bioinformatics review …“.jónsdóttir et al. fig. 1. number of papers in pubmed 1992–2004...
TRANSCRIPT

BIOINFORMATICS Review Vol 21 no 10 2005 pages 2145ndash2160doi101093bioinformaticsbti314
Structural bioinformatics
Prediction methods and databases within chemoinformaticsemphasis on drugs and drug candidatesSvava Oacutesk Joacutensdoacutettir1 Flemming Steen Joslashrgensen2 and Soslashren Brunak1lowast1Center for Biological Sequence Analysis BioCentrum-DTU Technical University of Denmark DK-2800 KongensLyngby Denmark and 2Department of Medicinal Chemistry Danish University of Pharmaceutical SciencesUniversitetsparken 2 DK-2100 Copenhagen Denmark
Received on February 11 2004 revised on February 4 2005 accepted on February 7 2005
Advance Access publication February 15 2005
ABSTRACTMotivation To gather information about available databases andchemoinformatics methods for prediction of properties relevant to thedrug discovery and optimization processResults We present an overview of the most important databaseswith 2-dimensional and 3-dimensional structural information aboutdrugs and drug candidates and of databases with relevant propertiesAccess to experimental data and numerical methods for selecting andutilizing these data is crucial for developing accurate predictive in silicomodels Many interesting predictive methods for classifying the suitab-ility of chemical compounds as potential drugs as well as for predictingtheir physico-chemical and ADMET properties have been proposed inrecent years These methods are discussed and some possible futuredirections in this rapidly developing field are describedContact svavacbsdtudk
INTRODUCTIONChemoinformatics (Blake 2000 Olsson and Oprea 2001 Kubinyi2003) is a rapidly growing field with a huge application poten-tial Chemoinformatics concerns the gathering and systematic useof chemical information and the use of those data to predict thebehavior of unknown compounds in silico
The word lsquochemoinformaticsrsquo is rather new but papers that fallunder this field date back to the mid-1960s where structurendashactivityrelationships (SAR) were proposed based on the work of Hanschand Fujita (1964) and Fujita et al (1964) and the earlier work ofHammett and Taft The first textbooks on chemoinformics have beenpublished recently (Leach and Gillet 2003 Gasteiger and Engel2003 Gasteiger 2003 Bajorath 2004)
The related field bioinformatics (Durbin et al 1998 Baldi andBrunak 2001 Mount 2001 Orengo et al 2002 Lengauer 2002)was fully established in the 1990s and has become an integratedactivity in most major pharmaceutical companies The basis behindthe success of bioinformatics was the access to a vast amount ofexperimental data together with the structured nature of geneticinformation Several authors have recently published reviews onthe use of bio- and chemoinformatics methods in drug discoveryprocesses (Stahura and Bajorath 2002 Hopkins and Groom 2002Golden 2003)
lowastTo whom correspondence should be addressed
The availability of experimental data relevant to chemoinformat-ics modeling is however much more restricted In fact most ofthe chemical information reside in the private domain If the suc-cess of the bioinformatics area should be mimicked the availabilityof new experimental data will be an absolute necessity for develop-ing efficient and robust models for prediction of various properties(Beresford et al 2002)
Implementing handling and searching chemical databases is acrucial aspect of chemoinformatics (Miller 2002) Chemical data-base techniques and data mining methods will improve as this fieldevolves also due to more implementation of new data structures(Miled et al 2003) Methods for full text data mining are likely tobe become very powerful in the years to come and will presumablyplay a highly important role in the general area of chemoinformaticsA new XML (extensible markup language) based approach for man-aging molecular information lsquochemical markup languagersquo (CML)was proposed by Murray-Rust and Rzespa (1999 httpwwwxml-cmlorg) Figure 1 shows the number of references to the wordslsquobioinformaticsrsquo lsquochemoinformaticsrsquo lsquochemogenomicsrsquo and lsquometa-bonomicsrsquo in PubMed from 1992 to 2004 It is seen that the presenttrend for chemoinformatics resembles the trend in bioinformaticsfive to ten years ago It should be mentioned that this graph is basedon one database only PubMed and is intended to give an idea aboutthe development in publishing frequency in these areas and not as acomplete overview
It is clear that the drug discovery and optimization process isundergoing very significant changes Many more hits are found thanpreviously especially due to the advances gained in combinator-ial chemistry (Gallop et al 1994 Gordon et al 1994) and highthroughput screening (HTS) The approach used in drug discoveryhas been linear with respect to various relevant properties but moreparallel approaches are evolving where not only the potency (activ-ity) and selectivity of the lead is examined at an early stage butalso other key properties Many of the compounds drawn out ofcombinatorial libraries may look promising at first but they fail atlater stages in the drug discovery process due to undesired proper-ties A compound can for example be feasible based on molecularstructure but due to aggregation limited solubility or limited uptakein the human organism it is not useful as a drug Many pharma-ceutical companies might even be repeating the same mistakesdue to these problems Methods for assessing these properties ata very early stage both experimentally and computationally are
copy The Author 2005 Published by Oxford University Press All rights reserved For Permissions please email journalspermissionsoupjournalsorg 2145
SOacuteJoacutensdoacutettir et al
Fig 1 Number of papers in PubMed 1992ndash2004 containing thekeywords lsquobioinformaticsrsquo (top left) lsquochemoinformaticsrsquo or lsquocheminform-aticsrsquo lsquochemogenomicsrsquo or lsquochemical genomicsrsquo and lsquometabonomicsrsquo orlsquometabolomicsrsquo
thus highly desirable This is expected to lower the cost of drugdiscovery and optimization significantly and hopefully provide anincreased number of useful leads In cases where a lead has suf-ficiently high activity but various properties need to be improvedchemoinformatics methods could be used to modify substructureswithin the lead space with minimal effect on the activity profile(Oprea et al 2001b)
Much improvement has occurred concerning techniques forin vitro measurements of various properties and in models usedfor accessing how well a given compound does absorb from thegastrointestinal tract (GIT) into the blood stream or its ability tocross the bloodndashbrain-barrier (BBB) (van de Waterbeemd et al2003) In most cases in vitro measurements are carried out usingdifferent cell models human or animal For measuring permeabilityand thus absorption the use of artificial membranes called PAMPA(parallel artificial membrane permeability assay) has become a pop-ular alternative to the CACO-2 (human colon carcinoma) cell line(van de Waterbeemd et al 2003) The cost of each measurementusing PAMPA is 120 of that of CACO-2 seemingly with com-parable accuracy (van de Waterbeemd et al 2001) PAMPA ishowever only useful for measuring passive permeability For invitro studies of liver toxicity the rat hepatoma cell line is a wellcharacterized and evaluated method which is often followed upby human hepatoma cell line studies (van de Waterbeemd et al2001) Toxicogenomic studies using microarray expression tech-niques have also become increasingly important Major concernsin toxicological studies are multiple endpoints dosendashresponse rela-tionships and selection of endpoints Other important aspects arepurity of drugs protein binding and metabolic stability amongothers
Likewise various computational methods are evolving rapidly atpresent Computational techniques used to search through chem-ical libraries and databases so-called virtual screening methodshave become increasingly popular in drug discovery (Walters et al1998 Boumlhm and Schneider 2000) A whole range of computa-tional techniques are used for searching for molecular similaritiesand dissimilarities (Sello 1998 Willett 2000 Bajorath 2002Gillet et al 2003) for extracting information about pharmaco-phores (structural models of targets or binding sites) from compound
libraries (Hopfinger and Duca 2000) for prediction of proper-ties for studying molecular interactions at the atomic level amongother things (Miller 2002) Chemoinformatics is strongly linkedto computational chemistry and molecular modeling (Burkert andAllinger 1982 Jensen 1999 Cramer 2002) Molecular modelingmethods are particularly useful for conducting conformational ana-lysis of molecules and for accessing the strength of intermolecularinteractions
Newly established fields like chemogenomics (or chemicalgenomics) metabonomics and metabolomics also play increasinglyimportant roles in modern drug discovery and development Chemo-genomics (Browne et al 2002) deals with interactions betweenchemical compounds and living systems in terms of induced gen-omic response In metabonomics (Nicholson and Wilson 2003)relatively low-molecular weight materials produced during genomicexpression within a cell are studied normally by use of 1H-NMRspectroscopy and multivariate data analysis (chemometrics) (Geladiand Kowalski 1986ba) It has been shown to be a useful tool forunderstanding drug efficacy and toxicity Metabolomics is similar tometabonomics but where metabonomics deals with integrated mul-ticellular biological systems metabolomics deals with simple cellsystems
This review presents an extensive and thorough overview of smallmolecule databases relevant to drug discovery and methods for clas-sifying chemical compounds as being drug-like andor lead-like Abrief overview of various methods for predicting ADMET (adsorp-tion distribution metabolism excretion and toxicity) propertiesBBB penetration and key physico-chemical properties is also givenbut it must be stressed that this overview is by no means completeReferences are given to more detailed review papers for the variousproperties
The fundamental behavior of substances is governed by inter-molecular interactions at various levels Physico-chemical propertiesof drug molecules are mostly governed by the interactions betweenthe drug molecules and the surrounding aqueous environment Thepotency of drugs depends on how well a given drug molecule (lig-and) fits into a target and how strong the interactions betweenthe ligand and the target are often studied with computationalmethods eg docking and scoring methods ADMET propertiesdepend on how the drug molecule interacts with a large num-ber of macromolecules in the human organism In cases wherea patient uses more than one drug drugndashdrug interactions arealso of importance and such interactions are unfortunately oftenignored The human organism is an immensely complicated sys-tem but modeling of properties can be accomplished by studyingvarious subsystems and their interactions using a broad range ofcomputational and experimental methods Recently Macchiaruloet al (2004) studied cross interactions between enzymes and smallmolecules in a cell that are caused by similarities in the molecularstructures of the metabolites (small molecules) and the flexibil-ity in binding at the active sites of the enzymes Based on theirresults they propose that HTS should not only involve a selec-tion of small molecules but also a panel of proteins to test forcross-reactivity
DATABASESThe availability of reliable experimental data and basic structuralinformation is crucial for successful modeling work In this section
2146
Prediction methods and databases within chemoinformatics
Table 1 Overview of small molecule databases and key informations provided in those
Database Structural Sub-structure Number of Number added Coverage Experimnetaldisplay search compounds per year data
General databasesACD SD-Files 2D 3D Yes 300000 NoCAPa Reagents SD-Files 2D3Db Yes 239219 NoSPRESIweb 20 SD-Files 2D Yes 4500000 1974ndash2002c Yes
Screening compoundsMDL SCD SD-Files 2D 3D Yes gt2000000 NoSPECSneta SD-Files 2D 3D Yes
Databases for medicinal agentsCMC SD-Files 2D 3D Yes 8473 250 1900ndashpresent YesMDDR SD-Files 2D 3D Yes 132726 10000 1988ndashpresent NoWDI SMILES 2D 3D Yes gt73000 3500 1983ndashpresent NoNCI 2D 3D 213628 NoMedChem SMILES 2D 3D 48500 YesWOMBAT SMILES 2D 3D 76165 1992ndash2004 YesBIOSTER SD-Files 2D3D Yes 9500 NoCHCRC Dictionary of Drugs Yes gt41000 Yes
Databases with ADMET propertiesMDL Metabolite SD-Files 2D 3D Yes gt10000e 1901ndashpresent YesAccelrysrsquo Metabolisma SD-Files 2D Yes 4101e 1970ndashpresent YesAccelrysrsquo Biotransformationad SD-Files 2D Yes 1744e 1987ndash1995 YesMDL Toxicity SD-Files 2D 3D Yes gt158000 1902ndashpresent YesDSSTox SD-Files SMILES 2D No 2002ndashpresent Yes
Databases with physico-chemical propertiesACDLabs physiochemical propf SD-Files 2D 3D No YesPHYSPROP from SRC SD-Files Yes 25250 YesAQUASOL dATAbASE No 6000 YesCrossFire Beilstein SD-Files 2D 3D 8000000 YesCRC Handbook 11000 Yes
aSimilarity search is also possible for these databasesb3D display only possible in the version interfaced with the Catalyst program and not in the Accord versioncThe Spresi database is being updated to presentdAccelrysrsquo Biotransformation database is a subset in the Accelrysrsquo Metabolism databaseeNumber of Parent compounds in the databasef A cluster of databases for a variety of properties provided with a free graphical program ACDChemSketch and can also be interfaced with ISISBase ISISDraw and ChemDrawNumber of compounds varies from one database to another see text for details
various databases relevant to drug discovery and development arediscussed including databases for available organic compoundsscreening compounds medicinal agents (drugs) as well as databaseswith ADMET properties and physico-chemical properties A fewprotein databases are also mentioned
An overview of the databases and key features is given in Table 1and the type of properties provided is listed in Table 2 Mostof the databases provide 1-dimensional (1D) 2-dimensional (2D)and 3-dimensional (3D) structural information (see below for fur-ther detail on molecular descriptors) The 1D coding is eithergiven in the SD-file format from MDL Information Systems or asSMILES (simplified molecular input line entry specification) strings(Weininger 1988 httpwwwdaylightcomsmilesf_smileshtml)In cases where only 1D and 2D information is given several pro-grams for generating 3D structures from 2D structures are availableOne should however bear in mind that due to conformational flex-ibility many 3D structures (equilibrium conformations) may existfor each molecule Examples of 1D 2D and 3D structures areshown in Figure 2
General small molecule and screening compounddatabasesThe best known database for synthetic organic and inorganiccompounds is the Available Chemicals Directory (ACD)1 whichcurrently includes approximately 300000 unique substances ACDis often used for representing non-drugs in model developmentand although some compounds within ACD may be biologicallyactive the majority of compounds are not The Chemicals Avail-able for Purchase (CAP) database contains similar informationwhere the CAP Reagents database currently has about 240000molecular structures and CAP Complete approximately 16 millioncompounds A new version of the Spresi database SPRESIwebcontains more than 45 million molecules including variousphysical properties and 35 million reactions The physical property
1Databases and computer programs are listed in alphabetical order beforethe references Some of the databases and programs are represented by anordinary reference and are not included in this list
2147
SOacuteJoacutensdoacutettir et al
Table 2 Overview of relevant experimental physico-chemical and ADMET properties provided in small molecule databases
Database Drug class Drug activity log P pKa Solubility Toxicity Metabolism
CMC Yes Yes Yes Yes No mdash mdashMDDR Yes Yes(ql)a mdash mdash mdash mdash mdashWDI Yes Yes(ql) mdash mdash mdash mdash mdashNCI Yes Yes(ql) mdash mdash mdash mdash mdashMedChem Yes Yes Yes Yes No mdash mdashWOMBAT Yes Yes Yes No Yes mdash mdashCHCRC Dictionary of Drugs mdash mdash Yes Yes Yes(ql) Yes mdashMDL Metabolite mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Metabolism mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Biotransformation mdash mdash mdash mdash mdash mdash YesMDL Toxicity mdash mdash mdash mdash mdash Yes mdashDSSTox mdash mdash mdash mdash mdash Yes mdashACDLabs physico-chemical prop mdash mdash Yes Yes Yes mdash mdashPHYSPROP from SRC mdash mdash Yes Yes Yes mdash mdashAQUASOL dATAbASE mdash mdash No No Yes mdash mdashCRC mdash mdash No No Yes(ql) No mdash
aql means that drug activity is given in qualitative terms
Fig 2 Structural information for a molecule can be given as a 1D SMILES string a 2D drawing or a 3D representation The SMILES string and the 2Ddrawing contain information about which atoms are bound to one another and the 3D representation shows how the atoms are located geometrically in relationto one another The atomic coordinates used are shown behind the 3D picture
information is however not well organized within the Spresidatabase
A number of databases have been developed especially for screen-ing purposes Three main catalogs previously being part of ACDthe SALOR (SigmandashAldrich Library of Rare Chemicals) Maybridgeand Bionet are now available through MDL Screening Com-pounds Directory (MDL SCD) The SPECSnet database contains
screening compounds building blocks and natural products providedby SPECS
Databases for medicinal agentsThe three main database collections of drug molecules are the Com-prehensive Medical Chemistry database (CMC) MDL Drug DataReport (MDDR) and the Derwent Word Drug Index (WDI) CMC
2148
Prediction methods and databases within chemoinformatics
contains presently 8473 drug compounds and is updated annuallywith compounds identified for the first time in the United StatesApproved Names (USAN) list The CMC database also includesinformation about drug class and measured or estimated valuesfor the acidndashbase dissociation constant (pKa) and the octanolwaterpartition coefficient (log P ) For log P 120 experimental and 8300calculated records are provided and for pKa 1200 measured recordsare given The MDDR contains 132726 drugs launched and underdevelopment collected from the patent literature and other relevantsources MDDR is updated monthly which adds up to approximately10000 new compounds per year and includes information aboutdrug class and drug activity in qualitative terms as well WDI con-tains around 73000 marketed drugs and drugs under developmentwith each record classified by drug activity mechanism of actiontreatment among other factors The Derwent Drug File is a highlyfocused database of selected journal articles and conference reportson all aspects of drug development
