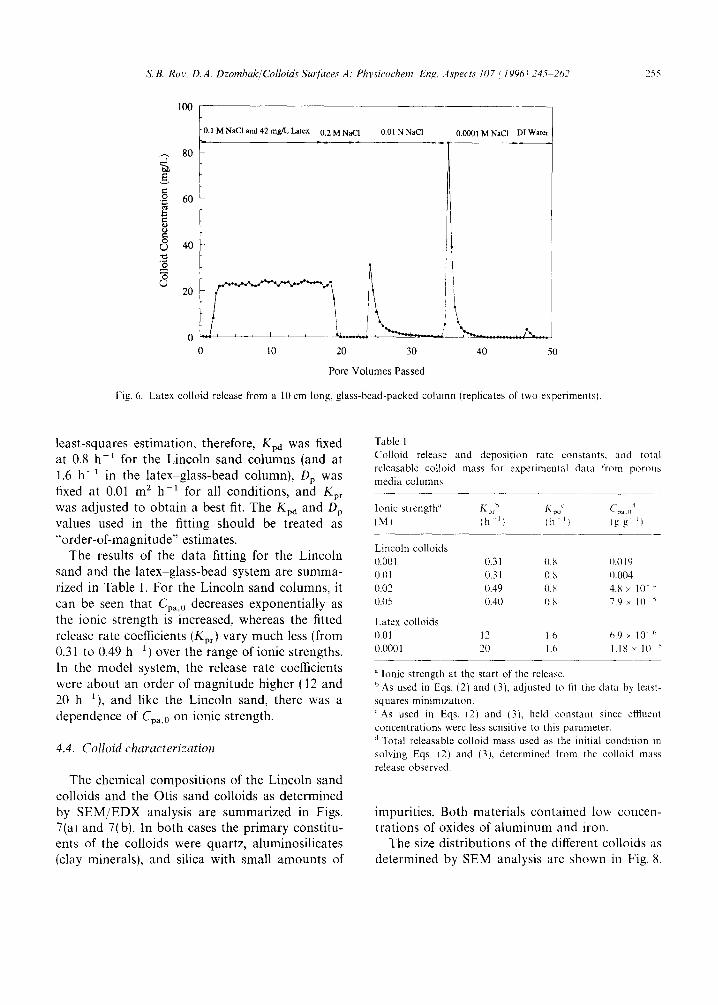
on the glass beads. When the inftuent concentration was reduced to 0.01 M, colloid release began. Only a part of the total colloids was released in this step, and when the influent concentration was reduced further (to 0.0001 M), there was another (larger) release of colloids. Finally, in a third step, when DI water was introduced into the column, there was almost no additional release. This implies that most of the releasable colloids had been removed from the column with the injection of
0.0001 M NaC1 solution, and that a further reduc- tion in ionic strength did not affect particle repul- sion significantly. The total colloids released over the three steps equaled only one-third of the col- loids initially deposited on the glass beads.
4.3. Modeling of colloid release
The solid lines shown in Figs. 3(a)-3(d) were produced by fitting Eqs. (2) and (3) to the Lincoln sand data using a procedure that included manual fitting for Dp and Kpd as well as least-squares minimization (using golden-section search [47]) for Kpr. In preliminary simulations with the solu- tion of the colloid transport model, it was seen that the effluent profile was most sensitive to values of Cp,.o, the initial mass of releasable colloids, and to Kpr, the release rate constant. The values of Kpa and Dp also affected the shape of the profile, although for the conditions investigated experi- mentally (i.e. with relatively large values of Cp,,0 ), the model was much less sensitive to these parame- ters. Larger values of Kpa and Dp resulted in effluent profiles that were flatter, with the area under the effluent curve remaining constant.
To describe experimental data for colloid release at different ionic strengths, it was found that pro- files similar to the data could not be obtained by keeping Cp,,o constant and by adjusting Kpr and Kpd alone. Lower values of Cpa,o were required at higher ionic strengths (as obtained by Ryan and Gschwend [19]). For this reason, Cpa,0 was set equal to the total mass concentration of colloids that were actually released in replicate experiments. This approximation is more accurate for the experi- ments where the effluent concentrations towards the end of the experiment were much smaller than the peak concentrations, i.e. the 0.001 and 0.01 M experiments.
Approximate values of parameters were initially estimated by varying Kpd , Kpr , and Dp over a wide range, and estimating best fits manually. Using this procedure, it was found that a small non-zero value of Kpd , and a very small value of Dp (in the vicinity of 0.01 m 2 h -x) was appropriate. These values were determined with less precision since the final effluent profile was not as sensitive to these values as to Kpr (for a fixed Cp~,o). For the
S.B. Roy,. D.A. Dzombak/Colloids Surlaces A: Phvsicochem.. Eng. Aspects 107 (1996) ,,+.'*~ =~,~." 2~5_
100
8O
g 60
e~ 0 ~ 40
o c,.)
