colloid-enhanced desorption of zinc in soil monoliths
TRANSCRIPT

This article was downloaded by: [134.117.10.200]On: 22 September 2013, At: 19:35Publisher: RoutledgeInforma Ltd Registered in England and Wales Registered Number: 1072954 Registeredoffice: Mortimer House, 37-41 Mortimer Street, London W1T 3JH, UK
International Journal of EnvironmentalStudiesPublication details, including instructions for authors andsubscription information:http://www.tandfonline.com/loi/genv20
Colloid-Enhanced Desorption Of Zinc InSoil MonolithsC.D. Barton a & A.D. Karathanasis aa Department of Forestry and Agronomy, University of Kentucky,respectively N-122K Ag. Science-North, Lexington, KY, 40546-0091,USAPublished online: 17 Sep 2010.
To cite this article: C.D. Barton & A.D. Karathanasis (2003) Colloid-Enhanced Desorption OfZinc In Soil Monoliths, International Journal of Environmental Studies, 60:4, 395-409, DOI:10.1080/00207230304728
To link to this article: http://dx.doi.org/10.1080/00207230304728
PLEASE SCROLL DOWN FOR ARTICLE
Taylor & Francis makes every effort to ensure the accuracy of all the information (the“Content”) contained in the publications on our platform. However, Taylor & Francis,our agents, and our licensors make no representations or warranties whatsoever as tothe accuracy, completeness, or suitability for any purpose of the Content. Any opinionsand views expressed in this publication are the opinions and views of the authors,and are not the views of or endorsed by Taylor & Francis. The accuracy of the Contentshould not be relied upon and should be independently verified with primary sourcesof information. Taylor and Francis shall not be liable for any losses, actions, claims,proceedings, demands, costs, expenses, damages, and other liabilities whatsoever orhowsoever caused arising directly or indirectly in connection with, in relation to or arisingout of the use of the Content.
This article may be used for research, teaching, and private study purposes. Anysubstantial or systematic reproduction, redistribution, reselling, loan, sub-licensing,systematic supply, or distribution in any form to anyone is expressly forbidden. Terms &Conditions of access and use can be found at http://www.tandfonline.com/page/terms-and-conditions

ISSN 0020-7233 print; ISSN 1029-0400 online © 2003 Taylor & Francis LtdDOI: 10.1080/0020723032000087952
Intern. J. Environ. Studies2003, Vol. 60, No. 4, August pp. 395–409
*Corresponding author. E-mail: [email protected]
COLLOID-ENHANCED DESORPTION OF ZINCIN SOIL MONOLITHS
C.D. BARTON and A.D. KARATHANASIS*
University of Kentucky, Department of Forestry and Agronomy, respectively N-122K Ag.Science-North, Lexington, KY 40546–0091, USA
(Received 3 August 2001)
The desorption and co-transport of metals by ex situ water dispersible soil colloids moving through macropores wasevaluated in a leaching experiment utilizing Zn-saturated soil monoliths. The monoliths were created byhydraulically driving steel pipe sections (50 cm diameter × 50 cm length) into a Loradale silt loam soil (fine, silty,mixed, mesic Typic Argiudolls). Each monolith was saturated in excess of the soil cation exchange capacity (CEC)with ZnCl2, then leached at a constant flux with a suspension of water-dispersible colloids fractionated from eitherthe Bt or O horizon of a Beasley (fine, montmorillonitic, mesic Typic Hapludalfs) or Rayne (fine-loamy, mixed,mesic, Typic Hapludults) soil, respectively. Eluents from the monoliths were collected and analysed periodically forcolloid and Zn concentration in the soluble and sorbed phase. Colloid and Zn concentration in the eluents variedgreatly with respect to colloid type. The Beasley colloid, which had a small particle diameter ( < 800 nm) and highsorptive affinity for Zn, showed considerable migrating ability through the soil matrix and enhanced Zn desorptionby up to 400 times over that exhibited by a distilled water flushing solution. Eluted Beasley colloids were saturatedwith Zn at concentrations greater than 50% of the soil’s CEC. Enhanced Zn desorption by colloids generated fromthe organic horizon of the Rayne soil was insignificant because the large colloid size ( > 1200 nm) limited transportthrough the soil matrix. Mineralogical analysis of the eluted colloids revealed that in situ colloid generation wasnegligible, thus confirming that nearly all of the desorbed Zn fraction was due to the ex situ applied colloid.
Keywords: Colloids; enhanced mobilization; metals; organic matter; zinc
1. INTRODUCTION
The mobility of colloids, cosolvents, and dissolved natural organic matter (NOM) throughsoils has gained considerable attention lately due to their possible enhancement ofcontaminant transport processes. Traditionally, many environmental contaminants wereassumed to be relatively immobile in soils because they are strongly sorbed to solid-phaseconstituents. More recently, consideration has been given to colloid-size particles, whichmay behave as a mobile solid phase carrying sorbed contaminants and thus, facilitatingcontaminant transport [1,2] Several researchers have documented the co-transport ofradionuclides [3], metals [4], and hydrophobic organic compounds [5] with colloids insubsurface environments. The dispersion and translocation of both organic [6] and
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

