cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein...
TRANSCRIPT

The FASEB Journal • Research Communication
Cigarette smoking affects oxidative protein folding inendoplasmic reticulum by modifying proteindisulfide isomerase
Harshavardhan Kenche,* Catherine J. Baty,‡ Kokilavani Vedagiri,* Steven D. Shapiro,§
and Anna Blumental-Perry*,†,1
*Anderson Cancer Institute, Memorial Health University Medical Center, and †Department ofBiomedical Sciences, Mercer University School of Medicine, Savannah, Georgia, USA; ‡Departmentof Cell Biology and §Division of Pulmonary, Allergy, and Critical Care Medicine, Department ofMedicine, University of Pittsburgh, Pittsburgh, Pennsylvania, USA
ABSTRACT The endoplasmic reticulum (ER) stressresponse (ERSR) and associated protein aggregation, isunder investigation for its role in human diseases,including chronic obstructive pulmonary disease (COPD)where cigarette smoking (CS) is a risk factor for diseasedevelopment. Our hypothesis states that CS-associatedoxidative stress interferes with oxidative protein fold-ing in the ER and elicits ERSR. We investigated ERSRinduction following acute CS exposure and delineatedmechanisms of CS-induced ERSR. Lung lysates frommice exposed or not to one cigarette were tested foractivation of the ERSR. Up to 4-fold increase in phos-phorylation of eIF2� and nuclear form of ATF6 wasdetected in CS-exposed animals. CS affected the for-mation of disulfide bonds through excessive posttrans-lational oxidation of protein disulfide isomerase (PDI).Increased amounts of complexes between PDI and itsclient proteins persisted in CS-exposed samples. BiPwas not a constituent of these complexes, demonstrat-ing the specificity of the early effects of CS exposureon ER. Disturbances in protein folding were accompa-nied by changes in the organization of ER network andER exit sites. Our results provide evidence that ERSR isinduced early in response to CS exposure and identifiesthe first known ER-resident target of CS PDI, demon-strating that CS affects oxidative protein folding.—
Kenche, H., Baty, C. J., Vedagiri, K., Shapiro, S. D.,Blumental-Perry, A. Cigarette smoking affects oxida-tive protein folding in endoplasmic reticulum bymodifying protein disulfide isomerase. FASEB J. 27,965–977 (2013). www.fasebj.org
Key Words: COPD � ER stress response
Chronic obstructive pulmonary disease (COPD) isa leading cause of morbidity and mortality. Much isknown about the pathogenesis of COPD, but a moredetailed picture has yet to emerge (1). The role ofstructural alveolar cells of the lung has only recentlybegun to be recognized. No treatments exist that haltthe progression of the disease; therefore, understand-ing the pathways contributing to the pathogenesis ofCOPD will have therapeutic implications. Cigarettesmoke (CS) exposure and the resultant oxidative stressare major risk factors for the development of COPD.The effects of oxidative stress have been extensivelyinvestigated, but it has only recently become evidentthat oxidative stress may have profound effects on thefunction of the endoplasmic reticulum (ER) by disturb-ing various aspects of protein folding, thus resulting ininduction of the ER stress response (ERSR) (2).
Involvement of ERSR in COPD pathogenesis is anarea of active investigation, since ERSR acts by alteringthe expression of genes involved in ER maintenance,antioxidant defense, inflammation, and apoptosis (3,4). Moreover, CS exposure and COPD are predisposingfactors for the development of lung cancer (5). Re-search in the field of cancer has revealed that ERSR ishighly induced in tumors, including those of the lung(6–9), and that ERSR induction is associated with
1 Correspondence: Memorial Health University MedicalCenter, Anderson Cancer Institute, Hoskins Research Bldg.,4700 Waters Ave, Savannah, GA 31405, USA. E-mail: [email protected]
doi: 10.1096/fj.12-216234This article includes supplemental data. Please visit http://
www.fasebj.org to obtain this information.
Abbreviations: ATF4, activation transcription factor 4;ATF6, activation transcription factor 6; BiP, immunoglobulinbinding protein; Chop, C/EBP homologous protein; COPD,chronic obstructive pulmonary disease; CS, cigarette smoke;CSE, cigarette smoke extract; DAB, 3,3-diaminobenzidine;DMPH, 2,4-dinitrophenylhydrazine; DSP, dithiobis(succin-imidyl propionate); eIF2�, eukaryotic translation-initiationfactor 2�; ER, endoplasmic reticulum; ERES, endoplasmicreticulum exit site; ERS, endoplasmic reticulum stress; ERSR,endoplasmic reticulum stress response; FBS, fetal bovineserum; HMWC, high-molecular-weight protein complex; IP,immunoprecipitation; IRE1, inositol-requiring protein 1;MLE12, mouse lung epithelial 12; NAC, N-acetyl-l-cysteine;NEM, N-ethylmaleimide; Nrf2, nuclear factor erythroid re-lated factor 2; PDI, protein disulfide isomerase; PERK, pro-tein kinase RNA (PKR)-like ER kinase; TLR, toll-like receptor;Tm, tunicamycin; XBP1, X-box binding protein 1
9650892-6638/13/0027-0965 © FASEB

cancer cell survival as well as resistance to anticancertreatments. Indeed, cancers appear to experience mul-tiple episodes of ER stress (ERS), triggered by hypoxiaand a lack of proper vascularization. The ERSR triggersa series of intracellular events that aim to help cellsovercome the consequences of stress and adapt to newconditions. If adaptation is unsuccessful and stressunresolved, ERSR signaling contributes to the elimina-tion of damaged cells through apoptosis (10). Cells thatsurvive become more resistant to a diverse range ofstressful challenges, which can aid in the developmentof tumors. Therefore, CS-induced ERSR may have adual role in smoke-related diseases in that it cancontribute to cell loss as seen in COPD and can alsoyield stress-resistant cells that are predisposed to malig-nant transformation. Three major sensors, inositol-requiring protein 1 (IRE1), protein kinase RNA (PKR)-like ER kinase (PERK), and activation transcriptionfactor 6 (ATF6), regulate ERSR through their respec-tive signaling cascades. On the accumulation of mis-folded proteins, PERK dimerizes and phosphorylateseukaryotic translation-initiation factor 2� (eIF2�), lead-ing to general translation inhibition; except in the caseof mRNAs whose upstream open reading frames allowthem to escape inhibition associated with the phos-phorylation of eIF2�, such as ATF4 mRNA (11). IRE1dimerizes to activate its kinase and RNase activities,initiating unconventional splicing of X-box bindingprotein 1 (XBP1) mRNA and creating a potent tran-scriptional activator, Xbp1. ATF6 commutes to theGolgi compartment, where it is cleaved by S1P and S2Pproteases to yield a cytosolic fragment that migrates tothe nucleus and activates transcription of ERS-respon-sive genes (12). The ERSR contributes to antioxidantdefense through PERK signaling. This condition isachieved by the activation of PERK, which consequentlyleads to enhanced function of nuclear factor erythroidrelated factor 2 (Nrf2), the transcription factor respon-sible for the induction of antioxidant defense genes(13). The transcription factor ATF4, a downstreamtarget of PERK, forms heterodimers with Nrf2 to coor-dinate and optimize both the ER and oxidative stressresponses (14).
All three branches of ERSR have been shown tocontribute to inflammation. Subsequently, correlationsbetween CS-induced ERSR and the inflammatory re-sponse in COPD have been suggested (4, 15, 16).Furthermore, it has been demonstrated that Toll-likereceptors (TLRs), which are pattern recognition recep-tors, play a significant role in the pathogenesis ofCOPD (17). TLR4 was found to have a protective roleagainst CS-induced apoptosis (18), and TLR4 andTLR9 were shown to be involved in CS-induced cyto-kine production (19). Recently, crosstalk between com-ponents of ERSR and components of both innate andadaptive immunity through TLR was established in livertissues (20); however, similar connections to ERSR inthe lungs are currently unclear.
To date, it has been demonstrated that CS exposureinduces ERSR in cell cultures (8, 21, 22); however, data
concerning the induction of ERSR in vivo is compli-cated. Korfei et al. (23) analyzed ERSR in patients withidiopathic pulmonary fibrosis (IPF) and used patientswith COPD, not grouped by the severity of the disease,as controls. The researchers did not detect a robustinduction of ERSR in tissues from patients with COPDas compared to patients with IPF. Nevertheless, Kelsenet al. (24) utilized the proteomic approach and demon-strated that several ER proteins [immunoglobulin bind-ing protein (BiP), calreticulin, and protein disulfideisomerase (PDI)] are up-regulated in smokers and mayhave a protective effect in the lungs. Min et al. (25)demonstrated that the magnitude of ERSR, measuredby the phosphorylation of eIF2�, the presence ofC/EBP homologous protein (Chop), and the overex-pression of retrograde translocation complexes, corre-lates with the severity of the disease in human smokers.
In COPD, as in many other conditions, diseaseprogression correlates with the severity of ERSR (26). Itis unclear whether in vivo CS is a primary cause of ERSRor whether ERSR only accompanies disease progres-sion. The purpose of this study was to see whether themurine model of one-time CS exposure results in theactivation of ERSR, as well to identify ER-relevant,downstream targets of CS that are currently unknown.