20
0.1 M NaCI and 42 mglL Latex 0.2 M NaCI 0.01 N NaCI 0.000l M NaC1 DI Wate~
0 I0 20 3O 40 50
Pore Volumes Passed
Fig. 6. Latex colloid release from a 10 cm long, glass-bead-packed column (replicates of two experimentsl.
least-squares estimation, therefore, Kpd w a s fixed at 0.8 h t for the Lincoln sand columns (and at 1.6 h-1 in the latex glass-bead column), Dp was fixed at 0.01 m 2 h -1 for all conditions, and Kpr
was adjusted to obtain a best fit. The Kpd and Dp Ionic strength" Kpr b /~pd c ('pa,O d
values used in the fitting should be treated as IMI (h ') (h ') ( gg '1
"order-of-magnitude" estimates. Lincoln colloids
The results of the data fitting for the Lincoln 0.001 0.31 0.8 0.019 sand and the latex-glass-bead system are summa- 001 0.31 0% 0.(t04 rized in Table 1. For the Lincoln sand columns, it 0.02 0.49 0.8 4.8 × 10 4
can be seen that Cpa,O decreases exponentially as 0.05 0.40 0.8 7,9 x 10 "
the ionic strength is increased, whereas the fitted Latex colloids release rate coefficients (Kpr) vary much less (from 0.01 12 J.~, 0.31 to 0.49 h t) over the range of ionic strengths. 0.0001 20 1.6 In the model system, the release rate coefficients were about an order of magnitude higher (12 and 20 h - l ) , and like the Lincoln sand, there was a dependence of Cp~.O on ionic strength.
4.4. Colloid characterization
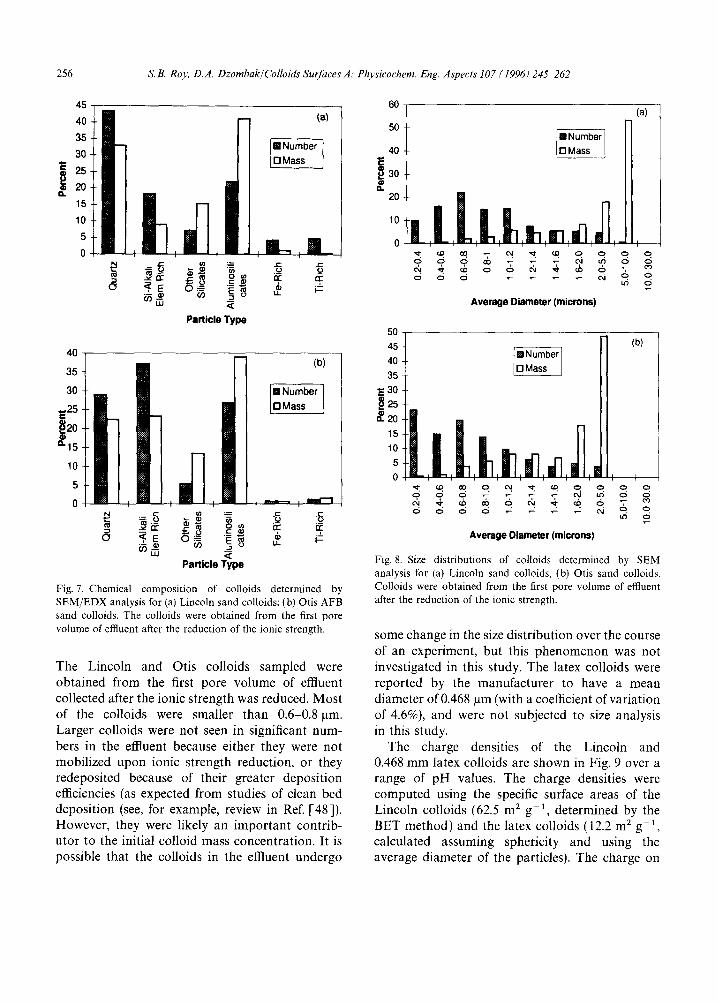
The chemical compositions of the Lincoln sand colloids and the Otis sand colloids as determined by SEM/EDX analysis are summarized in Figs. 7(a) and 7(b). In both cases the primary constitu- ents of the colloids were quartz, aluminosilicates (clay minerals), and silica with small amounts of
Table 1 Colloid release and deposition rate constants, and total releasable colloid mass for experimental data from porous media columns
6,9 > 10 ~ 1,18 ~'< 10 s
Ionic strength at the start of the release. b AS used in Eqs. (2} and (3), adjusted to fit the data by least- squares minimization. C As used in Eqs. (2) and (3), held constant since effluent concentrations were less sensitive to this parameter. d Total releasable colloid mass used as the initial condition in solving Eqs. (2) and (3), determined from the colloid mass release observed.
impurities. Both materials contained low concen- trations of oxides of aluminum and iron.
The size distributions of the different colloids as determined by SEM analysis are shown in Fig. 8.
256 S.B. Roy, D.A. Dzombak/Colloids Surfaces A: Physicochem. Eng. Aspects 107 (1996) 245 262
45 60 (a)
40 ~a/ 50 35
B Number I ~ M~'~7~ I 30 4O
g. 2o
5 0 , 1 1 ', . . . . : ,
0 • • " 7 . . . . .
