diagnostik und aktuelle therapie der myelofibrose · leukämie (cml). der „goldstandard“ der...
TRANSCRIPT

K. Wille, P. Sadjadian, P. Dargatz, M. Griesshammer.1 1Universitätsklinik für Hämatologie,
Onkologie, Hämostaseologie und Palliativmedizin, Johannes Wesling Klinikum Minden,
Universitätsklinikum der Ruhr-Universität Bochum
16. März 2018
Diagnostik und aktuelle Therapie der MyelofibroseDurch die Einführung der JAK1/2-Inhibitoren wurde die Therapie der Myelofibrose in den letzten Jahren revolutioniert. Diese neue Therapieoption hat sowohl die Lebensqualität als auch die Lebenserwartung der Patienten mit Myelofibrose deutlich verbessert. Lesen Sie im Folgenden, auf welche klinische Zeichen Sie achten müssen, welche diagnostischen Schritte einzuleiten sind und wie die Myelofibrose aktuell behandelt wird.
Die Myelofibrose ist eine seltene, klonale Erkrankung der pluripotenten hämatopoetischen Stamm- und Progenitorzellen. Durch übermäßige Freisetzung von verschiedenen Zytokinen und Wachstumsfaktoren, Dysregulation des JAK2-Signalwegs und die abnorme Proliferation der Hämatopoese kommt es letztendlich zu einer Veränderung des Knochenmarkstromas mit Faserbildung (1). Die Myelofibrose kann entweder de novo als Primäre Myelofibrose (PMF) oder sekundär aus einer Polycytaemia Vera (PV) oder einer Essentiellen Thrombozythämie (ET) als sog. Post-PV- bzw. Post-ET-Myelofibrose entstehen, die daher auch als sekundäre Myelofibrosen bezeichnet werden. Die Myelofibrose repräsentiert eine Subgruppe der sog. BCR-ABL1-negativen Myeloproliferativen Neoplasien (MPN), zu denen auch die ET und PV gehören. Es handelt sich bei den MPN um chronisch verlaufende, klonale Erkrankungen der hämatopoetischen Stammzelle im Knochenmark, die klinisch durch Hyperplasie einer oder mehrerer betroffener Zellreihen im Knochenmark mit Tendenz zur Faserbildung, thromboembolische Komplikationen und eine Splenomegalie charakterisiert sind. Zu den häufigsten Todesursachen bei Myelofibrose gehören die Transformation in eine akute myeloische Leukämie (20,1%), kardiovaskuläre Erkrankungen (12,3%) und Infektionen (10,4%) (2).
Klinik, Pathophysiologie und Molekularbiologie Die Trias aus Splenomegalie, Anämie und das Vorliegen sog. konstitutioneller Symptome (oder „B-Symptomatik“) wie Fatigue, Fieber, Knochenschmerzen, Nachtschweiß und/oder Gewichtsverlust ist das klassische Bild. Im initialen Stadium ist die PrimäreMyelofibrose (PMF), insbesondere wenn es sich um eine frühe fibrotische oder afibrotische Form handelt, häufig asymptomatisch. Im Blutbild ist dann oft eine ausgeprägte Thrombozythämie festzustellen. Häufig treten auch Mikrozirkulationsstörungen auf, wie z.B. eine Erythromelalgie, Sehstörungen oder Migräne-artige Kopfschmerzen, seltener kommt es zu thromboembolischen Ereignissen. Im Verlauf oder in fortgeschrittenen Stadien entwickeln sich Symptome der ineffektiven Hämatopoese

(progrediente Anämie, Thrombozytopenie, Leukozytopenie), Fatigue, Nachtschweiß, eine zunehmende Splenomegalie mit extramedullärer Hämatopoese, lebensbedrohliche Infektionen, Blutungen oder ein terminaler Blastenschub (3). Zu den verstärkt gebildeten inflammatorischen Zytokinen gehören z.B. TGF-beta, IL-1, IL-2 und IL-6 sowie VEGF, die für die chronische Inflammation bei der Myelofibrose im fortgeschrittenen Krankheitsstadium verantwortlich gemacht werden. Der hierdurch bedingte hypermetabolische Stoffwechsel führt sehr häufig zu den für Myelofibrose-Patienten typischen konstitutionellen Symptomen. Eine Mutation im JAK2-Gen tritt bei ca. 60% aller Patienten mit PMF auf; MPL-Mutationen finden sich in ca. 6%, CALR-Mutationen in ca. 25%, ca. 5-10% der PMF-Fälle sind triple-negativ. Tabelle 1 zeigt die Häufigkeit der „klassischen“ MPN-assoziierten genetischen Treibermutationen JAK2, MPL und CALR bei der PMF (4). Neben Mutationen, die zur Dysregulierung des JAK2-Signalwegs führen, gibt es auch Mutationen in epi-genetischen Regulator-Genen, wie z.B. ASXL1, SRSF2, EZH2 und IDH1/2.