MATERIALS AND METHODS
Reagents
N-ethylmaleimide (NEM) and N-acetyl-l-cysteine (NAC) werepurchased from Sigma-Aldrich (St. Louis, MO, USA). Dithio-bis(succinimidyl propionate) (DSP) was from Thermo Scien-tific (Waltham, MA, USA) and 3-(2,4 dioxocyclohexyl)propyl(DCP-Bio1) was from KeraFast (Boston, MA, USA). Thefollowing primary antibodies were used: eIF2�, eIF2�-P (CellSignaling Technologies, Danvers, MA, USA), ATF6 (U.S.Biologicals, Swampscott, MA, USA, and Abcam, Cambridge,MA, USA), monoclonal PDI (clone 1D3, Enzo Life Sciences,Farmingdale, NY, USA; and clone RL77, Thermo-Pierce,Rockford, IL, USA), polyclonal PDI (Thermo-Pierce), anti-Grp78 (Stressgen, Farmingdale, NY, USA), CHOP (AffinityBioreagents, Rockford, IL, USA), calreticulin (Epitomics,Burlingame, CA, USA), actin (Abcam). HRP-conjugated sec-ondary antibodies (anti-mouse, anti-rabbit) were fromThermo-Pierce. Alexa-594 and Alexa-488 were from Molecu-lar Probes (Eugene, OR, USA).
Mice
C57BL/6 female mice (8–10 wk old) from Taconic Farms(Germantown, NY, USA) were used for experiments. All micewere housed and used for the experiments under the direc-tion and approved protocols of the Mercer University Insti-tutional Animal Care and Use Committee (protocolA1009016).
Acute CS exposure in vivo
C57BL/6 female mice aged (8–10 wk) were exposed to oneunfiltered cigarette or a sham, using the smoking chamber asdescribed previously (27).
966 Vol. 27 March 2013 KENCHE ET AL.The FASEB Journal � www.fasebj.org

Tunicamycin (Tm) injection
C57BL/6 female mice (8–10 wk of age) were given a singleintraperitoneal injection (1 �g/g body weight) of 0.05 mg/mlsuspension of Tm in 150 mM dextrose (28).
Tissue processing
Animals were terminally anesthetized by a combination ofketamine/xylazine (100/5–10 mg/kg) followed by cervicaldislocation. Lungs were either flash-frozen or inflated asdescribed below.
Tissue processing for Western blot analysis
Lung and liver tissue homogenates were prepared by homog-enizing the tissue in lysis buffer (20 mM Tris, pH 7.4; 150 mMNaCl; and 0.5% Triton X-100) or RIPA buffer (150 mM NaCl;50 mM Tris, pH 8.0; 2 mM EDTA; 1% Nonidet P-40; and 0.1%SDS) supplemented with a protease inhibitory cocktail tablet(Roche, Indianapolis, IN, USA) and phosphatase inhibitorycocktails 2 and 3 (Sigma-Aldrich). Lysates were cleared bycentrifugation and analyzed by gel electrophoresis. Proteinconcentration was determined using Bradford protein assay(Bio-Rad, Hercules, CA, USA) with BSA as a standard, andverified using a Coomassie blue gel staining.
Tissue processing for immunohistochemistry (IHC) analysis
Lungs were inflated by instilling intratracheally a10% neutralbuffered formalin at 25 cmH2O for 10 min. They were thenligated and removed. Inflated lungs were fixed in the forma-lin solution for 24 h, followed by 48 h of cold PBS wash andparaffin embedding. Serial sagittal sections of 4 �m wereobtained for IHC.
Cell culture
The mouse lung epithelial 12 (MLE12) cell line was pur-chased from American Type Culture Collection (Manassas,VA, USA). MLE12 cells were cultured in RPMI1640 medium(Cellgro, Manassas, VA, USA) and supplemented with 4%fetal bovine serum (FBS; Serum International, Laval, QC,Canada), insulin-transferrin-selenous acid (ITS) premix (BDBiosciences, San Jose, CA, USA), 10 nM hydrocortisone(Sigma-Aldrich), 10 nM �-estradiol (Sigma-Aldrich), 10 mMHEPES, 2 mM glutamine (CellGro), 100 U/ml penicillin, and100 �g/ml streptomycin. All experiments were performed oncells between passages 2 and 16.
Preparation of cigarette smoke extract (CSE)
Research-grade 1R5F cigarettes (Kentucky Tobacco andHealth Research Institute, Lexington, KY, USA) were used toprepare the CSE. CSE was made fresh for each experiment bybubbling the smoke from 1 cigarette through 5 ml of serum-free medium in a 15-ml conical tube and filtering it througha 0.22-�m filter to remove large particles and maintainsterility. This solution was designated as 100% CSE anddiluted in culture medium to yield concentrations specifiedfor each experiment.
Cell lysate preparation
MLE12 cells were cultured to 75–90% confluence, incubatedwith or without CSE for the indicated time periods. Cells were
lysed on ice in 300 �l RIPA lysis buffer, supplemented withprotease and phosphatase inhibitors, passed 5 times througha 1-cc tuberculin syringe, and cleared by centrifugation.Protein concentration was determined by Bio-Rad Bradfordassay.
Nuclear extract preparation for ATF6 p50 detection
The nuclear form of ATF6 was detected by modification toScheiber et al. (29). Briefly, MLE12 cells were washed twicewith PBS, supplemented with proteasome inhibitor ALLN(40 �M); allowed to swell 15 min on ice in 10 mM Hepes (pH7.9), 10 mM KCl, 0.1 mM EDTA, and 0.1 mM EGTA supple-mented with 1 mM DTT, 10 �g/ml leupeptin, 313 �g/mlbenzamidine, 10 �g/ml pepstatin, and 10 �g/ml addition ofnonidet P-40 to the final concentration of 0.625%. Nuclearand postnuclear pellets were separated from the rest bycentrifugation. Pellets were resuspended in 20 mM Hepes(pH 7.9), 0.4 NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM PMSF,1 mM DTT, and 40 �M ALLN with rocking at 4°C for 5 min.Nuclear extracts were cleared by centrifugation and analyzedby Western blot.
PDI redox state and high-molecular-weight protein complex(HMWC) formation in cells
Procedures were performed as previously described (30).Briefly, cells were cultured to 75–90% confluence, incubatedwith or without CSE for indicated time periods, with orwithout 10 mM DTT or 5 mM diamide for 10 min at 37°C.Cells were washed twice with ice-cold PBS supplemented with20 mM NEM, to protect reduced disulfide bonds from furtheroxidation during lysis, then lysed in lysis buffer (20 mM Tris,pH 7.4; 150 mM NaCl; and 0.5% Triton X-100) for 30 min onice, and cleared by centrifugation. Protein concentration wasdetermined by Bio-Rad Bradford protein assay with BSA as astandard; 50 �g of the total protein was separated by nonre-ducing polyacrylamide gels (no DTT, not boiled). Gels (14%)were run at 4°C at 10 mA/gel to distinguish between redoxforms of PDI. Gels (8%) were used to detect complexesbetween PDI and its client proteins.
Crosslinking experiments
For in vivo crosslinking, MLE12 cells were cultured to 75–90%confluence, incubated with or without CSE for the indicatedtime periods. Crosslinking was performed as described byNawa et al. (31). Briefly, CSE cells were placed on ice, washedtwice with ice-cold PBS, and incubated at 4°C for 30 min with1.0 mM DSP in PBS (see Supplemental Fig. S2A for DSPconcentration optimization), then washed twice with coldPBS containing 0.04% EDTA. Cells were lysed in 350 �l of 50mM Tris (pH 7.4), 150 mM NaCl, 1% Triton-X, and 1 mMEDTA, supplemented with protease and phosphatase inhibi-tors by incubation on ice for 30 min. The lysates were clearedby centrifugation at 15,000 g for 10 min and analyzed byWestern blotting.
In vitro crosslinking was performed on frozen lung tissuesobtained from control and CS-exposed animals as describedin Oda et al. (32). Briefly, frozen lung tissues were homoge-nized in 500 �l of 50 mM bicine-NaOH (pH 8.0), 150 mMNaCl, 1% Nonidet P-40, and 5 mM iodoacetamide supple-mented with 0.4 mM DSP and protease and phosphataseinhibitors (see Supplemental Fig. S2B for DSP concentrationoptimization). Homogenates were vortexed for 10 s andincubated on ice for 30 min. Reaction was stopped byincubation with 5 mM glycine on ice for 10 min. Lysates were
967CIGARETTE SMOKING MODIFIES PDI AND CAUSES UPR

cleared by centrifugation at 15,000 g for 10 min and analyzedby Western blotting.
RNA extraction and XBP1 splicing analysis
RNA was extracted using TRIzol reagent (Life Technologies,Gaithersburg, MD, USA) followed by Qiagen RNeasy kit(Qiagen, Valencia, CA, USA). cDNA synthesis was carried outusing the High Capacity cDNA Reverse Transcription Kit(Applied Biosystems, Foster City, CA, USA).
XBP1 and actin RT-PCR
XBP1 PCR was performed using primers 5=-CACGCTTGGG-AATGGACACGCT-3= and 5=-TTGTCCAGAATGCCCAAAAGG-3=,95°C for 3 min, followed by 35 cycles of 95°C for 1 min, 60°Cfor 1 min, and 72°C for 1 min, followed by 10 min at 72°C.The PCR products were run on 2.3% agarose gels, showingbands of 231 and 205 bp corresponding to native and splicedforms of XBP1, respectively. Actin PCR was performed usingthe same amounts of cDNA as in XBP1 PCR. The primers5=-CCGCCCTAGGCACCAGGGTG-3= and 5=-GGCTGGGGT-GTTGAAGGTCTCAAA-3= were used, 95°C for 3 min, fol-lowed by 20 cycles of 95°C for 1 min, 55°C for 1 min and 72°Cfor 1 min, followed by 10 min at 72°C. The band intensitieswere quantified using Quantity-One software (Bio-Rad),XBP1 spliced band intensity was normalized to actin bandintensity.
