biomimetic macroporous hydrogels: protein ligand ... · modified), epoxy-containing, fg/phema and...
TRANSCRIPT

Journal of Biomaterials Science 20 (2009) 1781–1795www.brill.nl/jbs
Biomimetic Macroporous Hydrogels: Protein LigandDistribution and Cell Response to the Ligand
Architecture in the Scaffold
Irina N. Savina a,b, Maria Dainiak b,c, Hans Jungvid b, Sergey V. Mikhalovsky a and
Igor Yu. Galaev c,∗
a School of Pharmacy and Biomolecular Sciences, Brighton University, Brighton, UKb Protista Biotechnology AB, IDEON, SE 223 70 Lund, Sweden
c Department of Biotechnology, Lund University, P.O. Box 124, SE 221 00 Lund, Sweden
Received 7 August 2008; accepted 15 October 2008
AbstractMacroporous hydrogels (MHs), cryogels, are a new type of biomaterials for tissue engineering that canbe produced from any natural or synthetic polymer that forms a gel. Synthetic MHs are rendered bioac-tive by surface or bulk modifications with extracellular matrix components. In this study, cell response tothe architecture of protein ligands, bovine type-I collagen (CG) and human fibrinogen (Fg), immobilisedusing different methods on poly(2-hydroxyethyl methacrylate) (pHEMA) macroporous hydrogels (MHs)was analysed. Bulk modification was performed by cross-linking cryo-co-polymerisation of HEMA andpoly(ethylene glycol)diacrylate (PEGA) in the presence of proteins (CG/pHEMA and Fg/pHEMA MHs).The polymer surface was modified by covalent immobilisation of the proteins to the active epoxy (ep)groups present on pHEMA after hydrogel fabrication (CG–epHEMA and Fg–epHEMA MHs). The concen-tration of proteins in protein/pHEMA and protein–epHEMA MHs was 80–85 and 130–140 µg/ml hydrogel,respectively. It was demonstrated by immunostaining and confocal laser scanning microscopy that bulkmodification resulted in spreading of CG in the polymer matrix and spot-like distribution of Fg. On thecontrary, surface modification resulted in spot-like distribution of CG and uniform spreading of Fg, whichevenly coated the surface. Proliferation rate of fibroblasts was higher on MHs with even distribution ofthe ligands, i.e., on Fg–epHEMA and CG/pHEMA. After 30 days of growth, fibroblasts formed severalmonolayers and deposited extracellular matrix filling the pores of these MHs. The best result in terms ofcell proliferation was obtained on Fg–epHEMA. The ligands displayed on surface of these scaffolds werein native conformation, while in bulk-modified CG/pHEMA MHs most of the proteins were buried insidethe polymer matrix and were less accessible for interactions with specific antibodies and cells. The methodused for MH modification with bioligands strongly affects spatial distribution, density and conformation ofthe ligand on the scaffold surface, which, in turn, influence cell–surface interactions. The optimal type ofmodification varies depending on intrinsic properties of proteins and MHs.© Koninklijke Brill NV, Leiden, 2009
* To whom correspondence should be addressed. Tel.: (46-46) 222-0881; Fax: (46-46) 222-4713; e-mail:[email protected]
© Koninklijke Brill NV, Leiden, 2009 DOI:10.1163/156856208X386390

1782 I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795
KeywordsBiomimetic macroporous hydrogel, protein ligand distribution, ligand conformation, cell response
1. Introduction
Development of biomaterials for tissue engineering is a major area of biomedicalmaterials research. It is focused on developing three-dimensional (3D) scaffoldswith appropriate strength, porosity, degradation times, microstructure and abilityto elicit specific cellular responses [1–3]. Macroporous hydrogels (MHs) producedby cryotropic polymerisation represent an interesting group of biomaterials due totheir highly porous 3D structure, elasticity and easily modulated mechanical andbiochemical properties [4–8]. Cryotropic polymerisation takes place at sub-zerotemperatures when most of the solvent is frozen while the dissolved monomersor polymer precursors are concentrated in small non-frozen regions, where the gelformation proceeds. After melting the solvent crystals that perform like porogen,a system with large (10–200 µm) continuous interconnected pores is formed [9, 10].MHs prepared from natural sources, as well as synthetic MHs have high potentialas scaffolds for tissue engineering [11, 12].
In order to achieve biomolecular recognition of synthetic materials by cells thescaffolds need to be subjected to surface and bulk modification with bioactive mole-cules such as extracellular matrix (ECM) components or cell-adhesive peptidesderived from ECM proteins [13, 14]. Interactions of cells with various syntheticmaterials biocoated by means of surface or bulk modification has been a subject ofextensive studies. Bulk modification is performed by co-polymerisation or attach-ment of functional groups to the polymer chain before scaffold fabrication and canbe achieved through physical [15], chemical [4, 16, 17], photochemical [18] or ioniccross-linking [19]. Thus, recognition sites obtained by bulk modification are presentnot only on the surface but also in the bulk of the materials. Such modificationwith enzymatically degradable sequences can render biomaterials biodegradable byspecific proteases [20, 21]. On the other hand, bulk modification may affect the bi-ological activity of immobilised molecules due to their reduced mobility within thematerial [22]. It is more difficult to control the density of the ligand accessible forcells in bulk-modified materials than in surface-modified materials [23]. The rela-tionship between the bulk ligand concentration and the active ligand concentrationat the surface is dependent on the hydrogel composition [24].