There are a number of other available drug databases The NationalCancer Institute database (NCI) contains 213628 compounds andis made up of four publicly available NCI databases including boththe AIDS and the Cancer databases The MedChem database con-sists of 48500 compounds 67000 measured log P values 13700measured pKa values and 19000 pharmacological (drug) activit-ies According to the providers of the MedChem Bio-loom databaseall available data for log P and pKa have been gathered from theliterature In addition the database includes a sorted list contain-ing only those measurements carried out with high quality methodsThe MedChem database also contains a collection of biological andphysico-chemical data intended for QSPR (quantitative structurendashproperty relationship) modeling and software for calculating log P
values (Clog P ) The WOrld of Molecular BioAcivities database(WOMBAT) is a very comprehensive collection of biological activ-ity data and contains 76165 molecules 68543 SMILES 143000activities for 630 targets 1230 measured log P values and 527 meas-ured solubility values The biological activities in this database aremainly related to receptor antagonists and enzyme inhibitors whichare ranked by target class The BIOSTER is a database of bioanalog-ous pairs of molecules (bio-isosters) and contains over 9500 activemolecules including drugs agrochemicals and enzyme inhibitorsThe Chapman amp HallCRC Dictionary of Drugs contains over 41000drugs and available physico-chemical property and toxicity data
Databases for ADMET properties of drugsThe MDL Metabolite database contains information about meta-bolism pathways for xenobiotic compounds and biotransformations(primary medicinal agents) and experimental data from in vivoand in vitro studies The database includes more than 10000 par-ent compounds over 64000 biotransformations and over 40000molecules (parent compounds intermediates and final metabolites)Metabolism data are also found in the Accelrysrsquo Metabolism andBiotransformation databases The Metabolism database covers ver-tebrates (animals) invertebrates and plants This databases contains4101 parent compounds 30000 transformations and is being exten-ded to 40000 transformations The Accelrysrsquo Biotransformationdatabase on the other hand is a stand-alone database for verteb-rates only containing 1744 unique parent compounds and 9809transformations
The MDL Toxicity database includes more than 158000 chem-ical substances compiled from several major sources where around
65 are drugs and drug development compounds The database con-tains six categories of data ie acute toxicity mutagenicity skineyeirritation tumorigenicity reproductive effects and multiple doseeffects It also contains detailed information about how the in vivoand in vitro experiments were carried out including species of organ-ism or tissue studied dose etc The DSSTox project (DistributedStructure-Searchable Toxicity Public Database Network) (Richardand Williams 2002 httpwwwepagovnheerldsstox) is a forumfor publishing toxicity data which presently includes databases forcarcinogenicity in various animals as well as acute toxicity
The ToxExpress Reference Database is a toxicogenomic data-base which contains gene expression profiles from known toxicantsThis database is built on in vivo and in vitro studies of exposure totoxicants and around 110 compounds have been profiled
Other databases that might be of interest although primarilyfocused on environmental and occupational health issues are theTSCA93 database containing over 100000 substances and theTOXicology Data NETwork (TOXNET) database cluster
Other important small molecule databasesThe ACDLabs Physico-Chemical Property databases contain a vari-ety of physico-chemical data for organic substances includingexperimental log P for 18400 compounds over 31000 measuredpKa values for 16000 compounds and aqueous solubility (logS)for 5000 compounds Another collection of experimental data is thePhysical Properties Database (PHYSPROP) from Syracuse ResearchCorporation (SRC) which contains 13250 measured log P values1652 pKa values and 6340 records for aqueous solubility BothACDLabs and SRC market software for prediction of these and otherproperties A third database the AQUASOL dATAbASE containsaqueous solubilities for almost 6000 compounds
There are other important databases for small molecules Cross-Fire Beilstein contains more than 8 million organic compoundsover 9 million reactions a variety of properties including variousphysical properties pharmacodynamics and environmental toxicityThis database contains over 500000 bioactive compounds Otherimportant collections of physical properties of organic compoundsare CRC (Lide 2003 httpwwwhbcpnetbasecom) (11000 com-pounds) Chapman amp HallCRC Properties of Organic Compounds(29000 compounds) and the Design Institute of Physical PropertiesDatabase (DIPPR) (1743 compounds)
Protein databases and ligand informationIn this context we would also like to mention a few relevantmacromolecular and crystallographic databases with drug-discoveryrelevant information The Protein Data Bank (PDB) is the mainsource for available protein crystal structures and structural inform-ation obtained with NMR spectroscopy with currently more than28000 structures and a weekly growth of about 100 structures TheCambridge Structural Database (CSD) is the most comprehensivecollection of crystallographic data for small molecules containingaround 305000 structures Figure 3 shows the evolution in numberof structures within PDB and CSD over the last 12 years AlthoughCSD has a much larger number of entries the relative growth ratewithin PDB is higher which shows clearly the increased focus onprotein structure determination world wide (structural genomics)
PDB contains a number of useful sub-databases including aTarget Registration Database Relibase (Hendlich et al 2003Guumlnther et al 2003 httprelibaseccdccamacuk) is a powerful
2149
SOacuteJoacutensdoacutettir et al
Fig 3 Number of crystallographic structures in the CSD and the PDB inthe period 1992ndash2004 This figure can be viewed in colour onBioinformaticsonline
data mining tool which utilizes structural information aboutproteinndashligand complexes within PDB for comprehensive analysisof proteinndashligand interactions Relibase+ is an improved version ofRelibase with a number of additional features like proteinndashproteininteraction searching and a crystal packing module for studyingcrystallographic effects around ligand binding sites The LIGANDdatabase (Gotoet al 1998 2002 httpwwwgenomeadjpligand)is a chemical database for enzyme reactions including informationabout structures of metabolites and other chemical compounds theirreactions in biological pathways and about the nomenclature ofenzymes
Ji et al (2003) recently published an overview of various data col-lections for proteins associated with drug therapeutic effects adversereactions and ADME available over the internet
A number of databases containing proteinndashprotein interactiondata are available including Database of Interacting Proteins(DIP) (Xenearioset al 2002 httpdipdoe-mbiuclaedu) Bio-molecular Interaction Network Database (BIND) (Baderet al2003 httpbindca) Molecular Interaction Database (MINT)(Zanzoniet al 2002 httpcbmbiouniroma2itmint) Compre-hensive Yeast Protein Genome Database (MIPS) (Meweset al2002 httpmipsgsfdegenreprojyeast) Yeast Proteome Data-base (YPD) (Hodgeset al 1999 Costanzoet al 2000) the IntActdatabase (Hermjakobet al 2004 httpwwwebiacukintactindexhtml) and Human Protein Reference Database (HPRD) (Periet al2003 httpwwwhprdorg) among others Proteinndashprotein interac-tion databases are collections of information about actual proteinndashprotein contacts or complex associations within the proteome ofspecific organisms in particularly in yeast but also in humansfruitfly and other organisms
DESCRIPTORS USED FOR CHEMICALSTRUCTURESIn chemoinformatics the structural features of the individualmolecules are described by various parameters derived from themolecular structure the so-called descriptors (Todeschini andConsonni 2000) The simplest types are constitutional (1D) ortopological (2D) descriptors describing the types of atoms func-tional groups or types and order of the chemical bonds withinthe molecules Ghose atom types (Viswanadhanet al 1989)CONCORD atom types ISIS keys (MDL keys) (Durantet al 2002)and various other atombond indices discussed in the following
section are examples of such descriptors Physico-chemical proper-ties are often used as descriptors as well
The ISIS keys and CONCORD atom types are often referred toas fingerprint methods and were for example used in the training ofneural networks (Frimureret al 2000) for compound classificationISIS keys are a set of codes which represent the presence of certainfunctional groups and other structural features in the molecules andCONCORD atom types are generated by assigning up to six atom-type codes available within the CONCORD program (Pearlman2000) to each atom
In QSAR (quantitative structurendashactivity relationship) and QSPRmodeling constitutional and topological descriptors are combinedwith more complicated descriptors representing the 3D structureof the molecules This includes various geometrical descriptorsand most importantly a variety of electrostatic and quantum chem-ical descriptors The electrostatic descriptors are parameters whichdepend on the charge distribution within the molecule includ-ing the dipole moment Examples of quantum chemically deriveddescriptors are various energy values like ionization energiesHOMOndashLUMO gap etc Similarly descriptors derived from molecu-lar mechanics can be used (Dyekjaeligret al 2002) A variety ofdifferent types of descriptors used in QSAR and QSPR are discussedby Karelson (2000)
QSAR and QSPR models are empirical equations used forestimating various properties of molecules and have the form
P = a + b middot D1 + c middot D2 + d middot D3 + middot middot middot (1)
where P is the property of interestab c are regressioncoefficients andD1D2D3 are the descriptors
The so-called 3D QSAR methods differ somewhat from traditionalQSAR methods The CoMFA (comparative molecular field analysis)(Crameret al 1988 httpwwwtriposcomsciTechinSilicoDiscstrActRelationship) approach one of the most popular 3D QSARmethods uses steric and electrostatic interaction energies betweenprobe atoms and the molecules as descriptors In the VolSurf method(Crucianiet al 2000) a similar approach is used where descriptorsare generated from molecular interaction fields calculated by theGRID program (Goodford 1985)
REDUNDANCY OF DATA SETSWhen using data extracted from large databases to train and testdata driven prediction tools the redundancy completeness and rep-resentativeness of the underlying data sets are extremely importantThe redundancy affects strongly the evaluation of the predictive per-formance and also most importantly the bias of the method towardspecific over-represented subclasses of compounds
Within the general bioinformatics area this is an issue that has beendealt with in great detail It is well known that if a protein structureprediction algorithm is tested on sequences and structures whichare highly similar to the sequences and structures used to train itthe predictive performance is significantly overestimated (Sanderand Schneider 1991) Therefore methods have been constructed forcleaning data sets for examples that are lsquotoo easyrsquo such that theperformance of the algorithm on novel data will be estimated in amore reliable manner One needs obviously to define the similaritymeasure between the data objects under consideration for aminoacid sequences one typically uses alignment techniques such as themetric for quantitative pair-wise comparison (Hobohmet al 1992)
2150
Prediction methods and databases within chemoinformatics
Techniques have also been developed for redundancy reduction ofsequences containing functional sites such as signal peptide cleav-age sites in secreted proteins (Nielsen et al 1996) or translationinitiation sites in mRNA sequences (Pedersen and Nielsen 1997)
In this respect chemoinformatics is still in its infancy comparedto the thorough validation methods used in bioinformatics Forchemoinformatics applications redundancy within databases canaffect the learning capabilities of neural networks decision treesand other methods whereas redundancy between different databasesgenerally introduces noise and reduces the discriminating power ofthe model
Redundancy in this context means that the compounds in theirvectors component for component are similar Similarity of thechemical compounds within a data set can lead to over-fitting of amodel such that predictions for compounds similar to those used inthe training set are excellent but for different compounds the predic-tions are not accurate to the same level There are several examplesof compound clustering techniques where the Tanimoto coefficient(Patterson et al 1996) has been used as the metric quantifying thesimilarity between compounds normally with the goal of identifyingleads by similarity or for increasing the diversity of libraries used forscreening (Reynolds et al 1998 2001 Voigt et al 2001 Willett2003) The Tanimoto coefficient T compares binary fingerprintvectors and is defined as
T (x y) = Nxy(Nx + Ny minus Nxy) (2)
where Nxy is the number of 1 bits shared in the fingerprints ofmolecules x and y Nx the number of 1 bits in the fingerprintof molecule x and Ny the number of 1 bits in the fingerprint ofmolecule y A Tanimoto coefficient approaching zero indicates thatthe compounds being compared are very different while a Tanimotocoefficient approaching one indicates that they are very similar
In the drug-likeness predictor developed by Frimurer et al(2000) the concept of redundancy reduction normally used withinbioinformatics was transferred to compound data extracted fromMDDR and ACD In this case the compounds were representedby CONCORD atom types and the Tanimoto coefficient (Patter-son et al 1996) was used to calculate the similarity between anytwo compounds Using a given threshold for maximal similarity interms of a Tanimoto coefficient of say 085 the redundancy in a dataset can be reduced to a well-defined level This type of technique alsoallows for the creation of common benchmark principles and couldreplace frozen benchmark data sets which often become outdated asdatabases grow over time The performance of prediction techniquesdeveloped on different data sets can therefore be compared in a fairmanner despite the fact that the underlying data sets differ in size
In most of the chemoinformatics applications developed so far thiskind of data set cleaning has not been carried out Instead test setshave been selected randomly from large overall data sets therebyintroducing many cases where highly similar pairs of examplesare found both in the training and test parts of the data sets Theestimation of the predictive performance is therefore much moreconservative in the work of Frimurer et al while it may be too highfor some of the prediction tools where the split between training andtest data has been established just by random selection
Note that similarity to a neural network is somewhat different frommolecular similarity because the network can correlate all the vectorcomponents with each other in a nonlinear fashion Two very differ-ent compounds may appear very similar for a neural net in terms of
functionality while two quite similar compounds can be interpretedas having different properties A redundant data set will typically notconstrain the network weight structure as much as a nonredundantdata set Nonredundant data from complete and representative datasets will therefore in most cases lead to well-performing predictorseven if the weightstraining examples ratio is larger
METHODS FOR CLASSIFICATION OF DRUG-LIKESTRUCTURESSeveral methods have been developed to determine the suitability ofcompounds to be used as drugs (pharmaceuticals) based on know-ledge about the molecular structure and key physical propertiesReviews about a variety of methods have been published recently(Walters and Murcko 2002 Clark and Pickett 2000 Muegge 2003)
The task is to select compounds from combinatorial librariesin such a way that the probability of identifying a new drugthat will make it to the market is optimized To minimize theprobability of leaving out potential drugs in the HTS it is import-ant to ensure adequate diversity of the molecular structures used(Gillet et al 1999) A variety of methods for compound selec-tion have been proposed ranging from simple intuitive to advancedcomputational methods
Intuitive and graphical methodsMany researchers have developed simple counting methods and ahighly popular method is the lsquorule of fiversquo proposed by Lipinski et al(1997) Although the lsquorule of fiversquo originally addresses the possibil-ity or risk for poor absorption or permeation based on molecularweight (MW) the octanolndashwater partition coefficient (log P alsocalled lipophilicity) number of hydrogen bond donors (HBD) andacceptors (HBA) respectively it has been widely used to distinguishbetween drug-like and non-drug-like compounds A number of data-bases have included Lipinskisrsquo lsquorule of fiversquo data for each compoundusing calculated values for log P This is the case for SPECS MDLSCD ACD ACDLabs Physico-Chemical databases
Frimurer et al (2000) performed a detailed quantitative analysisassessment of the predictive value of Lipinskisrsquo lsquorule of fiversquo It wasfound that the correlation coefficient is close to zero in fact slightlynegative when evaluated on a nonredundant data set Oprea (2000)also discusses drug-related chemical databases and the performanceof the lsquorule of fiversquo He concludes that the lsquorule of fiversquo does not dis-tinguish between lsquodrugsrsquo and lsquonon-drugsrsquo because the distributionof the parameters used does not differ significantly between thesetwo groups of compounds The problem with the lsquorule of fiversquo isthat it represents conditions which are necessary but not sufficientfor a drug molecule Many (in fact most) of the molecules whichfulfill these conditions are not drug molecules The work of Lip-inski has had however a tremendous pioneering value concerningquantitative evaluation of lsquodrug-likenessrsquo of chemical structures
