double aromaticity and ring currents in all-carbon rings
TRANSCRIPT

DOI: 10.1002/chem.200900322
Double Aromaticity and Ring Currents in All-Carbon Rings
Patrick W. Fowler,*[a] Noriyuki Mizoguchi,[b] David E. Bean,[a] andRemco W. A. Havenith[c]
IntroductionCarbon clusters are known to exist in a rich variety of struc-tures. As cluster size increases, linear chains, cyclic struc-tures and three-dimensional cages are found. So far, themost extensive experimental and theoretical studies havebeen performed for small carbon clusters Cn.
[1–4] For exam-ple, Yang et al. measured photoelectron (PE) spectra on Cn
�
ions and observed an abrupt change in the electron affinityat C10
�, which was taken as evidence for a linear-to-monocy-clic transition.[5] This observation was supported by recentmeasurements by Handschuh et al.[6] They assigned the ob-served vibrational modes to four dominant structural types:linear chains for C5
� to C9� ; monocyclic rings for C10
� toC18
� ; bicyclic rings for C20�, C24
� and C28� ; and fullerenes for
even-numbered clusters larger than C30�. Magic number
peaks have been observed in the mass spectra of smallerCn
+ ions for odd-numbered clusters, especially at n= 3, 11,15 and 19.[7] In contrast, for small Cn
� ions, the magic num-bers include n=5, 10, 12, 16, 18 and 22,[6] or, under differentexperimental conditions, n=17, 21, 25 and 33.[8] For neu-trals, however, it has been found difficult to measure con-centrations without bias from ionisation and fragmentationprocesses. Recently, Kaizu et al.[9] and Wakabayashi et al.[10]
used single-photon (10.5 eV) ionisation of neutrals to obtainmass spectra of carbon clusters that exhibit the predomi-nance of C4m +2 (m =2, 3, 4). The presence of cyclic Cn (n=
6, 8, 10, 12, 14, 18, 22) clusters was confirmed in several ex-periments.[11–19]
Many theoretical studies have been performed for smallcarbon clusters Cn.
[20–38] It has long been recognised thatlinear isomers are strongly favoured for clusters smallerthan C10, but that clusters in the size range 10 to 20 existpredominantly as monocyclic rings. However, recent calcula-tions have revealed that, even for the small even-numberedclusters such as C6 and C8, ring isomers can be iso-energeticwith, or lower in energy than, the linear form.[29,30] It hasalso been shown theoretically that, in the range C10 to C30,the most stable structures predicted for cyclic C4m+2 clustersare of D(2m +1)h symmetry, with cumulenic bonding configura-tions, whereas cyclic C4m clusters take the form of bond-al-ternated planar rings of C(2m)h symmetry.[33–38]
There have been several attempts to understand the sta-bility of magic Cn clusters in terms of aromaticity. The spe-cial stability of cyclic carbon clusters could be considered to
Abstract: Double aromaticity of neu-tral, planar rings of carbon atoms isdemonstrated through visualisation ofthe induced ring currents, mapped atthe ipsocentric B3LYP/6-31G(d)//B3LYP/6-31G(d) level for species C6 toC30, with onset of delocalised current inthe in-plane p system at C10/C11. Bothin-plane and conventional out-of-plane
p systems have diatropic/paratropiccurrent in accordance with the H�ckelrule, with 4 m+ 2 occupation of the
out-of-plane p system taking prece-dence, as predicted by simple nestingof Frost–Musulin diagrams. The cur-rent-density maps show characteristicdouble-doughnut and double-track top-ographies for out-of-plane and in-planering currents, respectively, both gov-erned by a common framework of an-gular momentum rules.
Keywords: aromaticity · carbon ·cluster compounds · densityfunctional calculations · magneticproperties · ring currents
[a] Prof. P. W. Fowler, D. E. BeanDepartment of ChemistryUniversity of SheffieldSheffield, S3 7HF (UK)Fax: (+44) 0114-222-9346E-mail : [email protected]
[b] Dr. N. MizoguchiDepartment of PhysicsMeiji Pharmaceutical University2-522-1, Noshio, Kiyose, Tokyo 204-8588 (Japan)
[c] Dr. R. W. A. HavenithSchool of EducationWindesheim University of Applied SciencesCampus 2-6, 8016 CA Zwolle (The Netherlands)
Supporting information for this article is available on the WWWunder http://dx.doi.org/10.1002/chem.200900322.
� 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 6964 – 69726964

arise solely from the presence of 4m+2 p electrons, as inthe annulene analogues.[39] However, simple molecular orbi-tal considerations suggest another contribution to aromatici-ty for the bare carbon rings: one that arises from overlap ofthe outward-pointing, in-plane (hybrid) orbitals. Double aro-maticity, as defined by Chandrasekhar, Jemmis, and Schley-er,[40] arises from the presence of two orthogonal, cyclicallydelocalised electronic systems. Bare carbon rings possesstwo independent systems of p molecular orbitals (MOs), theconventional out-of-plane p (pout) MOs, which are antisym-metric with respect to reflection in the molecular plane, andin-plane p (pin) MOs composed of radially oriented spx orbi-tals (see Figure 1), which are symmetric with respect to re-
flection in the molecular plane.[41] Cyclic conjugation of pout
and pin electrons may make separate stabilising or destabilis-ing contributions to the overall energy of the Cn ring, andtherefore, when overlap within the in-plane p system be-comes significant, carbon rings may be expected to exhibitdouble aromaticity, double anti-aromaticity, pout aromaticitywith pin anti-aromaticity, or pout anti-aromaticity with pin ar-omaticity, as a function of electron count and orbital occu-pancy.