Immunohistochemical analysis of ATF6
Paraffin-embedded parasagittal sections of inflated lungswere subjected to antigen retrieval by steaming in 10 mM Trisand 1 mM EDTA buffer (pH 9.0). Endogenous peroxidaseactivity was quenched by incubation in 3% H2O2. Immuno-histochemistry for ATF6 was performed using the ABC kit(Vector Laboratories Inc., Burlingame, CA, USA) accordingto manufacturer instructions. Briefly, sections were blockedin Dako protein block supplemented with 5% goat serum andfollowed by avidin and biotin blocking (Vector Laboratories).The tissue sections were then incubated with rabbit anti-ATF6amino-terminal end (4 �g/ml; Abcam,) overnight at 4°C,followed by incubation with biotinylated secondary antibodyand avidin. 3,3-Diaminobenzidine (DAB; Vector Laborato-ries) was used as the chromogen; Mayer’s hematoxylin (Ther-moScientific) was used for counterstaining. All sections wereprocessed together. Images were acquired on an Olympusinverted scope IX51 (Olympus, Tokyo, Japan) using ImageJsoftware (U.S. National Institutes of Health, Bethesda, MD,USA). Acquisition setup was identical for all images. Images(6–12) of each animal were acquired and saved in a desig-nated folder. Intensity of DAB staining was analyzed using amacro written for ImageJ as described below.
Macro for DAB intensity analysis
The macro imported all pictures from a selected folder as astack and converted them to 8 bit, which converted theimages to greyscale, where original color values correspond todifferent intensities of the scale. Each stack of images wasthen separated into individual images and analyzed by count-ing the number of pixels in the image that corresponded toeach light intensity value (from 0 to 255) using the histogramfunction under the Analyze drop-down menu. The resultswere processed into a 2-column table, where “value” indicatesthe greyscale light intensity value and “count” indicates thenumber of pixels found at each light level. The resulting table
was saved as a separate file. The information in these tableswas further analyzed in Excel (Microsoft, Redmond. WA,USA). The Excel analysis tool used was built on the premisethat the brown DAB chromogen lies within the greyscalevalues of 0–71. The reason for this assumption was thatthe brown DAB chromogen staining is darker than the bluecounterstaining, so low light values should include only whathas been stained brown, and light values � 70–75 includeonly the blue-stained and unstained regions of the sample.Light values � 71 include most brown-stained regions whileavoiding the most blue-stained regions. This assumption wasconfirmed empirically. The macro creates a graphic presen-tation of the results, where light values are plotted on the xaxis, and the mean pixel count across all samples from thesame sample is plotted on the y axis.
Transient transfections of MLE12 cells and Luciferasereporter assays
Transfections and assays were performed using Effectenetransfection reagent (Qiagen) and the Dual-Glo LuciferaseAssay System (Promega, Madison, WI, USA), respectively.MLE12 cells were plated on 24-well plates at a density of 3 �103 cells/well and were allowed to adhere for 8 h andtransfected using Effectene transfection reagent according tothe manufacturer’s instructions. Briefly, cells were cotrans-fected with one of the reporter plasmids (0.4 �g/well),containing ATF6 binding sites, XBP1 binding site and BiPpromoter fused to firefly luciferase reporter, and Renillareporter plasmid (20 ng/well). Cells were incubated with thetransfection mixture overnight, washed with PBS, and ex-posed or not to CSE added to fresh MLE12 medium. Lu-ciferase and Renilla values were measured 6 h after CSEaddition using the Dual-Glo assay system according to themanufacturer’s instructions, using a standard luminometer.Luciferase values were normalized to Renilla values to accountfor transfection efficiency. Reporter plasmids were p5xATF6-GL3 (Addgene, Cambridge, MA, USA), Grp78 promoterreporter �169/LUC (gift from Amy Lee, University of South-ern California, Los Angeles, CA, USA), pEGFP-XBP1-Luc(Ron Prywes, Columbia University, New York, NY, USA), andpGL4.75 (CMV-Renilla; Promega).
PDI immunoprecipitation (IP)
Cells or lung tissues were lysed in 20 mM Tris (pH 7.4), 150mM NaCl, 0.5% Triton X-100 supplemented with a proteaseinhibitor cocktail (Roche), and phosphatase inhibitor cock-tails 2 and 3 (Sigma-Aldrich). Lysates (500–750 �g) wereprecleared with protein A/G plus-agarose (Thermo-Pierce).PDI was precipitated with a mixture of mouse anti-PDIantibodies (Enzo anti-PDI-1D3, 20 �g/500 �g protein; andPierce anti-PDI-RL77, 35 �g/500 �g). The RL77 was moreefficient in precipitating PDI from HMWCs at 4°C overnight.For electrophoresis, the samples were heated to 95°C for 10min in nonreducing conditions.
Detection of carbonylated proteins
Carbonylated proteins were detected using the OxyBlot Pro-tein Oxidation Detection Kit (Millipore, Bedford, MA, USA)according to the manufacturer’s instructions.
Detection of carbonylated PDI
Proteins (100 �g) from fresh cell lysates or lung tissuehomogenates were derivatized for 5 min using the OxyBlot kit
968 Vol. 27 March 2013 KENCHE ET AL.The FASEB Journal � www.fasebj.org

(Millipore). The reaction was stopped by the addition ofneutralization solution per the manufacturer instructions. Toget rid of 2,4-dinitrophenylhydrazine (DNPH), proteins werepelleted by ultracentrifugation (Beckman TL-100 ultracentri-fuge, TLA100.2 rotor; Beckman Coulter, Fullerton, CA, USA)at 100,000 g for 1 h at 4°C. Protein pellets were washed withIP lysis buffer and resuspended in 300 �l of fresh IP-lysisbuffer. PDI was immunoprecipitated as described above,resolved on nonreducing gels, followed by Western blottingprobed with anti-DNP rabbit antibodies according to OxyBlotkit instructions.
Real-time fluorescent microscopy
MLE12 cells (2�105) were plated on 35-mm glass-bottomeddishes (MatTek, Ashland, MA, USA) and cultured for 24–48h prior to imaging. To visualize the ER before and after theaddition of CS, MLE12 cells were washed with Hanks solution,fed with ER-trackergreen (Molecular Probes) for 10 min,rinsed with PBS, and incubated in MEM (Sigma-Aldrich)supplemented with 10 mM MES, 10 mM HEPES, 2.4 mMsodium bicarbonate (NaHCO3; pH 7.1), and 10% FBS at37°C on a temperature-controlled stage with or without 10%CSE. Images were acquired using an Olympus inverted mi-croscope equipped with a �60 1.4-N.A. oil-immersion objec-tive. Pairs of DIC and fluorescence images were collectedevery 15 min using time-lapse spinning video microscopy(Ultraview; Perkin Elmer, Wellesley, MA, USA). Shutter andfilter wheel timing and position were controlled by Meta-Morph Software (Molecular Devices Corp., Sunnyvale, CA.USA). Final images and movies were produced using Meta-Morph and Adobe Photoshop (Adobe Systems, San Jose, CA,USA).
Confocal microscopy
MLE12 cells were plated on coverslips, allowed to adhere for24 h, and incubated with or without 10% CSE, fixed with3.8% PFA at indicated time points after the addition of CSE,permeabilized by 0.05% saponin in PBS, and stained forcalreticulin and PDI. Confocal images were obtained using anOlympus Fluoview FV1000. Final images and movies wereproduced using MetaMorph and Adobe Photoshop.
Statistics
Data are expressed as means � sd. The blot intensities weremeasured in arbitrary densitometric units by Quantity-Onesoftware and then compared using Student’s t test to deter-mine the P value. Values of P �0.05 were considered statisti-cally significant.
RESULTS
Acute exposure to one cigarette is sufficient toactivate ERSR in mice
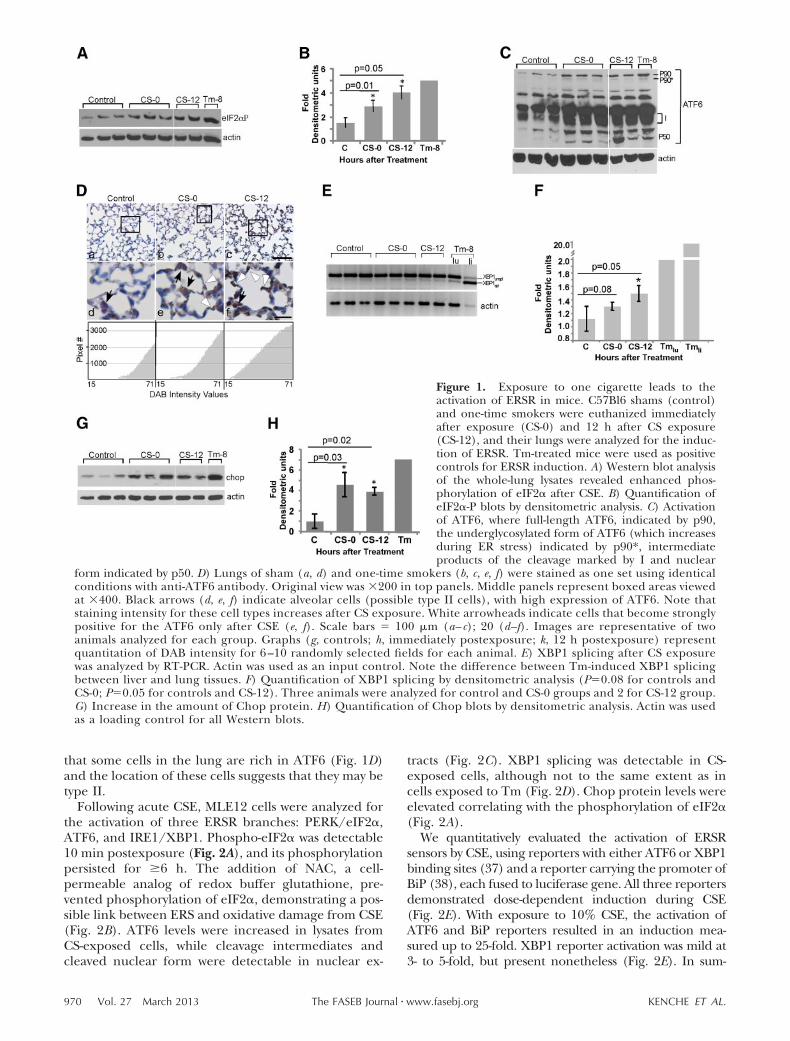
To determine whether activation of ERSR is an imme-diate result of CS exposure, wild-type mice were ex-posed to smoke from one cigarette and euthanized atvarious times postexposure. Unexposed mice were usedas controls. Total lung lysates were analyzed for activa-tion of the three branches of ERSR. An increase in theamount of phospho-eIF2� in CS-exposed mice wasobserved (Fig. 1A, B). Densitometric analysis of normal-
ized blots quantified the increase in the range of 2- to4-fold, comparable to the increase of phospho-eIF2�seen in lungs exposed to Tm.
