current medicinal chemistry, 199-212 199 medicinal ......current medicinal chemistry, 2007, 14,...
TRANSCRIPT

Current Medicinal Chemistry, 2007, 14, 199-212 199
Medicinal Chemistry of Fetal Hemoglobin Inducers for Treatment of-Thalassemia
Roberto Gambari*,1,2 and Eitan Fibach3
1ER-GenTech, Department of Biochemistry and Molecular Biology, Section of Molecular Biology, University of Ferrara,Ferrara, Italy2GenTech-for-Thal, Laboratory for the Development of Pharmacological and Pharmacogenomic Therapy of Thalassaemia,Biotechnology Centre, Ferrara, Italy3Department of Haematology, Hadassah - Hebrew University Medical Centre, Jerusalem, Israel
Abstract: In this review we summarize the achievements of medicinal chemistry in the field ofpharmacological approaches to the therapy of β-thalassemia using molecules able to stimulate the productionof fetal hemoglobin (HbF). We first describe the molecular basis of the pathology and the biochemical rationalof using HbF inducers for therapy; we then outlined the in vitro and in vivo experimental systems suitable forscreening of such potential drugs, and finally we describe the different classes of compounds with emphasison their advantages and disadvantages in the treatment. The results of these reviewed studies indicate that: (a)HbF inducers can be grouped in several classes based on their chemical structure and mechanism of action; (b)clinical trials with some of these inducers demonstrate that they are effective in ameliorating the symptoms ofβ-thalassemia; (c) a good correlation was found between HbF stimulation in vivo and in vitro indicating that invitro testing might be predictive of the in vivo response; (d) combined use of different inducers mightmaximize the effect, both in vitro and in vivo. However, (e) the response to HbF inducers, evaluated in vitro andin vivo, is variable, and some patients might be refractory to HbF induction by certain inducers; in addition, (f)several considerations call for caution, including the fact that most of the inducers exhibit in vitrocytotoxicity, predicting side effects in vivo following prolonged treatments.
Keywords: Fetal hemoglobin; β-thalassemia; Histone deacethylases; Hydroxyurea; DNA-binding drugs; Rapamycin.
This paper is dedicated to the memory of Professor Panos Ioannou.
INTRODUCTION acid). In addition, these clusters contain various sites that areresponsible for the regulation of the expression of each gene[1].The objective of this review is to summarize the
achievements of medicinal chemistry in the field ofpharmacological approaches to the therapy of β-thalassemia.We will shortly describe the molecular basis of thepathology and the experimental systems suitable for thescreening of potential therapeutic compounds. Majoremphasis will be dedicated to the description of the differentclasses of molecules employed in in vitro and in vivopreclinical studies, as well as recently performed clinicaltrials.
The expression of the globin genes is regulated duringontogeny. In humans, globin production is characterized bytwo major "switches" [2]. Production of embryonic Hbsswitches after the first two months of gestation into fetal Hb(HbF) (α2γ 2), and then again, before and immediately afterbirth, into adult Hb (HbA) (α2β2). Since both HbA and HbFcontain α chains, the switch from the former to the latterrepresents a decrease in the expression of the γ -globin genes,associated with an increase of β-globin gene expression. Theprevalence of HbF during embryonic life is explained by itshigh affinity to oxygen, a property that allows it to removeoxygen from HbA in the maternal red blood cells (RBCs)through the placenta.
1. HEMOGLOBINS AND THEIR SWITCH DURINGONTOGENY
Hemoglobin (Hb) is a tetramer of two α-like and two β-like globin polypeptide chains. In human, the genes for α-globins are clustered on chromosome 16, which contains onegene for ζ and two genes for α (α1 and α2, the proteins ofwhich are identical). The genes for β-like globins areclustered on chromosome 11, that contains genes for ε, βand δ, one gene for each, and two slightly different genes forγ (Gγ and Aγ , the proteins of which differ in one amino
Immediately after birth the newborn has 85-98% HbF,which gradually decreases to < 5% at the age of one year. Inadult life HbA is the major Hb, a small < 5% is HbA2(α2δ2) and the rest (<5%) is HbF which is concentrated in afew RBC [3-5].
2. -THALASSEMIAS
In β-thalassemias, mutations affecting the β-globin geneor its regulatory regions cause absence (β0) or reduced (β+)synthesis of β-globin chains [1-4]. This is associated with acorresponding excess of the complementary α-globin. The
*Address correspondence to this author at the Department of Biochemistryand Molecular Biology, Via L.Borsari n.46, 44100 Ferrara, Italy; Tel: +39-532-424443; Fax: +39-532-424500; E-mail: [email protected]
0929-8673/07 $50.00+.00 © 2007 Bentham Science Publishers Ltd.

200 Current Medicinal Chemistry, 2007, Vol. 14, No. 2 Gambari and Fibach
outcome of this unbalanced globin production is thedestruction by apoptosis of erythroid precursors in the bonemarrow and at extramedullary sites (ineffectiveerythropoiesis) and short survival of RBCs in the peripheralblood [5-10]. The disease is associated with morbidity andmortality due to severe chronic anemia and treatment-relatedcomplications (such as multi-organ failure due to ironoverload which is caused by increased absorption andtherapeutic blood transfusion).
recombinant DNA constructs in which the coding sequencesof two different luciferase reporter genes, firefly and renilla,were substituted for those of human Gγ -globin and β-globingenes, respectively. When HbF inducers were added tocultures of cells stably transfected with these constructs, theactivity of their human globin genes could be determined bya sensitive enzymatic assay of the two luciferases in the celllysates. The specificity of the inducers was determined bytheir ability to increase the activity γ -globin/firefly luciferasegene more than that of the β-globin/renilla luciferase.More than 200 mutations have been found to cause β-
thalassemia [10]. In contrast to α-thalassemia, wheredeletions in the α-globin gene cluster account for thedisease, the molecular defects causing β-thalassemia areusually point mutations involving only one or fewnucleotide(s) [10,11]. For instance, β°39-thalassemia iscaused by a stop codon mutation that leads to prematuretermination of β-globin chain synthesis [12,13]; the β°IVSI-1 mutation suppresses correct maturation of the β-globinRNA precursor [14], while in thalassemia with the β+IVSI-110 mutation normal and abnormal spliced β-globin RNAprecursor coexist [15].
Other groups introduced reporter genes within an intactβ-globin gene locus. Thus, Vadolas et al. [38] developed astable cellular genomic reporter assay based on a variant ofthe green fluorescence protein (EGFP) gene under the Gγ -globin promoter in the intact human β-globin locus. Theydemonstrated that K562 cells stably transfected with thisconstruct maintain a uniform basal level of EGFP expressionover long periods of continuous culture and that induction ofEGFP expression is associated with the induction of theendogenous γ -globin genes. Furthermore, they compared theGFP-induction potency of several agents, demonstrating thatthis system might help in identifying novel HbF inducers[38,39].
3. FETAL HEMOGLOBIN AMELIORATES THECLINICAL SYMPTOMS OF -THALASSEMIA Fig. (1) reports results obtained using cells stably
transfected with a Gγ -Aγ -EGFP β-globin locus construct[38] (Fig. 1A) or with reporter genes (coding green EGFPand far-red fluorescent protein, red-FP) under the control ofthe human γ -globin or β-globin promoters, respectively(Breveglieri, G.; Salvatori, F.; Feriotto, G.; Gambari, R.,manuscript in preparation) (Fig. 1B).
The proportion of HbF in postnatal life is influenced byvarious physiological [6] and genetic [7,8] factors. Anincrease in HbF may be acquired, e.g., in juvenile myelomo-nocytic leukemia [9] or during acute erythropoietic stress[10] and is frequently observed in β-hemoglobinopathies[11,12].
Human Erythroid-Like Cell LinesEpidemiological findings have shown that increased HbF
in β-thalassemia ameliorates the clinical symptoms [16,17].The most convincing finding was found in individuals withmutations associated with hereditary persistence of HbF(HPFH) in adults [22-25]. Coexistence of homozygous β-thalassemia with HPFH is asymptomatic. It seems that HbFcan functionally compensate for the absence of β-globinchains [16-21]. These findings have generated conside-rableinterest in identifying molecular and pharmacological waysto increase the production of HbF [26-36]. Indeed, severalgroups of compounds were found to reactivate the γ -globingenes in post-natal erythroid cells. However, many of thesedrugs have low efficacy and specificity, while some arepotentially toxic or carcinogenic. There is therefore an urgentneed for new agents that can induce HbF production withgreater efficiency and lower toxicity.
