collagénoses et vasculites systémiques: un...
TRANSCRIPT

Collagénoses et vasculitessystémiques: un aperçu
Patrick Liang
Service de rhumatologie
Université de Sherbrooke

Objectifs1. Reconnaître les situations cliniques évocatrices de
collagénoses et vasculites systémiques
2. Connaître et savoir prescrire et interpréter les principaux tests sérologiques associées
1. aux collagénoses et
2. aux vasculites à ANCA
3. Être en mesure de lancer une investigation à la recherche d’une collagénose ou vasculite systémique
4. Être en mesure de reconnaître les conditions pouvant mimer les collagénoses et les vasculites systémiques

Mise en situation Une femme de 44 ans vous est référée en raison d’un
polysymptomatologie comprenant des: polyarthralgies, myalgies, notion « d’allergie » à la
lumière, de phénomène de Raynaud, et
recherche d’anticorps anti nucléaires positive (1:80, granulaire)
On vous demande si cette patiente souffre d’une collagénose.
Votre opinion?

Collagénoses: définitionSynonymes: connectivites
Maladies inflammatoires, systémiques, caractérisées par
Atteinte de tissus riches en collagène,
Atteinte fréquente de vaisseaux sanguins
Atteinte multisystémique
Présence d’autoanticorps caractéristiques
Chevauchement des manifestations cliniques entre diverses collagénoses
Prédominance de femmes

Collagénoses: définition prodrome: symptômes habituellement non spécifiques
arthralgies, myalgies
fébricules
Raynaud
fatigue chronique
Perte de poids
Connectivites indifférenciées:
manifestations cliniques suggérant une collagénose mais ne permettant pas de la catégoriser: eg: Raynaud, arthrite, maladie pulmonaire intersitielle, sérosite,
éléments vasculitiques
Évolution vers entité clinique bien définie peut prendre des années, ou ne jamais se produire
Peut durer des mois ou des années

Collagénoses: définition Polyarthrite rhumatoide
Lupus érythémateux disséminé
Sclérodermie
Syndrome de Sjogren
Polymyosite
Connectivite mixte (MCTD)
Collagénoses indifférenciées

Critères diagnostiques:

Critères diagnostiques: limites Très sensibles et spécifiques, lorsqu’utilisés chez des
populations AVEC la maladie
Performance moyenne plus tôt durant la maladie
Jusqu’{ 50% des patients lupiques ne répondent pas aux critères diagnostiques de l’ACR durant les premières années de la maladie; il peut s’écouler plusieurs années avant qu’ils soient satisfaits

Lupus érythémateux disséminéMaladie autoimmune caractérisée par:
atteinte multisystémique (principaux: peau, articulations, hématologique, rénal: ad 50%))
production de nombreux autoanticorps
Distinguer de lupus cutané chronique 5% seulement auront des manifestations systémiques
Incidence la plus élevée durant les années reproductivesÂge Proportion Femme:Homme
16-55 65% 9:1
> 55 15% 2:1
< 16 20% 2:1

Lupus aigu

Lupus érythémateux disséminé Éruption cutanée:
malaire: maculeux, ou plaque, peu ou pas de squame, pas d’atrophie, épargne pli nasolabial.
Associé à maladie systémique active
DDx:
Dermatomyosite:
Distribution semblable à LCA
Peut y en avoir au scalp
Tend à être plus prurigineux
Épargne moins les plis nasolabiaux
Rash héliotrope et autres manifestations typiques de la dermatomyosite
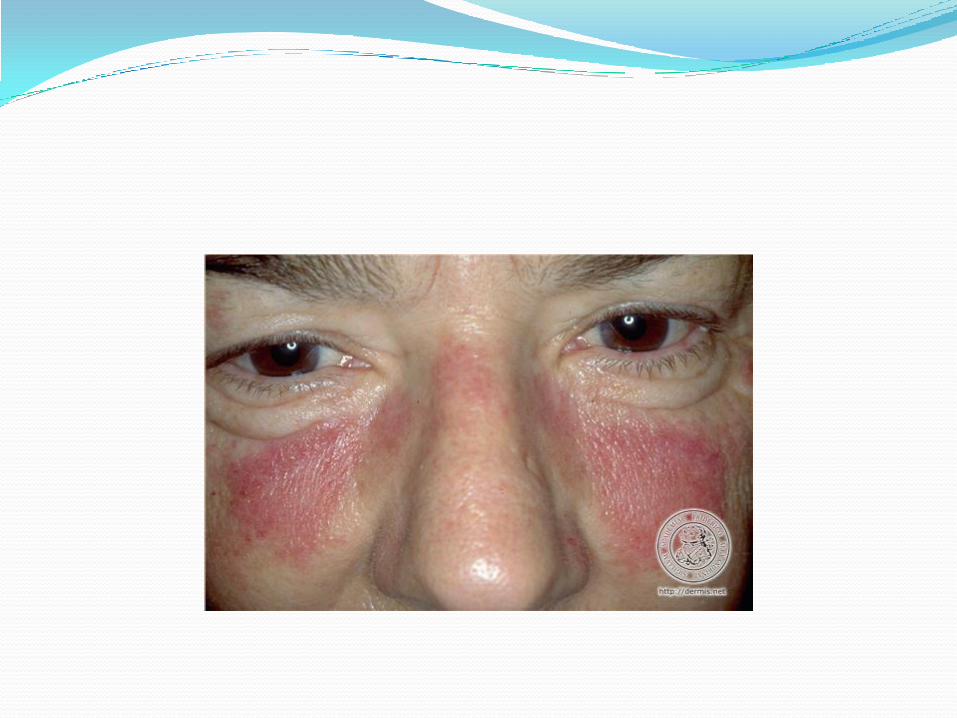




Lupus érythémateux disséminé Éruption cutanée suite: malaire DDx:
Rosacée: télangiectasies, pustules, pli nasolabial
Dermite séborrhéique: pli nasolabial, croûtes grasses, sourcils
Éruption polymorphe à la lumière: papules, plaques, zones photoexposées (visage moins souvent), très prurigineux, absence de manifestations systémiques, absence d’autoanticorps, histodifférente, durée: heures
Réaction médicamenteuse: ATCD d’exposition
Érysipèle: progressison rapide, T, croûtes