Tab. 1: Häufigkeit der MPN-assoziierten genetischen Aberrationen JAK2, MPL und CALR bei der PMF.
Gen Protein Mutation Frequenz bei PMF
JAK2 Januskinase 2 V617F Ca. 60%
MPL Thrombopoetin-Rezeptor
W515, seltenere,andere Mutationen
Ca. 6%
CALRCalreticulin Unterschiedlich, Exon 9
Ca. 25% allerPMF-Patienten,ca. 88% allerJAK2-negativenPMF-Patienten
Diagnostik und Diagnosekriterien Die Anamnese sollte speziell Fragen zu früheren Thrombosen/Thromboembolien, Mikrozirkulationsstörungen, Blutungsereignissen sowie abdominellen und konstitutionellen Symptomen beinhalten. Ein speziell für diese Patientengruppe entworfener Fragebogen ist MPN10, der insbesondere für die Steuerung einer Symptom-orientierten Therapie und zum

Monitoring der Krankheitsprogression geeignet ist (5). In der körperlichen Untersuchung sollte Wert gelegt werden auf die Palpation der Milz und der Leber sowie die Detektion von Anämie- und Blutungssymptomen. Wichtig ist auch die Durchführung eines abdominellen Ultraschalls zur quantitativen Bestimmung des Ausmaßes der Splenomegalie und der Lebergröße. In den Laboruntersuchungen zeigen sich häufig Zeichen des erhöhten Zellumsatzes wie eine Erhöhung der Alkalischen Phosphatase, der LDH sowie eine Hyperurikämie. Ein Differentialblutbild inklusive Retikulozyten, LDH, Ferritin, Leberenzymen, INR, Bilirubin, Haptoglobin sowie ein Coombs-Test sollten durchgeführt werden. Zum Ausschluss einer systemischen Mastozytose ist die Bestimmung der Serum-Tryptase entscheidend. Neben den laborchemischen Untersuchungen muss ein Blutausstrich unter dem Mikroskop begutachtet werden. Hier zeigen sich typischerweise ein leukerythroblastisches Blutbild mit Erythroblasten und Linksverschiebung der Granulopoese bis zum Myeloblasten, sowie Dakryozyten (sog. Tränentropfen-Erythrozyten). Des Weiteren erfolgt eine molekulargenetische Testung aus dem peripheren Blut: I.d.R. wird bei klinischem Verdacht auf eine PMF zunächst ein Screening auf eine JAK2-Mutation durchgeführt. Sollte das Ergebnis negativ sein, wird in einem zweiten Schritt auf eine Mutation im Calreticulin Gen (CALR) getestet, und nur wenn diese auch negativ ist, erfolgt ein abschließendes Screening auf eine mögliche MPLW515-Mutation. Das BCR-ABL-Fusionsgen muss auch bestimmt werden, wenn alle genannten Marker negativ sind und/ oder bei Verdacht auf eine chronische myeloische Leukämie (CML). Der „Goldstandard“ der Diagnostik der MPN ist und bleibt auch nach den neuesten Diagnose-Kriterien der WHO von 2016 die Knochenmarkpunktion inkl. Histologie und Zytologie (6). Insbesondere in der frühen, präfibrotischen Phase der Erkrankung ist die Durchführung einer Knochenmarkpunktion zur Abgrenzung einer frühen PMF gegenüber einer ET von entscheidender Bedeutung (7). Abbildung 1 zeigt das typische histologische Bild einer PMF in der HE-Färbung im bereits fortgeschrittenen Stadium (sog. „overt“ PMF nach WHO 2016). Zu sehen ist insbesondere das fast vollständige Fehlen der Hämatopoese.