A simple approach[41] to predicting the systematics of Cn
rings employs nested Frost–Musulin[42] diagrams (seeFigure 2), which have been shown to rationalise the resultsof more sophisticated calculations on carbon rings Cn (n= 6–30), recovering both the electronic configurations and theobserved trends in bond alternation.
In addition to geometric and energetic consequences, theanticipated variation in aromaticity also has implications forthe magnetic properties of all-carbon rings. For example,
modulation of magnetic properties with n is found in NICS(nucleus independent chemical shift[43]) calculations oncarbon rings.[44–46] The results of these calculations are com-patible with the conclusion that C4m+2 rings are doubly aro-matic, C4m rings are doubly anti-aromatic, C4m +1 and C4m+ 3
rings are p (pout) aromatic and s (pin) anti-aromatic.[41, 47]
In the ring-current model of magnetic response, aromatic-ity (anti-aromaticity) is associated with the ability of a cyclicsystem to support a diatropic (paratropic) ring current.[39] Inthis model, statements about double and mixed aromatici-ties correspond to definite predictions about the spatial dis-tribution of the induced current density. In conventional p
systems, these induced currents have a typical “doubledoughnut” distribution. The currents arising from a combi-nation of pout and pin electrons may be expected to exhibit amore complex topography.
In the present work, we use the established ipsocen-tric[48–50] technique for computation and mapping of currentdensity to visualise the currents in Cn rings over the wholerange from n=6 to n=30, and analyse orbital contributionsto identify the separate roles of the two orthogonal p sys-tems. In this way, a direct demonstration of the nature ofmixed aromaticity in some of these systems (those with n�11) is achieved.
Computational Methods
Structures were optimised with the B3LYP hybrid density functional and6-31G(d) basis, without symmetry constraints, for singlet states from C6
to C30. Vibrational frequencies were computed in order to characterisethe nature of the stationary points, and all but one of the rings presentedhere were characterised as planar minima. (For C9, the planar geometryis a transition state,[46] but its properties are included for purposes ofcomparison.) These calculations were performed with the GAUSSIAN 03program.[51]
The calculated geometries are listed in the Supporting Information. Insummary, the optimised structures for small C4m+2 (m =1–5) rings areequilateral, with D(2m+1)h symmetry; larger C4m+2 (m =6–7) rings adoptstructures of C(2m+1)h symmetry that have alternating bond lengths, pre-sumably as a consequence of second-order Jahn–Teller effects;[33–37] theoptimised structures for cyclic C4m (m= 1–7) species are of C(2m)h symme-
Figure 1. Illustration of pout (top) and pin (bottom) delocalised systemsarising from overlap of out-of-plane and radial orbitals, respectively.
Figure 2. Nested Frost–Musulin diagram for a planar C6 ring, giving aqualitative prediction of pout and pin orbital energies, constructed withbond resonance integrals jbin j < jbout j to reflect orbital overlap. In theparticular case illustrated, both pout and pin shells give closed-shell six-electron systems.
Chem. Eur. J. 2009, 15, 6964 – 6972 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 6965
FULL PAPER

try, again with alternating bond length; the stable structures for odd-membered carbon rings C4m+1 and C4m+3 have lower symmetries. Sub-stantial angle alternation is present in each ring Cn (n=6–30).
The current maps shown in this paper for the Cn rings (n=6–30) werecalculated using the ipsocentric[48–50] CTOCD-DZ (continuous transfor-mation of origin of current density—diamagnetic zero) method. Re-sponse calculations at the B3LYP/6-31G(d)//B3LYP/6-31G(d) level wereperformed with GAMESS-UK,[52] and the resulting perturbed Kohn–Sham orbitals were used in the calculation of the current density in theipsocentric approach with theSYSMO[53] package. For comparison,Coupled Hartree–Fock (CHF) calcu-lations were also performed for thesame geometries, with the same basisset. Current density maps, plotted ina plane at a distance of 1a0 above themolecular plane, show the currentdensity j(1), induced by a unit externalmagnetic field perpendicular to themagnetic plane. Anticlockwise circu-lations in the maps indicate diatropiccurrents, and clockwise circulationsindicate paratropic currents. Colourversions of all maps are included inthe Supporting Information. A usefulindication of current strength is jmax,the maximum of the modulus of thecurrent density taken over the plot-ting plane.
