carbamylated erythropoietin ameliorates the metabolic ... · chronic hypoxia carbamylated...
TRANSCRIPT

chronic hypoxia by severein vivoCarbamylated erythropoietin ameliorates the metabolic stress induced
Brines, and Michele Samaja Monica Fantacci, Paola Bianciardi, Anna Caretti, Thomas R. Coleman, Anthony Cerami, Michael
doi:10.1073/pnas.0608814103 published online Nov 7, 2006; PNAS
This information is current as of November 2006.
www.pnas.org#otherarticlesThis article has been cited by other articles:
E-mail Alerts. click hereat the top right corner of the article or
Receive free email alerts when new articles cite this article - sign up in the box
Rights & Permissions www.pnas.org/misc/rightperm.shtml
To reproduce this article in part (figures, tables) or in entirety, see:
Reprints www.pnas.org/misc/reprints.shtml
To order reprints, see:
Notes:

Carbamylated erythropoietin ameliorates themetabolic stress induced in vivo by severechronic hypoxiaMonica Fantacci*, Paola Bianciardi*, Anna Caretti*, Thomas R. Coleman†, Anthony Cerami†‡, Michael Brines†,and Michele Samaja*‡
*Department of Medicine, Surgery, and Dentistry, University of Milan, San Paolo Hospital, Milan 20142, Italy; and †The Kenneth S. WarrenInstitute and Warren Pharmaceuticals, Ossining, NY 10562
Contributed by Anthony Cerami, October 5, 2006 (sent for review September 15, 2006)
Ischemia and chronic hypoxia (CH) trigger a variety of adverseeffects arising from metabolic stress that injures cells. In responseto reduced O2, hypoxia-inducible factor 1� (HIF-1�) activates eryth-ropoietin (Epo) as well as many other target genes that counteractthe effects of O2 deficiency. Epo produced by the kidney stimulateserythrocyte production, leading to decreased HIF-1� production byimproved tissue O2 delivery. However, Epo is produced by manyother tissues, and it is currently unclear to what extent, if any,locally produced Epo modulates HIF-1� expression. Derivatives ofEpo that possess tissue-protective activities but do not stimulateerythropoiesis [e.g., carbamylated Epo (CEpo)] are useful tools withwhich to determine whether exogenous Epo modulates HIF-1� inthe absence of changes in hemoglobin concentration. We com-pared the effects of CH (6.5% O2 for 10 days) with or without CEpoadministered by daily s.c. injection (10 �g�kg of body weight).CEpo administration did not alter the survival rate, weight loss, orincreased hemoglobin concentration associated with CH. There-fore, CEpo does not directly suppress HIF-mediated erythropoiesis.CEpo does, however, prevent CH-induced neuronal increases ofHIF-1� and Epo receptor-associated immunoreactivity (a measureof stress) while reducing the apoptotic index. In contrast, themyocardium did not exhibit increased HIF-1� expression during CH,although CEpo did reduce the apoptotic index. These observationstherefore demonstrate that CEpo administration reduces the met-abolic stress caused by severe CH, resulting in improved cellularsurvival independent of erythrocyte production.
apoptosis � brain � hypoxia-inducible factor 1� � heart � Epo receptor
Chronic hypoxia (CH), a condition whereby the O2 supply totissues is inadequate with respect to need, occurs in several
settings encompassing physiological (high altitude and airlineflights) and pathological (congenital heart disease, anemia,carbon monoxide poisoning, and chronic lung diseases) situa-tions. A potentially lethal challenge, CH activates an array ofmolecular responses that enable the organism to compensate forthe lack of O2 at multiple levels. Among the most prominentadaptive responses, an increase in RBCs and Hb represents anattempt to maximize the blood O2-carrying capacity (1). Eryth-ropoiesis is under the control of the glycoprotein erythropoietin(Epo) that is primarily produced by the kidney to stimulatesurvival and differentiation of erythroid precursor cells intoRBCs (2). Hypoxia-inducible factor 1� (HIF-1�), the molecularlink between hypoxia and the induction of the Epo gene (3),regulates the transcription not only of the Epo gene but also ofmany other genes (4), including the Epo receptor (EpoR) (4) andproapoptotic proteins. In addition to its erythropoietic function,Epo is also produced locally, and it functions to protect a varietyof tissues from ischemic injury, including the central nervous(5–7) and cardiovascular systems (8, 9). Both endogenous (10)and exogenous Epo (11, 12) protect tissues by signaling pathwaysin common with ischemic preconditioning, a process whereby
repeated exposures to sublethal ischemia induce protectionagainst a major ischemic challenge (13).
Carbamylation of Epo lysine residues results in a derivative(CEpo) that lacks erythropoietic activity, yet it appears to haveidentical cytoprotective properties compared with Epo when eval-uated in a variety of cell lines in vitro and organs in vivo (14). It isnotable that both Epo (15) and CEpo (16) may have directmetabolic effects, e.g., by directly increasing the threshold of themitochondrial permeability transition pore to reactive oxygen spe-cies. Because the pharmacodynamics of CEpo are very similar torecombinant human Epo (rhEpo) (14), CEpo is under consider-ation as a candidate drug in various models of brain and heartischemia. Use of CEpo to protect from hypoxia-related injury isespecially promising because the lack of erythropoietic activity willavoid polycythemia, an undesirable specific consequence of CHthat leads to cardiac dysfunction (17), a situation only partlyovercome by increased in vivo nitric oxide production (18).
