dissolved organic matter in soil: challenging the paradigm of sorptive preservation
TRANSCRIPT

Dissolved organic matter in soil: challenging the
paradigm of sorptive preservation
Georg Guggenberger*, Klaus Kaiser
Lehrstuhl fur Bodenkunde und Bodengeographie, Universitat Bayreuth, 95440 Bayreuth, Germany
Received 7 January 2002; accepted 9 December 2002
Abstract
During the last decade, research in sedimentary systems led to the paradigm of sorptive
stabilization of organic matter (OM). Studies on soils also show that sorptive interactions between
dissolved organic matter (DOM) and mineral phases contribute to the preservation of soil OM. In the
first part of the paper, we summarize evidence for sorptive stabilization of OM in forest soils
including (a) pronounced retention of DOM in most subsoils, (b) strong chemisorptive binding
exhibiting strong hysteresis, and (c) similarity in the composition of DOM and OM in illuvial soil
horizons and clay-sized separates. However, the capacity of soils for sorption of DOM is not infinite.
In the second part of the paper, we present a case study where we relate the yearly retention of
dissolved organic carbon (DOC) in the mineral soil to the available sorption capacity of seven forest
soils. We estimate that the saturation of the sorption complex would occur within 4–30 years.
Assuming these soils are in steady-state equilibrium with respect to carbon cycling, this suggests a
mean residence time of the sorbed organic carbon (OC) of about the same time, therefore providing
little evidence for a long-term stabilization of sorbed OM.
One explanation for this discrepancy may be because in forest soils most surfaces are not
characterized by juvenile minerals but are covered with OM and colonized by microorganisms. This
is the case mainly in topsoil horizons but occurs also along preferential flow paths and on aggregate
surfaces. Biofilms develop particularly at sites receiving high input of nutrients and organic
substrates, i.e., DOM, such as preferential flow paths. The OM input enhances the heterotrophic
activity in the biofilm, converting the DOM into either organic compounds by microbial resynthesis
or inorganic mineralization products. Recent studies suggest that Fe hydrous oxides embedded
within the biofilms may serve as a sorbent and shuttle for dissolved organic compounds from the
surrounding aqueous media. We assume that sorption of DOM to the biofilm does not lead to a
stabilization of OM but is a prerequisite for its rapid turnover. Only when DOM is transported by
mass flow or diffusion to fresh, juvenile mineral surfaces, may sorption effectively stabilize OM.
0016-7061/02/$ - see front matter D 2002 Elsevier Science B.V. All rights reserved.
doi:10.1016/S0016-7061(02)00366-X
* Corresponding author. Present address: Institut fur Bodenkunde und Pflanzenernahrung, Martin-Luther-
Universitat Halle-Wittenberg, 06099 Halle, Germany. Tel.: +49-345-5522535; fax: +49-345-5527116.
E-mail address: [email protected] (G. Guggenberger).
www.elsevier.com/locate/geoderma
Geoderma 113 (2003) 293–310

This stabilization would involve complexation of functional groups, changed conformation, and
incalation in small pores.
D 2002 Elsevier Science B.V. All rights reserved.
Keywords: Organic matter; Organic carbon; Stabilization; Biofilms; Dissolved organic matter; Sorption
1. Introduction
Three types of pathways are commonly considered in the formation of stable organic
matter (OM) in soils (Christensen, 1996; Sollins et al., 1996). Selective enrichment of
organic compounds refers to the inherent recalcitrance of specific organic molecules
against degradation by microorganisms and enzymes. Chemical stabilization involves all
intermolecular interactions between organic substances and inorganic substances leading
to a decrease in availability of the organic substrate due to surface condensation and
changes in conformation, i.e., sorption to soil minerals and precipitation. Physical
stabilization is related to the decrease in the accessibility of the organic substrates to
microorganisms caused by occlusion within aggregates.
Recently, increasing evidence from studies in soils and sedimentary systems indicates
that sorptive protection of OM may be of particular importance, although chemisorption
of OM to clay-sized particles and physical protection of OM within organo-clay
aggregates often cannot be clearly distinguished. A first indication for the importance
of sorptive protection in soils is the frequently reported positive relationship between the
organic carbon (OC) content and the clay content (e.g., Burke et al., 1989; Hassink,
1997). Additional evidence comes from close relations between OC and BET surface
areas in coastal sediments (Mayer, 1994a; Keil et al., 1994) and subsoil horizons
(Mayer, 1994b), giving calculated surface loadings of 0.6–1.5 mg OC m� 2. These
loadings were considered to represent the ‘‘monolayer equivalent’’ (ME) range for OM
associated with mineral particles (Mayer, 1994a). Hedges and Keil (1995) assumed that
this finding is indicative of dissolved organic matter (DOM) sorption to mineral grains.
Keil et al. (1994) showed that simple desorption of OM from marine sediments with
water increased mineralization rates by up to five orders of magnitude. Similar results
were observed for soils by Nelson et al. (1994).
A reappraisal of the BET surface data using calculations of the reaction enthalpies
(Mayer, 1999; Mayer and Xing, 2001) and TEM studies (Ransom et al., 1997; Salmon
et al., 2000) showed that only a fraction of the mineral surface was covered with OM,
which occurred as patches and formed microaggregates with clay-sized particles. These
OM patches seem to be related to mineralogy. Ransom et al. (1998) reported that OM
appears to be preferentially sequestered in sediments, being rich in smectite but also in
metal oxyhydroxides, suggesting an influence of mineralogy on sorptive protection.
Likewise, Kaiser and Guggenberger (2000) calculated close correlations between
measures for Al and Fe hydrous oxides and OC concentrations in soils. In a
chronosequence study, Torn et al. (1997) identified the concentrations of short-range
ordered and noncrystalline minerals as the primary control for soil OC concentrations
and turnover.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310294