40
~N
if: if: o 6 d ,; ,-: ,-: ,-: e,i '5 6
~u ~ Average Diameter (microns) Particle Type
35
30
~25
o-15
10
5
o
50 45
(b) 40 35
B Number ] ,,.. 30
I nMass I .~ 25 t
~- 2o 15 10
0
Particle Type
Fig. 7. Chemical composition of colloids determined by SEM/EDX analysis for (a) Lincoln sand colloids; (b) Otis AFB sand colloids. The colloids were obtained from the first pore volume of effluent after the reduction of the ionic strength.
The Lincoln and Otis colloids sampled were obtained from the first pore volume of effluent collected after the ionic strength was reduced. Most of the colloids were smaller than 0.6-0.8 p.m. Larger colloids were not seen in significant num- bers in the effluent because either they were not mobilized upon ionic strength reduction, or they redeposited because of their greater deposition efficiencies (as expected from studies of clean bed deposition (see, for example, review in Ref. [48]). However, they were likely an important contrib- utor to the initial colloid mass concentration. It is possible that the colloids in the effluent undergo
(b)
Average Dlameter (microns)
e,i o o
Fig. 8. Size distributions of colloids determined by SEM analysis for (a) Lincoln sand colloids, (b) Otis sand colloids. Colloids were obtained from the first pore volume of effluent after the reduction of the ionic strength.
some change in the size distribution over the course of an experiment, but this phenomenon was not investigated in this study. The latex colloids were reported by the manufacturer to have a mean diameter of 0.468 p.m (with a coefficient of variation of 4.6%), and were not subjected to size analysis in this study.
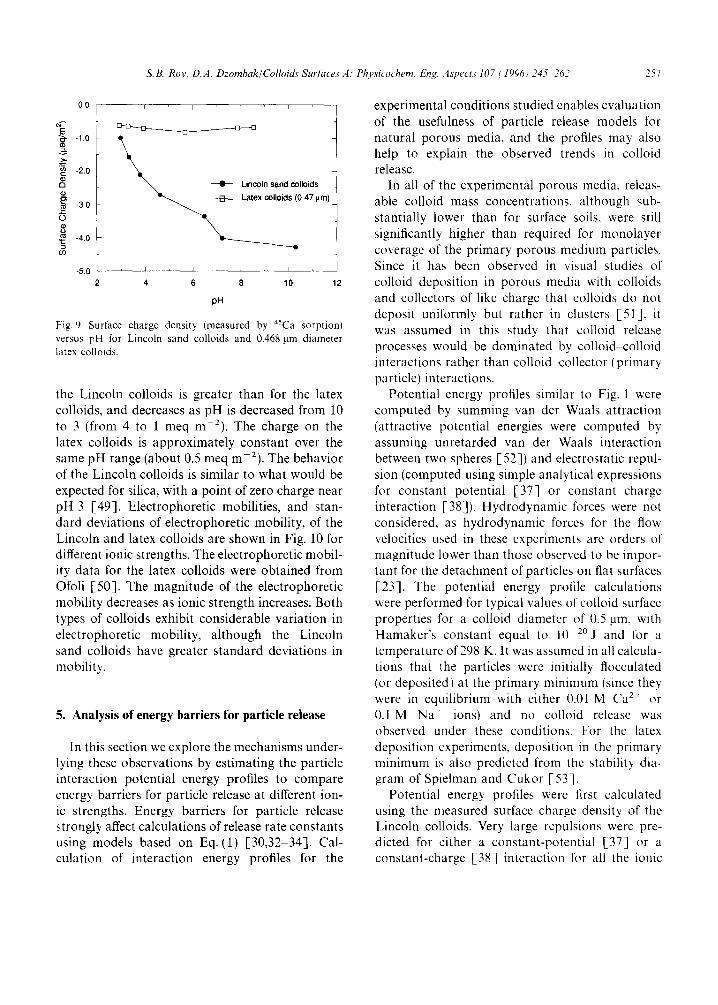
The charge densities of the Lincoln and 0.468 mm latex colloids are shown in Fig. 9 over a range of pH values. The charge densities were computed using the specific surface areas of the Lincoln colloids (62.5 m 2 g-a, determined by the BET method) and the latex colloids (12.2 m 2 g - i , calculated assuming sphericity and using the average diameter of the particles). The charge on
S. R Roy, D.A. Dzombak/Colloids SurJaces A: Physicochem. Eng. Aspects 107 (1996) 245 262 257
0.0
c~ -2.0 09 D - - 0 ~ Lincoln sand colloids -3.0 o.7.ml 6 8
-4,0 g,
-5,0 I I I I
4 6 8 10 12
pH
Fig. 9. Surface charge density (measured by 45Ca sorption) versus pH for Lincoln sand colloids and 0.468 gm diameter latex colloids.
the Lincoln colloids is greater than for the latex colloids, and decreases as pH is decreased from 10 to 3 (from 4 to 1 meq m 2), The charge on the latex colloids is approximately constant over the same pH range (about 0.5 meq m 2). The behavior of the Lincoln colloids is similar to what would be expected for silica, with a point of zero charge near pH 3 [49]. Electrophoretic mobilities, and stan- dard deviations of electrophoretic mobility, of the Lincoln and latex colloids are shown in Fig. 10 for different ionic strengths. The electrophoretic mobil- ity data for the latex colloids were obtained from Ofoli [50]. The magnitude of the electrophoretic mobility decreases as ionic strength increases. Both types of colloids exhibit considerable variation in electrophoretic mobility, although the Lincoln sand colloids have greater standard deviations in mobility.