396 C. D. BARTON AND A. D. KARATHANASIS
mineral [7] colloids from soil particles has also been documented. However, soil-contaminant-colloid partitioning and transport processes are still not well characterized orunderstood.
Improper disposal of waste materials has resulted in a global soil and water pollutionproblem. Heavy metals account for much of the contamination found at hazardous wastesites in the US, and have been detected in the soil and groundwater at approximately 65%of the US; EPA Superfund sites [8]. The mobility of metals in soils is controlled by severalchemical and physical reactions. Clays, silts, metal oxides, and carbonates have the abilityto retain metals through cation exchange (weak outer sphere complexation) and specificadsorption (strong inner sphere complexation) [9]. In cases of extensive contamination, themetal sorption capacity of the soil may be exceeded and the contaminant can form discretemetal–mineral phases [10]. These metal ions can be immobilized in the soil throughformation of insoluble precipitates, isomorphic substitution into the crystal lattice of clayminerals, and/or physical entrapment into immobile bulk water surrounding soil pores [11].Soil organic matter also plays an important role in metal retention due to its high charge,and the tendency for transitional metals to form stable complexes with organicligands [12].
Changes in the physicochemical state of the soil matrix may result in the alteration ofmetal solubility and sorption kinetics [13]. These changes could also result in the detachmentand dispersion of colloid size particles from the soil matrix [14,15]. Metals that were boundto an immobile solid-phase may become mobile through an association with detached anddispersed colloids that are capable of migrating through the porous network of a soil profile.Mobilization of retained metals may also be achieved through the formation of soluble metalcomplexes with organic acids, solvents or chelating agents into the soil [16,17]. Severalresearchers have documented enhanced metal solubility in the water-soluble fraction ofNOM [18,19]. Thus, macromolecular components of NOM, which fall within the size rangeof colloids [20], and mineral colloids are increasingly recognized for their ability to facilitatemetal transport. However, their ability to desorbe contaminants retained within the soilmatrix and enhance migration has not been established.
The generation of ex situ mineral and/or organic colloids dissimilar or similar to theirsurrounding soil environment may occur as the result of landscape disturbance or naturalsoil erosion processes; the breakdown of a clay liner; or the decomposition of mulch,compost or litter layers. These colloids can be mobilized and migrate through anunderlying or adjacent soil matrix, desorb retained contaminants and transport them to agreater depth. Owing to their large surface area (10–500 m2 g–1) [21] and potentially highsurface charge [22], colloids may exhibit an increased affinity for some contaminants, suchas heavy metals, over that of the soil matrix. In addition, ex situ colloids may include orbe enriched with material that is foreign to a particular soil matrix or aquifer, such asorganic matter, carbonates, clays, and/or Fe and Mn oxyhydroxides. Depending upon thecharge characteristics of the colloids and the nature of the foreign material associated withthem, partition coefficients and sorption energies of the colloid phase may be sufficientlyhigh to exhibit preferential adsorption of soluble metals over that of the immobile solidphase [23]. In highly contaminated sites, colloids may even strip metals from the soilmatrix to establish a new equilibrium between the two solid phases [1]. If a mobile colloidgenerated from an uncontaminated source exhibits a higher metal sorption coefficient thanthe contaminated soil matrix, the sorption energy difference may be sufficient enough topromote contaminant desorption in excess of that exhibited by water alone. Based uponthis hypothesis, this study evaluated the potential for ex situ mineral and organic soilcolloids to desorbe Zn from highly contaminated soil monoliths and enhance mobilizationto deeper soil layers.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

DESORPTION OF ZINC 397
2. MATERIALS AND METHODS
2.1 Soil Monoliths
Undisturbed soil monoliths from the Loradale soil were prepared in a pasture field at theUniversity of Kentucky Agricultural Experiment Station in Lexington, KY, USA. Ahydraulically driven sampling procedure was used to create the soil monoliths in an attemptto mimic natural flow conditions. Steel pipe sections with an inside diameter of 50 cm, wallthickness of 1 cm, and length of 1 m were used for the cores. The cylinders were bevelled atone end to facilitate penetration into the soil. Cylinders were placed vertically on a preparedarea in which the vegetation was removed and pushed � 0.5 m into the moist soil using a 30ton hydraulic drill rig. Due to the nature of this sampling technique, the final lengths differedslightly, measuring 0.54 and 0.5 m for the Beasley and Rayne monoliths, respectively. Themonoliths were carefully removed with a backhoe, transported to a nearby building andfitted, on the subsurface end, with a 52 cm diameter and 15 cm long PVC cap. A male hosefitting lined with a 325 mesh (45 �m) screen was screwed into a threaded nut, which waswelded into the bottom of the PVC cap. A polyethylene hose was attached to each fitting andthe monoliths were vertically repositioned approximately 30 cm above the floor for theleaching experiments.
2.2 Colloid Generation and Soil Characterization
Water dispersible colloids were fractionated from the Bt horizon of a Beasley silt loam soiland the O horizon of a Rayne sandy loam soil. These ex situ colloids were selected for theirhigh mineral and organic charge, respectively. The extraction of colloid fractions wasaccomplished by mixing 10 g of soil with 200 ml of D-H2O in plastic bottles, shakingovernight, centrifuging at × 130 g (750 rpm) for 3.5 minutes, and decanting. The colloidconcentration in the supernatant was determined gravimetrically. A subsample of the stockcolloid suspension was used for characterization of physicochemical and mineralogicalproperties.
Physicochemical properties of the soil and colloids used in the experiment weredetermined through methods established by the Natural Resources Conservation Service [24].Analyses were performed on air-dried colloid and soil samples that had been gently crushedand passed through a 0.23 mm mesh sieve. Extractable bases and CEC were analysed usingthe 1M ammonium acetate (NH4OAc), pH 7.0 (Buchner funnel) (5B1) (5A1b) methods,respectively [24]. Organic carbon was determined using a Leco Carbon Analyzer, Model CR-12 (Leco Corp., St. Joseph, MI). Soil pH was measured in a 1:1 soil–water suspension.Extractable Al was determined using the ammonium oxalate and sodium citrate-bicarbonate-dithionate procedure described by Shuman [25]. Mean colloid diameter was determined usinga Microscan Particle Size Analyzer (Quantachrome Corp., Boynton Beach, FL). Bulkdensity [24], saturated hydraulic conductivity [26], and particle size analysis [24] were alsodetermined for the Loradale soil. The physicochemical properties of the soil and colloidsincluded in this study are presented in Table I.
The mineralogical composition of stock and eluted colloids was determined by X-raydiffraction using 200 mg samples. A Phillips PW 1840 diffractometer and PW 1729 X-raygenerator (Mahwah, NJ) were utilized for X-ray analysis, according to procedures describedby Karathanasis and Hajek [27]. The diffractometer was equipped with a cobalt X-ray tubeoperated at 40 kV and 30 mA. The goniometer is of Bragg–Bretano design. A scanning rateof 0.05° 2� per min from 2° to 40° and a scattering slit of 0.1° was used for each sample.
Adsorption isotherms were generated to evaluate the affinity for Zn sorption by theLoradale soil matrix and the colloids used in the study. A 1 g air-dried colloid or soil sample
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