Surface modification is carried out after scaffold fabrication and is usuallyachieved by covalent binding of peptides or proteins on biomaterial surfaces usingdifferent coupling techniques [14]. A bi-functional cross-linker that has a spacerarm can be used for the immobilisation of peptides in order to ensure their flex-ibility [25]. The type or level of modification which is optimal for cell–surfaceinteraction varies depending on the specific cell types and intrinsic properties ofbiomaterials [14].

I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795 1783
Synthetic MHs have been modified and rendered bioactive by including ana-logues of cell-adhesive peptides during production of MHs, coupling proteins toreactive groups present on MHs either directly or via a spacer [26] and by graftingmacromolecules to MHs surface followed by derivatisation of the grafted polymerwith required functionality [6, 27, 28]. Despite the possibility of using a wide rangeof techniques for derivatisation of MH scaffolds, the dependence of conformation,density and distribution of the bioligand in bulk- and surface-modified MHs on themodification method has not been studied previously. Since cellular response is de-pendent on the density of the ligand [29], the spatial distribution of ligand [30] andits biological activity [31], considering these parameters is important for the designof biomimetic MH scaffolds.
In the present study biomimetic poly(2-hydroxyethyl methacrylate) (pHEMA)MHs modified by embedding protein during cryo-polymerisation and covalent im-mobilisation of two different proteins, bovine type-I collagen (CG) and fibrinogen(Fg) from human plasma, were studied in terms of protein spatial organisation andsurface density and their effect on cultured human fibroblasts. Previously, it wasdemonstrated that pHEMA MHs prepared by cross-linking polymerisation in thesemi-frozen state, are elastic sponge-like materials which have a unique structureof large interconnected pores with pore sizes up to 100 µm and total porosity of 94–97% [6]. pHEMA is inherently non-adhesive to cells, yet it can be rendered adhesiveby modification with peptides or proteins [32–34]. pHEMA hydrogels have foundwide application in medicine [35], in particular for production of contact lenses andartificial cornea [36, 37]. Here, the distribution of two different proteins, fibrinogenand collagen, introduced into pHEMA MHs by two different methods and responseof fibroblasts to the ligand architecture in the MHs has been studied. The potentialof pHEMA MHs subjected to surface and bulk modifications as scaffolds for tissueengineering, such as wound healing, has been evaluated.
2. Materials and Methods
2.1. Materials
N,N,N′,N′-Tetramethylethylenediamine (TEMED, 99%), bicinchoninic acid (BCA)protein assay kit, fluorescein isothiocyanate (FITC), bovine dermal type-I colla-gen (collagen from calf skin), fibrinogen from human plasma, monoclonal mouseantibody against bovine type-I collagen, monoclonal mouse antibody against hu-man fibrinogen, anti-mouse IgG (Fc-specific)-FITC antibody produced in goat anda 2,3-bis[2-methoxy-4-nitro-5-sulfophenyl]-2H-tetrazolium-5-carboxyanilide innersalt (XTT) based + 1% phenazine methosulfate (PMS) in vitro toxicology assay kitwere purchased from Sigma (St. Louis, MO, USA). Poly(ethylene glycol)diacrylate(PEGA, Mn approx. 258), 2-hydroxyethyl methacrylate (HEMA, 98%), allyl gly-cidyl ether (AGE, 99%), dinitrosalicylic acid (DNS) and ammonium persulfate(APS, 98%) were obtained from Aldrich (Steinheim, Germany). Dulbecco’s mod-ified Eagle’s medium (DMEM) with phenol red, glucose, glutamax and pyruvate,

1784 I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795
and DMEM without pyruvate and phenol red, Foetal Bovine Serum (FBS), trypsin-EDTA and kanamycine were obtained from Gibco-Invitrogen (Grand Island, NY,USA).
Human embryonic fibroblasts were provided by the Department of BiologicalSciences and Bioengineering, Indian Institute of Technology (Kanpur, India) as akind gift.
2.2. Preparation of Macroporous Hydrogels
The composition of the reaction mixtures used for production of plain (non-modified), epoxy-containing, Fg/pHEMA and CG/pHEMA MHs is presented inTable 1. For Fg/pHEMA and CG/pHEMA MHs preparation, collagen and fibrino-gen (0.5 mg/ml) were dissolved in water before the monomers were added. Thepreparation of MHs was carried out as follows. Each reaction mixture was de-gassed in vacuo for 10 min to eliminate dissolved oxygen and cooled on an icebath for 10 min. Free radical polymerisation was initiated by adding TEMED andAPS (1.0% each of the total co-monomers weight) and the mixture was stirredfor 10–15 s. Then 0.25 ml of the solution was quickly added into each glass tube(20 × 7 mm i.d.) closed at the bottom with a silicon cap. The solution in the tubeswas frozen within 20–30 min in the ARCTEST cooling chamber at −12◦C, incu-bated at that temperature overnight and thawed at room temperature. Caps wereremoved and MHs were washed thoroughly with deionised water to remove un-reacted monomers, initiator and cross-linker. Epoxy groups content was determinedby immobilising iminodiacetic acid to epoxy groups, equilibrating MHs with 0.2 M
Table 1.Characteristics of HEMA cryogels
Sample Composition for gel Swelling degree Volume of Protein concentrationpreparation ([Monomers]a/ (g H2O/g dried macro- by BCA assay[HEMA]:[PEGA]b/[AGE]: polymerd) pores (%)e (µg/ml hydrogel)[HEMA]b/proteinc)
HEMA 8/6:1/1:0/0 17 ± 0.5 89.0 ± 0.8 0epHEMA 8/6:1/1:5/0 16 ± 1.0 85.0 ± 1.0 0CG/HEMA 8/6:1/1:0/0.5 18 ± 0.8 87.9 ± 2.0 80–85Fg/HEMA 8/6:1/1:0/0.5 16 ± 1.0 88.7 ± 0.9 80–85CG–epHEMA 8/6:1/1:5/0 16 ± 1.0 85.0 ± 1.0 130–140Fg–epHEMA 8/6:1/1:5/0 16 ± 1.0 85.0 ± 1.0 130–140
a Total concentration of HEMA + PEGA + AGE in % (w/v).b Monomer concentration in mol%.c Protein concentration in mg/ml.d Swelling degree was estimated as ((mswollen gel − mdried gel)/mdried gel).e The volume of macropores in the swollen cryogel was roughly estimated by squeezing the free
water from the swollen gel matrix as follows: ((mswollen gel − msqueezed gel)/mswollen gel) × 100%.

