cd30 downregulation, mmae resistance, and mdr1 ...cd30 downregulation, mmae resistance, and mdr1...
TRANSCRIPT

Cancer Biology and Signal Transduction
CD30 Downregulation, MMAE Resistance, andMDR1 Upregulation Are All Associated withResistance to Brentuximab VedotinRobert Chen1, Jessie Hou2, Edward Newman2, Young Kim3, Cecile Donohue2,Xueli Liu4, Sandra H. Thomas1, Stephen J. Forman1, and Susan E. Kane2
Abstract
Brentuximab vedotin (BV) is an antibody–drug conjugatethat specifically delivers the potent cytotoxic drug monomethylauristatin E (MMAE) to CD30-positive cells. BV is FDAapproved for treatment of relapsed/refractory Hodgkin lym-phoma and anaplastic large cell lymphoma (ALCL); however,many patients do not achieve complete remission and devel-op BV-resistant disease. We selected for BV-resistant Hodgkinlymphoma (L428) and ALCL (Karpas-299) cell lines usingeither constant (ALCL) or pulsatile (Hodgkin lymphoma)exposure to BV. We confirmed drug resistance by MTS assayand analyzed CD30 expression in resistant cells by flowcytometry, qRT-PCR, and Western blotting. We also measureddrug exporter expression, MMAE resistance, and intracellularMMAE concentrations in BV-resistant cells. In addition, tissuebiopsy samples from 10 Hodgkin lymphoma and 5 ALCLpatients who had relapsed or progressed after BV treatment
were analyzed by immunohistocytochemistry for CD30expression. The resistant ALCL cell line, but not the Hodgkinlymphoma cell line, demonstrated downregulated CD30expression compared with the parental cell line. In contrast,the Hodgkin lymphoma cell line, but not the ALCL cell line,exhibited MMAE resistance and increased expression of theMDR1 drug exporter compared with the parental line. For bothHodgkin lymphoma and ALCL, samples from patients relapsed/resistant on BV persistently expressed CD30 by immunohistocy-tochemistry. One Hodgkin lymphoma patient sample expressedMDR1 by immunohistocytochemistry. Although loss of CD30expression is a possible mode of BV resistance in ALCL in vitromodels, this has not been confirmed in patients. MMAE resis-tance and MDR1 expression are possible modes of BV resistancefor Hodgkin lymphoma both in vitro and in patients. Mol CancerTher; 14(6); 1376–84. �2015 AACR.
IntroductionAbout 9,200 cases of Hodgkin lymphoma and 2,000 cases of
anaplastic large cell lymphoma (ALCL) are diagnosed in theUnited States annually (1). Although induction chemotherapyhas a high response rate, 30% of Hodgkin lymphoma and 40% to65% of ALCL patients will experience relapse (2, 3). Roughly halfof these patients can be salvaged with high-dose chemotherapyfollowed by autologous stem cell transplantation (ASCT; refs. 4,5). For the 50% of patients who relapse after ASCT, options arelimited. Hodgkin lymphoma is characterized by the presence ofReed–Sternberg cells, which comprise only a minority of cells inthe tumor mass and express CD30 surface antigen (6). Alterna-tively, ALCL is comprised of CD30-expressing lymphoma cells inthe majority of the tumor mass. Brentuximab vedotin (BV) is a
novel therapeutic in the class of antibody–drug conjugates (ADC)that consists of three components: the cAC10 chimeric IgG1antibody specific for CD30, the microtubule-disrupting agentmonomethyl auristatin E (MMAE), and a protease-cleavablelinker that covalently attaches MMAE to cAC10 (7). The entireADC is internalized upon binding to cell surface CD30, andlysosomal enzymes digest the protease cleavable linker, releasingMMAE, which disrupts the microtubule network and causes cellcycle arrest and apoptosis.
In a pivotal phase II trial for relapsed/refractory Hodgkinlymphoma, BV demonstrated an overall response rate (ORR) of75% and a complete response (CR) rate of 34% (8). In a phase IItrial in patients with relapsed/refractory ALCL, BV demonstratedan ORR of 86% and CR rate of 57% (9). Patients who achieve CRmay have durable remissions; however, those achieving onlypartial responses have relatively short response durations, withmedians of 3.5months inHodgkin lymphoma and 2.5months inALCL (8, 9). All patients who do not attain CR eventually developprogressive disease despite active treatment with BV. Given thatBV is theonly therapy approvedby the FDA for relapsed/refractoryHodgkin lymphoma in the last 20 years (10), and one of twoapproved therapies for ALCL, it is imperative that we understandits resistance mechanisms.
Currently, it is unknown whether BV-resistant tumors escapethrough alterations in surface expression of CD30 (resistance toantibody moiety), by development of resistance to the antimi-crotubule agent MMAE, or by expression of one or more trans-porters that export MMAE out of the cell. To explore possible BV
1Department of Hematology and Hematopoietic Cell Transplantation,City of Hope, Duarte, California. 2Department of Cancer Biology, Cityof Hope, Duarte, California. 3Department of Pathology, City of Hope,Duarte, California. 4Division of Biostatistics, Beckman Research Insti-tute of City of Hope, Duarte, California.
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Robert Chen, City of Hope, 1500 E. Duarte Rd., Duarte,CA 91010. Phone: 626-256-4673, ext. 65298; Fax: 626-301-8116; E-mail:[email protected]
doi: 10.1158/1535-7163.MCT-15-0036
�2015 American Association for Cancer Research.
MolecularCancerTherapeutics
Mol Cancer Ther; 14(6) June 20151376
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036

resistance mechanisms, we have selected cell lines for BV resis-tance and also have analyzed tumor samples from patients whoprogressed on BV therapy.
Materials and MethodsCell culture
The L428 (Hodgkin lymphoma) and Karpas-299 (ALCL) celllines were purchased from the Leibniz Institute DSMZ GermanCollection of Microorganisms and Cell Cultures, which authen-ticates cell lines using short tandem repeat DNA typing. Cells werepassaged in the laboratory for fewer than 6 months followingpurchase and original authentication. Cells were grown in RPMI-1640 (Cellgro Inc.) supplementedwith 10%heat inactivated FBS,2mmol/L glutamine, 100mg/mL streptomycin, and 100units/mLpenicillin. All cell lines were cultured at 37�C in a humidifiedwith5% CO2 atmosphere.
Selection of BV-resistant cell linesBVwas obtained fromCity of Hope Pharmacy. Selection of BV-
resistant cell lines used two different approaches. For the constantexposure approach, cells were incubated at sub-IC50 concentra-tions of BV and monitored for changes in cell number over 1month. BV concentration was then increased incrementally up tothe IC50 concentration as long as the cell numbers increased fromprior passage. For the pulsatile approach, cells were incubated atsupra-IC50 concentration until proliferation halted (as deter-mined by twice a week cell sampling and counting from theselection culture), and then cells were rescuedwith BV-freemedia.When consistent proliferation was seen, BVwas added back at thesame supra-IC50 concentration. Selection was deemed successfulwhen consistent proliferation was seen even in supra-IC50 con-centrations of BV.