Together, these results suggest that stabilization of OM by interactions with distinct
mineral matrices may be the single largest factor controlling OM preservation on the
Earth’s surface today (Keil et al., 1994). Because sorption is defined as the transfer of a
solute (the sorbate) from solution to an existing solid phase (the sorbent) (Sposito,
1984), a prerequisite for the sorptive stabilization of OM is that it must occur in a
dissolved state prior to sorption. Likewise, if precipitation (the accumulation of a solute
as a new solid phase) is considered as a process of OM stabilization (Boudot et al.,
1989), the precursor substance must be dissolved. Hence, the paradigm of sorptive
stabilization includes a significant fraction (if not all of the OM) must have passed
through the dissolved phase before undergoing sorption or precipitation (Hedges and
Keil, 1995). This gives rise to the investigation of DOM sorption to minerals not only in
rivers and sedimentary systems but also in soils.
In this discussion paper, we will first summarize results from DOM research in forest
soils to show that DOM sorption to soil minerals contributes to the formation of stable
soil OM. Then we will challenge on the paradigm of sorptive stabilization. Using a
combination of field data on dissolved organic carbon (DOC) fluxes in forest soils and
laboratory data on the soils’ sorption capacity for DOC, we will provide evidence that
the turnover of sorbed OC may be quite rapid. We will present a line of evidence
showing that, depending on the type of surface, DOM sorption can be a stabilizing
process but it may be also a prerequisite for OM mineralization.
2. Processes of dissolved organic matter retention in forest soils
In the last two decades, many studies have dealt with the dynamics of DOM in forest
ecosystems. It has been reported that about 10–40 g DOC m� 2 year� 1 is translocated
from the organic surface layer into the mineral soil horizons (summarized in Michalzik et
al., 2001; Fig. 1). This means that about 10–25% of total C input to the forest floor with
litter fall is leached from the organic surface layers (McDowell and Likens, 1988;
Guggenberger, 1992). In deeper mineral soil horizons, the DOC fluxes decline to about
1–10 g m� 2 year� 1 (Michalzik et al., 2001), suggesting pronounced DOM retention in
the subsoil horizons. Most authors consider that the DOC fluxes at a soil depth of about
90–100 cm represent the DOC export by leaching.
Sorption studies using disturbed soil samples (Kaiser and Zech, 1998) and intact soil
columns (Guggenberger and Zech, 1992) suggest that the DOC retention occurs rapidly.
Kaiser and Zech (1998) showed that 60–90% of the added DOC was retained by subsoil
horizons within 15 min of addition to the soil. On the other hand, Qualls and Haines
(1992) observed that only 14–33% of the DOC collected from litter layers and mineral
soil horizons decomposed during a 134-day incubation. Kalbitz et al. (2003), using a
double exponential model, found that 94% of DOM derived from a spruce Oa horizon
was stable and had a mean half-life of 8.6 years. Rapid sorption combined with slow
microbial decomposition of the larger part of DOM suggests that sorption processes are
primarily responsible for the pronounced DOC retention in the mineral subsoil. This is
corroborated by the fractionation of DOM occurring during its passage through the soil.
The strongly colored hydrophobic DOM fraction is preferentially retained by the soil
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 295

matrix (Kaiser and Zech, 1997), whereas the less colored hydrophilic DOM fraction is
relatively enriched in the mineral soil output (Vance and David, 1991; Kaiser et al.,
2002a,b). Because mineralization primarily affects the hydrophilic DOM fraction (Qualls
and Haines, 1992; Kalbitz et al., 2003), any considerable contribution of mineralization
to the DOM retention would induce the relative enrichment of the hydrophobic fraction
in the mineral subsoil.
One possible group of sorbent for DOM is phyllosilicates. In the presence of metal ions
such as Ca2 +, Al3 +, Fe2+/3 +, the anionic organic materials may be bound to the negatively
charged clay surfaces by formation of cation bridges (Theng, 1976; Baham and Sposito,
1994). This process may be particularly important in soils with neutral to slightly alkaline
pH or very acidic pH, where the solute concentration of Ca2 + or Al3 + and Fe2+/3 + are
high. However, recently it appeared that the primary sorbents for DOM are minerals of
variable charge. Using multiple regression analysis, Moore et al. (1992) and Kaiser et al.
(1996) related DOC sorption to the quantities of Fe and Al hydrous oxides of soils. This
statistical relationship was corroborated by sorption experiments carried out on soil
material of a spodic B horizon with different quantities of amorphous Al(OH)3 and
goethite (a-FeOOH) coatings (Kaiser and Guggenberger, 2000). The increasing DOC
sorption at higher concentrations of oxalate-extractable Al (Alox) and of dithionite–
citrate–bicarbonate extractable Fe (FeDCB) may be partly explained by the strong increase
of the surface area at higher loadings of the hydrous oxides (Kaiser and Guggenberger,
2000). However, the charge of the surface is also altered by the oxide coatings. Aluminum
and Fe hydrous oxides possess a net positive charge at slightly acidic soil pH (Theng and
Orchard, 1995). Such positively charged surfaces are a prerequisite for a strong sorption of
Fig. 1. Schematic illustration of the fate of dissolved organic carbon (DOC) in forest soils.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310296

the negatively charged organic polyelectrolytes. Indeed, the strong dependence of DOM
sorption on pH, the competition of DOM with specifically binding inorganic anions (e.g.,
phosphate), and the release of OH� during the sorption suggest that surface complexation
of functional groups via ligand exchange is the most important process in the sorption of
DOM on minerals and soils (Tipping, 1981; Gu et al., 1994; Weigand and Totsche, 1998).
Using DRIFT spectroscopy, Kaiser et al. (1997) provided direct evidence for a
chemisorptive bonding of DOM to hydrous oxides. Sorption of DOM to goethite and
amorphous Al(OH)3 resulted in a sharp decrease of the band at 1715 cm� 1 due to
protonated carboxylic groups. This was accompanied by a strong increase of the
carboxylate band together with a shift from 1625 to 1600–1605 cm� 1 and increasing
absorption at 1400 cm� 1. Such changes are due to complexation of carboxyl groups with
metals on the mineral surface resulting from ligand exchange reactions (Parfitt et al., 1977;
Gu et al., 1994). The same authors reported that the formation of bidentate complexes
between two organic ligands in ortho position (i.e., one carboxylic group and one phenolic
group) of an aromatic ring and a metal at the surfaces of oxides and hydroxides causes a
strong (innersphere) chemisorptive bonding. Indeed, increased absorption occurred also at
1270 cm� 1 indicating that phenolic groups are involved in the sorption of DOM on
hydrous oxide surfaces as well. Because the major part of the carboxylic groups participate
in the complexation reactions at the mineral surfaces, Kaiser et al. (1997) concluded that
each organic macromolecule is sorbed by many bonds. Sorption of DOM with multi-
dentate bondings per molecule, called the ‘‘octopus effect’’ (Podoll et al., 1987), might
alter strongly the conformation and electron distribution of the organic molecules (Stotzky
et al., 1996). Such changes may result in an inhibition of enzymatic decomposition of the
OM, caused by the inability of the enzymes either to detect or to react with the substrate
(Khanna et al., 1998). At high OM loadings, fewer carboxylic groups are involved in the
bonding than at low OM coverage, suggesting a weaker bonding and possibly less
conformational changes of the OM molecules.
The strong chemisorptive bonding coincides with a pronounced hysteresis of the
sorption. Gu et al. (1994) reported that about 60–90% of DOC sorbed to Fe oxides was
irreversibly bound. Likewise, Kaiser and Zech (1999) reported that 24 h after sorption,
less than 3% of sorbed OC could be released from goethite and amorphous Al(OH)3under solution conditions similar to those during the sorption step. Even at desorption
with H2PO4�, an anion that forms strong bondings on Al and Fe hydrous oxide surfaces
via complexation–ligand exchange, less than 50% of the sorbed hydrophobic DOC
could be desorbed (Kaiser and Zech, 1999). This proves the high affinity of the
hydrophobic DOM to Al and Fe hydrous oxide surfaces. In contrast, hydrophilic DOM
could be almost completely removed from the surface, suggesting weaker, and possibly
nonspecific bondings.
The strong and hardly reversible sorption of DOM to Al and Fe hydrous oxide
surfaces may help to explain the strong relationship between the concentration of
mineral-associated soil OM and the concentration of these mineral phases (Kaiser and
Guggenberger, 2000). Dissolved organic matter sorption is also considered to be
responsible for the strong resemblance in the structural composition of soil OM in an
alluvial B horizon as well as in the clay fraction of a topsoil with hydrophobic DOM
from the overlying forest floors (Kaiser and Guggenberger, 2000).
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 297