Muegge et al (2001) proposed a simple selection scheme fordrug-likeness based on the presence of certain functional groupspharmacophore points in the molecule This method and other meth-ods using functional group filters are significantly less accurate thanthe various machine learning methods discussed (Table 3)
Chemistry space or chemography methods are somewhat similarand are based on the hypothesis that drug-like molecules share certainproperties and thus cluster in graphical representations Analysis ofhow well molecular diversity is captured using a variety of descriptors
2151
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160
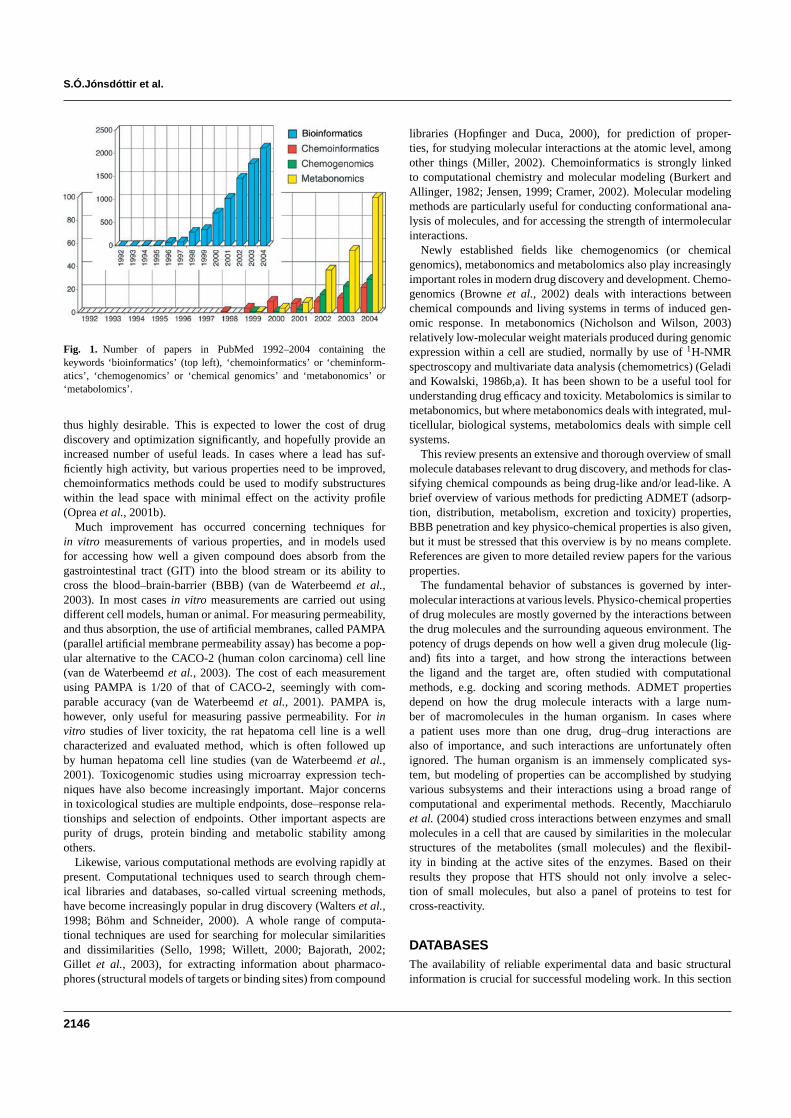
SOacuteJoacutensdoacutettir et al
Fig 1 Number of papers in PubMed 1992ndash2004 containing thekeywords lsquobioinformaticsrsquo (top left) lsquochemoinformaticsrsquo or lsquocheminform-aticsrsquo lsquochemogenomicsrsquo or lsquochemical genomicsrsquo and lsquometabonomicsrsquo orlsquometabolomicsrsquo
thus highly desirable This is expected to lower the cost of drugdiscovery and optimization significantly and hopefully provide anincreased number of useful leads In cases where a lead has suf-ficiently high activity but various properties need to be improvedchemoinformatics methods could be used to modify substructureswithin the lead space with minimal effect on the activity profile(Oprea et al 2001b)
Much improvement has occurred concerning techniques forin vitro measurements of various properties and in models usedfor accessing how well a given compound does absorb from thegastrointestinal tract (GIT) into the blood stream or its ability tocross the bloodndashbrain-barrier (BBB) (van de Waterbeemd et al2003) In most cases in vitro measurements are carried out usingdifferent cell models human or animal For measuring permeabilityand thus absorption the use of artificial membranes called PAMPA(parallel artificial membrane permeability assay) has become a pop-ular alternative to the CACO-2 (human colon carcinoma) cell line(van de Waterbeemd et al 2003) The cost of each measurementusing PAMPA is 120 of that of CACO-2 seemingly with com-parable accuracy (van de Waterbeemd et al 2001) PAMPA ishowever only useful for measuring passive permeability For invitro studies of liver toxicity the rat hepatoma cell line is a wellcharacterized and evaluated method which is often followed upby human hepatoma cell line studies (van de Waterbeemd et al2001) Toxicogenomic studies using microarray expression tech-niques have also become increasingly important Major concernsin toxicological studies are multiple endpoints dosendashresponse rela-tionships and selection of endpoints Other important aspects arepurity of drugs protein binding and metabolic stability amongothers
Likewise various computational methods are evolving rapidly atpresent Computational techniques used to search through chem-ical libraries and databases so-called virtual screening methodshave become increasingly popular in drug discovery (Walters et al1998 Boumlhm and Schneider 2000) A whole range of computa-tional techniques are used for searching for molecular similaritiesand dissimilarities (Sello 1998 Willett 2000 Bajorath 2002Gillet et al 2003) for extracting information about pharmaco-phores (structural models of targets or binding sites) from compound
libraries (Hopfinger and Duca 2000) for prediction of proper-ties for studying molecular interactions at the atomic level amongother things (Miller 2002) Chemoinformatics is strongly linkedto computational chemistry and molecular modeling (Burkert andAllinger 1982 Jensen 1999 Cramer 2002) Molecular modelingmethods are particularly useful for conducting conformational ana-lysis of molecules and for accessing the strength of intermolecularinteractions
Newly established fields like chemogenomics (or chemicalgenomics) metabonomics and metabolomics also play increasinglyimportant roles in modern drug discovery and development Chemo-genomics (Browne et al 2002) deals with interactions betweenchemical compounds and living systems in terms of induced gen-omic response In metabonomics (Nicholson and Wilson 2003)relatively low-molecular weight materials produced during genomicexpression within a cell are studied normally by use of 1H-NMRspectroscopy and multivariate data analysis (chemometrics) (Geladiand Kowalski 1986ba) It has been shown to be a useful tool forunderstanding drug efficacy and toxicity Metabolomics is similar tometabonomics but where metabonomics deals with integrated mul-ticellular biological systems metabolomics deals with simple cellsystems
This review presents an extensive and thorough overview of smallmolecule databases relevant to drug discovery and methods for clas-sifying chemical compounds as being drug-like andor lead-like Abrief overview of various methods for predicting ADMET (adsorp-tion distribution metabolism excretion and toxicity) propertiesBBB penetration and key physico-chemical properties is also givenbut it must be stressed that this overview is by no means completeReferences are given to more detailed review papers for the variousproperties
The fundamental behavior of substances is governed by inter-molecular interactions at various levels Physico-chemical propertiesof drug molecules are mostly governed by the interactions betweenthe drug molecules and the surrounding aqueous environment Thepotency of drugs depends on how well a given drug molecule (lig-and) fits into a target and how strong the interactions betweenthe ligand and the target are often studied with computationalmethods eg docking and scoring methods ADMET propertiesdepend on how the drug molecule interacts with a large num-ber of macromolecules in the human organism In cases wherea patient uses more than one drug drugndashdrug interactions arealso of importance and such interactions are unfortunately oftenignored The human organism is an immensely complicated sys-tem but modeling of properties can be accomplished by studyingvarious subsystems and their interactions using a broad range ofcomputational and experimental methods Recently Macchiaruloet al (2004) studied cross interactions between enzymes and smallmolecules in a cell that are caused by similarities in the molecularstructures of the metabolites (small molecules) and the flexibil-ity in binding at the active sites of the enzymes Based on theirresults they propose that HTS should not only involve a selec-tion of small molecules but also a panel of proteins to test forcross-reactivity
DATABASESThe availability of reliable experimental data and basic structuralinformation is crucial for successful modeling work In this section
2146
Prediction methods and databases within chemoinformatics
Table 1 Overview of small molecule databases and key informations provided in those
Database Structural Sub-structure Number of Number added Coverage Experimnetaldisplay search compounds per year data
General databasesACD SD-Files 2D 3D Yes 300000 NoCAPa Reagents SD-Files 2D3Db Yes 239219 NoSPRESIweb 20 SD-Files 2D Yes 4500000 1974ndash2002c Yes
Screening compoundsMDL SCD SD-Files 2D 3D Yes gt2000000 NoSPECSneta SD-Files 2D 3D Yes
Databases for medicinal agentsCMC SD-Files 2D 3D Yes 8473 250 1900ndashpresent YesMDDR SD-Files 2D 3D Yes 132726 10000 1988ndashpresent NoWDI SMILES 2D 3D Yes gt73000 3500 1983ndashpresent NoNCI 2D 3D 213628 NoMedChem SMILES 2D 3D 48500 YesWOMBAT SMILES 2D 3D 76165 1992ndash2004 YesBIOSTER SD-Files 2D3D Yes 9500 NoCHCRC Dictionary of Drugs Yes gt41000 Yes
Databases with ADMET propertiesMDL Metabolite SD-Files 2D 3D Yes gt10000e 1901ndashpresent YesAccelrysrsquo Metabolisma SD-Files 2D Yes 4101e 1970ndashpresent YesAccelrysrsquo Biotransformationad SD-Files 2D Yes 1744e 1987ndash1995 YesMDL Toxicity SD-Files 2D 3D Yes gt158000 1902ndashpresent YesDSSTox SD-Files SMILES 2D No 2002ndashpresent Yes
Databases with physico-chemical propertiesACDLabs physiochemical propf SD-Files 2D 3D No YesPHYSPROP from SRC SD-Files Yes 25250 YesAQUASOL dATAbASE No 6000 YesCrossFire Beilstein SD-Files 2D 3D 8000000 YesCRC Handbook 11000 Yes
aSimilarity search is also possible for these databasesb3D display only possible in the version interfaced with the Catalyst program and not in the Accord versioncThe Spresi database is being updated to presentdAccelrysrsquo Biotransformation database is a subset in the Accelrysrsquo Metabolism databaseeNumber of Parent compounds in the databasef A cluster of databases for a variety of properties provided with a free graphical program ACDChemSketch and can also be interfaced with ISISBase ISISDraw and ChemDrawNumber of compounds varies from one database to another see text for details
various databases relevant to drug discovery and development arediscussed including databases for available organic compoundsscreening compounds medicinal agents (drugs) as well as databaseswith ADMET properties and physico-chemical properties A fewprotein databases are also mentioned
An overview of the databases and key features is given in Table 1and the type of properties provided is listed in Table 2 Mostof the databases provide 1-dimensional (1D) 2-dimensional (2D)and 3-dimensional (3D) structural information (see below for fur-ther detail on molecular descriptors) The 1D coding is eithergiven in the SD-file format from MDL Information Systems or asSMILES (simplified molecular input line entry specification) strings(Weininger 1988 httpwwwdaylightcomsmilesf_smileshtml)In cases where only 1D and 2D information is given several pro-grams for generating 3D structures from 2D structures are availableOne should however bear in mind that due to conformational flex-ibility many 3D structures (equilibrium conformations) may existfor each molecule Examples of 1D 2D and 3D structures areshown in Figure 2
General small molecule and screening compounddatabasesThe best known database for synthetic organic and inorganiccompounds is the Available Chemicals Directory (ACD)1 whichcurrently includes approximately 300000 unique substances ACDis often used for representing non-drugs in model developmentand although some compounds within ACD may be biologicallyactive the majority of compounds are not The Chemicals Avail-able for Purchase (CAP) database contains similar informationwhere the CAP Reagents database currently has about 240000molecular structures and CAP Complete approximately 16 millioncompounds A new version of the Spresi database SPRESIwebcontains more than 45 million molecules including variousphysical properties and 35 million reactions The physical property
1Databases and computer programs are listed in alphabetical order beforethe references Some of the databases and programs are represented by anordinary reference and are not included in this list
2147
SOacuteJoacutensdoacutettir et al
Table 2 Overview of relevant experimental physico-chemical and ADMET properties provided in small molecule databases
Database Drug class Drug activity log P pKa Solubility Toxicity Metabolism
CMC Yes Yes Yes Yes No mdash mdashMDDR Yes Yes(ql)a mdash mdash mdash mdash mdashWDI Yes Yes(ql) mdash mdash mdash mdash mdashNCI Yes Yes(ql) mdash mdash mdash mdash mdashMedChem Yes Yes Yes Yes No mdash mdashWOMBAT Yes Yes Yes No Yes mdash mdashCHCRC Dictionary of Drugs mdash mdash Yes Yes Yes(ql) Yes mdashMDL Metabolite mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Metabolism mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Biotransformation mdash mdash mdash mdash mdash mdash YesMDL Toxicity mdash mdash mdash mdash mdash Yes mdashDSSTox mdash mdash mdash mdash mdash Yes mdashACDLabs physico-chemical prop mdash mdash Yes Yes Yes mdash mdashPHYSPROP from SRC mdash mdash Yes Yes Yes mdash mdashAQUASOL dATAbASE mdash mdash No No Yes mdash mdashCRC mdash mdash No No Yes(ql) No mdash
aql means that drug activity is given in qualitative terms
Fig 2 Structural information for a molecule can be given as a 1D SMILES string a 2D drawing or a 3D representation The SMILES string and the 2Ddrawing contain information about which atoms are bound to one another and the 3D representation shows how the atoms are located geometrically in relationto one another The atomic coordinates used are shown behind the 3D picture
information is however not well organized within the Spresidatabase
A number of databases have been developed especially for screen-ing purposes Three main catalogs previously being part of ACDthe SALOR (SigmandashAldrich Library of Rare Chemicals) Maybridgeand Bionet are now available through MDL Screening Com-pounds Directory (MDL SCD) The SPECSnet database contains
screening compounds building blocks and natural products providedby SPECS
Databases for medicinal agentsThe three main database collections of drug molecules are the Com-prehensive Medical Chemistry database (CMC) MDL Drug DataReport (MDDR) and the Derwent Word Drug Index (WDI) CMC
2148
Prediction methods and databases within chemoinformatics
contains presently 8473 drug compounds and is updated annuallywith compounds identified for the first time in the United StatesApproved Names (USAN) list The CMC database also includesinformation about drug class and measured or estimated valuesfor the acidndashbase dissociation constant (pKa) and the octanolwaterpartition coefficient (log P ) For log P 120 experimental and 8300calculated records are provided and for pKa 1200 measured recordsare given The MDDR contains 132726 drugs launched and underdevelopment collected from the patent literature and other relevantsources MDDR is updated monthly which adds up to approximately10000 new compounds per year and includes information aboutdrug class and drug activity in qualitative terms as well WDI con-tains around 73000 marketed drugs and drugs under developmentwith each record classified by drug activity mechanism of actiontreatment among other factors The Derwent Drug File is a highlyfocused database of selected journal articles and conference reportson all aspects of drug development
There are a number of other available drug databases The NationalCancer Institute database (NCI) contains 213628 compounds andis made up of four publicly available NCI databases including boththe AIDS and the Cancer databases The MedChem database con-sists of 48500 compounds 67000 measured log P values 13700measured pKa values and 19000 pharmacological (drug) activit-ies According to the providers of the MedChem Bio-loom databaseall available data for log P and pKa have been gathered from theliterature In addition the database includes a sorted list contain-ing only those measurements carried out with high quality methodsThe MedChem database also contains a collection of biological andphysico-chemical data intended for QSPR (quantitative structurendashproperty relationship) modeling and software for calculating log P
values (Clog P ) The WOrld of Molecular BioAcivities database(WOMBAT) is a very comprehensive collection of biological activ-ity data and contains 76165 molecules 68543 SMILES 143000activities for 630 targets 1230 measured log P values and 527 meas-ured solubility values The biological activities in this database aremainly related to receptor antagonists and enzyme inhibitors whichare ranked by target class The BIOSTER is a database of bioanalog-ous pairs of molecules (bio-isosters) and contains over 9500 activemolecules including drugs agrochemicals and enzyme inhibitorsThe Chapman amp HallCRC Dictionary of Drugs contains over 41000drugs and available physico-chemical property and toxicity data
Databases for ADMET properties of drugsThe MDL Metabolite database contains information about meta-bolism pathways for xenobiotic compounds and biotransformations(primary medicinal agents) and experimental data from in vivoand in vitro studies The database includes more than 10000 par-ent compounds over 64000 biotransformations and over 40000molecules (parent compounds intermediates and final metabolites)Metabolism data are also found in the Accelrysrsquo Metabolism andBiotransformation databases The Metabolism database covers ver-tebrates (animals) invertebrates and plants This databases contains4101 parent compounds 30000 transformations and is being exten-ded to 40000 transformations The Accelrysrsquo Biotransformationdatabase on the other hand is a stand-alone database for verteb-rates only containing 1744 unique parent compounds and 9809transformations
The MDL Toxicity database includes more than 158000 chem-ical substances compiled from several major sources where around
65 are drugs and drug development compounds The database con-tains six categories of data ie acute toxicity mutagenicity skineyeirritation tumorigenicity reproductive effects and multiple doseeffects It also contains detailed information about how the in vivoand in vitro experiments were carried out including species of organ-ism or tissue studied dose etc The DSSTox project (DistributedStructure-Searchable Toxicity Public Database Network) (Richardand Williams 2002 httpwwwepagovnheerldsstox) is a forumfor publishing toxicity data which presently includes databases forcarcinogenicity in various animals as well as acute toxicity
The ToxExpress Reference Database is a toxicogenomic data-base which contains gene expression profiles from known toxicantsThis database is built on in vivo and in vitro studies of exposure totoxicants and around 110 compounds have been profiled
Other databases that might be of interest although primarilyfocused on environmental and occupational health issues are theTSCA93 database containing over 100000 substances and theTOXicology Data NETwork (TOXNET) database cluster
Other important small molecule databasesThe ACDLabs Physico-Chemical Property databases contain a vari-ety of physico-chemical data for organic substances includingexperimental log P for 18400 compounds over 31000 measuredpKa values for 16000 compounds and aqueous solubility (logS)for 5000 compounds Another collection of experimental data is thePhysical Properties Database (PHYSPROP) from Syracuse ResearchCorporation (SRC) which contains 13250 measured log P values1652 pKa values and 6340 records for aqueous solubility BothACDLabs and SRC market software for prediction of these and otherproperties A third database the AQUASOL dATAbASE containsaqueous solubilities for almost 6000 compounds