The ipsocentric method allows thepartition of total current density intophysical non-redundant contributionsfrom occupied orbitals.[50] In contrastwith those arising from other distri-butions of origin, ipsocentric orbitalcontributions are not affected by spu-rious remixing of occupied orbitals inthe magnetic field.[54]
In order to tease out the orbitalorigin of aromaticity in the carbonrings using this technique, it was nec-essary to identify the molecular orbi-tals of pout and pin types. Symmetryunder reflection in the molecularplane distinguishes pout from otheroccupied orbitals, which comprisecarbon 1s cores, the 2-electroncarbon–carbon bonds, plus the pin
subsystem, easily identified from indi-vidual orbital plots. Those considera-tions allowed assignment of an elec-tron configuration in terms of pout and pin subsystems, and partition ofthe total current density into additive contributions from pout, pin, and un-derlying s framework orbitals. As inspection of the respective currentdensity maps reveals, only the pout, and pin sets of occupied orbitals leadto delocalised currents, and in each case the contribution from a givensubsystem is dominated by the highest occupied orbital (or orbital pair)within the set, that is, the “pout HOMO” or “pin HOMO”.
ResultsThe maps of pout and pin contributions to current density fallnaturally into two sets: those for small rings (C6–C10), forwhich there is evidence of conventional p ring current, butlittle or no in-plane p current, and those for the larger rings
(C11–C30), in which both subsystems give rise to delocalisedring currents.
Figures 3 and 4 present the current density arising frompout and pin HOMO orbitals, respectively, in C11 to C30. Eachfigure is divided into columns for systems with 4m+3, 4 m,4 m+ 1 and 4 m+ 2 carbon atoms. Individual maps are pre-sented in a uniform format in which all geometries aredrawn to scale and the largest current in each map is com-
pared in an inset to the “benzene standard”, that is, the p-HOMO current for benzene computed at a height of 1a0
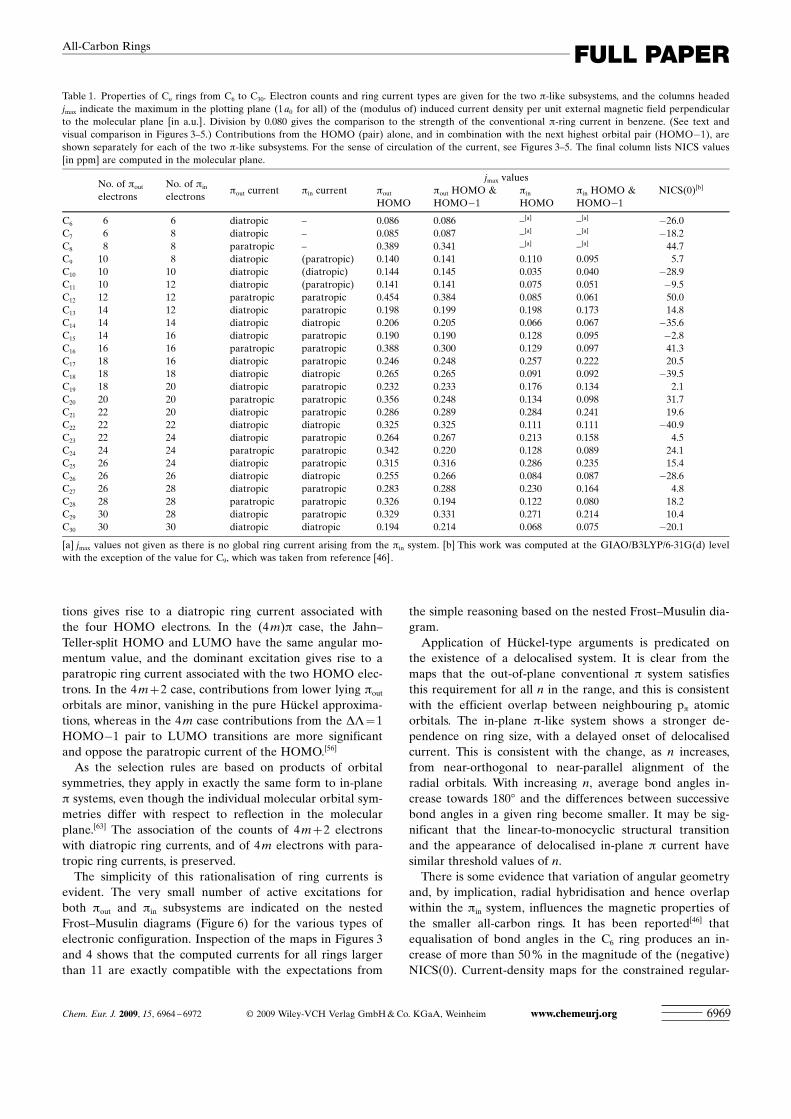
using the same functional, basis, and level of theory (inatomic units, this standard value is 0.080). For ease of com-parison, the overall sense of the ring current is also indicat-ed schematically. Figure 5 shows the maps for the remainingsmall rings, C6 to C10. Table 1 summarises the electron con-figurations, types, and strengths of currents for the full set ofsystems C6 to C30.