The solute composition strongly accentuate the sorption processes. Inorganic cations,
e.g., H+, Ca2 +, Al3 +, Ca2 +, highly influence the net electrical charge of DOM. Protonation
or binding of other cations to the acidic functional groups of OM reduces the negative
charge of the molecules and thus the water solubility (Tipping and Hurley, 1988; Tipping
and Woof, 1990). At high concentrations, cations may give rise to precipitation of DOM.
According to Dahlgren and Marrett (1991), precipitation of DOM is controlled by the
DOC/(Fe +Al) ratio and the pH in the soil solution. Critical ratios are 10 to 20 mol C
mol� 1 Fe plus Al (Buurman, 1985; Dahlgren and Marrett, 1991). Buurman (1985) argues
that in podzols more narrow ratios can occur due to buffer reactions of H+ with the
concurrent release of polyvalent metals and the mobilization of metals from the labile
metal pool in the course of the podzolization process. However, in subsoil horizons, such
narrow ratios may also be a product of DOC sorption leaving a solution behind that is
relatively enriched in polyvalent metals. Furthermore, at the given DOC concentrations in
the mineral soil input of about 20 to 50 mg l� 1, precipitation further requires a
concentration of polyvalent metals in the range of several micromoles per liter, which is
unlikely for most soils (Guggenberger, 1992).
In summary, there is ample evidence that sorptive preservation of OM is an important
process in the OM accumulation and stabilization in soil. One has to bear in mind,
however, that sorption experiments usually were carried out on juvenile minerals or
subsoil samples with little OM and showing little, if any, microbial activity. Furthermore,
the timescale of sorption experiments is usually in the range of several minutes to days.
This is too short to assess mineralization processes of the sorbed OM. Thus, the role of the
decomposer community in soil is neglected in these studies. For this reason, it is difficult
to scale up the laboratory results about sorptive stabilization of OM on free mineral
surfaces to the situation in the field.
3. Capacity of soils to sorb organic matter
In the previous section it was shown that relatively large quantities of DOM enter the
mineral horizons. Biodegradation of organic matter in the dissolved phase is too slow to
remove a large portion of the DOM percolating through the soil (Qualls and Haines, 1992;
Kalbitz et al., 2003), whereas sorption to mineral phases is an efficient sink for DOM in
subsoil horizons (Kaiser and Zech, 1998; Kaiser and Guggenberger, 2000). However,
sorption of DOM to mineral phases and mineral soil is not infinite but approaches sorption
maxima at large DOM additions (Kaiser and Zech, 1997). This enables an estimation of
the sorption capacity of soils for DOM.
To do this, we carried out sorption experiments as outlined by Kaiser et al. (1996) for
all mineral soil horizons of seven forest soils, including cambisols, podzols, an arenosol,
and a leptosol. Briefly, 75 ml of an artificial soil solution containing 0 to 101 mg OC l� 1
was added to 3 g of soil material. The solutions were zero-tension extracted from the
organic layers under spruce, pine, or beech, depending on the type of forests growing on
the soils. For the organic layer under spruce, the major cations in the solutions were NH4+
(0.26 mmol l� 1) and K+ (0.18 mmol l� 1); the major inorganic anions were SO42� (0.21
mmol l� 1) and H2PO4� (0.06 mmol l� 1). The ionic strength of the solutions obtained from
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310298
the organic layers under pine was 0.002 M with the major inorganic constituents NH4+, K+,
and SO42�. In the solutions extracted from beech litter, the ionic strength was 0.008 M; the
major inorganic solutes were Ca2 + and SO42�. Hence, in all cases it was assured that no
precipitation reactions took place during the sorption experiment. Suspensions were
shaken gently for 24 h, then passed through 0.45-Am membrane filters, and the filtrates
were analyzed for DOC. The sorbed amount of OC was calculated by the difference
between the DOC concentration in the initial solution and in the filtrate. Maximum
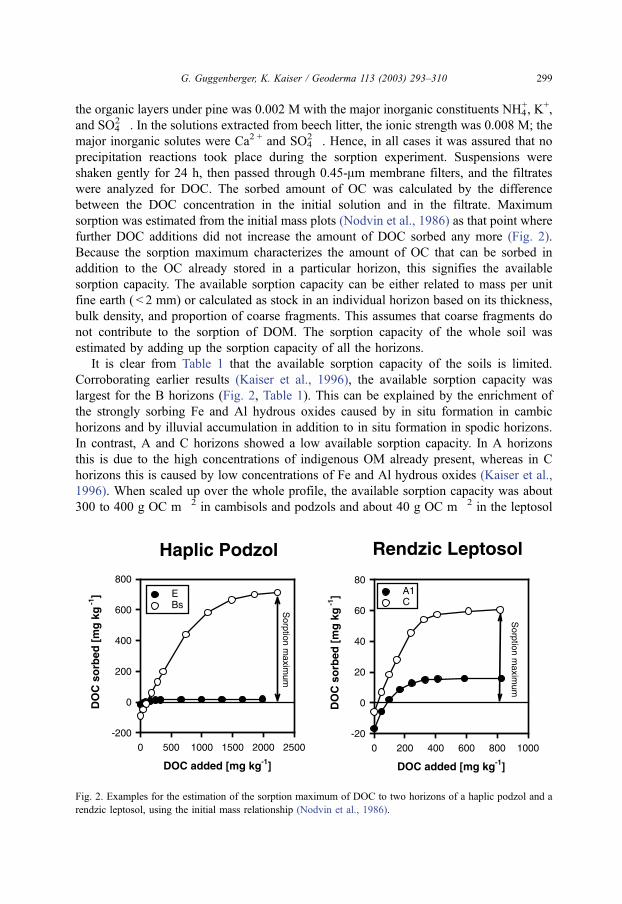
sorption was estimated from the initial mass plots (Nodvin et al., 1986) as that point where
further DOC additions did not increase the amount of DOC sorbed any more (Fig. 2).
Because the sorption maximum characterizes the amount of OC that can be sorbed in
addition to the OC already stored in a particular horizon, this signifies the available
sorption capacity. The available sorption capacity can be either related to mass per unit
fine earth ( < 2 mm) or calculated as stock in an individual horizon based on its thickness,
bulk density, and proportion of coarse fragments. This assumes that coarse fragments do
not contribute to the sorption of DOM. The sorption capacity of the whole soil was
estimated by adding up the sorption capacity of all the horizons.
It is clear from Table 1 that the available sorption capacity of the soils is limited.
Corroborating earlier results (Kaiser et al., 1996), the available sorption capacity was
largest for the B horizons (Fig. 2, Table 1). This can be explained by the enrichment of
the strongly sorbing Fe and Al hydrous oxides caused by in situ formation in cambic
horizons and by illuvial accumulation in addition to in situ formation in spodic horizons.
In contrast, A and C horizons showed a low available sorption capacity. In A horizons
this is due to the high concentrations of indigenous OM already present, whereas in C
horizons this is caused by low concentrations of Fe and Al hydrous oxides (Kaiser et al.,
1996). When scaled up over the whole profile, the available sorption capacity was about
300 to 400 g OC m� 2 in cambisols and podzols and about 40 g OC m� 2 in the leptosol
Fig. 2. Examples for the estimation of the sorption maximum of DOC to two horizons of a haplic podzol and a
rendzic leptosol, using the initial mass relationship (Nodvin et al., 1986).
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 299

