calcium–phosphate treatment of contaminated soil for arsenic immobilization
TRANSCRIPT

Applied Geochemistry 28 (2013) 145–154
Contents lists available at SciVerse ScienceDirect
Applied Geochemistry
journal homepage: www.elsevier .com/ locate/apgeochem
Calcium–phosphate treatment of contaminated soil for arsenic immobilization
Ghanashyam Neupane a,b,⇑, Rona J. Donahoe a
a Department of Geological Sciences, University of Alabama, Box 870338, Tuscaloosa, AL 35487, United Statesb University of Idaho-Idaho Falls, 1776 Science Center Drive, Suite 306, Idaho Falls, ID 83402, United States
a r t i c l e i n f o
Article history:Received 26 December 2011Accepted 12 October 2012Available online 26 October 2012Editorial handling by J. Routh
0883-2927/$ - see front matter � 2012 Elsevier Ltd. Ahttp://dx.doi.org/10.1016/j.apgeochem.2012.10.011
⇑ Corresponding author. Tel.: +1 208 282 7842; faxE-mail address: [email protected] (G. Neupan
a b s t r a c t
The application of As-based herbicides at several industrial sites has resulted in numerous localized areas ofAs-contaminated soil. In this study, an As-contaminated soil (As = 278 mg/kg) collected from an industrialsite located in the southeastern USA was subjected to inorganic phosphate (Pi) treatments. Although Pi
treatments have been previously used for flushing As from contaminated soils, in this study, contaminatedsoil was amended with Pi to study the possible immobilization of As through a co-precipitation mechanism.Specifically, the Pi amendment was aimed at simultaneous flushing of As from the soil with orthophos-phoric acid and co-precipitating it as Ca–phosphate–arsenate phases. Bench-scale Pi treatment experi-ments were performed at different pH conditions, with and without the addition of Ca. Sorption of Pi onBH soil in the presence or absence of additional Ca was determined, along with the associated mobilizationof As from the soil. A significant amount of the HNO3-digestible As (up to 55% at pH 4, 10–15% at pH 8, and�30% at pH 11) was released from the contaminated soil during the Pi sorption experiments. This increasedmobility of As after the addition of Pi resulted from the competitive desorption of As from the soil. AlthoughPi sorption at high pH (>8) was largely controlled by precipitation, As did not co-precipitate with Pi. Aque-ous geochemical modeling indicated that the lack of As co-precipitation during Pi-only treatment primarilyresulted from the deficiency of Ca in the system. When additional Ca (16.9 mmol) was supplied along withPi (3.38 mmol), the mobility of As decreased significantly at circum-neutral to high solution pH. Geochem-ical modeling suggested that the leachable As in the soil was potentially precipitated as As-bearing Ca–Pi
phases. X-ray diffraction analysis of precipitates separated from the treated soil and from the syntheticleachate confirmed that the formation of a poorly crystalline carbonate apatite phase occurred as a conse-quence of the treatment. The results of this study support the potential application of Ca–Pi treatment forremediation of As-contaminated soil at environmentally relevant pH conditions.
� 2012 Elsevier Ltd. All rights reserved.
1. Introduction
Arsenic is a toxic element, an acute dose of 50–300 mg As beingconsidered to be lethal for humans due to gastrointestinal, respira-tory, cardiovascular, neurological or other body system failures(ATSDR, 2000). Similarly, chronic ingestion of As, either throughfood or water, is responsible for several types of cancer (Jacksonand Grainge, 1975; Bates et al., 1992; Karagas et al., 2002). Elevatedconcentrations of As in the environment can occur by naturalprocesses as well as anthropogenic activities (Smedley andKinniburgh, 2002). Groundwater As-enrichment from naturalsources has adversely affected the health of millions of people inseveral areas of the world (Nordstrom, 2002; Hossain, 2006;Bundschuh et al., 2012). Arsenic contamination has occurredthrough the use of As-based pesticides (Simcox et al., 1995; Robinsonet al., 2007), herbicides (Yang and Donahoe, 2007) and wood pre-servatives (Morrell et al., 2003), and through mining and smeltingactivities (Carbonell-Barrachina et al., 2004).
ll rights reserved.
: +1 208 282 7929.e).
In the USA, As-based compounds have been used for severalpurposes such as agricultural pesticides and herbicides, wood pre-servatives and glass production (Welch et al., 2000). Although thewidespread use of different arsenical compounds gradually de-clined after the 1960s when the adverse environmental effects ofAs were realized, their legacy in environmental pollution is stillpronounced in many areas of the USA (Welch et al., 2000). Severalindustrial sites in the southeastern USA which received intensiveapplications of arsenical herbicides during the 1950s continue toleach As into surface water and groundwater and pose consider-able danger (Yang and Donahoe, 2007; Qi and Donahoe, 2008).Regulatory agencies such as the United States Environmental Pro-tection Agency (USEPA) have developed, and are enforcing morestringent standards for As levels in drinking water. For example,the USEPA lowered the maximum contaminant level (MCL) for Asin drinking water from 50 lg/L to 10 lg/L in 2006. This requiredthe development and implementation of As remediation strategiesat several contaminated sites to avoid potential contamination oflocal potable water sources.
Several techniques such as isolation and separation (Mulliganet al., 2001), solidification and stabilization (Yang et al., 2007),

146 G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154
vitrification (Mulligan et al., 2001), soil flushing (Alam et al., 2001),electrokinetic treatment (Yuan and Chiang, 2007), and bioremedi-ation (Ma et al., 2001) have been proposed for remediation ofAs-contaminated soils. Previous bench-scale As-remediationexperiments performed in the authors’ laboratory (Yang et al.,2007; Bhattacharyya et al., 2009; Neupane et al., 2010) havesuccessfully demonstrated the in situ chemical fixation of As incontaminated soil and coal fly ash with ferrous sulfate solutions.Flushing of As from contaminated soil and soil constituents withsolutions containing competing ligands such as inorganic phos-phate (Pi) and OH� has been studied extensively (Johnson andBarnard, 1979; Peryea and Kammereck, 1997; Wasay et al., 2000;Alam et al., 2001; Tokunaga and Hakuta, 2002; Kaplan and Knox,2004). Because of its similar chemical characteristics, Pi has a highpotential to desorb As(V) from soil through ion exchange reactions(Woolson et al., 1973; Jackson and Miller, 2000; Wasay et al., 2000;Alam et al., 2001; Kaplan and Knox, 2004). In particular, phospho-ric acid is very effective in rapidly displacing As from contaminatedsoil by providing Pi to compete with As(V) for adsorption sites, andby dissolving As-rich metal oxides present in soils (Tokunaga andHakuta, 2002). A few studies (e.g., Grisafe and Hummel, 1970;Twidwell et al., 1994) have also demonstrated co-precipitation ofAs with Pi minerals. In these studies, As(V) substituted for Pi to formcontinuous solid solutions of fluor- and chlor-apatites (Grisafe andHummel, 1970). Subsequently, Mahapatra et al. (1987) success-fully synthesized arsenate hydroxyapatite. Similarly, Twidwellet al. (1994) and others (Wilson, 1998; Orser, 2001) indicated re-moval of As from solution by precipitation of Ca–Pi–As(V) mineralsat pH 8–10. Their study indicated that Pi–As(V) hydroxyapatite witha P:As ratio P7 has a low solubility (<10 lg/L) and greater stabilityagainst atmospheric CO2 compared to other Pi–As(V) apatites.
In this study, an As-contaminated soil collected from an indus-trial site located in the southeastern USA was subjected to a Ca–Pi
treatment. The objective of this study is to test the potential appli-cation of phosphate-based treatment for remediation of As-con-taminated soils. The remediation method evaluated in this studyaims at flushing As from contaminated soil with phosphoric acid,and immobilizing the same through precipitation as Ca–Pi–As(V)
minerals.
2. Materials and methods
2.1. Contaminated site and soil sample collection
An As-contaminated soil sample was collected from an indus-trial site (BH) located near the Gulf Coast in the southeasternUSA. The soil had developed from Quaternary undifferentiatedmarine and fluvial sediments. With increasing depth, four sub-surface units: an unconfined sandy aquifer, a silty peat semi-confining bed, a semi-confined sand aquifer, and a confining silt/clay layer, have been reported at the site (Yang and Donahoe,2007). The regional climate of the contaminated area is humidand semi-tropical with an annual rainfall of about 162 cm (Yangand Donahoe, 2007). The rainy season at the sampling site beginsin May and ends in November, and accounts for ca. 60% of theannual rainfall (Black, 1993). Temperature is moderate, with anaverage high of 28 �C in summer and an average low of 10 �C inwinter (Yang and Donahoe, 2007).