There are some differences between the DFT mapsshown in this paper and CHF maps plotted with the samegeometries (see the Supporting Information for examples).In general, there is good qualitative agreement between thetwo methods, but DFT predicts an earlier onset of the pin
current (the bilobal pin current first appears at n= 11 or 12
Figure 3. Maps of current density, calculated at the ipsocentric B3LYP/6-31G(d) level, for carbon rings C11–C30,showing the orbital contribution from the pout HOMO orbital or pair of orbitals. Diatropic/paratropic circula-tions are indicated schematically for each ring by the circular arrows. The largest arrow in each map is alsocompared with the largest arrow from the p current-density map of benzene.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 6964 – 69726966
P. W. Fowler et al.

for the DFT maps, but at n=13 for the CHF maps). Overthe entire range, DFT maps show stronger currents than theCHF maps, in both the pout and pin systems. This differenceis typical of comparisons between calculations of inducedcurrent using DFT and CHF methods, and is usually attrib-uted to the smaller HOMO–LUMO gaps given by the DFTtreatment.[55]
Discussion
The significant trends, and the main results of this work, arereadily apparent from the collection of maps in Figures 3–5.All the systems treated in the calculations show strong out-of-plane ring currents, with the sense of circulation expectedfrom the electron count of the pout subsystem. This count iseasily established: as Table 1 shows, the filling of pout and pin
orbital manifolds of energy levels is consistent with Aufbaupredictions from the simple nested Frost–Musulin diagrams.For rings of 4 m+2 atoms, pout and pin subsystems each con-tain 4 m+2 electrons, and for rings of 4 m atoms the subsys-tems each contain 4m electrons, whether or not allowance ismade for Jahn–Teller distortion to bond-alternated planar
geometries. For rings with an odd number of atoms, 4m+ 1and 4 m +3, the pout system is preferentially occupied, with4 m+ 2 electrons in each case, leaving the residual 4m/4 m+
1 electrons in the pin system. From the maps we can see thatthe out-of-plane p systems have diatropic/paratropic current,in accordance with the H�ckel 4m +2/4 m rule. In this re-spect, the resemblance between the conventional p systemsof these all-carbon rings and their hydrocarbon analogues isexact.
Perhaps less evident is that the same correlation shouldbe found for the in-plane p electrons, which occupy orbitalsthat would correspond to localised C�H bonds in the hydro-carbon analogue. The maps show that, beyond a thresholdaround n= 11, the all-carbon rings exhibit diatropic/para-tropic currents in accordance with the numbers of electronsin the pin subsystem. Thus, the association of 4m+2 elec-trons with diatropic ring currents and 4m electrons with par-atropic ring currents is preserved. The underlying explana-tion for this parallelism relies on an extension of the angularmomentum arguments used to account for currents in annu-lenes,[56] as discussed below.
To use the language of double aromaticity, the ring-cur-rent maps indicate that the C4m+2 rings are doubly aromatic
Figure 4. Maps of current density, calculated at the ipsocentric B3LYP/6-31G(d) level, for carbon rings C11–C30, showing the orbital contribution from thepin HOMO orbital or pair of orbitals. Diatropic/paratropic circulations are indicated schematically for each ring by the circular arrows. The largest arrowin each map is also compared with the largest arrow from the p current-density map of benzene.
Chem. Eur. J. 2009, 15, 6964 – 6972 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 6967
FULL PAPERAll-Carbon Rings

as the ring currents in the pout and pin systems are both dia-tropic, the C4m rings are doubly anti-aromatic as both ringcurrents are paratropic, and the C4m+1 and C4m+ 3 rings havemixed aromaticity, with the aromatic component arisingfrom the pout electrons. In another context, these odd-num-bered rings might be called p aromatic and s anti-aromatic.
Table 1 also includes NICS values calculated at the ringcentres. The C4m +2 rings have large and negative NICSvalues; the C4m rings have large and positive NICS values;the C4m +1 rings have significant positive NICS values; andthe C4m+3 rings have smaller and generally positive NICSvalues. In the “doubly aromatic” 4m+2 and “doubly anti-aromatic” 4 m cases, in which the pout and pin ring currentsreinforce, the NICS values are as expected from the sense ofcirculation. In simple scatter plots of NICS values against(signed) maximum currents for both pout and pin subsystems(Figure S3, Supporting Information), the rings with electroncounts of 4 m/4 m +2 appear in opposite quadrants, demon-strating at least this basic level of agreement between thetwo magnetically-based indices. For the odd-numbered rings,the association between NICS and current direction is lessclear-cut; even so, the scatter plot gives some indication that
the positive NICS values, at least for the 4m+ 1 rings, arereflections of the paratropic nature of the pin current.
Questions about the interpretation of NICS(0) values andthe degree to which this quantity predicts ring current con-tributions have been aired in the literature on many occa-sions. There are difficulties in defining physically non-redun-dant orbital contributions to this isotropic property, as twoof its components necessarily mix s and p orbitals.[54] Vari-ous alternative schemes for disentangling s/p contributionshave been proposed,[57] amongst them NICS(0)pzz
[58] and theNICS-scan method.[59] Clearly, as they all rely on solutionsof the same Schrçdinger equation, information from differ-ent magnetic indices must ultimately be compatible, but cal-culation of integral properties rather than maps of spatialdistribution of currents necessarily involves some loss ofdetail, which may lead to ambiguities in interpretation.