Lupus érythémateux disséminé Éruption cutanée suite:
Lupus cutané subaigu: forme papulosquameuse ou annulaire
Épaules, partie supérieure du dos, thorax, face des extenseurs avant-bras; visage et scalp: moins.
Association avec maladie systémique: 50% Rechercher: Rx: HCT, IECA, BCC, statines, IFN a, b, sulfa DDx: Lyme et RAA (annulaire)
Psoriasis (papulosquameux) Lupus cutané chronique: scalp (50%), oreilles, visage, mains, autres plaques, croûtes, follicular plugs, atrophie, hyper ou
hypopigmentation Association avec maladie systémique: 5-10% DDx: éruption polymorphe à la lumière; séborrhée, rosacée, tinea,
sarcoidose

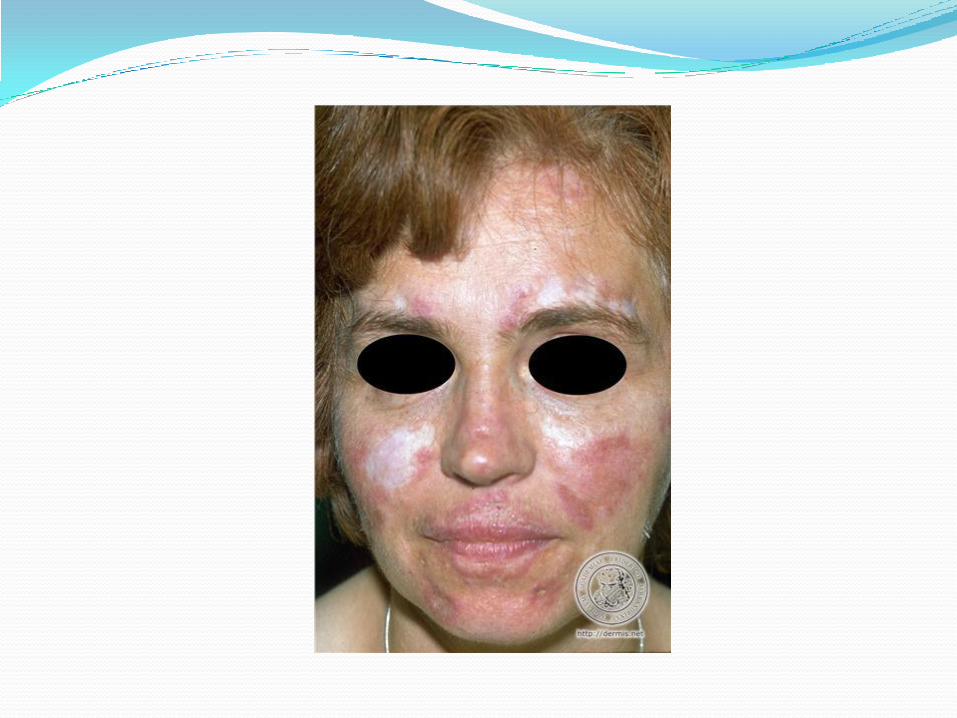

Photosensibilité Éruption cutanée à la lumière
érythémateux, maculopapuleux
Peut ulcérer
Zones photoexposées
Peut être prurigineux
Durée: jours
Étiologies: Lupus, dermatomyosite, rosacée, réaction médicamenteuse, réaction polymorphe à la lumière, porphyrie

Ulcères buccaux bénin:
chronique, intermittent sans changement de fréquence
suspect:
apparition récente, fréquent > 1/ mois
LED: tendance palais dur, moins symptomatique; association avec maladie systémique
Crohn, Behcet: ulcères superficiels, très douloureux, palais mou, joues, langue, groupées
DDx: Sprue, hémopathies, sx myélodysplasiques, neutropénie, VIH, herpes
Ulcère isolé chronique: infection, néoplasie, corps étranger

Exploration d’un symptôme musculosquelettique
Localisation: Articulaire vs péri-articulaire (vs douleur référée)
Inflammatoire vs non-inflammatoire
Aigu vs chronique (vs intermittent)
Distribution de l’atteinte
Manifestations extra-articulaires.

Mécanique Inflammatoire
Raideur matinale > 45-60 minutes
Éveils nocturnes
Amélioration par le repos
Examen: Gonflements
Manifestations extra-articulaires

Mécanique Inflammatoire
Raideur matinale > 45-60 minutes
√
Éveils nocturnes
Amélioration par le repos
Examen: Gonflements
Manifestations extra-articulaires

Mécanique Inflammatoire
Raideur matinale > 45-60 minutes
√
Éveils nocturnes √
Amélioration par le repos
Examen: Gonflements
Manifestations extra-articulaires

Mécanique Inflammatoire
Raideur matinale > 45-60 minutes
√
Éveils nocturnes √
Amélioration par le repos √
Examen: Gonflements
Manifestations extra-articulaires

Mécanique Inflammatoire
Raideur matinale > 45-60 minutes
√
Éveils nocturnes √
Amélioration par le repos √
Examen: Gonflements Indurés, osseux: arthrose Changements inflammatoires
Manifestations extra-articulaires

Mécanique Inflammatoire
Raideur matinale > 45-60 minutes
√
Éveils nocturnes √
Amélioration par le repos √
Examen: Gonflements Indurés, osseux: arthrose Changements inflammatoires
Manifestations extra-articulaires
√

Aigu vs chroniqueAigu Chronique
Inflammatoire Septique P.R.
Microcristallin Collagénoses
Traumatique SpA séronégatives
Viral Infections chroniques
Réactif, RAA Microcristallin
Pathologie chronique
Non-inflammatoire Trauma-Fx Syndrome d’accrochage
Rupture tendineuse Arthrose
Ostéonécrose Néoplasie
Hémarthrose Corps étranger
Ostéonécrose

Incidence
(par année)
Prévalence Âge
PR H: 0.2/1000
F: 0.4/1000
0.5-1% 40-60
SLE 2-5/100 000 1/2000 –
1/10 000
16-55: >65%
Sclérodermie 5-20/1 000
000
150/1 000 000 30-50
Sjogren 0.2% 0.5-2% 30-50
PM/DM 2-7/ 1 000
000
20-60/1 000000 10-15
40-60
MCTD 15-20/1 000
000
10/ 100 000 30-50

Collagénoses Conclusion # 1:
La plupart des collagénoses sont des maladies peu fréquentes; en général, donc, considérer d’autres possibilités diagnostiques avant les collagénoses, dans le diagnostic différentiel.