The soil at this site was contaminated during the 1950s throughthe one-time application of arsenic trioxide (arsenolite) rich herbi-cide produced at Anaconda smelter in Montana (Yang andDonahoe, 2007). Originally, arsenolite was applied as a 1-in.(2.5 cm) thick layer on top of the natural soil surface, and coveredwith a 5–10 cm thick layer of limestone gravel. During the last fivedecades, the arsenolite layer has weathered and dispersed into the
soil, creating a potential risk for As exposure to the local population(Yang and Donahoe, 2007). Because earlier studies (e.g., Yang andDonahoe, 2007) had indicated that As was mostly confined to thevadose zone, soil samples were collected at a single location from10 to 60 cm below the surface and placed in two 19 L-buckets. Thesoil was air-dried, passed through a 2 mm sieve, homogenized, andstored at room temperature for chemical/mineralogical character-izations and Pi treatments.
2.2. Characterization of BH soil
Arsenic and other elements in the BH soil samples were ex-tracted by microwave-assisted HNO3 partial digestion (MWD)(USEPA, 2007), and determined with a Perkin Elmer Optima3000DV inductively coupled plasma-optical emission spectrome-ter (ICP-OES). The soil pH was measured according to USEPA Meth-od 9045D (USEPA, 2004). The single-point BET specific surface areaof the soil sample was determined using ASTM D4567-03 method(ASTM, 2003). Minerals present in the bulk soil, the soil clay frac-tion, and experimental precipitates were determined by X-ray dif-fraction (XRD) using a Brüker D8 Advance powder diffractometer.The bulk soil sample was powdered in an iron Shatterbox� millfor 2 min for XRD analysis. The clay-sized soil fraction was sepa-rated by flotation according to Stoke’s Law. The XRD data wereanalyzed with DIFRACplus EVA version 11.0.03 (XRD data evalua-tion and presentation software provided by Bruker AXS), usingthe International Center for Diffraction Data, ICDD PDF-2 Release2005 database.
2.3. Experiments
2.3.1. Mobilization of As from BH soil as a function of pHMobilization of As from BH soil as a function pH was studied in
batch experiments. For each experimental setup, 3.0 g of soil wasweighed in a 50 mL centrifuge tube and then 45 mL of 0.1 mol NaClsolution was added. A series of similar experimental sets were pre-pared and agitated on an orbital platform shaker for 24 h. The pH ofthe soil mixture was set initially and adjusted periodically whenneeded in the range of 3–12 using 0.1 mol or 1.0 mol HCl andNaOH solutions. The supernatant solutions were filtered through0.2 lm nylon syringe filters after 20 min centrifugation at13,000g, acidified to 2% HNO3 with OPTIMA� ultrapure HNO3,and stored at 4 �C in a refrigerator until chemical analysis by ICP-OES.
The mobilization of As and Ca from soil was calculated using Eq.(1) and expressed as % mobilized.
% Mobilized ¼ Amount leached to solutionMWD extracted amount in soil
� 100% ð1Þ
2.3.2. Pi sorption and mobilization of As from BH soilThe sorption of Pi on BH soil was evaluated by constructing sorp-
tion isotherms, and by conducting sorption experiments as a func-tion of pH. The term ‘sorption’ is used to encompass all processes(adsorption, absorption, and precipitation) which transfer aqueoussorbate (e.g., Pi) to the solid phase (Sposito, 1986). Each sorptionexperiment was performed at a 1:15 solid:liquid ratio by mixing3.0 g of soil with 45 mL of 0.1 mol NaCl electrolyte in a 50 mL centri-fuge tube. The experiments were performed in duplicate, but eachexperiment was treated as an individual sample, rather than calcu-lating an average value for the duplicate samples.
The phosphate stock solution was prepared from 85% H3PO4;the Pi concentration was determined by ICP-OES analysis. Furtherdilutions of the Pi stock solution were performed with respect toits analyzed concentration. Isotherms for phosphate sorption on

G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154 147
BH soil at pH 4, 8, and 11 were constructed using 0.15–13.24 mmolinitial Pi concentrations. Sorption as a function of pH was deter-mined for 0.57 mmol and 3.38 mmol initial Pi concentrations inthe pH range from 3 to 12. The pH of the soil mixture was adjustedat the beginning of the experiment using HCl and NaOH solutions.Furthermore, pH was checked intermittently and adjusted, if nec-essary. The tubes were agitated for 24 h, and the samples werecentrifuged, filtered, acidified and stored for chemical analysis.During ICP-OES analyses, quality control check standards wererun after analysis of each batch of 10 samples. The relative stan-dard deviations of As, Ca and P concentrations in the check stan-dards were generally within ±5%, and never exceeded ±10%. Thesorption of Pi on BH soil was determined by calculating the differ-ence in concentrations of Pi in initial solution and equilibrium solu-tion. Finally, the mobilization of As and Ca from soil during Pi
sorption experiments was calculated using Eq. (1).
2.3.3. Sorption isotherm modelingExperimental sorption data were fitted with the Freundlich iso-
therm model (Eq. (2)):
qe ¼ KCne ð2Þ
where qe is the amount of sorbate (Pi) sorbed on BH soil (mmol/kg),Ce is the equilibrium sorbate concentration (mmol), K (L/kg) and n(dimensionless) are constants. Freundlich isotherm model parame-ters were obtained by converting Eq. (1) into linear form (Eq. (3)):
log qe ¼ log K þ n log C ð3Þ
where values of n and K can be determined from the slope and y-intercept of a graph obtained by plotting logCe along the x-axisand logqe along y-axis (Limousin et al., 2007). For each isotherm,the goodness of model fit was determined by calculating the coeffi-cient of determination (R2) with:
R2 ¼Pn
i¼1ðqm � �qeÞ2Pn
i¼1ðqm � �qeÞ2 þPn
i¼1ðqm � qeÞ2 ð4Þ
where qe is as defined in Eq. (2), qm is the model-derived maximumadsorbed amount (mmol/kg), and �qe is the average experimentalsorbed amount (mmol/kg). The closer the value of R2 is to 1.0, thebetter the model is in predicting sorption. The R2 value for each iso-therm was obtained by using the experimental and correspondingmodel datasets.
2.3.4. Ca–Pi treatment of BH soilThe contaminated soil was treated with Ca and Pi in batch
experiments. For each experiment, 3.0 g of soil was weighed in a50 mL centrifuge tube and 45 mL of a Ca–Pi treatment solutionwas added to it. The Ca treatment solutions were prepared fromdry, analytical grade CaCl2, and the Ca concentrations were deter-mined by ICP-OES analysis. Two sets of batch treatment experi-ments were performed over the pH range from 3 to 12. The firstset of experiments was conducted with Ca and Pi concentrationsof 5.63 mmol and 3.38 mmol, respectively, yielding a 1:67 initialCa:Pi ratio in the soil mixture. The second set of experiments usedthe same concentration of Pi, but a much higher concentration ofCa (16.90 mmol), produced an initial Ca:Pi ratio of 5:1. The pH ofthe soil mixture was adjusted with HCl and NaOH solutions andagitated for 24 h. Finally, the tubes were centrifuged and the super-natant solutions were separated by filtration and stored in a refrig-erator until chemical analysis. The precipitate in the treated soilwas analyzed by X-ray diffraction to identify the mineral phases.
The % mobilization of As during soil treatments was calculatedusing Eq. (1). However, Eq. (1) was modified to Eq. (5) so as to in-clude the externally supplied Ca while calculating % mobilizationof Ca during the treatment:
% Mobilized¼Ca in equilibrium solution�externally supplied CaMWD extracted amount of Ca
�100%
ð5Þ
According to Eq. (5), 0% mobilization indicates neither release ofCa from soil to solution nor transfer of Ca from solution to soil.Therefore, a soil solution with 0% mobilization of Ca contains onlythe externally supplied aqueous Ca. Positive % mobilization valuesindicate that the equilibrium solution is enriched in Ca due to therelease of Ca from soil, while negative % mobilization values meanthat some of the externally supplied Ca is transferred to the soil so-lid phase by sorption, mostly by precipitation mechanism. DuringCa–Pi treatments, 45 mL of 5.63 mmol or 16.9 mmol Ca, along with3.38 mmol Pi, were reacted with 3 g of BH soil. Therefore, completetransfer of Ca from the treatment solution to the solid phase wouldresult in calculated Ca mobilities of about �14% (for 5.63 mmoladded Ca) and �42% (for 16.9 mmol added Ca), with respect tothe soil’s 592 mmol/kg of HNO3-digestible Ca. For As, however,the calculated mobilization should be always P0%. A 0% mobilizedvalue indicates that no As is released from soil to solution duringthe Ca–Pi treatment.
