conjugated hyperbilirubinemia in children david brumbaugh ... · conjugated hyperbilirubinemia in...
TRANSCRIPT

DOI: 10.1542/pir.33-7-2912012;33;291Pediatrics in Review
David Brumbaugh and Cara MackConjugated Hyperbilirubinemia in Children
http://pedsinreview.aappublications.org/content/33/7/291located on the World Wide Web at:
The online version of this article, along with updated information and services, is
Pediatrics. All rights reserved. Print ISSN: 0191-9601. Boulevard, Elk Grove Village, Illinois, 60007. Copyright © 2012 by the American Academy of published, and trademarked by the American Academy of Pediatrics, 141 Northwest Pointpublication, it has been published continuously since 1979. Pediatrics in Review is owned, Pediatrics in Review is the official journal of the American Academy of Pediatrics. A monthly
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

Conjugated Hyperbilirubinemia in ChildrenDavid Brumbaugh, MD,*
Cara Mack, MD*
Author Disclosure
Drs Brumbaugh and
Mack have disclosed
no financial
relationships relevant
to this article. This
commentary does not
contain a discussion of
an unapproved/
investigative use of
a commercial product/
device.
Education Gaps
1. Awareness of telltale signs and performance of appropriate diagnostic testing can
help clinicians identify neonatal cholestasis in time to ameliorate its potentially
catastrophic outcomes.
2. The success of the Kasai procedure to restore bile flow is directly related to patient age:
at less than 60 days after birth, two-thirds of patients benefit from the procedure;
however, at 90 days after birth, chances for bile drainage diminish markedly.
Objectives After completing this article, readers should be able to:
1. Understand the metabolism of bilirubin, the differences between conjugated and
unconjugated bilirubin, and the relationship of conjugated hyperbilirubinemia to
cholestasis.
2. Delineate the causes of cholestasis in the newborn and know how to evaluate the
cholestatic neonate.
3. Manage the infant who has prolonged cholestasis.
4. Understand the causes of conjugated hyperbilirubinemia in the older child and
adolescent and know how to assess children who have conjugated hyperbilirubinemia.
IntroductionCentral to human digestive health are both the production of bile by hepatocytes and chol-angiocytes in the liver and the excretion of bile through the biliary tree. By volume, conju-gated bilirubin is a relatively small component of bile, the yellowish-green liquid that alsocontains cholesterol, phospholipids, organic anions, metabolized drugs, xenobiotics, and bileacids. In most cases, the elevation of serum-conjugated bilirubin is a biochemical manifesta-tion of cholestasis, which is the pathologic reduction in bile formation or flow.
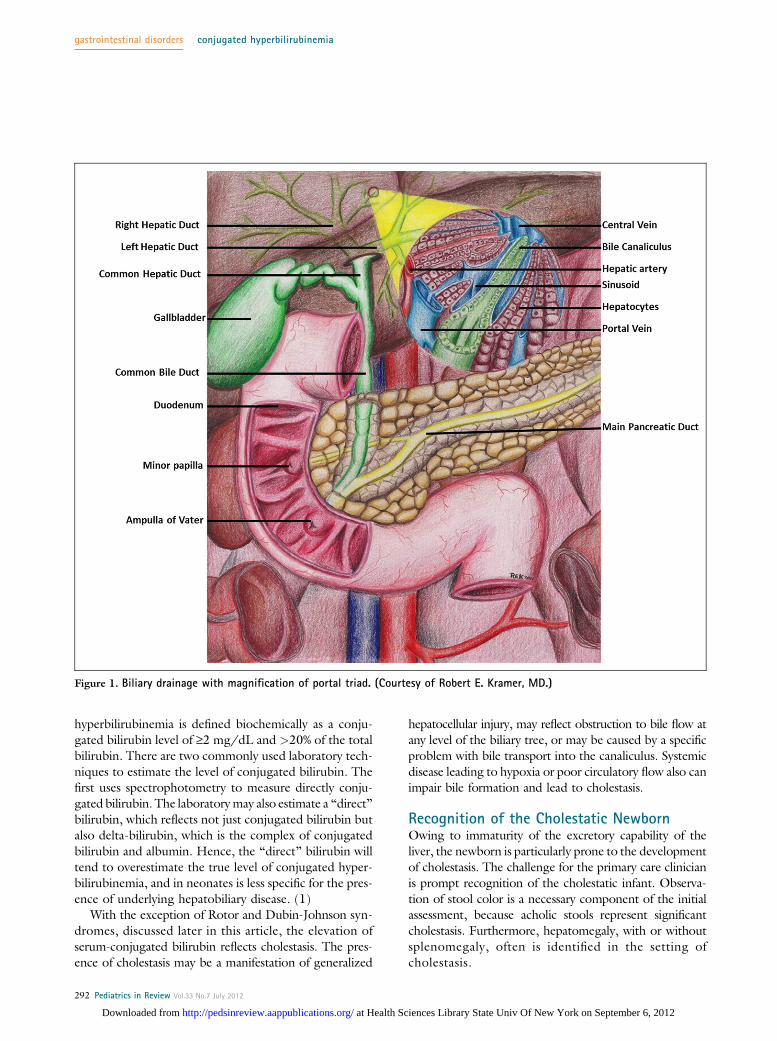
Complex mechanisms exist for the transport of bile com-ponents from serum into hepatocytes across the basolateralcell surface, for the trafficking of bile components throughthe hepatocyte, and finally for movement of these bile com-ponents across the apical cell surface into the bile canaliculus,which is the smallest branch of the biliary tree. From the bilecanaliculus, bile then flows into the extrahepatic biliary tree,including the common bile duct, before entering the duode-num at the ampulla of Vater (Fig 1). Isolated gene defectsin proteins responsible for trafficking bile components canlead to cholestatic diseases.
DiagnosisUnconjugated bilirubin is the product of heme breakdown,and this molecule, poorly soluble in water, is carried in thecirculation principally as a water-soluble complex joined withalbumin. Unconjugated bilirubin is then taken up into hep-atocytes, where a glucuronic acid moiety is added, ren-dering the conjugated bilirubin water soluble. Conjugated
Abbreviations
AIH: autoimmune hepatitisALT: alanine aminotransferaseAST: aspartate aminotransferaseA1AT: alpha-1 antitrypsinBA: biliary atresiaBRIC: benign recurrent intrahepatic cholestasisBSEP: bile salt excretory proteinCDC: choledochal cystERCP: endoscopic retrograde cholangiopancreatographyGGT: gamma glutamyltransferaseMCT: medium chain triglyceridesMRCP: magnetic resonance cholangiopancreatographyPFIC: progressive familial intrahepatic cholestasisPN: parenteral nutritionPSC: primary sclerosing cholangitis
*Digestive Health Institute, Children’s Hospital of Colorado, University of Colorado Anschutz Medical Campus, Denver, CO.