Exploration d’un symptôme musculosquelettique: collagénoses
Localisation: Articulaire vs péri-articulaire (vs douleur référée)
Inflammatoire vs non-inflammatoire
Aigu vs chronique (vs intermittent)
Distribution de l’atteinte: PR: IPP, MCP, poignets, coudes, épaules, genoux, chevilles,
MTPs, cou, (hanches)
Autres collagénoses: distribution rhumatoïde, plus souvent arthralgies; souvent sx distaux (mains, pieds), non érosif
Manifestations extra-articulaires.
Examens de laboratoire: Anticorps anti nucléaires

Autoanticorps et collagénoses La présence d’autoanticorps n’équivaut pas { un
diagnostic de collagénose.
Leur prescription doit être fondée sur une suspicion clinique raisonnable

Interpréter signification du test à la lumière de la clinique
Titre ANA Prévalence chez population normale
1 : 40 30%
1 : 80 10-12%
1 : 160 5-7%
1 : 320 3%
1 : 640 ≤ 1%

Facteur Antinucléaire
Interprétation:
Dilution de 1:160 et plus
En présence d’une clinique compatible
Faux positifs:
-Âge -Maladies immunes autres
-Infections que collagénoses
-Néoplasies -idiopathique

Facteur Antinucléaire
Faux positifs (suite):
Hydralazine ß-bloquants
Quinidine Sulfasalazine
Procainamide Phénothiazines
Phénytoine Tétracycline
INH Statines


FAN et collagénoses
Arbuckle MR. NEJM (2003);
349: 1526-1533

Anticorps antinucléaires Assez bonne sensibilité
Non spécifique
Donc, bon test de dépistage: Faire en 1er
Si positif et selon contexte clinique:
Faire recherche des ENA, et anti DNA
Autoanticorps Maladies associées
Ro, La Sjögren, LED, LCSA, photosensibilité, lupus néonatal
Sm LED (très spécifique mais infréquent)
u1RNP MCTD, LED, sclérodermie
Scl-70 (anti topoisomérase 1) Sclérodermie systémique diffuse
Anti ds DNA LED
Anti Jo-1 Polymyosite

Raynaud
Un homme de 45 ans consulte pour épisodes de blanchiment des doigts survenant principalement au froid
Une femme de 24 ans consulte pour épisodes de blanchiment des doigts survenant principalement au froid
Une femmde de 24 ans consulte pour sensation de froideur aux mains avec coloration bleutée (cyanose) de celles-ci
Quels éléments rechercher pour considérer un Raynaud secondaire?

Raynaud

Raynaud Primaire:
5-15% de la population; femmes surtout
Début: puberté; plus rare après 35-40 ans
Crises: symétriques
Pas d’événements ischémiques, ulcérations, atrophie
Pas d’anomalies des capillaires périunguéaux
Absence d’autoanticorps
Secondaire: y penser
Homme
après 40 ans
Un, deux doigts
crises asymétriques
Crises sévères
ulcérations
atrophie
anomalies lit capillaire périunguéal
Sclérodactylie
Symptômes systémiques
Autoanticorps

Raynaud secondaire

Raynaud TA aux 2 bras
Pouls périphériques
Recherche souffles cardiaques et vasculaires
Raynaud primaire?: pas autre test
Labos de base:
FSC
analyse d’urine
PT, PTT
VS
EPPs
FAN
FR
Selon suspicion clinique
kit sérologique
complément
anti topoisomérase 1, ACA
CPK
APL, Ac lupique
Cryoglobulines
Agglutinines froides
doppler artériel, angiographie (sx aigus, asymétrie de TA, pouls absents, ischémie sévère, atteinte d’un seul doigt)

ANA et sclérodermieSclérodermie diffuse Sclérodermie limitée (CREST)

Wigley. NEJM (2002);347: 1001

Sclérodermie Maladie systémique autoimmune
Femmes:hommes: 4-5:1
Age au Dx: moyenne: 50 ans
Manifestations les plus fréquentes: Raynaud
RGO
gonflement des doigts, sclérodactylie
Arthralgies
Absence de Raynaud et de sclérodactylie élimine pratiquement le Dx de sclérodermie

Sclérodermie: 2 grandes catégories
Limitée:
Raynaud de longue date
Sclérodactylie ou sclérodermie distale à coudes et/ou genoux
Pas d’atteinte du tronc
Atteintes systémiques moins fréquentes sauf
HTAP (10-20% des patients)
Télangiectasies: 80%
Diffuse:
Raynaud d’apparition récente
Atteinte cutanée plus extensive, incluant le tronc, et de progression rapide
Mx pulmonaire intersitielle (chez la plupart, mais slmt 10-20% ont mx progressive)
Crise rénale sclérodermique (10-20%)
atteinte GI plus basse, diarrhée chronique
Télangiectasies: 30%
Raynaudsclérodermie: initialement: oedème, puis, phase plus indurée, peau sèche, desquamation, Atteinte peau du visageRGO 75%

sclérodermie: investigation initiale Rechercher causes secondaires de Raynaud
FAN: 95% des patients
anti centromères: 20-30%: forme limitée
anti topoisomérase 1 (Scl-70): forme diffuse, fibrose pulmonaire
anti RNA polymérase I-III: atteinte diffuse, sévère, crise rénale, atteinte cardiaque
Dépistage d’organes cibles:
Études digestives: selon symptômes
ECG, echocardiaque
CT thorax, TFR
Biochimie de base, fonction rénale, analyse d’urine

Syndrome sec Vous voyez une femme de 62 ans, référée pour
impression de sécheresse buccale; ATCD: hypertension, hystérectomie il y a 25 ans, suite
ménorragies; elle croit avoir eu une transfusion à l’époque.
Sa médication: acetaminophène prn pour céphaléesamitriptyline au coucherhydrochlorotiazide 12.5 mg die
A-t-elle un syndrome de Sjögren?