and the arenosol. The latter soils are weakly developed with little accumulation of Fe
and Al hydrous oxides (Kaiser et al., 2002a,b).
When we now relate the available sorption capacity of the different horizons to the
amount of total C already stored in the horizons, it is obvious that in the topsoil horizons
Table 1
Available sorption capacity of seven soils for OC related to the storage of total OC and OC found in density
separates >1.6 g cm� 3 (i.e., the mineral-associated OC fraction)
Site/soil (FAO) Horizon Depth Available OC storage Available sorption capacity
(cm) sorption
capacity
(g m� 2)
Total OC
(g m� 2)
OC >1.6 g
cm� 3
(g m� 2)
% of
total OC
% of
OC >1.6
g cm� 3
Oberwarmensteinach/
cambic podzol
(Guggenberger and
Zech, 1993)
E
Bhs
Bw
C
0–10
10–35
35–75
75–90
6
110
211
42
2208
3840
1092
101
1632
3360
1092
101
0.3
2.9
19.3
41.0
0.4
3.3
19.3
41.0
Total 0–90 369 7241 6185 5.1 6.0
Wulfersreuth/
dystric cambisol
(Guggenberger and
Zech, 1993)
AE
Bsw
Bw1
Bw2
2BC
0–7
7–22
22–35
35–70
70–90
2
58
73
182
64
1940
2178
546
588
120
1490
1980
437
588
120
0.1
2.7
13.4
31.0
53.0
0.1
3.0
16.8
31.0
53.0
Total 0–90 380 5372 4615 7.1 8.2
Hohe Matzen/haplic
podzol (Guggenberger
and Zech, 1993)
Ah
E
Bh
Bs
Bws
C
0–10
10–20
20–26
26–36
36–68
68–90
1
4
9
36
145
139
1260
900
1320
2550
4066
924
300
360
1080
2150
3872
924
0.1
0.5
0.7
1.4
3.6
15.0
0.4
1.2
0.9
1.7
3.8
15.0
Total 0–90 334 11,020 8386 3.0 4.0
Betzenstein/rendzic
leptosol
(Kaiser et al., 2000)
Ah1
Ah2
C
0–10
10–25
25–90
2
8
35
9240
7452
4290
8003
7128
3900
0.0
0.1
0.8
0.0
0.1
0.9
Total 0–90 45 20,982 19,030 0.2 0.2
Sey bothenreuth/typic
arenosol
(Kaiser et al., 2000)
Ah
C1
C2
0–5
5–15
15–90
1
3
36
1050
160
1200
490
160
1200
0.1
2.0
3.0
0.2
2.0
3.0
Total 0–90 40 2410 1850 1.7 2.2
Waldstein/cambic
podzol (Kalbitz and
Matzner, unpublished;
Kaiser et al., 2002a)
AE
Bh
Bs
Bw
BwC
C
0–10
10–12
12–30
30–55
55–70
70–85
1
2
45
167
42
41
2660
911
4586
1960
144
144
878
765
4173
1823
137
137
0.0
0.2
1.0
8.5
29.0
28.5
0.1
0.3
1.0
9.2
30.7
29.9
Total 0–85 298 10,406 7913 2.9 3.7
Steigerwald/dystric
cambisol (Solinger
et al., 2001;
Kaiser et al.,
2002a)
Ah
Bw1
Bw2
BC
Total
0–5
5–24
24–50
50–80
0–80
2
150
127
133
411
3630
3663
832
510
7635
1271
2198
707
485
4660
0.0
5.6
15.3
26.0
5.4
0.2
6.8
18.0
27.4
8.8
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310300
the percentage of the total sorption capacity that is available is < 1% (Table 1). This
suggests that the capacity of these horizons to store chemically stabilized OC (Sollins et
al., 1996), i.e., the OC of the >1.6 g cm� 3 fraction, is almost exhausted and agrees well
with field data, showing that A horizons are a source rather than a sink for DOC (Kaiser et
al., 2002a,b). With increasing soil depth, the percentage of the available sorption capacity
increases by up to 50% in subsoil horizons of well-developed soils. From this it may be
concluded that the subsoil horizons have a high potential for sorptive retention of DOM.
The weakly developed rendzic leptosol and typic arenosol are the only exceptions. In these
soils, the contents of Fe and Al hydrous oxides are low throughout the whole profile
(Kaiser et al., 2002a,b) and this limited amount of sorbents appears to be almost
completely occupied by OC. Consequently, the DOC concentration in the mineral soil
output is quite high with 19 g l� 1 (rendzic leptosol) and 25 g l� 1 (typic arenosol) (Kaiser
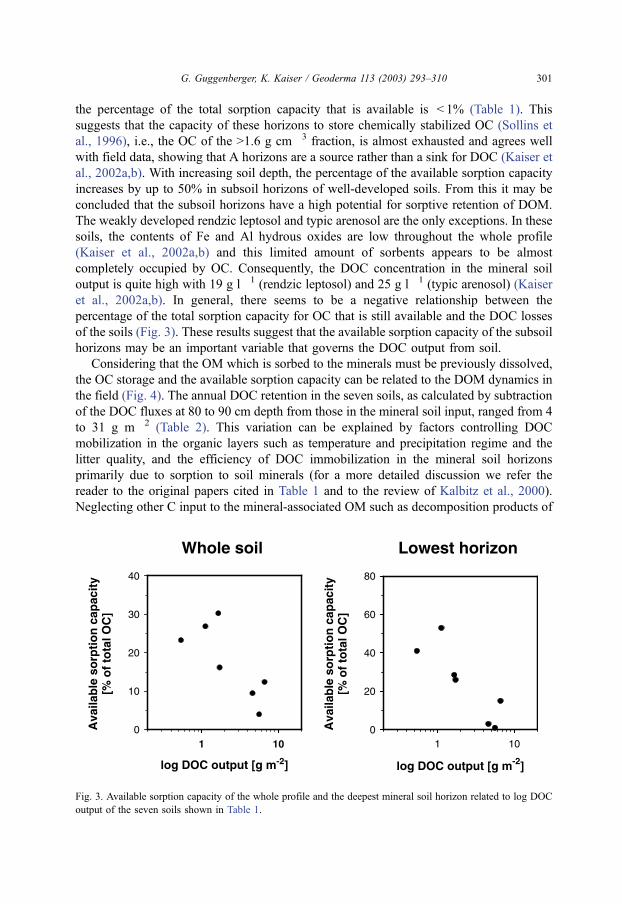
et al., 2002a,b). In general, there seems to be a negative relationship between the
percentage of the total sorption capacity for OC that is still available and the DOC losses
of the soils (Fig. 3). These results suggest that the available sorption capacity of the subsoil
horizons may be an important variable that governs the DOC output from soil.
Considering that the OM which is sorbed to the minerals must be previously dissolved,
the OC storage and the available sorption capacity can be related to the DOM dynamics in
the field (Fig. 4). The annual DOC retention in the seven soils, as calculated by subtraction
of the DOC fluxes at 80 to 90 cm depth from those in the mineral soil input, ranged from 4
to 31 g m� 2 (Table 2). This variation can be explained by factors controlling DOC
mobilization in the organic layers such as temperature and precipitation regime and the
litter quality, and the efficiency of DOC immobilization in the mineral soil horizons
primarily due to sorption to soil minerals (for a more detailed discussion we refer the
reader to the original papers cited in Table 1 and to the review of Kalbitz et al., 2000).
Neglecting other C input to the mineral-associated OM such as decomposition products of
Fig. 3. Available sorption capacity of the whole profile and the deepest mineral soil horizon related to log DOC
output of the seven soils shown in Table 1.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 301

