chemical kinetics: the rates of reactions 자연과학대학 화학과 박영동 교수 atkins,...
Post on 19-Dec-2015
231 views
TRANSCRIPT

Chemical kinetics:the rates of reactions
자연과학대학 화학과박영동 교수
Atkins, physical chemistry, 5th ed.
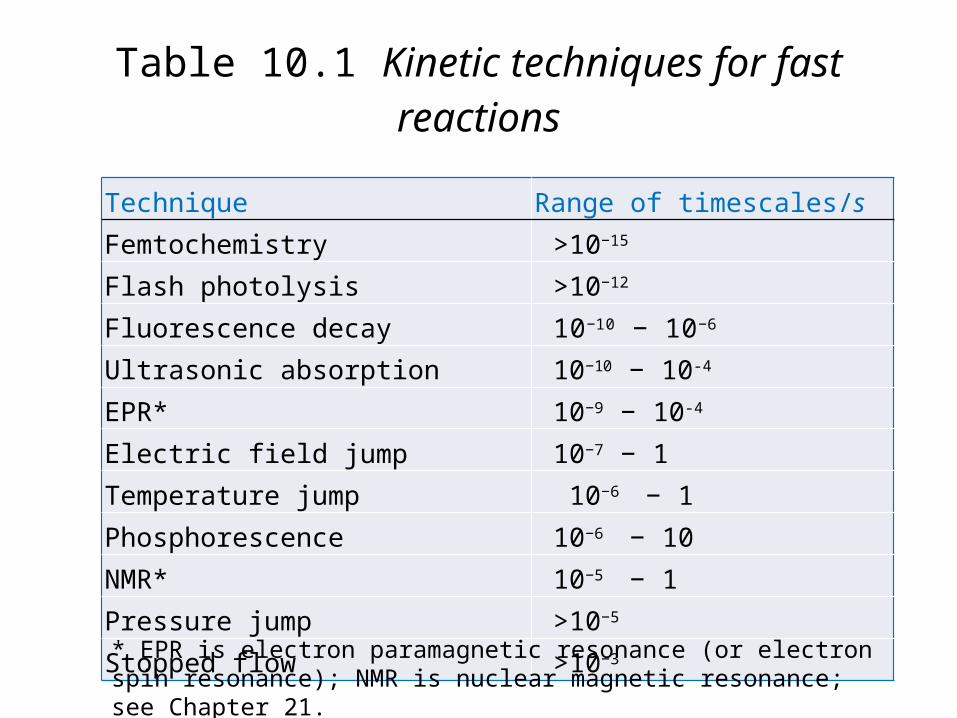
Technique Range of timescales/sFemtochemistry >10−15
Flash photolysis >10−12
Fluorescence decay 10−10 − 10−6
Ultrasonic absorption 10−10 − 10-4
EPR* 10−9 − 10-4
Electric field jump 10−7 − 1Temperature jump 10−6 − 1Phosphorescence 10−6 − 10NMR* 10−5 − 1Pressure jump >10−5
Stopped flow >10−3
Table 10.1 Kinetic techniques for fast reactions
* EPR is electron paramagnetic resonance (or electron spin reso-nance); NMR is nuclear magnetic resonance; see Chapter 21.
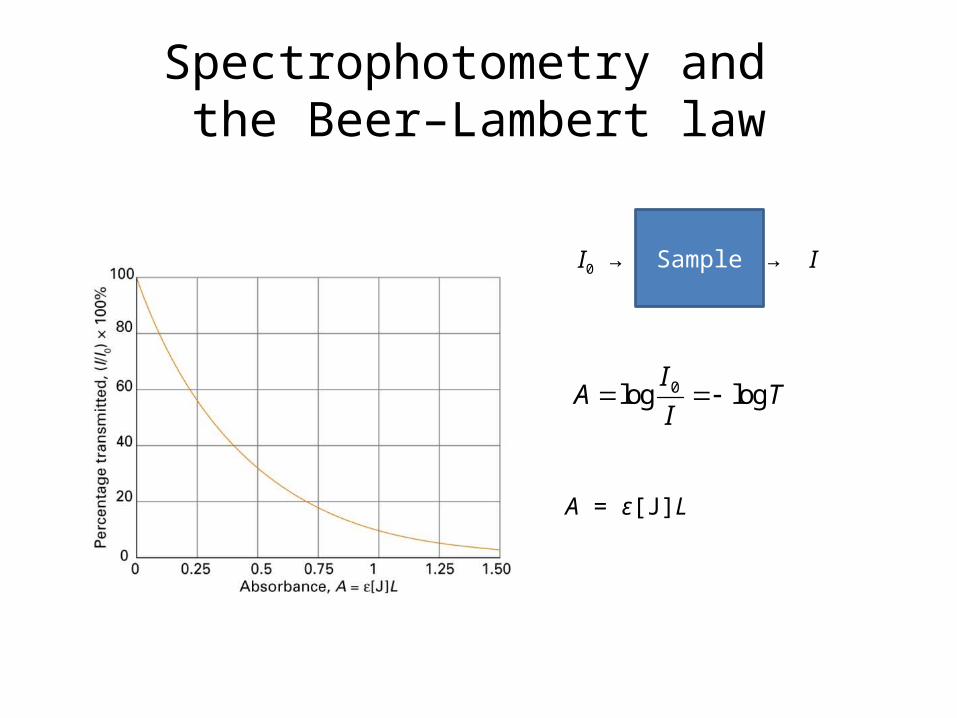
Spectrophotometry and the Beer–Lambert law
0log logI
A TI
A = ε[J]L
SampleI0 → → I
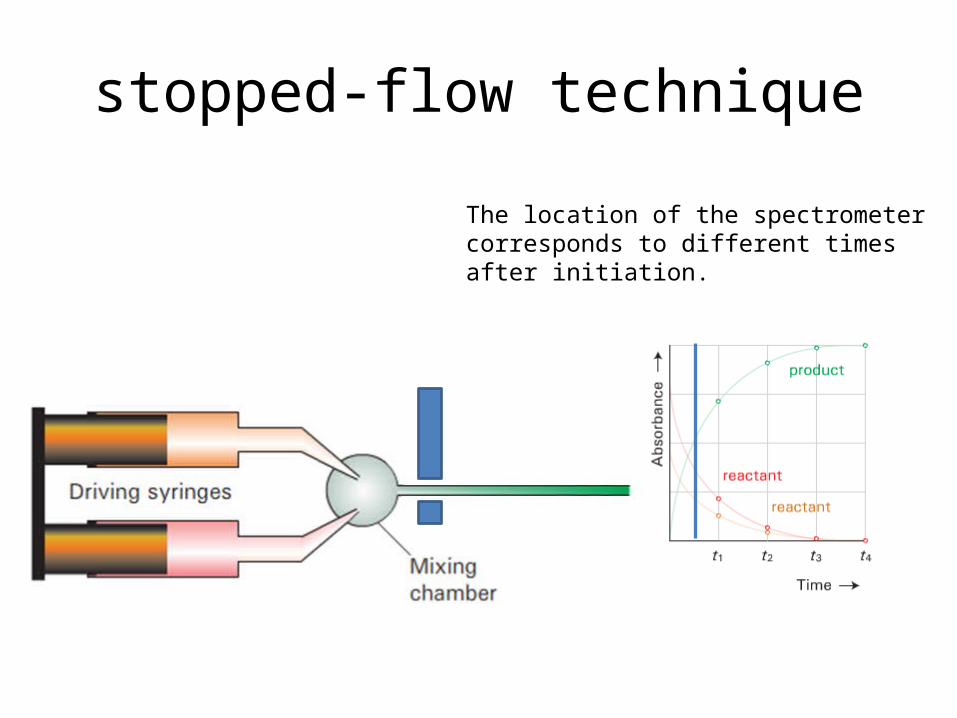
stopped-flow technique
The location of the spectrometer cor-responds to different times after initi-ation.