RNA extraction and quantitative real-time PCRTotal RNA was extracted using Trizol (Invitrogen), following
themanufacturer's protocol. ResidualDNAwasdigested using theDNA-free Kit following the manufacturer's instructions(Ambion). cDNA was obtained by reverse transcribing 10 mg ofRNA with SuperScript III reverse transcriptase and random pri-mers according to the manufacturer's instructions (Invitrogen).Expression of human mRNAs encoding CD30 (CD30-F: 50-CCAGGATCAAGTCACTCATCTCA-30, CD30-R: 50-AAGTCCCTG-GGCAAAGTAAAG-30), MRD1 (MRD1-F: 50-GCTCCTGACTATG-CCAAAGCC-30,MRD1-R,50-CTTCACCTCCAGGCTCAGTCCC-30),GAPDH(GAPDH-F: 50-CGCTCTCTGCTCCTCCTGTT-30, GAPDH-R:50-CCATGGTGTCTGAGCGATGT-30), andTBP(TBP-F:50-TGCA-CAGGAGCCAAGAGTGAA-30, TBP-R: 50-CACATCACAGCTCCC-CACCA-30)was determinedbyquantitative real-timePCR (CFX96;BIO-RAD) using 2X iQ SYBRGreenMastermix (BIO-RAD) accord-ing to themanufacturer's protocol. GAPDHexpressionwas used asan internal control for CD30 expression, and TBP expression wasused as an internal control for MDR1. Primers were ordered fromIntegrated DNA Technologies.
Protein extraction and immunoblottingCell lysates were collected in Trizol. Protein concentrations
were determinedwith the bicinchoninic acid (BCA) Protein Assay(Pierce) according to the manufacturer's instructions. Protein (10mg) was loaded onto a 7.5% SDS-PAGE gel, and proteins weretransferred to Hybond-LFP membrane (Amersham), followed by
primary and secondary antibody incubation. The following anti-bodies were used: rabbit monoclonal anti-CD30 (Abcam), rabbitmonoclonal anti-MDR1 (Cell Signaling), rabbitmonoclonal anti-GAPDH (Cell Signaling), and goat anti-rabbit-horseradish per-oxidase–conjugated secondary antibody (Abcam). Secondaryantibody was detected using an ECL Plus kit (Thermo FisherScientific). Signals were detected using BIO-RADMolecular Imag-er ChemiDoc XRSþ.
MTS proliferation assayCells were seeded in 96-well plates at 5,000 cells (L-428) and
10,000 cells (karpas-299) per well. Cells were incubated withincreasing amounts of BV (0.1 nmol/L to 100 mmol/L for Karpas-299; 1 mg/mL to 2,400 mg/mL for L428), in triplicate. Cell viabilitywas measured after 72 hours using the CellTiter 96R AqueousNon-Radioactive Cell Proliferation Assay (Promega) according tothemanufacturer's instructions. IC50 value is the concentration ofdrug which produced a 50% reduction in viability compared with0 drug control and was calculated from the dose–response curves.
Flow cytometryCells (2� 105) were incubated with 0.5 ng/mL of PE-conjugated
monoclonal antibodies at room temperature in the dark for 20minutes. Anti-CD30 antibody was purchased from Beckton Dick-inson andused at concentrations titrated for optimal staining. Flowcytometry was performed on a Beckman Coulter CyAn ADP 9colorCytometer. Lymphocytes or abnormal cell populations were gatedon CD45/side scatter (SSC) or forward scatter (FSC) dot plot.
ImmunohistochemistryTen Hodgkin lymphoma patient samples and 5 ALCL patient
sampleswere obtained at the timeof relapse orprogressive diseaseand analyzed by IHC. Samples were obtained from leftover tissueon aCity of Hope Institutional Review Board–approved protocol.For CD30 immunostaining, we used themonoclonalmouse anti-human CD30, clone Ber-H2. For MDR1 immunostaining, weused the monoclonal mouse anti-human MDR1, clone PG-M1.Both immunostainings were visualized with DAKO Envision/HRP kit (Dakocytomation). IHC staining was performed on 5-mm-thick paraffin-embedded tissue. Tissue sections were depar-affinized in xylene followed by 100% to 70% alcohol. Sampleswere then quenched in 3% hydrogen peroxide and pretreated topromote antigen retrieval with high pH. Slides were incubated inprimary antibody at 1/30 dilution for 20 minutes at roomtemperature. After rinsing in Dako Wash, slides were incubatedin EnVision FLEX/HRP (DAKO K-8000) for 20 minutes. Slideswere further washed in Dako buffer and then incubated withdiaminobenzidine tetrahydrochloride (DAB) from Dako, coun-terstained with hematoxylin, and mounted.
Intracellular MMAE accumulationKarpas-299 and Karpas-299R cells were treated with 16 ng/mL
of BV for 0, 2, 6, 24, and 48 hours. L428 and L428-R cells weretreated with 20 mg/mL of BV for 0, 2, 6, 24, and 48 hours. MMAEconcentration in cells was measured by LC-MS/MS according to amodification of a previously published method (11). Briefly,following addition of an internal standard, cell membranes weredisrupted by sonication and proteins were precipitated usingmethanol. MMAE concentrations in the extracted cell pellets werethen analyzed by gradient reversed phase HPLC separation andtandem mass spectrometric detection. The Waters Quattro
CD30 Expression and Brentuximab Vedotin Resistance
www.aacrjournals.org Mol Cancer Ther; 14(6) June 2015 1377
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036

Premier XEmass spectrometer (Milford) was operated in positiveelectrospray ionization mode, and quantitation was performedusing multiple reaction monitoring. MassLynx version 4.1 soft-ware was used for data acquisition and processing. Under opti-mized assay conditions, the lower limit of quantitation was 0.01ng/106 cells or 0.17 pg on column. Inter- and intra-day precisionand accuracy of the method were within �10% of target values.
Statistical considerationsCell proliferation/growth over time and across different con-
centrations are represented as means with 95% confidence inter-vals (error bars). For measurements over time, a repeated mea-sures ANOVAmodel was also used to examine possible time and
group interactions. For dose–response measurements, a four-parameter log-logistic model was fitted to the curves, and IC50
values were estimated accordingly. Statistical analyses were per-formed by two-tailed unpaired t test unless otherwise noted. Aneffect was considered statistically significant when the corre-sponding P value was less than 0.05. All statistical analysis wascarried out using both R software and Excel.