2.3.5. Precipitation of Ca–Pi–As(V) phases from synthetic leachateCalcium–Pi–As(V) solid phases were precipitated from synthetic
leachate prepared by mixing Ca, Pi, and As(V) stock solutions in0.1 mol NaCl. The calculated amount of vacuum desiccator driedNa2HAsO4�7H2O was used for preparation of the As(V) stock solu-tion. In addition, the concentration of As in the stock solutionwas determined by ICP-OES analysis. The initial synthetic leachatewith As(V) = 0.48 mmol, Pi = 3.38 mmol, and Ca = 6.44 mmol wasstirred for 24 h in a 250 mL beaker, while in contact with air, atroom temperature (ca. 21 �C) to precipitate Ca–Pi–As(V) solidphases. The As(V):Pi ratio was selected to be 1:7, as suggested byWilson (1998), whereas the Ca:(As(V) + Pi) ratio was fixed at 1.67.The pH of the synthetic leachate was adjusted to 8 with NaOHand HCl solutions. At the end of the 24 h period, the precipitateformed was separated by vacuum filtration and analyzed by XRD.
2.4. PHREEQC simulations and potential precipitation of Ca–Pi–As(V)
minerals
The aqueous chemical data were used to calculate the satura-tion index (SI) of several Ca, Ca–As(V), Ca–Pi, and Ca–Pi–As(V) min-erals with the PHREEQC geochemical code (Parkhurst andAppelo, 1999), using the llnl.dat database. Thermodynamic datafor several potential minerals and aqueous species (see Supple-mentary Tables S1 and S2) were collected from additional sources(Smith and Martell, 1976; Bothe and Brown, 1999; Wilson, 1998;Montastruc et al., 2003) and incorporated into the llnl.dat data-base. Most of thermodynamic data for different solid phases com-piled for this study were determined/estimated at 20–25 �C. Thelaboratory temperature during the experiments performed in thisstudy was in the range of 20–22 �C, and the thermodynamic datawere used as obtained without any temperature correction. Duringthe PHREEQC simulations, all solutions were allowed to equilibratewith atmospheric CO2 and O2.
The precipitation potential of a mineral was determined byevaluating its saturation state (undersaturation or oversaturation).The degree of undersaturation or oversaturation of an aqueoussample with respect to a particular mineral was determined interms of the SI, calculated using the following equation:
SI ¼ logðIAP=KspÞ ð6Þ
where IAP is the ion activity product and Ksp is the solubility con-stant for a particular mineral (Drever, 1997). If the SI for a mineral
148 G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154
is greater than 0, then the aqueous solution is considered to beoversaturated with respect to that particular mineral, and the solu-tion is likely to spontaneously precipitate that mineral, dependingon kinetic constraints.
Speciation modeling was performed by PHREEQC for differenttreatment scenarios. The chemical data obtained as a function ofpH from the batch experiments without external supply of Pi orCa were used as primary chemical data. Initially, the amounts ofAs and Ca mobilized as a function of pH were evaluated for poten-tial precipitation. Subsequently, the amounts of Pi and Ca suppliedto the soil during treatments were added to the primary chemicaldata, and the potential for precipitation of different minerals fromthe augmented solutions was evaluated.
3. Results
3.1. Characterization of BH soil
BH soil is a basic (pH � 8.4), medium dark gray (N4), sandy-clayloam. This soil mostly consists of quartz, calcite, and kaolinite withminor amount of gehlenite, montmorillonite, and muscovite. Theconcentration of As in BH soil is 278 mg/kg (3.71 mmol/kg). Thebackground concentration of As in soil near the sampling site hasbeen reported to be <3 mg/kg (Yang and Donahoe, 2007). BH soilhas high concentrations of Ca (23,700 mg/kg), Fe (1360 mg/kg),Al (5860 mg/kg), Mg (5600 mg/kg), K (765 mg/kg), and Na(280 mg/kg). The organic C content (loss-on-ignition) of this soilis 6.5 mg/g (Neupane et al., 2010) and the surface area of the bulksoil is 2.65 m2/g, as determined by the single point BET method.
Speciation of As in BH soil was studied in detail by Yang andDonahoe (2007), who used sequential chemical extraction fol-lowed by electron microprobe analysis, scanning electron micros-copy, l-XRD, and X-ray absorption spectroscopy (XAS). Yang andDonahoe (2007) reported that most of the As in BH soil remainssorbed to amorphous Al- and Fe-hydroxides. However, traceamounts of some As-rich minerals (e.g., phaunouxite) are alsopresent in the contaminated soil (Yang and Donahoe, 2007).Sequential chemical extraction and l-XANES studies indicated thatmost of the original As(III) arsenolite herbicide applied more than 5decades ago has been oxidized to As(V) by weathering processes(Yang and Donahoe, 2007).
3.2. Pi sorption on BH soil
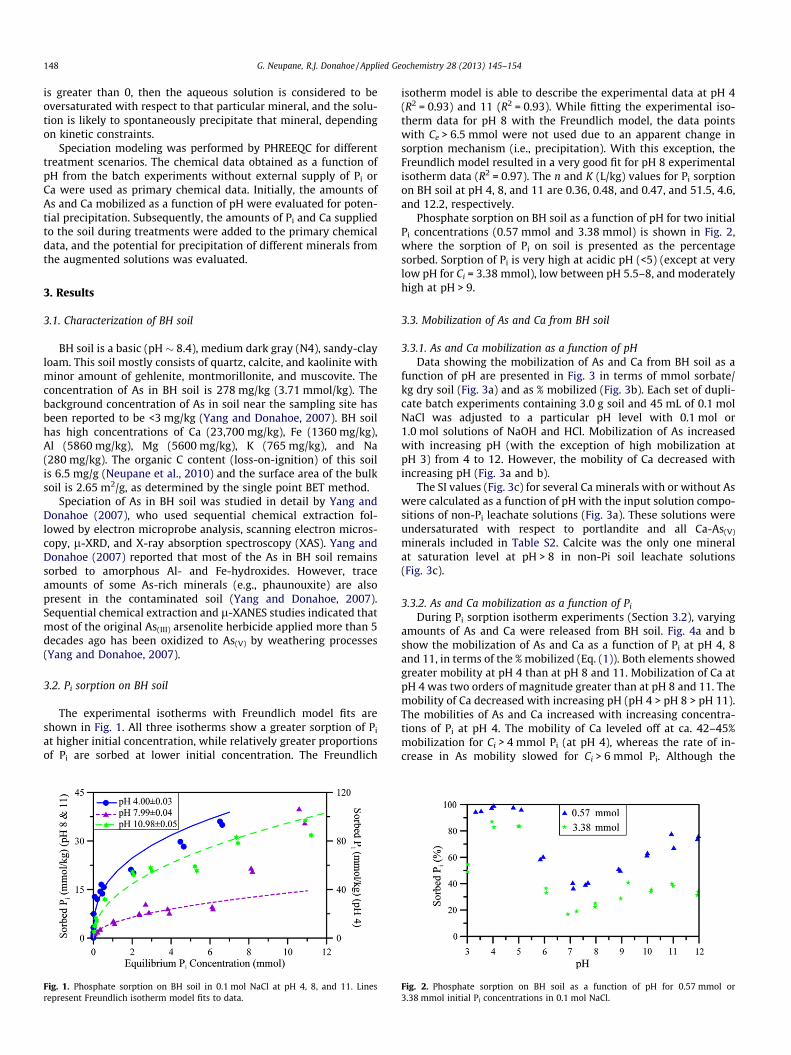
The experimental isotherms with Freundlich model fits areshown in Fig. 1. All three isotherms show a greater sorption of Pi
at higher initial concentration, while relatively greater proportionsof Pi are sorbed at lower initial concentration. The Freundlich
Fig. 1. Phosphate sorption on BH soil in 0.1 mol NaCl at pH 4, 8, and 11. Linesrepresent Freundlich isotherm model fits to data.
isotherm model is able to describe the experimental data at pH 4(R2 = 0.93) and 11 (R2 = 0.93). While fitting the experimental iso-therm data for pH 8 with the Freundlich model, the data pointswith Ce > 6.5 mmol were not used due to an apparent change insorption mechanism (i.e., precipitation). With this exception, theFreundlich model resulted in a very good fit for pH 8 experimentalisotherm data (R2 = 0.97). The n and K (L/kg) values for Pi sorptionon BH soil at pH 4, 8, and 11 are 0.36, 0.48, and 0.47, and 51.5, 4.6,and 12.2, respectively.