Article gastrointestinal disorders
Pediatrics in Review Vol.33 No.7 July 2012 291
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from
hyperbilirubinemia is defined biochemically as a conju-gated bilirubin level of ‡2 mg/dL and >20% of the totalbilirubin. There are two commonly used laboratory tech-niques to estimate the level of conjugated bilirubin. Thefirst uses spectrophotometry to measure directly conju-gated bilirubin. The laboratorymay also estimate a “direct”bilirubin, which reflects not just conjugated bilirubin butalso delta-bilirubin, which is the complex of conjugatedbilirubin and albumin. Hence, the “direct” bilirubin willtend to overestimate the true level of conjugated hyper-bilirubinemia, and in neonates is less specific for the pres-ence of underlying hepatobiliary disease. (1)
With the exception of Rotor and Dubin-Johnson syn-dromes, discussed later in this article, the elevation ofserum-conjugated bilirubin reflects cholestasis. The pres-ence of cholestasis may be a manifestation of generalized
hepatocellular injury, may reflect obstruction to bile flow atany level of the biliary tree, or may be caused by a specificproblem with bile transport into the canaliculus. Systemicdisease leading to hypoxia or poor circulatory flow also canimpair bile formation and lead to cholestasis.
Recognition of the Cholestatic NewbornOwing to immaturity of the excretory capability of theliver, the newborn is particularly prone to the developmentof cholestasis. The challenge for the primary care clinicianis prompt recognition of the cholestatic infant. Observa-tion of stool color is a necessary component of the initialassessment, because acholic stools represent significantcholestasis. Furthermore, hepatomegaly, with or withoutsplenomegaly, often is identified in the setting ofcholestasis.
Figure 1. Biliary drainage with magnification of portal triad. (Courtesy of Robert E. Kramer, MD.)
gastrointestinal disorders conjugated hyperbilirubinemia
292 Pediatrics in Review Vol.33 No.7 July 2012
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

In the early neonatal period, jaundice caused by phys-iologic unconjugated hyperbilirubinemia or human milkjaundice is impossible to distinguish from jaundice causedby cholestasis based on physical appearance alone. Indeed,physiologic unconjugated hyperbilirubinemia and chole-stasis can coexist in early infancy. A critical time pointfor establishing the diagnosis of cholestasis is at the 2-weekwell-child visit. Persistent jaundice at 2 weeks after birthshould alert the care provider to the possibility of cholesta-sis. The diagnosis is made by obtaining a conjugated biliru-bin level or “direct” bilirubin fraction, whichever is availablelocally. If the infant appears well otherwise, a second optionis to see the infant back in 1 week. If the jaundice persists at3 weeks after birth, laboratory evaluation is mandatory.
Expedient recognition of cholestasis is of great impor-tance in the neonatal period because early interventionmay improve outcome. For instance, in the case ofhypopituitarism, in which jaundice may be the presentingsymptom, early diagnosis may prevent catastrophic hypo-glycemia. Antimicrobial therapy in the cholestatic infantwho has an occult Gram-negative urinary tract infectionmay prevent bacteremia and sepsis. Avoidance of extendedfasting in an infant born with an underlying metabolic dis-order could prevent severe episodes of hypoglycemia andacidosis. Diagnosis of biliary atresia (BA) before 60 days ofage leads to earlier surgical intervention and improvedlong-term outcome.
Initial Approach to the Cholestatic InfantIn addition to conjugated hyperbilirubinemia, the serumaspartate aminotransferase (AST) and alanine aminotrans-ferase (ALT) levels typically are elevated to a variable de-gree, but are not specific for the cause of cholestasis. Thegamma glutamyltransferase (GGT) level usually is elevatedin cholestasis. Normal or low GGT levels in the setting ofcholestasis have been associated with bile acid synthesis de-fects, some cases of hypopituitarism, and progressive famil-ial intrahepatic cholestasis types 1 and 2 (PFIC1, PFIC2).
Abnormalities in hepatic synthetic function, such as aprolonged prothrombin time, elevated ammonia level, lowserum albumin concentration, or hypoglycemia, suggest ad-vanced hepatic injury and should prompt immediate referralto a pediatric tertiary care facility. A urinalysis and urine cul-ture will assess for urinary tract infection, and the presenceof reducing substances in the urine suggests galactosemia.
Newborn screens should be reviewed for the diagnosisof cystic fibrosis, hypothyroidism, galactosemia, and otherinborn errors of metabolism, all of which can present withneonatal cholestasis. Because of the broad differential diag-nosis for neonatal cholestasis, ultimately the diagnosis andtreatment of the cholestatic infant should be accomplished
in a center with expertise in pediatric gastroenterology andhepatology. Recent advances in molecular diagnostic tech-niques have led to targeted approaches to the identificationof genetic mutations that may cause neonatal cholestasis.A chip-based resequencingmethod allowed for identificationof suspected causative genemutations in 27% of subjects ina cohort of infants who have unexplained cholestasis. (2)
Differential Diagnosis of Neonatal CholestasisThe following should be considered in the differential di-agnosis of neonatal cholestasis (Table).
Extrahepatic Biliary ObstructionBA is the most common cause of neonatal cholestasis, ac-counting for w40% to 50% of all cases. (3) There are two
Table. Differential Diagnosis ofNeonatal Cholestasis
Congenital infection• Cytomegalovirus• Toxoplasmosis• Rubella• Herpes simplex virus• Syphilis• HIV
Acquired infection• Urinary tract infection• Sepsis
Metabolic• Alpha-1 antitrypsin deficiency• Cystic fibrosis• Galactosemia• Tyrosinemia• Defects in bile acid synthesis• Inborn errors of carbohydrate, fat, proteinmetabolism
Obstructive• Biliary atresia• Choledochal cyst• Inspissated bile syndrome• Spontaneous perforation of bile duct
Cholestatic syndromes• Alagille syndrome• Progressive familial intrahepatic cholestasis
Endocrinopathy• Hypothyroidism• Hypopituitarism
Drug or toxin induced• Parenteral nutrition• Drugs
Systemic disorder• Shock• Congenital heart disease/heart failure
gastrointestinal disorders conjugated hyperbilirubinemia
Pediatrics in Review Vol.33 No.7 July 2012 293
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

forms of BA. The embryonic form of BA, which is associ-ated with other congenital anomalies such as heterotaxysyndrome and polysplenia, accounts for w15% to 20%of BA.
The acquired form of BA is far more common (w85%);the etiology of this disease is unclear. The pathophysiologyof acquired BA is that of a brisk inflammatory response in-volving both the intra- and extrahepatic bile ducts. Theducts are destroyed gradually and replaced with fibrous scartissue. The lumen of the bile duct is eventually obliterated,and normal bile flow is impaired, leading to cholestasis.