398 C. D. BARTON AND A. D. KARATHANASIS
was added to 50 ml teflon test tubes with 35 ml adsorbate solution containing 0–20 mg l–1 Zn.Samples were shaken on a reciprocating shaker for 24 hours and centrifuged for one hour at× 2750 g (3500 rpm). Supernatants were collected and measured for Zn by atomic absorptionspectroscopy. The Freundlich isotherm equation was used to describe the experimentaladsorption data.
2.3 Leaching Experiments
The leaching experiment followed three phases: (a) Zn-saturation of the soil monoliths; (b)flushing excess Zn with D-H2O, and (c) flushing of the monoliths with colloid suspensions.During the Zn-saturation phase, 200 mg l–1 Zn solution was applied to the top of eachmonolith (downward vertical unsaturated flow) with a multi-channel peristaltic pump at aconstant flux (3.34 cm h–1), until measured eluent Zn concentrations were nearly equal to thatof the influent. The monoliths were subsequently leached with distilled water, at a constantflux (3.34 cm h–1), until the Zn breakthrough values were stabilized.
At that point, 300 mg l–1 suspensions of either Beasley or Rayne colloid were introducedinto separate monoliths at a constant flux (3.34 cm h–1). Colloid suspensions were preparedweekly and continuously agitated using a magnetic stirrer to prevent particle interaction andflocculation during the leaching procedure. Eluents from the monoliths were monitored withrespect to volume, colloid, and Zn concentration. Breakthrough curves (BTCs) weredeveloped by comparing changes in solute concentration (ratio of effluent concentration toinfluent concentration = C/Co) to increments of pore volume (flux-average volume ofsolution pumped per column pore volume).
Colloid concentrations were determined by filtration. A 50 ml portion of a shaken samplewas filtered through a dried and tared 0.2 �m microfilter. The filter and captured residue wereplaced in an oven at 100°C for five hours, removed and brought to room temperature in adesiccator, then reweighed. The colloid concentration was calculated as the differencebetween the initial and final weights divided by the volume of the sample. Another 50 ml
TABLE I Physicochemical and mineralogical properties of soils and colloids included in the study
Propertiesa Soil†
Loradale (Ap) Loradale (Bt)*
Colloids
Beasley Rayne
ElutedColloid
(Beasley)
Clay (%) 16 ± 2.0 28 ± 0.6 – –HC (cm min–1) 3.1 ± 1.7 0.7 ± 0.4 – –Bulk Density (g cm3) 1.2 ± 0.2 1.3 ± 0.1 – –OC (%) 3.4 ± 0.05 2.0 ± 0.01 0.7 ± 0.05 23.9 ± 0.8pH 5.5 ± 0.1 6.0 ± 0.1 6.1 ± 0.1 4.7 ± 0.3CEC (cmol kg–1 21.6 ± 0.2 18.5 ± 0.1 67.1 ± 0.5 50.4 ± 1.9TEB (cmol kg–1) 14.9 ± 1.1 13.2 ± 0.3 23.1 ± 0.2 2.6 ± 0.2Extractable Al (mg g–1) 1.5 ± 0.4 3.0 ± 0.3 5.4 ± 1.1 45.9 ± 3.3Mean Colloid Diameter (nm) – – 773 1241Smectite + Vermiculite (%) – – 55 – 52HIV (%) – 44 – 17 6Mica (%) – 15 23 – 20Kaolinite (%) – 35 14 8 13Quartz (%) – 6 8 75 9
a HC = hydraulic conductivity; OC = organic carbon; CEC = cation exchange capacity; TEB = total exchangeable bases; HIV =hydroxy-interlayered vermiculite.* Mineralogical analysis of Loradale soil was determined on clays extracted from Bt horizon.† Average and standard deviation of duplicate samples except for HC, which represents the average of five samples.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