Phosphate sorption on BH soil as a function of pH for two initialPi concentrations (0.57 mmol and 3.38 mmol) is shown in Fig. 2,where the sorption of Pi on soil is presented as the percentagesorbed. Sorption of Pi is very high at acidic pH (<5) (except at verylow pH for Ci = 3.38 mmol), low between pH 5.5–8, and moderatelyhigh at pH > 9.
3.3. Mobilization of As and Ca from BH soil
3.3.1. As and Ca mobilization as a function of pHData showing the mobilization of As and Ca from BH soil as a
function of pH are presented in Fig. 3 in terms of mmol sorbate/kg dry soil (Fig. 3a) and as % mobilized (Fig. 3b). Each set of dupli-cate batch experiments containing 3.0 g soil and 45 mL of 0.1 molNaCl was adjusted to a particular pH level with 0.1 mol or1.0 mol solutions of NaOH and HCl. Mobilization of As increasedwith increasing pH (with the exception of high mobilization atpH 3) from 4 to 12. However, the mobility of Ca decreased withincreasing pH (Fig. 3a and b).
The SI values (Fig. 3c) for several Ca minerals with or without Aswere calculated as a function of pH with the input solution compo-sitions of non-Pi leachate solutions (Fig. 3a). These solutions wereundersaturated with respect to portlandite and all Ca-As(V)
minerals included in Table S2. Calcite was the only one mineralat saturation level at pH > 8 in non-Pi soil leachate solutions(Fig. 3c).
3.3.2. As and Ca mobilization as a function of Pi
During Pi sorption isotherm experiments (Section 3.2), varyingamounts of As and Ca were released from BH soil. Fig. 4a and bshow the mobilization of As and Ca as a function of Pi at pH 4, 8and 11, in terms of the % mobilized (Eq. (1)). Both elements showedgreater mobility at pH 4 than at pH 8 and 11. Mobilization of Ca atpH 4 was two orders of magnitude greater than at pH 8 and 11. Themobility of Ca decreased with increasing pH (pH 4 > pH 8 > pH 11).The mobilities of As and Ca increased with increasing concentra-tions of Pi at pH 4. The mobility of Ca leveled off at ca. 42–45%mobilization for Ci > 4 mmol Pi (at pH 4), whereas the rate of in-crease in As mobility slowed for Ci > 6 mmol Pi. Although the
Fig. 2. Phosphate sorption on BH soil as a function of pH for 0.57 mmol or3.38 mmol initial Pi concentrations in 0.1 mol NaCl.

Fig. 3. Mobilized As and Ca as a function of pH during batch leaching of 3 g soil in45 mL 0.1 mol NaCl. (a) Mobilized As and Ca given in mmol/kg dry soil; (b) percentmobilized calculated with respect to HNO3-digestible soil concentrations(As = 3.71 mmol/kg; Ca = 592 mmol/kg); and (c) mineral SI calculated using PHRE-EQC. In (c) – 1: Ca3(AsO4)2, 2: Ca3(AsO4)2�3.66H2O, 3: Ca3(AsO4)2�4.25H2O, 4:Ca4(OH)2(AsO4)2�4H2O, 5: Ca5(AsO4)3OH, 6: CaHAsO4�H2O, 7: calcite, 8: Ferrarsite, 9:Guerinite, and 10: Portlandite. Red and black symbols (online version) representminerals with and without As, respectively. (For interpretation of the references tocolour in this figure legend, the reader is referred to the web version of this article.)
Fig. 4. Percent mobilized (a) As and (b) Ca as a function of the initial concentrationof Pi and calculated with respect to the HNO3-digestible concentrations of theseelements in BH soil (As = 3.71 mmol/kg; Ca = 592 mmol/kg). The inset in (b) showsthe expanded y-axis for pH 8 and 11.
G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154 149
mobilities of As and Ca were positively correlated at pH 4 forCi < 4 mmol Pi, their mobilities were not significantly correlatedat pH 8 and 11. At pH 8 and 11, the mobility of As was independentof Pi concentration, whereas the mobility of Ca gradually decreasedwith increasing Pi.
The SI values (Fig. S1) for several Ca–Pi–As(V) and Ca–Pi miner-als were calculated at pH 4, 8 and 11, with the input solution com-positions for these simulations having the As and Caconcentrations of the pH-specific non-Pi leachate, and the initialconcentrations of Pi used in the sorption experiments. Therefore,the As and Ca concentrations at each pH were the same for all Piconcentrations used. Because Pi was not allowed to sorb on the soilduring the PHREEQC simulations, the total Pi in solution was con-sidered to be freely available to interact with aqueous As and Ca.The mineral SI values varied with pH, but for most minerals theSI values at each pH did not change significantly with increasingPi. At pH 4 (Fig. S1a), PHREEQC simulations indicated that soil solu-tions were undersaturated with respect to all Ca–Pi–As(V) miner-als. Similarly, these solutions were also undersaturated withrespect to most of the Ca–Pi minerals (except amorphous Ca phos-phate, hydroxyapatite and octocalcium phosphate). The calculated
mineral SI values at pH 8 and 11 were significantly different fromSI values at pH 4. In particular, the simulated solutions at pH 8were supersaturated with respect to several Ca–Pi–As(V) and Ca–Pi minerals (Fig. S1b). At pH 11, the solutions were approximatelysaturated with respect to most of the As-bearing minerals acrossthe range of Pi concentrations used during the PHREEQCsimulations.
3.3.3. Mobilization of As and Ca as functions of pH and Pi
The mobilization of As and Ca as a function of pH in the pres-ence of Pi was evaluated using the chemical data obtained fromsorption experiments with initial Pi concentrations of 0.57 mmoland 3.38 mmol. Fig. 5 shows As and Ca concentrations when Pi re-acted with BH soil between pH 3–12. The concentrations of Camobilized as a function of pH for 0.57 mmol and 3.38 mmol Pi werevery similar; the highest mobility occurred at low pH. The mobilityof Ca decreased rapidly with increasing pH, and became essentiallyimmobile under circum-neutral to basic conditions. Arsenic alsoshowed similar mobilization for both initial concentrations of Pi
at pH > 6, but demonstrated significantly different mobilities forthe two Pi concentrations at pH < 6. Under acidic conditions, Asmobility was significantly higher for 3.38 mmol Pi. The lowest Asmobility occurred between pH 6–9.5. At pH > 9.5, As mobility againincreased until nearly 50% of the As in BH soil was leached at pH12.
PHREEQC modeling was also used to evaluate the potential pre-cipitation of Ca–Pi–As(V) minerals as a function of pH from the non-Pi soil leachate (Fig. 3) with the addition of 0.57 mmol and3.38 mmol of Pi (Fig. S2). Both concentrations of Pi resulted in sim-ilar variations in the SI values for several minerals. The SI of eachmineral increased with increasing pH and peaked between pH 7–9; the SI values then decreased slightly at pH > 9. Between pH 6–10, the simulated solutions were oversaturated with respect tomost of the Ca–Pi–As(V) minerals, some Ca–Pi phases (e.g., octocal-cium phosphate), and a few other minerals (e.g., hydroxylapatite)over the pH range from 3 to 12 (Fig. S2).

Fig. 5. Percent mobilized soil As and Ca as a function of pH in the presence of0.57 mmol or 3.38 mmol Pi. The percent mobilization was calculated with respect tothe HNO3-digestible concentrations of these elements in BH soil (As = 3.71 mmol/kg; Ca = 592 mmol/kg).
Fig. 7. Percent mobilized As and Ca as a function of pH in the presence of3.39 mmol Pi and 5.63 or 16.9 mmol added Ca. The percent mobilization wascalculated with respect to the HNO3-digestible concentrations of these elements inBH soil (As = 3.71 mmol/kg; Ca = 592 mmol/kg).