Infants who have acquired BA typically are asymptom-atic at birth and develop jaundice in the first weeks afterbirth. Typically, they feed well and thrive. As the bile flowdiminishes, the stool color loses its normal pigmentationand becomes acholic, or clay-colored. The finding ofacholic stools in the setting of a jaundiced newborn shouldprompt expedient evaluation for BA. Light-colored stoolsmay not be appreciated by the inexperienced parent,and the stool should be examined by the primary care pro-vider to assess pigmentation.
In Taiwan, which has one of the highest incidences ofBA in the world, a universal screening program providesparents with a stool color card on discharge from the new-born nursery (Fig 2). At 1month of age, the parents returnthe stool card to their provider after marking the picture ofthe stool color that most closely resembles the infant’s stool.The universal screening program has led to improvement ofearly detection of infants who have BA, which has resultedin dramatic improvement in surgical outcomes. (4)
Evaluation for BA includes abdominal ultrasonographyto rule out other anatomic abnormalities of the commonbile duct, such as choledochal cyst (CDC), and to identifyanomalies associated with the embryonic form of BA.A liver biopsy often is performed, and histopathologicfindings consistent with BA include bile ductular prolifer-ation, portal tract inflammation and fibrosis, and bile plugswithin the lumen of bile ducts. The gold standard in con-firming the diagnosis of BA is the intraoperative cholangio-gram, which shows obstruction of flow within a segmentor the entirety of the extrahepatic bile duct.
A Kasai portoenterostomy is then performed in an at-tempt to reestablish bile flow. This operation entails exci-sion of the fibrous bile duct followed by anastomosis ofa loop of jejunum to the base of the liver in a Roux-en-Yfashion. The success of this surgery, which is the restorationof bile flow to intestine, is directly related to the age of thepatient. Early Kasai procedure, defined as <60 days afterbirth, leads to initial biliary flow in approximately two-thirdsof patients; if performed after 90 days after birth the chanceof bile drainage is markedly diminished.
Even after restoration of bile flow with the Kasai oper-ation, BA continues to be a progressive disease in most pa-tients, with ongoing inflammatory injury to the intrahepaticbile ducts; 70% to 80% of patients who have BAwill developfibrosis, portal hypertension, and cirrhosis. BA continues tobe the most common reason for liver transplantation in pe-diatric patients. A recent nationwide study reported thatw50% of patients who have BA will require liver transplan-tation in the first 2 years after birth, with an overall incidenceof liver transplantation ofw80% in childhood. The progres-sive nature of this disease has led investigators to define BAas a chronic inflammatory disorder of the biliary tract.
Other causes of extrahepatic biliary obstruction includeCDC and spontaneous perforation of the common bileduct. These anatomic abnormalities can be diagnosed withabdominal ultrasonography. CDC may present with jaun-dice, acholic stools, and a palpable mass. Spontaneousperforation of the bile duct is a rare entity that usually oc-curs in the neonatal period. Infants present with jaundice,
Figure 2. English version of Infant Stool Color Card. Distributeduniversally to parents of newborns in Taiwan. (Reprinted withpermission from Chen SM, Chang MH, Du JC, et al. Screeningfor biliary atresia by infant stool color card in Taiwan.Pediatrics. 2006;117(4):1147–1154.)
gastrointestinal disorders conjugated hyperbilirubinemia
294 Pediatrics in Review Vol.33 No.7 July 2012
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

poor weight gain, ascites, acholic stools, and vomiting.Ultrasonography typically reveals ascites and fluid aroundthe gallbladder. Bile-stained ascitic fluid is a hallmarkfinding. The treatment of both of these conditions in-volves surgical intervention.
Stagnant flow of bile leading to cholestasis is seen oftenin the setting of intestinal disease and parenteral nutrition(PN) in the neonate. Precipitation of cholesterol and cal-cium salts within bile can result in the formation of sludge.Bile sludge can be detected by ultrasonography. Whensludge builds up and leads to biliary obstruction and the de-velopment of cholestasis, the patient is said to have inspis-sated bile syndrome. Inspissated bile can be managedconservatively with ursodeoxycholic acid, a bile salt that actsas a choleretic agent to promote bile flow. Because inspis-sated bile syndrome can mimic biliary atresia, the diagnosissometimes ismade at the time of intrahepatic cholangiogram,and saline flushes of the biliary tree by the surgeon canprovide the definitive therapy. The use of third-generationcephalosporin antibiotics, in particular ceftriaxone, has beenassociated with the formation of bile sludge in newborns.
InfectionsNeonatal cytomegalovirus infec-tion, vertically acquired from themother, is the most common con-genital infectious cause of neonatalcholestasis. Any of the conditionsformerly identified as the “TORCH”
family of infections (toxoplasmosis,rubella, cytomegalovirus, herpesvi-rus, syphilis) can lead to a similarpattern of cholestasis and growthrestriction. Acquired infections afterbirth can lead to cholestasis, inparticular Gram-negative infectionsassociated with urinary tract infec-tions and sepsis, because hepatic bileflow is very sensitive to circulatingendotoxins.
Genetic DisordersAlagille syndrome is an autosomaldominant mutation of the Jagged 1gene on chromosome 20. There isvariable penetrance of this mutation,which can lead to abnormalities ofthe liver (cholestasis), heart (periph-eral pulmonary stenosis), skeletal sys-tem (butterfly vertebrae), kidneys,and eyes (posterior embryotoxin).
The characteristic finding on liver histology is paucity ofbile ducts. The clinical course of liver disease in infantswho have alagille syndrome is highly variable, with somechildren experiencing a gradual improvement in cholestasisin childhood, whereas others progress to cirrhosis, requir-ing liver transplantation in childhood. Infants born withTrisomy 21 also are at increased risk for development ofa paucity of intrahepatic bile ducts; however, this situationusually is very mild, with resolution of cholestasis in in-fancy. Cystic fibrosis is another genetic disorder that canpresent with neonatal cholestasis and often is associatedwith meconium plug syndrome. Early diagnosis is aidedby the availability in all 50 states of newborn screeningfor cystic fibrosis by measurement of immunoreactivetrypsinogen levels.
Along the apical surface of the hepatocyte, there arespecific transporter proteins that are responsible for trafficof bile components into the bile canaliculus (Fig 3). (5)Defects in these proteins are associated with cholestaticdisease. For instance, a mutation in the gene coding forbile salt excretory protein (BSEP) interferes with bile salttrafficking into the canaliculus, leading to reduced bileflow and the toxic accumulation of hydrophobic bile acids
Figure 3. Canalicular membrane surface proteins, their substrates, and knownassociations with pediatric disease. (For a recent review, see Wagner M, Zollner G,Trauner M. New molecular insights into the mechanisms of cholestasis. J Hepatol.2009;51:565–580.)
gastrointestinal disorders conjugated hyperbilirubinemia
Pediatrics in Review Vol.33 No.7 July 2012 295
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

within hepatocytes. This mutation produces the clinicalphenotype of cholestasis and pruritis in the first year afterbirth, a condition known as PFIC2.