5. Analysis of energy barriers for particle release
In this section we explore the mechanisms under- lying these observations by estimating the particle interaction potential energy profiles to compare energy barriers for particle release at different ion- ic strengths. Energy barriers for particle release strongly affect calculations of release rate constants using models based on Eq.(1) [30,32-34]. Cal- culation of interaction energy profiles for the
experimental conditions studied enables evaluation of the usefulness of particle release models for natural porous media, and the profiles may also help to explain the observed trends in colloid release.
In all of the experimental porous media, releas- able colloid mass concentrations, although sub- stantially lower than for surface soils, were still significantly higher than required for monolayer coverage of the primary porous medium particles. Since it has been observed in visual studies of colloid deposition in porous media with colloids and collectors of like charge that colloids do not deposit uniformly but rather in clusters [51], it was assumed in this study that colloid release processes would be dominated by colloid-colloid interactions rather than colloid collector (primary particle) interactions.
Potential energy profiles similar to Fig. 1 were computed by summing van der Waals attraction (attractive potential energies were computed by assuming unretarded van der Waals interaction between two spheres [52]) and electrostatic repul- sion (computed using simple analytical expressions for constant potential [37] or constant charge interaction [38]). Hydrodynamic forces were not considered, as hydrodynamic forces for the flow velocities used in these experiments are orders of magnitude lower than those observed to be impor- tant for the detachment of particles on flat surfaces [23]. The potential energy profile calculations were performed for typical values of colloid surface properties for a colloid diameter of 0.5 ~tm, with Hamaker's constant equal to 10 20j and for a temperature of 298 K. It was assumed in all calcula- tions that the particles were initially flocculated (or deposited) at the primary minimum (since they were in equilibrium with either 0.01 M Ca z+ or 0.1 M Na ~ ions) and no colloid release was observed under these conditions. For the latex deposition experiments, deposition in the primary minimum is also predicted from the stability dia- gram of Spielman and Cukor [53].
Potential energy profiles were first calculated using the measured surface charge density of the Lincoln colloids. Very large repulsions were pre- dicted for either a constant-potential [37] or a constant-charge [38] interaction for all the ionic
258 S.B. Roy, D.A. Dzombak /Colloids Surfaces A: Physicochem. Eng. Aspects 107 (1996) 245-262
-5.0
o
'u~ -4.0 "T >
'~ -3.0 o
o
o
~ -2.0
1.1.1
• Lincoln colloids at pH 9
• 0.47 p.m Latex colloids ( D a t a f rom Ofoli, 1994)
- 1 . 0 i , , ~ i , ,
0.00 0.04 0.08 0.12
Ion ic S t r e n g t h (M)
Fig. 10. Electrophoretic mobility of Lincoln sand colloids (at pH 9) and 0.468 p,m latex colloids (pH in water without the addition of acid or base; data obtained from Ofoli [50]) for different ionic strengths. The error bars represent one standard deviation in mobility.
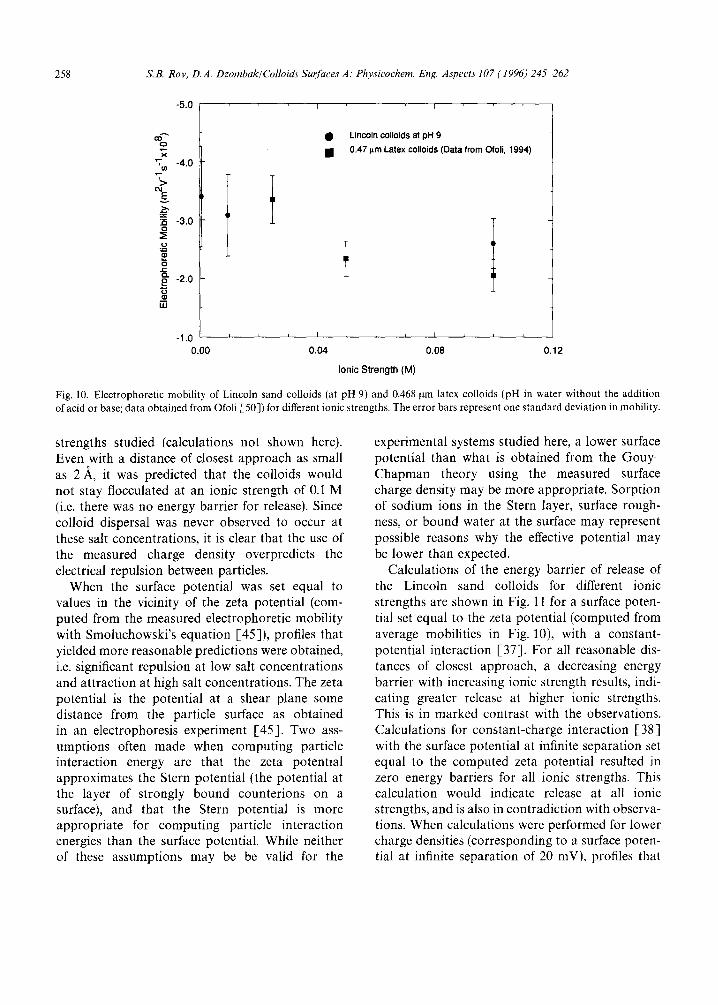
strengths studied (calculations not shown here). Even with a distance of closest approach as small as 2 ,~, it was predicted that the colloids would not stay flocculated at an ionic strength of 0.1 M (i.e. there was no energy barrier for release). Since colloid dispersal was never observed to occur at these salt concentrations, it is clear that the use of the measured charge density overpredicts the electrical repulsion between particles.