Recent work by Bultinck and co-workers[60,61] has demon-strated that it is possible to reconcile NICS values, ring de-localisation indices, and ring-current maps for polycyclic ar-omatic hydrocarbons within a common statistical approach,even though it is not possible to deduce all the detail ofcomplex current distributions from isolated NICS values. Asimilar approach may be expected to bring insight in caseswhere the complexity arises from competition between elec-tronic subsystems associated with a single cycle. As the pres-ent results show, the mapping approach allows the compet-ing effects to be separated. Compatibility between ring de-localisation indices and current-density maps has alreadybeen demonstrated for co-existing s and p ring currents inthe monocycle C6I6
2+ .[62] Although the nature of the s
system supporting the current is different, it could provefruitful to make a similar analysis of the delocalisation forthe present all-carbon rings, at least for the small cases inwhich the combinatorics of the multicentre indices willremain manageable.
Returning to the present results, the observations fromcurrent-density mapping are clear, in that both pout and pin
subsystems show a well-defined H�ckel-like association be-tween electron count and ring current. It remains now toshow why this correlation holds. Detailed arguments basedon angular momentum selection rules show that patterns oforbital occupancy have direct consequences for the ring-cur-rent aromaticity. In the ipsocentric formulation,[48–50] diatrop-ic currents arise from virtual excitations of translationalsymmetry, that is, in which the product of symmetries of theoccupied orbital and the target virtual orbital contains thesymmetry of an in-plane translation. Conversely, paratropiccurrents arise from virtual excitations of rotational symme-try, in which the product of occupied and target orbital sym-metries contains the symmetry of the unique in-plane rota-tion.
Electron counting and the magnetic criterion of aromatic-ity run together for conventional delocalised p monocycles,for which the selection rules distinguish between systemswith 4 m+ 2 and 4 m p electrons. In the (4 m+2)p case, theHOMO–LUMO frontier separates orbitals that differ byone unit of angular momentum, and the dominant excita-
Figure 5. Maps of current density, calculated at the B3LYP/6-31G(d)level, for carbon rings C6–C10, showing the orbital contribution from thepout HOMO orbital or pair of orbitals (left) and the pin HOMO orbital orpair of orbitals (right). All structures and arrows for these smaller mole-cules are plotted at double scale with respect to Figures 3 and 4, but oth-erwise the same plotting conventions are retained.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 6964 – 69726968
P. W. Fowler et al.
tions gives rise to a diatropic ring current associated withthe four HOMO electrons. In the (4 m)p case, the Jahn–Teller-split HOMO and LUMO have the same angular mo-mentum value, and the dominant excitation gives rise to aparatropic ring current associated with the two HOMO elec-trons. In the 4 m +2 case, contributions from lower lying pout
orbitals are minor, vanishing in the pure H�ckel approxima-tions, whereas in the 4 m case contributions from the DL= 1HOMO�1 pair to LUMO transitions are more significantand oppose the paratropic current of the HOMO.[56]
As the selection rules are based on products of orbitalsymmetries, they apply in exactly the same form to in-planep systems, even though the individual molecular orbital sym-metries differ with respect to reflection in the molecularplane.[63] The association of the counts of 4m+2 electronswith diatropic ring currents, and of 4m electrons with para-tropic ring currents, is preserved.
The simplicity of this rationalisation of ring currents isevident. The very small number of active excitations forboth pout and pin subsystems are indicated on the nestedFrost–Musulin diagrams (Figure 6) for the various types ofelectronic configuration. Inspection of the maps in Figures 3and 4 shows that the computed currents for all rings largerthan 11 are exactly compatible with the expectations from
the simple reasoning based on the nested Frost–Musulin dia-gram.
Application of H�ckel-type arguments is predicated onthe existence of a delocalised system. It is clear from themaps that the out-of-plane conventional p system satisfiesthis requirement for all n in the range, and this is consistentwith the efficient overlap between neighbouring pp atomicorbitals. The in-plane p-like system shows a stronger de-pendence on ring size, with a delayed onset of delocalisedcurrent. This is consistent with the change, as n increases,from near-orthogonal to near-parallel alignment of theradial orbitals. With increasing n, average bond angles in-crease towards 1808 and the differences between successivebond angles in a given ring become smaller. It may be sig-nificant that the linear-to-monocyclic structural transitionand the appearance of delocalised in-plane p current havesimilar threshold values of n.
There is some evidence that variation of angular geometryand, by implication, radial hybridisation and hence overlapwithin the pin system, influences the magnetic properties ofthe smaller all-carbon rings. It has been reported[46] thatequalisation of bond angles in the C6 ring produces an in-crease of more than 50 % in the magnitude of the (negative)NICS(0). Current-density maps for the constrained regular-
Table 1. Properties of Cn rings from C6 to C30. Electron counts and ring current types are given for the two p-like subsystems, and the columns headedjmax indicate the maximum in the plotting plane (1 a0 for all) of the (modulus of) induced current density per unit external magnetic field perpendicularto the molecular plane [in a.u.] . Division by 0.080 gives the comparison to the strength of the conventional p-ring current in benzene. (See text andvisual comparison in Figures 3–5.) Contributions from the HOMO (pair) alone, and in combination with the next highest orbital pair (HOMO�1), areshown separately for each of the two p-like subsystems. For the sense of circulation of the current, see Figures 3–5. The final column lists NICS values[in ppm] are computed in the molecular plane.