I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795 1785
CuSO4 and assessing the amount of bound Cu(II) as described elsewhere [38]. Theepoxy group content for epHEMA MH was 17 µmol epoxy group/ml gel.
2.3. Cryogel Characterisation
The total volume of macropores in the swollen cryogel was roughly estimated asfollows: the weight of the sample (msqueezed gel) was determined after squeezingthe free water from the swollen gel matrix, and the porosity was calculated asfollows: ((mswollen gel − msqueezed gel)/mswollen gel) × 100% [39]. The measurementswere done in triplicate and average value was shown. The swelling degree was esti-mated as amount of water in swollen cryogel per gram dried polymer. The MHs wasdried in oven at 60◦C until constant weight. The swelling capacity was calculatedas follows: ((mswollen gel − mdried gel)/mdried gel) × 100%.
2.4. Surface Modification via Protein Coupling to Epoxy-Containing MHs
Epoxy-containing MHs were placed into a 96-minicolumn well plate (Nunc,Roskilde, Denmark), a microtitre plate with open drop-forming units at the bot-tom of each well. The pH of collagen and fibrinogen solutions (1 mg/ml in 10 mMacetic acid and deionised water, respectively) was adjusted with 0.1 M NaOH to8.5–9.0. Epoxy–pHEMA MHs were equilibrated with a protein solution by passing0.5 ml of the solution through each well and incubated for 24 h at room temper-ature. Non-bound proteins were removed by washing each MH with 2 ml sterilephosphate-buffered saline (PBS, pH 7.4; 0.26 g KH2PO4,1.44 g Na2HPO4 · 2H2Oand 8.71 g NaCl per litre).
2.5. Determination of Protein Content in Modified MHs
The amount of protein incorporated into MHs was determined by the bicinchoninicacid (BCA) method [40]. An approach optimized by Kumar et al. for direct deter-mination of protein on the cryogel was used [41].
2.6. FITC Labelling of MHs
Collagen and fibrinogen in the CG–epHEMA, CG/HEMA, Fg–epHEMA andFg/HEMA were labelled by covalently linking a fluorescent marker, fluoresceineisothiocyanate (FITC). Labelling was achieved by the following procedure: MHsdiscs (0.25 ml) were washed with 0.1 M sodium phosphate buffer (pH 9.0), and20 µl FITC solution in DMSO (1 mg/ml) was added to each disk in 1 ml of sodiumphosphate buffer. The samples were incubated for 2 days and thoroughly washedwith water. A sheet of 1 mm thickness was cut from each MH and examinedusing confocal laser scanning microscopy (CLSM, Leica TCS SP5, Leica, Wet-zlar, Germany). pHEMA and epHEMA MHs without proteins were modified withFITC using the same method and used as control. Prior to modification with FITC,epHEMA MH was incubated in a buffer solution (pH 8.5–9.0) for 24 h, to hydrolysethe epoxy groups in a manner similar to modification with proteins. Control sam-ples demonstrated that there was some non-specific FITC sorption to the polymer

1786 I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795
matrix, which resulted in a staining of the hydrogel matrix as well. However, thisstaining has low intensity and could be eliminated by decreasing the intensity of theemitted light. All CLSM images were adjusted to represent the protein distributionrather than the structure of hydrogel matrix.
2.7. Immunostaining of MHs
Hydrated CG–epHEMA, CG/pHEMA, Fg–epHEMA and Fg/HEMA plugs(0.25 ml) were cut with a blade into 1–2-mm-thick discs. Two or three disks wereused for modification. Samples were thoroughly washed with deionised water, equi-librated with 10 mM PBS (pH 7.4) and gently dried with a cotton pad to removePBS from the macropores. The discs were equilibrated and incubated with primaryIgG (monoclonal mouse anti-collagen and anti-fibrinogen antibodies, dilution 1:50in PBS) for 3–4 h with gentle shaking at room temperature. Then the discs wererepeatedly washed with PBS to remove unbound IgG and gently dried with a cottonpad. Secondary FITC-labelled IgG solution (anti-mouse IgG (Fc-specific)-FITCantibody produced in goat, dilution 1:25 in PBS) was added to the discs. The sam-ples were incubated overnight at room temperature, washed with PBS to removeunbound FITC-labelled IgG and examined with CLSM.