DESORPTION OF ZINC 399
subsample from the shaken eluent was extracted and analysed for total recoverable Zn, usingthe EPA (200.2) HNO3-HCI method [28]. Stock colloid suspensions not leached through themonoliths were also analysed for total Zn to correct for background concentrations. Theremaining eluent samples were centrifuged at 3000 rpm for one hour to separate the solubleZn fraction from the colloid-bound fraction. Centrifuged supernatants were analysed forsoluble Zn by atomic absorption spectrophotometry using standards of the same matrix.
In an effort to determine the origin of the eluted colloids, selected samples with � 200 mgof residue were resuspended in 50 ml distilled water, filtered on a 0.2 �m membrane filter andtransferred to a petrographic glass slide for X-ray analysis following the procedures ofKarathanasis and Hajek [27]. Half of the resuspended residue samples were saturated with 1NKCI before X-ray analysis to facilitate vermiculite and chlorite identification.
3. RESULTS AND DISCUSSION
Since both monoliths involved a Loradale soil, reference to the monoliths will be made on thebasis of the applied colloid. Thus, the Loradale monolith that received Beasley colloids will bereferred to as the “Beasley” monolith, and the Loradale monolith that received Rayne colloidswill be referred to as the “Rayne” monolith. The selection of Zn as the contaminant of concernwas chosen because earlier laboratory results showed that it could be sorbed and co-transportedby various water-dispersible colloid types through Loradale soil columns [29].
3.1 Zinc Isotherms
Equilibrium adsorption isotherms of Zn on Beasley and Rayne colloids and a composite soilsample representing A and B horizons of the Loradale monolith were prepared in order toassess sorption affinities. Logarithmic transformation of the sorption data enabled thecalculation of partition coefficients (Kf) through the use of the Freundlich equation (Table II).Graphical representation of the isotherms revealed that the sorption affinity of Zn increasedin the order Rayne colloid < Loradale soil < Beasley colloid (Fig. 1).
The elevated CEC of the Beasley colloid over that of the other materials is likelyresponsible for its high Kf value (Table II). This heightened sorptive affinity may enable theBeasley colloid to desorb weakly sorbed Zn, or water bonded Zn forms, associated with theexchange sites of the Loradale soil matrix. The Rayne colloid, which had a higher CEC thanthe Loradale and a much greater organic carbon content than either the Loradale or Beasley(Table I), exhibited the lowest Kf values. This is likely the result of higher acidity levelsdisplayed by the Rayne colloids, which reduced the sorption affinity for Zn due to pHdependency [30,31,32].
TABLE II Freundlich-isotherm constants for adsorp-tion of Zn to colloids and soils
Colloids Kf† N‡ r2
Beasley 11.75 0.62 0.998Rayne 3.68 0.34 0.949
SoilsLoradale 6.74 0.49 0.995
† Kf = sorption capacity.‡ N = intensity factor.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

400 C. D. BARTON AND A. D. KARATHANASIS
In addition, the low base saturation (5.1%) and high extractable A1 concentration shownby the Rayne colloid may have caused a low bonding energy between the colloid surface andions in solution [33,34].
3.2 Zinc Sorption During the Saturation Phase
Both monoliths exhibited a strong affinity for Zn, showing complete attenuation untilbreakthrough occurred at 60 pore volumes (Fig. 2). The sorption phase continued untilretardation of the metal was greatly reduced in the Beasley monolith (C/Co = 0.75) and nolonger observed within the Rayne monolith (C/Co = 1). At that stage, the corresponding Znsaturation level was estimated to be 55.6 and 61.3 cmol kg–1 for the Beasley and Raynemonoliths, respectively, which is more than twice the CEC of the Loradale soil. Under the pHconditions of the experiment (� 6.0), retention of Zn through metal hydrolysis ispossible [10]. This hydrolysis may be accompanied by precipitation of metal hydroxides ina manner not easily distinguishable from adsorption reactions. Geochemical modelling ofaqueous-phase chemical equilibria using the MINTEQA2 [35] program indicated thatconditions were favourable for the formation of Zn oxides (zincite) as well. Thus, zincretention in excess of the soil’s CEC may be attributed to precipitation and retention ofinsoluble Zn hydroxides and oxides within the monoliths.
Following the Zn saturation phase (Fig. 1), the monoliths were flushed with distilled waterat 75 (Beasley) and 83 (Rayne) pore volumes to displace any excess soluble Zn remainingin the soil pores. Leaching with water continued for approximately ten more pore volumes,and ended when the eluent Zn concentration was stabilized around 2 mg l–1 (Fig. 2). The totalquantity of Zn removed during the H2O flushing stage accounted for less than 3% (� 6.5 g)of that applied to the monoliths. Eluted Zn concentrations in the last collected samples were
FIGURE 1 Linearized equilibrium adsorption isotherms for zinc adsorbed by Rayne and Beasley colloids and theLoradale soil.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