A defect in the gene coding for FIC1, another cana-licular surface protein, produces the clinical phenotypePFIC1, which, in addition to cholestasis, can present withdiarrhea and growth failure. Pruritis, a dominant clinicalfeature of both PFIC1 and PFIC2, typically is not prob-lematic until after 6 months of age.
PFIC3 is a syndrome caused by a defect in the genecoding for the transporter MDR3, which is responsiblefor phosphatidylcholine secretion into the bile canaliculus.The onset of cholestasis is variable in PFIC3 but typicallyoccurs later than in PFIC1 and PFIC2. In contrast toPFIC1 and PFIC2, which are featured by a GGT levelin the low or normal range, the GGT in PFIC3 is elevated.
Metabolic DisordersA range of metabolic diseases can present initially as cho-lestasis in the newborn period and are associated withgene mutations in most cases (thus, these diseases couldalso fall under the category of genetic disorders). Persis-tent jaundice in the newborn period is one of the earliestpotential clinical manifestations of alpha-1 antitrypsin(A1AT) deficiency, a defect in the “ATZ” molecule thatresults in abnormal accumulation of A1AT in the endo-plasmic reticulum of hepatocytes. The abnormal reten-tion of A1AT within the hepatocyte leads to abnormalbile formation and secretion.
Inborn errors of metabolism, which include disordersof fatty acid oxidation, tyrosinemia, and galactosemia,among others, can present in the neonatal period with aspectrum of liver disease that includes cholestasis. Finally,bile acid synthesis defects often present with neonatal cho-lestasis. As the production of bile acids from cholesteroland their subsequent export into the canaliculus are therate-limiting steps in bile flow, defects in a number of en-zymatic steps within this pathway result in abnormal bileacid synthesis and cholestasis. Bile acid synthesis defectsgenerally can be treated effectively by oral bile acidsupplementation.
EndocrinopathiesCongenital endocrinopathies must be considered in thedifferential diagnosis of neonatal conjugated hyperbi-lirubinemia. Neonatal cholestasis is a well-recognizedmanifestation of congenital hypothyroidism. Congenitalpanhypopituitarism is manifested by deficiencies in cortisol,growth hormone, and thyroid hormone. These hormoneshave been shown to promote bile formation or secretionand chronic deficiencies lead to cholestasis. Other clinical
findings associated with panhypopituitarism include opticnerve hypoplasia, septo-optic dysplasia, and, in male pa-tients, microphallus. In contrast to most of the cholestaticdiseases, which lead to an elevation in serum GGT concen-trations, the GGT level in hypopituitarism typically is nor-mal. Hypoglycemia can complicate prolonged fasts in theseinfants.
Drug HepatotoxicityDependent on the maturity of the neonate, there is vari-ability in the activity of members of the drug-metabolizingcytochrome P450 family in the newborn period. Thus, thenewborn infant may be particularly susceptible to drug-induced hepatotoxicity, which can take a predominantlycholestatic form. The most common drug-induced liverinjury is caused by PN, used in the newborn period for avariety of indications. The liver injury caused by PN ismultifactorial, but in particular the phytosterol present insoy-based lipid formulations is a known antagonist of thenuclear receptor FXR, which is a regulator of the BSEP,an essential protein involved in bile acid transport. (6)
Initial experience using fish oil–based sources of intra-venous lipids has been promising, but larger clinical trialsin neonates have not yet been performed. There are casereports of neonatal cholestasis associated withmaternal useof prescribed medications (carbamazepine) and illicitdrugs (methamphetamine). Postnatal infant exposureto antimicrobial agents, particularly ceftriaxone, flucona-zole, and micafungin, has been associated with the devel-opment of cholestasis.
Management of the Infant Who HasProlonged CholestasisFailure to thrive is found commonly in infants who havechronic cholestatic conditions. The cause of poor weightgain is multifactorial. Reduced bile flow to the intestine re-sults in poor solubilization of dietary fats in mixed micelles,leading to fat malabsorption and steatorrhea. Medium-chain triglycerides (MCT) do not require bile for intestinalabsorption and thus are preferred in infants who have cho-lestasis. Several commercially available formulas have highlevels ofMCT as their fat source, and there are supplementscontaining exclusivelyMCT that can be used for delivery ofadditional calories.
Infants who have chronic liver disease may have an in-creased baseline caloric need coupled with demands for ad-ditional calories for catch-up growth. Unfortunately, manyof these infants are anorexic, justifying the use of nasogastricfeeds for caloric delivery. Fat-soluble vitamin deficienciesare pervasive in infants who have chronic cholestasis andshould be managed aggressively with frequent monitoring
gastrointestinal disorders conjugated hyperbilirubinemia
296 Pediatrics in Review Vol.33 No.7 July 2012
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

of serum vitamin levels and use of oral fat-soluble vitaminsupplements.
Ursodeoxycholic acid is a hydrophilic bile acid that isuseful in managing many cholestatic conditions. This bileacid has two purported benefits. First, it can stimulate bileflow and reduce cholestasis; second, it may displace more-toxic bile acids from the hepatocyte, thus potentially less-ening the hepatocyte injury associated with cholestasis. Forsevere pruritis seen in cholestasis, which is caused by thedeposition of bile acids in the skin, oral antihistamines pro-vide no benefit. Ursodeoxycholic acid can be helpful, andthe oral antibiotic rifampin often is added for refractorypruritis. The action of this agent in reducing itching is stillincompletely understood; but rifampin has been shown toprovide dramatic relief for affected infants.
Approach to the Child and Adolescent WhoHas Conjugated HyperbilirubinemiaOutside of infancy, conjugated hyperbilirubinemia is amuch less common laboratory finding. Depending onthe cause of the hyperbilirubinemia, clinical manifestationswill vary and can include scleral icterus, jaundice, fatigue,pruritis, abdominal pain, and nausea. Chronicity of diseasecan be assessed by the history, keeping in mind that in thesetting of hepatobiliary disease, a nonspecific symptom,such as fatigue, may be present months before the develop-ment of more objective symptoms of cholestasis, such asjaundice and pruritis.
The physical examination should include assessment ofliver size and texture. A firm, nodular liver suggests chronichepatobiliary disease and the development of cirrhosis.Physical stigmata of portal hypertension and cirrhosis in-clude splenomegaly, ascites, palmar erythema, caput me-dusae, and spider angioma. Normal metabolism of thesteroid intermediate androstenedione to testosterone typi-cally occurs in the liver. In end-stage liver disease, moreandrostenedione is eventually converted to estradiol, lead-ing to the development of gynecomastia in male patients.In female adolescents, secondary amenorrhea may resultfrom chronic liver disease.