The rate of a reaction
J
1 [J]drate
dt

Reaction order
[A]arv k ath-order in A.
second-order in NO2
1/2[A] [B]rv k half order in A, first order in B, and 3/2-th order over-all.
for the gas-phase reac-tion H2(g) + Br2(g) →2 HBr(g)
a rate law is established experimentally, and cannot in general be inferred from the chemical equation for the reaction.
1/2 3/22 2
2
[H ] [Br ]1
[Br ] [HBr]2
krv
kr
[E][S]
[S] M
krv
K
Enzyme kinetics
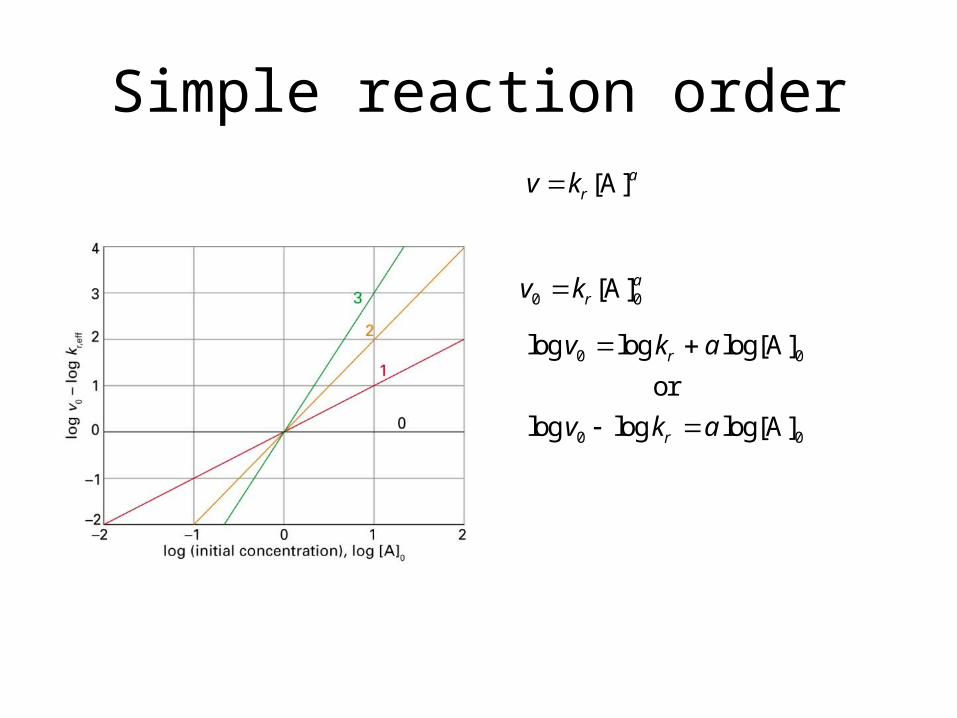
Simple reaction order[A]a
rv k
0 0[A]arv k
0 0
0 0
log log log[A]
or
log log log[A]
r
r
v k a
v k a
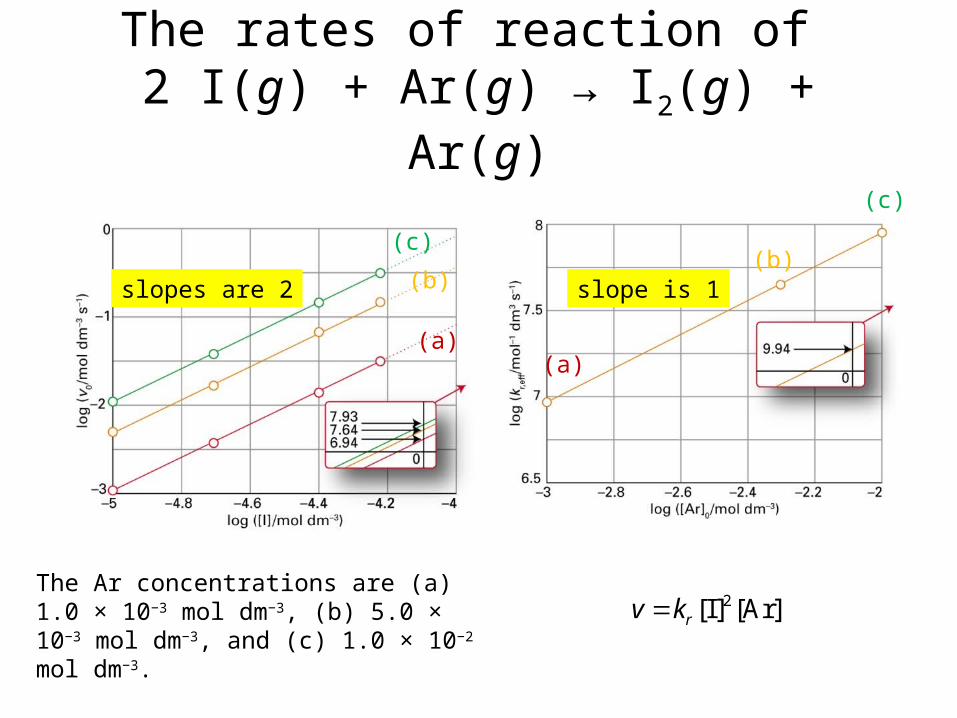
The rates of reaction of 2 I(g) + Ar(g) → I2(g) + Ar(g)
The Ar concentrations are (a) 1.0 × 10−3 mol dm−3, (b) 5.0 × 10−3 mol dm−3, and (c) 1.0 × 10−2 mol dm−3.
(c)
(b)
(a)
slopes are 2 slope is 1
2[I] [Ar]rv k
(c)
(b)
(a)
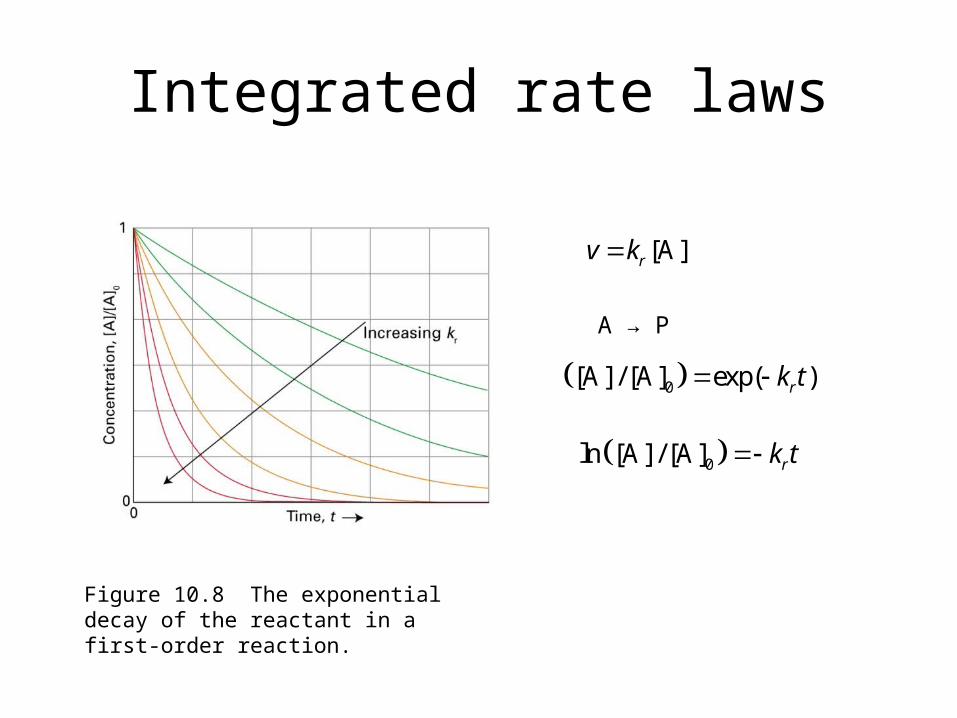
Integrated rate laws
Figure 10.8 The exponential decay of the reactant in a first-order reaction.
[A]rv k
A → P