Western blot demonstrated an increase in under-glycosylated intermediate and nuclear forms of ATF6in smoker lungs (Fig. 1C). IHC showed that there wasa small subset of cells that exhibited strong ATF6staining in control animals (Fig. 1D, black arrows). InCS-exposed animals, the staining was of greater in-tensity, not restricted to one cell type (Fig. 1Db, c, e, f), andnuclear localization was often visible. Notably, cellsthat form the walls of alveolar units were clearlypositive for ATF6 staining (Fig. 1D, white arrows).Both phospho-eIF2� and activated ATF6 were de-tected immediately after and up until 12 h afterexposure.
Activation of the IRE1 branch was assessed by XBP1splicing. Very low splicing was detectable in the lungs ofcontrol animals, while mild enhancement of the bandcorresponding to the spliced form of XBP1 was detect-able in the lungs of smokers (Fig. 1E, F; comparecontrols and CS-12). Semiquantitative RT-PCR foundthat an increase in XBP1 splicing was less than 2-fold 12h postexposure (Fig. 1F), supporting previous findingsthat CS has an inhibitory effect on XBP1 splicing in vitro(8, 21). However, it is possible that this inhibitionlessens at later times. A comparison of XBP1 splicing inthe lungs and livers of animals exposed to Tm revealedmild splicing in lungs (2-fold) and robust splicing inlivers (20-fold, Fig. 1E, F). Therefore, the significance ofthis 2-fold increase in the spliced form of XBP1 and itspresence after repeated smoking events requires fur-ther clarification.
Chop is a downstream protein in ERSR, and itsinduction correlates with the recovery of the ER fromstress or the switch to a proapoptotic arm of ERSR(33). Analysis of CHOP mRNA and protein expres-sion revealed that minute amounts of Chop protein ispresent in the total lung without the challenge of CS.CS exposure caused no statistically significantchanges in CHOP mRNA (not shown), but didincrease Chop protein in smokers (Fig. 1G, H). Thismay reflect post-transcriptional and translational sta-bilization of the already existing Chop protein inCS-exposed cells. In summary, CS exposure leads toERSR induction via phosphorylation of eIF2� andactivation of ATF6.
ERSR induced in MLE12 cells in vitro
MLE12, an epithelial cell line isolated from mouse lungtumors used to model alveolar epithelial cells, waschosen for analysis (34). MLE12 cells are type IIsurfactant-producing cells that would be greatly af-fected if proper protein folding in ER were disrupted.Type II cells are important for the repair of CS-induceddamage due to their ability to specialize into type I cells(35). Moreover, acute CS exposure predominantly im-poses oxidative stress on alveolar type II cells andbronchiolar epithelial cells (36). IHC staining shows
969CIGARETTE SMOKING MODIFIES PDI AND CAUSES UPR
that some cells in the lung are rich in ATF6 (Fig. 1D)and the location of these cells suggests that they may betype II.
Following acute CSE, MLE12 cells were analyzed forthe activation of three ERSR branches: PERK/eIF2�,ATF6, and IRE1/XBP1. Phospho-eIF2� was detectable10 min postexposure (Fig. 2A), and its phosphorylationpersisted for �6 h. The addition of NAC, a cell-permeable analog of redox buffer glutathione, pre-vented phosphorylation of eIF2�, demonstrating a pos-sible link between ERS and oxidative damage from CSE(Fig. 2B). ATF6 levels were increased in lysates fromCS-exposed cells, while cleavage intermediates andcleaved nuclear form were detectable in nuclear ex-
tracts (Fig. 2C). XBP1 splicing was detectable in CS-exposed cells, although not to the same extent as incells exposed to Tm (Fig. 2D). Chop protein levels wereelevated correlating with the phosphorylation of eIF2�(Fig. 2A).
We quantitatively evaluated the activation of ERSRsensors by CSE, using reporters with either ATF6 or XBP1binding sites (37) and a reporter carrying the promoter ofBiP (38), each fused to luciferase gene. All three reportersdemonstrated dose-dependent induction during CSE(Fig. 2E). With exposure to 10% CSE, the activation ofATF6 and BiP reporters resulted in an induction mea-sured up to 25-fold. XBP1 reporter activation was mild at3- to 5-fold, but present nonetheless (Fig. 2E). In sum-
Figure 1. Exposure to one cigarette leads to theactivation of ERSR in mice. C57Bl6 shams (control)and one-time smokers were euthanized immediatelyafter exposure (CS-0) and 12 h after CS exposure(CS-12), and their lungs were analyzed for the induc-tion of ERSR. Tm-treated mice were used as positivecontrols for ERSR induction. A) Western blot analysisof the whole-lung lysates revealed enhanced phos-phorylation of eIF2� after CSE. B) Quantification ofeIF2�-P blots by densitometric analysis. C) Activationof ATF6, where full-length ATF6, indicated by p90,the underglycosylated form of ATF6 (which increasesduring ER stress) indicated by p90*, intermediateproducts of the cleavage marked by I and nuclear
form indicated by p50. D) Lungs of sham (a, d) and one-time smokers (b, c, e, f) were stained as one set using identicalconditions with anti-ATF6 antibody. Original view was �200 in top panels. Middle panels represent boxed areas viewedat �400. Black arrows (d, e, f) indicate alveolar cells (possible type II cells), with high expression of ATF6. Note thatstaining intensity for these cell types increases after CS exposure. White arrowheads indicate cells that become stronglypositive for the ATF6 only after CSE (e, f). Scale bars 100 �m (a– c); 20 (d–f). Images are representative of twoanimals analyzed for each group. Graphs (g, controls; h, immediately postexposure; k, 12 h postexposure) representquantitation of DAB intensity for 6 –10 randomly selected fields for each animal. E) XBP1 splicing after CS exposurewas analyzed by RT-PCR. Actin was used as an input control. Note the difference between Tm-induced XBP1 splicingbetween liver and lung tissues. F) Quantification of XBP1 splicing by densitometric analysis (P0.08 for controls andCS-0; P0.05 for controls and CS-12). Three animals were analyzed for control and CS-0 groups and 2 for CS-12 group.G) Increase in the amount of Chop protein. H) Quantification of Chop blots by densitometric analysis. Actin was usedas a loading control for all Western blots.
970 Vol. 27 March 2013 KENCHE ET AL.The FASEB Journal � www.fasebj.org

mary, MLE12 cells reflect findings in the total lung andrepresent a good model for an in-depth understanding ofmechanisms of CS-induced ERSR.
CSE affects formation of disulfide bonds duringoxidative protein folding
Knowing that CS directly induces ERSR in exposed cellsand animals, aspects of ER function compromised byCS had to be determined. The protective effect ofglutathione suggests that oxidation of an unknowntarget is important (Fig. 2B and refs. 3, 8), The ER is aconsumer of glutathione, which is imported from thecytosol and utilized in the disulfide bond formationduring oxidative protein folding (39). PDI, which trans-fers oxidative equivalents to newly synthesized proteins,is one of the central proteins in oxidative proteinfolding (40). We analyzed the redox state of PDI inCS-exposed cells and found that PDI became hyperoxi-dized, with no reduced form detected (Fig. 3A, top gel).