Initial laboratory assessment will include the measure-ment of serum aminotransferases (AST, ALT), GGT,and bilirubin (including conjugated, or direct bilirubin),as well as performing tests of liver synthetic function, in-cluding prothrombin time and serum albumin level. Pa-tients who have poor liver synthetic function, manifestedas an elevated prothrombin time or low serum albuminlevel, should be referred urgently to a center with expertisein pediatric hepatology.
If the physical examination and initial laboratory find-ings do not support chronic liver disease, but there is an
elevated direct bilirubin fraction, consider a defect in thecanalicular transport of bilirubin. (7) Dubin-Johnson syn-drome is a defect in the anion transporter gene ABCC2,inherited in an autosomal recessive fashion, which leadsto elevation both of unconjugated and conjugated biliru-bin levels. Rotor syndrome has a similar presentation tothat of Dubin-Johnson syndrome, but the underlying ge-netic defect is unknown. These syndromes involve prob-lems in the storage/excretion of conjugated bilirubin andpresent with normal aminotransferase levels and the absenceof pruritis. The principal clinical objective is to distinguishthese benign conditions from the serious hepatobiliary dis-eases discussed later in this article.
The initial evaluation of a child or adolescent who hasconjugated hyperbilirubinemia should include abdominalultrasonography to assess for obstruction of the biliary tree.Typical symptoms reported with biliary obstruction includejaundice, abdominal pain (reliably reported as right upperquadrant or epigastric pain in older children and adoles-cents), nausea, and vomiting. A more acute presentationis seen when biliary obstruction is accompanied by cholan-gitis, which is a bacterial infection of the biliary tree causedby stasis of bile upstream from the obstruction. Patients af-flicted with cholangitis usually will have fever and leukocy-tosis and can develop bacterial sepsis.
Gallstone DiseaseThe most common cause of biliary obstruction in olderchildren and adolescents is gallstone disease (termed cho-lelithiasis). Little is known about the epidemiology ofgallstone disease in pediatrics. The pigmented stone isthe most commonly identified type of gallstone in chil-dren; however, overweight adolescent girls are at particularrisk of developing cholesterol stones. Identified risk factorsfor the development of gallstones in children include he-molytic disease, existing hepatobiliary disease, cystic fibro-sis, Crohn disease, chronic PN exposure, and obesity. Ifa gallstone becomes lodged within the common bile duct(termed choledocholithiasis), obstructive jaundice will re-sult and anticipated laboratory findings include elevationsin conjugated bilirubin, alkaline phosphatase, and GGT.AST and ALT levels may or may not be elevated. Shouldthe gallstone impact distally at the junction of the commonbile duct and pancreatic duct, the patient may be symp-tomatic with both obstructive jaundice and pancreatitis.
Plain abdominal radiographs and computed tomogra-phy are poor tests for the detection of gallstones becausemost stones are not calcified and therefore will not be vis-ible using these techniques. Ultrasonography is highly sen-sitive and specific for the detection of gallstones >1.5 mmin diameter within the lumen of the gallbladder; however,
gastrointestinal disorders conjugated hyperbilirubinemia
Pediatrics in Review Vol.33 No.7 July 2012 297
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from
the sensitivity drops off substantially for the detection ofgallstones within the common bile duct. The common bileduct will dilate in the setting of obstruction, and the diam-eter of the bile duct is readily measured by the ultrasonog-rapher. The combination of a dilated common bile ductwith clinical and laboratory evidence of obstructive jaun-dice is highly suspicious for a common bile duct stone.
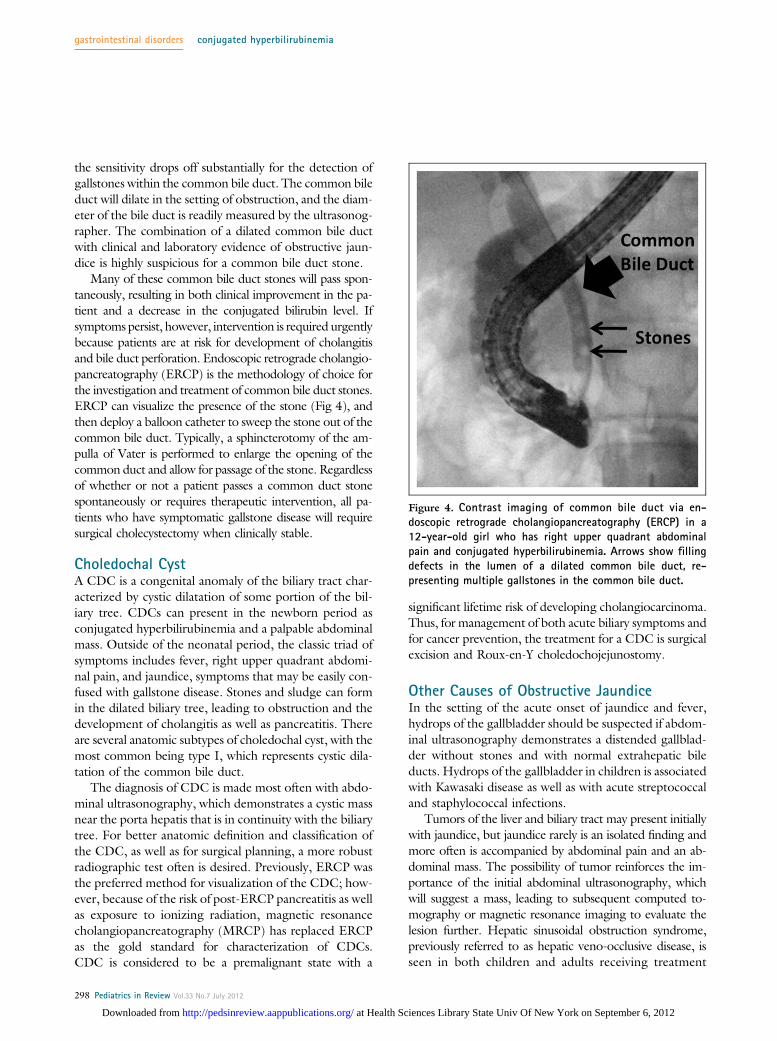
Many of these common bile duct stones will pass spon-taneously, resulting in both clinical improvement in the pa-tient and a decrease in the conjugated bilirubin level. Ifsymptoms persist, however, intervention is required urgentlybecause patients are at risk for development of cholangitisand bile duct perforation. Endoscopic retrograde cholangio-pancreatography (ERCP) is the methodology of choice forthe investigation and treatment of common bile duct stones.ERCP can visualize the presence of the stone (Fig 4), andthen deploy a balloon catheter to sweep the stone out of thecommon bile duct. Typically, a sphincterotomy of the am-pulla of Vater is performed to enlarge the opening of thecommon duct and allow for passage of the stone. Regardlessof whether or not a patient passes a common duct stonespontaneously or requires therapeutic intervention, all pa-tients who have symptomatic gallstone disease will requiresurgical cholecystectomy when clinically stable.