When the surface potential was set equal to values in the vicinity of the zeta potential (com- puted from the measured electrophoretic mobility with Smoluchowski's equation [-45]), profiles that yielded more reasonable predictions were obtained, i.e. significant repulsion at low salt concentrations and attraction at high salt concentrations. The zeta potential is the potential at a shear plane some distance from the particle surface as obtained in an electrophoresis experiment [45]. Two ass- umptions often made when computing particle interaction energy are that the zeta potential approximates the Stern potential (the potential at the layer of strongly bound counterions on a surface), and that the Stern potential is more appropriate for computing particle interaction energies than the surface potential. While neither of these assumptions may be be valid for the
experimental systems studied here, a lower surface potential than what is obtained from the Gouy- Chapman theory using the measured surface charge density may be more appropriate. Sorption of sodium ions in the Stern layer, surface rough- ness, or bound water at the surface may represent possible reasons why the effective potential may be lower than expected.
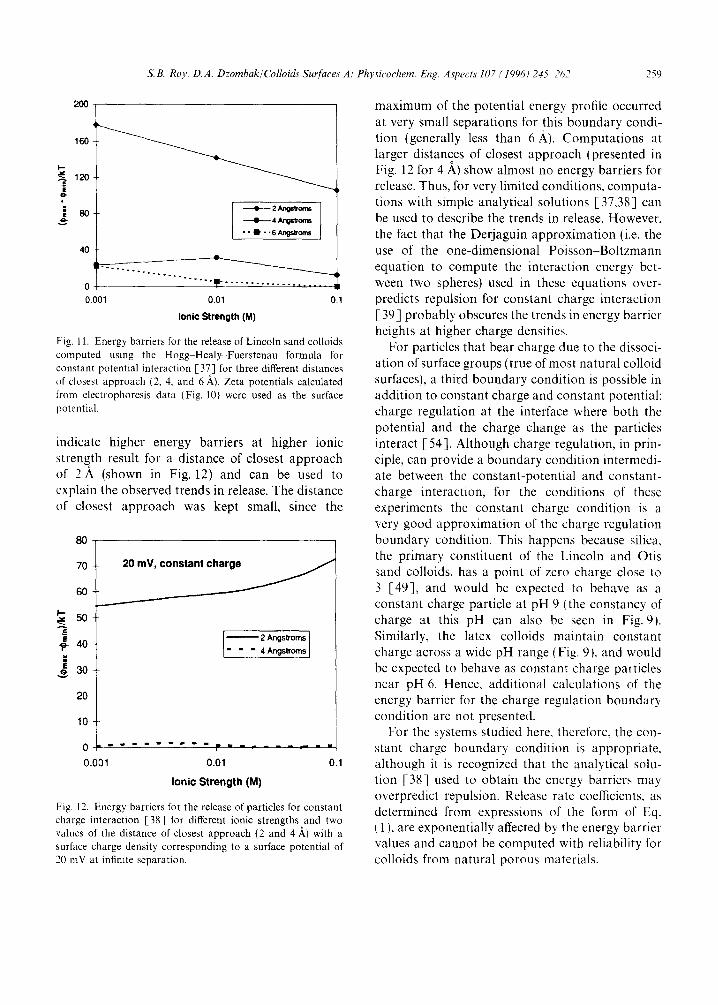
Calculations of the energy barrier of release of the Lincoln sand colloids for different ionic strengths are shown in Fig. 11 for a surface poten- tial set equal to the zeta potential (computed from average mobilities in Fig. 10), with a constant- potential interaction [37]. For all reasonable dis- tances of closest approach, a decreasing energy barrier with increasing ionic strength results, indi- cating greater release at higher ionic strengths. This is in marked contrast with the observations. Calculations for constant-charge interaction [38] with the surface potential at infinite separation set equal to the computed zeta potential resulted in zero energy barriers for all ionic strengths. This calculation would indicate release at all ionic strengths, and is also in contradiction with observa- tions. When calculations were performed for lower charge densities (corresponding to a surface poten- tial at infinite separation of 20 mV), profiles that
S.B. Roy, D.A. Dzombak/Colloids Surfaces A: Physicochem. Eng..4spects 107 (1996) 245 262 259
2OO
160
1-
12o E 4
=- 8o
40
2 Ange~onw 4 Angsb 'oe~
" " • - • 6 Angsb'om~
0 - - - m
0001 0.01 0,1
" ° . . . . . . . " ' ° ' ° ° ° " . . . . . ~11 . . . . . . . . . . . . . . . . . . . . .