150 G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154
3.4. Phosphate and Ca treatment of soil
3.4.1. Sorption of Pi during Ca–Pi treatmentPhosphate sorption on BH soil in the presence of externally sup-
plied Ca was also evaluated for a Ci of 3.38 mmol Pi. For one set ofexperiments, 5.63 mmol Ca was supplied to yield a Ca:Pi ratio of1.67:1 in solution; in a second set of experiments, excess Ca(16.9 mmol) was supplied, producing a Ca:Pi ratio of 5:1 in solu-tion. Comparison of Figs. 6 and 2 shows that sorption of Pi on BHsoil at pH < 5 was not significantly affected by external supply ofCa. At pH < 5, the sorption of Pi decreased with decreasing pH.Interestingly, the addition of 16.9 mmol Ca slightly decreased thesorption of Pi at pH < 5. However, the addition of external Ca signif-icantly increased Pi sorption by the soil between pH 5–7, comparedto Pi sorption in the same pH range without additional Ca. More-over, the additional Ca resulted in a large increase in removal ofPi from solution at pH > 8. In the absence of external Ca (Fig. 2), in-creased, but incomplete, transfer of Pi to the solid phase was ob-served at pH > 7. However, almost 100% of the Pi was transferredfrom the soil solution to the solid phase under basic conditionswhen additional Ca was supplied (Fig. 6).
3.4.2. Mobilization of As and Ca during Ca–Pi treatmentsFig. 7 shows the calculated mobilities of As and Ca as a function
of pH when both Ca and Pi were supplied to the soil. The amount ofCa mobilized from BH soil was slightly higher across the pH rangewhen less external Ca was supplied during treatment. In general,Ca mobility decreased with increasing solution pH (Fig. 7). No Cawas transferred from solution to the solid phase at pH < 5.8 andpH < 6.8 for 16.9 mmol and 5.63 mmol Ca, respectively. But atpH > 5.8 and pH > 6.8, respectively, for 16.9 mmol and 5.63 mmol
Fig. 6. Phosphate sorption on BH soil as a function of pH and initial Caconcentration. Initial Pi concentration was fixed at 3.38 mmol in all experiments,while the initial Ca concentration was either 5.63 mmol or 16.9 mmol.
of externally supplied Ca, net transfer of Ca from solution to thesoil occurred. At pH 8, nearly 45% and 60% of the externally sup-plied Ca was transferred to the soil, resulting in calculated Camobilities of �20% and �9% for 16.9 mmol and 5.63 mmol, respec-tively, of added Ca. With further increase in pH, a successivelyhigher proportion of the externally supplied Ca was transferredto the solid phase. Almost 85% and 98% of the added Ca was trans-ferred to the solid phase at pH 12 during Ca–Pi treatments using16.9 mmol and 5.63 mmol Ca, respectively.
The mobility of As also decreased with increasing pH (Fig. 7). AtpH 8, about 3% and 10% of the HNO3-digestible As was releasedfrom BH soil after Pi treatments using 16.9 mmol and 5.63 mmolCa, respectively. The Pi treatment with 5.63 mmol Ca resulted ina 4–6% mobilization of As at pH > 8. However, the Pi treatment
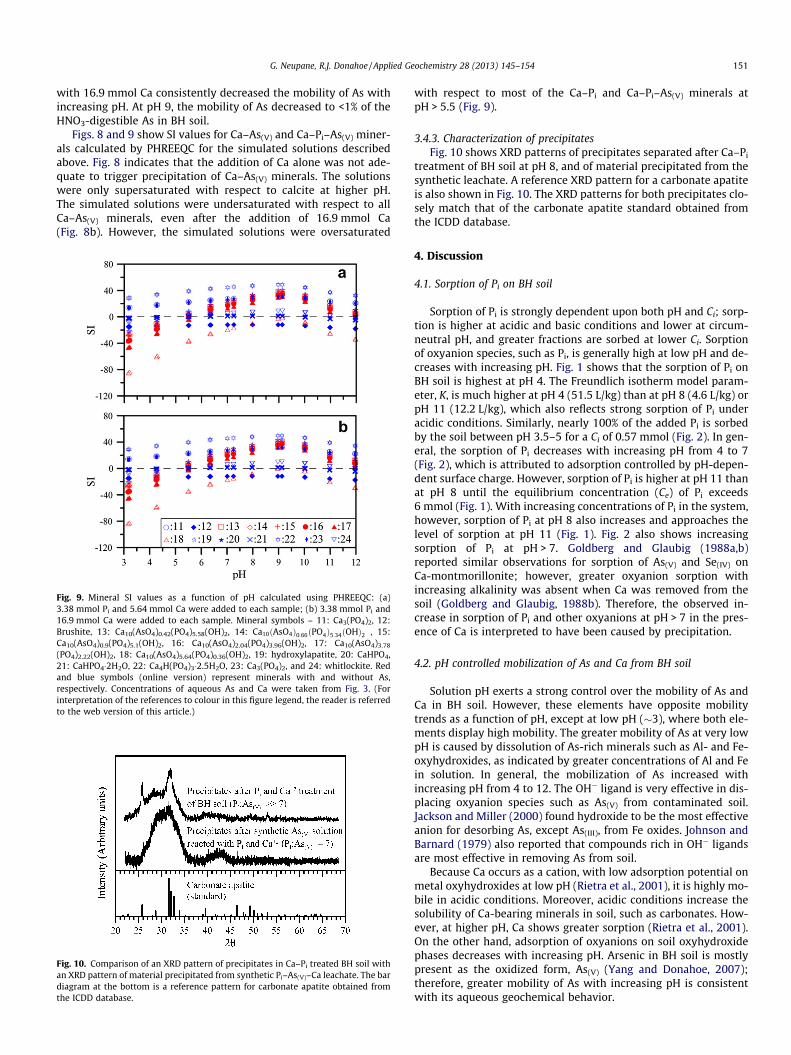
Fig. 8. Mineral SI values as a function of pH calculated using PHREEQC: (a)3.38 mmol Pi and 5.64 mmol Ca were added to each sample; (b) 3.38 mmol Pi
and 16.9 mmol Ca were added to each sample. Mineral symbols – 1: Ca3(AsO4)2,2: Ca3(AsO4)2�3.66H2O, 3: Ca3(AsO4)2�4.25H2O, 4: Ca4(OH)2(AsO4)2�4H2O, 5:Ca5(AsO4)3OH, 6: CaHAsO4�H2O, 7: calcite, 8: Ferrarsite, 9: Guerinite, and 10:Portlandite. Red and black symbols (online version) represent minerals with andwithout As, respectively. Concentrations of aqueous As and Ca were taken fromFig. 3. (For interpretation of the references to colour in this figure legend, the readeris referred to the web version of this article.)
G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154 151
with 16.9 mmol Ca consistently decreased the mobility of As withincreasing pH. At pH 9, the mobility of As decreased to <1% of theHNO3-digestible As in BH soil.
Figs. 8 and 9 show SI values for Ca–As(V) and Ca–Pi–As(V) miner-als calculated by PHREEQC for the simulated solutions describedabove. Fig. 8 indicates that the addition of Ca alone was not ade-quate to trigger precipitation of Ca–As(V) minerals. The solutionswere only supersaturated with respect to calcite at higher pH.The simulated solutions were undersaturated with respect to allCa–As(V) minerals, even after the addition of 16.9 mmol Ca(Fig. 8b). However, the simulated solutions were oversaturated
Fig. 9. Mineral SI values as a function of pH calculated using PHREEQC: (a)3.38 mmol Pi and 5.64 mmol Ca were added to each sample; (b) 3.38 mmol Pi and16.9 mmol Ca were added to each sample. Mineral symbols – 11: Ca3(PO4)2, 12:Brushite, 13: Ca10(AsO4)0.42(PO4)5.58(OH)2, 14: Ca10ðAsO4Þ0:66ðPO4Þ5:34ðOHÞ2 , 15:Ca10(AsO4)0.9(PO4)5.1(OH)2, 16: Ca10(AsO4)2.04(PO4)3.96(OH)2, 17: Ca10(AsO4)3.78
(PO4)2.22(OH)2, 18: Ca10(AsO4)5.64(PO4)0.36(OH)2, 19: hydroxylapatite, 20: CaHPO4,21: CaHPO4�2H2O, 22: Ca4H(PO4)3�2.5H2O, 23: Ca3(PO4)2, and 24: whitlockite. Redand blue symbols (online version) represent minerals with and without As,respectively. Concentrations of aqueous As and Ca were taken from Fig. 3. (Forinterpretation of the references to colour in this figure legend, the reader is referredto the web version of this article.)