Human erythroid-like cell lines, such as K562 [40], HEL[41] and UT-7 [42], were derived from cells explanted frompatients with various forms of myeloid leukemia. The cellsgrow in suspension cultures as single, undifferentiated cells,with low production of Hbs. When stimulated by variousagents, they respond within few days with a significantincrease in the production of Hbs and the expression of othererythroid-specific differentiation markers. Among theseerythroid-like cell lines, K562 cells, isolated andcharacterized by Lozzio & Lozzio [40] from a patient withchronic myelogenous leukemia in blast crisis, have beenextensively employed as a useful in vitro model to study themolecular mechanism(s) regulating the expression ofembryonic and fetal human globin genes [43-46], as well asfor screening of new differentiation-inducing compounds[47-55]. Rutherford et al. first reported in 1979 [47] thathemin induces a reversible accumulation of embryonic andfetal Hbs without affecting the rate of cell growth; the cellscan be indefinitely subcultured in the presence of theinducer. Thus, this cell line appears to be committed alongthe embryo-fetal erythroid differentiation pathway and, wheninduced by hemin, produces Hb independently of terminaldifferentiation. Since then, many inducers of erythroiddifferentiation were described, such as cytosine arabinoside(ara-C) [54], butyrates [56], 5-azacytidine [57], chromomycinand mithramycin [51], tallimustine [49,58], cisplatin andcisplatin analogs [50]. Unlike hemin, most of these inducersinhibit cell growth and activate terminal cell division. WhenK562 cells committed to differentiation by these inducers
4. IN VITRO MODEL SYSTEMS FOR SCREENINGOF POTENTIAL INDUCERS OF FETAL HEMO-GLOBIN
Cells Expressing Reporter Genes Under the Control ofthe -Globin Promoter
In order to screen for HbF inducers, several groups haveused cells transfected with reporter genes that are under thetranscriptional control of the Gγ -globin gene promoter. Themain advantage of these systems is that they can potentiallybe automated and thus serve for high-throughput screeningof HbF inducers. For instance, Skarpidi et al. [37] used
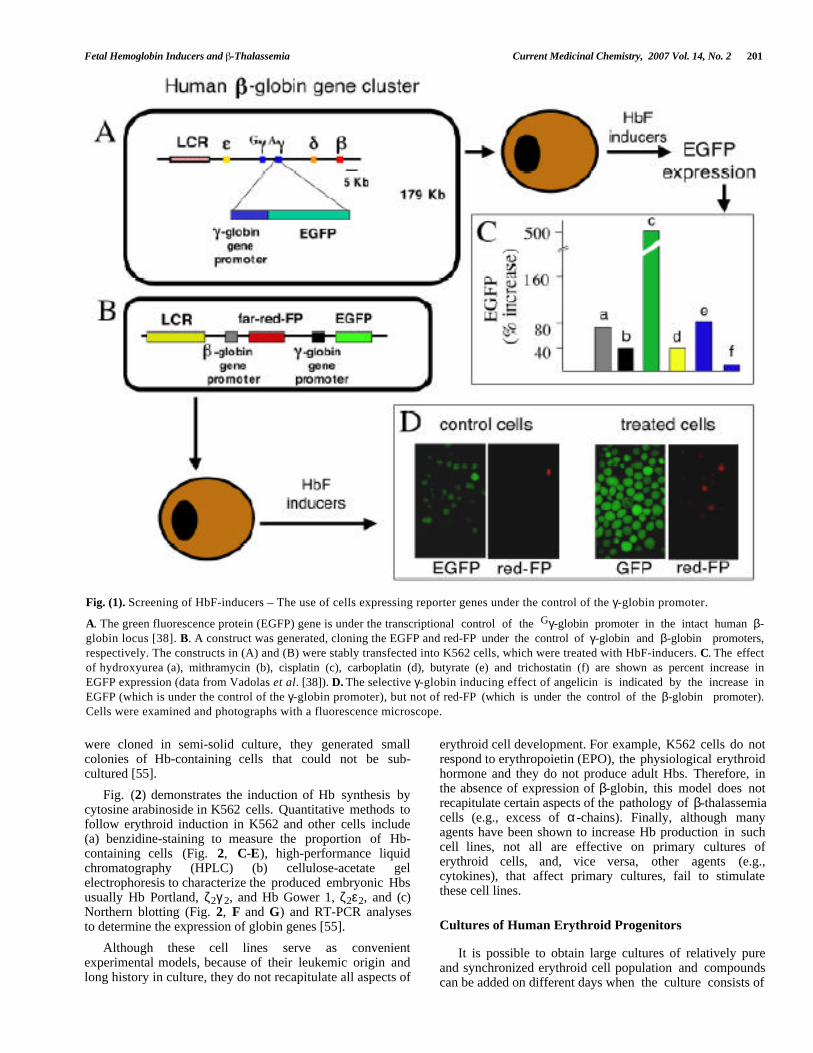
Fetal Hemoglobin Inducers and -Thalassemia Current Medicinal Chemistry, 2007 Vol. 14, No. 2 201
Fig. (1). Screening of HbF-inducers – The use of cells expressing reporter genes under the control of the γ-globin promoter.
A. The green fluorescence protein (EGFP) gene is under the transcriptional control of the Gγ-globin promoter in the intact human β-globin locus [38]. B. A construct was generated, cloning the EGFP and red-FP under the control of γ-globin and β-globin promoters,respectively. The constructs in (A) and (B) were stably transfected into K562 cells, which were treated with HbF-inducers. C. The effectof hydroxyurea (a), mithramycin (b), cisplatin (c), carboplatin (d), butyrate (e) and trichostatin (f) are shown as percent increase inEGFP expression (data from Vadolas et al. [38]). D. The selective γ-globin inducing effect of angelicin is indicated by the increase inEGFP (which is under the control of the γ-globin promoter), but not of red-FP (which is under the control of the β-globin promoter).Cells were examined and photographs with a fluorescence microscope.
were cloned in semi-solid culture, they generated smallcolonies of Hb-containing cells that could not be sub-cultured [55].
erythroid cell development. For example, K562 cells do notrespond to erythropoietin (EPO), the physiological erythroidhormone and they do not produce adult Hbs. Therefore, inthe absence of expression of β-globin, this model does notrecapitulate certain aspects of the pathology of β-thalassemiacells (e.g., excess of α-chains). Finally, although manyagents have been shown to increase Hb production in suchcell lines, not all are effective on primary cultures oferythroid cells, and, vice versa, other agents (e.g.,cytokines), that affect primary cultures, fail to stimulatethese cell lines.
Fig. (2) demonstrates the induction of Hb synthesis bycytosine arabinoside in K562 cells. Quantitative methods tofollow erythroid induction in K562 and other cells include(a) benzidine-staining to measure the proportion of Hb-containing cells (Fig. 2, C-E), high-performance liquidchromatography (HPLC) (b) cellulose-acetate gelelectrophoresis to characterize the produced embryonic Hbsusually Hb Portland, ζ2γ 2, and Hb Gower 1, ζ2ε2, and (c)Northern blotting (Fig. 2, F and G) and RT-PCR analysesto determine the expression of globin genes [55]. Cultures of Human Erythroid Progenitors
Although these cell lines serve as convenientexperimental models, because of their leukemic origin andlong history in culture, they do not recapitulate all aspects of
It is possible to obtain large cultures of relatively pureand synchronized erythroid cell population and compoundscan be added on different days when the culture consists of
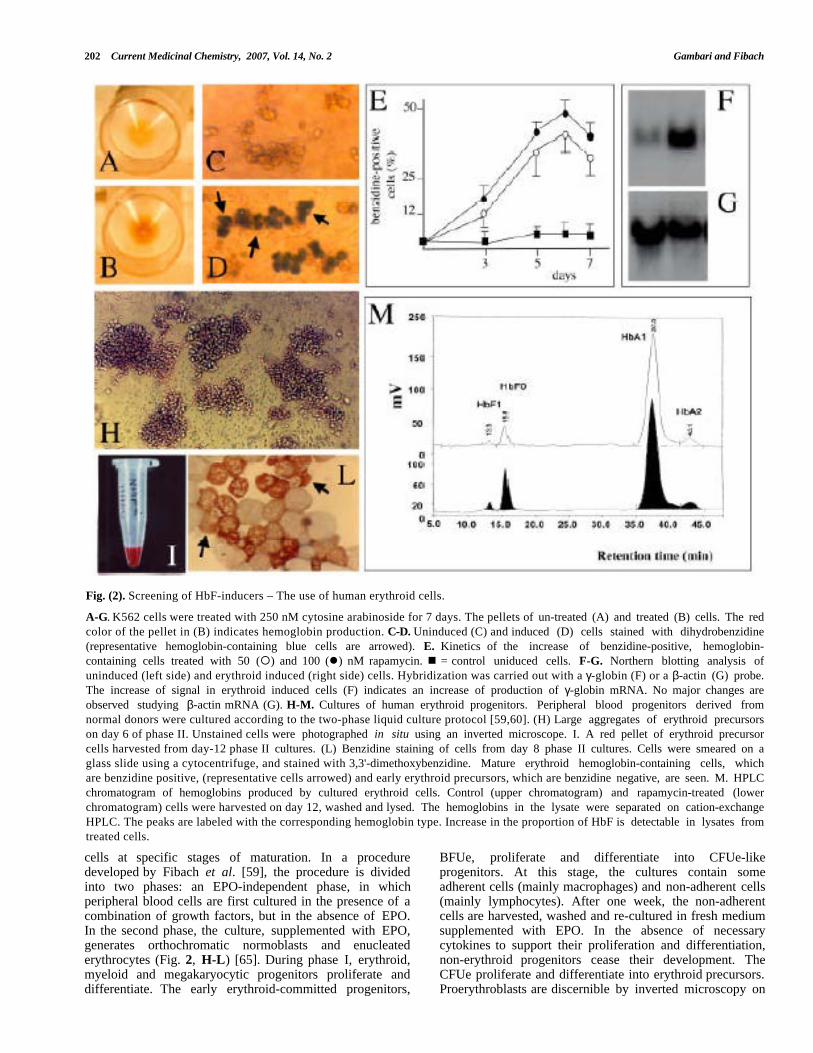
202 Current Medicinal Chemistry, 2007, Vol. 14, No. 2 Gambari and Fibach
Fig. (2). Screening of HbF-inducers – The use of human erythroid cells.