Abb. 1: Knochenmarkhistologie HE, 10x vergrößert (Quelle: Dr. med. Kai Wille).
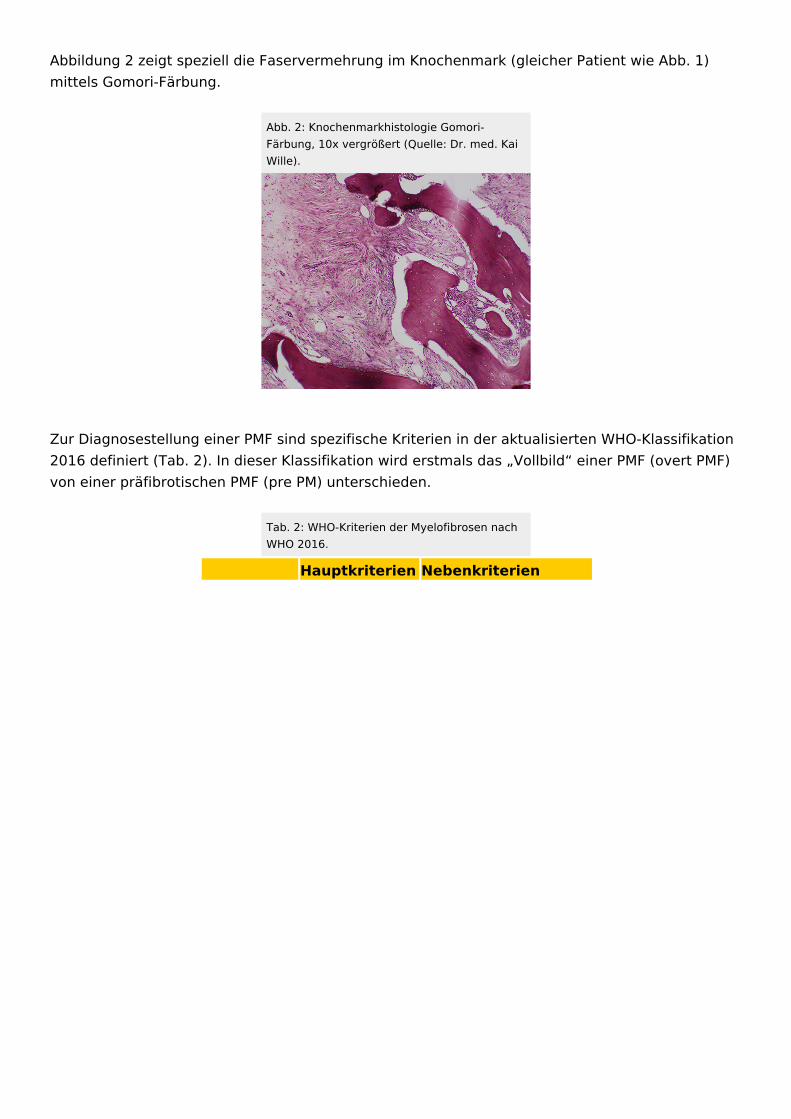
Abbildung 2 zeigt speziell die Faservermehrung im Knochenmark (gleicher Patient wie Abb. 1) mittels Gomori-Färbung.
Abb. 2: Knochenmarkhistologie Gomori-Färbung, 10x vergrößert (Quelle: Dr. med. Kai Wille).
Zur Diagnosestellung einer PMF sind spezifische Kriterien in der aktualisierten WHO-Klassifikation 2016 definiert (Tab. 2). In dieser Klassifikation wird erstmals das „Vollbild“ einer PMF (overt PMF) von einer präfibrotischen PMF (pre PM) unterschieden.