There are other important databases for small molecules Cross-Fire Beilstein contains more than 8 million organic compoundsover 9 million reactions a variety of properties including variousphysical properties pharmacodynamics and environmental toxicityThis database contains over 500000 bioactive compounds Otherimportant collections of physical properties of organic compoundsare CRC (Lide 2003 httpwwwhbcpnetbasecom) (11000 com-pounds) Chapman amp HallCRC Properties of Organic Compounds(29000 compounds) and the Design Institute of Physical PropertiesDatabase (DIPPR) (1743 compounds)
Protein databases and ligand informationIn this context we would also like to mention a few relevantmacromolecular and crystallographic databases with drug-discoveryrelevant information The Protein Data Bank (PDB) is the mainsource for available protein crystal structures and structural inform-ation obtained with NMR spectroscopy with currently more than28000 structures and a weekly growth of about 100 structures TheCambridge Structural Database (CSD) is the most comprehensivecollection of crystallographic data for small molecules containingaround 305000 structures Figure 3 shows the evolution in numberof structures within PDB and CSD over the last 12 years AlthoughCSD has a much larger number of entries the relative growth ratewithin PDB is higher which shows clearly the increased focus onprotein structure determination world wide (structural genomics)
PDB contains a number of useful sub-databases including aTarget Registration Database Relibase (Hendlich et al 2003Guumlnther et al 2003 httprelibaseccdccamacuk) is a powerful
2149
SOacuteJoacutensdoacutettir et al
Fig 3 Number of crystallographic structures in the CSD and the PDB inthe period 1992ndash2004 This figure can be viewed in colour onBioinformaticsonline
data mining tool which utilizes structural information aboutproteinndashligand complexes within PDB for comprehensive analysisof proteinndashligand interactions Relibase+ is an improved version ofRelibase with a number of additional features like proteinndashproteininteraction searching and a crystal packing module for studyingcrystallographic effects around ligand binding sites The LIGANDdatabase (Gotoet al 1998 2002 httpwwwgenomeadjpligand)is a chemical database for enzyme reactions including informationabout structures of metabolites and other chemical compounds theirreactions in biological pathways and about the nomenclature ofenzymes
Ji et al (2003) recently published an overview of various data col-lections for proteins associated with drug therapeutic effects adversereactions and ADME available over the internet
A number of databases containing proteinndashprotein interactiondata are available including Database of Interacting Proteins(DIP) (Xenearioset al 2002 httpdipdoe-mbiuclaedu) Bio-molecular Interaction Network Database (BIND) (Baderet al2003 httpbindca) Molecular Interaction Database (MINT)(Zanzoniet al 2002 httpcbmbiouniroma2itmint) Compre-hensive Yeast Protein Genome Database (MIPS) (Meweset al2002 httpmipsgsfdegenreprojyeast) Yeast Proteome Data-base (YPD) (Hodgeset al 1999 Costanzoet al 2000) the IntActdatabase (Hermjakobet al 2004 httpwwwebiacukintactindexhtml) and Human Protein Reference Database (HPRD) (Periet al2003 httpwwwhprdorg) among others Proteinndashprotein interac-tion databases are collections of information about actual proteinndashprotein contacts or complex associations within the proteome ofspecific organisms in particularly in yeast but also in humansfruitfly and other organisms
DESCRIPTORS USED FOR CHEMICALSTRUCTURESIn chemoinformatics the structural features of the individualmolecules are described by various parameters derived from themolecular structure the so-called descriptors (Todeschini andConsonni 2000) The simplest types are constitutional (1D) ortopological (2D) descriptors describing the types of atoms func-tional groups or types and order of the chemical bonds withinthe molecules Ghose atom types (Viswanadhanet al 1989)CONCORD atom types ISIS keys (MDL keys) (Durantet al 2002)and various other atombond indices discussed in the following
section are examples of such descriptors Physico-chemical proper-ties are often used as descriptors as well
The ISIS keys and CONCORD atom types are often referred toas fingerprint methods and were for example used in the training ofneural networks (Frimureret al 2000) for compound classificationISIS keys are a set of codes which represent the presence of certainfunctional groups and other structural features in the molecules andCONCORD atom types are generated by assigning up to six atom-type codes available within the CONCORD program (Pearlman2000) to each atom
In QSAR (quantitative structurendashactivity relationship) and QSPRmodeling constitutional and topological descriptors are combinedwith more complicated descriptors representing the 3D structureof the molecules This includes various geometrical descriptorsand most importantly a variety of electrostatic and quantum chem-ical descriptors The electrostatic descriptors are parameters whichdepend on the charge distribution within the molecule includ-ing the dipole moment Examples of quantum chemically deriveddescriptors are various energy values like ionization energiesHOMOndashLUMO gap etc Similarly descriptors derived from molecu-lar mechanics can be used (Dyekjaeligret al 2002) A variety ofdifferent types of descriptors used in QSAR and QSPR are discussedby Karelson (2000)
QSAR and QSPR models are empirical equations used forestimating various properties of molecules and have the form
P = a + b middot D1 + c middot D2 + d middot D3 + middot middot middot (1)
where P is the property of interestab c are regressioncoefficients andD1D2D3 are the descriptors
The so-called 3D QSAR methods differ somewhat from traditionalQSAR methods The CoMFA (comparative molecular field analysis)(Crameret al 1988 httpwwwtriposcomsciTechinSilicoDiscstrActRelationship) approach one of the most popular 3D QSARmethods uses steric and electrostatic interaction energies betweenprobe atoms and the molecules as descriptors In the VolSurf method(Crucianiet al 2000) a similar approach is used where descriptorsare generated from molecular interaction fields calculated by theGRID program (Goodford 1985)
REDUNDANCY OF DATA SETSWhen using data extracted from large databases to train and testdata driven prediction tools the redundancy completeness and rep-resentativeness of the underlying data sets are extremely importantThe redundancy affects strongly the evaluation of the predictive per-formance and also most importantly the bias of the method towardspecific over-represented subclasses of compounds
Within the general bioinformatics area this is an issue that has beendealt with in great detail It is well known that if a protein structureprediction algorithm is tested on sequences and structures whichare highly similar to the sequences and structures used to train itthe predictive performance is significantly overestimated (Sanderand Schneider 1991) Therefore methods have been constructed forcleaning data sets for examples that are lsquotoo easyrsquo such that theperformance of the algorithm on novel data will be estimated in amore reliable manner One needs obviously to define the similaritymeasure between the data objects under consideration for aminoacid sequences one typically uses alignment techniques such as themetric for quantitative pair-wise comparison (Hobohmet al 1992)
2150
Prediction methods and databases within chemoinformatics
Techniques have also been developed for redundancy reduction ofsequences containing functional sites such as signal peptide cleav-age sites in secreted proteins (Nielsen et al 1996) or translationinitiation sites in mRNA sequences (Pedersen and Nielsen 1997)
In this respect chemoinformatics is still in its infancy comparedto the thorough validation methods used in bioinformatics Forchemoinformatics applications redundancy within databases canaffect the learning capabilities of neural networks decision treesand other methods whereas redundancy between different databasesgenerally introduces noise and reduces the discriminating power ofthe model
Redundancy in this context means that the compounds in theirvectors component for component are similar Similarity of thechemical compounds within a data set can lead to over-fitting of amodel such that predictions for compounds similar to those used inthe training set are excellent but for different compounds the predic-tions are not accurate to the same level There are several examplesof compound clustering techniques where the Tanimoto coefficient(Patterson et al 1996) has been used as the metric quantifying thesimilarity between compounds normally with the goal of identifyingleads by similarity or for increasing the diversity of libraries used forscreening (Reynolds et al 1998 2001 Voigt et al 2001 Willett2003) The Tanimoto coefficient T compares binary fingerprintvectors and is defined as
T (x y) = Nxy(Nx + Ny minus Nxy) (2)
where Nxy is the number of 1 bits shared in the fingerprints ofmolecules x and y Nx the number of 1 bits in the fingerprintof molecule x and Ny the number of 1 bits in the fingerprint ofmolecule y A Tanimoto coefficient approaching zero indicates thatthe compounds being compared are very different while a Tanimotocoefficient approaching one indicates that they are very similar
In the drug-likeness predictor developed by Frimurer et al(2000) the concept of redundancy reduction normally used withinbioinformatics was transferred to compound data extracted fromMDDR and ACD In this case the compounds were representedby CONCORD atom types and the Tanimoto coefficient (Patter-son et al 1996) was used to calculate the similarity between anytwo compounds Using a given threshold for maximal similarity interms of a Tanimoto coefficient of say 085 the redundancy in a dataset can be reduced to a well-defined level This type of technique alsoallows for the creation of common benchmark principles and couldreplace frozen benchmark data sets which often become outdated asdatabases grow over time The performance of prediction techniquesdeveloped on different data sets can therefore be compared in a fairmanner despite the fact that the underlying data sets differ in size
In most of the chemoinformatics applications developed so far thiskind of data set cleaning has not been carried out Instead test setshave been selected randomly from large overall data sets therebyintroducing many cases where highly similar pairs of examplesare found both in the training and test parts of the data sets Theestimation of the predictive performance is therefore much moreconservative in the work of Frimurer et al while it may be too highfor some of the prediction tools where the split between training andtest data has been established just by random selection
Note that similarity to a neural network is somewhat different frommolecular similarity because the network can correlate all the vectorcomponents with each other in a nonlinear fashion Two very differ-ent compounds may appear very similar for a neural net in terms of
functionality while two quite similar compounds can be interpretedas having different properties A redundant data set will typically notconstrain the network weight structure as much as a nonredundantdata set Nonredundant data from complete and representative datasets will therefore in most cases lead to well-performing predictorseven if the weightstraining examples ratio is larger
METHODS FOR CLASSIFICATION OF DRUG-LIKESTRUCTURESSeveral methods have been developed to determine the suitability ofcompounds to be used as drugs (pharmaceuticals) based on know-ledge about the molecular structure and key physical propertiesReviews about a variety of methods have been published recently(Walters and Murcko 2002 Clark and Pickett 2000 Muegge 2003)
The task is to select compounds from combinatorial librariesin such a way that the probability of identifying a new drugthat will make it to the market is optimized To minimize theprobability of leaving out potential drugs in the HTS it is import-ant to ensure adequate diversity of the molecular structures used(Gillet et al 1999) A variety of methods for compound selec-tion have been proposed ranging from simple intuitive to advancedcomputational methods
Intuitive and graphical methodsMany researchers have developed simple counting methods and ahighly popular method is the lsquorule of fiversquo proposed by Lipinski et al(1997) Although the lsquorule of fiversquo originally addresses the possibil-ity or risk for poor absorption or permeation based on molecularweight (MW) the octanolndashwater partition coefficient (log P alsocalled lipophilicity) number of hydrogen bond donors (HBD) andacceptors (HBA) respectively it has been widely used to distinguishbetween drug-like and non-drug-like compounds A number of data-bases have included Lipinskisrsquo lsquorule of fiversquo data for each compoundusing calculated values for log P This is the case for SPECS MDLSCD ACD ACDLabs Physico-Chemical databases
Frimurer et al (2000) performed a detailed quantitative analysisassessment of the predictive value of Lipinskisrsquo lsquorule of fiversquo It wasfound that the correlation coefficient is close to zero in fact slightlynegative when evaluated on a nonredundant data set Oprea (2000)also discusses drug-related chemical databases and the performanceof the lsquorule of fiversquo He concludes that the lsquorule of fiversquo does not dis-tinguish between lsquodrugsrsquo and lsquonon-drugsrsquo because the distributionof the parameters used does not differ significantly between thesetwo groups of compounds The problem with the lsquorule of fiversquo isthat it represents conditions which are necessary but not sufficientfor a drug molecule Many (in fact most) of the molecules whichfulfill these conditions are not drug molecules The work of Lip-inski has had however a tremendous pioneering value concerningquantitative evaluation of lsquodrug-likenessrsquo of chemical structures
Muegge et al (2001) proposed a simple selection scheme fordrug-likeness based on the presence of certain functional groupspharmacophore points in the molecule This method and other meth-ods using functional group filters are significantly less accurate thanthe various machine learning methods discussed (Table 3)
Chemistry space or chemography methods are somewhat similarand are based on the hypothesis that drug-like molecules share certainproperties and thus cluster in graphical representations Analysis ofhow well molecular diversity is captured using a variety of descriptors
2151
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160
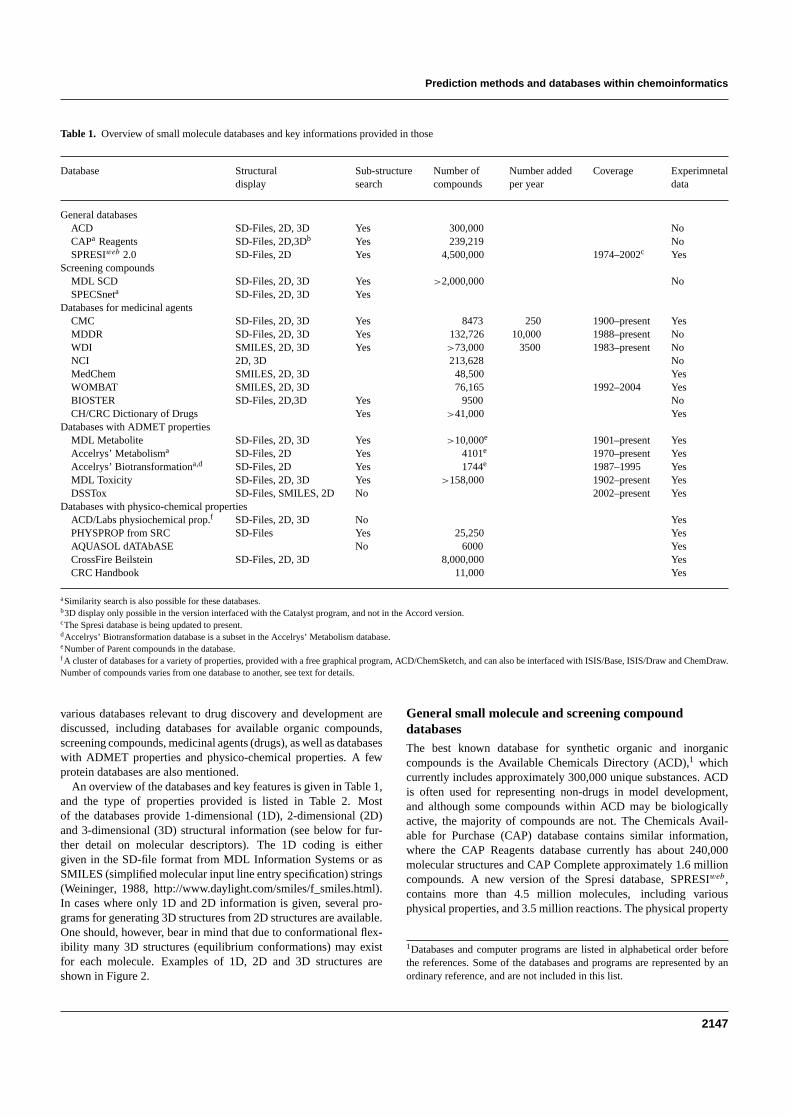
Prediction methods and databases within chemoinformatics
Table 1 Overview of small molecule databases and key informations provided in those
Database Structural Sub-structure Number of Number added Coverage Experimnetaldisplay search compounds per year data
General databasesACD SD-Files 2D 3D Yes 300000 NoCAPa Reagents SD-Files 2D3Db Yes 239219 NoSPRESIweb 20 SD-Files 2D Yes 4500000 1974ndash2002c Yes
Screening compoundsMDL SCD SD-Files 2D 3D Yes gt2000000 NoSPECSneta SD-Files 2D 3D Yes
Databases for medicinal agentsCMC SD-Files 2D 3D Yes 8473 250 1900ndashpresent YesMDDR SD-Files 2D 3D Yes 132726 10000 1988ndashpresent NoWDI SMILES 2D 3D Yes gt73000 3500 1983ndashpresent NoNCI 2D 3D 213628 NoMedChem SMILES 2D 3D 48500 YesWOMBAT SMILES 2D 3D 76165 1992ndash2004 YesBIOSTER SD-Files 2D3D Yes 9500 NoCHCRC Dictionary of Drugs Yes gt41000 Yes
Databases with ADMET propertiesMDL Metabolite SD-Files 2D 3D Yes gt10000e 1901ndashpresent YesAccelrysrsquo Metabolisma SD-Files 2D Yes 4101e 1970ndashpresent YesAccelrysrsquo Biotransformationad SD-Files 2D Yes 1744e 1987ndash1995 YesMDL Toxicity SD-Files 2D 3D Yes gt158000 1902ndashpresent YesDSSTox SD-Files SMILES 2D No 2002ndashpresent Yes
Databases with physico-chemical propertiesACDLabs physiochemical propf SD-Files 2D 3D No YesPHYSPROP from SRC SD-Files Yes 25250 YesAQUASOL dATAbASE No 6000 YesCrossFire Beilstein SD-Files 2D 3D 8000000 YesCRC Handbook 11000 Yes
aSimilarity search is also possible for these databasesb3D display only possible in the version interfaced with the Catalyst program and not in the Accord versioncThe Spresi database is being updated to presentdAccelrysrsquo Biotransformation database is a subset in the Accelrysrsquo Metabolism databaseeNumber of Parent compounds in the databasef A cluster of databases for a variety of properties provided with a free graphical program ACDChemSketch and can also be interfaced with ISISBase ISISDraw and ChemDrawNumber of compounds varies from one database to another see text for details
various databases relevant to drug discovery and development arediscussed including databases for available organic compoundsscreening compounds medicinal agents (drugs) as well as databaseswith ADMET properties and physico-chemical properties A fewprotein databases are also mentioned
An overview of the databases and key features is given in Table 1and the type of properties provided is listed in Table 2 Mostof the databases provide 1-dimensional (1D) 2-dimensional (2D)and 3-dimensional (3D) structural information (see below for fur-ther detail on molecular descriptors) The 1D coding is eithergiven in the SD-file format from MDL Information Systems or asSMILES (simplified molecular input line entry specification) strings(Weininger 1988 httpwwwdaylightcomsmilesf_smileshtml)In cases where only 1D and 2D information is given several pro-grams for generating 3D structures from 2D structures are availableOne should however bear in mind that due to conformational flex-ibility many 3D structures (equilibrium conformations) may existfor each molecule Examples of 1D 2D and 3D structures areshown in Figure 2
General small molecule and screening compounddatabasesThe best known database for synthetic organic and inorganiccompounds is the Available Chemicals Directory (ACD)1 whichcurrently includes approximately 300000 unique substances ACDis often used for representing non-drugs in model developmentand although some compounds within ACD may be biologicallyactive the majority of compounds are not The Chemicals Avail-able for Purchase (CAP) database contains similar informationwhere the CAP Reagents database currently has about 240000molecular structures and CAP Complete approximately 16 millioncompounds A new version of the Spresi database SPRESIwebcontains more than 45 million molecules including variousphysical properties and 35 million reactions The physical property