0[A] / [A] exp( )rk t
0ln [A] / [A] rk t
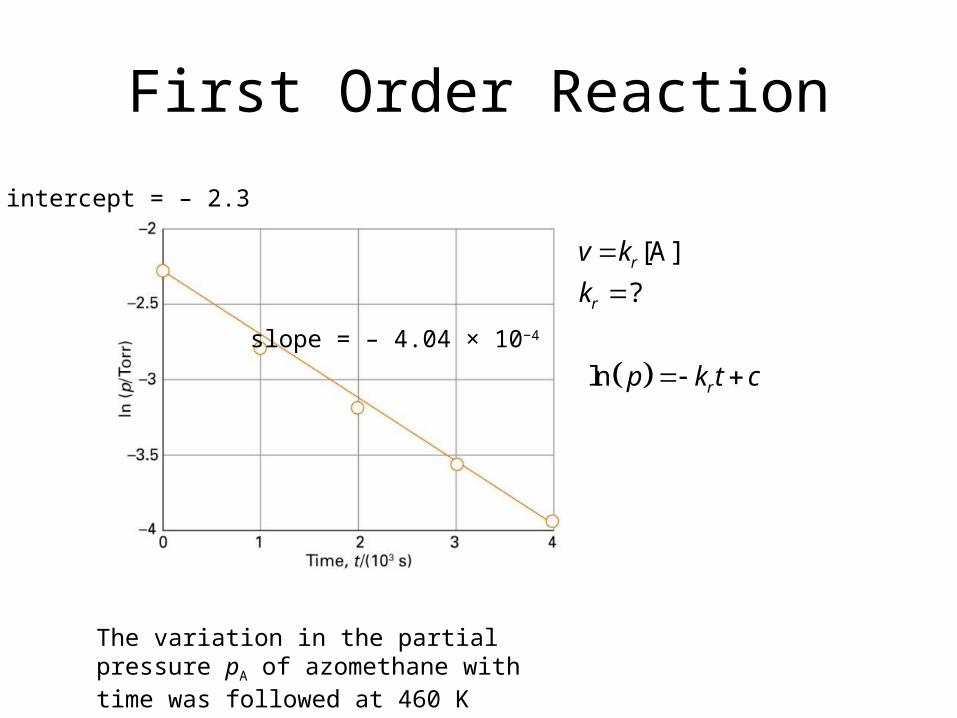
First Order Reaction
The variation in the partial pressure pA of azomethane with time was followed at 460 K
slope = – 4.04 × 10−4
[A]
?r
r
v k
k
intercept = – 2.3
ln rp k t c
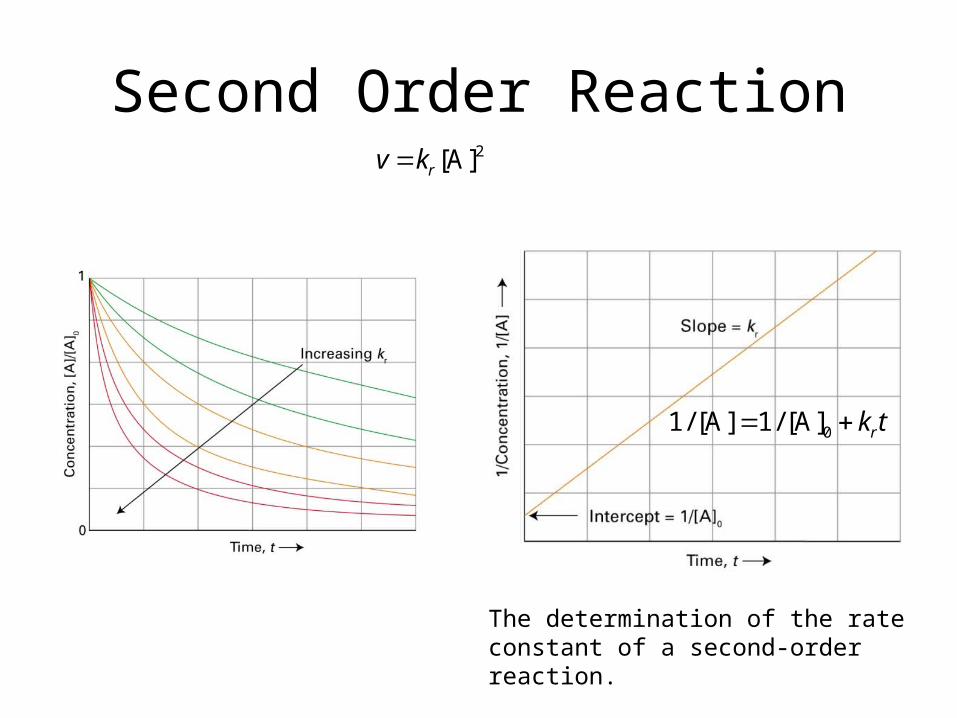
Second Order Reaction
The determination of the rate con-stant of a second-order reaction.
2[A]rv k
01/ [A] 1/ [A] rk t
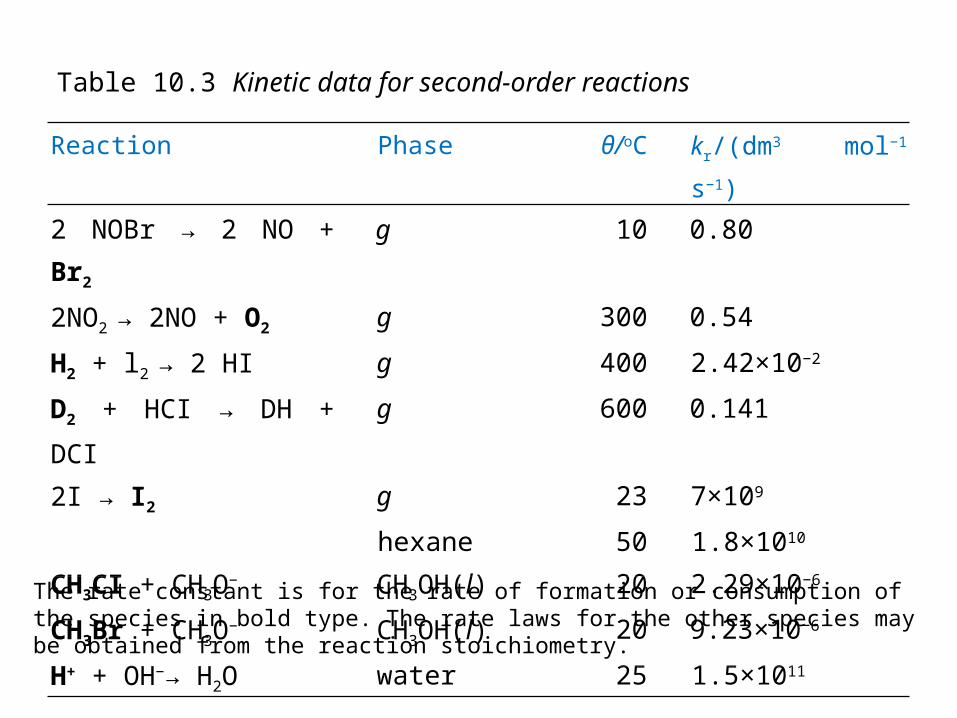
Reaction Phase θ/oC kr/(dm3 mol−1 s−1)
2 NOBr → 2 NO + Br2 g 10 0.80
2NO2 → 2NO + O2 g 300 0.54
H2 + l2 → 2 HI g 400 2.42×10−2
D2 + HCI → DH + DCI g 600 0.141
2I → I2 g 23 7×109
hexane 50 1.8×1010
CH3CI + CH3O− CH3OH(l) 20 2.29×10−6
CH3Br + CH3O− CH3OH(l) 20 9.23×10−6
H+ + OH−→ H2O water 25 1.5×1011
Table 10.3 Kinetic data for second-order reactions
The rate constant is for the rate of formation or consumption of the species in bold type. The rate laws for the other species may be obtained from the reaction stoichiometry.