2.8. Confocal Laser Scanning Microscopy
The samples were examined with a CLSM using a regular 20× objective. Confocalmicroscopy was carried out as follows. A disc of approx. 1 mm in height was cutfrom the wet MHs. The excitation and emission wavelengths were 488 and 530 nm,respectively. All images were generated by optical sectioning in the z-direction. Forthis purpose, 80 optical sections were taken along with a z-distance of 80 µm.
2.9. Maintenance of Cell Cultures
Human embryonic fibroblasts were maintained in DMEM media, supplementedwith 10% foetal bovine serum and 0.1% kanamycine monosulfate at 37◦C in thepresence of 5% CO2 in a humidified incubator. Cells were trypsinised (0.25%trypsin-EDTA) from tissue-culture plates, washed and split as they approached 80%confluence every 2–3 days.
2.10. Culturing of Cells on MHs
Cell seeding and culturing in MHs in a 96-minicolumn plate format are describedin details in [4]. Briefly, MHs were inserted into the wells of a 96-minicolumn plateand sterilised by incubation with 70% ethanol for 2 h. After washing with sterilewater to remove ethanol, MHs were equilibrated and incubated with the medium forat least 12 h prior to cell seeding. Cell seeding was performed by applying 0.3 mlcell suspension (2 × 105 cells/ml) to the top of each MH (0.25 ml). The cultureplate was aligned on top of a 96-well collector plate, closed with a lid and placedin a 95% air/5% CO2 incubator at 37◦C. Growth medium was changed by applying0.3 ml fresh medium to each MH every 24 h. The medium displaced from MHs wascollected in the collector plate and analysed by dinitrosalicylic acid glucose assay.

I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795 1787
2.11. Measurement of Glucose Consumption Rate
The sample (0.07 ml) was mixed with 0.1% 3,5-dinitrosalicylic acid in 0.05 MNaOH containing 4% potassium sodium tartrate in a capped test tube. Total volumeof the reaction mixture was 1.5 ml. The mixture was incubated at 95◦C for 5 min todevelop a red-brown colour. After cooling to room temperature on an ice bath, theabsorbance was recorded at 540 nm.
Glucose consumption was calculated as:
([glucosestart] − [glucose24 h]) × V,
where [glucosestart] is the initial concentration of glucose in the growth medium (thevalue given in the data sheet), [glucose24 h] is the concentration of glucose in theeffluent (used medium) displaced from MHs during medium exchange procedureand V is the volume of the effluent.
2.12. Scanning Electron Microscopy
MHs were fixed in 2.5% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.2)overnight, and post-fixed in 1% osmium tetroxide for 1 h, dehydrated in ethanol(30, 50, 75 and 99.5%) and critical-point-dried. The dried samples were coatedwith gold/palladium (40:60) and examined using a Jeol JSM-5600LV scanning mi-croscope.
3. Results and Discussion
3.1. Biomimetic MHs Preparation, Ligand Distribution
pHEMA MH scaffolds were prepared using a cryogelation technique which pro-vides preparation of highly porous gels with large interconnected pores and sponge-like morphology [6, 9]. Synthetic scaffolds are rendered bioactive by modificationwith extracellular matrix ligands. Collagen and fibrinogen are the extracellular ma-trix proteins which are widely used for improving cell adhesion and proliferationon the polymer surfaces [3, 43, 44]. These two proteins were used for biocoatingof MHs using two different strategies, i.e., surface and bulk modification. Sur-face modification involved preparation of MHs containing epoxy groups (epHEMAMH), which were then modified with the proteins. Thus, the proteins were cova-lently immobilised on the surface of MHs via interaction with epoxy groups presentin the matrix (CG–epHEMA and Fg–epHEMA MHs) after scaffold fabrication.Bulk modification involved adding the proteins to the reaction mixture before hy-drogel formation. Thus, collagen and fibrinogen were incorporated in the polymermatrix directly during the polymerisation, forming the pore walls together withHEMA polymer (CG/pHEMA and Fg/pHEMA MHs, respectively).
Epoxy group-containing pHEMA MHs had a pore size of 5–100 µm (Fig. 1).Fg–pHEMA and CG–pHEMA had similar structure (the images are not presented).The total volume of the macropores was 85–89% (Table 1). The presence of pro-tein in the reaction media did not affect the porous structure of CG/pHEMA and

1788 I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795
Figure 1. Scanning electron microscopy images of cross-section of epHEMA MH.
Fg/pHEMA formed (Table 1). The swelling degree of pHEMA MHs was 16–18 gH2O/g dried polymer (Table 1). The amount of protein in MHs obtained by di-rect monomer cryo-co-polymerisation with the protein present in solution and inpHEMA MHs containing proteins coupled via epoxy groups was 80–85 and 130–140 µg/ml hydrated cryogel, respectively (Table 1). It is noteworthy that MHs usedfor protein assay were thoroughly washed with ethanol and water prior to the analy-sis in the same way as those MHs which were used in cell-culture experiments.