roots or microbial metabolites, a simple relation of the annual DOC retention (g OC m� 2)
in the mineral soil horizons to the mineral-associated OC (i.e., OC in the fraction >1.6 g
cm� 2) stored in the soil (g OC m� 2) provides an estimate for the residence time of the
indigenous mineral-associated OC in the range of 186 to 1730 years (Table 2). From this,
it can be concluded that the soils store and stabilize a large amount of the incoming OC
very efficiently.
A different picture emerges when the DOC retention is related to the available sorption
capacity. The estimated number of years until exhaustion of the sorption capacity for OC
of the seven soils ranged from 4 to 30. When we now consider that these forest soils are
close to a steady-state equilibrium with respect to carbon sorption and mineralization, the
amount of new OC sorbed within this period of time is in the order of the amount of
sorbed OC being mineralized. This means a surprisingly low mean residence time of the
sorbed OC of < 50 years, especially in the biologically active rendzic leptosol. Even if we
consider that not all of the DOC retained is sorbed but that it is partly mineralized from the
solution, this will add only a few years to the estimated mean turnover time of the sorbed
OC. Of course, a complete saturation of the soils with OC is not possible. Depending on
the distribution coefficient, at a given solution concentration of OC there is a unique
concentration of sorbed OC. Thus, the available sorption capacity will never be filled up
completely. Consequently, the number of years for the estimated mean turnover time of the
sorbed OC must be reduced. Although considering a quite large error in the estimates of
the mean turnover time of the currently sorbed OC, it is obvious that it is in the range of
decades or less.
Summarizing the results, strong discrepancies on the sorptive stabilization of OM
in forest soils are observed. Sorption experiments on minerals and subsoil horizons
Fig. 4. Annual DOC sorption in the mineral soil horizons of a haplic podzol related to the available sorption
capacity.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310302

suggest strong chemisorptive retention of OM. This is corroborated by the high
residence time of indigenous OC already sorbed to the minerals as calculated from
the relation of the yearly DOC input and the stocks of mineral-associated OC. On
the other hand, the available sorption capacity of forest soils is limited, in particular
in the topsoil and upper subsoil horizons. Our OC budget estimates suggest a
relatively short mean residence time of the OC currently entering the mineral soil as
DOM.
As mentioned above, sorption experiments that have been carried out may not be well
suited to assess mineralization processes. This is further complicated by the spatial
heterogeneity of soil with respect to microbial biomass and activity. In all soils, there
are horizons or microsites having high microbial biomass and activities that may affect the
sorptive retention of DOM differently than locations with low microbial activity and large
amounts of juvenile mineral surfaces.
Table 2
Available sorption capacity of seven soils for OC as measured in the laboratory related to the published annual
DOC retention in the mineral soil horizons as determined in the field
Site/soil (FAO)/
reference for annual
DOC retention
Horizon Depth
(cm)
Available
sorption
capacity
Annual
DOC
retentiona
Estimated
residence
timeb
Time until
exhaustion
of sorption
g kg� 1 g m� 2 (g m� 2) (years) capacityc (years)
Oberwarmensteinach/ Total 0–90 n.d. 369 16 386 23
cambic podzol
(Guggenberger
and Zech (1993)
(E to C)
Wulfersreuth/dystric Total 0–90 n.d. 380 14 330 27
cambisol (Guggenberger
and Zech, 1993)
(AE to 2BC)
Hohe Matzen/haplic podzol Total 0–90 n.d. 334 31 270 11
Guggenberger
and Zech, 1993)
(Ah to C)
Betzenstein/rendzic leptosol Total 0–90 n.d. 45 11 1730 4
(Kaiser et al., 2000) (Ah1 to C)
Sey bothenreuth/typic arenosol Total 0–90 n.d. 40 4 463 10
(Kaiser et al., 2000) (Ah to C2)
Waldstein/cambic podzol Total 0–85 n.d. 298 10 791 30
(Michalzik and
Matzner, 1999)
(AE to C)
Steigerwald/dystric cambisol Total 0–80 n.d. 411 25 186 16
(Solinger et al., 2001) (Ah to BC)
n.d. = not determined.a Annual DOC retention by the soil is based on the assumption that DOC fluxes in the soil are primarily
controlled by sorption. It is calculated by subtracting the DOC output from the soil measured at a 80 to 90 cm
depths from the DOC fluxes entering the mineral soil. Data are taken from the references shown in the first
column.b Estimated residence time is calculated as OC (>1.6 g cm� 3) storage divided by annual DOC retention.c Time until exhaustion of sorption capacity is calculated as available sorption capacity divided by annual
DOC retention.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 303