First Order vs Second Order
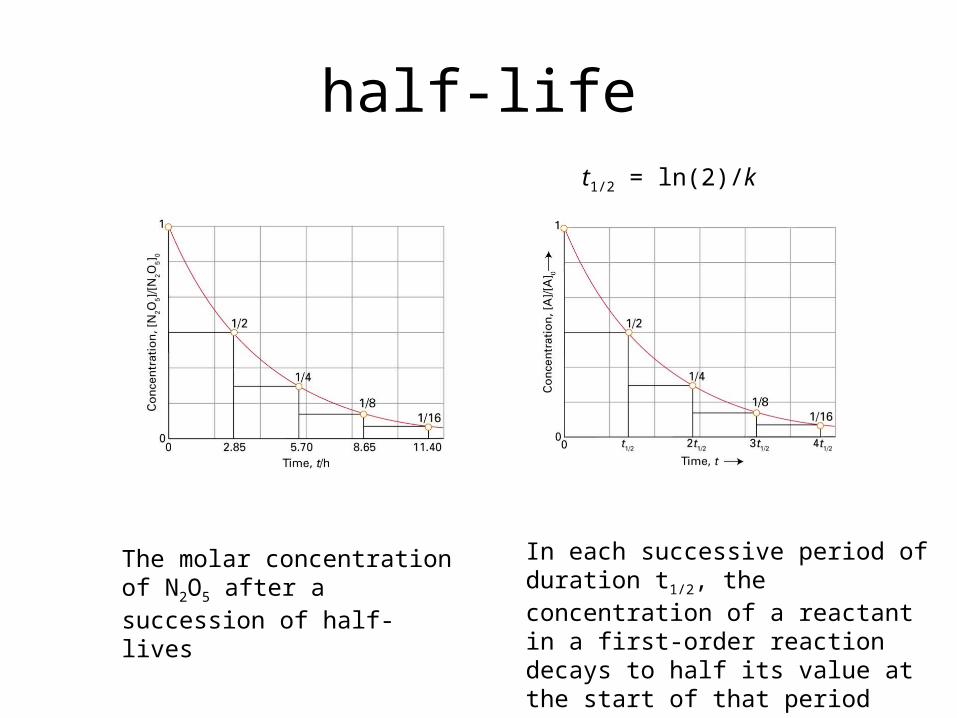
half-life
The molar concentration of N2O5 after a succession of half-lives
In each successive period of du-ration t1/2, the concentration of a reactant in a first-order reaction decays to half its value at the start of that period
t1/2 = ln(2)/k
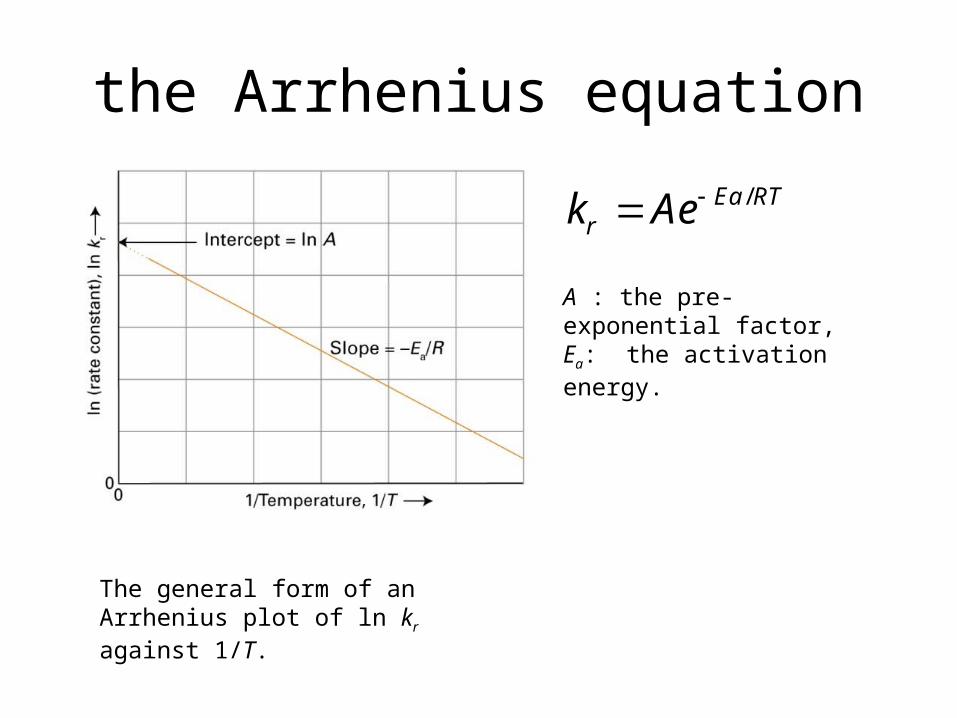
the Arrhenius equation
The general form of an Arrhenius plot of ln kr against 1/T.
A : the pre-exponential factor, Ea: the activation energy.
/Ea RTrk Ae
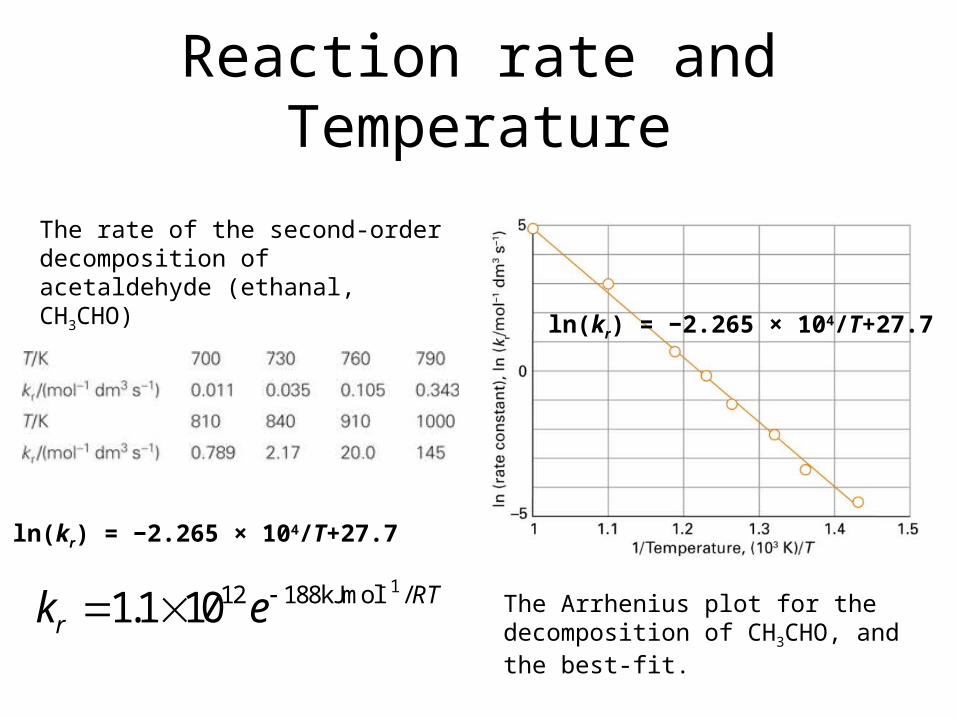
Reaction rate and Tempera-ture
The Arrhenius plot for the de-composition of CH3CHO, and the best-fit.
The rate of the second-order de-composition of acetaldehyde (ethanal, CH3CHO)
ln(kr) = −2.265 × 104/T+27.7
ln(kr) = −2.265 × 104/T+27.7
112 188kJmol /1.1 10 RTrk e
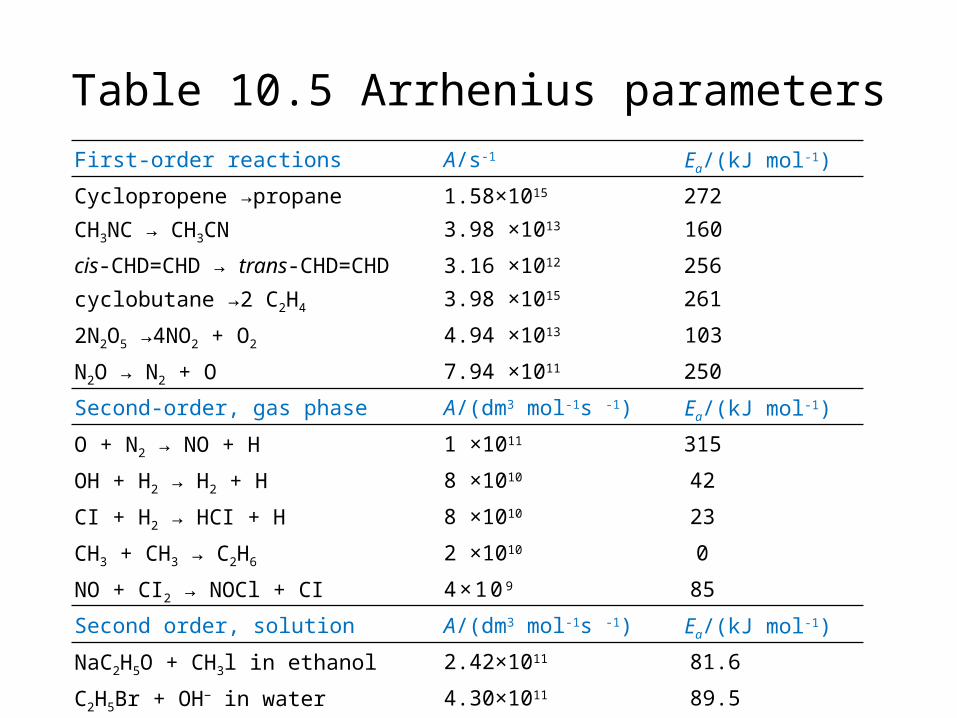
First-order reactions A/s-1 Ea/(kJ mol-1)
Cyclopropene →propane 1.58×1015 272
CH3NC → CH3CN 3.98 ×1013 160
cis-CHD=CHD → trans-CHD=CHD 3.16 ×1012 256
cyclobutane →2 C2H4 3.98 ×1015 261
2N2O5 →4NO2 + O2 4.94 ×1013 103
N2O → N2 + O 7.94 ×1011 250
Second-order, gas phase A/(dm3 mol-1s -1) Ea/(kJ mol-1)
O + N2 → NO + H 1 ×1011 315
OH + H2 → H2 + H 8 ×1010 42
CI + H2 → HCI + H 8 ×1010 23
CH3 + CH3 → C2H6 2 ×1010 0
NO + CI2 → NOCl + CI 4×10 9 85
Second order, solution A/(dm3 mol-1s -1) Ea/(kJ mol-1)