Spatial distribution of proteins in FITC-labelled MHs was studied using CLSM.The protein distribution during surface modification could depend on the reactiv-ity and distribution of the functional (epoxy) groups available for modification, aswell as on protein solubility in the reaction medium and its interaction with thefunctional groups. As the same epoxy-containing matrix has been used for mod-ification with two different proteins, the main factor leading to the difference inprotein conformation and density on the pore wall surface of epHEMA MHs is,therefore, the protein solubility in the reaction medium and its interaction with thereactive groups. In bulk modification protein distribution depends on the miscibility(compatibility) of protein with a monomer/polymer solution. One can expect that,when introduced during the gel formation, the protein will build up the gel wallstogether with the polymer. Such incorporation could take place partially due to in-volvement of proteins in chain termination during free radical co-polymerisationand by physical entrapment during the formation of polymeric network.
Spatial distribution of collagen and fibrinogen in MH matrix varied in differenttypes of MHs depending on the nature of the protein and the method used for gelmodification (Fig. 2). Collagen attached to pHEMA MH surface through epoxygroups had a spot-like discrete distribution in the hydrogel (Fig. 2a), whereas fib-rinogen was spread out evenly on the MH surface (Fig. 2b). In contrast, when theproteins were incorporated in the MHs by bulk modification, collagen was evenlydistributed in the MHs matrix (Fig. 2c), whereas fibrinogen appeared in the form ofseparated spots or aggregates distributed along the gel matrix (Fig. 2d).

I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795 1789
Figure 2. CLSM images of spatial distribution of collagen and fibrinogen on the surface-modified(a, CG–epHEMA; b, Fg–epHEMA) and bulk-modified (c, CG/pHEMA; d, Fg/pHEMA) MHs. Thesamples were labelled with FITC (see Materials and Methods). This figure is published in colour inthe online edition that can be accessed via http://www.brill.nl/jbs
Such a different behaviour of collagen and fibrinogen could be caused by a com-bination of the following factors: solubility of the protein in the reaction mediumand its interaction with the surface in the case of surface modification and by solu-bility/compatibility of proteins with HEMA solution, when protein was introducedby bulk modification. Human plasma fibrinogen (340 kDa) is a very hydrophilicprotein which consists of three different chains arranged in a dimer, (Aα, Bβ , γ )2,and has a pI of 5.8 [45–47]. The electrical charge and hydrophobicity of bioma-terial surfaces influence fibrinogen structure. Fibrinogen molecules spread out onhydrophobic and positively charged surfaces, as opposed to hydrophilic and nega-tively charged surfaces [48].
The fundamental structural unit of collagen is a long (300 nm) and thin (1.5 nmdiameter) molecule that consists of three coiled subunits. The isoelectric point ofcollagen is near 10 [49]. Thus, basic conditions (pH 9.0) used for surface modifi-cation of epHEMA MHs enhanced solubility of fibrinogen, while collagen, whichhas poor solubility in aqueous solutions, especially at high pH, was partially in theaggregated form. This resulted in spot-like distribution of collagen aggregates onthe surface, while fibrinogen was completely solubilised at pH 9.0, spread out onepHEMA surface evenly without forming aggregates.
On the other hand, during bulk modification the behaviour of protein in the re-action medium could be different due to differences in compatibility of proteins

1790 I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795
with HEMA monomer and HEMA polymer formed. Apparently, compatibility ofcollagen with HEMA was better and it was uniformly distributed in the polymersolution building into polymer walls of MHs (Fig. 2c). The spot-like distributionof fibrinogen molecules in the bulk of the MHs matrix could be a result of poorcompatibility of fibrinogen with HEMA solution (Fig. 2d). Moreover, the pH ofthe reaction mixture used for cryo-polymerisation was close to or lower than thepI of fibrinogen, decreasing its solubility. Thus, fibrinogen was embedded in anaggregated form during MH production.
3.2. Immunostaining of MHs
The biomolecules should retain biological activity and be accessible to interactionswith cells. To maintain their activity, immobilised proteins should be flexible, ex-perience minimal steric hindrance and have intact recognition sites available forinteraction with cells. It is essential to consider the surface chemistry of syntheticmaterials in relation to the biological function of the attached biomimetic ligand.The mobility and accessibility of the ligand strongly influence the cell attachmentand proliferation [31].
Immunostaining with monoclonal anti-collagen and anti-fibrinogen antibodieswas used to indentify the native form of collagen and fibrinogen on the sur-face of MHs. After labelling with monoclonal anti-collagen and monoclonal anti-fibrinogen antibodies, MHs were labelled with secondary antibodies conjugatedwith FITC. Unlike staining with FITC, a small molecule (389 Da) which interactswith nucleophilic functional groups of protein molecules immobilised both on thesurface and in the bulk of MH, regardless of their conformation, immunostainingtargets only surface bound proteins which retain the native conformation. MHs pro-duced by cryotropic polymerization have thick, non-porous polymer walls [39] andsuch large molecules as IgG (150 kDa) cannot access protein molecules distributedin polymer matrix of the pore walls.