Choledochal CystA CDC is a congenital anomaly of the biliary tract char-acterized by cystic dilatation of some portion of the bil-iary tree. CDCs can present in the newborn period asconjugated hyperbilirubinemia and a palpable abdominalmass. Outside of the neonatal period, the classic triad ofsymptoms includes fever, right upper quadrant abdomi-nal pain, and jaundice, symptoms that may be easily con-fused with gallstone disease. Stones and sludge can formin the dilated biliary tree, leading to obstruction and thedevelopment of cholangitis as well as pancreatitis. Thereare several anatomic subtypes of choledochal cyst, with themost common being type I, which represents cystic dila-tation of the common bile duct.
The diagnosis of CDC is made most often with abdo-minal ultrasonography, which demonstrates a cystic massnear the porta hepatis that is in continuity with the biliarytree. For better anatomic definition and classification ofthe CDC, as well as for surgical planning, a more robustradiographic test often is desired. Previously, ERCP wasthe preferred method for visualization of the CDC; how-ever, because of the risk of post-ERCP pancreatitis as wellas exposure to ionizing radiation, magnetic resonancecholangiopancreatography (MRCP) has replaced ERCPas the gold standard for characterization of CDCs.CDC is considered to be a premalignant state with a
significant lifetime risk of developing cholangiocarcinoma.Thus, for management of both acute biliary symptoms andfor cancer prevention, the treatment for a CDC is surgicalexcision and Roux-en-Y choledochojejunostomy.
Other Causes of Obstructive JaundiceIn the setting of the acute onset of jaundice and fever,hydrops of the gallbladder should be suspected if abdom-inal ultrasonography demonstrates a distended gallblad-der without stones and with normal extrahepatic bileducts. Hydrops of the gallbladder in children is associatedwith Kawasaki disease as well as with acute streptococcaland staphylococcal infections.
Tumors of the liver and biliary tract may present initiallywith jaundice, but jaundice rarely is an isolated finding andmore often is accompanied by abdominal pain and an ab-dominal mass. The possibility of tumor reinforces the im-portance of the initial abdominal ultrasonography, whichwill suggest a mass, leading to subsequent computed to-mography or magnetic resonance imaging to evaluate thelesion further. Hepatic sinusoidal obstruction syndrome,previously referred to as hepatic veno-occlusive disease, isseen in both children and adults receiving treatment
Figure 4. Contrast imaging of common bile duct via en-doscopic retrograde cholangiopancreatography (ERCP) in a12-year-old girl who has right upper quadrant abdominalpain and conjugated hyperbilirubinemia. Arrows show fillingdefects in the lumen of a dilated common bile duct, re-presenting multiple gallstones in the common bile duct.
gastrointestinal disorders conjugated hyperbilirubinemia
298 Pediatrics in Review Vol.33 No.7 July 2012
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

for cancer, particularly hematologic malignancy. Throughmechanisms that are not fully understood, the develop-ment of microthrombi in hepatic sinusoids leads to hepaticdysfunction that often is severe. Patients present with jaun-dice, hepatomegaly, ascites, and laboratory evidence ofhepatic synthetic dysfunction in addition to conjugatedhyperbilirubinemia and elevation of aminotransferase lev-els. Diagnosis is suggested by abdominal ultrasonographywith Doppler measurement, which shows a decrease or re-versal of portal venous blood flow.
Infectious HepatitisThe acute onset of jaundice, typically associated with rightupper quadrant pain, hepatomegaly, nausea, and malaise,with variable fever, is suggestive of an infectious hepatitis.Elevation of AST and ALT levels, usually at least 2 to 3times the upper limit of normal, is always seen in an infec-tious hepatitis, although the degree of hyperbilirubinemiacan be variable. A broad range of viral agents can lead toinfectious hepatitis. The incidence of hepatitis A infectionin the United States has plummeted drastically since theuniversal implementation of vaccination. Both hepatitisB and hepatitis C can cause jaundice at the time of acuteinfection, and thus it is important to measure serologicmarkers for hepatitis A, B, and C viruses in any child oradolescent who have hepatitis. Epstein-Barr virus and cy-tomegalovirus both can cause hepatitis and cholestasis inthe context of a mononucleosislike illness. Adenovirus, in-fluenza virus, parvovirus, members of the enterovirus fam-ily, herpes simplex virus, and varicella virus also can lead tohepatic involvement, usually in the context of other clinicalsymptoms typical of the individual virus.
Autoimmune Disease of the Liverand Biliary SystemAutoimmune hepatitis (AIH) is characterized by a chronicactive hepatitis and non–organ-specific autoantibodies. (8)Without treatment, this chronic hepatitis progresses to cir-rhosis and end-stage liver disease over time. AIH can presentat any age in children and adults, although the incidence in-creases with age during childhood and adolescence. AIH ismore common in girls, and the spectrum of clinical presen-tation is wide. AIH can present insidiously as fatigue, ma-laise, and recurrent fevers or fulminantly as acute liverfailure. Depending on the chronicity of disease, physicalfindings of portal hypertension may be present at diagnosis.Typically, there is elevation of AST and ALT levels, al-though with considerable variation in the degree of el-evation. Conjugated hyperbilirubinemia, a low albuminlevel, and an elevated prothrombin time indicate extensive
chronic disease. The serum immunoglobulin G (IgG) levelusually is elevated, and 90% of patients will test positive forat least one of the associated autoantibodies: antinuclearantibody (ANA), anti–smooth muscle antibody (ASMA),and anti-liver, kidney microsomal antibody (LKM).
Two distinct subtypes of AIH have been described andcan be distinguished by serologic autoantibody tests. Thefirst, AIH type I, is the most common, includes 80% of allpatients who have AIH, and is characterized by positiveANA or ASMA or both. AIH type 2 is more prevalentin younger children and is characterized by anti-LKM pos-itivity. Children who have AIH type 2 are more likely topresent with acute hepatic failure. AIH can be associatedwith the autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy syndrome, one of the polyglandularautoimmune syndromes characterized by mucocutaneouscandidiasis, hypothyroidism, and adrenal insufficiency.
Liver biopsy remains the gold standard in the diagnosisof AIH. Histologic features include a dense inflammatoryinfiltrate in the liver, consisting of mononuclear andplasma cells, that begins in the portal areas and extends be-yond the limiting plate into the parenchyma of the liver.Piecemeal necrosis of hepatocytes also is observed fre-quently. The treatment of AIH involves the use of immu-nosuppressive agents. Conventionally, remission (definedas the normalization of AST and ALT levels) is inducedby using corticosteroids with a taper over several months.Corticosteroid-sparing agents, in particular the immuno-modulator azathioprine, are given long term to maintainremission of AIH.