A-G. K562 cells were treated with 250 nM cytosine arabinoside for 7 days. The pellets of un-treated (A) and treated (B) cells. The redcolor of the pellet in (B) indicates hemoglobin production. C-D. Uninduced (C) and induced (D) cells stained with dihydrobenzidine(representative hemoglobin-containing blue cells are arrowed). E. Kinetics of the increase of benzidine-positive, hemoglobin-containing cells treated with 50 (¡) and 100 (l) nM rapamycin. n = control uniduced cells. F-G. Northern blotting analysis ofuninduced (left side) and erythroid induced (right side) cells. Hybridization was carried out with a γ-globin (F) or a β-actin (G) probe.The increase of signal in erythroid induced cells (F) indicates an increase of production of γ-globin mRNA. No major changes areobserved studying β-actin mRNA (G). H-M. Cultures of human erythroid progenitors. Peripheral blood progenitors derived fromnormal donors were cultured according to the two-phase liquid culture protocol [59,60]. (H) Large aggregates of erythroid precursorson day 6 of phase II. Unstained cells were photographed in situ using an inverted microscope. I. A red pellet of erythroid precursorcells harvested from day-12 phase II cultures. (L) Benzidine staining of cells from day 8 phase II cultures. Cells were smeared on aglass slide using a cytocentrifuge, and stained with 3,3'-dimethoxybenzidine. Mature erythroid hemoglobin-containing cells, whichare benzidine positive, (representative cells arrowed) and early erythroid precursors, which are benzidine negative, are seen. M. HPLCchromatogram of hemoglobins produced by cultured erythroid cells. Control (upper chromatogram) and rapamycin-treated (lowerchromatogram) cells were harvested on day 12, washed and lysed. The hemoglobins in the lysate were separated on cation-exchangeHPLC. The peaks are labeled with the corresponding hemoglobin type. Increase in the proportion of HbF is detectable in lysates fromtreated cells.
cells at specific stages of maturation. In a proceduredeveloped by Fibach et al. [59], the procedure is dividedinto two phases: an EPO-independent phase, in whichperipheral blood cells are first cultured in the presence of acombination of growth factors, but in the absence of EPO.In the second phase, the culture, supplemented with EPO,generates orthochromatic normoblasts and enucleatederythrocytes (Fig. 2, H-L) [65]. During phase I, erythroid,myeloid and megakaryocytic progenitors proliferate anddifferentiate. The early erythroid-committed progenitors,
BFUe, proliferate and differentiate into CFUe-likeprogenitors. At this stage, the cultures contain someadherent cells (mainly macrophages) and non-adherent cells(mainly lymphocytes). After one week, the non-adherentcells are harvested, washed and re-cultured in fresh mediumsupplemented with EPO. In the absence of necessarycytokines to support their proliferation and differentiation,non-erythroid progenitors cease their development. TheCFUe proliferate and differentiate into erythroid precursors.Proerythroblasts are discernible by inverted microscopy on

Fetal Hemoglobin Inducers and -Thalassemia Current Medicinal Chemistry, 2007 Vol. 14, No. 2 203
days 4-5 of phase II as large, round and smooth cells. Atthis stage they may be purified from remaining lymphocytesand erythrocytes on Percoll gradient and re-cultured in thesame medium. As the proerythroblasts continue to multiply,they form clusters and then large aggregates which, whenundisturbed, can reach hundreds of cells (Fig. 2H). As thesecells differentiate, they decrease in size and accumulate Hb(Fig. 2L, arrowed cells), and the aggregates assume areddish color (Fig. 2I).
Since the erythroid cells in phase II are grown insuspension, samples of cells can be withdrawn at any timewithout disturbing the cultures and assayed for morphology,size, number, cell viability and apoptosis, cell cycle orexpression of surface antigens. Hemoglobinization can beeasily followed by staining the cells with benzidine.
The Hb content of the developing erythroid cells can bemeasured by a variety of techniques, such as alkalinedenaturation, benzidine staining, cation-exchange HPLC forhemoglobins (Fig. 2M) and reverse-phase HPLC for globinchains. Using the HPLC techniques, Hb is measurable inculture as early as 5 days in phase II. On day 12, one mlculture is sufficient for multiple measurements. The meancellular Hb or HbF concentrations of erythroid cells arecalculated from the values of the HPLC determinationsdivided by the number of benzidine-positive cells. Thedistribution of the erythroid cell population with respect tointracellular content of HbF can be analyzed by flowcytometry using monoclonal antibodies directed specificallyagainst HbF [60]. Dual/triple staining with thecorresponding antibodies can be used for simultaneousanalysis of both HbA and HbF, or Hb and another marker ofinterest such as glycophorin A or CD34 surface antigens[60].
Peripheral blood cells are employed in this procedure forthe following reasons: (a) the availability of blood fromnormal individuals and patients; (b) the homogeneity of theperipheral blood erythroid progenitors, namely early BFUe,as opposed to the bone marrow which contains progenitorsat various developmental stages. Good results can beobtained with cells derived from other sources, includingCD34+ cells purified by immuno-magnetic beadtechnologies, but, in these cases, some modifications of theprocedure are required.
This system recapitulates many aspects of in vivoerythropoesis including globin RNA metabolism, cell cyclekinetics, expression of cell surface antigens, iron and ferritinmetabolism and transcription factors [60-62].
Several research groups have used this system to studythe effects of hundreds of compounds, including butyratederivatives [63,19], hemin [65], EPO [64]. For studyingtheir potential to enhance HbF production, compounds canbe added to phase I, phase II or both. Non-toxic drugs, suchas cytokines and hemin, may be added to the cultures at anytime. With cytotoxic drugs, such as hydroxyurea (HU) and5-azacytidine, because of their cytotoxic/cytostatic effects,they are usually added on day 4-8 of phase II.
5. MOUSE MODELS FOR -THALASSEMIA
Murine models mimicking β°-thalassemia are verydifficult to be obtained [66]. This is due to the fact that themouse β-globin locus contains four functional β-globingenes: βh1 and εy, which are transcribed only during theembryonic development and become silenced in 14-15 day-old embryos, and the b1 (βmajor) and b2 (βminor) genes,
Table 1. In Vivo Experimental Model Systems for Studying the Effects of HbF Inducers
Genotype Phenotype Reference
HbbTh1/Th1 (a) Mild anemia Skow et al., 1983 [68]
HbbTh2/Th2 (b) Severe anemia Sheehee et al., 1993 [70]
HbbTh3/- (c) Thalassemia intermedia Yang et al., 1995 [71]
HbbTh3/Th3(c) Death in utero Yang et al., 1995 [71]
Irradiated mouse transplanted with
HbbTh3/Th3 fetal liver cells Adult thalassemia major Rivella et al., 2005 [73]
HbbTh3/- carrying human globin locus Recapitulates β-IVS-110
with a β-IVSI-110 globin gene splicing mutation Vadolas et al., 2006 [74]
HbbTh3/- carrying human globin locus Recapitulates β°39 Jamsai et al., 2005 [75]
with a β°39 globin gene stop mutation
HbbTh3/- carrying human globin locus Recapitulates HbE Jamsai et al., 2006 [76]
with a β-thal/HbE globin gene phenotype
(a)Deletion including the b1 gene.(b)Targeted deletions of b1 gene with the insertion of a non-globin promoter.(c)Deletion of both b1 and b2 genes.

204 Current Medicinal Chemistry, 2007, Vol. 14, No. 2 Gambari and Fibach
Fig. (3). Strategies for screening and developing of HbF-inducers for treatment of β-thalassemia.
which are activated around 11 days after conception and inadults produce 80% and 20%, respectively, of the total (13-15 g/dl) Hb [66]. Notably, unlike humans, mice lack γ -likeglobin genes, and the embryonic to adult Hb switch occursbefore birth, rather than during the first 6 months after birth.Accordingly, no naturally occurring mutations thatcompletely inhibit expression of the b genes have ever beenobserved, as mice homozygous for such β° mutations areexpected to die prenatally of severe anemia [66].
do not survive more than a few hours after birth, whileheterozygous mice show a very mild phenotype [69]. Theth3 model was generated by deletion of both the βmajor andβminor genes [70,71]. Mice homozygous for this deletion dielate in gestation, as expected by the absence of the b1 and b2gene products. Heterozygotes (Hbbth3/+), however, are viablebut exhibit severe anemia (7 to 9 g/dL of Hb), abnormalRBC morphology, splenomegaly and hepatic irondeposition similar to that found in patients with β-thalassemia intermedia.However, a few adult mouse models of β-thalassemia
intermedia have been described (Table 1). In the th1 model,a spontaneous DNA deletion includes the b1 gene and itsadjacent upstream sequence, including the promoter [67].Mice that are homozygous (Hbbth1/th1) have reduced Hblevels but only a mild anemia, possibly due to atranslational compensatory mechanism that increases b2(βminor) globin synthesis [66,68]. Another mouse model(th2) was generated by targeted deletion of the b1 gene,introducing a non-globin promoter into the disrupted b1locus. Homozygous Hbbth2/th2 mice are severely anemic and
Recently, an adult mouse model of β0-thalassemia wasgenerated by engrafting wild-type mice, after myelo-ablation,with β-globin-null fetal liver cells harvested from 14.5- dayHbbth3/th3 embryos which lack both the βmajor and βminor
genes. After 6-7 weeks, these mice exhibited a severe anemia(2-4 g Hb/dL), low RBC and reticulocyte counts andhematocrit values. The profound anemia settled in after 50days, consistent with the clearance rate of the recipient’snormal RBCs, and the mice succumbed to ineffectiveerythropoiesis within 60 days [72]. Post mortem

Fetal Hemoglobin Inducers and -Thalassemia Current Medicinal Chemistry, 2007 Vol. 14, No. 2 205
examination demonstrated severe body mass reduction withmassive splenomegaly due to erythroid hyperplasia as wellas extensive hepatic extra-medullary hematopoiesis and ironoverload [66,72].
steps for screening, characterization and developing of auseful clinical HbF inducer.
Hypomethylating Agents
Since the work by Tariq et al. [73], showing the crucialrole of the human Locus Control Region (LCR) inreproducing the correct switch from γ -globin to β-globingene expression, β-thalassemic mouse models were generatedwhich, in addition to deletion of the mouse β-like globingenes, carry a mutated human β-globin gene cluster. Forinstance, Vadolas et al. [74] generated a "humanized" mousemodel carrying the common thalassemic β+-IVSI-110splicing mutation on a bacterial artificial chromosome thatcontains the human β-globin locus. They examinedheterozygous murine β-globin knock-out mice carrying eitherthe IVSI-110 or the normal human β-globin locus. A 90%decrease in human β-globin chain synthesis was observed inmice with the IVSI-110 mutation compared to mice withnormal human β-globin cluster. This difference has beenattributed to aberrant splicing; therefore, the humanizedIVSI-110 mice recapitulate the splicing defect found incomparable β-thalassemia patients. This model can thereforeserve as a platform for testing strategies for restoration ofnormal splicing. Other examples of “humanized” transgenicβ-thalassemia mice are listed in Table (1) [75,76].