1Databases and computer programs are listed in alphabetical order beforethe references Some of the databases and programs are represented by anordinary reference and are not included in this list
2147
SOacuteJoacutensdoacutettir et al
Table 2 Overview of relevant experimental physico-chemical and ADMET properties provided in small molecule databases
Database Drug class Drug activity log P pKa Solubility Toxicity Metabolism
CMC Yes Yes Yes Yes No mdash mdashMDDR Yes Yes(ql)a mdash mdash mdash mdash mdashWDI Yes Yes(ql) mdash mdash mdash mdash mdashNCI Yes Yes(ql) mdash mdash mdash mdash mdashMedChem Yes Yes Yes Yes No mdash mdashWOMBAT Yes Yes Yes No Yes mdash mdashCHCRC Dictionary of Drugs mdash mdash Yes Yes Yes(ql) Yes mdashMDL Metabolite mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Metabolism mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Biotransformation mdash mdash mdash mdash mdash mdash YesMDL Toxicity mdash mdash mdash mdash mdash Yes mdashDSSTox mdash mdash mdash mdash mdash Yes mdashACDLabs physico-chemical prop mdash mdash Yes Yes Yes mdash mdashPHYSPROP from SRC mdash mdash Yes Yes Yes mdash mdashAQUASOL dATAbASE mdash mdash No No Yes mdash mdashCRC mdash mdash No No Yes(ql) No mdash
aql means that drug activity is given in qualitative terms
Fig 2 Structural information for a molecule can be given as a 1D SMILES string a 2D drawing or a 3D representation The SMILES string and the 2Ddrawing contain information about which atoms are bound to one another and the 3D representation shows how the atoms are located geometrically in relationto one another The atomic coordinates used are shown behind the 3D picture
information is however not well organized within the Spresidatabase
A number of databases have been developed especially for screen-ing purposes Three main catalogs previously being part of ACDthe SALOR (SigmandashAldrich Library of Rare Chemicals) Maybridgeand Bionet are now available through MDL Screening Com-pounds Directory (MDL SCD) The SPECSnet database contains
screening compounds building blocks and natural products providedby SPECS
Databases for medicinal agentsThe three main database collections of drug molecules are the Com-prehensive Medical Chemistry database (CMC) MDL Drug DataReport (MDDR) and the Derwent Word Drug Index (WDI) CMC
2148
Prediction methods and databases within chemoinformatics
contains presently 8473 drug compounds and is updated annuallywith compounds identified for the first time in the United StatesApproved Names (USAN) list The CMC database also includesinformation about drug class and measured or estimated valuesfor the acidndashbase dissociation constant (pKa) and the octanolwaterpartition coefficient (log P ) For log P 120 experimental and 8300calculated records are provided and for pKa 1200 measured recordsare given The MDDR contains 132726 drugs launched and underdevelopment collected from the patent literature and other relevantsources MDDR is updated monthly which adds up to approximately10000 new compounds per year and includes information aboutdrug class and drug activity in qualitative terms as well WDI con-tains around 73000 marketed drugs and drugs under developmentwith each record classified by drug activity mechanism of actiontreatment among other factors The Derwent Drug File is a highlyfocused database of selected journal articles and conference reportson all aspects of drug development
There are a number of other available drug databases The NationalCancer Institute database (NCI) contains 213628 compounds andis made up of four publicly available NCI databases including boththe AIDS and the Cancer databases The MedChem database con-sists of 48500 compounds 67000 measured log P values 13700measured pKa values and 19000 pharmacological (drug) activit-ies According to the providers of the MedChem Bio-loom databaseall available data for log P and pKa have been gathered from theliterature In addition the database includes a sorted list contain-ing only those measurements carried out with high quality methodsThe MedChem database also contains a collection of biological andphysico-chemical data intended for QSPR (quantitative structurendashproperty relationship) modeling and software for calculating log P
values (Clog P ) The WOrld of Molecular BioAcivities database(WOMBAT) is a very comprehensive collection of biological activ-ity data and contains 76165 molecules 68543 SMILES 143000activities for 630 targets 1230 measured log P values and 527 meas-ured solubility values The biological activities in this database aremainly related to receptor antagonists and enzyme inhibitors whichare ranked by target class The BIOSTER is a database of bioanalog-ous pairs of molecules (bio-isosters) and contains over 9500 activemolecules including drugs agrochemicals and enzyme inhibitorsThe Chapman amp HallCRC Dictionary of Drugs contains over 41000drugs and available physico-chemical property and toxicity data
Databases for ADMET properties of drugsThe MDL Metabolite database contains information about meta-bolism pathways for xenobiotic compounds and biotransformations(primary medicinal agents) and experimental data from in vivoand in vitro studies The database includes more than 10000 par-ent compounds over 64000 biotransformations and over 40000molecules (parent compounds intermediates and final metabolites)Metabolism data are also found in the Accelrysrsquo Metabolism andBiotransformation databases The Metabolism database covers ver-tebrates (animals) invertebrates and plants This databases contains4101 parent compounds 30000 transformations and is being exten-ded to 40000 transformations The Accelrysrsquo Biotransformationdatabase on the other hand is a stand-alone database for verteb-rates only containing 1744 unique parent compounds and 9809transformations
The MDL Toxicity database includes more than 158000 chem-ical substances compiled from several major sources where around
65 are drugs and drug development compounds The database con-tains six categories of data ie acute toxicity mutagenicity skineyeirritation tumorigenicity reproductive effects and multiple doseeffects It also contains detailed information about how the in vivoand in vitro experiments were carried out including species of organ-ism or tissue studied dose etc The DSSTox project (DistributedStructure-Searchable Toxicity Public Database Network) (Richardand Williams 2002 httpwwwepagovnheerldsstox) is a forumfor publishing toxicity data which presently includes databases forcarcinogenicity in various animals as well as acute toxicity
The ToxExpress Reference Database is a toxicogenomic data-base which contains gene expression profiles from known toxicantsThis database is built on in vivo and in vitro studies of exposure totoxicants and around 110 compounds have been profiled
Other databases that might be of interest although primarilyfocused on environmental and occupational health issues are theTSCA93 database containing over 100000 substances and theTOXicology Data NETwork (TOXNET) database cluster
Other important small molecule databasesThe ACDLabs Physico-Chemical Property databases contain a vari-ety of physico-chemical data for organic substances includingexperimental log P for 18400 compounds over 31000 measuredpKa values for 16000 compounds and aqueous solubility (logS)for 5000 compounds Another collection of experimental data is thePhysical Properties Database (PHYSPROP) from Syracuse ResearchCorporation (SRC) which contains 13250 measured log P values1652 pKa values and 6340 records for aqueous solubility BothACDLabs and SRC market software for prediction of these and otherproperties A third database the AQUASOL dATAbASE containsaqueous solubilities for almost 6000 compounds
There are other important databases for small molecules Cross-Fire Beilstein contains more than 8 million organic compoundsover 9 million reactions a variety of properties including variousphysical properties pharmacodynamics and environmental toxicityThis database contains over 500000 bioactive compounds Otherimportant collections of physical properties of organic compoundsare CRC (Lide 2003 httpwwwhbcpnetbasecom) (11000 com-pounds) Chapman amp HallCRC Properties of Organic Compounds(29000 compounds) and the Design Institute of Physical PropertiesDatabase (DIPPR) (1743 compounds)
Protein databases and ligand informationIn this context we would also like to mention a few relevantmacromolecular and crystallographic databases with drug-discoveryrelevant information The Protein Data Bank (PDB) is the mainsource for available protein crystal structures and structural inform-ation obtained with NMR spectroscopy with currently more than28000 structures and a weekly growth of about 100 structures TheCambridge Structural Database (CSD) is the most comprehensivecollection of crystallographic data for small molecules containingaround 305000 structures Figure 3 shows the evolution in numberof structures within PDB and CSD over the last 12 years AlthoughCSD has a much larger number of entries the relative growth ratewithin PDB is higher which shows clearly the increased focus onprotein structure determination world wide (structural genomics)
PDB contains a number of useful sub-databases including aTarget Registration Database Relibase (Hendlich et al 2003Guumlnther et al 2003 httprelibaseccdccamacuk) is a powerful
2149
SOacuteJoacutensdoacutettir et al
Fig 3 Number of crystallographic structures in the CSD and the PDB inthe period 1992ndash2004 This figure can be viewed in colour onBioinformaticsonline
data mining tool which utilizes structural information aboutproteinndashligand complexes within PDB for comprehensive analysisof proteinndashligand interactions Relibase+ is an improved version ofRelibase with a number of additional features like proteinndashproteininteraction searching and a crystal packing module for studyingcrystallographic effects around ligand binding sites The LIGANDdatabase (Gotoet al 1998 2002 httpwwwgenomeadjpligand)is a chemical database for enzyme reactions including informationabout structures of metabolites and other chemical compounds theirreactions in biological pathways and about the nomenclature ofenzymes
Ji et al (2003) recently published an overview of various data col-lections for proteins associated with drug therapeutic effects adversereactions and ADME available over the internet
A number of databases containing proteinndashprotein interactiondata are available including Database of Interacting Proteins(DIP) (Xenearioset al 2002 httpdipdoe-mbiuclaedu) Bio-molecular Interaction Network Database (BIND) (Baderet al2003 httpbindca) Molecular Interaction Database (MINT)(Zanzoniet al 2002 httpcbmbiouniroma2itmint) Compre-hensive Yeast Protein Genome Database (MIPS) (Meweset al2002 httpmipsgsfdegenreprojyeast) Yeast Proteome Data-base (YPD) (Hodgeset al 1999 Costanzoet al 2000) the IntActdatabase (Hermjakobet al 2004 httpwwwebiacukintactindexhtml) and Human Protein Reference Database (HPRD) (Periet al2003 httpwwwhprdorg) among others Proteinndashprotein interac-tion databases are collections of information about actual proteinndashprotein contacts or complex associations within the proteome ofspecific organisms in particularly in yeast but also in humansfruitfly and other organisms
DESCRIPTORS USED FOR CHEMICALSTRUCTURESIn chemoinformatics the structural features of the individualmolecules are described by various parameters derived from themolecular structure the so-called descriptors (Todeschini andConsonni 2000) The simplest types are constitutional (1D) ortopological (2D) descriptors describing the types of atoms func-tional groups or types and order of the chemical bonds withinthe molecules Ghose atom types (Viswanadhanet al 1989)CONCORD atom types ISIS keys (MDL keys) (Durantet al 2002)and various other atombond indices discussed in the following
section are examples of such descriptors Physico-chemical proper-ties are often used as descriptors as well
The ISIS keys and CONCORD atom types are often referred toas fingerprint methods and were for example used in the training ofneural networks (Frimureret al 2000) for compound classificationISIS keys are a set of codes which represent the presence of certainfunctional groups and other structural features in the molecules andCONCORD atom types are generated by assigning up to six atom-type codes available within the CONCORD program (Pearlman2000) to each atom
In QSAR (quantitative structurendashactivity relationship) and QSPRmodeling constitutional and topological descriptors are combinedwith more complicated descriptors representing the 3D structureof the molecules This includes various geometrical descriptorsand most importantly a variety of electrostatic and quantum chem-ical descriptors The electrostatic descriptors are parameters whichdepend on the charge distribution within the molecule includ-ing the dipole moment Examples of quantum chemically deriveddescriptors are various energy values like ionization energiesHOMOndashLUMO gap etc Similarly descriptors derived from molecu-lar mechanics can be used (Dyekjaeligret al 2002) A variety ofdifferent types of descriptors used in QSAR and QSPR are discussedby Karelson (2000)
QSAR and QSPR models are empirical equations used forestimating various properties of molecules and have the form
P = a + b middot D1 + c middot D2 + d middot D3 + middot middot middot (1)
where P is the property of interestab c are regressioncoefficients andD1D2D3 are the descriptors
The so-called 3D QSAR methods differ somewhat from traditionalQSAR methods The CoMFA (comparative molecular field analysis)(Crameret al 1988 httpwwwtriposcomsciTechinSilicoDiscstrActRelationship) approach one of the most popular 3D QSARmethods uses steric and electrostatic interaction energies betweenprobe atoms and the molecules as descriptors In the VolSurf method(Crucianiet al 2000) a similar approach is used where descriptorsare generated from molecular interaction fields calculated by theGRID program (Goodford 1985)
REDUNDANCY OF DATA SETSWhen using data extracted from large databases to train and testdata driven prediction tools the redundancy completeness and rep-resentativeness of the underlying data sets are extremely importantThe redundancy affects strongly the evaluation of the predictive per-formance and also most importantly the bias of the method towardspecific over-represented subclasses of compounds
Within the general bioinformatics area this is an issue that has beendealt with in great detail It is well known that if a protein structureprediction algorithm is tested on sequences and structures whichare highly similar to the sequences and structures used to train itthe predictive performance is significantly overestimated (Sanderand Schneider 1991) Therefore methods have been constructed forcleaning data sets for examples that are lsquotoo easyrsquo such that theperformance of the algorithm on novel data will be estimated in amore reliable manner One needs obviously to define the similaritymeasure between the data objects under consideration for aminoacid sequences one typically uses alignment techniques such as themetric for quantitative pair-wise comparison (Hobohmet al 1992)
2150
Prediction methods and databases within chemoinformatics
Techniques have also been developed for redundancy reduction ofsequences containing functional sites such as signal peptide cleav-age sites in secreted proteins (Nielsen et al 1996) or translationinitiation sites in mRNA sequences (Pedersen and Nielsen 1997)
In this respect chemoinformatics is still in its infancy comparedto the thorough validation methods used in bioinformatics Forchemoinformatics applications redundancy within databases canaffect the learning capabilities of neural networks decision treesand other methods whereas redundancy between different databasesgenerally introduces noise and reduces the discriminating power ofthe model
Redundancy in this context means that the compounds in theirvectors component for component are similar Similarity of thechemical compounds within a data set can lead to over-fitting of amodel such that predictions for compounds similar to those used inthe training set are excellent but for different compounds the predic-tions are not accurate to the same level There are several examplesof compound clustering techniques where the Tanimoto coefficient(Patterson et al 1996) has been used as the metric quantifying thesimilarity between compounds normally with the goal of identifyingleads by similarity or for increasing the diversity of libraries used forscreening (Reynolds et al 1998 2001 Voigt et al 2001 Willett2003) The Tanimoto coefficient T compares binary fingerprintvectors and is defined as
T (x y) = Nxy(Nx + Ny minus Nxy) (2)
where Nxy is the number of 1 bits shared in the fingerprints ofmolecules x and y Nx the number of 1 bits in the fingerprintof molecule x and Ny the number of 1 bits in the fingerprint ofmolecule y A Tanimoto coefficient approaching zero indicates thatthe compounds being compared are very different while a Tanimotocoefficient approaching one indicates that they are very similar
In the drug-likeness predictor developed by Frimurer et al(2000) the concept of redundancy reduction normally used withinbioinformatics was transferred to compound data extracted fromMDDR and ACD In this case the compounds were representedby CONCORD atom types and the Tanimoto coefficient (Patter-son et al 1996) was used to calculate the similarity between anytwo compounds Using a given threshold for maximal similarity interms of a Tanimoto coefficient of say 085 the redundancy in a dataset can be reduced to a well-defined level This type of technique alsoallows for the creation of common benchmark principles and couldreplace frozen benchmark data sets which often become outdated asdatabases grow over time The performance of prediction techniquesdeveloped on different data sets can therefore be compared in a fairmanner despite the fact that the underlying data sets differ in size