Ionic Strength (M)
Fig. I l. Energy barriers for the release of Lincoln sand colloids computed using the Hogg-Healy-Fuerstenau formula for constant potential interaction [37] for three different distances of closest approach (2, 4, and 6 A). Zeta potentials calculated from electrophoresis data (Fig. 10) were used as the surface potential.
indicate higher energy barriers at higher ionic strength result for a distance of closest approach of 2A (shown in Fig. 12) and can be used to explain the observed trends in release. The distance of closest approach was kept small, since the
80
70
60
30
20
10
0
0.001
20 mV, constant charge J
. 2 Angstroms " " 4 Angstroms
0.01 0.1
Ionic Strength (M)
Fig. 12. Energy barriers for the release of particles for constant charge interaction [38] for different ionic strengths and two values of the distance of closest approach (2 and 4 A) with a surface charge density corresponding to a surface potential of 20 mV at infinite separation.
maximum of the potential energy profile occurred at very small separations for this boundary condi- tion (generally less than 6 ~.). Computations at larger distances of closest approach (presented in Fig. 12 for 4 ,~) show almost no energy barriers for release. Thus, for very limited conditions, computa- tions with simple analytical solutions [37,38] can be used to describe the trends in release. However, the fact that the Derjaguin approximation (i.e. the use of the one-dimensional Poisson-Boltzmann equation to compute the interaction energy bet- ween two spheres) used in these equations over- predicts repulsion for constant charge interaction [ 39 ] probably obscures the trends in energy barrier heights at higher charge densities.
For particles that bear charge due to the dissoci- ation of surface groups (true of most natural colloid surfaces), a third boundary condition is possible in addition to constant charge and constant potential: charge regulation at the interface where both the potential and the charge change as the particles interact [54]. Although charge regulation, in prin- ciple, can provide a boundary condition intermedi- ate between the constant-potential and constant- charge interaction, for the conditions of these experiments the constant charge condition is a very good approximation of the charge regulation boundary condition. This happens because silica, the primary constituent of the Lincoln and Otis sand colloids, has a point of zero charge close to 3 [49], and would be expected to behave as a constant charge particle at pH 9 (the constancy of charge at this pH can also be seen in Fig. 91. Similarly, the latex colloids maintain constant charge across a wide pH range (Fig. 9), and would be expected to behave as constant charge particles near pH 6. Hence, additional calculations of the energy barrier for the charge regulation boundary condition are not presented.
For the systems studied here, therefore, the con- stant charge boundary condition is appropriate, although it is recognized that the analytical solu- tion [38] used to obtain the energy barriers may overpredict repulsion. Release rate coefficients, as determined from expressions of the form of Eq. ( 1 ), are exponentially affected by the energy barrier values and cannot be computed with reliability for colloids from natural porous materials.

260 S.R Roy, D.A. Dzombak/Colloids Surfaces A." Physicochem. Eng. Aspects 107 (1996) 245 262
6. Discussion
The colloid release processes from the three experimental systems studied were similar in that the effluent concentration profiles obtained were similar in shape: they contained a sharp peak, followed by a long "tail". For all three systems, the peak concentrations and the mass of colloids released increased for greater reductions in ionic strength. The release process in the Lincoln sand and the latex-glass-bead systems could be described with Eqs. (2) and (3), with one primary adjustable parameter (Kpr) and with Cp,,0 set equal to the observed mass concentration of releasable colloids.
From the first-order model given by Eqs. (2) and (3), it would be expected that when the release rate is higher, a given mass of colloids would be released from a packed bed in a shorter time than when the release rate is lower, the total mass of colloids released not being dependent on the release rate constant. However, in this study, and in previous studies (see, for example, Refs. [ 14,19]), it has been demonstrated clearly that the mass of colloids released over reasonable experimental time frames does vary with changes in conditions (e.g. ionic strength) that affect the release rate. Therefore, when a release constant is obtained by fitting column data, such as those obtained in this study, one of the parameters that changes from experiment to experiment is the total mass of releasable colloids (Cpa,o) (see Table 1). The fitted rate constants that are obtained, therefore, explain only a part of the colloid release process. In our fitting procedure, no clear trend was found between the release rate constants and ionic strength caus- ing release in Lincoln sand.
The variation in electrophoretic mobilities in the Lincoln and the 0.468 p.m latex colloids (Fig. 10) indicates the presence of significant charge hetero- geneities in these colloids. These charge hetero- geneities would strongly affect the values of energy barriers of detachment for individual particles, and can explain the dependence of Cp,.O on ionic strength. For release at higher ionic strengths, only the colloids with the highest charge densities can escape, whereas for release at lower ionic strengths, colloids with lower charge densities can escape as
well. Thus, Cpa,o can increase with decreasing ionic strength. In studying the deposition of latex col- loids on glass beads, Song et al. [55] found that allowing for heterogeneity in the glass bead sur- faces could account for much greater deposition than predicted by theory. Ofoli [50] found that differences between theory and experiment in the slow flocculation of latex colloids could be partly accounted for by assuming a distribution of surface potential. It would appear from this work and prior colloid release studies (see, for example, Refs. [14,19]) that assuming similar distributions in surface properties of the collectors or colloids (or both) may also be needed to explain the different numbers (or masses) of releasable colloids for different ionic strengths.