Decreased HbF production after birth is associated withDNA methylation at the γ -globin gene promoter by DNAmethyltransferases [77-79]. These enzymes are inhibited bythe cytosine analogs, 5-azacytidine and 5-aza-2'-deoxycytidine (decitabine) [80]. In early studies, 5-azacytidine significantly increased HbF in thalassemiapatients, but clinical development of these analogs washalted after a poorly controlled animal study that suggestedthat 5-azacytidine might be carcinogenic. However, themajority of the preclinical studies with decitabine havesuggested a chemopreventive rather than carcinogenic effect[81]. Furthermore, decitabine, unlike 5-azacytidine, is notincorporated into RNA and is a more directed DNA-hypomethylating agent [82]. Accordingly, this class of HbFinducers should be carefully reexamined for their therapeuticpotential.
Hydroxyurea
One of the first studies showing a clear effect ofhydroxyurea [HU] was published in 1993 by Fibach et al.[63] using the two-phase liquid culture in which, aspreviously described, human peripheral blood-derivedprogenitor cells undergo proliferation and differentiation. HUwas found to have multiple effects on these cultured cells:(a) an increase in the proportion of HbF produced; (b) adecrease in cell number due to inhibition of cell
6. INDUCERS OF FETAL HEMOGLOBIN
HbF inducers can be grouped in several classes accordingto their chemical structure and mechanism of action (Table2) [77-115]. Fig. (3) shows a flow-chart of the necessary
Table 2. Inducers of Fetal Hemoglobin in Erythroid Precursor Cells from Human Donors
Inducer Mechanism of action References(a)
Hydroxyurea Inhibition of DNA synthesis Fibach et al., 2003 [63]
5-azacytidine Hypomethylation of DNA De Simone, 2004 [82]
Citarabine Hypomethylation of DNA Sauthararajah et al, 2003 [80]
Butyrates HDAC inhibitors Cao et al., 2004 [86]
Trichostatin HDAC inhibitor Marianna et al., 2001[84]
Apicidin HDAC inhibitors Witt et al., 2003 [88]
Scriptaid HDAC inhibitors Johnson et al., 2005 [90]
Mithramycin DNA-binding drug Fibach et al., 2003 [108]
Cisplatin and analogues DNA-binding drugs Bianchi et al., 2000 [50]
Tallimustine and analogues DNA-binding drugs Bianchi et al., 2001 [49]
Angelicin DNA-binding drug Lampronti et al., 2003 [107]
Rapamycin FRAP-mTOR signal transduction Mischiati et al., 2003 [111]
Fibach et al., 2006 [113]
Triple-helix oligodeoxynucleotides Activation of γ-globin
gene promoter Xu et al., 2000 [118]
Peptiole nucleic acids (PNAs) Artificial promoters Wang et al., 1999 [122]
(a)In some cases, only a representative reference in given.
206 Current Medicinal Chemistry, 2007, Vol. 14, No. 2 Gambari and Fibach
proliferation; (c) an increase in Hb content per cell (meancorpuscular hemoglobin, MCH); and (d) an increase in cellsize (mean corpuscular volume). The extent of these effectswas related to the HU dose and time of addition. Whenadded to cell cultures from normal individuals 4 daysfollowing their exposure to EPO, HU caused a 1.3- to 3.5-fold increase in the proportion of HbF, from 0.4% to 5.2%(mean 1.6%) in untreated to 1.5% to 8.2% (mean 3.1%) inHU-treated cultures. Addition of HU to cells isolated fromfour patients with β-thalassemia showed a 1.3- to 6.2-foldincrease.
increased expression of the γ -globin genes [83-92]. HDACinhibition leads to hyperacetylation of ε-amino groups oflysine residues in histones [92]. This in turn causes adecreased association of basic core histone proteins with theDNA, rendering certain genes more accessible to thetranscriptional machinery. Among HDAC inhibitors,trichostatin was found to possess high HbF-inducingactivity in human and mouse erythroleukemia cells. In arecent report, Witt et al. [64] have showed that, amongseveral specific HDAC inhibitors tested, apicidin was by farthe most efficient HbF-inducer (at nM to µM concentrations)in K562 cells [88], and that its effect involved, in additionto HDAC inhibition, p38 mitogen-activated protein (MAP)kinase signaling [64]. Scriptaid, a novel HDAC inhibitor,was shown to induce γ -globin in K562 cells and humanerythroid progenitors in vitro . Treatment with scriptaid of β-YAC transgenic mice, where the γ -globin gene iscompletely silenced, caused reticulocytosis and synthesis ofhuman γ -globin mRNA [90]. In a recent report, Johnson etal. [90] demonstrated that scriptaid induces γ -globinexpression via the p38 MAPK signaling; the p38-selectiveinhibitor SB203580 completely reversed the ability toinduce HbF.
When HU was administered to thalassemic mice itimproved the β-thalassemic phenotype. Sauvage te al.reported [93] that the hematocrit rose from 29 ± 3% at day 0to 37 ± 4% at day 30, despite myelo-suppression anddecreased reticulocyte counts. The βminor/α ratio of globinchain synthesis increased from 0.78 at day 0 to 0.97 at day30. Membrane defects improved: the proportion of bound αchains decreased, the proportion of spectrin and ankyrinincreased as did the deformability of RBC.
Several reports confirmed the HbF augmenting effect ofHU both in vitro and in vivo [94-104]. For example,Yavarian et al. [105] reported the treatment with HU of 133patients with transfusion-dependent β-thalassemia. After oneyear of treatment these patients were classified into: goodresponders (61%) who shifted from monthly bloodtransfusion dependency to a stable transfusion-free condition;moderate responers (23%) who remained transfusiondependent but at longer intervals (6 months or more), andnon responders, who remained at the same level oftransfusion dependency.
Further HDAC inhibitors were recently characterized fortheir effect on human γ -globin gene expression in transgenicmice. Among the hydroxamic acid derivatives of short-chainfatty acids studied, butyryl and propionyl hydroxamate weremost effective, increasing the human γ /murine α-globinmRNA ratios by 33.9% and 71%, respectively. This wasassociated with an increase in reticulocytes hematocrit, andthe in vivo levels of BFU-E [91].
DNA-Binding DrugsTable (3) summarizes the clinical studies with HU in β-
thalassemia patients. The results indicate that a significantproportion of the patients became transfusion-free aftertreatment. Figs. (4 and 5) show the molecular structures and
representative results of two DNA-binding drugs (DBDs)(mithramycin and angelicin) found to increase HbF inerythroid precursor cells. Interestingly, several DBDs are orhave been used in therapy. Chromomycin and mithramycinwere used in cancer and in hypercalcemia; tallimustine and
Inhibitors of Histone Deacetylases
Several findings suggest that inhibition of the activity ofhistone deacetylases (HDACs) is associated with an
Table 3. Clinical Trials Employing Hydroxyurea as In Vivo Inducer of Fetal Hemoglobin
Number of patients treated(a) Genotype/phenotype(b) Number of patients responding tothe treatment(c)
References
5 β-thalassemia intermedia 3 (60%) Hoppe et al., 1999 [123]
7 β-thalassemia intermedia 3 (43%) de Paula et al., 2003 [124]
4 β-thalassemia major 1 (25%) de Paula et al., 2003 [124]
2 β-thalassemia intermedia 2 (100%) Bradai et al., 2003 [125]
5 β-thalassemia major 5 (100%) Bradai et al., 2003 [125]
36 β-thalassemia major 25 (69%) Alebouyeh et al., 2004 [126]
133 β-thalassemia major 81 (61%) Yavarian et al., 2004 [105]
37 H-β-thalassemia intermedia 26 (70%) Dixit et al., 2005 [127]
166 β-thalassemia intermedia 83 (50%) Karimi et al., 2005 [128]
42 HbE/β°-thalassemia 50% Singer et al., 2005 [132](a)Dosages were between 5-20 mg/kg/day.(b)Only transfusion-dependent patients are included.(c)Transfusion independence or reduction of the transfusion frequency.
Fetal Hemoglobin Inducers and -Thalassemia Current Medicinal Chemistry, 2007 Vol. 14, No. 2 207
tallimustine analogues are anticancer and antiviral agents;angelicin and the analogue bergaptene are used in PUVA(psoralen plus UVA) therapy. Many reports demonstratedthat some DBDs display DNA sequence selectivity, and thateven similar DBDs differ with respect to stability of theircomplexes with DNA. In any case, DBDs interacting withthe major groove of DNA are expected to inhibit complexformation between transcription factors and target DNAelements [106].
Since, among the DBDs studied, mithramycin displayedthe lowest cytotoxicity, we compared it to HU on HbFproduction by thalassemic erythroid precursors [108]. Theresults demonstrated that in cultures derived from 12patients, mithramycin increased HbF production in all cases,while HU was not effective in two cases and was toxic inone. In the majority of cases the activity of mithramycin washigher than HU. In all cases, HU strongly inhibited cellproliferation, while, at concentrations able to induce HbFproduction, mithramycin had minimal effect on cell growth.Our group has recently demonstrated that tallimustine
[49-58] and some cisplatin analogues [50,107] as well as theGC-rich binders chromomycin and mithramycin [51]
are
powerful inducers of differentiation of K562 cells,suggesting that the expression of crucial genes involved inerythroid differentiation of these cells are influenced byDBDs. Several DBDs, such as tallimustine, mithramycin,cisplatin and angelicin, increase HbF production in erythroidprecursor cells from normal human subjects. The extent ofinduction was found to be higher than that of HU.