The majority of PDI usually exists as part of amultiprotein complex that is formed during proteinfolding by the association of client proteins with ERchaperones and isomerases (41). These associations aretransient and reversible. The ER quality-controlmachinery senses when a client protein achieves itsnative conformation and releases it from the chaper-one-PDI complex for export from the ER (42). Whenfolding is compromised, client proteins that do notassume the mature conformation persist in these mul-tiprotein complexes, called HMWCs. We testedwhether formation of HMWCs is enhanced by CSE byanalyzing complexes formed by PDI on nonreducinggels. HMWCs were more prominent in CS-exposed cells
than in untreated cells (Fig. 3A, middle gel, nonre-duced). The effect of CS exposure on their persistencewas dose-dependent (Fig. 3A). Persistence of HMWCsmay eventually lead to the formation of insolublemultiprotein complexes, which correlates with per-turbed ER function and predisposes cells to undergo-ing apoptosis (33). Figure 3A (arrow) demonstrates theinsolubility of HMWCs in CS-exposed cells by theirresistance to boiling and detergent. The formation ofHWMCs was prevented by the addition of NAC, imply-ing that oxidative damage to PDI is responsible forHWMC formation (Fig. 3B). Notably, NAC also pre-vented activation of ERSR in CS-exposed cells, linkingHWMC formation to the induction of ERSR (Fig. 2B).Levels of HMWCs were directly correlated with theduration of CS exposure (Fig. 3C, top gel). The ERchaperone, BiP, is a prominent constituent of abnor-mal HMWCs in some systems (33); however, it does notparticipate in disulfide bond formation but ratherassists in other aspects of protein folding. Its presencein HMWCs correlates with a poor prognosis for cellrecovery. We found that very little BiP is associated withHMWCs in CS-exposed MLE12 cells even after pro-longed exposure at all tested conditions (Fig. 3C,middle gel, and Supplemental Fig. S2 for optimizationof calcium and ATP levels to ensure continued BiPinteraction). To test whether our in vitro finding thatCS exposure does not increase BiP association withHMWCs reflects the in vivo situation, in vivo crosslink-ing of proteins was performed on cells exposed or notto CSE. A very minor increase in the association of BiPwith HMWCs was found in CS-exposed cells, while anincrease in PDI associated with HMWCs was 6- to10-fold (Fig. 3D, E).
Figure 2. CS exposure leads to theactivation of ERSR in MLE12 cells.A) MLE12 cells exposed or not toCSE for the indicated periods oftime were analyzed for the pres-ence of phosphorylated eIF2� andfor an increase in Chop protein.B) MLE12 cells were incubatedwith or without 1 mM NAC for 30min prior to addition of CSE, ex-posed to 10% CSE for 3 and 6 h,and analyzed for the presence ofphosphorylated eIF2�. C) MLE12
cells were exposed to 10% CSE for 6 h, and nuclear lysates were prepared and analyzed for the presence offull length (p90) and nuclear form (p50) of ATF6. D) XBP1 splicing was analyzed in MLE12 cells exposedto 10% CSE or Tm for 2 h, by RT-PCR. Note that in CSE cells, the unspliced band is weaker than in controlcells, while the spliced is stronger. E) Cells were cotransfected in triplicate with indicated ERS reportersmixed with Renilla reporter for 16 h. Amount of luciferase was normalized to the amount of Renilla activityafter 6 h of exposure to CSE and Tm. Experiments were performed 3 times.
971CIGARETTE SMOKING MODIFIES PDI AND CAUSES UPR
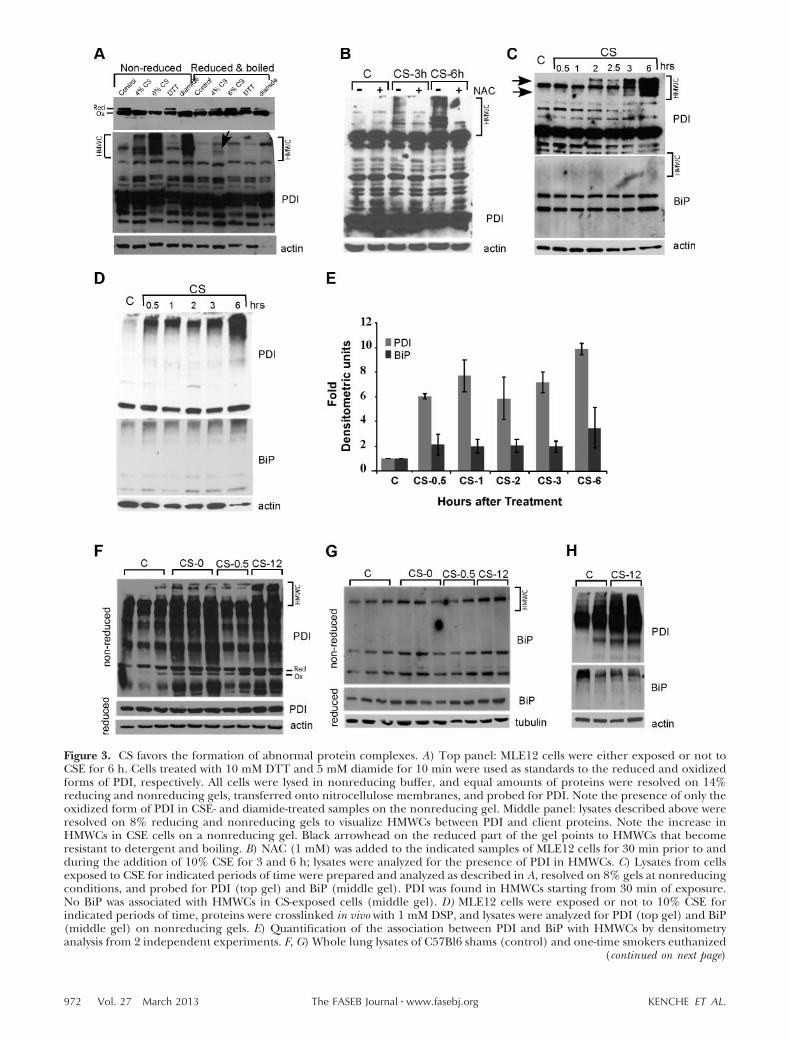
Figure 3. CS favors the formation of abnormal protein complexes. A) Top panel: MLE12 cells were either exposed or not toCSE for 6 h. Cells treated with 10 mM DTT and 5 mM diamide for 10 min were used as standards to the reduced and oxidizedforms of PDI, respectively. All cells were lysed in nonreducing buffer, and equal amounts of proteins were resolved on 14%reducing and nonreducing gels, transferred onto nitrocellulose membranes, and probed for PDI. Note the presence of only theoxidized form of PDI in CSE- and diamide-treated samples on the nonreducing gel. Middle panel: lysates described above wereresolved on 8% reducing and nonreducing gels to visualize HMWCs between PDI and client proteins. Note the increase inHMWCs in CSE cells on a nonreducing gel. Black arrowhead on the reduced part of the gel points to HMWCs that becomeresistant to detergent and boiling. B) NAC (1 mM) was added to the indicated samples of MLE12 cells for 30 min prior to andduring the addition of 10% CSE for 3 and 6 h; lysates were analyzed for the presence of PDI in HMWCs. C) Lysates from cellsexposed to CSE for indicated periods of time were prepared and analyzed as described in A, resolved on 8% gels at nonreducingconditions, and probed for PDI (top gel) and BiP (middle gel). PDI was found in HMWCs starting from 30 min of exposure.No BiP was associated with HMWCs in CS-exposed cells (middle gel). D) MLE12 cells were exposed or not to 10% CSE forindicated periods of time, proteins were crosslinked in vivo with 1 mM DSP, and lysates were analyzed for PDI (top gel) and BiP(middle gel) on nonreducing gels. E) Quantification of the association between PDI and BiP with HMWCs by densitometryanalysis from 2 independent experiments. F, G) Whole lung lysates of C57Bl6 shams (control) and one-time smokers euthanized
(continued on next page)
972 Vol. 27 March 2013 KENCHE ET AL.The FASEB Journal � www.fasebj.org

To confirm that PDI HMWCs formed in vivo, micewere either exposed or not to the smoke of onecigarette and euthanized at various times postexposure.Figure 3F demonstrates that there was an increase inPDI-containing HMWCs in lungs in response to smoke(Fig. 3F, top gel). CS-induced damage to oxidativeprotein folding was long lasting; PDI-containingHMWCs were most prominent 12 h postexposure andno BiP was found in HMWC in vivo at any time (Fig. 3G,upper gel). Figure 3H confirms our findings in cross-linked samples. This indicates that PDI may be themain ER-resident target of CS during initial exposure.BiP’s absence from HMWCs may attest to the nonapo-ptotic nature of the effects of CS at early times postex-posure.
CS-induced oxidation of PDI is responsible forinduction of ERSR
PDI is a conformation and redox-sensitive protein (43).In a few pathological conditions, post-translationalmodifications of PDI by abnormal oxidation, nitrosyla-tion, or glutathionylation render it inactive throughpossible changes in the 3D conformation of the protein(44, 45). CS exposure, a known source of ROS, cancause post-translational oxidation and affect properfunction of PDI. Figure 3A demonstrates that PDI inCS-exposed cells runs predominantly as an oxidizedprotein. To distinguish between physiologically or ab-normally oxidized PDI, we further analyzed post-trans-lational modifications of PDI. Although we detectedsome nitrosylated and glutathionylated proteins in
HMWCs after CS exposure, immunoprecipitated PDIshowed no nitrosylation or glutathionylation patterndifferent from that of controls (Supplemental Fig. S3).
To test whether CS exposure leads to the carbonyla-tion of PDI, freshly prepared lysates from control andCS-exposed cells were derivatized using the OxyBlotprotein oxidation detection kit; PDI was immunopre-cipitated, and its carbonylation was visualized usingWestern blot. PDI was found excessively carbonylatedin CS-exposed cells, with most of the oxidized PDIcorresponding to the fraction residing within HMWCs(Fig. 4A). Our in vitro findings reflect happenings invivo, where CS exposure causes marked carbonylationof PDI (Fig. 4B). Similar to in vitro findings, themajority of oxidized PDI resided within the HMWCfraction in association with client proteins that fail tofold properly. We conclude that CS affects the redoxstate of ER and oxidative protein folding by affectingthe activity of PDI by post-translational oxidation.