Fig. 10. Comparison of an XRD pattern of precipitates in Ca–Pi treated BH soil withan XRD pattern of material precipitated from synthetic Pi–As(V)–Ca leachate. The bardiagram at the bottom is a reference pattern for carbonate apatite obtained fromthe ICDD database.
with respect to most of the Ca–Pi and Ca–Pi–As(V) minerals atpH > 5.5 (Fig. 9).
3.4.3. Characterization of precipitatesFig. 10 shows XRD patterns of precipitates separated after Ca–Pi
treatment of BH soil at pH 8, and of material precipitated from thesynthetic leachate. A reference XRD pattern for a carbonate apatiteis also shown in Fig. 10. The XRD patterns for both precipitates clo-sely match that of the carbonate apatite standard obtained fromthe ICDD database.
4. Discussion
4.1. Sorption of Pi on BH soil
Sorption of Pi is strongly dependent upon both pH and Ci; sorp-tion is higher at acidic and basic conditions and lower at circum-neutral pH, and greater fractions are sorbed at lower Ci. Sorptionof oxyanion species, such as Pi, is generally high at low pH and de-creases with increasing pH. Fig. 1 shows that the sorption of Pi onBH soil is highest at pH 4. The Freundlich isotherm model param-eter, K, is much higher at pH 4 (51.5 L/kg) than at pH 8 (4.6 L/kg) orpH 11 (12.2 L/kg), which also reflects strong sorption of Pi underacidic conditions. Similarly, nearly 100% of the added Pi is sorbedby the soil between pH 3.5–5 for a Ci of 0.57 mmol (Fig. 2). In gen-eral, the sorption of Pi decreases with increasing pH from 4 to 7(Fig. 2), which is attributed to adsorption controlled by pH-depen-dent surface charge. However, sorption of Pi is higher at pH 11 thanat pH 8 until the equilibrium concentration (Ce) of Pi exceeds6 mmol (Fig. 1). With increasing concentrations of Pi in the system,however, sorption of Pi at pH 8 also increases and approaches thelevel of sorption at pH 11 (Fig. 1). Fig. 2 also shows increasingsorption of Pi at pH > 7. Goldberg and Glaubig (1988a,b)reported similar observations for sorption of As(V) and Se(IV) onCa-montmorillonite; however, greater oxyanion sorption withincreasing alkalinity was absent when Ca was removed from thesoil (Goldberg and Glaubig, 1988b). Therefore, the observed in-crease in sorption of Pi and other oxyanions at pH > 7 in the pres-ence of Ca is interpreted to have been caused by precipitation.
4.2. pH controlled mobilization of As and Ca from BH soil
Solution pH exerts a strong control over the mobility of As andCa in BH soil. However, these elements have opposite mobilitytrends as a function of pH, except at low pH (�3), where both ele-ments display high mobility. The greater mobility of As at very lowpH is caused by dissolution of As-rich minerals such as Al- and Fe-oxyhydroxides, as indicated by greater concentrations of Al and Fein solution. In general, the mobilization of As increased withincreasing pH from 4 to 12. The OH� ligand is very effective in dis-placing oxyanion species such as As(V) from contaminated soil.Jackson and Miller (2000) found hydroxide to be the most effectiveanion for desorbing As, except As(III), from Fe oxides. Johnson andBarnard (1979) also reported that compounds rich in OH� ligandsare most effective in removing As from soil.
Because Ca occurs as a cation, with low adsorption potential onmetal oxyhydroxides at low pH (Rietra et al., 2001), it is highly mo-bile in acidic conditions. Moreover, acidic conditions increase thesolubility of Ca-bearing minerals in soil, such as carbonates. How-ever, at higher pH, Ca shows greater sorption (Rietra et al., 2001).On the other hand, adsorption of oxyanions on soil oxyhydroxidephases decreases with increasing pH. Arsenic in BH soil is mostlypresent as the oxidized form, As(V) (Yang and Donahoe, 2007);therefore, greater mobility of As with increasing pH is consistentwith its aqueous geochemical behavior.

152 G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154
The high mobility of Ca under acidic conditions is not accompa-nied by higher mobilization of As (except at pH 3), indicating thatthese elements are not associated with the same solid phase in BHsoil. Using a 7-step sequential chemical extraction procedure, Qiand Donahoe (2008) reported that more than 60% of theHNO3-digestible As in BH soil is associated with amorphous Al–Fe-oxyhydroxides, and another 20% is associated with crystallineAl–Fe oxides. The remaining 20% was reported to be associatedwith the water soluble, exchangeable, carbonate, Mn-oxide, andorganic soil fractions (Qi and Donahoe, 2008). Furthermore, the soilsolutions are also undersaturated with respect to all Ca–As miner-als (Fig. 3c). The relatively higher SI values calculated for all Ca–Asminerals between pH 6–9 are due to the presence of both elementsin soil leachate in this pH range. At pH < 6, the concentration of Asis very low, and at pH > 8 the concentration of Ca is low. Therefore,the highly undersaturated state of the soil solution with respect toCa–As minerals at low pH and high pH is controlled by low aque-ous concentrations of As and Ca, respectively.
4.3. Phosphate induced mobilization of As and Ca from BH soil
Although Pi solutions have been reported previously to be effi-cient agents for flushing As from soil (e.g., Peryea and Kammereck,1997; Alam et al., 2001; Tokunaga and Hakuta, 2002), the effective-ness is dependent upon solution pH. The mobility of As at pH 4 in-creases with increasing initial concentration of Pi; however, Asmobility is independent of Pi concentration in solutions at pH 8and 11. Moreover, the mobility of As at pH 8 and 11 with varyingamount Pi (Fig. 4a) is very similar to its mobility at these pH levelswithout Pi (Fig. 3). Therefore, the Pi-independent mobility of As atpH 8 and 11 can be attributed to pH-induced desorption of As asso-ciated with Al- and Fe-oxyhydroxides in the soil. The greatermobility of As at pH 4 is mostly related to Pi-induced competitivedesorption.
The Ca–Pi–As(V) and Ca–Pi mineral SI values calculated at pH 4,8, and 11 by PHREEQC for simulated solutions having compositionsobtained by adding the initial Pi concentrations to the non-Pi soilleachate, show different probabilities for mineral precipitation(Fig. S1). At pH 4 (Fig. S1a), the solutions are undersaturated withrespect to all Ca–Pi–As(V) minerals. At pH 8, the simulated solutionsare supersaturated with respect to several Ca–Pi–As(V) and Ca–Pi
minerals (Fig. S1b), because the non-Pi leachate (Fig. 3) is slightlyricher in Ca than the actual equilibrium solution (Fig. 4b). At pH11, the solutions are approximately saturated with respect to mostof the As-bearing minerals across the range of Pi concentrations. AtpH 11, the controlling factor for the degree of saturation is presum-ably the amount of aqueous Ca, rather than the amount of As or Pi
in solution. Hence, it is likely that the mobility of soil As can be de-creased through precipitation of Ca–Pi–As(V) minerals, if adequateCa is supplied along with Pi during soil treatment.
4.4. Mobilization of As and Ca as functions of pH and Pi
Mobilization of As and Ca as a function of pH in the presenceof Pi is different, particularly at pH > 7. The increase in aqueous Asconcentrations without mobilization of Ca under basic conditionsindicates that these two elements are not associated with a com-mon phase in BH soil. Similarly, the lower mobility of Ca withhigher As mobility, and the reversal in sorption of Pi at pH > 7(Figs. 2 and 5), indicate precipitation of a Ca–Pi phase withoutincorporation of As. Although sorption of Pi by BH soil at pH > 7is probably controlled by precipitation, only a portion of the totalPi added (up to 80% for 0.57 mmol and up to 40% for 3.38 mmolinitial Pi concentration) (Fig. 2) is removed from the soil solution.The complete removal of Pi from solution by precipitation at highpH is prevented primarily by the deficiency of dissolved Ca.
Similarly, As that leaches from BH soil does not precipitate, dueto the apparent deficiency of Ca at pH > 8. Supplying adequateCa in the treatment solution along with Pi would eventually leadto co-precipitation of As with Ca–Pi phases, and decrease itsmobility. Similarly, the undersaturated state of the simulatedsolutions with respect to Ca–Pi–As(V) minerals at lower pH iscaused by low concentrations of As, whereas at higher pH suchundersaturated conditions are due to low concentrations of Cain the non-Pi leachate.