Rapamycin
Rapamycin (Fig. 6) is a lipophilic macrolide also calledsirolimus, isolated from a strain of Streptomyceshygroscopicus found in a soil from Easter Island (known bythe inhabitants as Rapa Nui) [109,110]. We recentlydemonstrated that rapamycin induces erythroiddifferentiation of K562 cells and increases HbF production
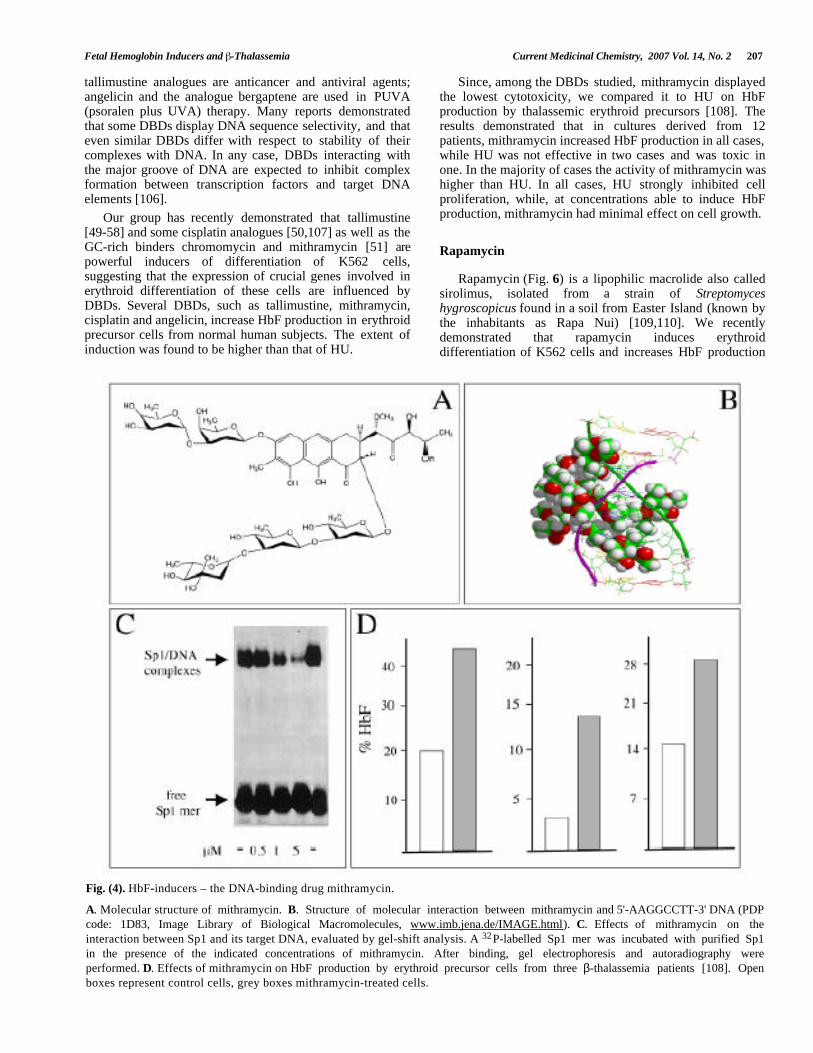
Fig. (4). HbF-inducers – the DNA-binding drug mithramycin.
A. Molecular structure of mithramycin. B. Structure of molecular interaction between mithramycin and 5'-AAGGCCTT-3' DNA (PDP
code: 1D83, Image Library of Biological Macromolecules, www.imb.jena.de/IMAGE.html). C. Effects of mithramycin on theinteraction between Sp1 and its target DNA, evaluated by gel-shift analysis. A 32P-labelled Sp1 mer was incubated with purified Sp1in the presence of the indicated concentrations of mithramycin. After binding, gel electrophoresis and autoradiography wereperformed. D. Effects of mithramycin on HbF production by erythroid precursor cells from three β-thalassemia patients [108]. Openboxes represent control cells, grey boxes mithramycin-treated cells.
208 Current Medicinal Chemistry, 2007, Vol. 14, No. 2 Gambari and Fibach
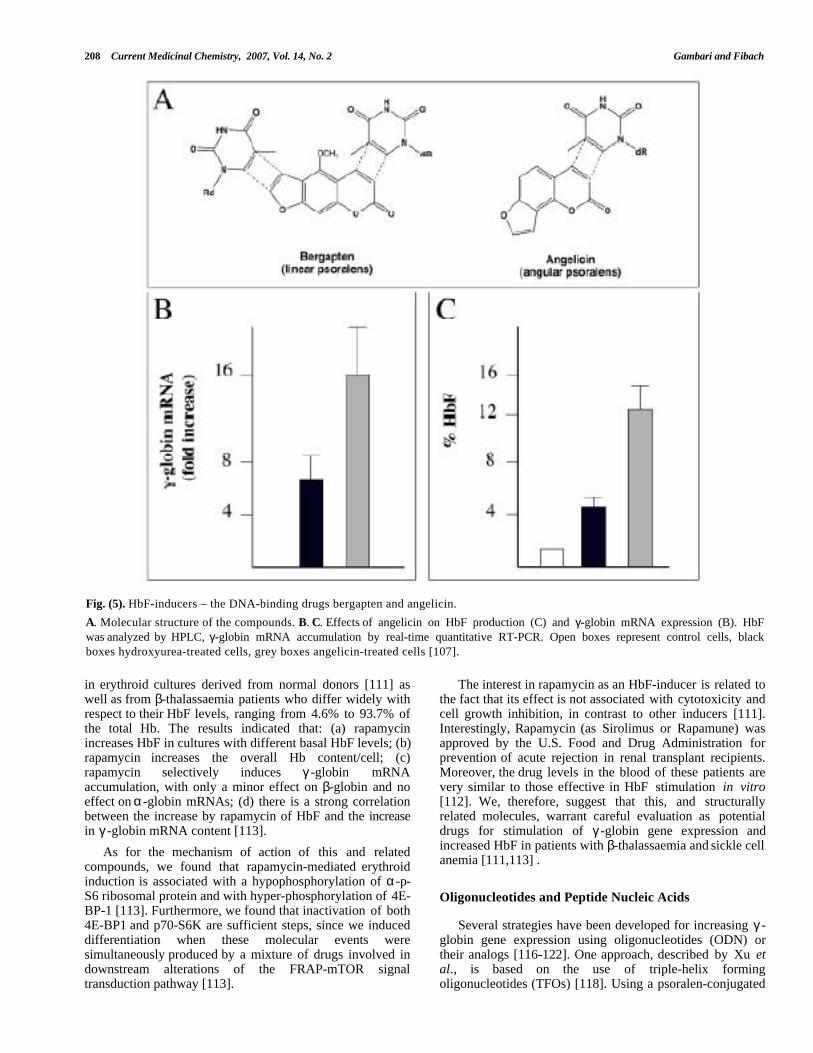
Fig. (5). HbF-inducers – the DNA-binding drugs bergapten and angelicin.
A. Molecular structure of the compounds. B. C. Effects of angelicin on HbF production (C) and γ-globin mRNA expression (B). HbFwas analyzed by HPLC, γ-globin mRNA accumulation by real-time quantitative RT-PCR. Open boxes represent control cells, blackboxes hydroxyurea-treated cells, grey boxes angelicin-treated cells [107].
in erythroid cultures derived from normal donors [111] aswell as from β-thalassaemia patients who differ widely withrespect to their HbF levels, ranging from 4.6% to 93.7% ofthe total Hb. The results indicated that: (a) rapamycinincreases HbF in cultures with different basal HbF levels; (b)rapamycin increases the overall Hb content/cell; (c)rapamycin selectively induces γ -globin mRNAaccumulation, with only a minor effect on β-globin and noeffect on α-globin mRNAs; (d) there is a strong correlationbetween the increase by rapamycin of HbF and the increasein γ -globin mRNA content [113].
The interest in rapamycin as an HbF-inducer is related tothe fact that its effect is not associated with cytotoxicity andcell growth inhibition, in contrast to other inducers [111].Interestingly, Rapamycin (as Sirolimus or Rapamune) wasapproved by the U.S. Food and Drug Administration forprevention of acute rejection in renal transplant recipients.Moreover, the drug levels in the blood of these patients arevery similar to those effective in HbF stimulation in vitro[112]. We, therefore, suggest that this, and structurallyrelated molecules, warrant careful evaluation as potentialdrugs for stimulation of γ -globin gene expression andincreased HbF in patients with β-thalassaemia and sickle cellanemia [111,113] .
As for the mechanism of action of this and relatedcompounds, we found that rapamycin-mediated erythroidinduction is associated with a hypophosphorylation of α-p-S6 ribosomal protein and with hyper-phosphorylation of 4E-BP-1 [113]. Furthermore, we found that inactivation of both4E-BP1 and p70-S6K are sufficient steps, since we induceddifferentiation when these molecular events weresimultaneously produced by a mixture of drugs involved indownstream alterations of the FRAP-mTOR signaltransduction pathway [113].
Oligonucleotides and Peptide Nucleic Acids
Several strategies have been developed for increasing γ -globin gene expression using oligonucleotides (ODN) ortheir analogs [116-122]. One approach, described by Xu etal., is based on the use of triple-helix formingoligonucleotides (TFOs) [118]. Using a psoralen-conjugated

Fetal Hemoglobin Inducers and -Thalassemia Current Medicinal Chemistry, 2007 Vol. 14, No. 2 209
Fig. (6). HbF-inducers – the DNA-binding drug rapamycin.
A. Molecular structure of rapamycin. B. Effects of rapamycin on HbF production in erythroid precursor cells from four β-thalassemicpatients with different starting levels of HbF. HbF was analyzed by HPLC [113]. Open boxes represent control cells, grey boxesrapamycin-treated cells.