In a model in which rats are exposed to normobaric CH for 15days, we observed marked deterioration of myocardial protec-tion (19) and sustained apoptosis in a number of organs (20).Although plasma Epo was not measured, an increase in blood Hbby �70% indicates a substantial Epo stimulation. Although it iswell known that Epo is made locally in many tissues in the settingof hypoxia (for review, see ref. 21), the increased apoptosis wehave observed suggests that endogenous Epo levels are notsufficient to provide protection during prolonged hypoxia. It isalso well known that administration of exogenous Epo providesprotection from a wide variety of injuries, including ischemia andhypoxia (21), but it is unknown whether these effects are throughan increased resistance to metabolic stress or a primary reduc-tion of metabolic stress. The objective of this work was todetermine whether CEpo provides antiapoptotic effects duringsevere CH in vivo without an increase in RBC mass, as happenswith treatment by rhEpo. Here, we demonstrate that CEpoadministration reduced metabolic stress as measured by in-creased brain and heart cell survival and decreased HIF-1� andEpoR-associated immunoreactivity after exposure of mice tosevere CH (6.5% O2, corresponding to an altitude of 8,500 m).
ResultsA total of 45 mice (30.1 � 0.3 g at the time of study initiation)were used, having baseline parameters as summarized in Table
Author contributions: M.S. designed research; M.F., P.B., A. Caretti, and M.S. performedresearch; T.R.C., A. Cerami, and M.B. contributed new reagents�analytic tools; M.F., P.B., A.Caretti, T.R.C., and M.B. analyzed data; and T.R.C., A. Cerami, M.B., and M.S. wrote thepaper.
Conflict of interest statement: T.R.C., A. Cerami, and M.B. are employees of WarrenPharmaceuticals, which is developing tissue-protective cytokines for potential clinical use.
Abbreviations: CEpo, carbamylated erythropoietin; CH, chronic hypoxia; Epo, erythropoietin;EpoR, erythropoietin receptor; HIF-1�, hypoxia-inducible factor 1�; NeuN, neuronal nuclei;rhEpo, recombinant human erythropoietin; TdT, terminal deoxynucleotidyltransferase.
‡To whom correspondence may be addressed. E-mail: [email protected] [email protected].
© 2006 by The National Academy of Sciences of the USA
www.pnas.org�cgi�doi�10.1073�pnas.0608814103 PNAS � November 14, 2006 � vol. 103 � no. 46 � 17531–17536
PHA
RMA
COLO
GY

1. Although the survival rate in the normoxic groups was 17 of17 (100%), exposure to hypoxia was associated with mortality inboth vehicle- (6 of 14 or 43%) and CEpo-treated (4 of 14 or 29%)groups. Deaths generally occurred within a few hours after theonset of hypoxia. Fisher’s exact test did not support any signif-icant association between the treatments (P not significant).Body weight decreased in both the CH groups, with no differ-ence between vehicle- and CEpo-treated mice. Likewise, theblood Hb concentration increased in response to CH in bothvehicle- and CEpo-treated mice. As expected, CEpo treatmentdid not increase the blood Hb concentration in normoxic animals(Table 1).
Brain tissue was obtained and frozen in liquid nitrogen within1 min after the animals were euthanized. Fig. 1 summarizes theexpression level of the proteins assessed and the results of theTUNEL measurements in the studied groups. Whereas TUNELpositivity and EpoR-associated immunoreactivity are expressedas the number of terminal deoxynucleotidyltransferase (TdT)-labeled nuclei and EpoR-associated immunoreactivity-positivecells per unit area, respectively, the levels of HIF-1� and Epoprotein expression are expressed as total pixel intensity per unitarea. To obtain a comparative evaluation that takes into accountthe possible occurrence of significant differences by chancealone, we first performed ANOVA of pooled data by one-wayANOVA separately for each parameter. Because the variationsamong the means were significantly greater than expected bychance alone for all parameters except Epo, the Bonferronimultiple comparison test could be used to detect differencesbetween selected pairs of groups for TdT-labeled nuclei, EpoR-associated immunoreactivity-positive cells, and HIF-1� expres-sion. Treatment with CEpo in normoxic animals did not induceappreciable differences in any parameter. CH increased thenumber of TdT-labeled nuclei, the number of EpoR-associatedimmunoreactivity-positive cells, and the expression level ofHIF-1�. Treatment with CEpo blunted the CH-induced increasein all of these parameters to a value close to that measured insamples from normoxic animals.
Next, we determined whether the observed increase in thenumber of TdT-labeled nuclei and EpoR-associated immuno-reactivity-positive cells could be attributed to neuronal cells. Tothat aim, we labeled the primary antibody against EpoR-associated immunoreactivity with rhodamine, which yields a redsignal, and the primary antibody against neuronal nuclei (NeuN)with fluorescein, which yields a green signal that was digitallyconverted to blue to increase visibility. After merging the images,the combination of the two dyes yields a bright purple color. Fig.2 Left shows sample images taken from hypoxic brains fromvehicle- and CEpo-treated mice. This procedure was performedin five sections per animal, and five fields were examined persection. Essentially all EpoR-associated immunoreactivity re-sides in neurons in either vehicle- (95 � 2%) or CEpo-treated(96 � 3%) animals (P not significant). In contrast, the propor-
tion of neurons that express EpoR-associated staining is dra-matically reduced in CEpo-treated animals (35 � 1.5% versus79 � 4%; P � 0.0001, two-tailed). Together, these data arguethat neurons respond to CH stress by increasing EpoR-associated expression and that CEpo treatment dramaticallyreduces this stress response. We could not comment on thecellular localization of HIF-1� because of diffuse labeling.