In most of the chemoinformatics applications developed so far thiskind of data set cleaning has not been carried out Instead test setshave been selected randomly from large overall data sets therebyintroducing many cases where highly similar pairs of examplesare found both in the training and test parts of the data sets Theestimation of the predictive performance is therefore much moreconservative in the work of Frimurer et al while it may be too highfor some of the prediction tools where the split between training andtest data has been established just by random selection
Note that similarity to a neural network is somewhat different frommolecular similarity because the network can correlate all the vectorcomponents with each other in a nonlinear fashion Two very differ-ent compounds may appear very similar for a neural net in terms of
functionality while two quite similar compounds can be interpretedas having different properties A redundant data set will typically notconstrain the network weight structure as much as a nonredundantdata set Nonredundant data from complete and representative datasets will therefore in most cases lead to well-performing predictorseven if the weightstraining examples ratio is larger
METHODS FOR CLASSIFICATION OF DRUG-LIKESTRUCTURESSeveral methods have been developed to determine the suitability ofcompounds to be used as drugs (pharmaceuticals) based on know-ledge about the molecular structure and key physical propertiesReviews about a variety of methods have been published recently(Walters and Murcko 2002 Clark and Pickett 2000 Muegge 2003)
The task is to select compounds from combinatorial librariesin such a way that the probability of identifying a new drugthat will make it to the market is optimized To minimize theprobability of leaving out potential drugs in the HTS it is import-ant to ensure adequate diversity of the molecular structures used(Gillet et al 1999) A variety of methods for compound selec-tion have been proposed ranging from simple intuitive to advancedcomputational methods
Intuitive and graphical methodsMany researchers have developed simple counting methods and ahighly popular method is the lsquorule of fiversquo proposed by Lipinski et al(1997) Although the lsquorule of fiversquo originally addresses the possibil-ity or risk for poor absorption or permeation based on molecularweight (MW) the octanolndashwater partition coefficient (log P alsocalled lipophilicity) number of hydrogen bond donors (HBD) andacceptors (HBA) respectively it has been widely used to distinguishbetween drug-like and non-drug-like compounds A number of data-bases have included Lipinskisrsquo lsquorule of fiversquo data for each compoundusing calculated values for log P This is the case for SPECS MDLSCD ACD ACDLabs Physico-Chemical databases
Frimurer et al (2000) performed a detailed quantitative analysisassessment of the predictive value of Lipinskisrsquo lsquorule of fiversquo It wasfound that the correlation coefficient is close to zero in fact slightlynegative when evaluated on a nonredundant data set Oprea (2000)also discusses drug-related chemical databases and the performanceof the lsquorule of fiversquo He concludes that the lsquorule of fiversquo does not dis-tinguish between lsquodrugsrsquo and lsquonon-drugsrsquo because the distributionof the parameters used does not differ significantly between thesetwo groups of compounds The problem with the lsquorule of fiversquo isthat it represents conditions which are necessary but not sufficientfor a drug molecule Many (in fact most) of the molecules whichfulfill these conditions are not drug molecules The work of Lip-inski has had however a tremendous pioneering value concerningquantitative evaluation of lsquodrug-likenessrsquo of chemical structures
Muegge et al (2001) proposed a simple selection scheme fordrug-likeness based on the presence of certain functional groupspharmacophore points in the molecule This method and other meth-ods using functional group filters are significantly less accurate thanthe various machine learning methods discussed (Table 3)
Chemistry space or chemography methods are somewhat similarand are based on the hypothesis that drug-like molecules share certainproperties and thus cluster in graphical representations Analysis ofhow well molecular diversity is captured using a variety of descriptors
2151
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160
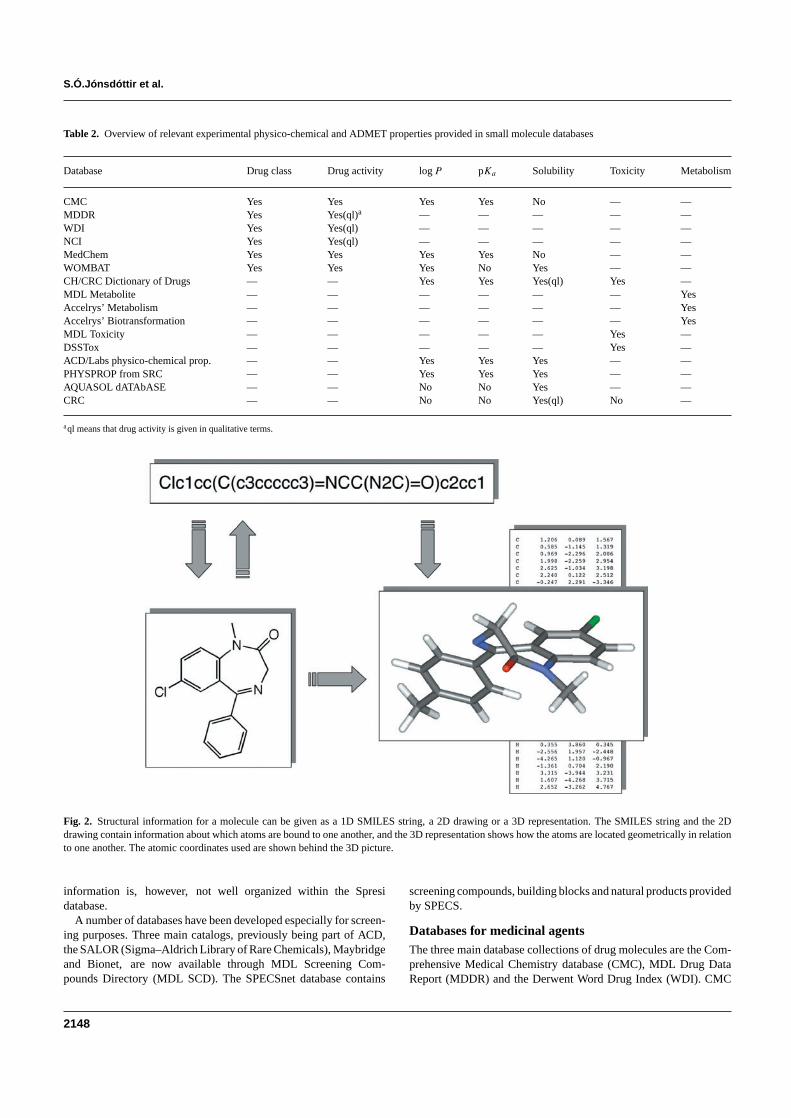
SOacuteJoacutensdoacutettir et al
Table 2 Overview of relevant experimental physico-chemical and ADMET properties provided in small molecule databases
Database Drug class Drug activity log P pKa Solubility Toxicity Metabolism
CMC Yes Yes Yes Yes No mdash mdashMDDR Yes Yes(ql)a mdash mdash mdash mdash mdashWDI Yes Yes(ql) mdash mdash mdash mdash mdashNCI Yes Yes(ql) mdash mdash mdash mdash mdashMedChem Yes Yes Yes Yes No mdash mdashWOMBAT Yes Yes Yes No Yes mdash mdashCHCRC Dictionary of Drugs mdash mdash Yes Yes Yes(ql) Yes mdashMDL Metabolite mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Metabolism mdash mdash mdash mdash mdash mdash YesAccelrysrsquo Biotransformation mdash mdash mdash mdash mdash mdash YesMDL Toxicity mdash mdash mdash mdash mdash Yes mdashDSSTox mdash mdash mdash mdash mdash Yes mdashACDLabs physico-chemical prop mdash mdash Yes Yes Yes mdash mdashPHYSPROP from SRC mdash mdash Yes Yes Yes mdash mdashAQUASOL dATAbASE mdash mdash No No Yes mdash mdashCRC mdash mdash No No Yes(ql) No mdash
aql means that drug activity is given in qualitative terms
Fig 2 Structural information for a molecule can be given as a 1D SMILES string a 2D drawing or a 3D representation The SMILES string and the 2Ddrawing contain information about which atoms are bound to one another and the 3D representation shows how the atoms are located geometrically in relationto one another The atomic coordinates used are shown behind the 3D picture
information is however not well organized within the Spresidatabase
A number of databases have been developed especially for screen-ing purposes Three main catalogs previously being part of ACDthe SALOR (SigmandashAldrich Library of Rare Chemicals) Maybridgeand Bionet are now available through MDL Screening Com-pounds Directory (MDL SCD) The SPECSnet database contains
screening compounds building blocks and natural products providedby SPECS
Databases for medicinal agentsThe three main database collections of drug molecules are the Com-prehensive Medical Chemistry database (CMC) MDL Drug DataReport (MDDR) and the Derwent Word Drug Index (WDI) CMC
2148
Prediction methods and databases within chemoinformatics
contains presently 8473 drug compounds and is updated annuallywith compounds identified for the first time in the United StatesApproved Names (USAN) list The CMC database also includesinformation about drug class and measured or estimated valuesfor the acidndashbase dissociation constant (pKa) and the octanolwaterpartition coefficient (log P ) For log P 120 experimental and 8300calculated records are provided and for pKa 1200 measured recordsare given The MDDR contains 132726 drugs launched and underdevelopment collected from the patent literature and other relevantsources MDDR is updated monthly which adds up to approximately10000 new compounds per year and includes information aboutdrug class and drug activity in qualitative terms as well WDI con-tains around 73000 marketed drugs and drugs under developmentwith each record classified by drug activity mechanism of actiontreatment among other factors The Derwent Drug File is a highlyfocused database of selected journal articles and conference reportson all aspects of drug development
There are a number of other available drug databases The NationalCancer Institute database (NCI) contains 213628 compounds andis made up of four publicly available NCI databases including boththe AIDS and the Cancer databases The MedChem database con-sists of 48500 compounds 67000 measured log P values 13700measured pKa values and 19000 pharmacological (drug) activit-ies According to the providers of the MedChem Bio-loom databaseall available data for log P and pKa have been gathered from theliterature In addition the database includes a sorted list contain-ing only those measurements carried out with high quality methodsThe MedChem database also contains a collection of biological andphysico-chemical data intended for QSPR (quantitative structurendashproperty relationship) modeling and software for calculating log P
values (Clog P ) The WOrld of Molecular BioAcivities database(WOMBAT) is a very comprehensive collection of biological activ-ity data and contains 76165 molecules 68543 SMILES 143000activities for 630 targets 1230 measured log P values and 527 meas-ured solubility values The biological activities in this database aremainly related to receptor antagonists and enzyme inhibitors whichare ranked by target class The BIOSTER is a database of bioanalog-ous pairs of molecules (bio-isosters) and contains over 9500 activemolecules including drugs agrochemicals and enzyme inhibitorsThe Chapman amp HallCRC Dictionary of Drugs contains over 41000drugs and available physico-chemical property and toxicity data
Databases for ADMET properties of drugsThe MDL Metabolite database contains information about meta-bolism pathways for xenobiotic compounds and biotransformations(primary medicinal agents) and experimental data from in vivoand in vitro studies The database includes more than 10000 par-ent compounds over 64000 biotransformations and over 40000molecules (parent compounds intermediates and final metabolites)Metabolism data are also found in the Accelrysrsquo Metabolism andBiotransformation databases The Metabolism database covers ver-tebrates (animals) invertebrates and plants This databases contains4101 parent compounds 30000 transformations and is being exten-ded to 40000 transformations The Accelrysrsquo Biotransformationdatabase on the other hand is a stand-alone database for verteb-rates only containing 1744 unique parent compounds and 9809transformations
The MDL Toxicity database includes more than 158000 chem-ical substances compiled from several major sources where around
65 are drugs and drug development compounds The database con-tains six categories of data ie acute toxicity mutagenicity skineyeirritation tumorigenicity reproductive effects and multiple doseeffects It also contains detailed information about how the in vivoand in vitro experiments were carried out including species of organ-ism or tissue studied dose etc The DSSTox project (DistributedStructure-Searchable Toxicity Public Database Network) (Richardand Williams 2002 httpwwwepagovnheerldsstox) is a forumfor publishing toxicity data which presently includes databases forcarcinogenicity in various animals as well as acute toxicity
The ToxExpress Reference Database is a toxicogenomic data-base which contains gene expression profiles from known toxicantsThis database is built on in vivo and in vitro studies of exposure totoxicants and around 110 compounds have been profiled
Other databases that might be of interest although primarilyfocused on environmental and occupational health issues are theTSCA93 database containing over 100000 substances and theTOXicology Data NETwork (TOXNET) database cluster
Other important small molecule databasesThe ACDLabs Physico-Chemical Property databases contain a vari-ety of physico-chemical data for organic substances includingexperimental log P for 18400 compounds over 31000 measuredpKa values for 16000 compounds and aqueous solubility (logS)for 5000 compounds Another collection of experimental data is thePhysical Properties Database (PHYSPROP) from Syracuse ResearchCorporation (SRC) which contains 13250 measured log P values1652 pKa values and 6340 records for aqueous solubility BothACDLabs and SRC market software for prediction of these and otherproperties A third database the AQUASOL dATAbASE containsaqueous solubilities for almost 6000 compounds
There are other important databases for small molecules Cross-Fire Beilstein contains more than 8 million organic compoundsover 9 million reactions a variety of properties including variousphysical properties pharmacodynamics and environmental toxicityThis database contains over 500000 bioactive compounds Otherimportant collections of physical properties of organic compoundsare CRC (Lide 2003 httpwwwhbcpnetbasecom) (11000 com-pounds) Chapman amp HallCRC Properties of Organic Compounds(29000 compounds) and the Design Institute of Physical PropertiesDatabase (DIPPR) (1743 compounds)
Protein databases and ligand informationIn this context we would also like to mention a few relevantmacromolecular and crystallographic databases with drug-discoveryrelevant information The Protein Data Bank (PDB) is the mainsource for available protein crystal structures and structural inform-ation obtained with NMR spectroscopy with currently more than28000 structures and a weekly growth of about 100 structures TheCambridge Structural Database (CSD) is the most comprehensivecollection of crystallographic data for small molecules containingaround 305000 structures Figure 3 shows the evolution in numberof structures within PDB and CSD over the last 12 years AlthoughCSD has a much larger number of entries the relative growth ratewithin PDB is higher which shows clearly the increased focus onprotein structure determination world wide (structural genomics)
PDB contains a number of useful sub-databases including aTarget Registration Database Relibase (Hendlich et al 2003Guumlnther et al 2003 httprelibaseccdccamacuk) is a powerful
2149
SOacuteJoacutensdoacutettir et al
Fig 3 Number of crystallographic structures in the CSD and the PDB inthe period 1992ndash2004 This figure can be viewed in colour onBioinformaticsonline
data mining tool which utilizes structural information aboutproteinndashligand complexes within PDB for comprehensive analysisof proteinndashligand interactions Relibase+ is an improved version ofRelibase with a number of additional features like proteinndashproteininteraction searching and a crystal packing module for studyingcrystallographic effects around ligand binding sites The LIGANDdatabase (Gotoet al 1998 2002 httpwwwgenomeadjpligand)is a chemical database for enzyme reactions including informationabout structures of metabolites and other chemical compounds theirreactions in biological pathways and about the nomenclature ofenzymes
Ji et al (2003) recently published an overview of various data col-lections for proteins associated with drug therapeutic effects adversereactions and ADME available over the internet
A number of databases containing proteinndashprotein interactiondata are available including Database of Interacting Proteins(DIP) (Xenearioset al 2002 httpdipdoe-mbiuclaedu) Bio-molecular Interaction Network Database (BIND) (Baderet al2003 httpbindca) Molecular Interaction Database (MINT)(Zanzoniet al 2002 httpcbmbiouniroma2itmint) Compre-hensive Yeast Protein Genome Database (MIPS) (Meweset al2002 httpmipsgsfdegenreprojyeast) Yeast Proteome Data-base (YPD) (Hodgeset al 1999 Costanzoet al 2000) the IntActdatabase (Hermjakobet al 2004 httpwwwebiacukintactindexhtml) and Human Protein Reference Database (HPRD) (Periet al2003 httpwwwhprdorg) among others Proteinndashprotein interac-tion databases are collections of information about actual proteinndashprotein contacts or complex associations within the proteome ofspecific organisms in particularly in yeast but also in humansfruitfly and other organisms
DESCRIPTORS USED FOR CHEMICALSTRUCTURESIn chemoinformatics the structural features of the individualmolecules are described by various parameters derived from themolecular structure the so-called descriptors (Todeschini andConsonni 2000) The simplest types are constitutional (1D) ortopological (2D) descriptors describing the types of atoms func-tional groups or types and order of the chemical bonds withinthe molecules Ghose atom types (Viswanadhanet al 1989)CONCORD atom types ISIS keys (MDL keys) (Durantet al 2002)and various other atombond indices discussed in the following
section are examples of such descriptors Physico-chemical proper-ties are often used as descriptors as well
The ISIS keys and CONCORD atom types are often referred toas fingerprint methods and were for example used in the training ofneural networks (Frimureret al 2000) for compound classificationISIS keys are a set of codes which represent the presence of certainfunctional groups and other structural features in the molecules andCONCORD atom types are generated by assigning up to six atom-type codes available within the CONCORD program (Pearlman2000) to each atom
In QSAR (quantitative structurendashactivity relationship) and QSPRmodeling constitutional and topological descriptors are combinedwith more complicated descriptors representing the 3D structureof the molecules This includes various geometrical descriptorsand most importantly a variety of electrostatic and quantum chem-ical descriptors The electrostatic descriptors are parameters whichdepend on the charge distribution within the molecule includ-ing the dipole moment Examples of quantum chemically deriveddescriptors are various energy values like ionization energiesHOMOndashLUMO gap etc Similarly descriptors derived from molecu-lar mechanics can be used (Dyekjaeligret al 2002) A variety ofdifferent types of descriptors used in QSAR and QSPR are discussedby Karelson (2000)
QSAR and QSPR models are empirical equations used forestimating various properties of molecules and have the form
P = a + b middot D1 + c middot D2 + d middot D3 + middot middot middot (1)
where P is the property of interestab c are regressioncoefficients andD1D2D3 are the descriptors
The so-called 3D QSAR methods differ somewhat from traditionalQSAR methods The CoMFA (comparative molecular field analysis)(Crameret al 1988 httpwwwtriposcomsciTechinSilicoDiscstrActRelationship) approach one of the most popular 3D QSARmethods uses steric and electrostatic interaction energies betweenprobe atoms and the molecules as descriptors In the VolSurf method(Crucianiet al 2000) a similar approach is used where descriptorsare generated from molecular interaction fields calculated by theGRID program (Goodford 1985)
REDUNDANCY OF DATA SETSWhen using data extracted from large databases to train and testdata driven prediction tools the redundancy completeness and rep-resentativeness of the underlying data sets are extremely importantThe redundancy affects strongly the evaluation of the predictive per-formance and also most importantly the bias of the method towardspecific over-represented subclasses of compounds