DESORPTION OF ZINC 401
1.63 and 2.12 mg l–1 for the Beasley and Rayne monoliths, respectively. These final valueswere used as the influent concentrations (Co) for the desorption phase of the experiment.
3.3 Colloid Flushing and Elution
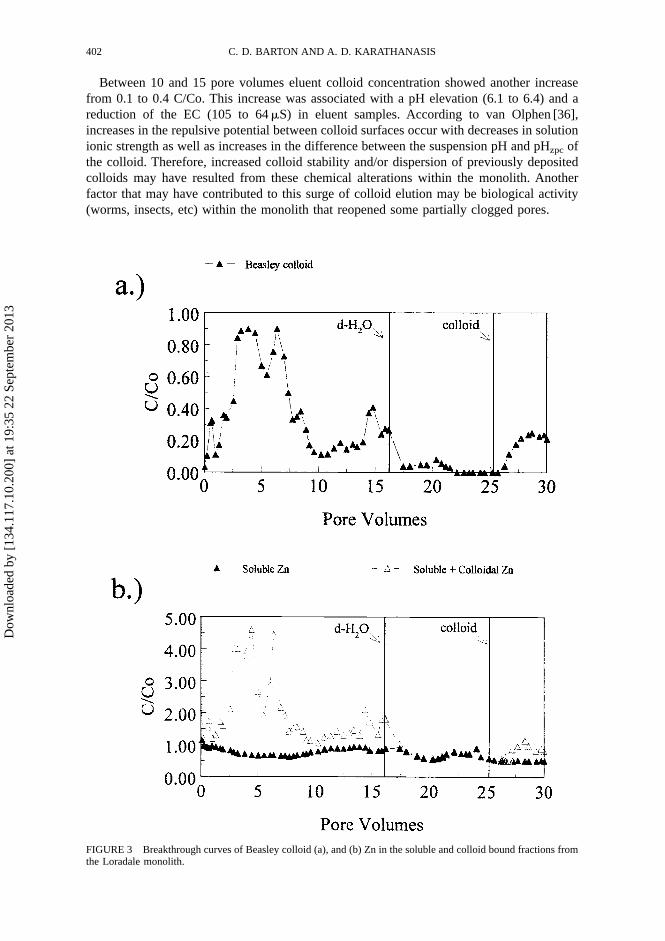
The Beasley colloid showed a gradual breakthrough to C/Co values greater than 0.8 at 2.5pore volumes (Fig. 3a). This gradual breakthrough indicates that adsorption and/or filtrationprocesses were responsible for some colloid retention within the monolith during earlyleaching stages [7]. Between 2.5 and 7.0 pore volumes, colloid concentration in the eluentwas maintained around 0.8, suggesting a significant decrease in colloid filtration. Colloidelution declined in the subsequent 3.0 pore volumes to C/Co values of 0.2. This reductionmay be the result of clogged macropore space, or a reduction of pore diameter by colloiddeposition on pore walls.
FIGURE 2 Breakthrough curves of Zn eluted in the absence of colloids from the Loradale monoliths.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13
402 C. D. BARTON AND A. D. KARATHANASIS
Between 10 and 15 pore volumes eluent colloid concentration showed another increasefrom 0.1 to 0.4 C/Co. This increase was associated with a pH elevation (6.1 to 6.4) and areduction of the EC (105 to 64 �S) in eluent samples. According to van Olphen [36],increases in the repulsive potential between colloid surfaces occur with decreases in solutionionic strength as well as increases in the difference between the suspension pH and pHzpc ofthe colloid. Therefore, increased colloid stability and/or dispersion of previously depositedcolloids may have resulted from these chemical alterations within the monolith. Anotherfactor that may have contributed to this surge of colloid elution may be biological activity(worms, insects, etc) within the monolith that reopened some partially clogged pores.
FIGURE 3 Breakthrough curves of Beasley colloid (a), and (b) Zn in the soluble and colloid bound fractions fromthe Loradale monolith.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

DESORPTION OF ZINC 403
As the input solution was switched to distilled water, a steep reduction in Beasleycolloid breakthrough to C/Co values less than 0.05 was evident (Fig. 3(a)). The relativeabsence of colloids in the eluent after elution of one pore volume of distilled water,suggests that desorption of retained colloids was minimal. Similar findings have beenreported by Seta and Karathanasis [7] and Ryan and Gschwend [37], who observedirreversible adsorption of mineral colloids on soils and aquifer materials, respectively.However, generation of in situ colloids as a result of reductions in the ionic strength ofthe input solution, was not observed during the water leaching phase of theexperiment [14,38].
Upon reverting back the input solution to the Beasley colloid suspension, at � 25 porevolumes, a smooth and gradual breakthrough of colloids to C/Co values of 0.3 occurred,which was maintained at that level for the remainder of the experiment. A comparison ofcolloid breakthrough patterns before and after the distilled water stage suggests that theporosity of the soil matrix was apparently more stable during the second colloid leachingcycle, and that the pores responsible for the early, more elevated, breakthrough werecompromised as colloid flow paths. This may have implications on the long-term colloidtransport since nearly 70% of the input colloids were being retained within the monolith after30 pore volumes of leaching.
Elution of the Rayne colloid through the Loradale monolith was much lower than thatobserved for the Beasley colloid (Fig. 4(a)). With the exception of the first pore volume,essentially no Rayne colloids were measured in eluent samples. Their maximum colloidbreakthrough in samples from the first pore volume was about 10 mg l–1 (C/Co = 0.03) anddropped to about 0 afterwards. In an effort to determine if colloid size may have inhibited theRayne colloid transport, mean particle size determinations were performed. The resultsindicated that the mean diameter of the Rayne colloid was much larger than that of theBeasley (1241 versus 773 nm). A decrease in colloid mobility through porous media withincreasing diameter has been documented by several researchers [22,39]. Seta andKarathanasis [7] noted that the mobility of kaolinitic soil colloids, with a mean colloiddiameter of 1050 nm, decreased considerably as the colloids passed through Loradale soilcolumns. According to filtration theory [40], colloids with diameters between 500 and1000 nm are likely to be transported through an uncharged porous medium of glass beads.Colloid size may be even more limiting in transport through a charged soil matrix. Therefore,the large particle size may be responsible for the total attenuation of the Rayne colloid withinthe soil monolith.
3.4 Zinc Elution During the Flushing Phase
Zinc desorption and elution from the monoliths, in the presence of colloids, was evaluated asa soluble fraction, and as soluble plus colloid-bound (total) fraction. The soluble Zn fraction,eluted in association with the Beasley colloid, exhibited a gradual decline from a C/Co valueof 1.2 in the initial sample to a C/Co value of 0.5 after 30 pore volumes (Fig. 3(b)). The totalZn fraction was considerably higher (up to 4.5 C/Co) than the soluble fraction during thecolloid application cycles, showing excellent correlation with colloid breakthrough trends. Inthe early stages of desorption, when colloid breakthrough exceeded a C/Co of 0.8, total Zneluted from the monoliths reached C/Co levels in excess of 4.0. This corresponds to a 400%increase in Zn desorption over that of distilled water alone. As the colloid breakthroughdecreased, so did the magnitude of Zn desorption. However, a relatively stable Zn desorptionof � 0.5 C/Co was maintained even in the later stages of the experiment, when colloids werestill eluted from the monolith.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