To determine whether TUNEL positivity, or apoptosis, wasassociated with neuronal cells, we performed the same proce-dure as described above, but we associated a red label withTdT-labeled nuclei and a blue label with NeuN (Fig. 2 Right). Itappears that in CH brain samples only 35 � 3% of neuronal cellscolocalize with TdT-labeled nuclei. In CEpo-treated animals,this value is 25 � 9% (P not significant), indicating that only a
Fig. 1. CEpo treatment moderates the CH-induced neuronal stress response.Epo (A), EpoR (B), HIF-1� (C), and TUNEL (D) positivity are expressed as,respectively, protein level (pixel intensity from immunofluorescence images),no. of cells with EpoR-associated immunoreactivity, protein level (pixel inten-sity from immunofluorescence images) and no. of TdT-positive nuclei in braintissue. All data (mean � SEM) refer to 0.037-mm2 unit area. (Insets) P value ofone-way ANOVA. §, P � 0.05 vs. respective value in normoxic animals; #, P �0.05 vs. respective value in vehicle-treated animals (Bonferroni post hoc test).
Table 1. Main characteristics (mean � SEM) of the tested animals
Normoxia Hypoxia
Vehicle CEpo Vehicle CEpo
Total, n 7 10 14 14Survived, n 7 10 8 10Body weight change, g 1.2 � 0.7 0.2 � 0.5 �12.2 � 0.4* �11.1 � 0.8*Hemoglobin, mM 6.61 � 0.70 5.86 � 0.33 9.43 � 0.48† 8.61 � 0.50†
Mice were exposed for 10 days to either normoxia (21% O2) or hypoxia (6.5% O2), and they were treated witheither vehicle (PBS) or CEpo (10 �g�kg), both injected s.c. once a day. When significant (P � 0.05), the one-wayANOVA was followed by the Bonferroni multiple-comparison test aimed to test normoxia vs. hypoxia or vehiclevs. CEpo differences. No differences were assessed between vehicle- and CEpo-treated animals under eitherhypoxia or normoxia. *, P � 0.0001 by ANOVA; †, P � 0.0002 by ANOVA.
17532 � www.pnas.org�cgi�doi�10.1073�pnas.0608814103 Fantacci et al.
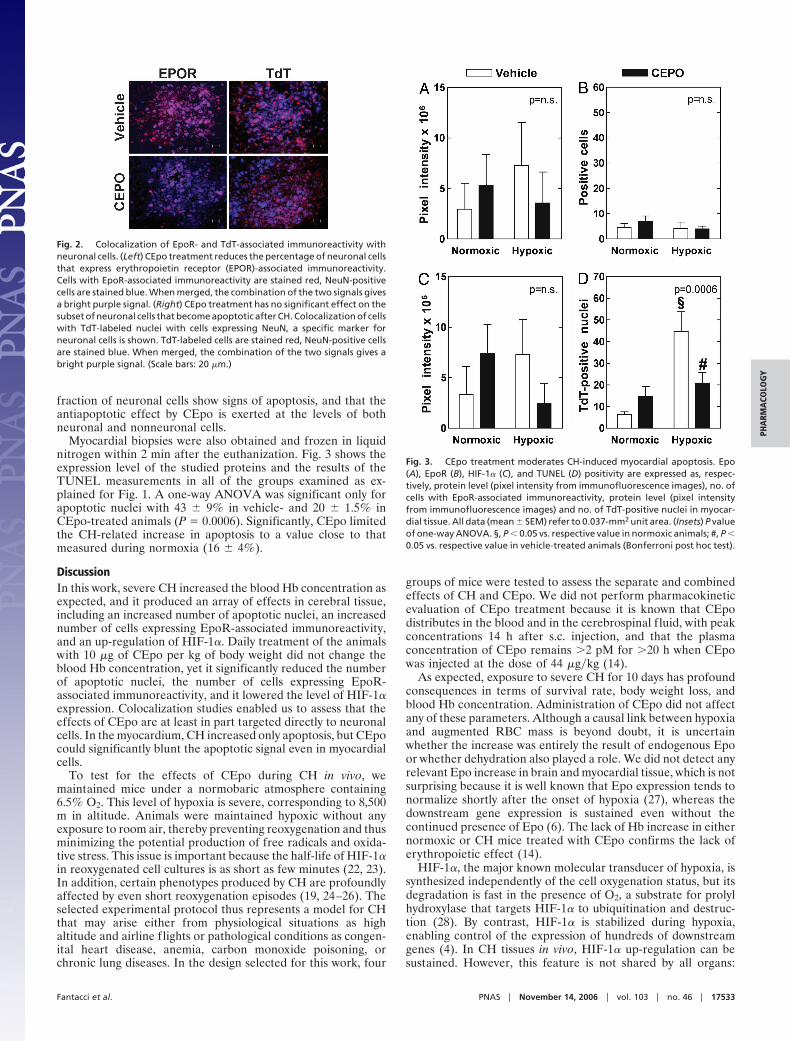
fraction of neuronal cells show signs of apoptosis, and that theantiapoptotic effect by CEpo is exerted at the levels of bothneuronal and nonneuronal cells.