The pattern of protein distribution in modified MHs revealed by immunostainingwas similar to the one provided by staining with FITC, demonstrating that proteinsretained native binding sites after modification. The spot-like distribution of colla-gen in CG–epHEMA and fibrinogen in Fg/pHEMA (Fig. 3a and 3d) and more evenspreading of the proteins along the polymer matrix on the surface in Fg–epHEMAand CG–pHEMA (Fig. 3b and 3c) was observed. However, not all of the protein in-troduced in MH by bulk modification and detected by FITC staining was observedby CLSM after immunostaining (Figs 2 and 3). Most of the protein was ‘buried’in the polymer matrix and was not accessible for interaction with the antibodies.From the immunostaining images, the amount of fibrinogen accessible on the sur-face appeared to be larger than that of collagen. Fibrinogen introduced in MHs bysurface modification was evenly distributed on the polymer surface and retained itsnative structure with most of the sites available for interactions with the antibodies(Fig. 3b).
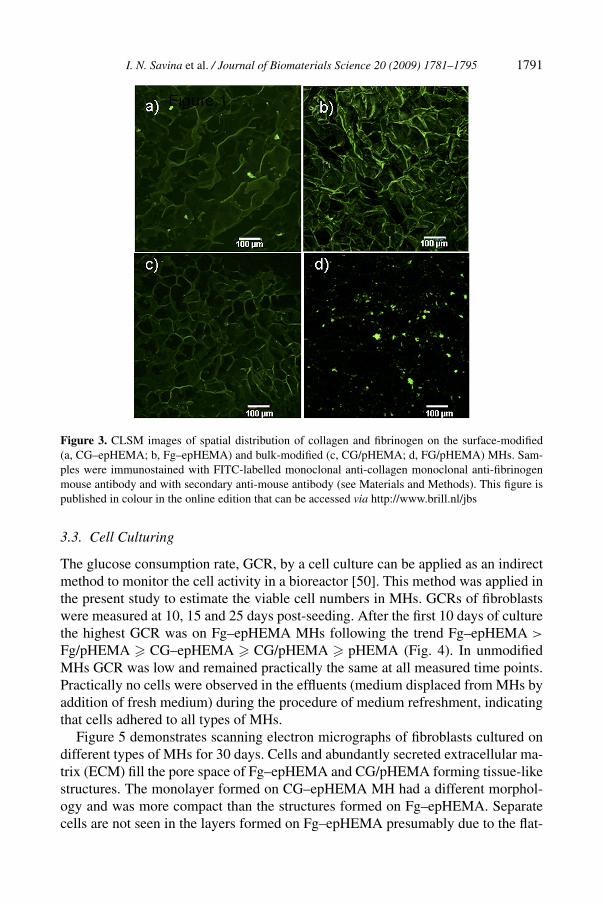
I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795 1791
Figure 3. CLSM images of spatial distribution of collagen and fibrinogen on the surface-modified(a, CG–epHEMA; b, Fg–epHEMA) and bulk-modified (c, CG/pHEMA; d, FG/pHEMA) MHs. Sam-ples were immunostained with FITC-labelled monoclonal anti-collagen monoclonal anti-fibrinogenmouse antibody and with secondary anti-mouse antibody (see Materials and Methods). This figure ispublished in colour in the online edition that can be accessed via http://www.brill.nl/jbs
3.3. Cell Culturing
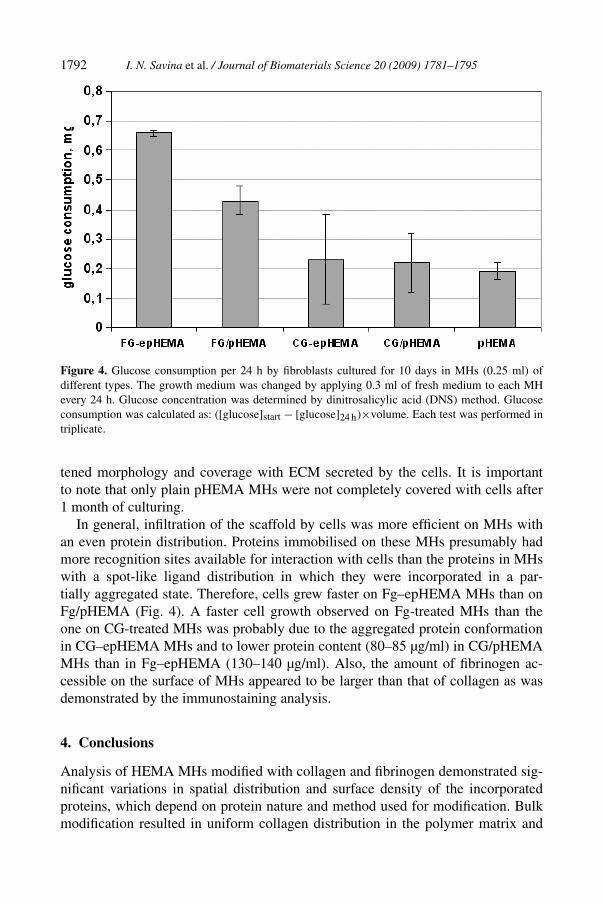
The glucose consumption rate, GCR, by a cell culture can be applied as an indirectmethod to monitor the cell activity in a bioreactor [50]. This method was applied inthe present study to estimate the viable cell numbers in MHs. GCRs of fibroblastswere measured at 10, 15 and 25 days post-seeding. After the first 10 days of culturethe highest GCR was on Fg–epHEMA MHs following the trend Fg–epHEMA >
Fg/pHEMA � CG–epHEMA � CG/pHEMA � pHEMA (Fig. 4). In unmodifiedMHs GCR was low and remained practically the same at all measured time points.Practically no cells were observed in the effluents (medium displaced from MHs byaddition of fresh medium) during the procedure of medium refreshment, indicatingthat cells adhered to all types of MHs.