Syndrome sec Xérostomie:
sécheresse buccale
sensation de soif
dysphagie
difficultés du port de dentiers
sensation de brûlure
complications: candidase, caries, cheilite, dysgueusie
Étiologies
Déshydratation
Respiration par la bouche
Médicaments:
antihistaminiques, TCA, neuroleptiques, clonidine, benzo, diurétiques
Obstruction conduits: radiation, lithiase
infiltrations: Sjogren, sarcoïdose, lymphome, GVHD, VIH, hépatite C, amyloïdose
Causes autres: oreillons, Coxackie

Syndrome sec Xérophtalmie:
impression de sécheresse occulaire
sensation de corps étranger
utlisation de gouttes
complications: conjonctivite, ulcération cornéenne
Étiologies
Production:
Déshydratation
Médicaments
Obstruction des conduits
Infiltrations
Évaporation:
Dysfn glandes de Meibomius
Verres de contact
anomalies de clignement des yeux (travail devant écran, Parkinson, Hyperthyroïdie,...)

Syndrome de Sjogren Maladie autoimmune systémique dont la principale
manifestation en est une d’exocrinopathieprincipalement des yeux et de la bouche.
primaire: absence d’évidence d’autre maladie immune systémique
secondaire: associé à autre maladie immune systémique
Atteintes extra-glandulaires relativement fréquentes
Caractéristique histopathologique: infiltrats lymphocytaires des glandes exocrines.

syndrome de Sjogren Évolution:
Habituellement lente, sans détérioration rapide de l’état
Manifestations extra glandulaires:
lésions caractérisées par des infiltrats lymphocytaires péri épithéliaux: pneumopathies, hépatite, néphrite intersitielle: indolent
lésions extra-épithéliales: glomérulonéphrite, neuropathie périphérique, purpura vasculaire: évolution peut être plus rapide, plus morbide.
fil directeur: présence de cryoglobulines

Syndrome de SjogrenCritères diagnostiques: subjectifs :
1) xérostomie (gonflement parotidien présent ad 50%)
2)xérophtalmie objectifs :
3)test de Schirmer ou rose Bengale 4) sialographie, scintigraphie des glandes salivaires 5) biopsie glandes salivaires mineures : utile pour exclure
autres maladies (sarcoïdose) ou si hôte atypique (homme avec VIH en autres)
6) anti-Ro/La diagnostic si ≥ 4 critères dont 5) ou 6) et exclusion conditions
associées à syndrome sec (irradiation cou, VIH, Hép C, lymphome, sarcoïdose, GVH, Rx anticholineriques, diurétiques…)

Sjogren: quand y penser contexte clinique évocateur
Exclusion causes iatrogènes (responsables ad 60% des cas)
Recherche atteinte autres glandes exocrines (trachée: toux, Estomac: gastrite atrophique, sécheresse vaginale, sécheresse cutanée (xérose)
Recherche de manifestations extraglandulaires compatibles: purpura, neuropathies périphériques, pneumopathie interstitielle, enzymites hépatiques, arthralgies/arthrite, Raynaud, atteinte rénale (tubulointerstitielle, glomérulaire)

Bilan initial Évaluation ophtalmologique: Schirmer, rose bengal, tear break up time
Évaluation ORL: ?Scintigraphie, Bx selon contexte
Labos:
FSC, VS
Électrolytes
EPPs
Analyse d’urine, créat.
CXR
FAN, recherche d’anti Ro et anti La
FR
enzymes hépatiques
TSH
Cryoglobulines

Syndrome de Sjögren Le risque de survenue d’un syndrome
lymphoprolifératif est accru environ 4% à 10 ans surtout: lymphome B, zone marginale, extranodal, bas
grade
Question: le risque relatif en comparaison à la population non atteinte est de 44
1. Vrai2. Faux3. Ça dépend...
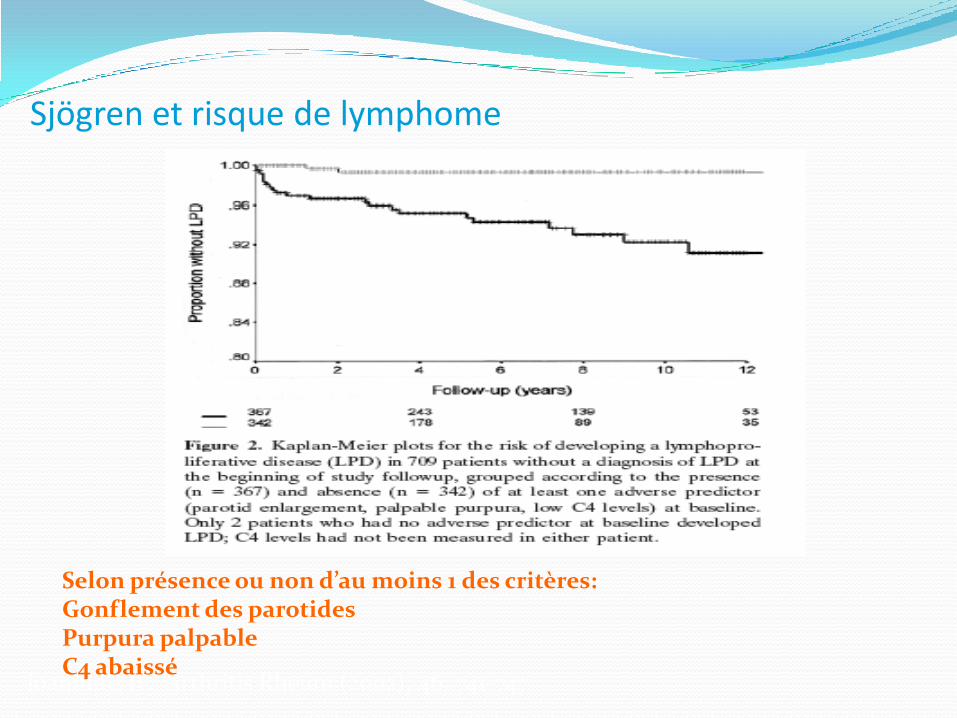
Sjögren et risque de lymphome
Ioannidis JP. Arthritis Rheum (2002); 46: 741-747
Selon présence ou non d’au moins 1 des critères: Gonflement des parotidesPurpura palpableC4 abaissé