TFO designed to bind to a site overlapping with an Oct-1binding site at the -280 region of the γ -globin gene resultedin targeted mutagenesis with a frequency of 20%. In vitroprotein binding assays indicated that these mutationsreduced Oct-1 binding to its target site. In vivo geneexpression assays in mouse erythroleukemia cellsdemonstrated activation of γ -globin gene expression bythese mutations. These findings suggest that this sitenegatively regulates γ -globin gene expression, and thatmutations at the site can activate the γ -globin gene.
In summary, these molecular technologies provide novelapproaches for induction of γ -globin gene expression andtreatment of β-thalassemia.
Erythropoietin
The rationale for in vivo treatment with recombinanthuman EPO in thalassemia comes from studies in baboons,thalassemic mice and in erythroid cultures of β-thalassemiapatients. The results demonstrated an increase in γ -globinsynthesis and consequently in HbF, resulting inimprovement in erythropoietic parameters. Studies withEPO, mainly in patients with β-thalassemia intermedia,showed a significant, dose-related increase in thalassemiaerythropoiesis without changes in the proportion of HbF, inMCV and in MCH, and no major side effects during 9months of continuous treatment. Accordingly, the use ofEPO in combination with true HbF inducers is expected tocause both increased erythropoiesis and preferential inductionof HbF. Indeed, EPO was reported to be required tomaximize HbF induction by inducers such as HU andHDAC inhibitors [114,115].
We attempted to apply a transcription factor decoystrategy for targeting putative repressor factors. One decoyoligonucleotide was able to induce erythroid differentiationin K562 cells, and increased γ -globin mRNA expression andHbF production in erythroid precursor cells from normaldonors [119].
Activation of γ -globin genes was recently proposed withthe aid of “artificial promoters”, as recently reviewed[120,121]. Using peptide-nucleic acids (PNAs) designed tobind to the 5' flanking region of the γ -globin gene,induction of expression of a reporter gene construct wasdemonstrated both in vitro and in vivo. In this case, PNAsare designed for a stable triple helix targeting the sensestrand of DNA, allowing RNA polymerase to starttranscription [122]. Interestingly, PNA-mediated inductionof endogenous γ -globin gene expression was alsodemonstrated in K562 cells.
7. COMBINED USE OF VARIOUS INDUCERS OFFETAL HEMOGLOBIN
Several recent reports show that a combined use of HbFinducers might maximize the effect on HbF with lower

210 Current Medicinal Chemistry, 2007, Vol. 14, No. 2 Gambari and Fibach
toxicity than each individual inducer. For example,Marianna et al. [131] investigated the effects of the butyrateanaolg valproic acid and the HDAC inhibitor trichostatin incombination with hemin. They showed that the combinationhad a significant higher γ -globin enhancing capacity withlower toxicity in human erythroid liquid cultures. Thesefindings suggest that combination of these drugs (all FDA-approved) might be helpful for the treatment ofhemoglobinopathies. This and similar studies are veryimportant, in consideration of the concerns about the dose-limiting myelotoxicity and potential carcinogenicity of HbFinducing agents.
9. CONCLUSIONS AND FUTURE PERSPECTIVES
Several conclusions should be drawn following thecomparative analysis of the data found in the literature onHbF inducers as potential drugs for pharmacologicaltreatment of β-thalassemia.
1 The approach is reasonable, on the basis of theclinical parameters exhibited by HPFH patients.
2 Clinical trials (even if still limited) employing HbFinducers were effective in ameliorating in vivo clinicalparameters of β-thalassemia patients.
8. CLINICAL TRIALS3 Good correlation of in vivo and in vitro results of
HbF synthesis and globin mRNA accumulationindicates that in vitro testing might be predictive ofin vivo responses.Clinical trials aimed at increasing HbF synthesis in β-
thalassemia have included administration of cell-cyclespecific agents (e.g., HU), hematopoietic growth factors(e.g., EPO) and short-chain fatty acids (e.g., butyrate and itsanalogues), all of which stimulate γ -globin synthesis bydifferent mechanisms. HU is the most studied drug and it iscurrently the only HbF-inducing drug in routine use [105,125-134] (Table 3).
4 A combined use of HbF inducers might be useful tomaximize HbF induction, both in vitro and in vivo.
However, several considerations introduce cautions.
1 Most of the HbF inducers exhibit in vitrocytotoxicity, predicting side effects in vivo duringprolonged treatment (as expected for the therapy ofthe β-thalassemia).For example, Dixit et al. studied the response to HU of
thirty-seven patients with β-thalassemia intermedia [127].Major response was defined as transfusion independence orHb rise of more than 20g/l, and minor response as rise in Hbof 10-20 g/l or reduction in transfusion frequency by 50%.Twenty-six patients (70.2%) showed response to HUtherapy. Seventeen patients (45.9%) were major responders,and nine patients (24.3%) showed minor response. MeanHbF levels rose on HU therapy. Older age, low baseline Fcell percent, and low baseline HbF levels (below 10%) werepredictors of poor response. Response was evident within 1month of starting HU therapy in the majority of responders.Thus, a short trial of HU therapy can predict durableresponse [127].
2 The response to HbF inducers, evaluated in vitro andin vivo, is variable, and some β-thalassemia patients(and the erythroid cells derived from them) might berefractory to induction; the reasons for thisphenomenon are largely unknown.
Accordingly, several approaches need to be followed,including gene expression profiling and proteomic studies tofully characterize the pharmacogenomic effects of HbFinducers. In addition, medicinal chemistry will continue toplay a pivotal role in the development of novel HbFinducers, with the aim to identify agents acting throughdifferent mechanisms and exhibiting low toxic effects.Finally, a strict collaboration between clinicians andresearchers is required to bring the most interesting HbFinducers from the laboratory to the clinic.
Singer et al. [132] reported a multicenter trial of 42patients treated with HU for two years; almost half thepatients demonstrated a significant increase in steady-stateHb level. Combined treatment of HU with EPO benefitedselected patients, but the addition of sodium phenyl butyratewas ineffective. After 5 years of follow-up, a subset ofpatients remained off transfusions.
ACKNOWLEDGEMENTS
R.G. is granted by AIRC, Fondazione Cassa diRisparmio di Padova e Rovigo, Cofin-2005, by UEobiettivo 2, by UE ITHANET Project and by the STAMINAProject of Ferrara University. This research is also supportedby Regione Emilia-Romagna (Spinner Project) and byAssociazione Veneta per la Lotta alla Talassemia, Rovigo(AVLT).
As for demethylatying agents, in phase I/II studies,decitabine, at DNA hypomethylating but noncytotoxicdoses, was well tolerated and effective in increasing HbF andtotal Hb levels both in patients who had and had notresponded to prior HU therapy [81,82]. Therapy withbutyrates has been also reported [135,136].
ABBREVIATIONSAs for the possibility to predict in vivo response, the data
suggest a correlation between the in vitro results onerythroid precursor cells isolated from β-thalassemia patientsand the patients' response to treatment (e.g., [134]). If thiscorrelation is confirmed, it will be possible by testingcultures of cells derived from the patient's peripheral bloodto design the optimal treatment for each patient. This willprevent both expensive and potentially risky treatment frompatients who do no respond to treatment and suggest analternative treatment with other agents.
Hb = Hemoglobin
HbF = Fetal hemoglobin
HPFH = High persistence of fetal hemoglobin
EPO = Erythropoietin
GFP = Green fluorescence protein
RFP = Red fluorescence protein

Fetal Hemoglobin Inducers and -Thalassemia Current Medicinal Chemistry, 2007 Vol. 14, No. 2 211
HDAC = Histone deacethylase [44] Mitsuma, A.; Asano, H.; Kinoshita, T.; Murate, T.; Saito, H.;Stamatoyannopoulos, G.; Naoe, T. Biochim Biophys Acta, 2005,1727, 125.HU = Hydroxyurea
[45] Addya, S.; Keller, M.A.; Delgrosso, K.; Ponte, C.M.; Vadigepalli,R.; Gonye, G.E.; Surrey, S. Physiol. Genomics., 2004, 19, 117.HPLC = High performance liquid chromatography
[46] Kim, A.; Dean, A. Proc. Natl. Acad. Sci. USA , 2004,101, 7028.CML = Chronic myelogenous leukemia [47] Rutherford, T.; Clegg, J.B.; Higgs, D.R.; Jones, R.W.; Thompson,J.; Weatherall, D.J. Proc. Natl. Acad. Sci. USA , 1981, 78, 348.DNMT = DNA methyltransferases
[48] Rutherford, T.R.; Clegg, J.B.; Weatherall, D.J. Nature, 1979, 280,164.RBC = Red blood cell
[49] Bianchi, N.; Chiarabelli, C.; Borgatti, M.; Mischiati, C.; Fibach, E.;Gambari, R. Br. J. Haematol. , 2001, 113, 951.
REFERENCES [50] Bianchi, N.; Ongaro, F.; Chiarabelli, C.; Gualandi, L.; Mischiati,C.; Bergamini, P.; Gambari, R. Biochem. Pharmacol., 2000, 60,31.
[1] Steinberg, M.H.; Forget, B.G.; Higgs, D.R.; Nagel, R.L. Disordersof Hemoglobin: Genetics, Pathophysiology and ClinicalManagement, Cambridge University Press, Cambridge, UK, 2001.
[51] Bianchi, N.; Osti F.; Rutigliano, C.; Corradini, FG.; Borsetti, E.;Tomassetti, M.; Mischiati, C.; Feriotto, G.; Gambari, R. Br. J.Haematol., 1999, 104, 258.
[2] Thein, S.L. Br. J. Haematol., 2004, 124, 264. [52] Cortesi, R.; Gui, V.; Osti, F.; Nastruzzi, C.; Gambari, R. Eur. J.Haematol., 1998, 61, 295.[3] Old, J.M. Blood Rev ., 2003, 17, 43.