Tab. 2: WHO-Kriterien der Myelofibrosen nach WHO 2016.
Hauptkriterien Nebenkriterien

PMF(overt PMF)
1. Vorliegen einer atypischen und gesteigerten Megakaryopoesesowie Reticulin und/oder Kollagen-Fibrose Grad 2 oder 3.2. WHO-Kriterien für PV, ET, BCR-ABL-positive CML,MDS oder andere myeloischen Neoplasien nicht erfüllt.3. JAK2-Mutation oder MPLW515-Mutation oderCALR-Mutation, oder falls keine dieser Mutationen vorliegt,Vorliegen eines anderen klonalen Markers oder kein Hinweisauf eine reaktive Myelofibrose.
1. LeukoerythroblastischesBlutbild2. Erhöhte LDH3. Anämie4. Palpable Splenomegalie5. Leukozytose ≥ 11 G/L

PräfibrotischePrimäre Myelofibrose (pre PM)
1. Vorliegen einer atypischen und gesteigertenMegakaryopoese ohne Reticulin-Fibrose > Grad 1.2. WHO-Kriterien für PV, ET, BCR-ABL-positive CML,MDS oder andere myeloischen Neoplasien nicht erfüllt3. JAK2-Mutation oder MPLW515-Mutation oder CALR-Muation,oder falls keine dieser Mutationen vorliegt, Vorliegen einesanderen klonalen Markers oder kein Hinweis auf eine reaktive Myelofibrose
1. Erhöhte LDH2. Anämie3. Palpable Splenomegalie4. Leukozytose ≥ 11 G/L
Für die Diagnose overt PMF oder pre PMF üssen jeweils alle 3 Hauptkriterien und mindestens eines der Nebenkriterien erfüllt sein.
Differentialdiagnose Die Differentialdiagnose umfasst grundsätzlich alle Erkrankungen, die mit einer Markraumfibrose einhergehen oder in deren Folge diese auftreten kann. Für die Diagnosestellung Post-ET- und Post-PV-MF gelten weiterhin die 2008 publizierten Kriterien der International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) (8).

Eine Knochenmarkfibrose kann auch durch eine Infiltration des Knochenmarks durch einen maligen Tumor (z.B. Prostata-, Bronchialkarzinom etc.) mit sekundärer Faservermehrung entstehen. Eine Fibrose des Knochenmarks liegt aber auch bei anderen hämatologischen Erkrankungen vor, die dann in die Differentialdiagnose der PMF mit einbezogen werden müssen. Hier sind neben der Haarzellleukämie insbesondere die Myelodysplasien mit Fibrose und die systemische Mastozytose zu erwähnen. Letztendlich können Fibrosen des Knochenmarks auch als Folge einer interstitiellen Myelitis und lokal nach Strahlenbehandlung und bei anderen internistischen Erkrankungen wie Kollagenosen oder Tuberkulosen des Knochenmarks auftreten. Prognose und Risikostratifizierung Der individuelle Verlauf und die Prognose von Patienten mit Myelofibrose sind z.T. sehr unterschiedlich. Um die Prognose des einzelnen Patienten zum Diagnosezeitpunkt und im Verlauf besser einzuschätzen, wurden Risiko-Scores entwickelt. Aktuell sind 3 prognostische Scores am besten etabliert.
IPSS-Prognose-Score Dieser bereits 2009 publizierte Score der IWG-MRT unterscheidet 4 Risiko-Gruppen bei Erstdiagnose PMF (9). Insgesamt 5 Variablen gehen in die Berechnung ein (Tab. 3).
Tab. 3: Prognose-Gruppen nach IPSS.
PrognosegruppePunktzahl mOS(Monate)
Niedrigrisiko 0 135Intermediär-1 1 95Intermediär-2 2 48Hochrisiko ≥ 3 27Jeweils 1 Punkt für 1.) Alter < 65 Jahre,2.) Konstitutionelle Symptome (Fieber,Gewichtsverlust, Nachtschweiß), 3.) Hb< 10 g/dl , 4.) Leukozyten < 25 G/l und5.) Blasten im peripheren Blut ≥ 1%.