7. Conclusions
In the three porous media studied here (Lincoln sand, Otis sand, and latex in glass beads), colloid release could be induced by lowering the ionic strength of a univalent, symmetric electrolyte. The rate of colloid release, and the total mass of colloids released were both larger for greater reductions in ionic strength. Colloids in the effluent from the sand columns were analyzed for size distribution and chemistry by SEM/EDX analysis. The major components of the colloids were pure and impure forms of silica, and most of the colloids were smaller than 1 p.m.
The column effluent data were fitted with an advection-dispersion transport equation for col- loids with terms for release and deposition. Release rate coefficients and total mass of releasable col- loids were used as the adjustable parameters in fitting the effluent colloid concentrations versus time. The differences in the total masses of colloids released for different reductions in ionic strength could not be explained merely by the changing release rate coefficients in the advection-dispersion equation. Different values of the total colloid mass available for release had to be used for each ionic strength. The differences in total releasable colloids for each ionic strength may be a result of hetero- geneities in the colloids, an effect that was not explicitly accounted for in the models employed.

s .R Roy, D.A. Dzornbak/Colloids SurJaces A: Physicochem. Eng. Aspects 107 (1996) 245 262 261
The trends of colloid release observed in the co lumn experiments could be explained quali ta-
tively in terms of the magni tude of energy barriers for particle de tachment for a constant-charge inter- action (albeit with a charge density much lower than that measured). In terac t ion potent ial energy profiles computed by using surface potentials in
the range of the particle zeta potentials [38] showed lower energy barriers for colloid detach-
ment at lower ionic strengths for a cons tant charge
b o u n d a r y condi t ion (using a fixed distance of mini- mum approach). In teract ion energy profiles com- puted using a cons tant potent ial b o u n d a r y [37] condi t ion or by using the measured charge density
showed trends that were opposite to those observed, or predicted de tachment at all ionic strengths.
In na tura l porous media, quali tat ive predictions of colloid release may be made with available particle in teract ion theories which use easily obta ined zeta potent ial information. Quant i ta t ive
predictions of release rates are not likely to be reliable, however, because of the exponential sensi-
tivity of theoretical rate constants to potent ial energy barriers for detachment , values of which cannot be determined accurately for most colloids.
Acknowledgments
We gratefully acknowledge the assistance of John Wilson, Robert Puls and Robert Powell of the USEPA R.S. Kerr Labora tory in ob ta in ing the Lincoln sand and the Cape Cod aquifer materials.
We thank Gary Casuccio, Steve Schlaegle, and John Johns of RJ Lee Group , Inc., who provided access to and help with using the "Personal SEM" inst rument ; A n n a Morfesis of the P P G Fiberglass Research Center for access to the Pen Kem System 3000 ins t rument ; and Dennis Prieve for several
helpful discussions. We also thank Joseph Ryan and an a n o n y m o u s reviewer for helpful comments on the manuscript .
This study was supported by the U.S. Envi ronmenta l Protect ion Agency (grant no. R819266-01-0), the Nat iona l Science F o u n d a t i o n (PYI grant no. BCS-9157086), and the A l u m i n u m C o m p a n y of America (TC 943095).
References
[1] J.F. McCarthy and J.M. Zachara, Environ. Sci. Technol., 23 (19891 496.
[2] J.F. McCarthy and F.J. Wobber, Manipulation of Groundwater Colloids for Enviromnental Restoration, Lewis Publishers, 1993.
[3] B.L. McNeal and N.T. Coleman. Soil Sci. Soc. Am. Proc., 20 11966~ 308.
[4] H.J. Frenkel, J.O. Goertzen and J.D. Rhoades, Soil Sci. Soc. Am. J., 42 (1978} 32.
[5] D.L. Suarez, J.D. Rhoades, R. Lavado and C.M. (irievc, Soil Sci. Soc. Am. J., 48 119841 50.
[6] N. Alperovitch, 1. Shainberg, R. Keren and M,J. Singer, Clays Clay Miner., 33 (1985i 443.
[7] R. Keren and M.J. Singer. Soil Sci. Soc. Am..I., 54 ( 19901 1310.
[8] R. Keren and M.J. Singer, Soil Sci, Soc. Am. J.. 55 11991 ) 61.
[9] J.N. Ryan and P.M. Gschwend, Environ. Sci. Technol., 28 11994i 1717.
[10] K.C, Khilar and H.S. Fogler. J. ('olloid Interface Sci., 101 11984) 214.
[11] S.F. Kia, H.S. Fogler and M.G. Reed, ,I. Colloid lnlerfilce Sci., 118 (1987) 158.
[12] S.F. Kia, H.S. Fogler, M.G. Reed and R.N. Vaidya, SPE Prod. Eng., November (1987) 277.
[13] R.N. Vaidya and H.S. Fogler, Colloids Surtkiccs, 50 11991)) 215.
[14] R.J. Kuo and E. Matijevid, J. Colloid Interface Sci., 78 ( 1980~ 4(17.
[15] N. Kallay and E. Matijevic, J. Colloid lntertiice Sci., 83 11981 ) 289.
[16] JD. Nelligan, N. Kallay and F. Maliievid. J. Colloid Interface Sci., 89 ( 19821 9.