Myocardial biopsies were also obtained and frozen in liquidnitrogen within 2 min after the euthanization. Fig. 3 shows theexpression level of the studied proteins and the results of theTUNEL measurements in all of the groups examined as ex-plained for Fig. 1. A one-way ANOVA was significant only forapoptotic nuclei with 43 � 9% in vehicle- and 20 � 1.5% inCEpo-treated animals (P � 0.0006). Significantly, CEpo limitedthe CH-related increase in apoptosis to a value close to thatmeasured during normoxia (16 � 4%).
DiscussionIn this work, severe CH increased the blood Hb concentration asexpected, and it produced an array of effects in cerebral tissue,including an increased number of apoptotic nuclei, an increasednumber of cells expressing EpoR-associated immunoreactivity,and an up-regulation of HIF-1�. Daily treatment of the animalswith 10 �g of CEpo per kg of body weight did not change theblood Hb concentration, yet it significantly reduced the numberof apoptotic nuclei, the number of cells expressing EpoR-associated immunoreactivity, and it lowered the level of HIF-1�expression. Colocalization studies enabled us to assess that theeffects of CEpo are at least in part targeted directly to neuronalcells. In the myocardium, CH increased only apoptosis, but CEpocould significantly blunt the apoptotic signal even in myocardialcells.
To test for the effects of CEpo during CH in vivo, wemaintained mice under a normobaric atmosphere containing6.5% O2. This level of hypoxia is severe, corresponding to 8,500m in altitude. Animals were maintained hypoxic without anyexposure to room air, thereby preventing reoxygenation and thusminimizing the potential production of free radicals and oxida-tive stress. This issue is important because the half-life of HIF-1�in reoxygenated cell cultures is as short as few minutes (22, 23).In addition, certain phenotypes produced by CH are profoundlyaffected by even short reoxygenation episodes (19, 24–26). Theselected experimental protocol thus represents a model for CHthat may arise either from physiological situations as highaltitude and airline flights or pathological conditions as congen-ital heart disease, anemia, carbon monoxide poisoning, orchronic lung diseases. In the design selected for this work, four
groups of mice were tested to assess the separate and combinedeffects of CH and CEpo. We did not perform pharmacokineticevaluation of CEpo treatment because it is known that CEpodistributes in the blood and in the cerebrospinal f luid, with peakconcentrations 14 h after s.c. injection, and that the plasmaconcentration of CEpo remains �2 pM for �20 h when CEpowas injected at the dose of 44 �g�kg (14).
As expected, exposure to severe CH for 10 days has profoundconsequences in terms of survival rate, body weight loss, andblood Hb concentration. Administration of CEpo did not affectany of these parameters. Although a causal link between hypoxiaand augmented RBC mass is beyond doubt, it is uncertainwhether the increase was entirely the result of endogenous Epoor whether dehydration also played a role. We did not detect anyrelevant Epo increase in brain and myocardial tissue, which is notsurprising because it is well known that Epo expression tends tonormalize shortly after the onset of hypoxia (27), whereas thedownstream gene expression is sustained even without thecontinued presence of Epo (6). The lack of Hb increase in eithernormoxic or CH mice treated with CEpo confirms the lack oferythropoietic effect (14).
HIF-1�, the major known molecular transducer of hypoxia, issynthesized independently of the cell oxygenation status, but itsdegradation is fast in the presence of O2, a substrate for prolylhydroxylase that targets HIF-1� to ubiquitination and destruc-tion (28). By contrast, HIF-1� is stabilized during hypoxia,enabling control of the expression of hundreds of downstreamgenes (4). In CH tissues in vivo, HIF-1� up-regulation can besustained. However, this feature is not shared by all organs:
Fig. 2. Colocalization of EpoR- and TdT-associated immunoreactivity withneuronal cells. (Left) CEpo treatment reduces the percentage of neuronal cellsthat express erythropoietin receptor (EPOR)-associated immunoreactivity.Cells with EpoR-associated immunoreactivity are stained red, NeuN-positivecells are stained blue. When merged, the combination of the two signals givesa bright purple signal. (Right) CEpo treatment has no significant effect on thesubset of neuronal cells that become apoptotic after CH. Colocalization of cellswith TdT-labeled nuclei with cells expressing NeuN, a specific marker forneuronal cells is shown. TdT-labeled cells are stained red, NeuN-positive cellsare stained blue. When merged, the combination of the two signals gives abright purple signal. (Scale bars: 20 �m.)
Fig. 3. CEpo treatment moderates CH-induced myocardial apoptosis. Epo(A), EpoR (B), HIF-1� (C), and TUNEL (D) positivity are expressed as, respec-tively, protein level (pixel intensity from immunofluorescence images), no. ofcells with EpoR-associated immunoreactivity, protein level (pixel intensityfrom immunofluorescence images) and no. of TdT-positive nuclei in myocar-dial tissue. All data (mean � SEM) refer to 0.037-mm2 unit area. (Insets) P valueof one-way ANOVA. §, P � 0.05 vs. respective value in normoxic animals; #, P �0.05 vs. respective value in vehicle-treated animals (Bonferroni post hoc test).