Primary sclerosing cholangitis (PSC) is a progressive,autoimmune-mediated disease targeting both the intra-and extrahepatic bile ducts, resulting in significant scarringof the biliary tree. Patients present with laboratory evi-dence of bile duct injury, having elevations of GGT andalkaline phosphatase levels. AST, ALT, and conjugated bil-irubin concentration may be elevated as well. Imaging ofbile ducts, either withMRCP or ERCP, shows evidence ofstricturing and dilation of affected portions of the biliarytree.
PSC usually is associated with inflammatory bowel dis-ease, particularly ulcerative colitis, and can progress slowlyto cirrhosis. Several features distinguish PSC in childrenfrom adult disease. (9) In a subgroup of children who havePSC, there is elevation of IgG levels, autoantibody titers,and histologic features on liver biopsy similar to AIH,known as “overlap syndrome.” These children may favor-ably respond to immunosuppressive therapy. However, formost children and adults with PSC, there is a disconcertinglack of immunomodulatory therapies that can reverse thecourse of PSC.
gastrointestinal disorders conjugated hyperbilirubinemia
Pediatrics in Review Vol.33 No.7 July 2012 299
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

Drug- and Toxin-Induced CholestasisDrug hepatotoxicity can manifest in many forms, rangingfrom a systemic drug hypersensitivity syndrome to isolatedcholestasis. Although some forms of hepatotoxicity are pre-dictable, most are idiosyncratic owing to genetic variabilityin drug metabolism, making it difficult to understand thepathogenesis of hepatotoxicity in a given patient. (10)Some drug-induced cholestasis can present as an isolatedelevation of conjugated bilirubin; however, often there isa mixed hepatitic-cholestatic reaction with elevation of ami-notransferases and conjugated bilirubin. Commonly usedmedications in pediatrics that potentially can present withcholestatic liver injury include amoxicillin/clavulanic acid,oral contraceptives, and erythromycin. In any patientwho has cholestasis of unknown origin, it is critical to obtaina comprehensive medication history that includes recrea-tional use of drugs. Ecstasy, in particular, has been associatedwith hepatotoxicity and the development of jaundice. Usein adolescents of anabolic steroids for bodybuilding has beenreported to cause cholestasis. Questioning also should be di-rected at therapies that are not regulated by the Food andDrug Administration, such as nutritional supplements andhomeopathic treatments, because hepatotoxic metabolitesof these substances have been described.
Wilson DiseaseWilson disease is caused by an autosomal recessive inheriteddefect in the ATP7B gene, which codes for a hepatocyteprotein responsible for trafficking of copper into bile.(11) If the liver cannot excrete copper, the metal accumu-lates in the liver, brain, kidneys, and eyes. Copper toxicitythen produces the end-organ dysfunction seen in Wilsondisease. Wilson disease rarely presents before 5 years ofage, but its age of presentation and clinical manifestationsvary.With age, the likelihood of liver involvement at presen-tation decreases, whereas the likelihood of neuropsychiatricdisease increases.
The spectrum of the hepatic presentation of Wilsondisease includes an acute syndrome with nausea, fatigue,and elevated aminotransferases, mimicking infectious hepa-titis. Long-standing liver injury may present with jaundiceand conjugated hyperbilirubinemia. Other common hepaticpresentations of Wilson disease include chronic hepatitis,cirrhosis with portal hypertension, and fulminant hepaticfailure. A clue to Wilson disease in the laboratory evaluationis a low alkaline phosphatase level in the setting of elevationof serum aminotransferase and conjugated bilirubin levels.
Wilson disease also can affect the kidneys, manifesting asproximal tubular dysfunction with urinary loss of uric acidand subsequent low serum uric acid levels. Wilson disease
affects the hematologic system, leading in some patients toa direct antibody test (Coombs)-negative hemolytic ane-mia. Because of the varied presentation of this disease,a high degree of suspicion for Wilson disease must be keptin every school-age child or adolescent presenting with anytype of hepatic injury.
The practitioner must rely on interpretation of a num-ber of diagnostic studies in the evaluation for Wilson dis-ease. The sensitivity and specificity of these tests can varydepending on the clinical presentation. Diagnostic testsinclude measurement of serum ceruloplasmin, which istypically low (<20 mg/dL), and ophthalmologic exami-nation for Kayser-Fleischer rings, which are the cornealdeposition of copper seen on slit-lamp examination. Kayser-Fleischer rings are present in 95% of patients who manifesta neuropsychiatric presentation but are seen less frequentlyin patients who have a hepatic presentation of disease. Serumcopper level is a poor screening test for Wilson disease, buta quantitative 24-hour urine copper measure of >40 mg issuggestive of the disorder. Liver tissue can be sent for quan-titative copper measurement, and genetic testing is available.Prompt diagnosis of Wilson disease is important because theinstitution of copper chelation therapy can halt progressionof the disease, which is uniformly fatal if untreated.
Benign Recurrent Intrahepatic CholestasisAutosomal recessive mutations in canalicular transportproteins FIC1 and BSEP produce the phenotypes PFIC1and PFIC2, respectively, which typically present in infancyor childhood and may progress to liver failure early in life.Less severe mutations in these genes can produce the dis-ease known as benign recurrent intrahepatic cholestasis(BRIC). Importantly, BRIC does not lead to progressiveliver disease, cirrhosis, or hepatic dysfunction. BRIC is anepisodic disorder and presents in the first or second decadeafter birth with pruritis, often severe, and jaundice. Epi-sodes may be precipitated by viral illnesses and typicallyare heralded by the onset of pruritis, followed weeks laterby the development of jaundice. Nausea and steatorrheaalso may be present. Laboratory tests of liver function re-veal normal or mildly elevated serum AST and ALT, withelevation of both conjugated bilirubin and alkaline phos-phatase. The GGT concentration typically is normal ormildly elevated. The prothrombin time may be mildly pro-longed because of vitamin K malabsorption and deficiencyin the setting of cholestasis. Episodes can last weeks tomonths, and patients are completely well with normal livertesting in the intermediary periods. Treatment is directedtoward relief of pruritis, typically with ursodeoxycholicacid and rifampin, and correction of any fat-soluble vita-min deficiencies.
gastrointestinal disorders conjugated hyperbilirubinemia
300 Pediatrics in Review Vol.33 No.7 July 2012
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

With the initial episode of pruritis and jaundice, ana-tomic and histologic tests may be required to distinguishBRIC from PSC and other causes of intrahepatic cholesta-sis. Detailed imaging of the biliary tree, with either ERCPor MRCP, will be normal. During an episode, the dom-inant histologic finding in the liver is centrilobular cho-lestasis. Hepatic lobular or portal inflammation is anunusual finding in the liver. In contrast to other inflam-matory diseases of the liver, such as AIH and PSC, liverhistology in BRIC will return to normal in asymptom-atic periods.