Facteurs de risque de lymphome chez patients avec Sjögren
Gonflement parotides
Adénopathies
Neuropathie périphérique
Vasculite cutanée
Glomérulonéphrite
Hypocomplémentémie
Cryoglobulines
Anémie/lymphopénie
Voulgarelis M. Arthritis Rheum (1999); 42: 1765-1772Ioannidis JP. Arthritis Rheum (2002); 46: 741-747

Distinction des collagénoses Est-ce important?
Différents organes cibles
Différents traitements
Différents pronostics
Distinguer des imitateurs (infection, néo, médicaments…)
Comment?
Clinique
Labos, sérologies

Caractéristiques des collagénoses et imitateursMaladie Manifestations cliniques Labos / sérologies
Polyarthrite rhumatoïde Polysynovites, manifestations extraarticulaires moins fréquentes (sauf Sjögren)
FR, anti CCPSérologies autres moins fréquent (anti Ro, La) ou absentes (Sm, RNP, dsDNA)
Sclérodermie Raynaud et sclérodactylie chez 95% pts
Sérologies typiques: anti centromères, anti topoisoméraseI, anti RNA pol I, III
Sjögren Syndrome sec domine le tableau clinique, arthrite moins marquée/sévère, habituellement non érosive; vasculite cryo, infiltrlymphocytaire (pulm, foie, rein)
Anti Ro: 60%Anti La: 30%
PM/DM Faiblesse proximale dominante; rash évocateur; syndrome anti synthéthase
Anti Jo-1: 20-25%
MCTD Raynaud, doigts boudinés,sclérodactylie, myosite, arthrite
ANA positif haut titreAnti RNP positifs, exclusivement
UCTD Manifestations diverses évocatrices, mais ne correspondant pas une collagénose définie
ANA positifs
Chevauchement Clinique correspondant à plus d’un Dx
FR, anti CCP, ANA, ENA, autres

Caractéristiques des collagénoses et imitateurs
Maladie Caractéristiques cliniques Labos / sérologies
Vasculites systémiques Patterns cliniques évocateurs (voir plus loin)
Absence d’ANA et FR (*sauf vasculite cryo)ANCAHépatite B, C, VIHHistopathologie suggestive
Syndrome antiphospholipide Épisodes thrombotiques gros vaisseaux (petits vaisseaux aussi si CAPS), hxobstétricale
Anti ß2 GP1, anticoagulant lupique, anticardiolipines
Behcet Ulcères buccaux douloureux, génitaux avec cicatrice, uvéite, pseudofolliculite, panniculites
Absence d’autoanticorpsBiopsie: surtout infiltrats neutrophiliques, immunofluorescence négative
Still Fièvre intermittente,arthralgies/-ites, rash chez la majorité
Absence d’autoanticorps, ferritine typiquement très élevée
Lupus médicamenteux Arthralgie/-ites, rash, sérosites; rarement + sévère
ANA positifs 99% (homogène); anti histone

Groupe de maladies inflammatoires chroniques au cours desquelles la paroi des vaisseaux sanguins est la cible d’une réaction immune.
Elles peuvent être primaires:
Aucune étiologie connue ne peut être démontrée
ou secondaires:
Induction par : infection, néoplasie, collagénoses,
médicaments

CHAPEL HILL NOMENCLATURE
Gracieuseté de Loïc Guillevin

selon le type de vaisseau touché de façon prédominante
Gros vaisseaux: aorte et ses principales branches
Moyens vaisseaux: artères viscérales principales et leurs branches (peut inclure des artères moyennes et petites)
Petits vaisseaux: artérioles, capillaires, veinules

1. Manifestations multisystémiques inexpliquées
Exclusion de: Infections (bactériennes (endocardite), mycoses, viralesNéo: carcinomes, lymphomes, myélodysplasie,
leucémieEmboliques: valvulopathie: myxome, endocardite marantique, Libman-Sacks, cholesterolCoagulopathies, PTTMédicamentsCalciphylaxieSarcoïdose
Vascularites secondaires: causes ci-haut, collagénoses
2. Syndrome clinique caractérisé

Large Moyen petit
Claudication Livedo purpura
souffle nécrose digitale vésicules
Pouls ulcère papules nécrotiques
∆ TA érythème noueux Hémorragie alvéolaire
Anévrysme ischémie mésentérique Glomérulonéphrite
mononévrite multiplex splinters,
épisclérite/sclérite
microanévrysmes
3. Classification selon la taille de vaisseau touché de façon prédominante:



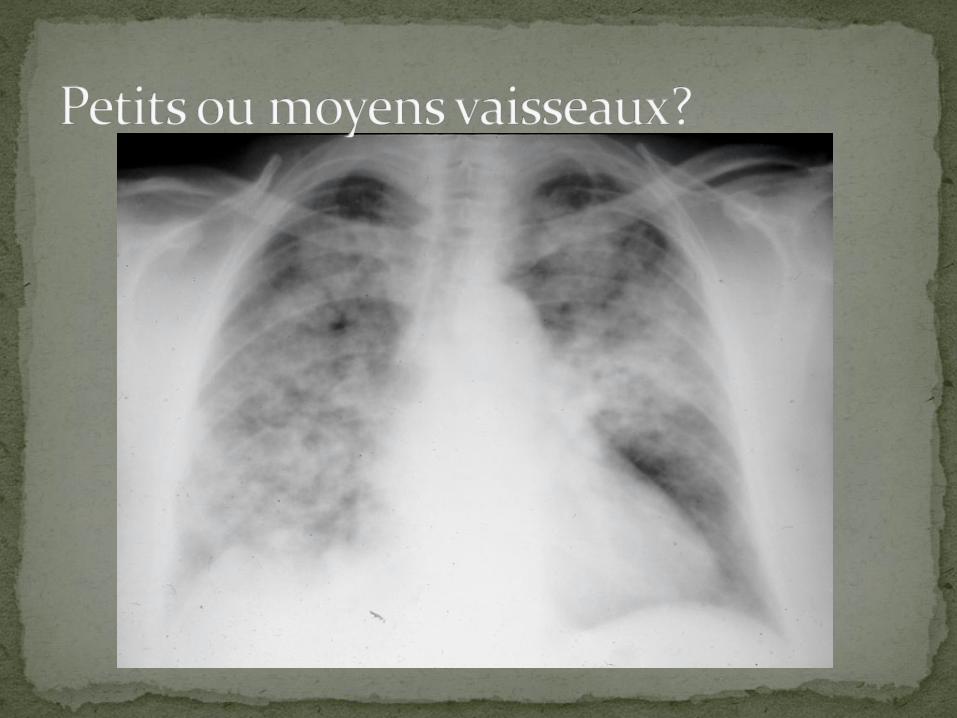


Vascularite nécrosante n’atteignant que les vaisseaux de moyen calibre, i.e., petites et moyennes artères
Étiologie inconnue
Dans certains cas, associée { infections par virus de l’hépatite B, et parfois hépatite C et VIH
Rares cas de vascularites nécrosantes avec: EBV, CMV, B19
Lésions histopathologiques caractérisées par régions d’inflammation focales, exsudatives, d’aspect nodulaire, le long des artères.