Fantacci et al. PNAS � November 14, 2006 � vol. 103 � no. 46 � 17533
PHA
RMA
COLO
GY

whereas brain, muscle, and kidney cortex exhibit sustainedHIF-1� response, HIF-1� is barely detectable in CH myocar-dium and liver (20), a pattern confirmed in this work. In theheart, it is likely that hypoxia-induced vasodilatation, e.g., pro-duced by local metabolites (29), leads to increases in coronaryflow, thereby maintaining adequate O2 delivery without a needto recruit hypoxia-related mechanisms. By contrast, in braintissue, hypoxia-induced vasodilatation may not represent animportant event as in the myocardium. This difference results ina sustained up-regulation of HIF-1�, especially in the brain (20).
In this work, we show that CEpo daily injections (10 �g�kg)depress hypoxia-induced HIF-1� up-regulation. This observa-tion is consistent with a reduction of metabolic stress associatedwith CEpo administration. Similar observations have been ob-tained by using human ovarian cancer cells exposed to rhEpo,which inhibited HIF-1� stabilization and hypoxia-induced tran-scription of VEGF without affecting the growth rate of thetumor. This effect was presumably at the posttranscriptionallevel because HIF-1� mRNA levels were not affected (30).Several independent observations favor the hypothesis thatCEpo acts directly by destabilizing HIF-1�. First, both rhEpo(15) and CEpo (16) limit mitochondrial permeability transitionpore opening in cardiac myocytes, thereby preventing mitochon-dria swelling and release of substances such as reactive oxygenspecies and cytochrome c. Because mitochondria are consideredO2 sensors, the increase in threshold for reactive oxygen speciesproduced within mitochondria during hypoxia may destabilizeHIF-1� (31, 32). Second, this hypothesis fits into the proposalthat one of the signaling pathways involved in mediating Epotissue protection is the activation of the survival kinases pathway(21), which controls the phosphorylation of prosurvival mole-cules, e.g., phosphatidylinositol-3 kinase�Akt and p42�p44 ex-tracellular signal-regulated kinases (ERK1�2) (33). BecauseERK1�2 is increased during chronic hypoxia (10% O2 in rats)(34), this mechanism may be one by which CEpo returns HIF-1�levels to near normoxic values during CH.
The specificity of the TUNEL method has been questionedbecause TUNEL positivity often reflects a range of cellularconditions rather than only apoptosis (35), and thus it may failto discriminate among apoptosis, necrosis, and autolytic celldeath (36). Also, although generally recognized as a hallmark ofapoptosis, DNA fragmentation may be a late event that does notnecessarily imply apoptosis, such that we cannot rule out thatTUNEL positivity might also indicate necrosis. Despite theselimitations, we report here that both hypoxic brain and myocar-dial tissue exhibited a strong apoptotic signal, which is markedlyreduced by CEpo treatment. These data are only in part con-sistent with previously published data, which did not report anincreased number of TdT-stained nuclei in the hypoxic brain(20). Perhaps the different model (mice vs. rats) and the moresevere hypoxia conditions in the present study (6.5% vs. 10% O2)might account for the difference. In either case, we found thatCEpo treatment markedly reduced TUNEL positivity, consis-tent with the claimed antiapoptotic effects of rhEpo and CEpo(for a review, see ref. 37). Indeed, rhEpo prevents apoptosis incultured adult rat cardiac myocytes exposed to 3% O2 hypoxiafor 28 h (8), and it decreases the number of TdT-positive neuronsafter focal cerebral ischemia and hypoxia-induced neuronaldeath through activation of ERK1�2 (6).
Two distinct receptors for Epo and CEpo exist. Whereas thehomodimeric Epo receptor binds only Epo and mediates pri-marily its erythropoietic function, the heterodimer composed bythe common � receptor (�cR) subunit also known as CD131 canbind both Epo and CEpo, and it is associated with tissueprotection. The two receptors are coexpressed in Epo-sensitivecells, but CEpo signals only through �cR (38). In this work, wefocused on EpoR-associated immunoreactivity because it isamong the genes regulated by HIF-1� (4).
Binding of Epo or rhEpo to the EpoR initiates a variety ofsignaling pathways, including JAK2 (autophosphorylation ofEpoR-associated immunoreactivity), STAT5 (gene expression),MAPK, NO production, and KATP
� channels, which turn out tomediate the erythropoietic effect of Epo in the bone marrowcells (39). In the present study, we observed that EpoR-associated immunoreactivity expression in brain tissue is up-regulated by hypoxia in vivo, consistently with the finding thatEpo stimulates EpoR-associated immunoreactivity transcriptionfactor in erythroid progenitor cells (40). In addition, we foundthat increased EpoR-associated immunoreactivity is specificallylocalized in neuronal cells. Although CEpo treatment does notresult in any change of EpoR-associated immunoreactivity innormoxic cells, it blunts up-regulation of EpoR-associated im-munoreactivity in CH neuronal cells.
In contrast, we found no up-regulation of EpoR-associatedimmunoreactivity in the myocardium, and consequently therewas no effect of CEpo treatment on EpoR-associated immuno-reactivity in this tissue. This observation is partly consistent withthe lack of EpoR-associated immunoreactivity in adult hearts,although its presence during embryogenesis is critical for heartdevelopment (41). Nevertheless, CEpo treatment reduced apo-ptosis, in agreement with other observations that treatment withrhEpo reduced myocardial infarction and TdT-labeled nuclei inthe myocardial area at risk (9, 16, 42).