4. Sorption of organic matter to surfaces differing in the microbial activity
In podzols, sorption of DOM has been considered an important process in the formation
of OM in subsoil horizons. The Bh horizon is generally thought to result from DOM
sorption. Often, Bh horizons have large OC concentrations, which is a prerequisite for the
high density of microorganisms found in these horizons (McKeague et al., 1986; Fig. 5).
Concurrently, the 14C age of OM shows a minimum in the Bh horizon and is only about
100 years (Rumpel et al., 2002; Fig. 5). This is about the same order of magnitude as the
mean residence time of OM estimated in Table 2, and suggests a rapid mineralization of
the OM in the Bh horizon. The substrate utilized by the microbial community can be
efficiently replenished by the large DOM input. At large OM loadings, which are typical
for Bh horizons, not all functional groups are involved in the chemisorptive bonding, and
steric changes of the substrate are probably not as pronounced than at smaller OM loadings
(Kaiser et al., 1997). Hence, the stabilizing effect of sorption on microbial degradability
may not be as efficient than at lower surface loadings. In the deeper soil horizons, the
percentage of available sorption sites is larger (Table 1). Accordingly, almost all functional
groups are involved in the bonding (Kaiser et al., 1997). Recent results further suggest that
at small loadings most OM is associated with micropores and/or other high-surface sites
(Kaiser and Guggenberger, 2003). This strong sorption in the horizons beneath the Bh
horizon leads to a more pronounced stabilization of OM, which is illustrated by the high14C age of the OM (Fig. 5).
Such compartmentalization is not only important between different horizons but also
with respect to the pathway of water, ions, and DOM fluxes in soil. Preferential flow paths
have been identified as biological hot spots (Bundt et al., 2001a,b). Compared to the soil
Fig. 5. Microbial density (left; from McKeague et al., 1986) and 14C age (right; from Rumpel et al., 2002) in a
podzol. The measured 14C activity was corrected for isotope fractionation and given in percent modern carbon
(pMC) according to the international standard. The radiocarbon age in years BP was calculated according to
Stuiver and Polach (1977). Please note that both analyses were carried out at two different soil profiles.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310304
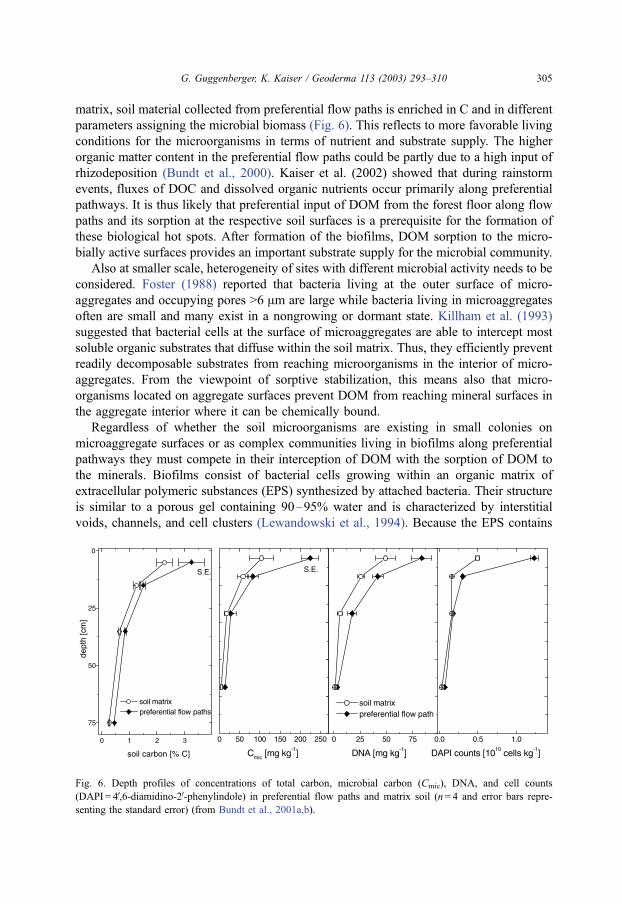
matrix, soil material collected from preferential flow paths is enriched in C and in different
parameters assigning the microbial biomass (Fig. 6). This reflects to more favorable living
conditions for the microorganisms in terms of nutrient and substrate supply. The higher
organic matter content in the preferential flow paths could be partly due to a high input of
rhizodeposition (Bundt et al., 2000). Kaiser et al. (2002) showed that during rainstorm
events, fluxes of DOC and dissolved organic nutrients occur primarily along preferential
pathways. It is thus likely that preferential input of DOM from the forest floor along flow
paths and its sorption at the respective soil surfaces is a prerequisite for the formation of
these biological hot spots. After formation of the biofilms, DOM sorption to the micro-
bially active surfaces provides an important substrate supply for the microbial community.
Also at smaller scale, heterogeneity of sites with different microbial activity needs to be
considered. Foster (1988) reported that bacteria living at the outer surface of micro-
aggregates and occupying pores >6 Am are large while bacteria living in microaggregates
often are small and many exist in a nongrowing or dormant state. Killham et al. (1993)
suggested that bacterial cells at the surface of microaggregates are able to intercept most
soluble organic substrates that diffuse within the soil matrix. Thus, they efficiently prevent
readily decomposable substrates from reaching microorganisms in the interior of micro-
aggregates. From the viewpoint of sorptive stabilization, this means also that micro-
organisms located on aggregate surfaces prevent DOM from reaching mineral surfaces in
the aggregate interior where it can be chemically bound.
Regardless of whether the soil microorganisms are existing in small colonies on
microaggregate surfaces or as complex communities living in biofilms along preferential
pathways they must compete in their interception of DOM with the sorption of DOM to
the minerals. Biofilms consist of bacterial cells growing within an organic matrix of
extracellular polymeric substances (EPS) synthesized by attached bacteria. Their structure
is similar to a porous gel containing 90–95% water and is characterized by interstitial
voids, channels, and cell clusters (Lewandowski et al., 1994). Because the EPS contains
Fig. 6. Depth profiles of concentrations of total carbon, microbial carbon (Cmic), DNA, and cell counts
(DAPI = 4V,6-diamidino-2V-phenylindole) in preferential flow paths and matrix soil (n= 4 and error bars repre-
senting the standard error) (from Bundt et al., 2001a,b).
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 305

large concentrations of sugar acids, biofilms usually possess a negative charge (Morgan et
al., 1990). The net negative surface charge of biofilms is likely to decrease sorption of
negatively charged OM to the biofilm and its diffusion within the biofilm (Carlson and
Silverstein, 1998). Recently, Lunsdorf et al. (2000) identified microgranular clusters of
Fe–O material and phyllosilicates directly associated with the bacterial cell envelope in
electron spectroscopic images. Within the biofilms, the authors also found granules of
storage OM and concluded that the minerals within the biofilm may remove OM from the
surrounding aqueous media and transport the organic substrate to the decomposers.
Sorption of DOM to positively charged Fe hydrous oxides associated with biofilms
may thus be an efficient pathway for OM uptake into biofilms, where the organic substrate
can be utilized by the microbial consortium. Interestingly, the active mineral compound
involved in the sorption into biofilms appears to be the same as for whole soils, i.e., Fe
hydrous oxides of low crystallinity.
5. Conclusion
Based on these findings, we propose a model shown in Fig. 7 where the location of the
OM is decisive for its fate. In forest soils, organic matter in the soil solution is mineralized
at rates that are much slower than the mean residence time of DOM in the mineral soil
(e.g., Qualls and Haines, 1992). This suggests that mineralization cannot be responsible
for the DOM retention in the mineral soil. Kalbitz et al. (2003) identified that the stable
DOM component, which dominates OM in the solution percolating into the mineral soil, is
associated with high UV absorbance and high aromaticity. According to Guggenberger et
al. (1994), such structures can be best described as lignocellulose-degradation products.
Fig. 7. Conceptual model of the fate of OM in the soil solution and sorbed to soil surfaces differing in their
biological activity.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310306

For a complete degradation of such compounds, a consortium of microorganisms is
necessary (Shevchenko and Bailey, 1996). However, the density of bacteria in the soil
solution is low and individuals are physically separated. Hence, there is little possibility
for degradation of complex organic molecules in the soil solution.
However, these DOM components have a large affinity for sorption to positively
charged mineral surfaces, i.e., Fe and Al hydrous oxides (Kaiser et al., 1997). The sorption
is predominantly a chemisorptive process that goes along with changes in conformation. In
case of juvenile surfaces, part of the sorbed OM seems to be located in small pores that are
inaccessible for microorganisms (Kaiser and Guggenberger, 2003). Hence, OM sorbed to
juvenile surfaces is efficiently stabilized against microbial mineralization and sorptive
preservation is certainly taking place. According to Table 1, such conditions rarely occur
in topsoil horizons but primarily in subsoil horizons.
With time, more OM is sorbed to the mineral surfaces. The sorption capacity is almost
saturated and the bondings are not as strong as at smaller OM loadings (Kaiser et al., 1997).
This attracts heterotrophic microorganisms to colonize the OM-loaded mineral surfaces.
Cell colonies and biofilms develop at such sites of high nutrient and organic substrate
availability. These are topsoil horizons in general, but may also include preferential flow
paths and surfaces adjacent to interaggregate pores. Biofilms compete with inorganic
surfaces for OM sorption and are probably an effective sink for DOM in the mineral soils.
Iron hydrous oxides within the biofilms may serve as sorbents and OM shuttles to provide
substrate to the microbial community (Lunsdorf et al., 2000). Efficient sorption of OM into
the biofilms leads to a concentration of OM at this microsite. Likewise, the diverse
communities living in biofilms enable a more rapid mineralization of complex organic
molecules than of the same organic compounds in the soil solution. Sorption of OM into
biofilms may thus be a prerequisite for a quite rapid OM mineralization, and we conclude
that this is largely responsible for the short mean residence time estimated for the DOM
retained in the seven mineral soils shown in Table 2.
Therefore, to predict the fate of DOM in soil, the organic compounds not only need to
be classified into pools of different physicochemical and biochemical properties. It seems
even more important to address the probability of whether DOM is sorbed to juvenile
mineral surfaces or into biofilms. This requires detailed research on the flow paths of
DOM, the location and activity of biofilms, the sorption of DOM into and mineralization
by biofilms, and the location of active Fe and Al hydrous oxides in the natural soil
environment.
References
Baham, J., Sposito, G., 1994. Adsorption of dissolved organic carbon extracted from sewage sludge on mont-
morillonite and kaolinite in the presence of metal ions. J. Environ. Qual. 23, 147–153.
Boudot, J.P., Bel Dadj Brahim, A., Steiman, R., Seigle-Murandi, F., 1989. Biodegradation of synthetic organo-
metallic complexes of iron and aluminium with selected metal to carbon ratios. Soil Biol. Biochem. 21,
961–966.
Bundt, M., Albrecht, A., Froidevaux, P., Blaser, P., Fluhler, H., 2000. Impact of preferential flow on radionuclide
distribution in soil. Environ. Sci. Technol. 34, 3895–3899.
Bundt, M., Widmer, F., Pesaro, M., Zeyer, J., Blaser, P., 2001a. Preferential flow paths: biological ‘hot spots’ in
soils. Soil Biol. Biochem. 33, 729–738.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 307