NaC2H5O + CH3l in ethanol 2.42×1011 81.6
C2H5Br + OH− in water 4.30×1011 89.5
CH3I + S2O32− in water 2.19×1012 78.7
Sucrose + H2O in acidic water 1.50×1015 107.9
Table 10.5 Arrhenius parameters
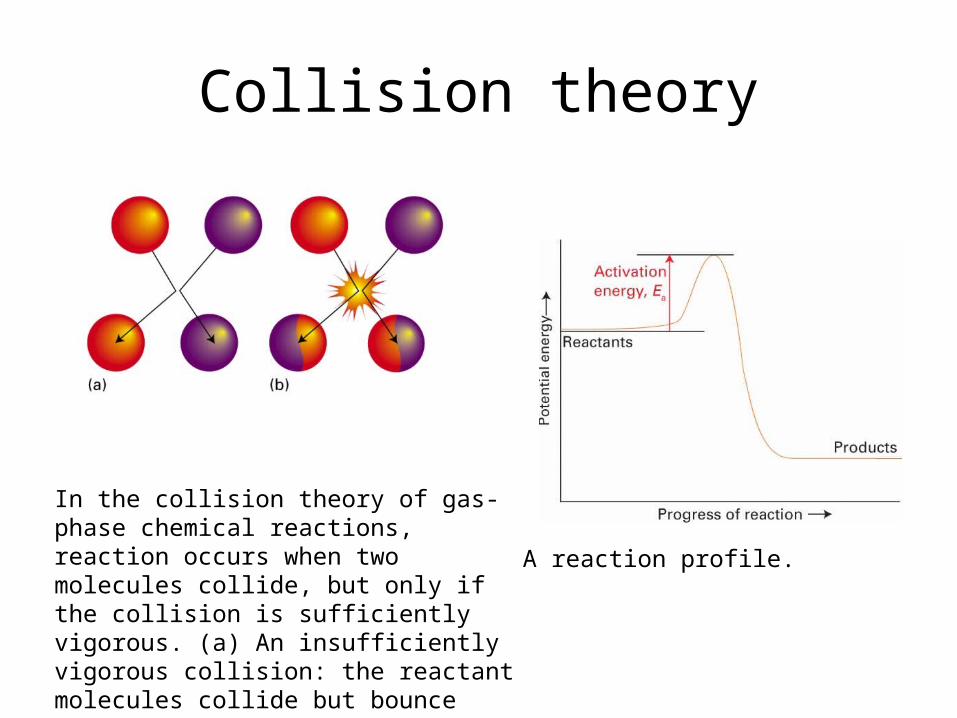
Collision theory
In the collision theory of gas-phase chemical reactions, reaction occurs when two molecules collide, but only if the collision is sufficiently vigorous. (a) An insufficiently vigorous colli-sion: the reactant molecules collide but bounce apart unchanged. (b) A sufficiently vigorous collision results in a reaction.
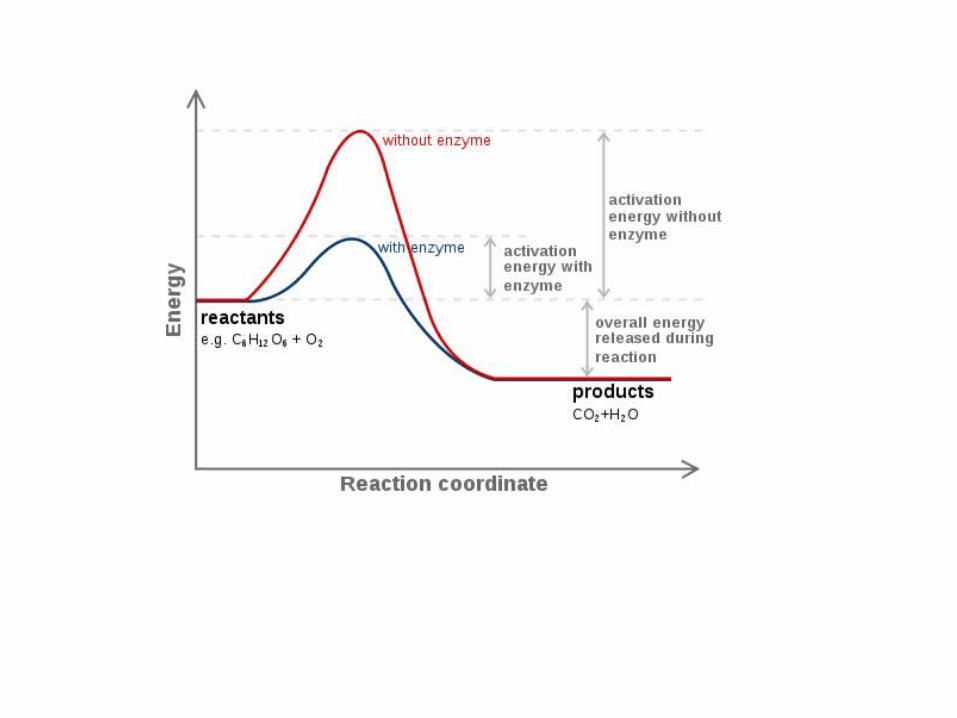
A reaction profile.
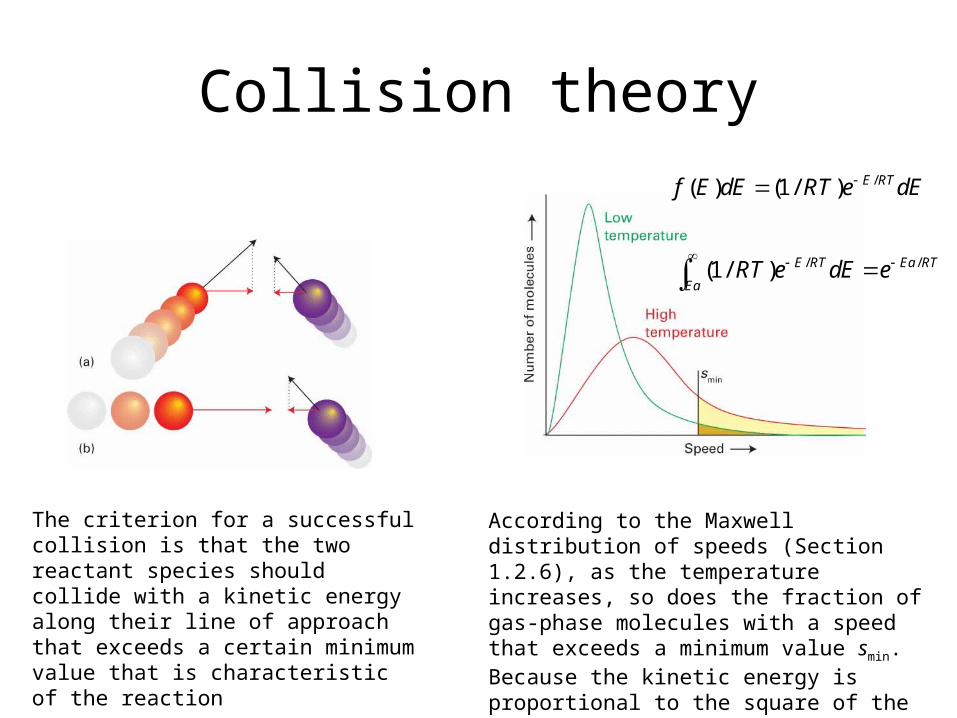
Collision theory
According to the Maxwell distribution of speeds (Section 1.2.6), as the temperature increases, so does the fraction of gas-phase molecules with a speed that exceeds a minimum value smin. Because the kinetic energy is proportional to the square of the speed, it follows that more molecules can collide with a mini-mum kinetic energy Ea (the activation energy) at higher temperatures.
The criterion for a successful collision is that the two reactant species should collide with a kinetic energy along their line of approach that exceeds a certain minimum value that is characteristic of the reaction
/ /(1/ ) E RT Ea RT
EaRT e dE e
/( ) (1/ ) E RTf E dE RT e dE