No. of pout
electronsNo. of pin
electronspout current pin current
jmax valuesNICS(0)[b]pout
HOMOpout HOMO &HOMO�1
pin
HOMOpin HOMO &HOMO�1
C6 6 6 diatropic – 0.086 0.086 –[a] –[a] �26.0C7 6 8 diatropic – 0.085 0.087 –[a] –[a] �18.2C8 8 8 paratropic – 0.389 0.341 –[a] –[a] 44.7C9 10 8 diatropic (paratropic) 0.140 0.141 0.110 0.095 5.7C10 10 10 diatropic (diatropic) 0.144 0.145 0.035 0.040 �28.9C11 10 12 diatropic (paratropic) 0.141 0.141 0.075 0.051 �9.5C12 12 12 paratropic paratropic 0.454 0.384 0.085 0.061 50.0C13 14 12 diatropic paratropic 0.198 0.199 0.198 0.173 14.8C14 14 14 diatropic diatropic 0.206 0.205 0.066 0.067 �35.6C15 14 16 diatropic paratropic 0.190 0.190 0.128 0.095 �2.8C16 16 16 paratropic paratropic 0.388 0.300 0.129 0.097 41.3C17 18 16 diatropic paratropic 0.246 0.248 0.257 0.222 20.5C18 18 18 diatropic diatropic 0.265 0.265 0.091 0.092 �39.5C19 18 20 diatropic paratropic 0.232 0.233 0.176 0.134 2.1C20 20 20 paratropic paratropic 0.356 0.248 0.134 0.098 31.7C21 22 20 diatropic paratropic 0.286 0.289 0.284 0.241 19.6C22 22 22 diatropic diatropic 0.325 0.325 0.111 0.111 �40.9C23 22 24 diatropic paratropic 0.264 0.267 0.213 0.158 4.5C24 24 24 paratropic paratropic 0.342 0.220 0.128 0.089 24.1C25 26 24 diatropic paratropic 0.315 0.316 0.286 0.235 15.4C26 26 26 diatropic diatropic 0.255 0.266 0.084 0.087 �28.6C27 26 28 diatropic paratropic 0.283 0.288 0.230 0.164 4.8C28 28 28 paratropic paratropic 0.326 0.194 0.122 0.080 18.2C29 30 28 diatropic paratropic 0.329 0.331 0.271 0.214 10.4C30 30 30 diatropic diatropic 0.194 0.214 0.068 0.075 �20.1
[a] jmax values not given as there is no global ring current arising from the pin system. [b] This work was computed at the GIAO/B3LYP/6-31G(d) levelwith the exception of the value for C9, which was taken from reference [46].
Chem. Eur. J. 2009, 15, 6964 – 6972 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 6969
FULL PAPERAll-Carbon Rings

hexagonal structure (Figure 7) show a conventional p cur-rent of almost identical strength (jmax =0.086) and a moreuniform distribution of pin current compared with that in theD3h equilibrium structure, though in both cases the pin cur-rent is a superposition of atom-centred circulations ratherthan a true global ring current.
Figures 8–10 present current-density maps for three typicalcases of rings Cn in order to il-lustrate in more detail thepoints made above. All the in-duced ring currents have bilo-bal distributions, but with es-sentially different topographiesfor out-of-plane and in-planesystems. The conventional pout
ring current follows the circuitof atoms above and below theplane, whereas the two lobesof the pin current lie inside andoutside the atomic circuit. Inthe 4 m +2 systems, such as C18
(see Figure 8), interior and ex-terior pin ring currents are ofcomparable strength, but inother cases the interior currentis more intense, as would beexpected on the grounds of or-bital overlap.
Figure 9 illustrates another difference between 4m+ 2and 4 m systems, which was alluded to earlier. The currentdensity maps for both pout and pin of 4 m C20 are dominatedby the intense paratropic two-electron contribution of therespective HOMO orbitals, with partial cancellation by thefour-electron diatropic contributions from the HOMO�1, aspredicted by angular momentum arguments, and in exactanalogy with the conventional paratropic ring current of theanti-aromatic monocyclic hydrocarbons.[56]
Figure 6. Nested Frost–Musulin diagrams showing the general patterns of frontier molecular orbitals for thefour types of Cn rings, in idealised Dnh symmetry.
Figure 7. Current-density maps showing the current arising from the fourelectrons in the pout HOMO (top) and the four electrons in the pin
HOMO (bottom) of the C6 ring with D6h symmetry.
Figure 8. Current-density maps showing the current arising from the fourelectrons in the pout HOMO (left) and the four electrons in the pin
HOMO (right) of the C18 ring. Side views of the currents show how theyare distributed around the molecular plane. These side views are plottedin a plane perpendicular to the molecular plane, outside of the ring, at adistance of 1 a0 away from a carbon-carbon s bond.
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 6964 – 69726970
P. W. Fowler et al.

Figure 10 illustrates the mixed character of the aromatici-ty of rings with odd numbers of carbon atoms, as in the caseof C19, in which a single external field produces opposed cir-culations in different regions of the same molecule.
These examples confirm the general utility of current-den-sity mapping as a way of visualising the concept of double
aromaticity, and explaining its physical origins by a frontier-orbital “spectroscopic” picture of ring current. The availabil-ity of a robust scheme for defining orbital contributionsmakes it possible to distinguish between classes of systemsin which the two aromatic components reinforce, and thosein which they compete.