Perte de poids de plus de 4 kg Livedo reticularis Douleur testiculaire Myalgies, faiblesses, ou douleurs aux jambes Mononeuropathie ou polyneuropathie Pression diastolique > 90 mm Hg ↑ BUN ou créatinine HBsAg ou Ac + Anomalies artériographiques suggestives (anévrysmes,
occlusions) Biopsie: petite ou moyenne artère avec infiltrat de PMNs
≥3 critères: sensibilité: 82%spécificité: 86.6%
Inflammation nécrosante des petites ou moyennes artères, sans glomérulonéphrite ni vasculite des artérioles, capillaires ou veinules
Chapel Hill: PAN

30-60 ans Touche les hommes autant que les femmes Rare: incidence d’environ 1-3/1 000 000 Hyperthermie: 50-60% Perte de poids: 60-70% Manifestations musculosquelettiques: 70-80%
arthralgies, myalgies, arthrite Neuropathie périphérique: 50-70% Manifestations abdominales: 25-50% Hypertension: 20-25% Insuffisance rénale: 25% Lésions cutanées: 50%

Absence de glomérulonéphrite
Absence de manifestations pulmonaires
Pas associé à ANCA (présents chez au plus 5% des patients)

Granulomatose de Wegener (WG)
Polyangiite microscopique (PAM)
Syndrome de Churg-Strauss (CSS)
Glomérulonéphrite pauci-immune
Vasculites médicamenteuses à ANCA

vasculites à ANCA PAN
Taille des vaisseaux Petits et moyens:
artérioles, capillaires et
veinules post capillaires
moyens:
petites et moyennes artères
Manifestations
pulmonaires
OUI NON
Glomérulonéphrite OUI NON
Circulation artérielle rénale NON OUI
Peau Purpura palpable, macules, Nodules sous-cutanés,
ulcères, ischémie/gangrène
digitale
ANCA OUI
(30-90%)
NON
(0-5%)

Churg-Strauss Wegener PAM
rhinite
allergique/asthme/
atopie
++ - -
sinusites ++ ++ -
Poumons:
infiltrats
nodules
+ + +
+ + (cavitation) -
Eosinophilie +++ + -
Inflamm.
granulomateuse
+ + -
Infiltrats
rétroorbitaires
- + -
Neuropathies ++ + +
Glomérulonéphrite + ++ ++

Atteinte principalement des petits vaisseaux Souvent associée (jusqu’{ 90%) { l’infection par le virus
de l’hépatite C NB: 50% des patients avec hépatite C auront une
cryoglobulinémie. Seulement une petite proportion développera une vasculite cryoglobulinémique
Principaux symptômes: purpura Arthralgies/arthrite fatigue Polyneuropathie périphérique sensitive› motrice Glomérulonéphrite (dont évolution habituellement plus
bénigne que vasculites à ANCA)

4. Évaluation de l’étendue de l’atteinte
• Labos
• Imagerie
5. Sérologies
• ANA/ENA, ANCA, cryoglobulines, anti GBM, VIH, hépatite B, C
6. Biopsie/immunofluorescence, lorsque possible

Clinique: histoire et examen complets
Laboratoires
Imagerie
Biopsie lorsque possible
Exclusion de maladies infectieuses ou néoplasiques ou encore d’effets secondaires médicamenteux

Un homme de 45 ans, par ailleurs en bonne santé, sinon pour une hyperthyroïdie traitée, est hospitalisé pour hémorragie alvéolaire, et protéinurie à 2 g/24 heure.
Quels examens demander?

Buts: 1) définir l’extension de la maladie2) poser le bon diagnostic

De base: Permet d’évaluer sommairement l’extension de l’atteinte
FSC
VS/CRP
AST/ALT
EPP
Analyse d’urine et sédiment, si anormale.
Créatinine
Coagulogramme
Radiographie pulmonaire
Exclusion d’infection
Anémie: possible avec toutesVasculites à ANCA: leucocytose, thrombocytose
Rarement pénie. Si oui:Considérer: collagénose, néo, médicaments, désordre hémato
1/3 des Wegener ont uneatteintes pulmonaire asymptomatique
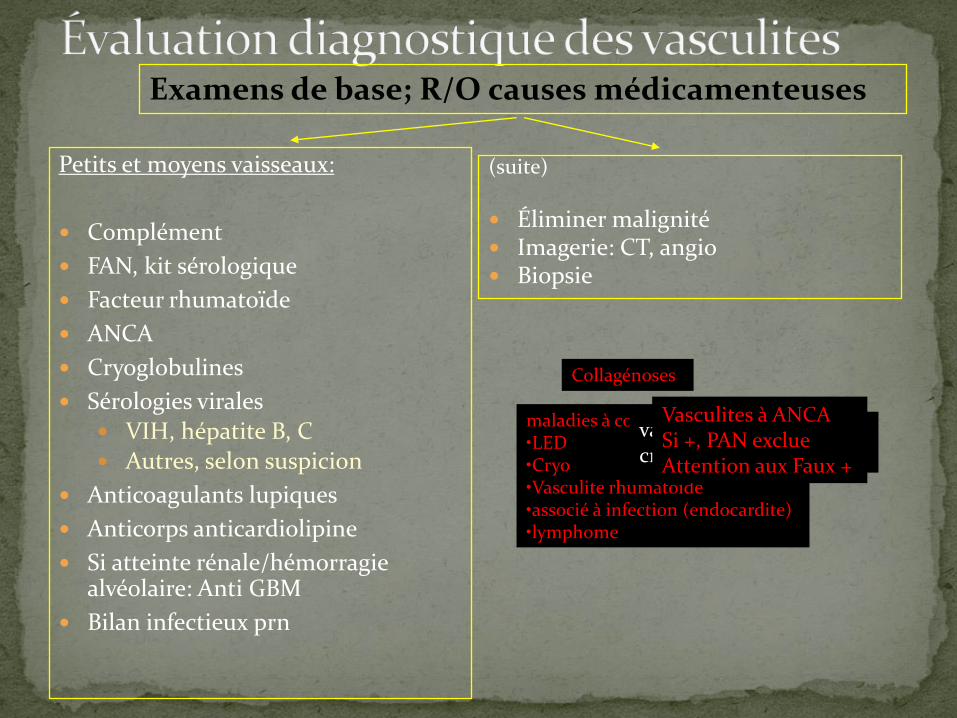
Petits et moyens vaisseaux:
Complément
FAN, kit sérologique
Facteur rhumatoïde
ANCA
Cryoglobulines
Sérologies virales VIH, hépatite B, C Autres, selon suspicion
Anticoagulants lupiques
Anticorps anticardiolipine
Si atteinte rénale/hémorragie alvéolaire: Anti GBM
Bilan infectieux prn
(suite)
Éliminer malignité Imagerie: CT, angio Biopsie
maladies à complexes immuns:•LED•Cryo•Vasculite rhumatoïde•associé à infection (endocardite)•lymphome
Examens de base; R/O causes médicamenteuses
Collagénoses
vasculite rhumatoïdecryoglobulinémie
Vasculites à ANCASi +, PAN exclueAttention aux Faux +