collision frequency
Z : the collision frequencyρ : the steric factor
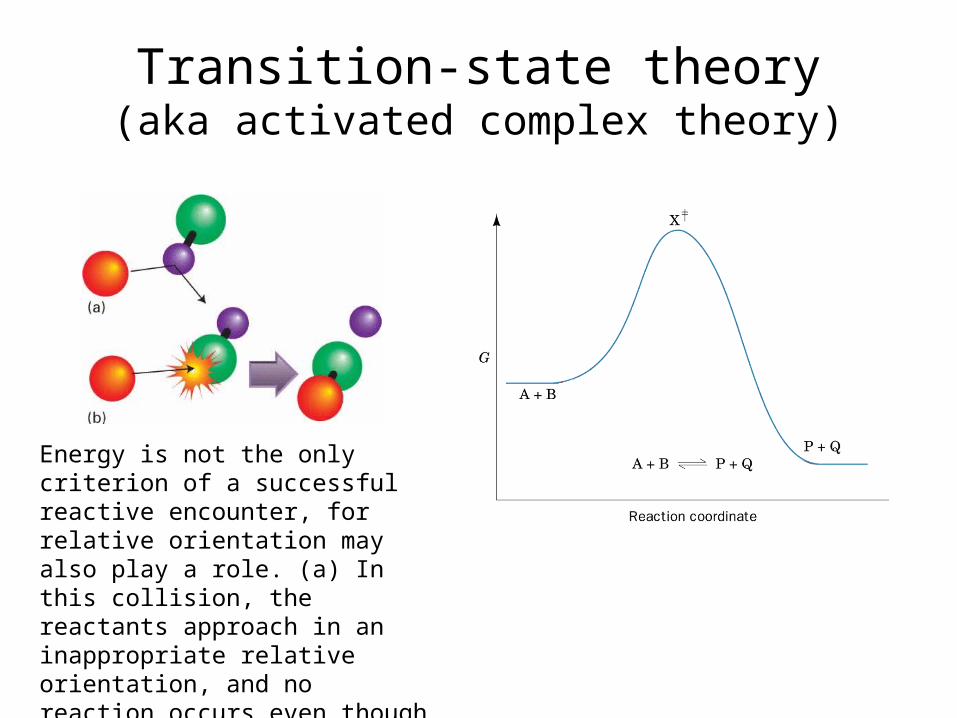
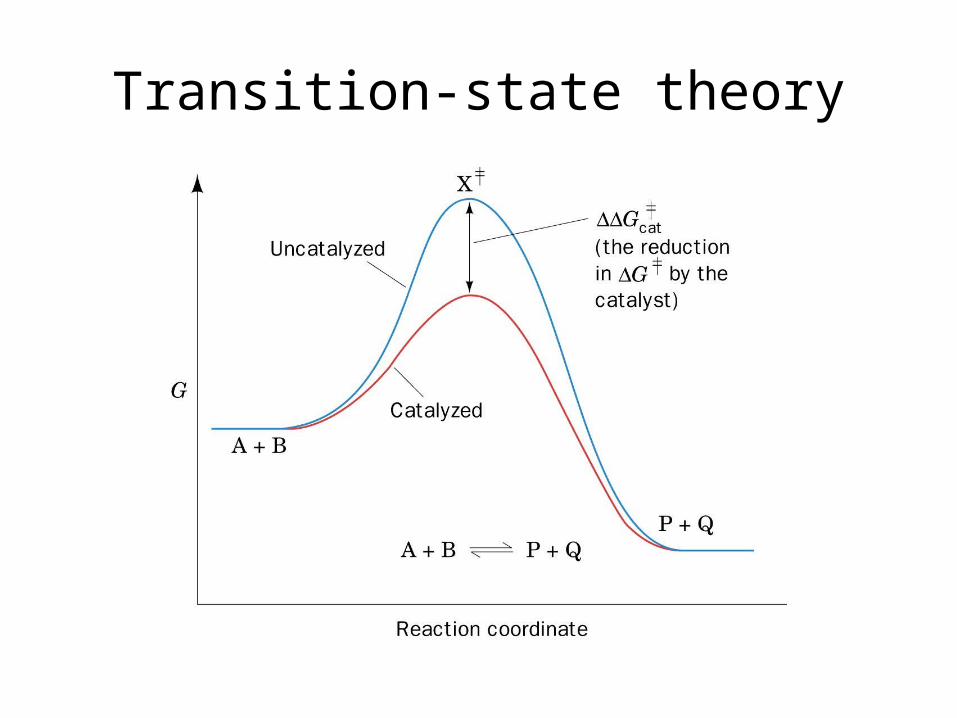
Transition-state theory(aka activated complex theory)
Energy is not the only criterion of a suc-cessful reactive encounter, for relative orientation may also play a role. (a) In this collision, the reactants approach in an inappropriate relative orientation, and no reaction occurs even though their en-ergy is sufficient. (b) In this encounter, both the energy and the orientation are suitable for reaction.
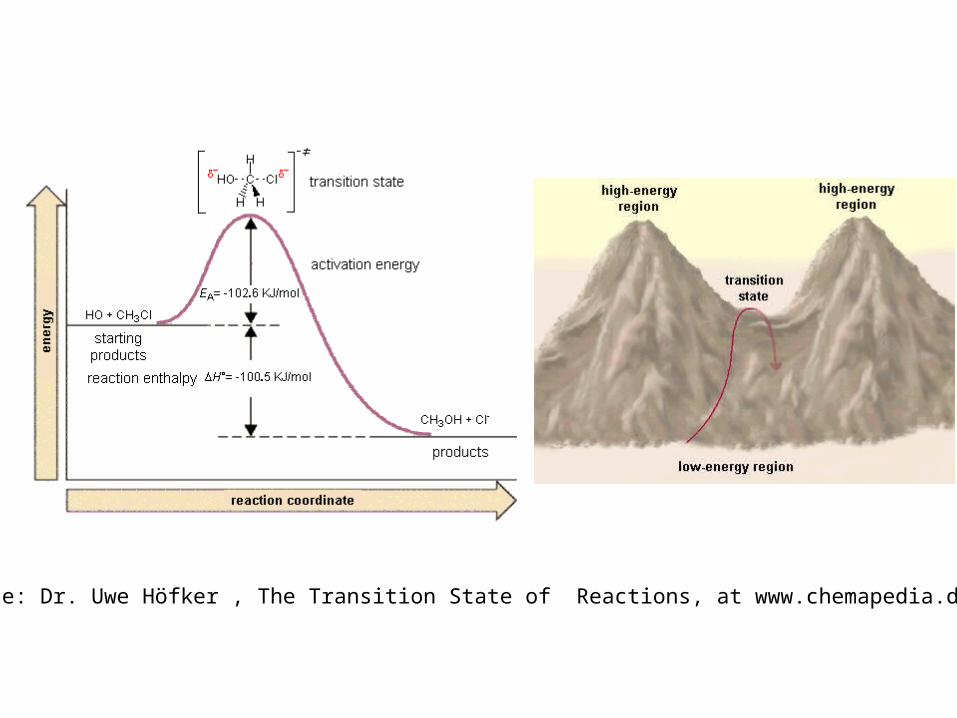
source: Dr. Uwe Höfker , The Transition State of Reactions, at www.chemapedia.de
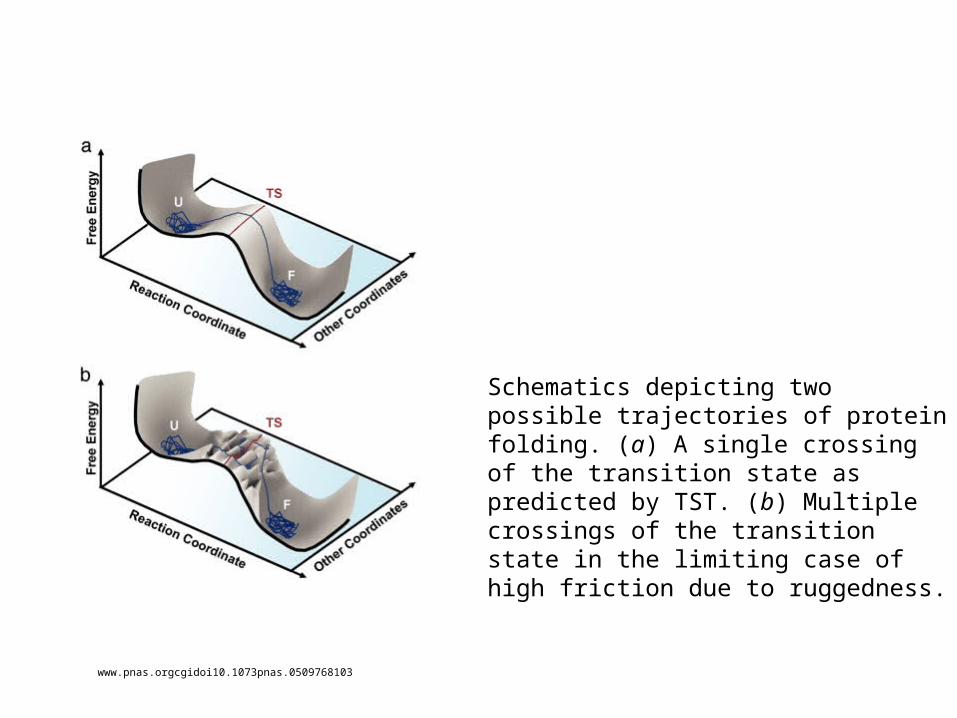
www.pnas.orgcgidoi10.1073pnas.0509768103
Schematics depicting two possible trajectories of protein folding. (a) A single crossing of the transition state as predicted by TST. (b) Multiple crossings of the transition state in the limiting case of high friction due to ruggedness.