[4] Bank, A. Blood, 2006,107, 435. [53] Osti, F.; Corradini, F.G.; Hanau, S.; Matteuzzi, M.; Gambari, R.Haematologica , 1997, 82, 395.[5] Stamatoyannopoulos, G. Exp. Hematol., 2005, 33, 259.
[6] Thein, S.L. Int. J. Hematol., 2002, 76, 96. [54] Bianchi-Scarrà, G.; Fiorentini, P.; Gambari, R.; Nastruzzi, C.;Barbieri, R.; Sessarego, M.; Ravazzolo, R.; Garre, C. Exp.Hematol., 1989, 17, 859.
[7] Fucharoen, S.; Winichagoon, P. Int. J. Hematol., 2002, 76, 83.[8] Schrier, S.L. Curr. Opin. Hematol., 2002, 9, 123.[9] Wonke, B. Semin. Hematol ., 2001, 38, 350. [55] Gambari, R. Minerva Biotecnologica, 2003, 15, 123.[10] Globin Gene Server, http://globin.bx.psu.edu/. [56] Davis, M.G.; Kawai, Y.; Arinze, I.J. Biochem. J., 2000, 346, 455.[11] Tuzmen, S.; Schechter, A.N. Blood Rev ., 2001, 15, 19. [57] Gambari, R.; del Senno, L.; Barbieri, R.; Viola, L.; Tripodi, M.;
Raschella, G.; Fantoni, A. Cell Differ., 1984, 14, 87.[12] Orkin, S.H.; Goff, S.C. J. Biol. Chem., 1981, 256, 9782.[13] Hall, G.W.; Thein. S. Blood, 1994, 83, 2031. [58] Chiarabelli, C.; Bianchi, N.; Borgatti, M.; Prus, E.; Fibach, E.;
Gambari, R. Haematologica , 2003, 88, 826.[14] Naja, R.P.; Kaspar, H.; Shbaklo, H.; Chakar, N.; Makhoul, N.J.;Zalloua, P.A. Am. J. Hematol., 2004, 75, 220. [59] Fibach, E.; Manor, D.; Oppenheim, A.; Rachmilewitz, E.A. Blood,
1989, 73, 100.[15] Piyamongkol, W.; Harper, J.C.; Delhanty, J.D.; Wells, D. Prenat.Diagn., 2001, 21, 753. [60] Amoyal, I.; Goldfarb, A.; Fibach, E. Hemoglobin, 2003, 27, 77.
[16] Olivieri, N.F. Semin. Hematol ., 1996, 33, 24. [61] Fibach, E.; Premakala, P.; Rodgers, G.P.; Samid, D. Blood ,1993,82, 2203.[17] Rochette, J.; Craig, J.E.; Thein, S.L. Blood Rev ., 1994, 8, 213.
[18] Blau, C.A.; Stamatoyannopoulos, G. Curr. Opin. Hematol., 1994,1, 136.
[62] Fibach E. Minerva Biotecnologica, 2003, 15, 129.[63] Fibach, E.; Burke, L.P.; Schechter, A.N.; Noguchi, C.T.; Rodgers,
G.P. Blood, 2003, 81, 1630.[19] Witt, O.; Schmejkal, S.; Pekrun, A. Am. J. Hematol., 2000, 64,319. [64] Fibach, E.; Rachmilewitz, E.A. Exp. Hematol., 1993, 21, 184.
[20] Rees, D.C. J. Pediatr. Hematol. Oncol ., 2000, 22, 567. [65] Fibach, E.; Kollia, P.; Schechter, A.N.; Noguchi, C.T.; Rodgers,G.P. Blood, 1995, 85, 2967.[21] Weatherall, D.J. Harvey Lect., 1998, 94, 1.
[22] Forget, B.G. Ann. N.Y. Acad. Sci. USA, 1998, 850, 38. [66] Breda, L.; Rivella, S. Minerva Biotecnologica, 2003, 15, 107.[23] Bhardwaj, U.; McCabe, E.R. Mol. Diagn., 2005, 9, 151. [67] Skow, L.C.; Burkhart, B.A.; Johnson, F.M.; Popp, R.A.; Popp,
D.M.; Goldberg, S.Z. Cell, 1983, 34, 1043.[24] Liu, L.R.; Du, Z.W.; Zhao, H.L.; Liu, X.L.; Huang, X.D.; Shen, J.;Ju, L.M.; Fang, F.D.; Zhang, J.W. J. Biol. Chem., 2005, 280, 7452. [68] Curcio, M.J.; Kantoff, P.; Schafer, M.P.; Anderson, W.F.; Safer,
B. J. Biol. Chem., 1986, 261, 16126.[25] Garner, C.; Dew, T.K.; Sherwood, R.; Rees, D.; Thein, S.L. Br. J.Haematol., 2003, 123, 353. [69] Shehee, W.R.; Oliver, P.; Smithies, O. Proc. Natl. Acad. Sci. USA,
1993, 90, 3177.[26] Lal, A.; Vichinsky, E. Semin. Hematol ., 2004, 41, 17.[27] Rodgers, G.P.; Dover, G.J.; Uyesaka. N.; Noguchi, C.T.;
Schechter, A.N.; Nienhuis, A.W. N. Engl. J. Med ., 1993, 328, 73.[70] Yang, B.; Kirby, S.; Lewis, J.; Detloff, P.J.; Maeda, N.; Smithies,
O. Proc. Natl. Acad. Sci. USA , 1995, 92, 11608.[28] Rochette, J.; Craig, J.E.; Thein, S.L. Blood Reviews, 1994, 8, 213. [71] Ciavatta, D.J.; Ryan, T.M.; Farmer, S.C.; Townes, T.M. Proc. Natl.
Acad. Sci USA, 1995, 92, 9259.[29] Rodgers, G.P.; Rachmilewitz, E.A. Br. J. Haematol., 1995, 91,263. [72] Rivella, S.; May, C.; Chadburn, A.; Riviere, I.; Sadelain, M. Blood,
2003, 101, 2932.[30] Steinberg, M.H.; Lu, L.Z.; Barton, F.B.; Terrin, M.L.; Charache,S., Dover, G.J. Blood, 1997, 89,1078. [73] Enver, T.; Ebens, A.J.; Forrester, W.C.; Stamatoyannopoulos, G.
Proc. Natl. Acad. Sci. USA , 1989, 86, 7033.[31] Olivieri, N.F.; Rees, D.C.; Ginder, G.D.; Thein, S.L., Waye, J.S.;Chang, L.; Brittenham, G.M.; Weatherall, D.J. Ann. N.Y. Acad.Sci. USA, 1998, 850, 100.
[74] Vadolas, J.; Nefedov, M.; Wardan, H.; Mansooriderakshan, S.;Voullaire, L.; Jamsai, D.; Williamson, R.; Ioannou, P.A. J. Biol.Chem., 2006, 281, 7399.[32] Swank, R.A.; Stomatoyannopoulos, G. Curr. Opin. Genet. Dev.,
1998, 8, 366. [75] Jamsai, D.; Zaibak, F.; Vadolas, J.; Voullaire, L.; Fowler, K.J.;Gazeas, S.; Peters, H.; Fucharoen, S.; Williamson, R.; Ioannou,P.A. Genomics, 2006, 88, 309.
[33] Cao, H. Hematology , 2004, 9, 223.[34] Lo, L.; Singer, S.T. Pediatr. Clin. North Am., 2002, 49, 1165.[35] Atweh, G.F.; Loukopoulos, D. Semin. Hematol ., 2001, 38, 367. [76] Jamsai, D.; Williamson, R.; Ioannou, P.A. Genomics, 2005, 85,
453.[36] Olivieri, N.F,; Weatherall, D.J. Hum. Mol. Genet., 1998, 7, 1655.[37] Skarpidi, E.; Vassilopoulos, G.; Li, Q.; Stamatoyannopoulos, G.
Blood, 2000, 96, 321.[77] Oppenheim, A.; Katzir, Y.; Fibach, E.; Goldfarb, A.;
Rachmilewitz, E. Blood, 1985, 66, 1202.[38] Vadolas, J.; Wardan, H.; Orford, M.; Williamson, R.; Ioannou,
P.A. Hum. Mol. Genet., 2004, 13, 223.[78] Stamatoyannopoulos, G. Exp. Hematol., 2005, 33, 259.[79] Poncz, M.; Sutton, M.; Delgrosso, K.; Schwartz, E.; Surrey, S.
Nucleic Acids Res., 1987, 15, 5169.[39] Vadolas, J.; Wardan, H.; Orford, M.; Voullaire, L.; Zaibak, F.;Williamson, R.; Ioannou, P.A. Blood, 2002, 100, 4209. [80] Saunthararajah, Y.; Hillery, C.A.; Lavelle, D.; Molokie, R.; Dorn,
L.; Bressler, L.; Gavazova, S.; Chen, Y.H.; Hoffman, R.;DeSimone, J. Blood, 2003, 102, 3865.
[40] Lozzio, C.B.; Lozzio, B.B. Blood, 1975, 45, 321.[41] Martin, P.; Papayannopoulou, T. Science, 1982, 216, 1233.[42] Vaisman, B.; Meyron-Holtz, E.G.; Fibach, E.; Krichevsky, A.M.;
Konijn, A.M. Br. J. Haematol ., 2000, 110, 394.[81] Fathallah, H.; Atweh, G.F. Blood Rev ., 2006, 20, 227.[82] DeSimone, J. Semin. Hematol ., 2004, 41, 1.
[43] Zhao, Q.; Zhou, W.; Rank, G.; Sutton, R.; Wang, X.; Cumming, H.;Cerruti, L.; Cunningham, J.M.; Jane, S.M. Blood, 2006, 107, 2138.
[83] McCaffrey, P.G.; Newsome, D.A.; Fibach, E.; Yoshida, M.; Su,M.S. Blood, 1997, 90, 2075.