References1. Davis AR, Rosenthal P, Escobar GJ, Newman TB. Interpretingconjugated bilirubin levels in newborns. J Pediatr. 2011;158(4):562–565. e12. Matte U, Mourya R, Miethke A, et al. Analysis of genemutations in children with cholestasis of undefined etiology.J Pediatr Gastroenterol Nutr. 2010;51(4):488–4933. Hartley JL, Davenport M, Kelly DA. Biliary atresia. Lancet.2009;374(9702):1704–17134. Lien TH, Chang MH, Wu JF, et al; Taiwan Infant Stool ColorCard Study Group. Effects of the infant stool color card screeningprogram on 5-year outcome of biliary atresia in Taiwan.Hepatology.2011;53(1):202–2085. Wagner M, Zollner G, Trauner M. New molecular insights intothe mechanisms of cholestasis. J Hepatol. 2009;51(3):565–5806. Carter BA, Taylor OA, Prendergast DR, et al. Stigmasterol, a soylipid-derived phytosterol, is an antagonist of the bile acid nuclearreceptor FXR. Pediatr Res. 2007;62(3):301–3067. Strassburg CP.Hyperbilirubinemia syndromes (Gilbert-Meulengracht,Crigler-Najjar, Dubin-Johnson, and Rotor syndrome). Best PractRes Clin Gastroenterol. 2010;24(5):555–5718. Mieli-Vergani G, Heller S, Jara P, et al. Autoimmune hepatitis.J Pediatr Gastroenterol Nutr. 2009;49(2):158–1649. Mieli-Vergani G, Vergani D. Unique features of primarysclerosing cholangitis in children. Curr Opin Gastroenterol. 2010;26(3):265–26810. Navarro VJ, Senior JR. Drug-related hepatotoxicity. N EnglJ Med. 2006;354(7):731–73911. Roberts EA, Schilsky ML; Division of Gastroenterology andNutrition, Hospital for Sick Children, Toronto, Ontario, Canada.A practice guideline on Wilson disease. Hepatology. 2003;37(6):1475–1492
Suggested ReadingSuchy FJ, Sokol RJ, Balistreri WF, eds. Liver Disease in Children.
3rd ed. New York, NY: Cambridge University Press; 2007
Summary
• A variety of anatomic, infectious, autoimmune, andmetabolic diseases can lead to conjugatedhyperbilirubinemia, both in the newborn period andlater in childhood.
• The pediatric practitioner is most likely to encounterconjugated hyperbilirubinemia in the neonatalperiod.
• It is crucial to maintain a high degree of suspicion forcholestasis in the persistently jaundiced newborn. Thegoal is recognition of conjugated hyperbilirubinemiabetween 2 and 4 weeks after birth, allowing for theprompt identification and management of infants whohave biliary atresia, which remains the most commoncause of neonatal cholestasis.
PIR QuizThis quiz is available online at http://www.pedsinreview.aappublications.org. NOTE: Since January 2012, learners cantake Pediatrics in Review quizzes and claim credit online only. No paper answer form will be printed in the journal.
New Minimum Performance Level RequirementsPer the 2010 revision of the American Medical Association (AMA) Physician’s Recognition Award (PRA) and creditsystem, a minimum performance level must be established on enduring material and journal-based CME activities thatare certified for AMA PRA Category 1 CreditTM. To successfully complete 2012 Pediatrics in Review articles for AMAPRA Category 1 CreditTM, learners must demonstrate a minimum performance level of 60% or higher on thisassessment, which measures achievement of the educational purpose and/or objectives of this activity.
Starting with 2012 Pediatrics in Review, AMA PRA Category 1 CreditTM can be claimed only if 60% or more of thequestions are answered correctly. If you score less than 60% on the assessment, you will be given additionalopportunities to answer questions until an overall 60% or greater score is achieved.
gastrointestinal disorders conjugated hyperbilirubinemia
Pediatrics in Review Vol.33 No.7 July 2012 301
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

1. A 3-month-old boy is jaundiced and is found to have conjugated hyperbilirubinemia; however, his gammaglutamyltransferase level is in the low normal range. Which of the following conditions is most likely tobe present?
A. Alpha-1 antitrypsin deficiencyB. Biliary atresiaC. Cystic fibrosisD. Progressive familial intrahepatic cholestasisE. Rubella infection
2. A 14-year-old girl presents with a history of intermittent right upper quadrant pain over the last 2 months. Herlaboratory evaluation reveals a direct bilirubin of 2.3 mg/dL. Of the following, what is the most appropriatenext study?
A. Abdominal ultrasonographyB. Endoscopic retrograde cholangiopancreatographyC. Hepatobiliary iminodiacetic acid scanD. Liver biopsyE. Targeted mutation analysis of the uridine diphosphate glucuronosyltransferase 1-1 gene to assess for
Gilbert syndrome
3. You are examining a jaundiced 1-month-old girl and hear a heart murmur consistent with peripheral pulmonicstenosis. A blood test reveals conjugated hyperbilirubinemia, causing you to suspect this condition:
A. Alagille syndromeB. Biliary atresiaC. Cystic fibrosisD. HypothyroidismE. Progressive familial intrahepatic cholestasis
4. A toddler who has chronic cholestasis has pruritus that is refractory to ursodeoxycholic acid. Which of thefollowing medications may be helpful in reducing symptoms?
A. AmoxicillinB. DiphenhydramineC. OndansetronD. RifampinE. Sulfisoxazole
5. A 5-week-old boy has been found to have biliary atresia. His parents are hesitant to authorize surgery andprefer “to see how he progresses. If he does not do well, he can always have surgery later.” Which of thefollowing statements regarding Kasai portoenterostomy is true:
A. Age at the time of the Kasai procedure is not associated with surgical outcome.B. Approximately 50% of children with biliary atresia have spontaneous resolution of their disease and do not
require a Kasai procedure.C. The Kasai procedure involves insertion of an prosthetic bile duct.D. The Kasai procedure is curative and most patients do not require follow-up of their liver disease.E. The Kasai procedure, when performed at <60 days after birth, is associated with better outcome.
gastrointestinal disorders conjugated hyperbilirubinemia
302 Pediatrics in Review Vol.33 No.7 July 2012
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from

DOI: 10.1542/pir.33-7-2912012;33;291Pediatrics in Review
David Brumbaugh and Cara MackConjugated Hyperbilirubinemia in Children
ServicesUpdated Information &
http://pedsinreview.aappublications.org/content/33/7/291including high resolution figures, can be found at:
References
http://pedsinreview.aappublications.org/content/33/7/291#BIBLat: This article cites 11 articles, 0 of which you can access for free
Permissions & Licensing
/site/misc/Permissions.xhtmltables) or in its entirety can be found online at: Information about reproducing this article in parts (figures,
Reprints/site/misc/reprints.xhtmlInformation about ordering reprints can be found online:
at Health Sciences Library State Univ Of New York on September 6, 2012http://pedsinreview.aappublications.org/Downloaded from