ResultsBV-resistant Hodgkin lymphoma and ALCL cell line selection
MTS assays determined the IC50 of parental cell lines to be:L428 (Hodgkin lymphoma) 27 mg/mL � 4.8 mg/mL and Karpas-299 (ALCL) 29 ng/mL � 21 ng/mL (Table 1). BV-resistant cell
Table 1. Comparison of parental and BV-resistant cell lines
L428-P L428-R Karpas-P Karpas-R
IC50 to BV 27 � 4.8 mg/mL 236 � 22 mg/mL 29 � 21 ng/mL 19 � 1.9 mg/mLRelative resistance 1 8.7 1 655Cells CD30þ 98% 97% 96% 59%Median intensity CD30 280 � 10 265 � 5 592 � 51 78 � 17
0
100,000
200,000
300,000
400,000
500,000
600,000
10730
Live
cel
l cou
nt
BV treatment �me (days)
L428 parental vs. BV-resistant growthA
L428-R
L428-P0
500,000
1,000,000
1,500,000
2,000,000
2,500,000
3,000,000
3,500,000
9730
Live
cel
l cou
nt
BV treatment �me (days)
Karpas-299 parental vs. BV-resistant growthB
Karpas-R
Karpas-P
0
20
40
60
80
100
2,0001,00030010030100
Live
cel
ls (%
con
trol
)
BV concentra�on (μg/mL) BV concentra�on (μg/mL)
L428 parental vs. BV-resistant IC50C
L428-RL428-P
0
20
40
60
80
100
Live
cel
ls (%
con
trol
)
Karpas-299 parental vs. BV-resistant IC50D
Karpas-RKarpas-P
Figure 1.BV-resistant ALCL and Hodgkin lymphoma in vitro cell models. Proliferation experiments were performed in duplicate wells and averaged over 2 separateexperiments, and a repeatedmeasures ANOVAmodel is applied to show that time, group, and their interactions are all very significant (A and B). Viable cells countswere performed by hemocytometer with methylene blue. A, L428 and L428-R were seeded at 100,000 cells per well and incubated with BV at 80 mg/mL.L428-R cells are able to proliferate at this concentration whereas L428 cannot (P < 0.0001). B, Karpas-299 and Karpas-R were seeded at 40,000 cells per well andincubated with BV at 30 ng/mL. Karpas-R cells are able to proliferate at this concentration whereas Karpas-299 cannot (P < 0.0003). MTS assays wereperformed in triplicate wells and averaged over 3 separate experiments, and cells were seeded in 96-well plates at 5,000 cells (L428) and 10,000 cells (Karpas-299)per well. C, a four-parameter log-logistic model was fitted to assess inhibitory effect of L428-P and L428-R, respectively. The estimated IC50 SEs are 27.46(4.76) and 236.08 (22.15), respectively. D, a four-parameter log-logistic model was fitted to assess inhibitory effect of Karpas-P and Karpas-R, respectively. Theestimated IC50 SEs are 0.0029 (0.021) and 19.45 (1.87), respectively.
Chen et al.
Mol Cancer Ther; 14(6) June 2015 Molecular Cancer Therapeutics1378
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036
models were selected using two different approaches. For theconstant exposure approach, L428 and Karpas-299 cell lines wereincubated at sub-IC50 concentrations of BV (25 mg/mL for L428and 10 ng/mL for Karpas-299), and cell numbers weremonitoredover the course of 3 months in culture. The BV concentration wasincreased when consistent cell proliferation at the initial selectingconcentration was achieved. This approach was successful forselecting Karpas-299–resistant cells as they were able to grow inconcentrations of BV as high as 20 ng/mL, but we were unable toobtain resistant L428 cells bymeans of constant exposure to drug.We therefore used a pulsatile approach, in which L428 cells wereincubated in a supra-IC50 concentration of BV (50 mg/mL), andcell numbers were assessed twice weekly until no further prolif-erationwas seen. Cells were then rescuedwith BV-freemedia untilproliferation was again observed (increase in cell number for 3consecutive weeks), at which point 50 mg/mL BV was added backto the cells. This process was continued until consistent prolifer-ation in 50 mg/mL of BV was achieved.
We confirmed BV resistance in both cell lines using cell prolif-eration assays andMTS assays (Fig. 1). In cell proliferation assays,resistant cell lines were able to proliferate in the presence of BV at
concentrations above their respective parental line IC50s (Fig. 1Aand B). At the same concentrations, the parental cell lines quicklydied. In MTS assays, resistant cell lines demonstrated IC50s shiftedto higher BV concentrations: L428-R (236 mg/mL� 22 mg/mL, 8.7-fold relative resistance), Karpas-R (19 mg/mL � 1.9 mg/mL, 655-fold relative resistance; Fig. 1C and D; Table 1).
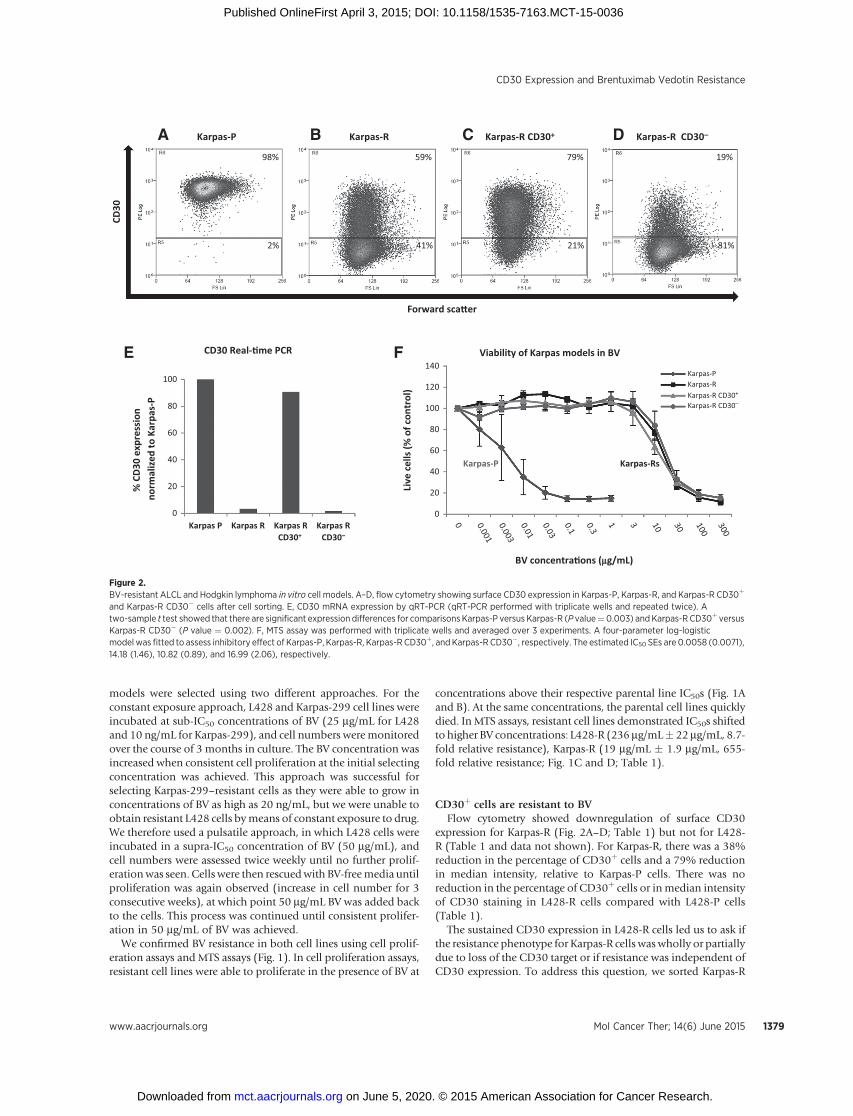
CD30þ cells are resistant to BVFlow cytometry showed downregulation of surface CD30
expression for Karpas-R (Fig. 2A–D; Table 1) but not for L428-R (Table 1 and data not shown). For Karpas-R, there was a 38%reduction in the percentage of CD30þ cells and a 79% reductionin median intensity, relative to Karpas-P cells. There was noreduction in the percentage of CD30þ cells or inmedian intensityof CD30 staining in L428-R cells compared with L428-P cells(Table 1).