Bundt, M., Zimmermann, S., Blaser, P., Hagedorn, F., 2001b. Sorption and transport of metals in preferential flow
paths and soil matrix after the addition of wood ash. Eur. J. Soil Sci. 52, 423–432.
Burke, I.C., Yonker, C.M., Parton, W.J., Cole, C.V., Flach, K., Schimel, D.S., 1989. Texture, climatic and
cultivation effects on soil organic matter content in U.S. grassland soils. Soil Sci. Soc. Am. J. 53, 800–805.
Buurman, P., 1985. Carbon/sesquioxide ratios in organic complexes and the transition of albic-spodic horizon.
J. Soil Sci. 36, 225–260.
Carlson, G., Silverstein, J., 1998. Effect of molecular size and charge on biofilm sorption of organic matter. Water
Res. 32, 1580–1592.
Christensen, B.T., 1996. Carbon in primary and secondary organomineral complexes. In: Carter, M.R., Stewart,
B.A. (Eds.), Structure and Organic Matter Storage in Agricultural Soils. Advances in Soil Science. CRC
Press, Boca Raton, FL, pp. 97–165.
Dahlgren, R.A., Marrett, D.J., 1991. Organic carbon sorption in arctic and subalpine Spodosol B horizons. Soil
Sci. Soc. Am. J. 55, 1382–1390.
Foster, R.C., 1988. Microenvironments of soil microorganisms. Biol. Fertil. Soils 6, 189–203.
Gu, B., Schmitt, J., Chen, Z., Liang, L., McCarthy, J.F., 1994. Adsorption and desorption of natural organic
matter on iron oxide: mechanisms and models. Environ. Sci. Technol. 28, 38–46.
Guggenberger, G., 1992. Eigenschaften und Dynamik geloster organischer Substanzen (DOM) unterschiedlich
immissionsbelasteten Fichtenstandorten. Bayreuth. Bodenkdl. Ber. 26, 1–164.
Guggenberger, G., Zech, W., 1992. Retention of dissolved organic carbon and sulfate in aggregated forest soils.
J. Environ. Qual. 21, 643–653.
Guggenberger, G., Zech, W., 1993. Dissolved organic carbon control in acid forest soils of the Fichtelgebirge
(Germany) as revealed by distribution patterns and structural composition analyses. Geoderma 59, 109–129.
Guggenberger, G., Zech, W., Schulten, H.-R., 1994. Formation and mobilization pathways of dissolved organic
carbon: evidence from chemical structural studies of organic carbon fractions in acid forest floor solutions.
Org. Geochem. 21, 51–66.
Hassink, J., 1997. The capacity of soils to preserve organic C and N by their association with clay and silt
particles. Plant Soil 191, 77–87.
Hedges, J.I., Keil, R.G., 1995. Sedimentary organic matter preservation: an assessment and speculative synthesis.
Mar. Chem. 49, 81–115.
Kaiser, K., Guggenberger, G., 2000. The role of DOM sorption to mineral surfaces in the preservation of organic
matter in soils. Org. Geochem. 31, 711–725.
Kaiser, K., Guggenberger, G., 2003. Mineral surfaces and soil organic matter. Eur. J. Soil Sci. (in press).
Kaiser, K., Zech, W., 1997. Competitive sorption of dissolved organic matter fractions to soils and related mineral
phases. Soil Sci. Soc. Am. J. 61, 64–69.
Kaiser, K., Zech, W., 1998. Rates of dissolved organic matter release and sorption in forest soils. Soil Sci. Soc.
Am. J. 61, 64–69.
Kaiser, K., Zech, W., 1999. Release of natural organic matter sorbed to oxides and a subsoil. Soil Sci. Soc. Am. J.
63, 1157–1166.
Kaiser, K., Guggenberger, G., Zech, W., 1996. Sorption of DOM and DOM fractions to forest soils. Geoderma
74, 281–303.
Kaiser, K., Guggenberger, G., Haumaier, L., Zech, W., 1997. Dissolved organic matter sorption on subsoils and
minerals studied by 13C-NMR and DRIFT spectroscopy. Eur. J. Soil Sci. 48, 301–310.
Kaiser, K., Guggenberger, G., Zech, W., 2000. Organically-bound nutrients in dissolved organic matter fractions
in seepage and pore water of weakly developed forest soils. Acta Hydrochim. Hydrobiol. 28, 411–419.
Kaiser, K., Eusterhues, K., Guggenberger, G., Kogel-Knabner, I., 2002a. Stabilisation of organic matter in the
subsoil. J. Plant Nutr. Soil Sci. 165, 451–459.
Kaiser, K., Guggenberger, G., Kaupenjohann, M., Zech, W., 2002b. Refractory organic substances in aggregated
forest soils—retention versus translocation. In: Frimmel, F.H., et al. (Ed.), Refractory Organic Substances in
Aqueous Systems. Wiley-VCH, Weinheim, Germany, pp. 413–436.
Kalbitz, K., Solinger, S., Park, J.-H., Michalzik, B., Matzner, E., 2000. Controls on the dynamics of dissolved
organic matter in soils: a review. Soil Sci. 165, 277–304.
Kalbitz, K., Schmerwitz, J., Schwesig, D., Matzner, E., 2003. Biodegradation of soil-derived dissolved organic
matter as related to its properties. Geoderma 113, 273–291 (this issue).
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310308