Figure 5 demonstrates scanning electron micrographs of fibroblasts cultured ondifferent types of MHs for 30 days. Cells and abundantly secreted extracellular ma-trix (ECM) fill the pore space of Fg–epHEMA and CG/pHEMA forming tissue-likestructures. The monolayer formed on CG–epHEMA MH had a different morphol-ogy and was more compact than the structures formed on Fg–epHEMA. Separatecells are not seen in the layers formed on Fg–epHEMA presumably due to the flat-
1792 I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795
Figure 4. Glucose consumption per 24 h by fibroblasts cultured for 10 days in MHs (0.25 ml) ofdifferent types. The growth medium was changed by applying 0.3 ml of fresh medium to each MHevery 24 h. Glucose concentration was determined by dinitrosalicylic acid (DNS) method. Glucoseconsumption was calculated as: ([glucose]start − [glucose]24 h)×volume. Each test was performed intriplicate.
tened morphology and coverage with ECM secreted by the cells. It is importantto note that only plain pHEMA MHs were not completely covered with cells after1 month of culturing.
In general, infiltration of the scaffold by cells was more efficient on MHs withan even protein distribution. Proteins immobilised on these MHs presumably hadmore recognition sites available for interaction with cells than the proteins in MHswith a spot-like ligand distribution in which they were incorporated in a par-tially aggregated state. Therefore, cells grew faster on Fg–epHEMA MHs than onFg/pHEMA (Fig. 4). A faster cell growth observed on Fg-treated MHs than theone on CG-treated MHs was probably due to the aggregated protein conformationin CG–epHEMA MHs and to lower protein content (80–85 µg/ml) in CG/pHEMAMHs than in Fg–epHEMA (130–140 µg/ml). Also, the amount of fibrinogen ac-cessible on the surface of MHs appeared to be larger than that of collagen as wasdemonstrated by the immunostaining analysis.
4. Conclusions
Analysis of HEMA MHs modified with collagen and fibrinogen demonstrated sig-nificant variations in spatial distribution and surface density of the incorporatedproteins, which depend on protein nature and method used for modification. Bulkmodification resulted in uniform collagen distribution in the polymer matrix and

I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795 1793
Figure 5. Scanning electron micrographs of fibroblasts cultured for 30 days on surface-modified(a, CG–epHEMA; b, Fg–epHEMA), bulk-modified (c, CG/pHEMA; d, Fg/pHEMA) and plain (e)MHs.
spot-like distribution of fibrinogen. In contrast, surface modification resulted inspot-like distribution of collagen and spreading out of fibrinogen, which evenlycoated the surface. Surface-modified MHs had more protein present on the surfaceand available for interaction with cells than in case of bulk modification of MHs, inwhich most of the immobilised protein was distributed in the polymer matrix andinaccessible for cell interaction. Surface modification with fibrinogen produced aneven coating of the surface with the protein retaining its natural conformation. Itexplains the best performance of Fg–epHEMA MHs in cell culturing.

1794 I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795
The coupling chemistry is also an important parameter in designing biomimeticMHs as it has major effect on the spatial distribution and density of the ligand inthe polymer matrix, which, in turn, influence cell–scaffold interactions.
Acknowledgements
This work was financially supported by Protista Biotechnology AB (Bjuv, Sweden),Swedish Research Council (project 621-2007-36-24), FP6 project MTKI-CT-2006-042768-MATISS, FP7 project (PIAP-GA-2008-218242) and the Royal Society (In-ternational Incoming Short Visit 2007/R3).
References
1. E. Cukierman, R. Pankov, D. R. Stevens and K. M. Yamada, Science 294, 1708 (2001).2. R. Langer, Mol. Ther. 1, 12 (2000).3. L. L. Hench and J. M. Polak, Science 295, 1014 (2002).4. M. B. Dainiak, I. Savina, I. Musolino, A. Kumar, B. Mattiasson and I. Galaev, Biotechnol. Progr.,
in press (2008).5. I. Bloch, V. I. Lozinsky, I. Y. Galaev, K. Yavriyanz, M. Vorobeychik, D. Azarov, L. G. Damshkaln,
B. Mattiasson and P. Vardi, J. Biomed. Mater. Res. 75A, 802 (2005).6. I. N. Savina, V. Cnudde, S. D’Hollander, L. VanHoorebeke, B. Mattiasson, I. Y. Galaev and F. Du
Prez, Soft Matter 3, 1176 (2007).7. M. B. Dainiak, A. Kumar, I. Y. Galaev and B. Mattiasson, Proc. Natl. Acad. Sci. USA 103, 849
(2006).8. I. Y. Galaev, M. B. Dainiak, F. M. Plieva and B. Mattiasson, Langmuir 23, 35 (2007).9. V. I. Lozinsky, I. Y. Galaev, F. M. Plieva, I. N. Savina, H. Jungvid and B. Mattiasson, Trends
Biotechnol. 21, 445 (2003).10. M. B. Dainiak, I. Y. Galaev, A. Kumar, F. M. Plieva and B. Mattiasson, Adv. Biochem. Engin.