The sustained CD30 expression in L428-R cells led us to ask ifthe resistance phenotype for Karpas-R cells waswholly or partiallydue to loss of the CD30 target or if resistance was independent ofCD30 expression. To address this question, we sorted Karpas-R
0
20
40
60
80
100
Karpas P Karpas R Karpas RCD30+
Karpas RCD30–
% C
D30
expr
essi
on
norm
aliz
ed to
Kar
pas-
P
CD30 Real-�me PCR E
Karpas-PA
Forward sca�er
CD30
98%
2%
B Karpas-R
59%
41%
C Karpas-R CD30+
79%
21%
Karpas-R CD30–D19%
81%
0
20
40
60
80
100
120
140
Live
cel
ls (%
of c
ontr
ol)
BV concentra�ons (mg/mL)
Viability of Karpas models in BVFKarpas-PKarpas-RKarpas-R CD30+
Karpas-R CD30–
Karpas-P Karpas-Rs
Figure 2.BV-resistant ALCL and Hodgkin lymphoma in vitro cell models. A–D, flow cytometry showing surface CD30 expression in Karpas-P, Karpas-R, and Karpas-R CD30þ
and Karpas-R CD30� cells after cell sorting. E, CD30 mRNA expression by qRT-PCR (qRT-PCR performed with triplicate wells and repeated twice). Atwo-sample t test showed that there are significant expression differences for comparisons Karpas-P versus Karpas-R (P value¼0.003) andKarpas-R CD30þ versusKarpas-R CD30� (P value ¼ 0.002). F, MTS assay was performed with triplicate wells and averaged over 3 experiments. A four-parameter log-logisticmodel was fitted to assess inhibitory effect of Karpas-P, Karpas-R, Karpas-R CD30þ, and Karpas-R CD30�, respectively. The estimated IC50 SEs are 0.0058 (0.0071),14.18 (1.46), 10.82 (0.89), and 16.99 (2.06), respectively.
CD30 Expression and Brentuximab Vedotin Resistance
www.aacrjournals.org Mol Cancer Ther; 14(6) June 2015 1379
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036
cells into CD30þ and CD30� subpopulations and examined therespective CD30 levels and BV resistance profiles of these sortedsubpopulations. The CD30� subpopulation had lower surfaceCD30 protein (Fig. 2A–D) and mRNA (Fig. 2E), compared withtheCD30þ subpopulation andwith parental Karpas-P cells. In theexperiment shown here, the sorted CD30þ subpopulation had79% CD30þ cells with a median intensity of 98, whereas theCD30� subpopulation had 19% CD30þ cells at a median inten-sity of 36 after 1week of culture in the absence of BV. The unsortedKarpas-R population was 59% CD30þwith a median intensity of74 under these same conditions, whereas the parental Karpas-Pcells were essentially 100% CD30þ with a median intensity of556. MTS assays showed that the unsorted Karpas-R population,the sorted CD30þ subpopulation, and the sorted CD30� sub-population were all equally resistant to BV (Fig. 2F). This exper-iment was performed in triplicate wells and repeated 3 times withessentially the same results.
BV resistance phenotype is not permanent in Karpas-R cellsWe also askedwhether downregulation of CD30 and resistance
were stable phenotypes in the Karpas-R cell line. Karpas-R cellswere cultured in BV-free growth medium for 26 weeks and thenreassayed for CD30 expression by flow cytometry and for BVresistance byMTS. After 26weekswithout drug selection, 100%ofthe Karpas-R cells were CD30þ and themedian intensity was 340,suggesting a partial reversion of the CD30 downregulation phe-notype (Supplementary Fig. S1). MTS assay showed that thesecells were 11-fold more resistant to BV, a partial reversion of thephenotype, but still with substantial resistance despite regainedCD30 expression (Supplementary Fig. S2).
CD30 is not downregulated in primary Hodgkin lymphomaand ALCL
Given the transient reduction of CD30 expression that weobserved in Karpas-R cells (but not in L428-R cells), we wantedto determine CD30 status in patients with acquired BV resistance.We previously reported that CD30 continued to be expressed in 2Hodgkin lymphomapatientswith BV resistance (12), andwenowreport the analysis of samples from 15 additional patients (10Hodgkin lymphoma, 5 ALCL) who initially achieved response to
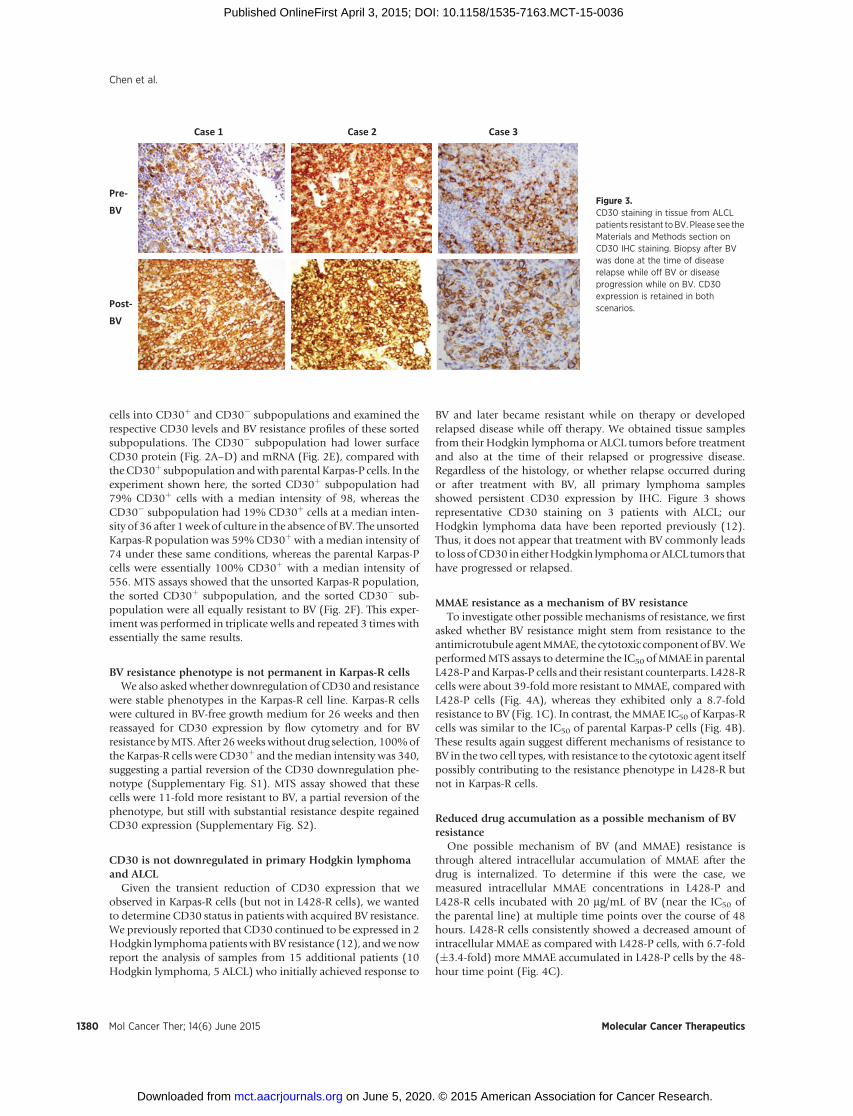
BV and later became resistant while on therapy or developedrelapsed disease while off therapy. We obtained tissue samplesfrom their Hodgkin lymphoma or ALCL tumors before treatmentand also at the time of their relapsed or progressive disease.Regardless of the histology, or whether relapse occurred duringor after treatment with BV, all primary lymphoma samplesshowed persistent CD30 expression by IHC. Figure 3 showsrepresentative CD30 staining on 3 patients with ALCL; ourHodgkin lymphoma data have been reported previously (12).Thus, it does not appear that treatment with BV commonly leadsto loss ofCD30 in eitherHodgkin lymphomaorALCL tumors thathave progressed or relapsed.