pANCAcANCA

Immunofluorescence indirecte:3 patrons
cANCA(rehaussement cytoplasmique)
Confirmation par ELISA ou RIA
Anti-PR3
pANCA(rehaussement périnucléaire
Confirmation par ELISA ou RIA
Anti-MPO
ELISA/RIA négatif
aANCA(ANCA atypiques)
granulomatose de WegenerEndocardite S aureus
PAMSyndrome de Churg StraussGN pauciimmune
vasculite médicamenteuse
InfectionsMx hépatiques immunesMII

Granulomatose de Wegener
Diffus: 90% (cANCA 75-85%)
Limité: 50-65%
Rémission: 30-35%
Polyangiite microscopique
50-80% (pANCA 60%)
Syndrome de Churg Strauss
30-70% (pANCA principalement)

Fonction de la probabilité pré-test
VPP
Manifestations ORL uniquement: 7-10%
Manifestations ORL-pulmonaires 45%
Manifestations ORL-pulmonaires-rein 98%
Besoin d’exclure les conditions mimant des vasculites et pouvant être ANCA +. (Infections, médicaments)
Le résultat d’ANCA + n’est en soi que rarement suffisant pour confirmer un diagnostic de vasculite: Bx lorsque possible

Radiographies:
Poumons: WG: Infiltrats interstitiels ou alvéolaires, parfois evanescents
Nodules pouvant devenir cavitaires
CSS: Infiltrats (surtout) evanescents chez 38-70%
Nodules: moins fréquents, moins tendance à cavitation
Hémorragie alvéolaire: rare
PAM: Infiltrats alvéolointerstitiels

Radiographies:
Sinus: Peuvent être anormales même chez pts asymptomatiques: épaississements muqueux
Mauvais examen:
Sensibilité mauvaise
Mauvaise définition spatiale
Radiation élevée
Avantageusement remplacé par CT scan sinus et orbites

Tomographie axiale (sinus, thoracique, yeux):
Sinus: Épaississement muqueuxdestruction, erosions osseuses, cartilagineuses (WG)
N.B. Les changements sont non-spécifiques et toute récidive a/n sinus, d’autant plus si isolée, devrait d’abord faire penser à une infection
Orbites (WG): Infiltrat rétroorbitaireInfiltrat muscles extra-oculaires
Thorax: Nodules, infiltrats interstitiels, hémorragie alvéolaire peuvent être présents, même en présence d’une radiographie pulmonaire normale
N.B. Adénopathies ne sont pas des trouvailles usuelles
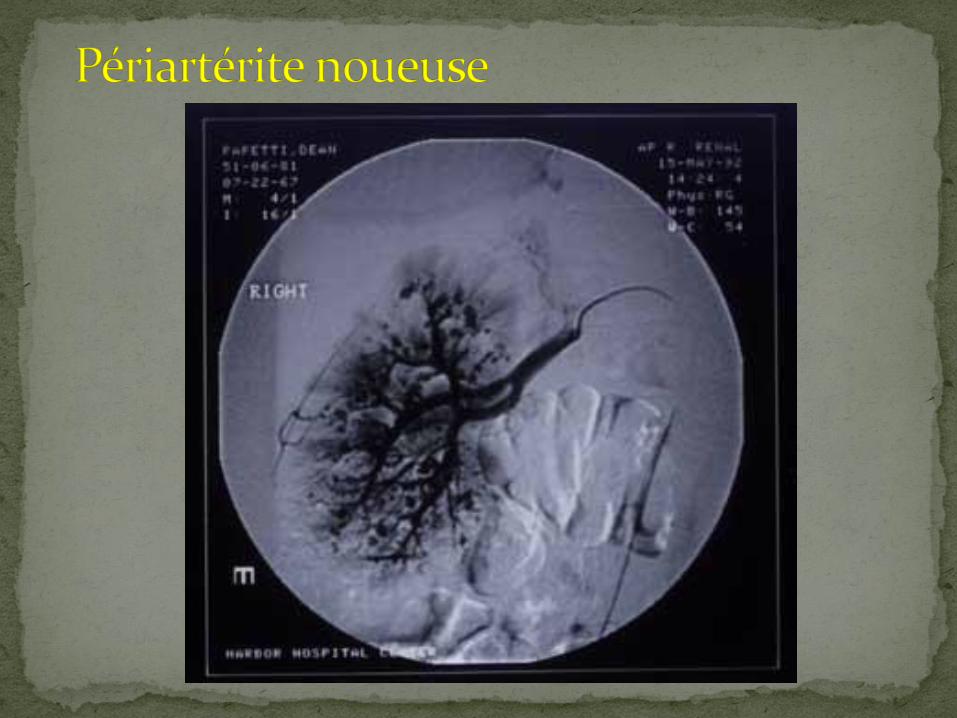