DIPSS-Prognose-Score Mit dem DIPPS (Tab. 4), der die gleichen Variablen wie der IPSS verwendet, kann auch im Verlauf der Erkrankung die Einteilung in 4 Risiko-Gruppen erfolgen, wobei der Anämie eine Punktzahl von 2 im Vergleich zum IPSS zugeordnet wird (10).Somit errechnen sich folgende 4 Risiko-Gruppen:• Niedrigrisiko: 0 Punkte• Intermediär-1-Risiko: 1-2 Punkte

• Intermediär-2-Risiko: 3-4 Punkte• Hochrisiko: 5-6 Punkte
Tab. 4: DIPPS-Prognose-Score.
FaktorenPunktzahl
(Score)0 1 2
Alter (in Jahren) ≤ 65
> 65
Leukozyten (G/l) ≤ 25
> 25
Hb (g/dl) ≥ 10 <
10Blasten imperipherenBlut (%)
< 1 ≥ 1
KonstitutionelleSymptome nein ja <
10
DIPPS-Plus-Score Diese Weiterentwicklung des DIPPS (Tab. 5) beinhaltet weitere Risikofaktoren, die sich in Studien als relevant bezüglich des Überlebens herausgestellt haben (11):• Transfusionsbedarf• Thrombozyten < 100 G/l• Ungünstiger Karyotyp (definiert als komplexer Karyotyp alleine oder 2 Aberrationen einschließlich -7/7q-, i(17q), inv(3), -5/5q- 12p- oder 11q23-) Jedem dieser Risikofaktoren wird 1 Punkt zugeschrieben (max. 8 Punkte). Mittlerweile sind weitere Prognose-Scores entwickelt worden, um die einzelnen Risiko-Gruppen noch besser zu diskriminieren, wie MIPSS70 oder MIPSS70+, sowie MYSEC-PM für die sekundären Myelofibrosen, die weitere molekulare Marker und /oder zytogenetische Risikofaktoren beinhalten. Diese müssen sich aber erst noch in der Praxis bewähren.
Tab. 5: DIPPS-Plus-Prognose-Score.
Prognosegruppe Punktzahl mOS(Jahre)
Niedrigrisiko 0 15,4Intermediär-1 1 6,5Intermediär-2 2-3 2,9Hochrisiko ≥ 4 1,3

Therapie der Myelofibrose Die Therapie umfasst eine Spannbreite von der allogenen Stammzelltransplantation als einziger kurativer Option bis hin zur rein supportiven (oder auch „Problem-orientieren“) Therapie. Der aktuelle Therapiealgorithmus der Deutschen Gesellschaft für Hämtologie und Onkologie (DGHO) (12) sieht ein Risiko-adaptiertes Vorgehen nach IPSS oder DIPSS vor (Abb. 3). Kurative Therapie Die einzige kurative Therapie istdie allogene Stammzelltransplantation (allo-SCT), die aufgrund der schlechten Prognose insbesondere bei fitten Patienten mit Intermediär-Risiko-2 oder Hochrisiko diskutiert werden sollte (13) (Abb. 3). Auch junge Patienten (< 65 Jahre) mit einem Intermediär-1-Risiko gelten als Kandidaten für eine alloSCT, wenn zusätzlich eine refraktäre, transfusionsabhängige Anämie und/oder ein Blastenanteil im peripheren Blut > 2% und/oder ein Hochrisiko-Karyotyp vorliegt (14). In aktuellen Studien wird ein möglicher Vorteil des Einsatzes des JAK-Inhibitors Ruxolitinib vor Transplantation überprüft, da ein positiver Effekt auf die hämatologische Regeneration nach der Transplantation durch die Reduktion der proinflammatorischen Zytokine und durch die Reduktion der Milzgröße vor der alloSCT gezeigt wurde (15).
Abb. 3: Therapiealgorithmus der PMF, Post-ET- oder Post-PV-Myelofibrose. 1Problem-orientierte Therapie=Erythropoetin, Hydroxyurea, Interferon, Steroide oder Androgene
Palliative/Symptomatische Therapie Mögliche Ziele einer palliativen/Symptom-orientierten Therapie sindhauptsächlich die Prävention von Komplikationen wie Thrombosen oderBlutungen, die Symptomkontrolle und die Reduktion des Risikos der Krankheitsprogression.