MMAE resistance as a mechanism of BV resistanceTo investigate other possible mechanisms of resistance, we first
asked whether BV resistance might stem from resistance to theantimicrotubule agentMMAE, the cytotoxic component of BV.WeperformedMTS assays to determine the IC50 ofMMAE in parentalL428-P andKarpas-P cells and their resistant counterparts. L428-Rcells were about 39-fold more resistant to MMAE, compared withL428-P cells (Fig. 4A), whereas they exhibited only a 8.7-foldresistance to BV (Fig. 1C). In contrast, theMMAE IC50 of Karpas-Rcells was similar to the IC50 of parental Karpas-P cells (Fig. 4B).These results again suggest different mechanisms of resistance toBV in the two cell types, with resistance to the cytotoxic agent itselfpossibly contributing to the resistance phenotype in L428-R butnot in Karpas-R cells.
Reduced drug accumulation as a possible mechanism of BVresistance
One possible mechanism of BV (and MMAE) resistance isthrough altered intracellular accumulation of MMAE after thedrug is internalized. To determine if this were the case, wemeasured intracellular MMAE concentrations in L428-P andL428-R cells incubated with 20 mg/mL of BV (near the IC50 ofthe parental line) at multiple time points over the course of 48hours. L428-R cells consistently showed a decreased amount ofintracellular MMAE as compared with L428-P cells, with 6.7-fold(�3.4-fold) more MMAE accumulated in L428-P cells by the 48-hour time point (Fig. 4C).
Pre-BV
Post-BV
Case 1 Case 2 Case 3
Figure 3.CD30 staining in tissue from ALCLpatients resistant toBV. Please see theMaterials and Methods section onCD30 IHC staining. Biopsy after BVwas done at the time of diseaserelapse while off BV or diseaseprogression while on BV. CD30expression is retained in bothscenarios.
Chen et al.
Mol Cancer Ther; 14(6) June 2015 Molecular Cancer Therapeutics1380
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036
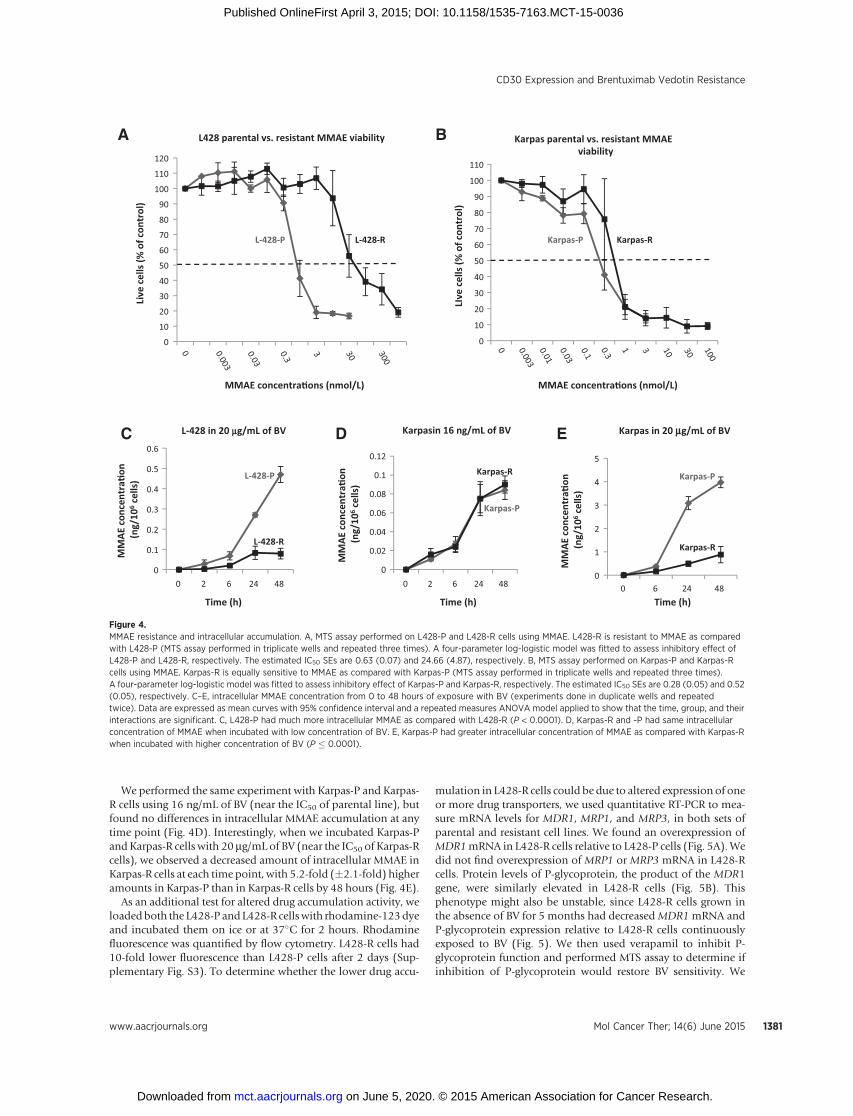
We performed the same experiment with Karpas-P and Karpas-R cells using 16 ng/mL of BV (near the IC50 of parental line), butfound no differences in intracellular MMAE accumulation at anytime point (Fig. 4D). Interestingly, when we incubated Karpas-Pand Karpas-R cells with 20 mg/mL of BV (near the IC50 of Karpas-Rcells), we observed a decreased amount of intracellular MMAE inKarpas-R cells at each time point, with 5.2-fold (�2.1-fold) higheramounts in Karpas-P than in Karpas-R cells by 48 hours (Fig. 4E).