404 C. D. BARTON AND A. D. KARATHANASIS
Evidently, the higher affinity of the Beasley colloid for Zn over that of the Loradale soil(Table II) resulted in competitive sorption between the two solid phases, which allowed Znto be stripped from the soil matrix and adsorbed onto the migrating clay colloid. Mills etal. [1] indicated that sorption exchanges of a metal between two solid phases may continueuntil a state of equilibrium is established. It is doubtful that such an equilibrium was reachedduring the short duration of this experiment or, if it was, it happened only in localized areaswithin the monolith. Therefore, these conditions allowed Zn to migrate through the soilmacropores attached to the ex situ colloid for the length of the experiment as long as thecolloids maintained their mobility. Since the number of interactive exchange sites availableon the eluting Beasley colloids is limited compared with the sites available within the matrixof the entire soil monolith, the extent of Zn desorption is controlled by the concentration of
FIGURE 4 Breakthrough curves of Rayne colloid (a), and (b) Zn in the soluble and colloid bound fractions fromthe Loradale monolith.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

DESORPTION OF ZINC 405
the colloid eluted through the soil monolith, and the accessibility of interactive sites withinthe monolith soil matrix.
The association of the eluted Zn with the eluted colloids was assessed with HNO3-HClextractions [28]. The results indicated that exchange sites on the eluted colloids were between60 and 100% saturated with Zn (Fig. 5). A few colloid eluents exhibited Zn saturation inexcess of 100% during the peak colloid breakthrough period, which may indicate some metalprecipitation on colloid surfaces or desorption of Zn from the matrix through in situ colloidgeneration. Regardless of the colloid concentration, however, high Zn saturation of themobile colloids was evident throughout the study. The slight decline in Zn saturationobserved during the distilled water leaching stage is attributed to dilution effects on readilyexchangeable Zn, present within the macropore space of the monolith.
The lack of colloid breakthrough in the Rayne monolith resulted in a drastically differentZn BTC (Fig. 4(b)). A limited release of colloids in the first pore volume led to anenhancement of Zn breakthrough of the order of 0.2 (C/Co) over that of soluble Zn. With theexception of two other samples, however, the total eluted Zn concentration was equivalent tothat of the soluble fraction. Even though the concentration of Rayne colloids in the eluent wasnot detectable in most samples, the application of the colloid to the soil had an influence onZn elution. A comparison of soluble Zn BTCs for each pore volume eluted from the twomonoliths revealed that the Rayne monolith consistently released more soluble Zn than theBeasley monolith (Fig. 6). Soluble Zn elution fluctuated between 0.5 and 1.3 C/Co, showing3–4 wavy cycles. After an initial decline to about 0.7 C/Co at 10 pore volumes, it peaked to1.3 at 18 pore volumes, before declining irregularly to about 0.5 C/Co during the D-H2Oflushing cycle. This irregular breakthrough pattern could have been caused by fluctuations inthe flow velocity of the Zn flushing solutions/suspensions [16]. Even though the input flowvelocity was maintained at a constant rate throughout the experiment, the accumulation ofcolloids within the matrix and eventual reduction of macroporosity due to partial clogging
FIGURE 5 Percentage CEC saturation of eluted colloids by Zn as influenced by eluted colloid concentration.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

406 C. D. BARTON AND A. D. KARATHANASIS
may have altered the solution flow path and soil hydraulic conductivity. It is also likely thatdecreases in flow velocity within the matrix may have enhanced the formation of solublemetal organic acid-metal complexes, due to increased contact time and reduced mass transferresistance for Zn dissolution.
A reduction in the eluted Zn was evident during the D-H2O leaching phase of theexperiment, but the elution pattern continued to be irregular with the peak and valley patternshown in the presence of colloids. Decreasing the ionic strength in the input solution(D-H2O) may have resulted in the detachment and remobilization of some filtered colloidsand reopening of previously clogged pore space. However, if the colloids were still too largeto pass through the entire length of the monolith, they eventually were redeposited within thematrix. This remobilization of colloids within the matrix without final elution, and theresulting dynamic changes in the porosity, may explain the irregular fluxes in Zn desorptionand elution observed.
FIGURE 6 Comparison of the soluble Zn fraction eluted from the Rayne and Beasley monoliths at equivalent porevolumes.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