212 Current Medicinal Chemistry, 2007, Vol. 14, No. 2 Gambari and Fibach
[84] Pace, B.S.; Qian, X.H.; Sangerman, J.; Ofori-Acquah, S.F.; Baliga,B.S.; Han, J.; Critz, S.D. Exp. Hematol., 2003, 31, 1089.
[111] Mischiati, C.; Sereni, A.; Lampronti, I.; Bianchi, N.; Borgatti, M.;Prus, E.; Fibach, E.; Gambari, R. Br. J. Haematol ., 2004, 126, 612.
[85] Haley, J.D.; Smith, D.E.; Schwedes, J.; Brennan, R.; Pearce, C.;Moore, C.; Wang, F.; Petti, F.; Grosveld, F.; Jane, S.M.; Noguchi,C.T.; Schechter, A.N. Biochem. Pharmacol., 2003, 66, 1755.
[112] Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Kidney Internat.,2001, 59, 3.
[113] Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.;Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Eur. J.Haematol., 2006, 77, 437.
[86] Cao, H.; Stamatoyannopoulos, G.; Jung, M. Blood, 2004, 103, 701.[87] Skarpidi, E.; Cao, H.; Heltweg, B.; White, B.F.; Marhenke, R.L.;
Jung, M.; Stamatoyannopoulos, G. Exp. Hematol., 2003, 31, 197. [114] Chaidos, A.; Makis, A.; Hatzimichael, E.; Tsiara, S.; Gouva, M.;Tzouvara, E.; Bourantas, K.L. Acta Haematol ., 2004, 111, 189.[88] Witt, O.; Monkemeyer, S.; Ronndahl, G.; Erdlenbruch, B.;
Reinhardt, D.; Kanbach, K.; Pekrun, A. Blood, 2003, 101, 2001. [115] Kohli-Kumar, M.; Marandi, H.; Keller, M.A.; Guertin, K.;Hvizdala, E. J. Pediatr. Hematol. Oncol., 2002, 24, 777.[89] Gore, S.D.; Weng, L.J.; Figg, W.D.; Zhai, S.; Donehower, R.C.;
Dover, G.; Grever, M.R.; Griffin, C.; Grochow, L.B.; Hawkins,A.; Burks, K.; Zabelena, Y.; Miller, C.B. Clin. Cancer Res., 2002,8, 963.
[116] Suwanmanee, T.; Sierakowska, H.; Fucharoen, S.; Kole, R. Mol.Ther., 2002, 6, 718.
[117] Mercatante, D.R.; Sazani, P.; Kole, R. Curr. Cancer Drug Targets ,2001, 1, 211.[90] Johnson, J.; Hunter, R.; McElveen, R.; Qian, X.H.; Baliga, B.S.;
Pace, B.S. Cell Mol. Biol. (Noisy-le-grand) , 2005, 51, 229. [118] Xu, X.S.; Glazer, P.M.; Wang, G. Gene, 2000, 242, 219.[91] Cao, H.; Jung, M.; Stamatoyannopoulos, G. Exp. Hematol., 2005,
33, 1443.[119] Bianchi, N.; Feriotto, G.; Gambari, C.; Mischiati, C.
PCT/EP01/02804, Synthetic oligonucleotides as inducers oferythroid differentiation, March 13, 2001.[92] Cao, H. Hematology , 2004, 9, 223.
[93] Sauvage, C.; Rouyer-Fessard, P.; Beuzard, Y. Br. J. Haematol.,1993, 84, 492.
[120] Wang, G.; Jing, K.; Balczon, R.; Xu, X. J. Mol. Biol., 2001, 313,933.
[94] Wang, M.; Tang, D.C.; Liu, W.; Chin, K.; Zhu, J.G.; Fibach, E.;Rodgers, G.P. Br. J. Haematol ., 2002, 119, 1098.
[121] Wang, G.; Xu, X.S. Cell Res., 2004, 14, 111.[122] Wang, G.; Xu, X.; Pace, B.; Dean, D.A.; Glazer, P.M.; Chan, P.;
Goodman, S.R.; Shokolenko, I. Nucleic Acids Res., 1999, 27, 2806.[95] Loukopoulos, D.; Voskaridou, E.; Stamoulakatou, A.;Papassotiriou, Y.; Kalotychou, V.; Loutradi, A.; Cozma, G.;Tsiarta, H.; Pavlides, N. Ann. N.Y. Acad. Sci., 1998, 850, 120.
[123] Hoppe, C.; Vichinsky, E.; Lewis, B.; Foote, D.; Styles, L. Am. J.Hematol., 1999, 62, 221.
[96] Choudhry, V.P.; Lal, A.; Pati, H.P.; Arya, L.S. Indian. J. Pediatr.,1997, 64, 395.
[124] de Paula, E.V.; Lima, C.S.; Arruda, V.R.; Alberto, F.L.; Saad, S.T.;Costa, F.F. Eur. J. Haematol ., 2003, 70, 151.
[97] Rigano, P.; Manfre, L.; La Galla, R.; Renda, D.; Renda, M.C.;Calabrese, A.; Calzolari, R.; Maggio, A. Hemoglobin, 1997, 21,219.
[125] Bradai, M.; Abad, M.T.; Pissard, S.; Lamraoui, F.; Skopinski, L.;de Montalembert, M. Blood, 2003, 102, 1529.
[126] Alebouyeh, M.; Moussavi, F.; Haddad-Deylami, H.; Vossough, P.Ann. Hematol., 2004, 83, 430.[98] Fucharoen, S.; Siritanaratkul, N.; Winichagoon, P.; Chowthaworn,
J.; Siriboon, W.; Muangsup, W.; Chaicharoen, S.; Poolsup, N.;Chindavijak, B.; Pootrakul, P.; Piankijagum, A.; Schechter, A.N.;Rodgers, G.P. Blood, 1996, 87, 887.
[127] Dixit, A.; Chatterjee, T.C.; Mishra, P.; Choudhry, D.R.;Mahapatra, M.; Tyagi, S.; Kabra, M.; Saxena, R.; Choudhry, V.P.Ann. Hematol., 2005, 84, 441.
[99] de Franceschi, L.; Rouyer-Fessard, P.; Alper, S.L.; Jouault, H.;Brugnara, C.; Beuzard, Y. Blood, 1996, 87, 1188.
[128] Karimi, M.; Darzi, H.; Yavarian, M. J. Pediatr. Hematol. Oncol.,2005, 27, 380.
[100] Letvin, N.L.; Linch, D.C.; Beardsley, G.P.; McIntyre, K.W.;Nathan, D.G. New Engl. J. Med ., 1984, 310, 869.
[129] Singer, S.T.; Kuypers, F.A.; Olivieri, N.F.; Weatherall, D.J.;Mignacca, R.; Coates, T.D.; Davies, S.; Sweeters, N.; Vichinsky,E.P. Br. J. Haematol ., 2005, 131, 378.[101] Loukopoulos, D.; Voskaridou, E.; Kalotychou, V.; Schina, M.;
Loutradi, A.; Theodoropoulos, I. Blood Cells Mol. Dis., 2000, 26,453.
[130] Watanapokasin, R.; Sanmund, D.; Winichagoon, P.; Muta, K.;Fucharoen, S. Ann. Hematol., 2006, 85, 164.
[102] Mancuso, A.; Maggio, A.; Renda, D.; Di Marzo, R.; Rigano, P. Br.J. Haematol ., 2006, 133, 105.
[131] Marianna, P.; Kollia, P.; Akel, S.; Papassotiriou, Y.;Stamoulakatou, A.; Loukopoulos, D. Haematologica , 2001, 86,700.[103] Panigrahi, I.; Dixit, A.; Arora, S.; Kabra, M.; Mahapatra, M.;
Choudhry, V.P.; Saxena, R. Hematology , 2005, 10, 61. [132] Singer, S.T.; Kuypers, F.A.; Olivieri, N.F.; Weatherall, D.J.;Mignacca, R.; Coates, T.D.; Davies, S.; Sweeters, N.; Vichinsky,E.P. Ann. N.Y, Acad. Sci., 2005, 1054, 250.
[104] Zargari, O.; Kimyai-Asadi, A.; Jafroodi, M. Pediatr Dermatol.,2004, 21, 633.
[105] Yavarian, M.; Karimi, M.; Bakker, E.; Harteveld, C.L.; Giordano,P.C. Haematologica , 2004, 89, 1172.
[133] Kohli-Kumar, M.; Marandi, H.; Keller, M.A.; Guertin, K.;Hvizdala, E. J. Pediatr. Hematol. Oncol., 2002, 24, 777.
[106] Gambari, R.; Fibach, E. Minerva Biotecnologica, 2003,15, 145. [134] Watanapokasin, Y.; Chuncharunee, S.; Sanmund, D.; Kongnium,W.; Winichagoon, P.; Rodgers, G.P.; Fucharoen, S. Exp. Hematol.,2005, 33, 1486.
[107] Lampronti, I.; Bianchi, N.; Borgatti, M.; Fibach, E.; Prus, E.;Gambari, R. Eur. J. Haematol ., 2003, 71, 189.
[108] Fibach, E.; Bianchi, N.; Borgatti, M.; Prus, E.; Gambari, R. Blood,2003, 102, 1276.
[135] Perrine, S.P.; Ginder, G.D.; Faller, D.V.; Dover, G.H.; Ikuta, T.;Witkowska, H.E.; Cai, S.P.; Vichinsky, E.P.; Olivieri, N.F. NewEngl. J. Med., 1993, 328, 81.[109] Sehgal, S.N. Transplant. Proc., 2003, 35, 7.
[110] Gummert, J.F.; Ikonen, T.; Morris, R.E. J. Am. Soc. Nephrol.,1999, 10, 1366.
[136] Perrine, S.P.; Dover, G.H.; Daftari, P.; Walsh, C.T.; Jin, Y.; Mays,A.; Faller, D.V. Br. J. Haematol., 1994, 88, 555.