The greater mobility of As at low pH (except at pH � 3) in thepresence of Pi is caused by competitive desorption, rather than dis-solution. However, the increased mobility of As at intermediateand higher pH is related to pH-induced desorption because Asmobility at pH 8 and 11 is independent of Pi. Therefore, at cir-cum-neutral to basic pH, the supply of additional Pi does not havea significant effect on the mobility of As. However, the addition ofPi increases the mobility of As under acidic soil conditions.
4.5. Phosphate and Ca treatments
The sorption of Pi on BH soil at pH > 7 is changed when externalCa is also simultaneously added to the system. In particular, theadditional Ca resulted in a large increase in removal of Pi fromsolution under alkaline conditions, and this nearly complete trans-fer of Pi from the treatment solution to soil is attributed primarilyto precipitation. Precipitation of Ca–Pi phases under alkaline pHconditions is also supported by the observed negative mobility ofCa. A large fraction (45–60%) of the externally supplied Ca is trans-ferred to the solid phase during soil treatment. The precipitation ofCa–Pi phases also helps decrease the mobility of soil As. In partic-ular, the observed decrease in As mobility after Ca–Pi soil treat-ments (Fig. 7) is remarkable when compared to the high mobilityof As during Pi-only soil treatments (Figs. 3a and b, 4a and 5).The decreased mobility of As, along with a large transfer of exter-nally supplied Ca and Pi, indicates precipitation of As-bearing Ca–Pi
mineral phases. PHREEQC simulations also showed that the solu-tions are oversaturated with respect to most of the Ca–Pi andCa–Pi–As(V) minerals at pH > 5.5. Although the addition of5.63 mmol and 16.9 mmol Ca with 3.38 mmol Pi resulted in verysimilar mineral SI values as a function of pH (Figs. 8 and 9), theBH soil treatment experiments using the lower Ca concentration(5.63 mmol) failed to completely immobilize the As. Therefore,the addition of excess Ca, along with Pi, is required for completeimmobilization of As in contaminated soil.
Similarly, speciation modeling indicated that the syntheticleachate (As: 0.48 mmol, Ca: 6.44 mmol, Pi: 3.38 mmol, pH: 8)equilibrated with atmospheric CO2 is oversaturated with respectto all of the Ca–Pi–As(V) minerals included in Table S2. However,the synthetic leachate is also undersaturated with respect to sev-eral Ca–As(V) minerals. Previous studies by Twidwell et al. (1994)and others (Wilson, 1998; Orser, 2001) have reported that removalof As from solution can be achieved by precipitation of apatite-likeminerals at pH 8–10. Their studies also demonstrated that expo-sure of the experimental system to air (i.e., CO2) could potentiallyresult in conversion of the precipitate into CaCO3, with subsequentrelease of As back into solution, if the Pi:As(V) ratio is < 7. However,if the Pi:As(V) ratio of Ca–Pi–As(V) minerals is P 7, the precipitatesbecome stable under atmospheric conditions (Wilson, 1998; Orser,2001). In the present study, the Ca–Pi–As(V) precipitate formed inBH soil after Ca–Pi treatment should have a Pi:As(V) ratio� 7, asdetermined by mass-balance calculations. The precipitation ofthe Ca–Pi–As(V) solid phase, along with the low mobility of As,the negative mobility of Ca, and the nearly complete removal ofPi from solution at pH > 8, suggest that Ca–Pi treatment success-fully immobilized As in BH soil.

G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154 153
5. Conclusions
In this study, the applicability of Pi treatment with and withoutadded Ca for immobilization of As in contaminated soil was testedthrough various batch experiments. An As-contaminated soil sam-ple collected from an industrial site was subjected to bench-scale Pi
and Ca–Pi treatments. Batch experiments showed that the soil hasa significant sorption capacity for Pi. Phosphate sorption as a func-tion of the initial concentration of Pi and pH indicated that the leastsorption of Pi, along with the lowest As mobility, occur at circum-neutral pH conditions. Arsenic mobility increased while increasingthe initial concentration of Pi at pH 4, but this trend was indepen-dent of Pi concentration at pH 8 and 11. Therefore, the mobilizationof As from BH soil at pH 4 is related to Pi-induced competitivedesorption, whereas mobilization of As at higher pH levels isattributed to pH-induced desorption.
Treatment of BH soil with Pi and Ca resulted in decreased mobil-ity of As. After Ca–Pi treatment, geochemical modeling indicatedthat soil solutions and synthetic leachate are oversaturated withrespect to several Ca–Pi–As(V) phases. The externally supplied Caand Pi are, therefore, most likely removed from solution by precip-itation of solid phases during soil treatment. XRD analysis indi-cated the existence of a poorly crystalline carbonate-apatitephase in the Ca–Pi treated soil. A similar phase was also identifiedin precipitate from synthetic Ca–Pi–As(V) leachate. Most impor-tantly, treatment of BH soil with 3.38 mmol Pi and 16.9 mmol Caalmost completely immobilized soil As from circum-neutral tohigh pH levels.
The results of this study demonstrate that Ca–Pi treatmentcould be potentially applicable for immobilization of As in contam-inated soils. In particular, this method would work best for Asremediation in Ca-rich soils, which do not respond as well aslow-Ca soils to other chemical fixation treatment methods. Thistreatment method could also be extended for remediation of As-contaminated wastewater. The low solubilities of many of theCa–Pi–As(V) minerals suggest that Ca–Pi treatment has promise asan effective, long-term method for in situ chemical fixation of Asin contaminated soils and wastewaters.
Acknowledgements
The authors would like to thank Dr. Sidhartha Bhattacharyya,Elizabeth Y. Graham, and Ziming Yue for their help during labora-tory experiments and chemical analyses. This research was partlyfunded by student research grants from the Geological Society ofAmerica, the Gulf Coast Association of Geological Societies, andthe W.G. Hooks Fund (UA Department of Geological Sciences). Sug-gestions from three anonymous reviewers and Associate Editor Dr.Joyanto Routh were very helpful in revising the originalmanuscript.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.apgeochem.2012.10.011.
References
Alam, M.G.M., Tokunaga, S., Maekawa, T., 2001. Extraction of arsenic in a syntheticarsenic-contaminated soil using phosphate. Chemosphere 43, 1035–1041.
ASTM, 2003. ASTM D4567-03: Standard Test Method for Single-pointDetermination of Specific Surface Area of Catalysts and Catalyst CarriersUsing Nitrogen Adsorption by Continuous Flow Method. ASTM International.
ATSDR, 2000. Case Studies in Environmental Medicine – Arsenic Toxicity. ATSDRPublication No.: ATSDR-HE-CS-2002-0003 U.S. Department of Health and
Human Services, Agency for Toxic Substances and Disease Registry, Divisionof Toxicology and Environmental Medicine.
Bates, M.N., Smith, A.H., Hopenhayn-Rich, C., 1992. Arsenic ingestion and internalcancers: a review. Am. J. Epidemiol. 135, 462–476.
Bhattacharyya, S., Donahoe, R.J., Patel, D., 2009. Experimental study of chemicaltreatment of coal fly ash to reduce the mobility of priority trace elements. Fuel88, 1173–1184.
Black, R.J., 1993. Florida Climate Data. Circular EES-5, Florida Cooperative ExtensionService, Institute of Food and Agricultural Sciences, University of Florida.
Bothe, J.V., Brown, P.W., 1999. The stabilities of calcium arsenates at 23 ± 1�C. J.Hazard. Mater. 69, 197–207.
Bundschuh, J., Litter, M.I., Parvez, F., Román-Ross, G., Nicolli, H.B., Jean, J., Liu, C.,López, D., Armienta, M.A., Guilherme, L.R.G., Cuevas, A.G., Cornejo, L., Cumbal, L.,Toujaguez, R., 2012. One century of arsenic exposure in Latin America: a reviewof history and occurrence from 14 countries. Sci. Total Environ. 429, 2–35.
Carbonell-Barrachina, A.A., Rocamora, A., García-Gomis, C., Martínez-Sánchez, F.,Burló, F., 2004. Arsenic and zinc biogeochemistry in pyrite mine waste from theAznalcóllar environmental disaster. Geoderma 122, 195–203.