Watch-and-wait-Strategie Patienten mit einem Niedrigrisiko oder Intermedär-Risiko-1 (nach IPSS oder DIPSS) ohne klinische Probleme sollten aufgrund der relativ guten Prognose einer Watch-and-wait-Strategie zugeführt oder in ein entsprechendesStudienkonzept aufgenommen werden (16) (Abb. 3). Ruxolitinib Mit dem oralen JAK1/2-InhibitorRuxolitinib steht seit 2012 die erste zugelassene Therapie zur Behandlung der Myelofibrose zur Verfügung. Durch Ruxolitinib werden insbesondere die konstitutionellen Symptome und die Splenomegalie positiv beeinflusst. Darüber hinaus ist in beiden Phase-III Zulassungsstudien (COMFORT I: Ruxolitinib verglichen mit Placebo und in COMFORT II verglichen mit der besten verfügbaren Therapie) in einer Post-hoc-Analyse auch ein signifikanter lebensverlängernder Effekt für Ruxolitinib und in Knochenmarkuntersuchungen sogar ein Rückgang der Fibrose festgestellt worden (17, 18). Ruxolitinib ist zur Behandlung von krankheitsbedingter Splenomegalie oder Symptomen bei Erwachsenen mit PMF, aber auch mit Post-PV MF oder Post-ET MF, zugelassen (Abb. 3). Die Dosis von Ruxolitinib zu Beginn orientiert sich in erster Linie an der Thrombozytenzahl. Die Dauer der Therapie mit Ruxolitib ist nicht begrenzt. I.d.R. sprechen die Patienten innerhalb der ersten 12 Behandlungswochen an (18). Allerdings wird auch ein späteres Ansprechen der Patienten beobachtet, sodass vor der definitiven Beurteilung des Ansprechens die Therapie über 6 Monate fortgesetzt werden sollte. Eine häufige unerwünschte Wirkung (AE) der Ruxolinitib-Therapie sind Zytopenien, wobei die Thrombozytopenie die häufigste ist. Patienten mit höhergradigen Thrombozytopenien unter50 G/l sollten kein Ruxolitinib erhalten. Bei moderaten Thrombozytopenien oder einer leichten Anämie (z.B. Thrombozytenzahlen zwischen 50 G/l bis 100 G/l oder Anämie < 10 G/l) sollte eine Startdosis von 2 x 5 mg Ruxolitib pro Tag zunächst nicht überschritten werden. Weitere nennenswerte AEs von Ruxolitinib sind eine erhöhte Infektneigung, Gewichtszunahme und das so-genannte „Ruxolitinib-Absetzsyndrom“; beim plötzlichen Absetzen der Therapie wurde eine Häufung an raschen Krankheitsprogressionen inklusive Milzschwellung, Zunahme der Zytopenien und sogar hämodynamische Dekompensationen beschrieben (19). Bei elektiven Eingriffen oder Operationen sollte Ruxolitinib daher möglichst nicht abgesetzt werden. Falls doch notwendig, wird ein Ausschleichen von Ruxolitinib über mindestens 7 Tagen empfohlen.
Problem-orientierte Strategien für Patienten mit PMF Hyperproliferation Zur Kontrolle einer Hyperproliferation (Thrombozytose, Leukozytose) der Blutbildung mit oder

ohne Splenomegalie kommt in erster Linie Hydroxyurea (=Hydroxycarbamid) zum Einsatz. Vor der Zulassung von Ruxolitinib wurde Hydroxyurea als die medikamentöse Standardtherapie für die Myelofibrose betrachtet (20). Obwohl die Effekte von Hydroxyurea auf die Myelofibrose nicht durch prospektiv randomisierte Studien gesichert sind, sprechen viele Beobachtungen für eine Verlangsamung der Progression, eine günstige Wirkung auf eine vorbestehende Anämie und eine zeitweise Verbesserung der Lebensqualität (21). Es gibt nun auch zunehmende Erfahrungen mit der Kombination von Hydroxyurea und Ruxolitinib. Insbesondere in Fällen mit ausgeprägter Leukozytose bei noch ausreichender Thrombozytenzahl kann mit dieser Kombination die Leukozytenzahl gut kontrolliert werden.