Within the general bioinformatics area this is an issue that has beendealt with in great detail It is well known that if a protein structureprediction algorithm is tested on sequences and structures whichare highly similar to the sequences and structures used to train itthe predictive performance is significantly overestimated (Sanderand Schneider 1991) Therefore methods have been constructed forcleaning data sets for examples that are lsquotoo easyrsquo such that theperformance of the algorithm on novel data will be estimated in amore reliable manner One needs obviously to define the similaritymeasure between the data objects under consideration for aminoacid sequences one typically uses alignment techniques such as themetric for quantitative pair-wise comparison (Hobohmet al 1992)
2150
Prediction methods and databases within chemoinformatics
Techniques have also been developed for redundancy reduction ofsequences containing functional sites such as signal peptide cleav-age sites in secreted proteins (Nielsen et al 1996) or translationinitiation sites in mRNA sequences (Pedersen and Nielsen 1997)
In this respect chemoinformatics is still in its infancy comparedto the thorough validation methods used in bioinformatics Forchemoinformatics applications redundancy within databases canaffect the learning capabilities of neural networks decision treesand other methods whereas redundancy between different databasesgenerally introduces noise and reduces the discriminating power ofthe model
Redundancy in this context means that the compounds in theirvectors component for component are similar Similarity of thechemical compounds within a data set can lead to over-fitting of amodel such that predictions for compounds similar to those used inthe training set are excellent but for different compounds the predic-tions are not accurate to the same level There are several examplesof compound clustering techniques where the Tanimoto coefficient(Patterson et al 1996) has been used as the metric quantifying thesimilarity between compounds normally with the goal of identifyingleads by similarity or for increasing the diversity of libraries used forscreening (Reynolds et al 1998 2001 Voigt et al 2001 Willett2003) The Tanimoto coefficient T compares binary fingerprintvectors and is defined as
T (x y) = Nxy(Nx + Ny minus Nxy) (2)
where Nxy is the number of 1 bits shared in the fingerprints ofmolecules x and y Nx the number of 1 bits in the fingerprintof molecule x and Ny the number of 1 bits in the fingerprint ofmolecule y A Tanimoto coefficient approaching zero indicates thatthe compounds being compared are very different while a Tanimotocoefficient approaching one indicates that they are very similar
In the drug-likeness predictor developed by Frimurer et al(2000) the concept of redundancy reduction normally used withinbioinformatics was transferred to compound data extracted fromMDDR and ACD In this case the compounds were representedby CONCORD atom types and the Tanimoto coefficient (Patter-son et al 1996) was used to calculate the similarity between anytwo compounds Using a given threshold for maximal similarity interms of a Tanimoto coefficient of say 085 the redundancy in a dataset can be reduced to a well-defined level This type of technique alsoallows for the creation of common benchmark principles and couldreplace frozen benchmark data sets which often become outdated asdatabases grow over time The performance of prediction techniquesdeveloped on different data sets can therefore be compared in a fairmanner despite the fact that the underlying data sets differ in size
In most of the chemoinformatics applications developed so far thiskind of data set cleaning has not been carried out Instead test setshave been selected randomly from large overall data sets therebyintroducing many cases where highly similar pairs of examplesare found both in the training and test parts of the data sets Theestimation of the predictive performance is therefore much moreconservative in the work of Frimurer et al while it may be too highfor some of the prediction tools where the split between training andtest data has been established just by random selection
Note that similarity to a neural network is somewhat different frommolecular similarity because the network can correlate all the vectorcomponents with each other in a nonlinear fashion Two very differ-ent compounds may appear very similar for a neural net in terms of
functionality while two quite similar compounds can be interpretedas having different properties A redundant data set will typically notconstrain the network weight structure as much as a nonredundantdata set Nonredundant data from complete and representative datasets will therefore in most cases lead to well-performing predictorseven if the weightstraining examples ratio is larger
METHODS FOR CLASSIFICATION OF DRUG-LIKESTRUCTURESSeveral methods have been developed to determine the suitability ofcompounds to be used as drugs (pharmaceuticals) based on know-ledge about the molecular structure and key physical propertiesReviews about a variety of methods have been published recently(Walters and Murcko 2002 Clark and Pickett 2000 Muegge 2003)
The task is to select compounds from combinatorial librariesin such a way that the probability of identifying a new drugthat will make it to the market is optimized To minimize theprobability of leaving out potential drugs in the HTS it is import-ant to ensure adequate diversity of the molecular structures used(Gillet et al 1999) A variety of methods for compound selec-tion have been proposed ranging from simple intuitive to advancedcomputational methods
Intuitive and graphical methodsMany researchers have developed simple counting methods and ahighly popular method is the lsquorule of fiversquo proposed by Lipinski et al(1997) Although the lsquorule of fiversquo originally addresses the possibil-ity or risk for poor absorption or permeation based on molecularweight (MW) the octanolndashwater partition coefficient (log P alsocalled lipophilicity) number of hydrogen bond donors (HBD) andacceptors (HBA) respectively it has been widely used to distinguishbetween drug-like and non-drug-like compounds A number of data-bases have included Lipinskisrsquo lsquorule of fiversquo data for each compoundusing calculated values for log P This is the case for SPECS MDLSCD ACD ACDLabs Physico-Chemical databases
Frimurer et al (2000) performed a detailed quantitative analysisassessment of the predictive value of Lipinskisrsquo lsquorule of fiversquo It wasfound that the correlation coefficient is close to zero in fact slightlynegative when evaluated on a nonredundant data set Oprea (2000)also discusses drug-related chemical databases and the performanceof the lsquorule of fiversquo He concludes that the lsquorule of fiversquo does not dis-tinguish between lsquodrugsrsquo and lsquonon-drugsrsquo because the distributionof the parameters used does not differ significantly between thesetwo groups of compounds The problem with the lsquorule of fiversquo isthat it represents conditions which are necessary but not sufficientfor a drug molecule Many (in fact most) of the molecules whichfulfill these conditions are not drug molecules The work of Lip-inski has had however a tremendous pioneering value concerningquantitative evaluation of lsquodrug-likenessrsquo of chemical structures
Muegge et al (2001) proposed a simple selection scheme fordrug-likeness based on the presence of certain functional groupspharmacophore points in the molecule This method and other meth-ods using functional group filters are significantly less accurate thanthe various machine learning methods discussed (Table 3)
Chemistry space or chemography methods are somewhat similarand are based on the hypothesis that drug-like molecules share certainproperties and thus cluster in graphical representations Analysis ofhow well molecular diversity is captured using a variety of descriptors
2151
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

Prediction methods and databases within chemoinformatics
contains presently 8473 drug compounds and is updated annuallywith compounds identified for the first time in the United StatesApproved Names (USAN) list The CMC database also includesinformation about drug class and measured or estimated valuesfor the acidndashbase dissociation constant (pKa) and the octanolwaterpartition coefficient (log P ) For log P 120 experimental and 8300calculated records are provided and for pKa 1200 measured recordsare given The MDDR contains 132726 drugs launched and underdevelopment collected from the patent literature and other relevantsources MDDR is updated monthly which adds up to approximately10000 new compounds per year and includes information aboutdrug class and drug activity in qualitative terms as well WDI con-tains around 73000 marketed drugs and drugs under developmentwith each record classified by drug activity mechanism of actiontreatment among other factors The Derwent Drug File is a highlyfocused database of selected journal articles and conference reportson all aspects of drug development
There are a number of other available drug databases The NationalCancer Institute database (NCI) contains 213628 compounds andis made up of four publicly available NCI databases including boththe AIDS and the Cancer databases The MedChem database con-sists of 48500 compounds 67000 measured log P values 13700measured pKa values and 19000 pharmacological (drug) activit-ies According to the providers of the MedChem Bio-loom databaseall available data for log P and pKa have been gathered from theliterature In addition the database includes a sorted list contain-ing only those measurements carried out with high quality methodsThe MedChem database also contains a collection of biological andphysico-chemical data intended for QSPR (quantitative structurendashproperty relationship) modeling and software for calculating log P
values (Clog P ) The WOrld of Molecular BioAcivities database(WOMBAT) is a very comprehensive collection of biological activ-ity data and contains 76165 molecules 68543 SMILES 143000activities for 630 targets 1230 measured log P values and 527 meas-ured solubility values The biological activities in this database aremainly related to receptor antagonists and enzyme inhibitors whichare ranked by target class The BIOSTER is a database of bioanalog-ous pairs of molecules (bio-isosters) and contains over 9500 activemolecules including drugs agrochemicals and enzyme inhibitorsThe Chapman amp HallCRC Dictionary of Drugs contains over 41000drugs and available physico-chemical property and toxicity data
Databases for ADMET properties of drugsThe MDL Metabolite database contains information about meta-bolism pathways for xenobiotic compounds and biotransformations(primary medicinal agents) and experimental data from in vivoand in vitro studies The database includes more than 10000 par-ent compounds over 64000 biotransformations and over 40000molecules (parent compounds intermediates and final metabolites)Metabolism data are also found in the Accelrysrsquo Metabolism andBiotransformation databases The Metabolism database covers ver-tebrates (animals) invertebrates and plants This databases contains4101 parent compounds 30000 transformations and is being exten-ded to 40000 transformations The Accelrysrsquo Biotransformationdatabase on the other hand is a stand-alone database for verteb-rates only containing 1744 unique parent compounds and 9809transformations
The MDL Toxicity database includes more than 158000 chem-ical substances compiled from several major sources where around
65 are drugs and drug development compounds The database con-tains six categories of data ie acute toxicity mutagenicity skineyeirritation tumorigenicity reproductive effects and multiple doseeffects It also contains detailed information about how the in vivoand in vitro experiments were carried out including species of organ-ism or tissue studied dose etc The DSSTox project (DistributedStructure-Searchable Toxicity Public Database Network) (Richardand Williams 2002 httpwwwepagovnheerldsstox) is a forumfor publishing toxicity data which presently includes databases forcarcinogenicity in various animals as well as acute toxicity
The ToxExpress Reference Database is a toxicogenomic data-base which contains gene expression profiles from known toxicantsThis database is built on in vivo and in vitro studies of exposure totoxicants and around 110 compounds have been profiled
Other databases that might be of interest although primarilyfocused on environmental and occupational health issues are theTSCA93 database containing over 100000 substances and theTOXicology Data NETwork (TOXNET) database cluster
Other important small molecule databasesThe ACDLabs Physico-Chemical Property databases contain a vari-ety of physico-chemical data for organic substances includingexperimental log P for 18400 compounds over 31000 measuredpKa values for 16000 compounds and aqueous solubility (logS)for 5000 compounds Another collection of experimental data is thePhysical Properties Database (PHYSPROP) from Syracuse ResearchCorporation (SRC) which contains 13250 measured log P values1652 pKa values and 6340 records for aqueous solubility BothACDLabs and SRC market software for prediction of these and otherproperties A third database the AQUASOL dATAbASE containsaqueous solubilities for almost 6000 compounds
There are other important databases for small molecules Cross-Fire Beilstein contains more than 8 million organic compoundsover 9 million reactions a variety of properties including variousphysical properties pharmacodynamics and environmental toxicityThis database contains over 500000 bioactive compounds Otherimportant collections of physical properties of organic compoundsare CRC (Lide 2003 httpwwwhbcpnetbasecom) (11000 com-pounds) Chapman amp HallCRC Properties of Organic Compounds(29000 compounds) and the Design Institute of Physical PropertiesDatabase (DIPPR) (1743 compounds)
Protein databases and ligand informationIn this context we would also like to mention a few relevantmacromolecular and crystallographic databases with drug-discoveryrelevant information The Protein Data Bank (PDB) is the mainsource for available protein crystal structures and structural inform-ation obtained with NMR spectroscopy with currently more than28000 structures and a weekly growth of about 100 structures TheCambridge Structural Database (CSD) is the most comprehensivecollection of crystallographic data for small molecules containingaround 305000 structures Figure 3 shows the evolution in numberof structures within PDB and CSD over the last 12 years AlthoughCSD has a much larger number of entries the relative growth ratewithin PDB is higher which shows clearly the increased focus onprotein structure determination world wide (structural genomics)
PDB contains a number of useful sub-databases including aTarget Registration Database Relibase (Hendlich et al 2003Guumlnther et al 2003 httprelibaseccdccamacuk) is a powerful
2149
SOacuteJoacutensdoacutettir et al
Fig 3 Number of crystallographic structures in the CSD and the PDB inthe period 1992ndash2004 This figure can be viewed in colour onBioinformaticsonline
data mining tool which utilizes structural information aboutproteinndashligand complexes within PDB for comprehensive analysisof proteinndashligand interactions Relibase+ is an improved version ofRelibase with a number of additional features like proteinndashproteininteraction searching and a crystal packing module for studyingcrystallographic effects around ligand binding sites The LIGANDdatabase (Gotoet al 1998 2002 httpwwwgenomeadjpligand)is a chemical database for enzyme reactions including informationabout structures of metabolites and other chemical compounds theirreactions in biological pathways and about the nomenclature ofenzymes
Ji et al (2003) recently published an overview of various data col-lections for proteins associated with drug therapeutic effects adversereactions and ADME available over the internet
A number of databases containing proteinndashprotein interactiondata are available including Database of Interacting Proteins(DIP) (Xenearioset al 2002 httpdipdoe-mbiuclaedu) Bio-molecular Interaction Network Database (BIND) (Baderet al2003 httpbindca) Molecular Interaction Database (MINT)(Zanzoniet al 2002 httpcbmbiouniroma2itmint) Compre-hensive Yeast Protein Genome Database (MIPS) (Meweset al2002 httpmipsgsfdegenreprojyeast) Yeast Proteome Data-base (YPD) (Hodgeset al 1999 Costanzoet al 2000) the IntActdatabase (Hermjakobet al 2004 httpwwwebiacukintactindexhtml) and Human Protein Reference Database (HPRD) (Periet al2003 httpwwwhprdorg) among others Proteinndashprotein interac-tion databases are collections of information about actual proteinndashprotein contacts or complex associations within the proteome ofspecific organisms in particularly in yeast but also in humansfruitfly and other organisms
DESCRIPTORS USED FOR CHEMICALSTRUCTURESIn chemoinformatics the structural features of the individualmolecules are described by various parameters derived from themolecular structure the so-called descriptors (Todeschini andConsonni 2000) The simplest types are constitutional (1D) ortopological (2D) descriptors describing the types of atoms func-tional groups or types and order of the chemical bonds withinthe molecules Ghose atom types (Viswanadhanet al 1989)CONCORD atom types ISIS keys (MDL keys) (Durantet al 2002)and various other atombond indices discussed in the following
section are examples of such descriptors Physico-chemical proper-ties are often used as descriptors as well
The ISIS keys and CONCORD atom types are often referred toas fingerprint methods and were for example used in the training ofneural networks (Frimureret al 2000) for compound classificationISIS keys are a set of codes which represent the presence of certainfunctional groups and other structural features in the molecules andCONCORD atom types are generated by assigning up to six atom-type codes available within the CONCORD program (Pearlman2000) to each atom
In QSAR (quantitative structurendashactivity relationship) and QSPRmodeling constitutional and topological descriptors are combinedwith more complicated descriptors representing the 3D structureof the molecules This includes various geometrical descriptorsand most importantly a variety of electrostatic and quantum chem-ical descriptors The electrostatic descriptors are parameters whichdepend on the charge distribution within the molecule includ-ing the dipole moment Examples of quantum chemically deriveddescriptors are various energy values like ionization energiesHOMOndashLUMO gap etc Similarly descriptors derived from molecu-lar mechanics can be used (Dyekjaeligret al 2002) A variety ofdifferent types of descriptors used in QSAR and QSPR are discussedby Karelson (2000)