Keil, R.G., Montluc�on, D.B., Prahl, F.G., Hedges, J.I., 1994. Sorptive preservation of labile organic matter in
marine sediments. Nature 370, 549–552.
Khanna, M., Yoder, M., Calamai, L., Stotzky, G., 1998. X-ray diffractometry and electron microscopy of DNA
from Bacillus subtilis bound on clay minerals. Sci. Soils 3, 1.
Killham, K., Amato, M., Ladd, J.N., 1993. Effect of substrate location in soil and soil pre-water regime on carbon
turnover. Soil Biol. Biochem. 25, 57–62.
Lewandowski, Z., Stoodley, P., Altobelli, S., Fukushima, E., 1994. Hydrodynamics and kinetics in biofilm
systems: recent advances and new problems. Water Sci. Technol. 29, 223–229.
Lunsdorf, H., Erb, R.W., Abraham, W.-R., Timmis, K.N., 2000. ‘Clay hutches’: a novel interaction between
bacteria and clay minerals. Environ. Microbiol. 2, 161–168.
Mayer, L.M., 1994a. Surface area control of organic carbon accumulation in continental shelf sediments. Geo-
chim. Cosmochim. Acta 58, 1271–1284.
Mayer, L.M., 1994b. Relationships between mineral surfaces and organic carbon concentrations in soils and
sediments. Chem. Geol. 114, 347–363.
Mayer, L.M., 1999. Extent of coverage of mineral surfaces by organic matter in marine sediments. Geochim.
Cosmochim. Acta 63, 207–215.
Mayer, L.M., Xing, B., 2001. Organic matter– surface relationships in acid soils. Soil Sci. Soc. Am. J. 65,
250–258.
McDowell, W.H., Likens, G.E., 1988. Origin, composition, and flux of dissolved organic carbon in the Hubbard
Brook valley. Ecol. Monogr. 58, 177–195.
McKeague, J.A., Cheshire, M.V., Andreux, F., Berthelin, J., 1986. Organo-mineral complexes in relation to
pedogenesis. In: Huang, P.M., Schnitzer, M. (Eds.), Interactions of Soil Minerals with Natural Organics
and Microbes. SSSA Spec. Publ., vol. 17. SSSA, Madison, WI, pp. 1–28.
Michalzik, B., Matzner, E., 1999. Dynamics of dissolved organic nitrogen and carbon in a Central European
Norway spruce ecosystem. Eur. J. Soil Sci. 50, 579–590.
Michalzik, B., Kalbitz, K., Park, J.H., Solinger, S., Matzner, E., 2001. Fluxes and concentrations of dissolved
organic carbon and nitrogen—a synthesis for temperate forests. Biogeochemistry 52, 173–205.
Moore, T.R., De Souza, W., Koprivnijak, J.F., 1992. Controls on the sorption of dissolved organic carbon on
soils. Soil Sci. 154, 120–129.
Morgan, J.W., Forster, C.F., Evinson, L., 1990. A comparative study of the nature of biopolymers extracted from
anaerobic and activated sludges. Water Res. 24, 743–750.
Nelson, P.N., Dictor, M.-C., Soulsa, G., 1994. Availability of organic carbon in soluble and particle-size fractions
from a soil profile. Soil Biol. Biochem. 26, 1549–1555.
Nodvin, S.C., Driscoll, C.T., Likens, G.E., 1986. Simple partitioning of anions and dissolved organic carbon in a
forest soil. Soil Sci. 142, 27–35.
Parfitt, R.L., Fraser, A.R., Farmer, V.C., 1977. Adsorption on hydrous oxides. III. Fulvic acid and humic acid on
geothite, gibbsite and imogolite. J. Soil Sci. 28, 289–296.
Podoll, R.T., Irwin, K.C., Brendinger, S., 1987. Sorption of water-soluble oligomers on sediments. Environ. Sci.
Technol. 21, 562–568.
Qualls, R.G., Haines, B.L., 1992. Biodegradability of dissolved organic matter in forest throughfall, soil solution
and stream waters. Soil Sci. Soc. Am. J. 56, 578–586.
Ransom, B., Bennett, R.H., Baerwald, R., Shea, K., 1997. TEM study of in situ organic matter continental
margins: occurrence and the ‘‘monolayer’’ hypothesis. Mar. Geol. 138, 1–9.
Ransom, B., Kim, D., Kastner, M., Wainwright, S., 1998. Organic matter preservation on continental slopes:
importance of mineralogy and surface area. Geochim. Cosmochim. Acta 62, 1329–1345.
Rumpel, C., Kogel-Knabner, I., Bruhn, F., 2002. Origin and chemical composition of organic carbon in two forest
soils of different pedogenesis. Org. Geochem. (submitted for publication).
Salmon, V., Derenne, S., Lallier-Verges, E., Largeau, C., Beaudoin, B., 2000. Protection of organic matter by
mineral matrix in a Cenomanian black shale. Org. Geochem. 31, 463–474.
Shevchenko, S.M., Bailey, G.W., 1996. Life after death: lignin–humic relationships reexamined. Crit. Rev.
Environ. Sci. Technol. 26, 95–153.
Solinger, S., Kalbitz, K., Matzner, E., 2001. Controls on the dynamics of dissolved organic carbon and nitrogen
in a Central European deciduous forest. Biogeochemistry 55, 327–349.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310 309

Sollins, P., Homann, P., Caldwell, B.A., 1996. Stabilization and destabilization of soil organic matter: mecha-
nisms and controls. Geoderma 74, 65–105.
Sposito, G., 1984. The Surface Chemistry of Soils. Oxford Univ. Press, New York, NY.
Stotzky, G., Gallori, E., Khanna, M., 1996. Transformation in soil. In: Akkermans, A.D.L., van Elsas, J.D., de
Bruijn, F.J. (Eds.), Molecular Microbial Ecology Manual. Kluwer, Dordrecht, The Netherlands, pp. 1–28.
Stuiver, M., Polach, H.A., 1977. Discussion: reporting of 14C data. Radiocarbon 19, 355–363.
Theng, B.K.G., 1976. Interactions between montmorillonite and fulvic acid. Geoderma 15, 243–251.
Theng, B.K.G., Orchard, V.A., 1995. Interactions of clays with microorganisms and bacterial survival in soil: a
physicochemical perspective. In: Huang, P.M., Berthelin, J., Bollag, J.-M., McGill, W.B., Page, A.L. (Eds.),
Environmental Impact of Soil Component Interactions, Vol II. Metals, Other Inorganics, and Microbial
Activities. CRC Lewis Publishers, Boca Raton, FL, pp. 123–143.
Tipping, E., 1981. The adsorption of aquatic humic substances by iron oxides. Geochim. Cosmochim. Acta 45,
191–199.
Tipping, E., Hurley, M.A., 1988. A model of solid– solution interactions in acid organic soils, based on the
complexation properties of humic substances. J. Soil Sci. 39, 505–519.
Tipping, E., Woof, C., 1990. Humic substances in acid organic soils: modelling their release to the soil solution in
terms of humic charge. J. Soil Sci. 41, 573–586.
Torn, M.S., Trumbore, S.E., Chadwick, O.A., Vitousek, P.M., Hendricks, D.M., 1997. Mineral control of soil
organic carbon storage and turnover. Nature 389, 170–173.
Vance, G.F., David, M.B., 1991. Chemical characteristics and acidity of soluble organic substances from northern
hardwood forest floor, central Maine, U.S.A. Geochim. Cosmochim. Acta 55, 3611–3625.
Weigand, H., Totsche, K.U., 1998. Flow and reactivity effects on dissolved organic matter transport in soil
columns. Soil Sci. Soc. Am. J. 62, 1268–1274.
G. Guggenberger, K. Kaiser / Geoderma 113 (2003) 293–310310