Acknowledgements
P.W.F. thanks the Royal Society/Wolfson Scheme for a Research MeritAward and D.E.B. thanks EPSRC and The University of Sheffield for fi-nancial support.
[1] W. Weltner, Jr., R. J. V. Zee, Chem. Rev. 1989, 89, 1713 – 1747.[2] A. Van Orden, R. J. Saykally, Chem. Rev. 1998, 98, 2313 –2357.[3] N. G. Gotts, G. von Helden, M. T. Bowers, Int. J. Mass. Spectrom.
Ion Processes 1995, 149, 217 –229.[4] R. O. Jones, J. Chem. Phys. 1999, 110, 5189 – 5200.[5] S. Yang, K. J. Taylor, M. J. Craycraft, J. Conceicao, C. L. Pettiette, O.
Cheshnovsky, R. E. Smalley, Chem. Phys. Lett. 1988, 144, 431 –436.[6] H. Handschuh, G. Gantefoer, B. Kessler, P. S. Bechthold, W. Eber-
hardt, Phys. Rev. Lett. 1995, 74, 1095 – 1098.[7] E. A. Rohlfing, D. M. Cox, A. Kaldor, J. Chem. Phys. 1984, 81,
3322 – 3330.[8] C. Q. Jiao, D. K. Phelps, S. Lee, Y. Huang, B. S. Freiser, Rapid
Commun. Mass Spectrom. 1994, 8, 404 –408.[9] K. Kaizu, M. Kohno, S. Suzuki, H. Shiromaru, T. Moriwaki, Y.
Achiba, J. Chem. Phys. 1997, 106, 9954 – 9956.[10] T. Wakabayashi, T. Momose, T. Shida, J. Chem. Phys. 1999, 111,
6260 – 6263.[11] J. D. Presilla-M�rquez, J. A. Sheehy, J. D. Mills, P. G. Carrick, C. W.
Larson, Chem. Phys. Lett. 1997, 274, 439 –444.[12] S. L. Wang, C. M. L. Rittby, W. R. M. Graham, J. Chem. Phys. 1997,
107, 6032 –6037.[13] S. L. Wang, C. M. L. Rittby, W. R. M. Graham, J. Chem. Phys. 1997,
107, 7025 –7033.[14] J. D. Presilla-M�rquez, J. Harper, J. A. Sheehy, P. G. Carrick, C. W.
Larson, Chem. Phys. Lett. 1999, 300, 719 –726.[15] A. K. Ott, G. A. Rechsteiner, C. Felix, O. Hampe, M. F. Jarrold, K.
Raghavachari, J. Chem. Phys. 1998, 109, 9652 –9655.[16] G. A. Rechtsteiner, C. Felix, A. K. Ott, O. Hampe, R. P. Van Duyne,
M. F. Jarrold, K. Raghavachari, J. Phys. Chem. A 2001, 105, 3029 –3033.
[17] M. Grutter, M. Wyss, E. Riaplov, J. P. Maier, S. D. Peyerimhoff, M.Hanrath, J. Chem. Phys. 1999, 111, 7397 –7401.
[18] A. E. Boguslavskiy, H. Ding, J. P. Maier, J. Chem. Phys. 2005, 123,034305.
[19] L. Belau, S. E. Wheeler, B. W. Ticknor, M. Ahmed, S. R. Leone,W. D. Allen, H. F. Schaefer III, M. A. Duncan, J. Am. Chem. Soc.2007, 129, 10229 –10243.
[20] K. S. Pitzer, E. Clementi, J. Am. Chem. Soc. 1959, 81, 4477 – 4485.[21] R. Hoffmann, Tetrahedron 1966, 22, 521 –538.[22] K. Raghavachari, R. A. Whiteside, J. A. Pople, J. Chem. Phys. 1986,
85, 6623 –6628.[23] M. S. Islam, A. K. Ray, J. Phys. B 1989, 22, 2071 –2079.[24] F. Diederich, Y. Rubin, C. B. Knobler, R. L. Whetten, K. E. Schriv-
er, K. N. Houk, Y. Li, Science 1989, 245, 1088 –1090.[25] C. Liang, H. F. Schaefer III, J. Chem. Phys. 1990, 93, 8844 –8849.[26] V. Parasuk, J. Almlof. M. W. Feyereisen, J. Am. Chem. Soc. 1991,
113, 1049 –1050.[27] J. D. Watts, R. J. Bartlett, Chem. Phys. Lett. 1992, 190, 19–24.[28] J. Hutter, H. P. Luethi, F. Diederich, J. Am. Chem. Soc. 1994, 116,
750 – 756.[29] V. Pless, H. U. Suter, B. Engels, J. Chem. Phys. 1994, 101, 4042 –
4048.
Figure 9. Current-density maps showing the currents in the C20 ring aris-ing from the two electrons in the pout HOMO (top left) and the pin
HOMO (top right), and from the four electrons in the pout HOMO�1(bottom left), and the pin HOMO�1 (bottom right).
Figure 10. Map of the total (all-electron) induced current density of theC19 ring, showing the inside track of the pin current and the upper lobe ofthe dominant pout current.