As an additional test for altered drug accumulation activity, weloadedboth the L428-P andL428-R cellswith rhodamine-123dyeand incubated them on ice or at 37�C for 2 hours. Rhodaminefluorescence was quantified by flow cytometry. L428-R cells had10-fold lower fluorescence than L428-P cells after 2 days (Sup-plementary Fig. S3). To determine whether the lower drug accu-
mulation in L428-R cells could be due to altered expression of oneor more drug transporters, we used quantitative RT-PCR to mea-sure mRNA levels for MDR1, MRP1, and MRP3, in both sets ofparental and resistant cell lines. We found an overexpression ofMDR1mRNA in L428-R cells relative to L428-P cells (Fig. 5A).Wedid not find overexpression of MRP1 or MRP3 mRNA in L428-Rcells. Protein levels of P-glycoprotein, the product of the MDR1gene, were similarly elevated in L428-R cells (Fig. 5B). Thisphenotype might also be unstable, since L428-R cells grown inthe absence of BV for 5 months had decreasedMDR1mRNA andP-glycoprotein expression relative to L428-R cells continuouslyexposed to BV (Fig. 5). We then used verapamil to inhibit P-glycoprotein function and performed MTS assay to determine ifinhibition of P-glycoprotein would restore BV sensitivity. We
0
10
20
30
40
50
60
70
80
90
100
110
LIve
cel
ls (%
of c
ontr
ol)
MMAE concentra�ons (nmol/L)
Karpas parental vs. resistant MMAEviability
Karpas-RKarpas-P
B
0
10
20
30
40
50
60
70
80
90
100
110
120
Live
cel
ls (%
of c
ontr
ol)
MMAE concentra�ons (nmol/L)
L428 parental vs. resistant MMAE viability
L-428-RL-428-P
A
0
0.02
0.04
0.06
0.08
0.1
0.12
4824620
MM
AEco
ncen
tra�
on(n
g/10
6ce
lls)
Time (h)
Karpasin 16 ng/mL of BVD
0
0.1
0.2
0.3
0.4
0.5
0.6
4824620
MM
AEco
ncen
tra�
on(n
g/10
6ce
lls)
Time (h)
L-428 in 20 mg/mL of BVC
L-428-R
L-428-P Karpas-R
Karpas-P
0
1
2
3
4
5
482460
MM
AEco
ncen
tra�
on(n
g/10
6ce
lls)
Time (h)
Karpas in 20 mg/mL of BVE
Karpas-R
Karpas-P
Figure 4.MMAE resistance and intracellular accumulation. A, MTS assay performed on L428-P and L428-R cells using MMAE. L428-R is resistant to MMAE as comparedwith L428-P (MTS assay performed in triplicate wells and repeated three times). A four-parameter log-logistic model was fitted to assess inhibitory effect ofL428-P and L428-R, respectively. The estimated IC50 SEs are 0.63 (0.07) and 24.66 (4.87), respectively. B, MTS assay performed on Karpas-P and Karpas-Rcells using MMAE. Karpas-R is equally sensitive to MMAE as compared with Karpas-P (MTS assay performed in triplicate wells and repeated three times).A four-parameter log-logistic model was fitted to assess inhibitory effect of Karpas-P and Karpas-R, respectively. The estimated IC50 SEs are 0.28 (0.05) and 0.52(0.05), respectively. C–E, intracellular MMAE concentration from 0 to 48 hours of exposure with BV (experiments done in duplicate wells and repeatedtwice). Data are expressed as mean curves with 95% confidence interval and a repeated measures ANOVA model applied to show that the time, group, and theirinteractions are significant. C, L428-P had much more intracellular MMAE as compared with L428-R (P < 0.0001). D, Karpas-R and -P had same intracellularconcentration of MMAE when incubated with low concentration of BV. E, Karpas-P had greater intracellular concentration of MMAE as compared with Karpas-Rwhen incubated with higher concentration of BV (P � 0.0001).
CD30 Expression and Brentuximab Vedotin Resistance
www.aacrjournals.org Mol Cancer Ther; 14(6) June 2015 1381
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036
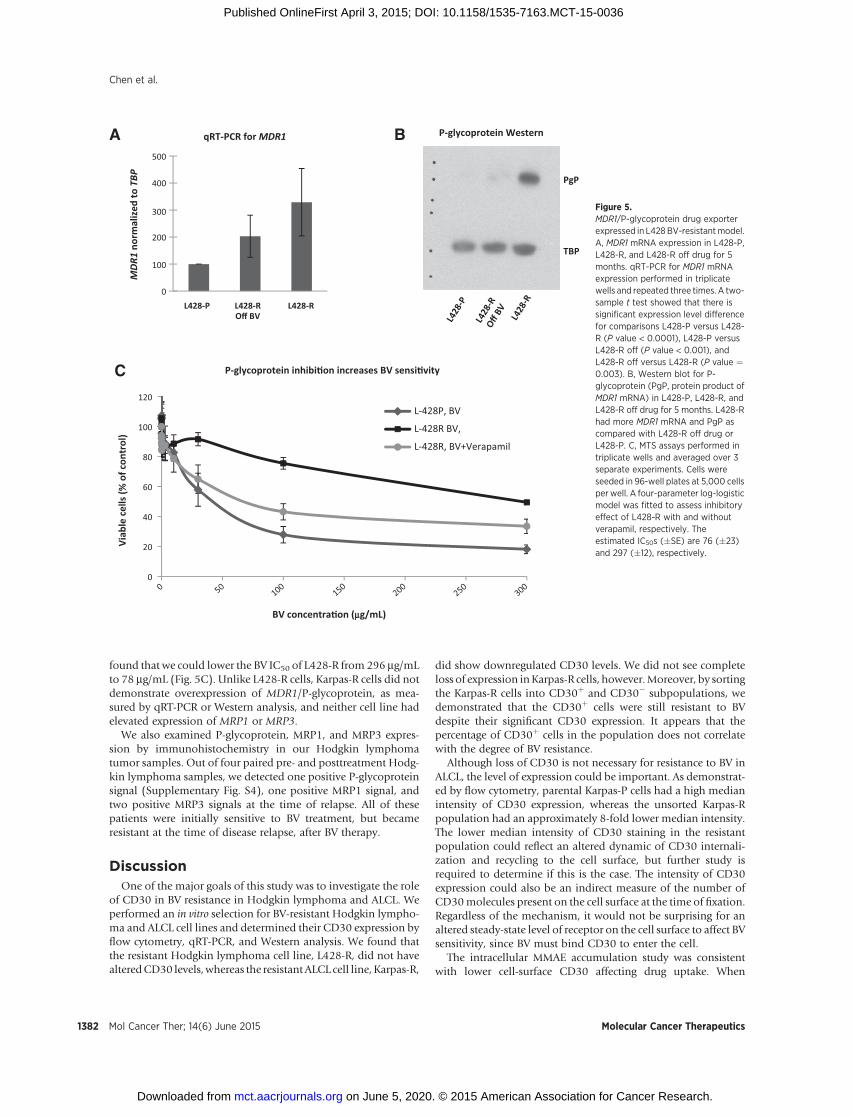
found that we could lower the BV IC50 of L428-R from 296 mg/mLto 78 mg/mL (Fig. 5C). Unlike L428-R cells, Karpas-R cells did notdemonstrate overexpression of MDR1/P-glycoprotein, as mea-sured by qRT-PCR or Western analysis, and neither cell line hadelevated expression of MRP1 or MRP3.
We also examined P-glycoprotein, MRP1, and MRP3 expres-sion by immunohistochemistry in our Hodgkin lymphomatumor samples. Out of four paired pre- and posttreatment Hodg-kin lymphoma samples, we detected one positive P-glycoproteinsignal (Supplementary Fig. S4), one positive MRP1 signal, andtwo positive MRP3 signals at the time of relapse. All of thesepatients were initially sensitive to BV treatment, but becameresistant at the time of disease relapse, after BV therapy.
DiscussionOne of the major goals of this study was to investigate the role
of CD30 in BV resistance in Hodgkin lymphoma and ALCL. Weperformed an in vitro selection for BV-resistant Hodgkin lympho-ma and ALCL cell lines and determined their CD30 expression byflow cytometry, qRT-PCR, and Western analysis. We found thatthe resistant Hodgkin lymphoma cell line, L428-R, did not havealteredCD30 levels,whereas the resistant ALCL cell line, Karpas-R,
did show downregulated CD30 levels. We did not see completeloss of expression inKarpas-R cells, however.Moreover, by sortingthe Karpas-R cells into CD30þ and CD30� subpopulations, wedemonstrated that the CD30þ cells were still resistant to BVdespite their significant CD30 expression. It appears that thepercentage of CD30þ cells in the population does not correlatewith the degree of BV resistance.