In summary, treatment with CEpo prevents in part the effectsinduced by severe chronic hypoxia without further increasinghypoxia-induced polycythemia. Specifically, in the brain, CEpoprevents overexpression of HIF-1�- and EpoR-associated im-munoreactivity, and it reduces DNA fragmentation. In the heart,HIF-1�- and EpoR-associated immunoreactivity do not appearto be a specific target of chronic hypoxia, but CEpo reducesDNA fragmentation nevertheless. These results suggest thatfurther studies need to be performed to evaluate CEpo as anexogenous agent that stimulates preconditioning against hyp-oxia, a potentially lethal situation occurring in a great number ofboth physiological and pathological situations.
Experimental ProceduresMaterials. CEpo was provided by Warren Pharmaceuticals(Ossining, NY) as a 2.3 mg�ml stock solution. We used thefollowing antibodies and dilutions: mouse anti-HIF-1� mono-clonal Ab (diluted 1:400 in 1.5% normal goat serum; ChemiconInternational, Temecula, CA), rabbit anti-Epo polyclonal Ab(H-162, diluted 1:200; Santa Cruz Biotechnology, Santa Cruz,CA), and rabbit anti-EpoR polyclonal Ab (H-194, diluted 1:100;Santa Cruz Biotechnology). Although a recent study has raisedquestions concerning the specificity of this last antibody for theEpoR per se, especially when used for immunocytochemistry(43), numerous studies (e.g., refs. 44–47) have noted thatmetabolically stressed cells increase expression of epitopes thatare recognized by this antibody (as well as by other polyclonaland monoclonal antibodies raised again the EpoR). Further, thecytoprotective effects of Epo are neutralized by the sameantibody (44). The cells that express those epitopes are in turnprotected by Epo (as well as nonerythropoietic tissue-protectivecytokines). Therefore, we will refer to the epitopes identified bythis antibody as ‘‘EpoR-associated immunoreactivity.’’ To iden-tify neuronal cells, we used mouse anti-NeuN monoclonal Ab(diluted 1:100; Chemicon International). The secondary anti-bodies included goat anti-mouse and anti-rabbit IgG fluorescein-conjugated Ab (diluted 1:200 in 1.5% normal goat serum; SantaCruz Biotechnology) and anti-rabbit IgG rhodamine-conjugatedAb (diluted 1:200; Santa Cruz Biotechnology). To detect DNAfragmentation, we used the TUNEL ApopTag Red in situapoptosis detection kit (Chemicon International).
17534 � www.pnas.org�cgi�doi�10.1073�pnas.0608814103 Fantacci et al.

Animals. The investigation conformed to the Guide for the Careand Use of Laboratory Animals published by the NationalInstitutes of Health (48). Male ICR CD-1 mice (5–6 weeks old,30 g body weight at entry into the study) were randomlydivided into four groups: normoxic, normoxic treated withCEpo, hypoxic, and hypoxic treated with CEpo. Animalsexposed to CH were caged in a previously described hypoxicchamber (24) with minor modifications. To achieve the desiredlevel of hypoxia, we used two separate cylinders containing airand N2. The f low from the cylinders was regulated indepen-dently by two precision glass tube f low meters (Key Instru-ments, Trevose, PA), then the outputs were mixed, and the O2content was measured by an AERO2-MAT 4100 Clark elec-trode (Syland Scientific, Heppenheim, Germany). The f lowmeters were adjusted to 1.7 � 0.1 and 0.8 � 0.1 liters per minof N2 and air, respectively, to yield a final O2 content in therange of 6.5–6.8%, with total f low rate fixed at 2.5 � 0.2 litersper min. Normoxic animals were caged in the same chamberas that described for hypoxic animals, except that the chamberwas supplied with air (21% O2) alone. All mice had free accessto water and conventional laboratory diet until 24 h beforebeing euthanized. Room temperature was kept at 21 � 2°C,and a 12-h light cycle was maintained.
Exposure to either normoxia or CH was for 10 days. Forinjection into mice, the stock CEpo solution was diluted in sterilePBS (1 �l of stock solution in 765 �l). One hundred microlitersof this solution, equivalent to 10 �g of CEpo per kg of bodyweight, was injected s.c. once a day during the exposure to eithernormoxia or CH. To administer CEpo, mice were first trans-ferred from the hypoxic environment into a procedure chamberthat was maintained at the same O2 content as the hypoxicchamber. CEpo was then injected, and mice were transferredback to the hypoxic chamber without any exposure to room air.In control experiments, mice were injected with PBS alone. Atthe end of the experiment, CH mice were transferred into theprocedure chamber, and they were anesthetized i.p. with sodiumthiopental (10 mg per 100 g of body weight) plus heparin (200units). Then blood was withdrawn by intracardiac puncture intoheparinized syringes, and the animals were euthanized by cer-vical dislocation. After being euthanized, mice were quicklytaken out of the procedure chamber, weighed, and dissected(�0.5 min). While one operator removed the brain, another oneexcised the heart. Blood was immediately assayed for total Hbconcentration.