Drever, J.I., 1997. The Geochemistry of Natural Waters: Surface and GroundwaterEnvironments, third ed. Prentice Hall, New Jersey.
Goldberg, S., Glaubig, R.A., 1988a. Anion sorption on a calcareous, montmorilloniticsoil–arsenic. Soil Sci. Soc. Am. J. 52, 1297–1300.
Goldberg, S., Glaubig, R.A., 1988b. Anion sorption on a calcareous, montmorilloniticsoil–selenium. Soil Sci. Soc. Am. J. 52, 954–958.
Grisafe, D.A., Hummel, F.A., 1970. Pentavalent ion substitutions in the apatitestructure. Part A: Crystal chemistry. J. Solid State Chem. 2, 160–166.
Hossain, M.F., 2006. Arsenic contamination in Bangladesh – an overview. Agric.Ecosyst. Environ. 113, 1–16.
Jackson, R., Grainge, J.W., 1975. Arsenic and cancer. Can. Med. Assoc. J. 113, 396–401.
Jackson, B.P., Miller, W.P., 2000. Effectiveness of phosphate and hydroxide fordesorption of arsenic and selenium species from iron oxides. Soil Sci. Soc. Am. J.64, 1616–1622.
Johnson, S.E., Barnard, W.M., 1979. Comparative effectiveness of fourteen solutionsfor extracting arsenic from four western New York soils. Soil Sci. Soc. Am. J. 43,304–308.
Kaplan, D.I., Knox, A.S., 2004. Enhanced contaminant desorption induced byphosphate mineral additions to sediment. Environ. Sci. Technol. 38, 3153–3160.
Karagas, M.R., Stukel, T.A., Tosteson, T.D., 2002. Assessment of cancer risk andenvironmental levels of arsenic in New Hampshire. Int. J. Hyg. Environ. Health205, 85–94.
Limousin, G., Gaudet, J.-P., Charlet, L., Szenknect, S., Barthès, V., Krimissa, M., 2007.Sorption isotherms: a review on physical bases, modeling and measurement.Appl. Geochem. 22, 249–275.
Ma, L.Q., Komar, K.M., Tu, C., Zhang, Z., Cai, Y., Kennelley, E.D., 2001. A fern thathyperaccumulates arsenic. Nature 409, 579.
Mahapatra, P.P., Mahapatra, L.M., Mishra, B., 1987. Arsenate hydroxyapatite: aphysico-chemical and thermodynamic investigation. Polyhedron 6, 1049–1052.
Montastruc, L., Azzaro-Pantel, C., Biscans, B., Cabassud, M., Domenech, S., Dibouleau,L., 2003. A general framework for pellet reactor modelling: application to P-recovery. Trans. Inst. Chem. Eng.: Part A: Chem. Eng. Res. Des. 81, 1271–1278.
Morrell, J.J., Keefe, D., Baileys, R.T., 2003. Copper, zinc, and arsenic in soilsurrounding douglas-fir poles treated with ammoniacal copper zinc arsenate(ACZA). J. Environ. Qual. 32, 2095–2099.
Mulligan, C.N., Yong, R.N., Gibbs, B.F., 2001. Remediation technologies for metal-contaminated soil and groundwater: an evaluation. Eng. Geol. 60, 193–207.
Neupane, G., Donahoe, R.J., Qi, Y., 2010. Fate of arsenic, iron, and lanthanum inarsenic-contaminated soils treated with ferrous sulfate and ferrous sulfate–lanthanum chloride, G-073. In: Fields, K.A., Wickramanayake, G.B. (Chairs),Remediation of Chlorinated and Recalcitrant Compounds—2010. 7th Internat.Conf. Remediation of Chlorinated and Recalcitrant Compounds (Monterey, CA;May 2010). Battelle Memorial Institute, Columbus, OH. <www.battelle.org/chlorcon>.
Nordstrom, D.K., 2002. Worldwide occurrences of arsenic in ground water. Science296, 2143–2145.
Orser, T., 2001. Removal of Arsenic from Waste Water Solutions as Storable StablePrecipitates. MS Thesis, Department of Metallurgical and Materials Engineering,Montana Tech of the University of Montana.
Parkhurst, D.L., Appelo, C.A.J., 1999. User’s Guide to PHREEQC (Version 2) – AComputer Program for Speciation, Batch-reaction, One-dimensional Transport,and Inverse Geochemical Calculations. U.S. Geol. Survey, Water Resour. Invest.Rep. 99-4259.
Peryea, F.J., Kammereck, R., 1997. Phosphate-enhanced movement of arsenic out oflead arsenate-contaminated topsoil and through uncontaminated subsoil.Water Air Soil Pollut. 93, 243–254.
Qi, Y., Donahoe, R.J., 2008. The environmental fate of arsenic in surface soilcontaminated by historical herbicide application. Sci. Total Environ. 405, 246–254.
Rietra, R.P.J.J., Hiemstra, T., van Riemsdijk, W.H., 2001. Interaction between calciumand phosphate adsorption on goethite. Environ. Sci. Technol. 35, 3369–3374.
Robinson Jr., G.R., Larkins, P., Boughton, C.J., Reed, B.W., Sibrell, P.L., 2007. Assessmentof contamination from arsenical pesticide use on orchards in the Great Valleyregion, Virginia and West Virginia, USA. J. Environ. Qual. 36, 654–663.
Simcox, N.J., Fenske, R.A., Wolz, S.A., Lee, I.C., Kalman, D.A., 1995. Pesticides inhousehold dust and soil: exposure pathways for children of agriculturalfamilies. Environ. Health Perspect. 103, 1126–1134.

154 G. Neupane, R.J. Donahoe / Applied Geochemistry 28 (2013) 145–154
Smedley, P.L., Kinniburgh, D.G., 2002. A review of the source, behaviour anddistribution of arsenic in natural waters. Appl. Geochem. 17, 517–568.
Smith, R.M., Martell, A.E., 1976. Critical Stability Constants. IV. Inorganic Complexes.Plenum Press, New York.
Sposito, G., 1986. On distinguishing adsorption from surface precipitation. In: Davis,J.A., Hayes, K.F. (Eds.), Geochemical Processes at Mineral Surfaces. Am. Chem.Soc. Symp. Series, vol. 323, pp. 217–228.
Tokunaga, S., Hakuta, T., 2002. Acid washing and stabilization of an artificialarsenic-contaminated soil. Chemosphere 46, 31–38.
Twidwell, L.G., Plessas, K.O., Comba, P.G., Dahnke, D.R., 1994. Removal of arsenicfrom wastewaters and stabilization of arsenic bearing waste solids: summary ofexperimental studies. J. Hazard. Mater. 36, 69–80.
USEPA, 2004. Method 9045D: Soil and Waste pH. Revision 4, November 2004, USEnvironmental Protection Agency, USA.
USEPA, 2007. Method 3051A: Microwave-assisted Acid Digestion of Sediments,Sludges, Soils and Oils. Revision 1, February 2007, US Environmental ProtectionAgency, USA.
Wasay, S.A., Parker, W., Van Geel, P.J., Barrington, S., Tokunaga, S., 2000. Arsenicpollution of a loam soil: retention form and decontamination. Soil Sed. Contam.9, 51–64.
Welch, A.H., Westjohn, D.B., Helsel, D.R., Wanty, R.B., 2000. Arsenic in groundwater of the United States: occurrence and geochemistry. Ground Water 38,589–604.
Wilson, S.R., 1998. Removal of Arsenic from ASARCO Acid Plant Blowdown Water asStable, Storable Precipitates. MS Thesis, Department of Metallurgical andMaterials Engineering, Montana Tech of the University of Montana.
Woolson, E.A., Axley, J.H., Kearney, P.C., 1973. Chemistry and phytotoxicity ofarsenic in soils: II. Effects of time and phosphorus. Soil Sci. Soc. Am. J. 37, 254–259.
Yang, L., Donahoe, R.J., 2007. The form, distribution and mobility of arsenic in soilscontaminated by arsenic trioxide, at sites in southeast USA. Appl. Geochem. 22,320–341.
Yang, L., Donahoe, R.J., Redwine, J.C., 2007. In situ chemical fixation ofarsenic-contaminated soils: an experimental study. Sci. Total Environ. 387,28–41.
Yuan, C., Chiang, T.S., 2007. The mechanisms of arsenic removal from soil byelectrokinetic process coupled with iron permeable reaction barrier.Chemosphere 67, 1533–1542.