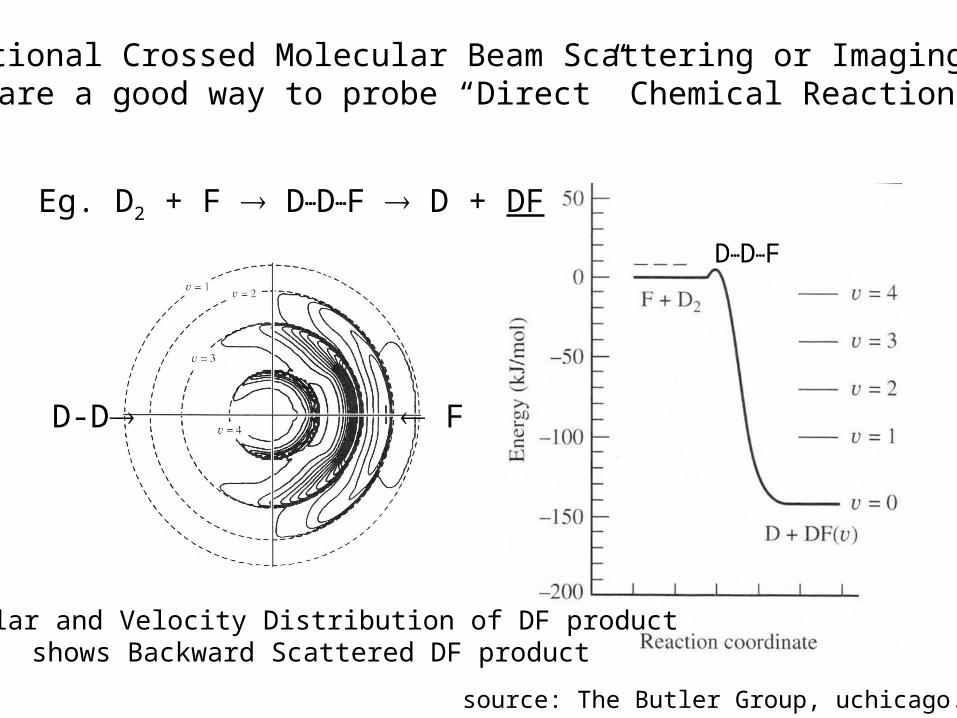
Traditional Crossed Molecular Beam Scattering or Imaging Exptsare a good way to probe “Direct” Chemical Reactions
Angular and Velocity Distribution of DF product shows Backward Scattered DF product
Eg. D2 + F D…D…F D + DF
D…D…F
D-D F
source: The Butler Group, uchicago.edu
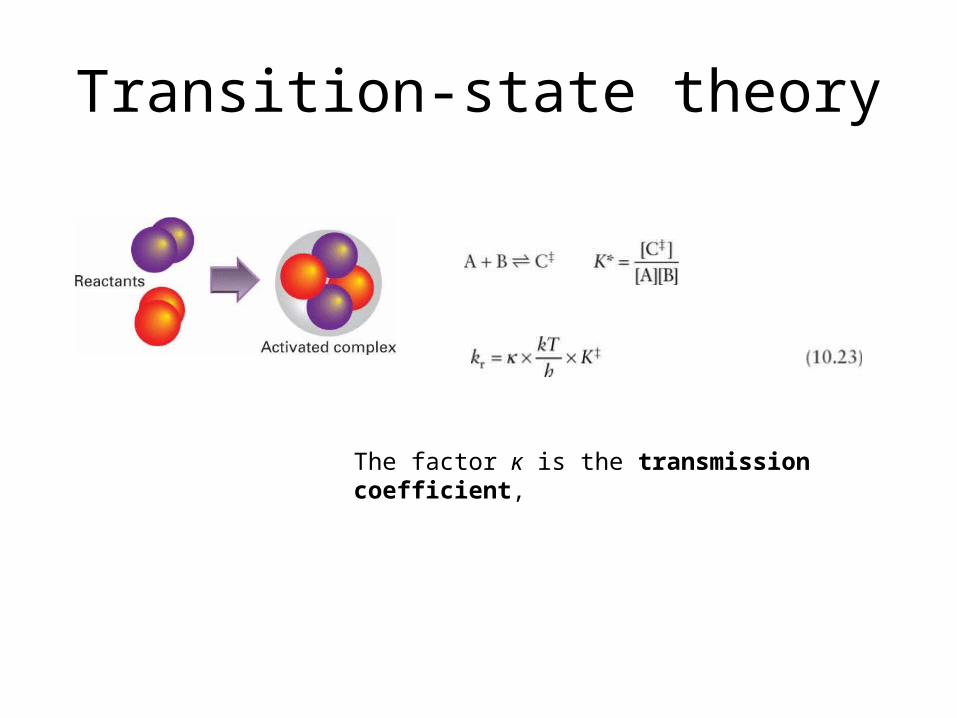
Transition-state theory
The factor κ is the transmission co-efficient,
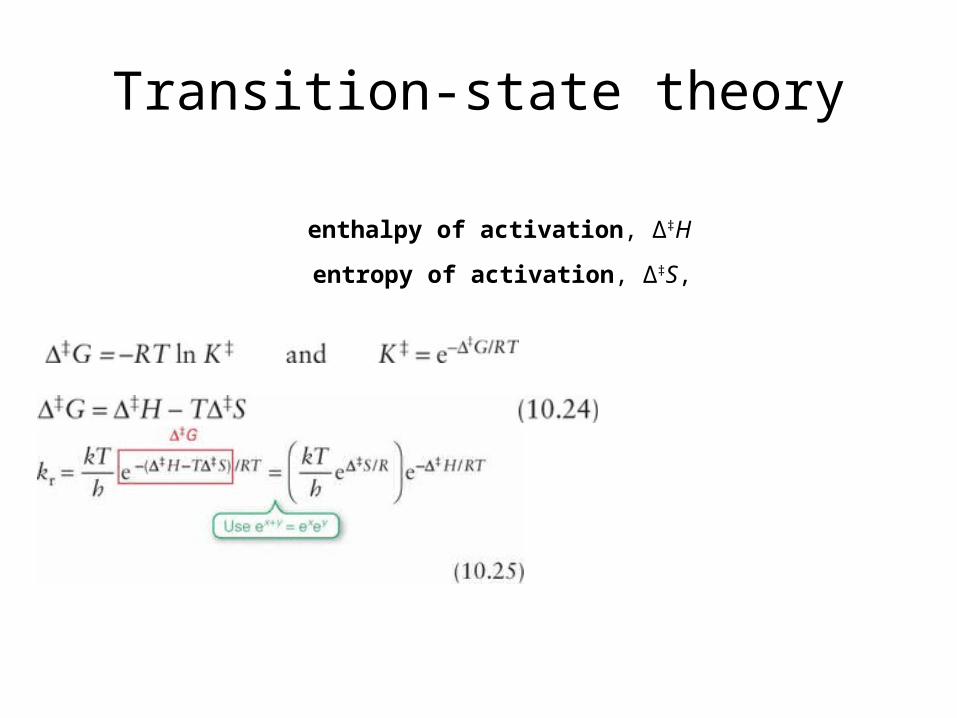
Transition-state theory
enthalpy of activation, Δ‡H
entropy of activation, Δ‡S,
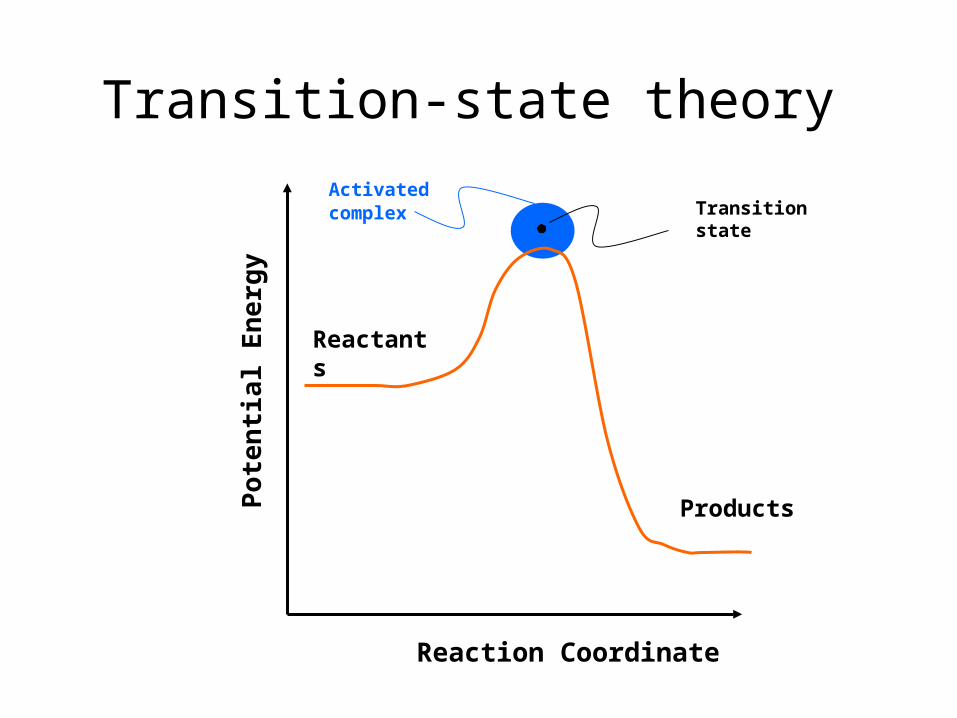
Reaction Coordinate
Pote
nti
al En
erg
y
Reac-tants
Prod-ucts
Activated complex Transition
state
Transition-state theory
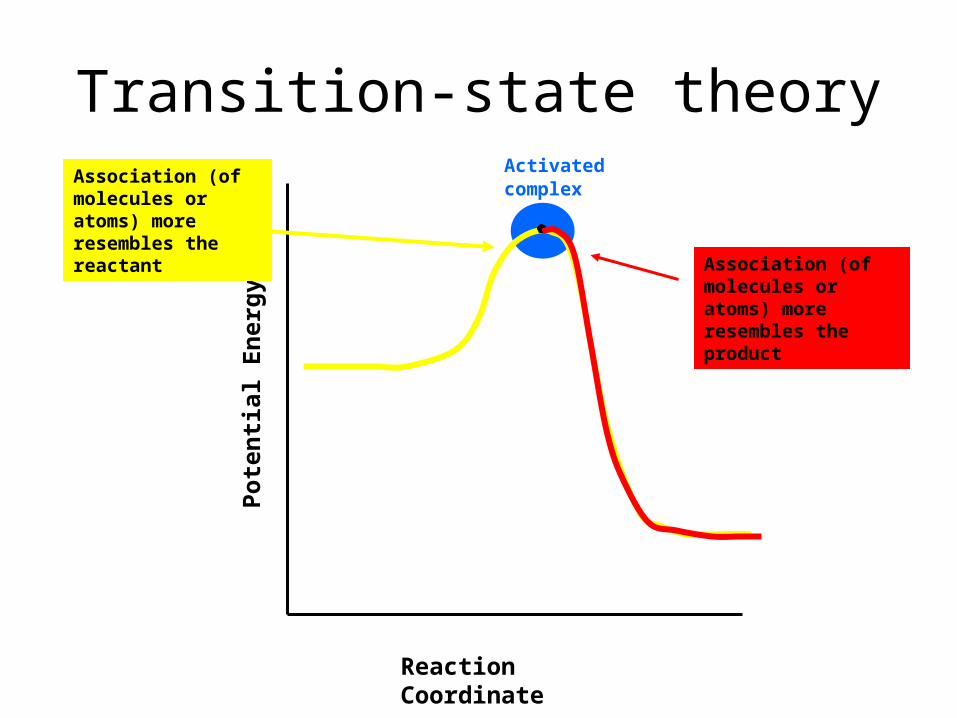
Reaction Coordinate
Po
ten
tial
En
erg
y
Activated complex
Association (of molecules or atoms) more re-sembles the reac-tant Association (of
molecules or atoms) more re-sembles the prod-uct
Transition-state theory