Anämie Zur Behandlung einer therapiebedürftigen Anämie werden insbesondere bei zusätzlicher Autoimmunhämolyse häufig mit Erfolg Kortikosteroide eingesetzt. Ca. 1/3 der Patienten spricht auf diese Therapie an, die meisten allerdings nur vorübergehend. In einigen publizierten Arbeiten wird die Wertigkeit der Erythropoetin-Behandlung in Hinblick auf die Knochenmarkfibrose-bedingte, hyporegeneratorische Anämie beschrieben (22). Bei einer initialen Gabe von 3x 10.000 I.E. pro Woche kann mit einem Ansprechen bei ca. der Hälfte der Patienten gerechnet werden. Es kann bis zu 3 Monate dauern, bis ein Ansprechen auftritt. Ein Serum-Erythropoetin-Spiegel < 125 U/l ist Voraussetzung für ein günstiges Ansprechen auf Erythropoetin, allerdings ist Vorsicht geboten, da die Splenomegalie hierunter auch zunehmen kann. Auch Androgene (z.B. Nandrolon) oder Gonadotropinhemmer (z.B. Danazol) sind in Einzelfall-Berichten bei transfusionspflichtiger Anämie eingesetzt worden. Die Wirksamkeit kann auch erst nach 2-3 Monaten beurteilt werden. Falls Androgene starke Nebenwirkungen (Anstieg der Leberwerte, Virilisierung bei Frauen) verursachen, müssen sie abgesetzt werden. Ein Ansprechen der Anämie kann in ca. 50% der behandelten Fälle erwartet werden (23).
Splenomegalie Bei Patienten mit im Vordergrundstehender Splenomegalie wird heute aufgrund der Effektivität und Zulassung Ruxolitinib eingesetzt. Nur wenn hier mangels Ansprechen oder AEs Probleme entstehen, kommen die Milzbestrahlung oder die Splenektomie in Diskussion. Die durchschnittliche Ansprechdauer nach Bestrahlung beträgt allerdings maximal 6 Monate. Wiederholte Bestrahlungen sind im Verlauf möglich, vor allem, wenn vorher nur kleinere Dosen eingesetzt wurden. Für die Splenektomie ist zu beachten, dass die Morbidität (ca. 27%) und Mortalität (ca. 6%) gerade für diese rein Problem-orientierte, palliative Therapie sehr hoch ist und bisher kein lebensverlängernder Effekt nachgewiesen wurde.
Interferon-alpha (vorzugsweise in pegylierter Form) Interferone sprechen bei der Myelofibrose am besten an, wenn die Splenomegalie nicht zu groß ist (< 6 cm unter Rippenbogen), die Thrombozytopenie und Transfusionsbedürftigkeit mit Erythrozyten nicht zu ausgeprägt ist und insbesondere eine frühe Form der Fibrose vorliegt (prePM). Empfohlene Anfangsdosis: PegIntron® 50 µg/Woche oder Pegasys® mit

durchschnittlicher Dosierung 90 µg/Woche. Interferon-alpha ist allerdings nicht für die Behandlung der Myelofibrose zugelassen (24).
Andere JAK-Hemmer, neuere Substanzen und Strategien Mittlerweile wurde außer Ruxolitinib noch eine Reihe anderer JAK-Hemmer bei der Myelofibrose getestet (25) (Tab. 6).