Vasculites primaires:
PAN
Kawasaki
Behcet
Wegener
Churg Strauss
PAN:
Pratiquement tous les patients avec angio positive sont symptomatiques
nausées, vomissements
diarrhée, rectorragie
douleur abdominale
symptômes systémiques
Angio:
lésions multiples
sténoses/occlusions, en plus des anévrysmes

Vasculites secondaires
infection
néo: tricholeucémie
PR
LED
syndrome anti phospholipide
Causes non vasculitiques:
drogues
athérosclérose
dysplasie fibromusculaire(surtout sténoses)
Maladies du collagène
Marfan
EDS (IV)
autres

Biopsie: Dx: Idéalement toujours, si possible
Où: Selon organe touché
ORL (Wegener): sinus: 55%
muqueuse nasale: 20%
larynx: 18%
Poumons: Bx ouverte: Meilleur rendement Dx: ad 91%
BTB: Mauvais rendement : 7%
Rein: glomérulonéphrite
Utile pour l’immunofluorescence (DDx)
inflammation granulomateuse: rare

Où: Selon organe touché
Peau: Toujours faire immunofluorescence
Nerf: Si présence de symptôme dans le territoire correspondant; de préférence aux membres inférieurs Prélever aussi muscle: augmenter rendement (Said G. Neurol Clin 1997)
Muscle: Si suggestion de myopathie, ou absence apparente de site à biopsier; peut être guidée par irm
musculaire

Un homme de 45 ans, par ailleurs en bonne santé, sinon pour une hyperthyroïdie traitée, est hospitalisé pour hémorragie alvéolaire, et protéinurie à 2 g/24 heure.
Pas d’évidence pour une collagénose; le bilan infectieux est négatif; les ANCA sont positifs, ainsi que les anticorps anti PR3 et MPO. Anti GBM -N’a pas eu de biopsie.Il s’améliore jusqu’{ l’obtention de la rémission clinique, sous
immunosuppresseurs.Il consulte à nouveau 3 semaines après son congé, pour récidive de
dyspnée et infiltrats alvéolaires.
Quoi faire?

Toute récidive de symptômes sous thérapie adéquate doit être considérée comme une infection (ou néoplasie ou effet secondaire médicamenteux) jusqu’{ preuve du contraire
Infections: tous les immunosuppresseurs
Pneumopathies interstitielles: Cy, MTX, AZA
Toxicité hépatique: Cy, MTX, AZA
Tocixité rénale: MTX, AZA

Peut-on prédire une rechute?
1) Le diagnostic est important:
1) Granulomatose de Wegener: environ 50%
1) Les formes limitées rechutent davantage
2) PAM: 10-35 %
3) Churg Strauss: 20-34 %
4) PAN:
HBV +: 12,2%
HBV -: 32,5%
5) Artérite temporale: ad 40-50%, selon les séries

La sévérité de la maladie initiale n’influence pas le risque de rechute
? risque accru si infection récente
ANCA et risque de rechute: Un titre élevé de façon persistante Titre négatif qui redevient positif Élévation du titre La plupart des rechutes s’accompagnent
d’une élévation des ANCAs* 29% des pts n’auront pas de rechute malgré élévation des
ANCAs
En conséquence, ont une utilité limitée dans le suivi: Suivre le patient de plus près si élévation du titrePas d’indication de modifier le traitement
Corrélation très imparfaite

Vasculites à ANCA: Induction:
FSC, VS/CRP, Biochimie, Créatinine, Analyse d’urine: Aux 2 semaines
Consolidation: FSC, VS/CRP, Biochimie, Créatinine, Analyse d’urine: Aux 4-6 semaines
Fin des médicaments: Aux 3 mois ou selon la clinique.
Radiographie pulmonaire: aux 6 mois pour les 2 premières années aux 6-12 mois par la suite
ANCA: ?

Combien de temps sera-t-il traité?
Quelle est sa probabilité de rechute?

Propylthiouracil
Allopurinol
Hydralazine
Minocycline
ciprofloxacine
Clozapine
Penicilline
Sulfasalazine
phenytoine
•Manifestations peuvent survenir quelques semaines à quelques mois après le début du médicament
•Peuvent être légères à sévères, indissociables de celles rencontrées dans la granulomatose de Wegener
•Notre expérience: manifestations cutanées, arthralgies/arthrite, uvéites représentent les manifestations les plus fréquentes

1) Exclure infections: Cultures, echocardiaque, sérologies virales (hépatites), VIH
2) Exclure néoplasies: selon âge et selon indices cliniques
3) Exclure coagulopathies (par exemple, si manifestation ischémiques)
4) Exclure collagénoses: selon manifestations cliniques (pas d’indication de demander toutes les sérologies en l’absence de faible probabilité pré test)
5) Exclure médications causales, y compris agents sympathomimétiques
6) Analyse et sédiment urinaire: chez tout patient avec mx multisystémique
7) Biopsie: toujours, lorsque possible
8) Imagerie: CT, angiographie, IRM: selon syndrome clinique


Sclérodermie: principes thérapeutiques
• Pas de traitement de fond• Traitement selon organes touchés• Peau: phase initiale, oedémateuse, inflammatoire
– immunosupression peut être suggérée– anti TNF?
• Rein:– éviter stéroïdes hautes doses si possible– AINS: attention– crise rénale: IECA
• Poumons:– Mx interstitielle: alvéolite: stéroïdes, cyclophosphamide– HTAP: anticoagulation, O2, prostaglandines IV, bosentan, ?BCC
• TGI:– manoeuvres anti reflux, IPP, prokinétiques– diarrhées persistantes: ATB empiriques pour pullulation bactérienne

Sjogren: principes thérapeutiques
• Hygiène oculaire• Hygiène dentaire• bonbons, gomme sans sucre• Éviter Rx responsables de syndrome sec• Agents muscariniques: pilocarpine• Biothérapies:
– anti TNF: pas efficace– IFN alpha: plus ou moins efficace– Rituximab: peu efficace– hydroxychloroquine