Transition-state theory• The Eyring Equation:
– Reaction between A and B proceeds through the formation of the activated complex (AB)‡ in a rapid pre-equilibrium.
A + B (AB)‡ → productK‡ k‡
• Rate, v = k‡[(AB)‡]
•Now, rate, v = k2[A][B]

Rate of decay of activated complex
Transition-state theory
Factors affecting the Activated Complex going through the Transi-tion State:
• – vibration frequency
• Centrifugal effect of rotation• It is supposed that k‡ and k‡ =
is the transmission coefficient (the prob-ability that the activated complex will pass through the transition state to form product).• Usually assume, 1

• If reaction is in gas phase, then:‡
Transition-state theoryConcentration of activated complex:
A + B (AB)‡
‡(AB)‡
A B
pK
p p
• Recall the Ideal Gas Law:
AA A A ; ; [A] A
np V n RT p RT p RT
V
• Substituting for gas pressure in K‡:‡ ‡
‡2
[(AB) ] [(AB) ]
[A][B]( ) [A][B]
RTK
RT RT

Transition-state theoryConcentration of activated complex (cont’d):
‡ ‡ [(AB) ] [A][B]K RT
Need to determine: k‡ and K‡
• Recall: rate, v = k‡[(AB)‡]
Therefore: rate, v = k‡ K‡RT[A][B]
For a bimolecular reaction
rate, v = k2[A][B]
Therefore: k2 = k‡ K‡RT

• K‡, the equilibrium constant between the reac-tants and the activated complex can be written in terms of the standard molar partition functions (qө) of the species involved.
Transition-state theory
‡(AB) /‡
A B
oA E RT
N qK e
q q Φ
ΦΦ
‡(AB ) (A) (B)o o o oE E E E
• Where:
• And Eo is the molar energy of the species involved
Obtain qө from spectroscopic data for A and B but not for (AB)‡

• Assume that a vibration of the activated complex (AB)‡ tips it through the transition state (TS). (i.e. a vibrational mode converts to translation and TS breaks a bond). The partition function for this vi-bration is:
Transition-state theory
Tkh Beq /1
1
• is the same frequency that determines k‡.
• is much less than a normal molecular vibration frequency, since the complex is falling apart (hence much weaker bond) and force constant is very low.

• For small values of , assume: 1/ Tkh B
• The partition function reduces to:
h
Tk
Tk
hq B
B
11
1
• We can therefore write:
‡ ‡(AB) (AB)Bk T
q qh
where denotes the partition function for all the other modes (trans, rot …) of the complex.
q
Transition-state theory

• K‡ can now be written as: ‡‡ Bk T
K Kh
• where is a modified equilibrium constant with one mode of vibration of (AB)‡ removed.
‡K
‡‡ (AB) /
A B
oA E RT
N qK e
q q
The rate constant:
Recall that: k2 = k‡ K‡RT where k‡ =
Substitution gives:
‡ ‡2 ( ) B Bk T k T
k K RT K RTh h
Transition-state theory

• In terms of molar concentration (and application of the Ideal Gas Law), it can be shown that:
‡
‡ CKK
RT
• Substitution gives:‡
‡‡2
CB B BC
Kk T k T k Tk K RT RT K
h h RT h
‡
2B
C
k Tk K
h Eyring Equation
Transition-state theory

• Assume = 1, and rearranging gives:
Eyring Equation(Thermodynamic Aspects)
‡2
CB
k hK
k T
• re-call:
• and:
‡‡ ln CG RT K ‡ ‡ ‡G H T S
• ‡ ‡ ‡ln CRT K H T S
• Substitution and rearrangement gives:
‡ ‡2 1
lnB
k h H S
k T R T R
Plot LHS vs 1/T
Slope = -H‡ / R
Intercept = S‡ / R
Another form of Eyring Equation
Transition-state theory