Sample Preparation. Biopsies from the frozen organs were pro-cessed by embedding them in OCT (Optimum Cutting Temper-ature Compound; Leica Instruments, Nussloch, Germany), andserial 5-�m-thick sections were obtained by using a cryomic-rotome (CM1510; Leica), and they were placed on silanized glassslides. The sections were dried at room temperature for 3 min,fixed in 4% buffered formalin for 45 min at 4°C, rinsed twice for5 min in PBS, postfixed with ethanol�acetic acid 2:1 (vol�vol) at�20°C for 5 min, rinsed twice for 5 min in PBS, boiled for 10 minin 10 mM citrate buffer (pH 6.0), washed once in distilled water
and three times in PBS, and finally used for immunofluorescenceor DNA fragmentation staining.
Immunofluorescence. The sections were immersed in 10% normalgoat serum for 1 h with gentle agitation, incubated overnight at4°C with the primary antibody, washed in PBS, and incubated atroom temperature for 45 min with the secondary antibody. Anegative control was prepared for each section by replacing theprimary antibody with 1.5% normal goat serum. The slides wereexamined at a magnification of �40 in an inverted fluorescencemicroscope (Axiovert 25 CFL; Zeiss, Gottingen, Germany),equipped with a filter for the detection of either fluorescein(filter set 9, excitation bandpass 450–490 nm, emission low-pass515 nm) or rhodamine (filter set 15, excitation bandpass 546 �12 nm, emission low-pass 590 nm). The images were acquired byeither a CCD camera (AxioCam czv CD 4.0; Zeiss) or a digitalcamera system for microscopy (DS-2Mv; Nikon, Tokyo, Japan),and stored in a personal computer. For the quantification of thesignals, the images were analyzed by IPlab Software (Scanalytics,Fairfax, VA) and split into RGB channels. Either the green orthe red channel was used to calculate the color intensity as thesum of the pixel intensity values. At least five random fields persection were selected, and the total color intensity was measuredand subtracted from the signal detected by using the negativecontrols. The color intensity in the image is expressed as the sumof pixel intensity � 106�0.037 mm2.
In Situ TdT Assay for Detection of Apoptosis. The TUNEL techniquewas used to identify apoptotic nuclei by the TdT-mediated dUTP-rhodamine nick end-labeling technique. After blocking, the sec-tions were incubated for 60 min at 37°C with TdT to label free 3�OH DNA termini with digoxigenin nucleotides, and the reactionwas stopped by immersing the slide in stop�wash buffer for 10 minat room temperature. After three PBS washes, the sections wereincubated with antidigoxigenin rhodamine-conjugated antibody inthe dark for 15 min at 37°C and then for 15 min at roomtemperature, and finally they were washed four times in PBS. Anegative control was established for each biopsy by replacing TdTwith PBS. After the mounting as described above, images wereacquired by using a rhodamine detection filter. Two operatorscounted the number of TdT-labeled nuclei by examining at leastfive random fields in a blinded procedure. Results are expressed asnumber of TdT-labeled nuclei�0.037 mm2.
Hemoglobin Concentration. The Hb concentration (in mM) wasdetermined by the Drabkin assay using the extinction coefficient11.05 mM�1�cm�1.
Statistics. All data are presented as mean � SEM. To assess thesignificance of the differences among the various groups, weperformed one-way ANOVA, followed by Bonferroni’s multiplecomparison test if significant (P � 0.05). To estimate the effectof CEpo on the survival rate, Fisher’s exact test was used.
This work was supported in part by the Ministero Universita e Ricerca,Rome, Italy (Grant 2004054720�002, PRIN2004 to M.S.) and by Fonda-zione Cariplo, Milan, Italy (Grant 5201001-55 to M.S.).
1. Hurtado A, Merino C, Delgado E (1945) Arch Int Med 75:284–323.2. Jelkmann W (1992) Physiol Rev 72:449–489.3. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE (1991) Proc Natl Acad Sci USA
88:5680–5684.4. Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG,
Semenza GL (2005) Blood 105:659–669.5. Sasaki R (2003) Intern Med 42:142–149.6. Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, Keenan
S, Gleiter C, Pasquali C, Capobianco A, et al. (2001) Proc Natl Acad Sci USA98:4044–4049.
7. Brines M, Ghezzi P, Keenan S, Agnello D, de Lanerolle N, Cerami C, Itri L,Cerami A (2000) Proc Natl Acad Sci USA 97:10526–10531.
8. Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P, Salio M, CeramiA, Brines M (2003) Proc Natl Acad Sci USA 100:4802–4806.
9. Moon C, Krawczyk M, Ahn D, Ahmet I, Paik D, Lakatta EG, Talan MI (2003)Proc Natl Acad Sci USA 100:11612–11617.
10. Prass K, Scharff A, Ruscher K, Lowl D, Muselmann C, Victorov I, Kapinya K,Dirnagl U, Meisel A (2003) Stroke 34:1981–1986.
11. Baker JE (2005) Vascul Pharmacol 42:233–241.12. Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL,
Semenza GL (2003) Circulation 108:79–85.13. Reimer K, Murry C, Jennings R (1990) Circulation 82:2266–2268.14. Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, Savino C, Bianchi
M, Nielsen J, Gerwien J, et al. (2004) Science 305:239–242.