CS affects ER morphology and ER exit site (ERES)maintenance and distribution
We used live-cell fluorescent microscopy of CS-exposedcells to evaluate the kinetics and implications of CSinterference with protein folding in the ER by usingER-Trackergreen. In cells exposed to CSE, the formationof large aggregates was observed 15 min after CSEaddition (Fig. 5A, botttom panel, and SupplementalMovie S2), demonstrating that the effect of CSE onprotein folding in ER is rapid and occurs with exposureto a single cigarette. Aggregates became more defined
immediately (CS-0), 30 min (CS-0.5), and 12 h (CS-12) after CS exposure were analyzed for the presence of PDI (F) and BiP(G) in HMWCs on nonreducing (top panels) and reducing gels (middle panels). H) In vitro crosslinking of proteins wasperformed in total lung lysates obtained from control and CS-exposed animals. Lysates were checked for the presence of PDI(top gel) and BiP (lower gel) in HMWCs. Note the accumulation of PDI in HMWCs in smokers, especially 12 h after CSexposure. No BiP was associated with HMWCs in CSE animals. Either actin or tubulin was used as a loading control.
Figure 4. CS-induced oxidation of PDI. A) Leftpanel: MLE12 cells were either exposed or notto 10% CSE for 6 h, lysed, and immediatelyderivatized with OxyBlot detection kit for 5min. The extensive carbonylation of proteins isvisible in CS-exposed cells. The carbonylatedproteins tend toward high-molecular-weightproteins. Right panel: lysates from control andCS-exposed cells were derivatized using Oxy-Blot detection kit, and PDI was immunoprecipi-tated from derivatized lysates. IP of PDI was runon a nonreducing gel and analyzed by Westernblotting using anti-DNP antibody from OxyBlotkit. The main 57-kDa band corresponding toPDI is not significantly different between con-trols and CSE cells. PDI in HMWCs from CS-exposed cells shows extensive carbonylation.B) Fresh lysates from C57Bl6 shams (control)
and one-time smokers euthanized immediately (CS-0), 30 min (CS-30=), and 12 h (CS-12) postexposure were derivatized withDNPH, and PDI was immunoprecipitated as described in A. PDI, which persisted in HMWCs (unresolved, long-lastingcomplexes between PDI and its client proteins), was found to be excessively carbonylated. Note that the main 57-kDa band isweaker in CS-30= and CS-12 as compared to the control animals, while the PDI fraction in HMWCs is higher in smokers.
973CIGARETTE SMOKING MODIFIES PDI AND CAUSES UPR

with time and persisted during the entire imagingperiod (2 h for CSE cells). No aggregates were seen incontrols throughout 3 h of imaging (Fig. 5A, top panel,and Supplemental Movie S1). Therefore, even acute CSexposure interferes with ER morphology, and as aconsequence, likely interferes with proper functioningof the ER.
Having shown that CS has immediate negative effectson protein folding and ER morphology, we checkedwhether CS affects the exit of proteins from ERthrough evaluation of ERESs. ERESs are specializeddomains of ER where the exit of properly foldedproteins from ER occurs (46). ERESs are usually long-lived structures, and their quantity stays constant foreach particular cell (47). We followed ERESs in CS-exposed cells by calreticulin immunolabeling. ERESs incontrol cells appeared as dotted structures spreadthroughout the cell, with significant concentration inperinuclear regions (Fig. 5Ba, white arrows) while PDImarked reticular ER network (Fig. 5Bb). After CSEaddition, the number of both peripheral and perinu-clear ERES was diminished (Fig. 5Bc, white arrows),and PDI was found in dense aggregates (Fig. 5Bd,arrowheads). We concluded that the architecture ofERESs was altered after acute CSE.
DISCUSSION
We have found that acute CS exposure in vivo modifiesPDI and affects oxidative protein folding in ER, whichin turn leads to perturbations in ER homeostasis andinduction of ERSR through PERK (eIF2�) and ATF6.The third branch of ERSR, demonstrates a statistically
nonsignificant increase in the amount of spliced XBP1at later times post-CSE. This exclusion of the XBP1branch in response to CS, especially at early timespostexposure, correlates with a previously reported CSvapor inhibitory effect on XBP1 splicing in vitro (8, 21).Nevertheless, an increase in the total and spliced formsof XBP1 mRNA was seen in patients with COPD (48).Exposure to Tm that resulted in robust XBP1 splicingin livers caused a marginal 2-fold increase in XBP1splicing in lungs. Therefore, mild XBP1 activation mayalso reflect tissue- and cell type-specific differences.Type II MLE12 cells demonstrated moderate XBP1splicing after CSE. The implications of cell type-specificinhibition of XBP1 splicing to the resistance or suscep-tibility of cells to CSE must be investigated.
It is particularly intriguing because the absence ofXBP1 splicing may mimic a deletion in XBP1. It hasbeen demonstrated that gut specific deletions of XBP1in mice lead to diseases characterized by chronic in-flammation (49, 50). Can CS inhibition of XBP1 splic-ing in certain cell types contribute to chronic inflam-mation seen in COPD? Moreover, it has been foundthat stimulation of murine macrophages with TLR4ligand lipopolysaccharide (LPS) results in XBP1 splic-ing (17). Does CS exposure lead to the inhibition ofXBP1 splicing in alveolar macrophages, and will thisaffect the functioning of alveolar macrophages in smok-ers?
It has been shown that inhibition of XBP1 splicingfavors the activation of autophagy in murine models ofneurodegenerative disorders (51). Activation of au-tophagy halts the progression of some neurodegenera-tive diseases by reducing the accumulation of mis-folded, aggregated proteins. Autophagy is strongly
Figure 5. CS affects ER morphology and maintenance of ER exit sites. A) For live-cell imaging. ER of MLE12 cells was visualizedusing ER-trackergreen, cells were placed on temperature-controlled stage and exposed or not to 10% CSE as described inMaterials and Methods. Images were collected every 15 min. Top panel: control cells imaged for 3 h without the addition of CSE.Bottom panel: images acquired at 15-min intervals after addition of pH-balanced 10% CSE. B) Cells were grown on cover slidesfor 24–48 h, exposed or not to 10% CSE for 30 min. Cells were fixed in 3.8% PFA and stained for PDI (b, c) and calreticulin(CRT; a, d). White arrows point to perinuclear ERESs, which disintegrate in CSE cells; arrowheads point to dense PDI-containingaggregates seen in CSE cells.
974 Vol. 27 March 2013 KENCHE ET AL.The FASEB Journal � www.fasebj.org

activated by CS exposure in vitro and in vivo; however,this activation was connected to the activation of anextrinsic apoptotic pathway involving Fas-ligand, not tothe clearance of protein aggregates (52). Does CS-induced inhibition of XBP1 splicing contribute torobust activation of CS-induced autophagy, deregula-tion of which is an important player in COPD patho-genesis?
Activation of ERSR sensors was accompanied byinduction of some aspects of the downstream ERSRprogram, as demonstrated by the phosphorylation ofeIF2� and an increase in Chop protein. We did notdetect a robust induction of classical ERSR genes at thelevel of mRNA following acute CSE; however, 25 of 84genes on UPR array were mildly up-regulated (notshown). Up-regulation of several ER-resident proteins,such as BiP, PDI, and calreticulin, was demonstrated inthe lungs of chronic smokers, but not in patients withCOPD (24, 48). Up-regulation of Chop protein inmultiple human lung cell types, especially in alveolarmacrophages, was reported after acute exposure, butthe response weakened during chronic exposure (53).Therefore, the fine-tuning of CS-induced ERSR pro-gram and its interactions with other stress signalingpathways activated by CS exposure are important forthe understanding of the pathogenesis of COPD.
We tested the effects of CS exposure on oxidativeprotein folding and identified PDI, a redox-sensitivekey component in the formation of disulfide bonds andprotein, up-regulated in lungs of smokers (24, 43) as anER-resident target of CS. In CS-exposed cells andtissues, PDI persisted in HMWCs in association with itsclient proteins, meaning that PDI-associated proteinswere unable to achieve mature conformation and werenot released from the chaperone. BiP was not found inthese complexes, indicating that disulfide bond forma-tion was primarily affected at early times postexposure.Post-translational modifications of PDI became recentlya known cause and upstream signaling event of ERSR invarious diseases accompanied by oxidative stresses. Itwas shown to undergo abnormal nitrosylation in neuro-degenerative disorders and abnormal glutathionylation incells treated with the chemotherapeutic agent O(2)-[2,4-dinitro-5-(N-methyl-N-4-carboxyphenylamino)phenyl]1-(N,N-dimethylamino)diazen-1-ium-1,2-diolate (PABA/NO). Abnormal post-translational modifications of PDIdisrupt the 3D structure and lead to inhibition of itsisomerase activity (44, 45). We found that after CSexposure, PDI was extensively carbonylated in bothcells and lung extracts, and was found predominantlyin complexes with client proteins, facts that attest toPDI’s inability to assist in disulfide bond formation.Oxidation of PDI may also contribute to antioxidantdefense and have a protective function. Indeed, PDIhas two active sites, each containing two conservedcysteine residues important for the activity of the pro-tein. Modifications of active cysteines in redox-sensitiveproteins may have a regulatory role in triggering anti-oxidant defenses (54). This can explain the signifi-cance of PDI up-regulation in non-COPD smokers (24).
We found that another ER-resident chaperone, BiP, wasless affected during initial exposure to CS. This doesnot rule out more delayed effects of CS exposure on thefunction of BiP and the involvement of BiP in COPDpathogenesis, but it does show that BiP is not a primarytarget of CS. We conclude that during the early stagesof exposure to CS, abnormal oxidation of PDI byCS-derived ROS is a primary cause of ERSR through itseffect on oxidative protein folding in ER.