Chem. Eur. J. 2009, 15, 6964 – 6972 � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemeurj.org 6971
FULL PAPERAll-Carbon Rings

[30] J. M. L. Martin, P. R. Taylor, J. Phys. Chem. 1996, 100, 6047 –6056.[31] D. A. Plattner, K. N. Houk, J. Am. Chem. Soc. 1995, 117, 4405 –
4406.[32] M. S. Deleuze, M. G. Giuffreda, J.-P. FranÅois, L. S. Cederbaum, J.
Chem. Phys. 2000, 112, 5325 – 5338.[33] E. J. Bylaska, R. Kawai, J. H. Weare, J. Chem. Phys. 2000, 113,
6096 – 6106.[34] M. Saito, Y. Okamoto, Phys. Rev. B 1999, 60, 8939 – 8942.[35] T. Torelli, L. Mitas, Phys. Rev. Lett. 2000, 85, 1702 –1705.[36] E. J. Bylaska, J. H. Weare, R. Kawai, Phys. Rev. B 1998, 58, R7488 –
R7491.[37] S. Sen, P. Seal, S. Chakrabarti, Phys. Rev. B 2006, 73, 245401.[38] S. Arulmozhiraja, T. Ohno, J. Chem. Phys. 2008, 128, 114301.[39] V. I. Minkin, M. N. Glukhovtsev, B. Y. Simkin, Aromaticity and Anti-
aromaticity, Wiley, New York, 1994.[40] J. Chandrasekhar, E. D. Jemmis, P. von R. Schleyer, Tetrahedron
Lett. 1979, 20, 3707 –3710.[41] N. Mizoguchi, Chem. Ind. 2007, 58, 102 – 106, in Japanese.[42] A. A. Frost, B. Musulin, J. Chem. Phys. 1953, 21, 572 –573.[43] P. von R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao, N. J. R. van
Eikema Hommes, J. Am. Chem. Soc. 1996, 118, 6317 – 6318.[44] S. Mart�n-Santamar�a, H. Rzepa, Chem. Commun. 2000, 1503 – 1504.[45] S.-H. Xu, M.-Y. Zhang, Y.-Y. Zhao, B.-G. Chen, J. Zhang, C.-C. Sun,
Chem. Phys. Lett. 2006, 421, 444 –447.[46] M. D. Wodrich, C. Corminboeuf, S. S. Park, P. von R. Schleyer,
Chem. Eur. J. 2007, 13, 4582 – 4593.[47] N. Mizoguchi, 12th International Symposium on Novel Aromatic
Compounds, Programme and Abstracts, 2007, p. 240.[48] T. A. Keith, R. F. W. Bader, Chem. Phys. Lett. 1993, 210, 223 –231.[49] S. Coriani, P. Lazzeretti, M. Malagoli, R. Zanasi, Theor. Chim. Acta.
1994, 89, 181 –192.[50] E. Steiner, P. W. Fowler, J. Phys. Chem. A 2001, 105, 9553 –9562.[51] Gaussian 03, Revision B.01, M. J. Frisch, G. W. Trucks, H. B. Schle-
gel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgom-ACHTUNGTRENNUNGery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S.Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani,N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K.Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda,
O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian,J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann,O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y.Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg,V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O.Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Fores-man, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski,B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L.Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Na-nayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen,M. W. Wong, C. Gonzalez, J. A. Pople, Gaussian, Inc., PittsburghPA, 2004.
[52] M. F. Guest, I. J. Bush, H. J. J. Van Dam, P. Sherwood, J. M. H.Thomas, J. H. Van Lenthe, R. W. A. Havenith, J. Kendrick, Mol.Phys. 2005, 103, 719 – 747.
[53] P. Lazzeretti, R. Zanasi, SYSMO Package, University of Modena,1980. Additional routines by P. W. Fowler, E. Steiner, R. W. A. Ha-venith, A. Soncini.
[54] E. Steiner, P. W. Fowler, Phys. Chem. Chem. Phys. 2004, 6, 261 – 272.[55] R. W. A. Havenith, P. W. Fowler, Chem. Phys. Lett. 2007, 449, 347 –
353.[56] E. Steiner, P. W. Fowler, Chem. Commun. 2001, 2220 –2221.[57] Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta, P. von R.
Schleyer, Chem. Rev. 2005, 105, 3842 – 3888.[58] H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R.
Puchta, P. v. R. Schleyer, Org. Lett. 2006, 8, 863 –866.[59] A. Stanger, Chem. Eur. J. 2006, 12, 2745 –2751.[60] S. Fias, S. Van Damme, P. Bultinck, J. Comput. Chem. 2008, 29, 358 –
366.[61] S. Fias, P. W. Fowler, J. L. Delgado, U. Hahn, P. Bultinck, Chem.
Eur. J. 2008, 14, 3093 –3099.[62] R. W. A. Havenith, P. W. Fowler, S. Fias, P. Bultinck, Tetrahedron
Lett. 2008, 49, 1421 –1424.[63] R. W. A. Havenith, A. Rassat, P. W. Fowler, J. Chem. Soc. Perkin
Trans. 2 2002, 723 – 727.
Received: February 5, 2009Published online: June 5, 2009
www.chemeurj.org � 2009 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim Chem. Eur. J. 2009, 15, 6964 – 69726972
P. W. Fowler et al.