Fantacci et al. PNAS � November 14, 2006 � vol. 103 � no. 46 � 17535
PHA
RMA
COLO
GY

15. Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, Ziman BD,Wang S, Ytrehus K, Antos CL, Olson EN, Sollott SJ (2004) J Clin Invest113:1535–1549.
16. Moon C, Krawczyk M, Paik D, Coleman T, Brines M, Juhaszova M, Sollott SJ,Lakatta EG, Talan MI (2006) J Pharmacol Exp Ther 316:999–1005.
17. Wagner K, Katschinski D, Hasegawa J, Schumacher D, Meller B, GembruchU, Schramm U, Jelkmann W, Gassmann M, Fandrey J (2001) Blood 97:536–542.
18. Ruschitzka F, Wenger R, Stallmach T, Quasching T, de Wit C, Wagner K,Labugger R, Kelm M, Noll G, Rulicke T, et al. (2000) Proc Natl Acad Sci USA97:11609–11613.
19. Milano G, Corno A, Lippa S, von Segesser L, Samaja M (2002) Exp Biol Med227:389–397.
20. Bianciardi P, Fantacci M, Caretti A, Ronchi R, Milano G, Morel S, vonSegesser L, Corno A, Samaja M (2006) Biochem Biophys Res Commun342:875–880.
21. Brines M, Cerami A (2006) Kidney Int 70:246–250.22. Jiang B, Semenza G, Bauer C, Marti H (1996) Am J Physiol 271:C1172–C1180.23. Wiesener M, Turley H, Allen W, William C, Eckardt K, Talks K, Wood S,
Gatter K, Harris A, Pugh C, et al. (1998) Blood 92:2260–2268.24. Corno A, Milano G, Samaja M, Tozzi P, von Segesser L (2002) J Thorac
Cardiovasc Surg 124:105–112.25. Milano G, Bianciardi P, Corno AF, Raddatz E, Morel S, von Segesser LK,
Samaja M (2004) Exp Biol Med 229:1196–1205.26. Corno AF, Milano G, Morel S, Tozzi P, Genton CY, Samaja M, von Segesser
LK (2004) J Thorac Cardiovasc Surg 127:1301–1308.27. Eckardt KU, Dittmer J, Neumann R, Bauer C, Kurtz A (1990) Am J Physiol
258:F1432–F1437.28. Lando D, Peet D, Whelan D, Gorman J, Whitelaw M (2002) Science 295:858–
861.29. Berne R (1980) Circ Res 47:807–813.30. Hale SA, Wong C, Lounsbury KM (2006) Gynecol Oncol 100:14–19.31. Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC,
Hammerling U, Schumacker PT (2005) Cell Metab 1:401–408.32. Chandel N, McClintock D, Feliciano C, Wood T, Melendez J, Rodriguez A,
Schumacker P (2000) J Biol Chem 275:25130–25138.
33. Armstrong SC (2004) Cardiovasc Res 61:427–436.34. Morel S, Milano G, Ludunge KM, Corno AF, Samaja M, Fleury S, Bonny C,
Kappenberger L, von Segesser LK, Vassalli G (2006) Basic Res Cardiol101:336–345.
35. Yaoita H, Ogawa K, Maehara K, Maruyama Y (2000) Cardiovasc Res 45:630–641.
36. Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W,Schulte-Hermann R (1995) Hepatology 21:1465–1468.
37. Ghezzi P, Brines M (2004) Cell Death Differ 11(Suppl 1):S37–S44.38. Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, Latini R,
Xie QW, Smart J, Su-Rick CJ, et al. (2004) Proc Natl Acad Sci USA101:14907–14912.
39. Bartesaghi S, Marinovich M, Corsini E, Galli CL, Viviani B (2005) Neurotoxi-cology 26:923–928.
40. Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT, Noguchi CT(2000) J Biol Chem 275:39754–39761.
41. Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B,Lin CS, Nikodem VM, Hempstead B, Flanders KC, Costantini F, Noguchi CT(2002) Development (Cambridge, UK) 129:505–516.
42. Fiordaliso F, Chimenti S, Staszewsky L, Bai A, Carlo E, Cuccovillo I, Doni M,Mengozzi M, Tonelli R, Ghezzi P, et al. (2005) Proc Natl Acad Sci USA102:2046–2051.
43. Elliott S, Busse L, Bass MB, Lu H, Sarosi I, Sinclair AM, Spahr C, Um M, VanG, Begley CG (2006) Blood 107:1892–1895.
44. Liu R, Suzuki A, Guo Z, Mizuno Y, Urabe T (2006) J Neurochem 96:1101–1110.45. Shein NA, Horowitz M, Alexandrovich AG, Tsenter J, Shohami E (2005)
J Cereb Blood Flow Metab 25:1456–1465.46. Siren AL, Knerlich F, Poser W, Gleiter CH, Bruck W, Ehrenreich H (2001)
Acta Neuropathol 101:271–276.47. Spandou E, Papoutsopoulou S, Soubasi V, Karkavelas G, Simeonidou C,
Kremenopoulos G, Guiba-Tziampiri O (2004) Brain Res 1021:167–172.48. Committee on Care and Use of Laboratory Animals (1985) Guide for the Care
and Use of Laboratory Animals (Natl Inst Health, Bethesda), DHHS Publ No(NIH) 85-23.
17536 � www.pnas.org�cgi�doi�10.1073�pnas.0608814103 Fantacci et al.