We saw that oxidized PDI persisted in cells for �12 hpostexposure. PDI is a relatively stable protein with ahalf-life of 96 h; therefore, CS exposure resulted in thepresence of inefficiently functioning PDI in cells (55).A decline in the ability to fold proteins because of theinadequate functioning of chaperones is one character-istic of an aging cell (56, 57). Therefore, post-transla-tional oxidation of PDI may represent one of themechanisms by which CS exposure leads to prematurecellular senescence: by creating chaperones with lessthan optimal capacities.
Moreover, individual genetic differences in the effi-ciency of the ER to carry out its cellular functions andto mount an efficient ERSR not resulting in apoptoticcell death both underlie human diseases such as auto-immune and neurodegenerative disorders (58). There-fore, genetic differences in ERSR or in the ability ofindividual cells to function with inefficient chaperonesmay be one of the determinants that confer the suscep-tibility of a smoker to COPD.
What is the possible fate of PDI-complexes? Torelieve stress and restore organelle homeostasis, theERSR is accompanied by the retrotranslocation ofunwanted proteins to the cytosol for proteasomal deg-radation and the up-regulation of the exit of matureproteins from the ER through ERESs. Min et al. (25)demonstrated that in mice that were exposed to CS andin patients with COPD, proteins are exported from theER to the cytosol for proteasomal degradation, withoverexpression of the retrograde translocation com-plex (VCP-Rma1-Grp78). PDI complexes may retro-translocate through VCP-Rma1-Grp78 for proteasomaldegradation as a cellular attempt to restore ER homeo-stasis. Another way to cope with unwanted proteins andcounterbalance an expanded ER is autophagy (59, 60),a hallmark of COPD (61). Whether there is any con-nection between autophagy and ERSR during COPDdevelopment is unknown, but both PERK and IRE1branches of ERSR have been shown to regulate au-tophage (62). Due to the large size of PDI-proteincomplexes, some may be too large for retrotransloca-tion to the cytosol and require clearance by mecha-nisms other than proteasomal degradation.
Our data indicate that ERES maintenance and distri-bution are affected by CS exposure. Disintegration ofperinuclear and peripheral ERESs seen after CS expo-sure is likely to lead to a disturbance in the traffickingof proteins that achieved a mature conformation, be-cause an alteration of ERES morphology was shown tocause a functional defect in ER-to-Golgi trafficking(63). All of these small, but cumulative changes can
975CIGARETTE SMOKING MODIFIES PDI AND CAUSES UPR

have profound effects on the homeostasis, senescenceand survival of CS-exposed cells.
In summary, strong evidence supports that an imbal-ance of cellular proteostasis may occur early in re-sponse to CS exposure. The ERSR is a modifier ofproteostasis imbalance with an important, currentlyemerging role in the development of COPD. A preciseunderstanding of the underlying causes of ERSR induc-tion and its consequences in the progression of COPDwill lead to the development of new approaches in thetreatment of this deadly disease.
The authors thank Celia Reynolds for editorial andtechnical help, Jenny Karlsson for help with live imaging,and Jeremy Brown for help with the ImageJ macro foranalysis of intensity of IHC staining. This research wasfunded by an ATS/Alpha-1 Foundation partnership grantin �1-antitrypsin deficiency (A.B.-P.), a Flight AttendantMedical Research Institute Young Clinical Scientist Award(grant 092207; A.B.-P.), and by the Welcome Fund fromthe Curtis and Elizabeth Anderson Cancer Institute.
REFERENCES
1. Shapiro, S. D., and Ingenito, E. P. (2005) The pathogenesis ofchronic obstructive pulmonary disease. Am. J. Respir. Cell Mol.Biol. 32, 367–372
2. Schrçder, M. (2008) Endoplasmic reticulum stress responses.Cell. Mol. Life Sci. 65, 862–894
3. Marcinak, S. J., and Ron, D. (2010) The unfolded proteinresponse in lung disease. Proc. Am. Thorac. Soc. 7, 356–362
4. Blumental-Perry, A. (2012) Unfolded protein response inchronic obstructive pulmonary disease: smoking, aging anddisease: a SAD trifecta. Curr. Mol. Med. 12, 883–898
5. Wang, H., Yang, L., Zou, L., Huang, D., Guo, Y., Pan, M., Tan,Y., Zhong, H., Ji, W., Ran, P., Zhong, N., and Lu, J. (2012)Association between chronic obstructive pulmonary disease andlung cancer: a case-control study in southern Chinese and ameta-analysis. PLoS One 7, e46144
6. Blumental-Perry, A. (2012) Endoplasmic reticulum stress, thefuture of cancer research and a new designated journal. Endo.Reticu. Stress Cancers 1, 1–3
7. Healy, S. J., Gorman, A. M., Mousavi-Shafaei, P., Gupta, S., andSamali, A. (2009) Targeting the endoplasmic reticulum-stressresponse as an anticancer strategy. Eur. J. Pharmacol. 625,234–246
8. Jorgensen, E., Stinson, A., Shan, L., Yang, J., Gietl, D., andAlbino, A. P. (2008) Cigarette smoke induces endoplasmicreticulum stress and the unfolded protein response in normaland malignant human lung cells. BMC Cancer 8, 229
9. Kim, K. M., Yu, T. K., Chu, H. H., Park, H. S., Jang, K. Y., Moon,W. S., Kang, M. J., Lee, D. G., Kim, M. H., Lee, J. H., and Chung,M. J. (2012) Expression of ER stress and autophagy-relatedmolecules in human non-small cell lung cancer and premalig-nant lesions. Int. J. Cancer 131, E362–E370
10. Wu, J., and Kaufman, R. (2006) From acute ER stress tophysiological roles of the unfolded protein response. Cell DeathDiffer. 13, 374–384
11. Vattem, K. M., and Wek, R. C. (2004) Reinitiation involvingupstream ORFs regulates ATF4 mRNA translation in mamma-lian cells. Proc. Natl. Acad. Sci. U. S. A. 101, 11269–11274
12. Xu, C., Bailly-Maitre, B., and Reed, J. C. (2005) Endoplasmicreticulum stress: cell life and death decisions. J. Clin. Invest. 115,2656–2664
13 Cantin, A. M. Cellular response to cigarette smoke and oxidants:adapting to survive. Proc. Am. Thorac. Soc. 7, 368–375
14. He, C. H., Gong, P., Hu, B., Stewart, D., Choi, M. E., Choi,A. M. K., and Alam, J. (2001) Identification of activatingtranscription factor 4 (ATF4) as an Nrf2-interacting protein. J.Biol. Chem. 276, 20858–20865
15. Kelsen, S. G. (2012) Respiratory epithelial cell responses tocigarette smoke: the unfolded protein response. [E-pub aheadof print] Pulm. Pharmacol. Ther. doi: 10.1016/j.pupt.2012.07.005
16. Ribeiro, C. M., and O’Neal, W. K. (2012) Endoplasmic reticu-lum stress in chronic obstructive lung diseases. Curr. Mol. Med.12, 872–882
17. Lawless, M. W., and Greene, C. M. (2012) Toll-like receptorsignalling in liver disease: ER stress the missing link? Cytokine 59,195–202
18. An, C. H., Wang, X. M., Lam, H. C., Ifedigbo, E., Washko, G. R.,Ryter, S. W., and Choi, A. M. (2012) TLR4 Deficiency promotesautophagy during cigarette smoke-induced pulmonary emphy-sema. Am. J. Physiol. 303, L748–L757
19. Mortaz, E., Henricks, P. A., Kraneveld, A. D., Givi, M. E.,Garssen, J., and Folkerts, G. (2011) Cigarette smoke induces therelease of CXCL-8 from human bronchial epithelial cells viaTLRs and induction of the inflammasome. Biochim. Biophys. Acta1812, 1104–1110
20. Bezemer, G. F., Sagar, S., van Bergenhenegouwen, J., Georgiou,N. A., Garssen, J., Kraneveld, A. D., and Folkerts, G. (2012) Dualrole of Toll-like receptors in asthma and chronic obstructivepulmonary disease. Pharmacol. Rev. 64, 337–358
21. Hengstermann, A., and Muller, T. (2008) Endoplasmic reticu-lum stress induced by aqueous extracts of cigarette smoke in3T3 cells activates the unfolded-protein-response-dependentPERK pathway of cell survival. Free Radic. Biol. Med. 44, 1097–1107
22. Tagawa, Y., Hiramatsu, N., Kasai, A., Hayakawa, K., Okamura,M., Yao, J., and Kitamura, M. (2008) Induction of apoptosis bycigarette smoke via ROS-dependent endoplasmic reticulumstress and CCAAT/enhancer-binding protein-homologous pro-tein (CHOP). Free Radic. Biol. Med. 45, 50–59
23. Korfei, M., Ruppert, C., Mahavadi, P., Henneke, I., Markart, P.,Koch, M., Lang, G., Fink, L., Bohle, R. M., Seeger, W., Weaver,T. E., and Guenther, A. (2008) Epithelial endoplasmic reticu-lum stress and apoptosis in sporadic idiopathic pulmonaryfibrosis. Am. J. Respir. Crit. Care. Med. 178, 838–846