Although loss of CD30 is not necessary for resistance to BV inALCL, the level of expression could be important. As demonstrat-ed by flow cytometry, parental Karpas-P cells had a high medianintensity of CD30 expression, whereas the unsorted Karpas-Rpopulation had an approximately 8-fold lower median intensity.The lower median intensity of CD30 staining in the resistantpopulation could reflect an altered dynamic of CD30 internali-zation and recycling to the cell surface, but further study isrequired to determine if this is the case. The intensity of CD30expression could also be an indirect measure of the number ofCD30molecules present on the cell surface at the time of fixation.Regardless of the mechanism, it would not be surprising for analtered steady-state level of receptor on the cell surface to affect BVsensitivity, since BV must bind CD30 to enter the cell.
The intracellular MMAE accumulation study was consistentwith lower cell-surface CD30 affecting drug uptake. When
0
20
40
60
80
100
120
Viab
le c
ells
(% o
f con
trol
)
BV concentra�on (mg/mL)
P-glycoprotein inhibi�on increases BV sensi�vity
L-428P, BV
L-428R BV,
L-428R, BV+Verapamil
MDR
1 no
rmal
ized
to T
BPqRT-PCR for MDR1 P-glycoprotein Western
PgP
TBP
L428-P L428-ROff BV
L428-R
BA500
400
300
200
100
0
C
L428
-P
L428
-ROff
BV L428
-R
Figure 5.MDR1/P-glycoprotein drug exporterexpressed in L428BV-resistantmodel.A, MDR1 mRNA expression in L428-P,L428-R, and L428-R off drug for 5months. qRT-PCR for MDR1 mRNAexpression performed in triplicatewells and repeated three times. A two-sample t test showed that there issignificant expression level differencefor comparisons L428-P versus L428-R (P value < 0.0001), L428-P versusL428-R off (P value < 0.001), andL428-R off versus L428-R (P value ¼0.003). B, Western blot for P-glycoprotein (PgP, protein product ofMDR1 mRNA) in L428-P, L428-R, andL428-R off drug for 5 months. L428-Rhad more MDR1 mRNA and PgP ascompared with L428-R off drug orL428-P. C, MTS assays performed intriplicate wells and averaged over 3separate experiments. Cells wereseeded in 96-well plates at 5,000 cellsper well. A four-parameter log-logisticmodel was fitted to assess inhibitoryeffect of L428-R with and withoutverapamil, respectively. Theestimated IC50s (�SE) are 76 (�23)and 297 (�12), respectively.
Mol Cancer Ther; 14(6) June 2015 Molecular Cancer Therapeutics1382
Chen et al.
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036

incubated in a low concentration of BV, Karpas-R cells appeared tohave sufficient CD30 expression to allow efficient drug entry. In ahigher concentration of BV, however, Karpas-R had 5.2-fold lowerintracellular MMAE accumulation than Karpas-P cells. The dif-ference in intracellular MMAE accumulation between Karpas-Pand Karpas-R at the high BV concentration, and lack of differenceat the low BV concentration, could suggest a saturation effect ofCD30. Limited BV cellular entry due to lower cell-surface CD30might explain a 3-fold reduction in MMAE sensitivity, but it doesnot match the 1,100-fold reduction in BV IC50 that we measuredfor Karpas-R cells. Indeed, resistance in our cell lines was notcorrelatedwith either the percentage ofCD30þ cells or themedianintensity of cell-surface CD30 signal in a given cell population,suggesting that the resistance phenotype must involve anothermechanism that is not related to CD30 accessibility.
We also found that CD30 downregulation in Karpas-R cells wasnot permanent. The resistant Karpas-R population was able to re-express CD30 and regain partial sensitivity to BV after a prolongedperiod of growth in the absence of drug. This phenomenon hassignificant clinical correlations. Although IHC analysis of primaryALCL specimens did not show CD30 downregulation, the major-ity of our patient samples were obtained from patients who hadnot been exposed toBV forweeks ormonths.Given thatCD30 canbe re-expressed in cell lines during a periodwithout drug exposurein cell lines, it is perhaps not surprising that our patient sampleswere all CD30 positive as well. It remains to be determinedwhether CD30 downregulation occurs in ALCL patients duringor immediately after BV therapy, as in our cell line model, andlikewise whether patients with regained or persistent CD30 pos-itivity after a prolonged period off of therapy are neverthelessresistant to BV.
There are other targets on the surface of Hodgkin lymphomatumors, such as CD70 or CD25, that can be used as a way toefficiently deliver potent cytotoxic molecules such as MMAE orrelated agents (13–15). SGN-75, which has been tested in renalcell carcinoma and B-cell lymphomas, can deliver a derivative ofMMAF (monomethyl auristatin F) directly to CD70-expressingcells (16). Another drug, SGN-CD70A, which is currently under-going phase I testing, can deliver a pyrrolobenzodiazepine (PBD)dimer directly to CD70-expressing cells (17). It is possible thatboth of these drugs could be utilized in Hodgkin lymphomapatients who are resistant to BV.
In terms of Hodgkin lymphoma, our MTS assays showed thatL428-R cells were resistant to unconjugated MMAE and thatsurface expression of CD30 was not altered relative to L428-Pcells. We also discovered that L428-R cells overexpressed MDR1.Although this is the first report of MDR1 overexpression in anHodgkin lymphoma cell line resistant to an ADC, MDR1 and
other drug exporters have been implicated in drug resistance formany other tumor types (18–21). This overexpression was con-firmed by qRT-PCR and Western analysis. Drug exporter activitywas confirmed by a rhodamine efflux assay, and inhibition of P-glycoprotein function was able to reverse the resistance pheno-type.We also found that 3 of 4Hodgkin lymphomapaired patientsamples stained positive for one of the class of drug transporters atthe time of relapse, and those patients were resistant to BVtreatment. This suggests that drug transporters can play a role inBV drug resistance in Hodgkin lymphoma. Although MMAE canbe actively pumped out of the cell by P-glycoprotein or othertransporters, there are other cytotoxic agents that can be linked toantibody drug conjugates that are not substrates for transport, andthese could be utilized as CD30 immunoconjugates in the future.
Disclosure of Potential Conflicts of InterestR. Chen has received speakers bureau honoraria from Seattle Genetics, Inc.
No potential conflicts of interest were disclosed by the other authors.
Authors' ContributionsConception and design: R. Chen, J. Hou, E. Newman, Y. Kim, S.J. Forman,S.E. KaneDevelopment of methodology: R. ChenAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): R. Chen, J. Hou, E. Newman, Y. Kim, C. DonohueAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): R. Chen, J. Hou, E. Newman, Y. Kim, X. Liu,S.H. Thomas, S.J. Forman, S.E. KaneWriting, review, and/or revision of the manuscript: R. Chen, J. Hou,E. Newman, Y. Kim, S.H. Thomas, S.J. Forman, S.E. KaneAdministrative, technical, or material support (i.e., reporting or organizingdata, constructing databases): R. ChenStudy supervision: R. Chen, E. Newman, S.J. Forman
AcknowledgmentsThe authors thank the Flow Cytometry Core and the Analytical Pharmacol-
ogy Core facilities of the City of HopeComprehensive Cancer Center (grant NCICA33572). R. Chen is an NCI K12 Calabresi Career Development Scholar.