Biotechnol. 106, 101 (2007).11. M. B. Dainiak, B. Mattiasson and I. Galaev, BIOforum Eur. 12, 18 (2007).12. P. Dubruel, R. Unger, S. VanVlierberghe, V. Cnudde, P. J. S. Jacobs, E. Schacht and C. J. Kirk-
patrick, Biomacromolecules 8, 338 (2007).13. P. X. Ma, Mater. Today 7, 30 (2004).14. H. Shin, S. Jo and A. G. Mikos, Biomaterials 24, 4353 (2003).15. R. A. Stile and K. E. Healy, Biomacromolecules 2, 185 (2001).16. D. L. Elbert and J. A. Hubbell, Biomacromolecules 2, 430 (2001).17. J. S. Mao, H. F. Liu, Y. J. Yin and K. D. Yao, Biomaterials 24, 1621 (2003).18. M. Borkenhagen, J.-F. Clemence, H. Sigrist and P. Aebischer, J. Biomed. Mater. Res. 40, 392
(1999).19. J. A. Rowley, G. Madlambayan and D. J. Mooney, Biomaterials 20, 45 (1999).20. J. L. West and J. A. Hubbell, Macromolecules 32, 241 (1999).21. S. Halstenberg, A. Panitch, S. Rizzi, H. Hall and J. A. Hubbell, Biomacromolecules 3, 710 (2002).22. D. L. Hern and J. A. Hubbell, J. Biomed. Mater. Res. 39, 266 (1998).23. A. D. Cook, J. S. Hrkach, N. N. Gao, I. M. Johnson, U. B. Pajvani, S. M. Cannizzaro and
R. Langer, J. Biomed. Mater. Res. 35, 513 (1997).24. E. Behravesh, V. I. Sikavitsas and A. G. Mikos, Biomaterials 24, 4365 (2003).

I. N. Savina et al. / Journal of Biomaterials Science 20 (2009) 1781–1795 1795
25. A. Rezania, R. Johnson, A. R. Lefkow and K. E. Healy, Langmuir 15, 6931 (1999).26. A. Kumar, V. Bansal, K. S. Nandakumar, I. Y. Galaev, P. K. Roychoudhury, R. Holmdahl and
B. Mattiasson, Biotechnol. Bioeng. 93, 636 (2006).27. I. N. Savina, B. Mattiasson and I. Y. Galaev, Polymer 46, 9596 (2005).28. I. N. Savina, B. Mattiasson and I. Y. Galaev, J. Polymer Sci. Part A: Polym. Chem. 44, 1952 (2006).29. S. P. Palecek, J. C. Loftus, M. H. Ginsberg, D. A. Lauffenburger and A. F. Horwitz, Nature 385,
537 (1997).30. S. P. Massia and J. A. Hubbell, J. Cell Biol. 114, 1089 (1991).31. D. Thid, M. Bally, K. Holm, S. Chessari, S. Tosatti, M. Textor and J. Gold, Langmuir 23, 11693
(2007).32. T. T. Yu and M. S. Shoichet, Biomaterials 26, 1507 (2005).33. M. P. Lutolf, F. E. Weber, H. G. Schmoekel, J. C. Schense, T. Kohler, R. Muller and J. A. Hubbell,
Nature Biotechnol. 21, 513 (2003).34. L. Aucoin, C. M. Griffith, G. Pleizier, Y. Deslandes and H. Sheardown, J. Biomater. Sci. Polymer
Edn 13, 447 (2002).35. A. S. Hoffman, Adv. Drug Deliv. Rev. 54, 3 (2002).36. P. C. Nicolson and J. Vogt, Biomaterials 22, 3273 (2001).37. C. R. Hicks, A. B. Clayton, S. Vijayasekaran, G. J. Crawford, T. V. Chirila and I. J. Constable,
Ophthalmol. Plast. Reconstr. Surg. 15, 326 (1999).38. P. Arvidsson, F. M. Plieva, V. I. Lozinsky, I. Y. Galaev and B. Mattiasson, J. Chromatogr. A 986,
275 (2003).39. F. M. Plieva, M. Karlsson, M.-R. Aguilar, D. Gomez, S. Mikhalovsky and I. Y. Galaev, Soft Matter
1, 303 (2005).40. P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano,
E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem. 150, 76 (1985).41. A. Kumar, F. M. Plieva, I. Y. Galaev and B. Mattiasson, J. Immunol. Methods 283, 185 (2003).42. M. B. Dainiak, I. Y. Galaev and B. Mattiasson, Enzyme Microb. Technol. 40, 688 (2007).43. L. G. Griffith and G. Naughton, Science 295, 1009 (2002).44. B.-S. Kim and D. J. Mooney, Trends Biotechnol. 16, 224 (1998).45. A. Henschen, F. Lottspeich, M. Kehl and C. Southan, Ann. N.Y. Acad. Sci. 408, 28 (1998).46. J. L. Ortega-Vinuesa, P. Tengvall and I. Lundström, Thin Solid Films 324, 257 (1998).47. C. J. van Oss, J. Protein Chem. 9, 487 (1990).48. C. Fuss, J. C. Palmaz and E. A. Sprague, J. Vasc. Interv. Radiol. 12, 677 (2001).49. A. L. Andrade, J. M. F. Ferreira and R. Z. Domingues, Mater. Res. 7, 631 (2004).50. F. Meuwly, F. Papp, P.-A. Ruffieux, A. R. Bernard, A. Kadouri and U. von Stockar, J. Biotechnol.
122, 122 (2006).