DESORPTION OF ZINC 407
3.5 Mineralogy of Eluted Colloids
In an effort to determine whether there was some contamination of the eluted colloidsgenerated within the monolith during the leaching experiment, the mineralogy of the elutedBeasley colloids was compared with the mineralogy of the clay fraction of the Loradale soil.The Rayne colloids were also analysed, but there was not enough colloid elution to makecomparisons. The main mineralogical differences between the Loradale and Beasley samplesincluded less mica, more kaolinite, and lack of smectite in the Loradale (Table I).
The nearly identical mineralogical composition of the eluted Beasley colloid comparedwith that of the input Beasley colloid, suggested that in situ colloid generation wasinsignificant within the soil monoliths. Diffraction patterns from eluted colloids at differentstages of the experiment were also found to be nearly identical. Some minor mineralogicaldifferences between eluted and applied colloids may be indicative of the preferentialadsorption of specific minerals within the matrix as described by Kaplan et al. [22]. However,the compositional differences were not significant enough to suggest a major change in thecolloid composition that could possibly alter its mobility or sorptive capacity. Therefore, poreclogging, rather than in situ particle generation, was probably responsible for the reducedbreakthrough in the Beasley colloid observed at 7 pore volumes (Fig. 3(a)). Themineralogical findings strongly suggest that colloid elution during the entire leachingexperiment was exclusively due to the Beasley colloid. Moreover, these findings offeradditional verification for the suggested enhancement of Zn desorption and mobilization bythe ex situ colloids used in the flushing solutions.
4. CONCLUSIONS
The desorption of Zn from a contaminated soil onto ex situ mobile mineral colloids wasdemonstrated in this experiment. The magnitude of desorption and transportability of themetal-enriched colloid was dependent upon the physicochemical characteristics of the colloidand the soil matrix. The high sorptive affinity for Zn and the small particle size diameter ofthe Beasley colloids contributed to significant enhancement in metal desorption and transportcompared with that generated by a distilled water flushing solution. Desorption and co-transport by the Rayne colloid was inhibited due to its lower sorptive capacity for Zncompared with that of the soil matrix, and its high particle size diameter, which preventedmigration through the monolith’s macropore system. Soluble organic matter associated withthe Rayne colloid, however, may have contributed to some Zn desorption through the metalcomplex formation at leaching stages of low flow velocity within the monolith, whenretention time was sufficiently long to achieve solute-matrix equilibrium.
Considering the number of metal contaminated waste disposal sites worldwide, and thecommon practice of covering these areas with a clay cap, the results from this experimentmay have significant ramifications. The breakdown of these caps and similar clay linerscould result in the mobilization of colloids that have the potential to strip metals from anunderlying contaminated soil and enhance their migration to deeper depths. Therefore,consideration should be given toward additional research to test this theory on a suite ofmetals and colloid types (mineral and organic).
References
[1] W.B. Mills, S. Liu and F.K. Fong, “Literature review and model (COMET) for colloid/metal transport in porousmedia”, Ground Water 29, 199–208 (1991).
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