Grant SupportThis work was supported by the NIH NCI K12 CA01727 (PI: J. Mortimer)
awarded to R. Chen, the NCI Lymphoma SPORE (CA107399) with Develop-ment Research Program award to R. Chen, NCI CA33572 (PI: S. Rosen) withTeamLeadership Supplement to R. Chen,NCICA062505 (PI: E.Newman),NCICA186717 (PI: R. Morgan), and the Tim Nesvig Lymphoma Research Fund(awarded to R. Chen).
The costs of publication of this articlewere defrayed inpart by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received January 14, 2015; revisedMarch 24, 2015; acceptedMarch 26, 2015;published OnlineFirst May 1, 2015.
References1. American Cancer Society. Cancer facts and figures 2014. Atlanta, Georgia:
American Cancer Society; 2014. Available from: http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2014/index.
2. Quddus F, Armitage JO. Salvage therapy forHodgkin's lymphoma.Cancer J2009;15:161–3.
3. Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, et al. ALK�anaplastic large-cell lymphoma is clinically and immunophenotypicallydifferent from both ALKþ ALCL and peripheral T-cell lymphoma, nototherwise specified: report from the International Peripheral T-Cell Lym-phoma Project. Blood 2008;111:5496–504.
4. MoskowitzCH,Nimer SD, Zelenetz AD, Trippett T,Hedrick EE, FilippaDA,et al. A 2-step comprehensive high-dose chemoradiotherapy second-lineprogram for relapsed and refractory Hodgkin disease: analysis by intent totreat and development of a prognostic model. Blood 2001;97:616–23.
5. Kuruvilla J, Nagy T, Pintilie M, Tsang R, Keating A, Crump M. Similarresponse rates and superior early progression-free survival with gemcita-bine, dexamethasone, and cisplatin salvage therapy compared with car-mustine, etoposide, cytarabine, and melphalan salvage therapy prior toautologous stem cell transplantation for recurrent or refractory Hodgkinlymphoma. Cancer 2006;106:353–60.
www.aacrjournals.org Mol Cancer Ther; 14(6) June 2015 1383
CD30 Expression and Brentuximab Vedotin Resistance
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036

6. Pileri SA, Ascani S, Leoncini L, Sabattini E, Zinzani PL, Piccaluga PP, et al.Hodgkin's lymphoma: the pathologist's viewpoint. J Clin Pathol 2002;55:162–76.
7. Francisco JA, Cerveny CG, Meyer DL, Mixan BJ, Klussman K, Chace DF,et al. cAC10-vcMMAE, an anti-CD30-monomethyl auristatin E conju-gate with potent and selective antitumor activity. Blood 2003;102:1458–65.
8. Younes A, Gopal AK, Smith SE, Ansell SM, Rosenblatt JD, Savage KJ,et al. Results of a pivotal phase II study of brentuximab vedotin forpatients with relapsed or refractory Hodgkin's lymphoma. J Clin Oncol2012;30:2183–9.
9. Pro B, Advani R, Brice P, Bartlett NL, Rosenblatt JD, Illidge T, et al.Brentuximab vedotin (SGN-35) in patients with relapsed or refractorysystemic anaplastic large-cell lymphoma: results of a phase II study. J ClinOncol 2012;30:2190–6.
10. de Claro RA, McGinn K, Kwitkowski V, Bullock J, Khandelwal A, Habte-mariam B, et al. U.S. Food and Drug Administration approval summary:brentuximab vedotin for the treatment of relapsed Hodgkin lymphoma orrelapsed systemic anaplastic large-cell lymphoma. Clin Cancer Res2012;18:5845–9.
11. Boswell CA, Mundo EE, Zhang C, Bumbaca D, Valle NR, Kozak KR, et al.Impact of drug conjugation onpharmacokinetics and tissue distribution ofanti-STEAP1 antibody-drug conjugates in rats. Bioconjug Chem 2011;22:1994–2004.
12. Nathwani N, Krishnan AY, Huang Q, Kim Y, Karanes C, Smith EP, et al.Persistence of CD30 expression in Hodgkin lymphoma following bren-tuximab vedotin (SGN-35) treatment failure. Leuk Lymphoma 2012;53:2051–3.
13. Shaffer DR, Savoldo B, Yi Z, Chow KK, Kakarla S, Spencer DM, et al. T cellsredirected against CD70 for the immunotherapy of CD70-positive malig-nancies. Blood 2011;117:4304–14.
14. O'Malley DP, Chizhevsky V, Grimm KE, Hii A, Weiss LM. Utility ofBCL2, PD1, and CD25 immunohistochemical expression in the diag-nosis of T-cell lymphomas. Appl Immunohistochem Mol Morphol2014;22:99–104.
15. Berkowitz JL, Janik JE, Stewart DM, Jaffe ES, Stetler-Stevenson M, Shih JH,et al. Safety, efficacy, and pharmacokinetics/pharmacodynamics of dacli-zumab (anti-CD25) in patients with adult T-cell leukemia/lymphoma.Clin Immunol 2014;155:176–87.
16. TannirNM, Forero-Torres A, Ramchandren R, Pal SK, Ansell SM, Infante JR,et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renalcell carcinoma. Invest New Drugs 2014;32:1246–57.
17. ClinicalTrials.gov [homepage on the Internet]. NCT02216890: safety studyof SGN-CD70A in Cancer Patients 2014. Bothell, WA: Seattle Genetics Inc.[cited 2015 Apr 14]. Available from: https://clinicaltrials.gov/ct2/show/NCT02216890.
18. O'Brien C, Cavet G, Pandita A, Hu X, Haydu L, Mohan S, et al. Functionalgenomics identifies ABCC3 as a mediator of taxane resistance in HER2-amplified breast cancer. Cancer Res 2008;68:5380–9.
19. Greaves W, Xiao L, Sanchez-Espiridion B, Kunkalla K, Dave KS, Liang CS,et al. Detection of ABCC1 expression in classical Hodgkin lymphoma isassociated with increased risk of treatment failure using standard chemo-therapy protocols. J Hematol Oncol 2012;5:47.
20. Marchetti S, Pluim D, Beijnen JH, Mazzanti R, van Tellingen O, SchellensJH. Effect of the drug transporters ABCB1, ABCC2, and ABCG2 on thedisposition and brain accumulation of the taxane analog BMS-275,183.Invest New Drugs 2014;32:1083–95.
21. Du Y, Su T, Zhao L, Tan X, Chang W, Zhang H, et al. Associations ofpolymorphisms in DNA repair genes and MDR1 gene with chemotherapyresponse and survival of non-small cell lung cancer. PLoS One 2014;9:e99843.
Mol Cancer Ther; 14(6) June 2015 Molecular Cancer Therapeutics1384
Chen et al.
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036

2015;14:1376-1384. Published OnlineFirst April 3, 2015.Mol Cancer Ther Robert Chen, Jessie Hou, Edward Newman, et al. Are All Associated with Resistance to Brentuximab Vedotin
UpregulationMDR1CD30 Downregulation, MMAE Resistance, and
Updated version
10.1158/1535-7163.MCT-15-0036doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2015/04/04/1535-7163.MCT-15-0036.DC1
Access the most recent supplemental material at:
Cited articles
http://mct.aacrjournals.org/content/14/6/1376.full#ref-list-1
This article cites 19 articles, 9 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/14/6/1376.full#related-urls
This article has been cited by 10 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/14/6/1376To request permission to re-use all or part of this article, use this link
on June 5, 2020. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst April 3, 2015; DOI: 10.1158/1535-7163.MCT-15-0036