[17] J.E. Kolakowski and E. Mai[jevid, J. Chem. Soc., Faraday Trans. 1, 75 119791 65.
[18] N. Kalla~, B. Biskup, M. Tomid and 1!. Matije\'id, J. Colloid Interface Sci., 114 119861 357.
[19] J.N. Ryan and P.M. Gschwend, J. Colloid lnterlitce Sci.. 164 (19c~4) 21.
[20] L. McDowell-Boyer, Environ. Sci. Technol., 2611992) 586. [21] M. Hubbe, Colloids Surfaces, 16 11985~ 227. [22] M. ttubbe, Colloids Surfaces, 16 t 1985) 249. [23] S.K. Das, R.S. Schechter and M.M. Sharma, J. Colloid
Interface Sci.. /64 (1994j 63. [24] M.M. Sharma, H. Chamoun, I).H.S.R Sarma and R.S.
Schechier. J. Colloid Interface Sci., 149 ( 19921 121. [25] F.M. Dunnivant, P.M. Jardinc. D.L. Taylor and J.F.
McCarlhy, Soil Sci. Soc. Am. J.. 56 i 1992) 437. [26] J.F. McCarthy, T.M. Williams, L. Liang, P.M. ]ardine,
L.W. Jolley, D.L. Taylor, A.V. Pahlmbo and L.W. I~ooper, Environ. Sci. Technol., 27 119931 667.
[27] D. Backhus, J.N. Ryan. D.M. Groher..IK. MacFarlane, and P.M. Gschwend, Ground Water. 31 119931 466.
[28] tt. van Olphen, Clay Colloid Chemistry, 2nd cdn., John Wiley, New York, 1977

262 S.B. Roy, D.A. Dzombak/Colloids Surfaces A." Physicochem. Eng. Aspects 107 (1996) 245~62
[29] G. Frens and J.Th.G. Overbeek, J. Colloid Interface Sci., 36 (19713 286.
[30] E. Ruckenstein and D.C. Prieve, AIChE J., 22 ( 19763 276. [-31] G. Frens and J.Th.G. Overbeek, J. Colloid Interface Sci.,
38 (1972) 376. [32] S. Chandrasekhar, Rev. Mod. Phys., 15(1) (19433 3-89.
Reprinted in N. Wax (Ed.), Selected Papers on Noise and Stochastic Processes, Dover Publications, New York, 1954.
[33] B. Dahneke, J. Colloid Interface Sci., 50 (1975) 89. [34] B. Dahneke, J. Colloid Interface Sci., 50 (1975) 194. [35] L.A. Spielman and S.K. Friedlander, J. Colloid Interface
Sci., 46 (19743 22. [-36] R. Rajagopalan and J.S. Kim, J. Colloid Interface Sci.,
83 (19813 428. [37] R. Hogg, T.W. Healy and D.W. Fuerstenau, Trans.
Faraday Soc., 62 (1966) 1638. [38] G.R. Wiese and T.W. Healy, Trans. Faraday Soc., Vol. 66
( 19703 490. [-39] A.B. Glendinning and W.B. Russel, J. Colloid and
Interface Sci., 93 (19833 95. [40] M.Y. Corapcioglu and S. Jiang, Water Resour. Res., 29
(19933 2215. [41] J.E. Saiers, G.M. Hornberger and L. Liang, Water Resour.
Res., 30 (1994) 2499. [42] G.S. Casuccio, P.B. Janocko, R.J. Lee, J.F. Kelly, S.L.
Dattner and J.S. Mgebroff, Air Pollut. Control Assoc. J., 33 (19833 937.
[43] B.C. Henderson, I.M. Stewart and G.S. Casuccio, Am. Lab., November (19893.
[44] W. Stumm, and J.J, Morgan, Aquatic Chemistry, 2nd edn., John Wiley, New York, 198l.
[45] R.J. Hunter, The Zeta Potential in Colloid Science, Academic Press, London, 1981.
[46] D.A. Dzombak and R.J.M. Hudson, in C.P. Huang, C.R. O'Melia and J.J. Morgan (Eds.), Aquatic Chemistry: Interracial and Interspecies Processes, American Chemical Society, Washington, DC, 1995.
[47] W.H. Press, S.A. Teukolsky, W.T. Vetterling and B.P. Flannery, Numerical Recipes in C, 2nd edn., Cambridge University Press, Cambridge, 1992.
[48] C.R. O'Melia, Environ. Sci. Technol., 14 (19803 1052. [49] G. Sposito, The Chemistry of Soils, Oxford University
Press, New York, 1989. [50] R.Y. Ofoli, Ph. D. Thesis, Dept. of Chemical Engineering,
Carnegie Mellon University, 1994. [51] A.C. Payatakes, H.Y. Park and J. Petrie, Chem. Eng.
Sci., 36 (1981) 1319. [52] H.C. Hamaker, Physica, 4 (19373 1058. [53] L.A. Spielman and P.M. Cukor, J. Colloid Interface Sci.,
43 (1973) 51. [54] T.W. Healy, D. Chan and L.R. White, Pure Appl. Chem.,
52 (19803 1207. [-55] L. Song, P.R. Johnson and M. Elimelech, Environ. Sci.
Technol., 28 (1994) 1164.