408 C. D. BARTON AND A. D. KARATHANASIS
[2] J.F. McCarthy and J.M. Zachara, “Subsurface transport of contaminants”, Environmental Science Technology23, 496–503, (1989).
[3] W.R. Penrose, W.L. Polzer, E.H. Essington, D.M. Nelson and K.A. Orlandini, “Mobility of plutonium andamericium through a shallow aquifer in a semiarid region”, Environmental Science Technology 24, 228–234(1990).
[4] D.I. Kaplan, P.M. Bertsch and D.C. Adriano, “Facilitated transport of contaminant metals through an acidifiedaquifer”, Ground Water 33, 708–717 (1995).
[5] A.T. Kan and M.B. Tomson, “Ground water transport of hydrophobic organic compounds in the presence ofdissolved organic matter”, Environmental Toxicology Chemistry 9, 253–263 (1990).
[6] W.P. Johnson and G.L. Amy, “Facilitated transport and enhanced desorption of polycyclic aromatichydrocarbons by natural organic matter in aquifer sediments”, Environmental Science Technology 29, 807–817(1995).
[7] A.K. Seta and A.D. Karathanasis, Stability and transportability of water-dispersible soil colloids, Soil ScienceSociety America Journal 61, 604–611, (1997).
[8] US EPA, “Recent developments for in-situ treatment of metal contaminated soils. EPA-542-R-97–004”,Washington, DC, USA (1997).
[9] G. Sposito, The Surface Chemistry of Soils (Oxford University Press, New York, 1984).[10] W.L. Lindsay, Chemical Equilibria in Soils (Wiley, New York, 1979).[11] D.L. Sparks, “Kinetics of metal sorption reactions”, in: H.E. Allen, C.P., Huang, G.W. Bailey and A.R. Bowers
(eds.), Metal Speciation and Contamination of Soil (Lewis Boca Raton, FL, USA 1995) pp. 35–56.[12] H.A. Elliott, M.R. Liberati and C.P. Huang, “Competitive adsorption of heavy metals by soils”, Journal
Environmental Quality 15, 214–219 (1986).[13] D.L. Sparks, Kinetics of Soil Chemical Processes, (Academic Press, New York, 1989).[14] S.B. Roy and D.A. Dzombak, “Chemical factors influencing colloid-facilitated transport of contaminants in
porous media”, Environmental Science Technology 37, 656–664 (1997).[15] D.A. Dzombak and F.M. Morel, Surface Complexation Modeling – Hydrous Ferric Oxide (Wiley, New York,
1990).[16] A. Davis and I. Singh, “Washing of Zinc (II) from contaminated soil column”, Journal Environmental
Engineering 121, 174 (1995).[17] H.A. Elliott and G.A. Brown, “Comparative evaluation of NTA and EDTA for extractive decontamination of
Pb-polluted soils”, Water Air Soil Pollution 45, 361–367 (1989).[18] P.L. Giusquiani, G. Gigliotti and D. Businelli, Mobility of heavy metals in urban-waste amended soils, Journal
Environmental Quality 21, 330–335 (1992).[19] G. Sposito, L.J. Lund and A.C. Chang, Trace metal chemistry in arid-zone field soils amended with sewage
sludge: I. Fractionation of Ni. Cu, Zn, Cd, and Pb in solid phases, Soil Science Society America Journal 46,260–264 (1982).
[20] W. Stumm and J.J. Morgan, Aquatic Chemistry 2nd edn (Wiley, New York, 1981).[21] L. Liang and J.F. McCarthy, Colloidal transport of metal contaminants in groundwater, in: H.E. Allen, C.P.
Huang, G.W. Bailey and A.R. Bowers (Eds.), Metal Speciation and Contamination of Soil (Lewis Publishers,Boca Raton, FL, USA, 1995) pp 87–112.
[22] D.I. Kaplan, P.M. Bertsch and D.C. Adriano, “Mineralogical and physicochemical differences between mobileand nonmobile colloidal phases in reconstructed pedons”, Soil Science Society America Journal 61, 641–649(1997).
[23] A. Tessier, R. Carignan and N. Belzile, “Reactions of trace metals near the sediment-water interface in lakes”,in: J.V. DePinto, V. Lick, J.F. Paul (eds.), Transport and Transformations of Contaminants Near the Sediment-Water Interface, (Lewis Publishers, Chelsea, MI, USA 1999) pp 129–152.
[24] Natural Resources Conservation Service, Soil Survey Laboratory Methods Manual. Soil Survey Investigations,Report No. 42. United States Department of Agriculture, Washington, DC, USA (1996).
[25] L.M. Shuman, “Fractionation method for soil microelements”, Soil Science 140, 11–22 (1985).[26] A. Klute, and C. Dirksen, Hydraulic conductivity and diffusivity: Laboratory methods, in: A. Klute (Ed.)
Methods of Soil Analysis, Part 1. Physical and Mineralogical Methods-Agronomy Monograph No. 9, 2nd edn,(American Society of Agronomy, Madison, WI, USA, 1986) pp. 687–735.
[27] A.D. Karathanasis and B.F. Hajek, “Revised methods for quantitative determination of minerals in soil clays”,Soil Science Society America Journal 46, 419–425 (1982).
[28] US EPA., Methods for the determination of metals in environmental samples. Method 200.2 EPA/600/R-94/111. Washington, DC, USA (1994).
[29] A.D. Karathanasis, “Subsurface migration of Cu and Zn mediated by soil colloids”, Soil Science SocietyAmerican Journal 63, 830–838 (1999).
[30] R.D. Harter, Effect of soil pH on adsorption of Pb, Cu, Zn and Ni, Soil Science Society America Journal 47,47–51 (1983).
[31] S. Kuo and A.S. Baker, Sorption of copper, zinc and cadmium by some acid soils, Soil Science Society AmericaJournal 44, 969–974 (1980).
[32] L.M. Shuman, The effect of soil properties on zinc adsorption by soils, Soil Science Society AmericaProceedings 39, 454–458 (1975).
[33] G.L. Ji and H.Y. Li, “Electrostatic adsorption of cations”, in: T.R. Yu (Ed), Chemistry of Variable Charged Soils(Oxford University Press, Oxford, UK 1997) pp 64–112.
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13

DESORPTION OF ZINC 409
[34] C.E. Marshall, The Physical Chemistry and Mineralogy of Soils, Vol 1 (Wiley, New York, 1964).[35] J.D. Allison, D.S. Brown, and K.J. Novo-Gradac, MINTEQA2/PRODEFA2, a geochemical assessment model
for environmental systems: Version 3.0 user’s manual. USEPA, Environmental Research Laboratory, Athens,GA, USA (1990).
[36] H. van Olphen, An Introduction to Clay Colloid Chemistry, 2nd edn (Wiley, New York, 1977).[37] J.N. Ryan and P.M. Geschwend, “Effect of solution chemistry on clay colloid release from an iron oxide-coated
aquifer sand”, Environmental Science Technology 28, 1717–1726 (1994).[38] C.H. Swartz and P.M. Gschwend. “Mechanisms controlling release of colloids in groundwater in a southeastern
coastal plain aquifer sand”, Environmental Science Technology 32, 1779–1785 (1998).[39] D. Ronen, M. Magaritz, U. Weber, A.J. Amiel and E. Klein. “Characterization of suspended particles collected
in groundwater under natural gradient flow conditions”, Water Resources Research 28, 1279–1291 (1992).[40] K.M. Yao, M.T. Habibian and C.R. O’Melia, “Water and wastewater filtration: Concepts and applications”,
Environmental Science Technology 5, 1105–1112 (1971).
Dow
nloa
ded
by [
134.
117.
10.2
00]
at 1
9:35
22
Sept
embe
r 20
13