24. Kelsen, S. G., Duan, X., Ji, R., Perez, O., Liu, C., and Merali, S.(2008) Cigarette smoke induces an unfolded protein responsein the human lung: a proteomic approach. Am. J. Respir. CellMol. Biol. 38, 541–550
25. Min, T., Bodas, M., Mazur, S., and Vij, N. (2011) Critical role ofproteostasis-imbalance in pathogenesis of COPD and severeemphysema. J. Mol. Med. (Berl.) 89, 577–593
26. Hetz, C., and Glimcher, L. H. (2011) Protein homeostasisnetworks in physiology and disease. Curr. Opin. Cell Biol. 23,123–125
27. Hautamaki, R. D., Kobayashi, D. K., Senior, R., and Shapiro, S.(1997) Requirement for macrophage elastase for cigarettesmoke-induced emphysema in mice. Science 277, 2002–2004
28. Wu, J., Rutkowski, D. T., Dubois, M., Swathirajan, J., Saunders,T., Wang, J., Song, B., Yau, G. D.-Y., and Kaufman, R. J. (2007)ATF6a optimizes long-term endoplasmic reticulum function toprotect cells from chronic stress. Dev. Cell 13, 351–364
29. Schreiber, E., Matthias, P., Müller, M., and Schaffner, W. (1989)Rapid detection of octamer binding proteins with “mini-ex-tracts”, prepared from a small number of cells. Nucleic Acids Res.17, 6419
30. Molteni, S. N., Fassio, A., Ciriolo, M. R., Filomeni, G., Pasqual-etto, E., Fagioli, C., and Sitia, R. (2004) Glutathione limitsEro1-dependent oxidation in the endoplasmic reticulum. J. Biol.Chem. 279, 32667–32673
31. Nawa, D., Shimada, O., Kawasaki, N., Matsumoto, N., andYamamoto, K. (2007) Stable interaction of the cargo receptorVIP36 with molecular chaperone BiP. Glycobiology 17, 913–921
32. Oda, K., Wada, I., Takami, N., Fujiwara, T., Misumi, Y., andIkehara, Y. (1996) Bip/GRP78 but not calnexin associates witha precursor of glycosylphosphatidylinositol-anchored protein.Biochem. J. 316, 623–630
33. Marciniak, S. J., Yun, C. Y., Oyadomari, S., Novoa, I., Zhang, Y.,Jungreis, R., Nagata, K., Harding, H. P., and Ron, D. (2004)CHOP induces death by promoting protein synthesis andoxidation in the stressed endoplasmic reticulum. Genes Dev. 18,3066–3077
34. Wikenheiser, K., Vorbroker, D., Rice, W., Clark, J., Bachurski,C., Oie, H., and Whitsett, J. (1993) Production of immortalizeddistal respiratory epithelial cell lines from surfactant protein
976 Vol. 27 March 2013 KENCHE ET AL.The FASEB Journal � www.fasebj.org

C/simian virus 40 large tumor antigen transgenic mice. Proc.Natl. Acad. Sci. U. S. A. 90, 11029–11033
35. Henson, P. M., Vandivier, R. W., and Douglas, I. S. (2006) Celldeath, remodeling, and repair in chronic obstructive pulmonarydisease? Proc. Am. Thorac. Soc. 3, 713–717
36. Aoshiba, K., Koinuma, M., Yokohori, N., and Nagai, A. (2003)Immunohistochemical evaluation of oxidative stress in murinelungs after cigarette smoke exposure. Inhalat. Toxicol. 15, 1029–1038
37. Wang, Y., Shen, J., Arenzana, N., Tirasophon, W., Kaufman,R. J., and Prywes, R. (2000) Activation of ATF6 and an ATF6DNA binding site by the endoplasmic reticulum stress response.J. Biol. Chem. 275, 27013–27020
38. Mao, C., Tai, W.-C., Bai, Y., Poizat, C., and Lee, A. S. (2006) Invivo regulation of Grp78/BiP transcription in the embryonicheart: role of endoplasmic reticulum stress response elementand GATA-4. J. Biol. Chem. 281, 8877–8887
39. Chakravarthi, S., Jessop, C. E., and Bulleid, N. J. (2006) The roleof glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 7, 271–275
40. Frand, A. R., and Kaiser, C. A. (1999) Ero1p oxidizes proteindisulfide isomerase in a pathway for disulfide bond formation inthe endoplasmic reticulum. Mol. Cell 4, 469–477
41. Ma, Y., and Hendershot, L. M. (2004) ER chaperone functionsduring normal and stress conditions. J. Chem. Neuro. 28, 51–65
42. Anelli, T., and Sitia, R. (2008) Protein quality control in theearly secretory pathway. EMBO J. 27, 315–327
43. Lumb, R. A., and Bulleid, N. J. (2002) Is protein disulfideisomerase a redox-dependent molecular chaperone? EMBO J.21, 6763–6770
44. Townsend, D. M., Manevich, Y., He, L., Xiong, Y., Bowers, R. R.,Jr., Hutchens, S., and Tew, K. D. (2009) Nitrosative stress-induced s-glutathionylation of protein disulfide isomerase leadsto activation of the unfolded protein response. Cancer Res. 69,7626–7634
45. Uehara, T., Nakamura, T., Yao, D., Shi, Z. Q., Gu, Z., Ma, Y.,Masliah, E., Nomura, Y., and Lipton, S. A. (2006) S-nitrosylatedprotein-disulphide isomerase links protein misfolding to neuro-degeneration. Nature 441, 513–517
46. Budnik, A., and Stephens, D. (2009) ER exit sites–localizationand control of COPII vesicle formation. FEBS Lett. 583, 3796–3803
47. Stephens, D., Lin-Marq, N., Pagano, A., Pepperkok, R., andPaccaud, J. (2000) COPI-coated ER-to-Golgi transport com-plexes segregate from COPII in close proximity to ER exit sites.J. Cell Sci. 113, 2177–2185
48. Aksoy, R. J., Ji, R., Katamreddy, S. R., Barrero, C. A., Merali, S.,Muniswamy, M., Mallilankaraman, K., and Kelsen, S. G. (2011)The unfolded protein response (UPR) to heightened endoplas-mic reticulum (ER) stress is impaired in COPD lung. Am. J.Respir. Crit. Care Med. 183, A4094
49. Biswas, S. K., and Rahman, I. (2009) Environmental toxicity,redox signaling and lung inflammation: the role of glutathione.Mol. Aspects Med. 30, 60–76
50. Kaser, A., Lee, A. H., Franke, A., Glickman, J. N., Zeissig, S., Tilg,H., Nieuwenhuis, E. E., Higgins, D. E., Schreiber, S., Glimcher,L. H., and Blumberg, R. S. (2008) XBP1 links ER stress tointestinal inflammation and confers genetic risk for humaninflammatory bowel disease. Cell 134, 743–756
51. Vidal, R. L., Figueroa, A., Court, F. A., Thielen, P., Molina, C.,Wirth, C., Caballero, B., Kiffin, R., Segura-Aguilar, J., Cuervo,A. M., Glimcher, L. H., and Hetz, C. (2012) Targeting the UPRtranscription factor XBP1 protects against Huntington’s diseasethrough the regulation of FoxO1 and autophagy. Hum. Mol.Genet. 21, 2245–2262
52. Haspel, J. A., and Choi, A. M. (2011) Autophagy: a core cellularprocess with emerging links to pulmonary disease. Am. J. Respir.Crit. Care Med. 184, 1237–1246
53. Geraghty, P., Wallace, A., and M. D’Armiento, J. (2011) Induc-tion of the unfolded protein response by cigarette smoke isprimarily an activating transcription factor 4-C/EBP homolo-gous protein mediated process. Int. J. COPD 6, 309–219
54. Jacob, C., Battaglia, E., Burkholz, T., Peng, D., Bagrel, D., andMontenarh, M. (2012) Control of oxidative posttranslationalcysteine modifications: from intricate chemistry to widespreadbiological and medical applications. Chem. Res. Toxicol. 25,588–604
55. Terada, K., Manchikalapudi, P., Noiva, R., Jauregui, H. O.,Stockert, R. J., and Schilsky, M. L. (1995) Secretion, surfacelocalization, turnover, and steady state expression of proteindisulfide isomerase in rat hepatocytes. J. Biol. Chem. 270, 20410–20416
56. Roth, D. M., Balch, William E (2011) Modeling general proteo-stasis: proteome balance in health and disease. Curr. Opin. CellBiol. 23, 126–134
57. Balch, W. E., Morimoto, R. I., Dillin, A., and Kelly, J. W. (2008)Adapting proteostasis for disease intervention. Science 319, 916–919
58. Dombroski, B. A., Nayak, R. R., Ewens, K. G., Ankener, W.,Cheung, V. G., and Spielman, R. S. (2010) Gene expression andgenetic variation in response to endoplasmic reticulum stress inhuman cells. Am. J. Hum. Genet. 86, 719–729
59. Bernales, S., McDonald3, K. L., and Walter, P. (2006) Au-tophagy counterbalances endoplasmic reticulum expansionduring the unfolded protein response. PLoS Biol. 4, 2311–2323
60. Bernales, S., Schuck, S., and Walter, P. (2007) Selective au-tophagy of the endoplasmic reticulum. Autophagy 3, 285–287
61. Ryter, S. W., and Choi, A. M. (2010) Autophagy in the lung. Proc.Am. Thorac. Soc. 7, 13–21
62. Weston, R. t., and Puthalakath, H. (2010) Endoplasmic reticu-lum stress and BCL-2 family members. Adv. Exp. Med. Biol. 687,65–77
63. Wilhelm, J., Buszczak, M., and Sayles, S. (2005) Efficient proteintrafficking requires trailer hitch, a component of a ribonucleo-protein complex localized to the ER in Drosophila. Dev. Cell 9,675–685
Received for publication July 5, 2012.Accepted for publication November 5, 2012.
977CIGARETTE SMOKING MODIFIES PDI AND CAUSES UPR