Neue Ansätze kombinieren Ruxolitinib mit Pomalidomid (Phase-Ib-Studie der deutschen MPN-Studiengruppe GSG-MPN) oder Thalidomid bzw. Ruxo-litinib mit Azacytidin, vor allem in den akzelerierten Stadien der Myelofibrose (26). Sotatercept, ein „Activin Rezeptor Typ 2A IgG-Fc“-Fusionsprotein, zeigt in ersten Studien ermutigende Daten bei Myelofibrose-assoziierter Anämie oder Transfusionsabhängigkeit. -Darüber hin-aus werden derzeit Histon-Deacetylase-Inhibitoren (HDAC) sowie Inhibitoren des mTOR-Signalwegs im Rahmen klinischer Studien bei der Myelofibrose auf Sicherheit und Wirksamkeit geprüft.
Tab. 6: JAK-Inhibitoren, die bei Myelofibrose in Studien getestet wurden.
Inhibitor Target Profil
Ruxolitinib JAK1/JAK2- Zugelassen für Myelofibrose (2012) und PV (2015) in Europa
Pacritinib JAK2/FLT3
- Untersucht in PERSIST-1 & -2 Phase-III-Studien, auch gut wirksambzgl. Reduktion der Splenomegalie und Kontrolle der Symptome beiThrombozyten < 100 G/l- Allerdings transienter FDA-Stopp 1/2016 bis 2/2017 wegen verstärkterZNS-Blutungen und unklarer Myokardinfarkte, jetzt wieder anlaufendeStudien (PAC 203-Studie)
Momelotinib JAK1/JAK2
- In SYMPLIFY I & II Phase-III-Studien untersucht und effektiv,aber Ruxolitinib nicht überlegen- Effektiv bei Anämie und Transfusionsbedürftigkeit- Nebenwirkung Polyneuropathie
Icacitinib (INCB039110) JAK1/selektiv - Effektiv in Phase-II-
Studie bei Myelofibrose
Fedratinib JAK2/FLT3
- Effektiv in JAKARTA Phase-III-Studie bei Myelofibrose- Aber Programmstopp wegen Wernicke Encephalopathiein einigen Fällen
NS-01872 JAK2/Src
- Effektiv in Phase-I bis II-Studien bei Myelofibrose- weniger myelosuppressiv
Fazit Die Diagnostik und Therapie der PMF stellt weiterhin eine große Herausforderung für die Behandler dar. Goldstandard der Diagnostik bleibt die Knochenmarkhistologie ergänzt um klinische, laborchemische sowie molekulare- und zytogenetische Parameter. Die Therapie richtet sich nach der Eingruppierung in etablierte Risiko-Scores und nach dem Performance-Status des Patienten und umfasst ein Spektrum reichend von einer Watch-and-wait-Strategie bis hin zur alloSCT. Die Einführung neuer Substanzen (insbesondere des JAK-HemmersRuxolitinib) hat die Therapie der PMF in den letzten Jahren revolutioniert.
Interessenkonflikte: Es besteht kein Interessenkonflikt. Zum Artikel „Diagnostik und aktuelle Therapie der Myelofibrose“ ist auch ein CME-Test verfügbar – hier kommen Sie direkt zur Teilnahme.(verfügbar bis zum 19.03.2020)

Dr. med. Kai Wille
Universitätsklinik für Hämatologie, Onkologie, Hämostaseologie und PalliativmedizinJohannes Wesling Klinikum MindenUniversitätsklinikum d. Ruhr-Universität BochumHans-Nolte-Str. 132429 Minden Tel.: 0571/7904201E-Mail: [email protected]
ABSTRACT
K. Wille, P. Sadjadian, P. Dargatz, M. Griesshammer 1, 1Universitätsklinik für Hämatologie, Onkologie, Hämostaseologie und Palliativmedizin,Johannes Wesling Klinikum Minden, Universitätsklinikum der Ruhr-Universität Bochum
In myelofibrosis, diagnostic pathways and treatment options still are challenges for the practitioners. Up to now, the diagnostic “gold standard” is the bone marrow biopsy complemented by clinical, laboratory and molecular-/cytogenetic parameters. Relevant criteria in selecting the individual treatment are risk classifications and performance status and do range from “watch and wait” to allogeneic stem cell transplantation. Fortunately, the introduction of novel drugs (like JAK inhibitors) reordered the therapy in the past few years.
Keywords: Primary myelofibrosis, bone marrow biopsy, risk classification, JAK inhibitors