QSAR and QSPR models are empirical equations used forestimating various properties of molecules and have the form
P = a + b middot D1 + c middot D2 + d middot D3 + middot middot middot (1)
where P is the property of interestab c are regressioncoefficients andD1D2D3 are the descriptors
The so-called 3D QSAR methods differ somewhat from traditionalQSAR methods The CoMFA (comparative molecular field analysis)(Crameret al 1988 httpwwwtriposcomsciTechinSilicoDiscstrActRelationship) approach one of the most popular 3D QSARmethods uses steric and electrostatic interaction energies betweenprobe atoms and the molecules as descriptors In the VolSurf method(Crucianiet al 2000) a similar approach is used where descriptorsare generated from molecular interaction fields calculated by theGRID program (Goodford 1985)
REDUNDANCY OF DATA SETSWhen using data extracted from large databases to train and testdata driven prediction tools the redundancy completeness and rep-resentativeness of the underlying data sets are extremely importantThe redundancy affects strongly the evaluation of the predictive per-formance and also most importantly the bias of the method towardspecific over-represented subclasses of compounds
Within the general bioinformatics area this is an issue that has beendealt with in great detail It is well known that if a protein structureprediction algorithm is tested on sequences and structures whichare highly similar to the sequences and structures used to train itthe predictive performance is significantly overestimated (Sanderand Schneider 1991) Therefore methods have been constructed forcleaning data sets for examples that are lsquotoo easyrsquo such that theperformance of the algorithm on novel data will be estimated in amore reliable manner One needs obviously to define the similaritymeasure between the data objects under consideration for aminoacid sequences one typically uses alignment techniques such as themetric for quantitative pair-wise comparison (Hobohmet al 1992)
2150
Prediction methods and databases within chemoinformatics
Techniques have also been developed for redundancy reduction ofsequences containing functional sites such as signal peptide cleav-age sites in secreted proteins (Nielsen et al 1996) or translationinitiation sites in mRNA sequences (Pedersen and Nielsen 1997)
In this respect chemoinformatics is still in its infancy comparedto the thorough validation methods used in bioinformatics Forchemoinformatics applications redundancy within databases canaffect the learning capabilities of neural networks decision treesand other methods whereas redundancy between different databasesgenerally introduces noise and reduces the discriminating power ofthe model
Redundancy in this context means that the compounds in theirvectors component for component are similar Similarity of thechemical compounds within a data set can lead to over-fitting of amodel such that predictions for compounds similar to those used inthe training set are excellent but for different compounds the predic-tions are not accurate to the same level There are several examplesof compound clustering techniques where the Tanimoto coefficient(Patterson et al 1996) has been used as the metric quantifying thesimilarity between compounds normally with the goal of identifyingleads by similarity or for increasing the diversity of libraries used forscreening (Reynolds et al 1998 2001 Voigt et al 2001 Willett2003) The Tanimoto coefficient T compares binary fingerprintvectors and is defined as
T (x y) = Nxy(Nx + Ny minus Nxy) (2)
where Nxy is the number of 1 bits shared in the fingerprints ofmolecules x and y Nx the number of 1 bits in the fingerprintof molecule x and Ny the number of 1 bits in the fingerprint ofmolecule y A Tanimoto coefficient approaching zero indicates thatthe compounds being compared are very different while a Tanimotocoefficient approaching one indicates that they are very similar
In the drug-likeness predictor developed by Frimurer et al(2000) the concept of redundancy reduction normally used withinbioinformatics was transferred to compound data extracted fromMDDR and ACD In this case the compounds were representedby CONCORD atom types and the Tanimoto coefficient (Patter-son et al 1996) was used to calculate the similarity between anytwo compounds Using a given threshold for maximal similarity interms of a Tanimoto coefficient of say 085 the redundancy in a dataset can be reduced to a well-defined level This type of technique alsoallows for the creation of common benchmark principles and couldreplace frozen benchmark data sets which often become outdated asdatabases grow over time The performance of prediction techniquesdeveloped on different data sets can therefore be compared in a fairmanner despite the fact that the underlying data sets differ in size
In most of the chemoinformatics applications developed so far thiskind of data set cleaning has not been carried out Instead test setshave been selected randomly from large overall data sets therebyintroducing many cases where highly similar pairs of examplesare found both in the training and test parts of the data sets Theestimation of the predictive performance is therefore much moreconservative in the work of Frimurer et al while it may be too highfor some of the prediction tools where the split between training andtest data has been established just by random selection
Note that similarity to a neural network is somewhat different frommolecular similarity because the network can correlate all the vectorcomponents with each other in a nonlinear fashion Two very differ-ent compounds may appear very similar for a neural net in terms of
functionality while two quite similar compounds can be interpretedas having different properties A redundant data set will typically notconstrain the network weight structure as much as a nonredundantdata set Nonredundant data from complete and representative datasets will therefore in most cases lead to well-performing predictorseven if the weightstraining examples ratio is larger
METHODS FOR CLASSIFICATION OF DRUG-LIKESTRUCTURESSeveral methods have been developed to determine the suitability ofcompounds to be used as drugs (pharmaceuticals) based on know-ledge about the molecular structure and key physical propertiesReviews about a variety of methods have been published recently(Walters and Murcko 2002 Clark and Pickett 2000 Muegge 2003)
The task is to select compounds from combinatorial librariesin such a way that the probability of identifying a new drugthat will make it to the market is optimized To minimize theprobability of leaving out potential drugs in the HTS it is import-ant to ensure adequate diversity of the molecular structures used(Gillet et al 1999) A variety of methods for compound selec-tion have been proposed ranging from simple intuitive to advancedcomputational methods
Intuitive and graphical methodsMany researchers have developed simple counting methods and ahighly popular method is the lsquorule of fiversquo proposed by Lipinski et al(1997) Although the lsquorule of fiversquo originally addresses the possibil-ity or risk for poor absorption or permeation based on molecularweight (MW) the octanolndashwater partition coefficient (log P alsocalled lipophilicity) number of hydrogen bond donors (HBD) andacceptors (HBA) respectively it has been widely used to distinguishbetween drug-like and non-drug-like compounds A number of data-bases have included Lipinskisrsquo lsquorule of fiversquo data for each compoundusing calculated values for log P This is the case for SPECS MDLSCD ACD ACDLabs Physico-Chemical databases
Frimurer et al (2000) performed a detailed quantitative analysisassessment of the predictive value of Lipinskisrsquo lsquorule of fiversquo It wasfound that the correlation coefficient is close to zero in fact slightlynegative when evaluated on a nonredundant data set Oprea (2000)also discusses drug-related chemical databases and the performanceof the lsquorule of fiversquo He concludes that the lsquorule of fiversquo does not dis-tinguish between lsquodrugsrsquo and lsquonon-drugsrsquo because the distributionof the parameters used does not differ significantly between thesetwo groups of compounds The problem with the lsquorule of fiversquo isthat it represents conditions which are necessary but not sufficientfor a drug molecule Many (in fact most) of the molecules whichfulfill these conditions are not drug molecules The work of Lip-inski has had however a tremendous pioneering value concerningquantitative evaluation of lsquodrug-likenessrsquo of chemical structures
Muegge et al (2001) proposed a simple selection scheme fordrug-likeness based on the presence of certain functional groupspharmacophore points in the molecule This method and other meth-ods using functional group filters are significantly less accurate thanthe various machine learning methods discussed (Table 3)
Chemistry space or chemography methods are somewhat similarand are based on the hypothesis that drug-like molecules share certainproperties and thus cluster in graphical representations Analysis ofhow well molecular diversity is captured using a variety of descriptors
2151
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

SOacuteJoacutensdoacutettir et al
Fig 3 Number of crystallographic structures in the CSD and the PDB inthe period 1992ndash2004 This figure can be viewed in colour onBioinformaticsonline
data mining tool which utilizes structural information aboutproteinndashligand complexes within PDB for comprehensive analysisof proteinndashligand interactions Relibase+ is an improved version ofRelibase with a number of additional features like proteinndashproteininteraction searching and a crystal packing module for studyingcrystallographic effects around ligand binding sites The LIGANDdatabase (Gotoet al 1998 2002 httpwwwgenomeadjpligand)is a chemical database for enzyme reactions including informationabout structures of metabolites and other chemical compounds theirreactions in biological pathways and about the nomenclature ofenzymes
Ji et al (2003) recently published an overview of various data col-lections for proteins associated with drug therapeutic effects adversereactions and ADME available over the internet
A number of databases containing proteinndashprotein interactiondata are available including Database of Interacting Proteins(DIP) (Xenearioset al 2002 httpdipdoe-mbiuclaedu) Bio-molecular Interaction Network Database (BIND) (Baderet al2003 httpbindca) Molecular Interaction Database (MINT)(Zanzoniet al 2002 httpcbmbiouniroma2itmint) Compre-hensive Yeast Protein Genome Database (MIPS) (Meweset al2002 httpmipsgsfdegenreprojyeast) Yeast Proteome Data-base (YPD) (Hodgeset al 1999 Costanzoet al 2000) the IntActdatabase (Hermjakobet al 2004 httpwwwebiacukintactindexhtml) and Human Protein Reference Database (HPRD) (Periet al2003 httpwwwhprdorg) among others Proteinndashprotein interac-tion databases are collections of information about actual proteinndashprotein contacts or complex associations within the proteome ofspecific organisms in particularly in yeast but also in humansfruitfly and other organisms
DESCRIPTORS USED FOR CHEMICALSTRUCTURESIn chemoinformatics the structural features of the individualmolecules are described by various parameters derived from themolecular structure the so-called descriptors (Todeschini andConsonni 2000) The simplest types are constitutional (1D) ortopological (2D) descriptors describing the types of atoms func-tional groups or types and order of the chemical bonds withinthe molecules Ghose atom types (Viswanadhanet al 1989)CONCORD atom types ISIS keys (MDL keys) (Durantet al 2002)and various other atombond indices discussed in the following
section are examples of such descriptors Physico-chemical proper-ties are often used as descriptors as well
The ISIS keys and CONCORD atom types are often referred toas fingerprint methods and were for example used in the training ofneural networks (Frimureret al 2000) for compound classificationISIS keys are a set of codes which represent the presence of certainfunctional groups and other structural features in the molecules andCONCORD atom types are generated by assigning up to six atom-type codes available within the CONCORD program (Pearlman2000) to each atom
In QSAR (quantitative structurendashactivity relationship) and QSPRmodeling constitutional and topological descriptors are combinedwith more complicated descriptors representing the 3D structureof the molecules This includes various geometrical descriptorsand most importantly a variety of electrostatic and quantum chem-ical descriptors The electrostatic descriptors are parameters whichdepend on the charge distribution within the molecule includ-ing the dipole moment Examples of quantum chemically deriveddescriptors are various energy values like ionization energiesHOMOndashLUMO gap etc Similarly descriptors derived from molecu-lar mechanics can be used (Dyekjaeligret al 2002) A variety ofdifferent types of descriptors used in QSAR and QSPR are discussedby Karelson (2000)
QSAR and QSPR models are empirical equations used forestimating various properties of molecules and have the form
P = a + b middot D1 + c middot D2 + d middot D3 + middot middot middot (1)
where P is the property of interestab c are regressioncoefficients andD1D2D3 are the descriptors
The so-called 3D QSAR methods differ somewhat from traditionalQSAR methods The CoMFA (comparative molecular field analysis)(Crameret al 1988 httpwwwtriposcomsciTechinSilicoDiscstrActRelationship) approach one of the most popular 3D QSARmethods uses steric and electrostatic interaction energies betweenprobe atoms and the molecules as descriptors In the VolSurf method(Crucianiet al 2000) a similar approach is used where descriptorsare generated from molecular interaction fields calculated by theGRID program (Goodford 1985)
REDUNDANCY OF DATA SETSWhen using data extracted from large databases to train and testdata driven prediction tools the redundancy completeness and rep-resentativeness of the underlying data sets are extremely importantThe redundancy affects strongly the evaluation of the predictive per-formance and also most importantly the bias of the method towardspecific over-represented subclasses of compounds
Within the general bioinformatics area this is an issue that has beendealt with in great detail It is well known that if a protein structureprediction algorithm is tested on sequences and structures whichare highly similar to the sequences and structures used to train itthe predictive performance is significantly overestimated (Sanderand Schneider 1991) Therefore methods have been constructed forcleaning data sets for examples that are lsquotoo easyrsquo such that theperformance of the algorithm on novel data will be estimated in amore reliable manner One needs obviously to define the similaritymeasure between the data objects under consideration for aminoacid sequences one typically uses alignment techniques such as themetric for quantitative pair-wise comparison (Hobohmet al 1992)
2150
Prediction methods and databases within chemoinformatics
Techniques have also been developed for redundancy reduction ofsequences containing functional sites such as signal peptide cleav-age sites in secreted proteins (Nielsen et al 1996) or translationinitiation sites in mRNA sequences (Pedersen and Nielsen 1997)
In this respect chemoinformatics is still in its infancy comparedto the thorough validation methods used in bioinformatics Forchemoinformatics applications redundancy within databases canaffect the learning capabilities of neural networks decision treesand other methods whereas redundancy between different databasesgenerally introduces noise and reduces the discriminating power ofthe model
Redundancy in this context means that the compounds in theirvectors component for component are similar Similarity of thechemical compounds within a data set can lead to over-fitting of amodel such that predictions for compounds similar to those used inthe training set are excellent but for different compounds the predic-tions are not accurate to the same level There are several examplesof compound clustering techniques where the Tanimoto coefficient(Patterson et al 1996) has been used as the metric quantifying thesimilarity between compounds normally with the goal of identifyingleads by similarity or for increasing the diversity of libraries used forscreening (Reynolds et al 1998 2001 Voigt et al 2001 Willett2003) The Tanimoto coefficient T compares binary fingerprintvectors and is defined as
T (x y) = Nxy(Nx + Ny minus Nxy) (2)
where Nxy is the number of 1 bits shared in the fingerprints ofmolecules x and y Nx the number of 1 bits in the fingerprintof molecule x and Ny the number of 1 bits in the fingerprint ofmolecule y A Tanimoto coefficient approaching zero indicates thatthe compounds being compared are very different while a Tanimotocoefficient approaching one indicates that they are very similar
In the drug-likeness predictor developed by Frimurer et al(2000) the concept of redundancy reduction normally used withinbioinformatics was transferred to compound data extracted fromMDDR and ACD In this case the compounds were representedby CONCORD atom types and the Tanimoto coefficient (Patter-son et al 1996) was used to calculate the similarity between anytwo compounds Using a given threshold for maximal similarity interms of a Tanimoto coefficient of say 085 the redundancy in a dataset can be reduced to a well-defined level This type of technique alsoallows for the creation of common benchmark principles and couldreplace frozen benchmark data sets which often become outdated asdatabases grow over time The performance of prediction techniquesdeveloped on different data sets can therefore be compared in a fairmanner despite the fact that the underlying data sets differ in size
In most of the chemoinformatics applications developed so far thiskind of data set cleaning has not been carried out Instead test setshave been selected randomly from large overall data sets therebyintroducing many cases where highly similar pairs of examplesare found both in the training and test parts of the data sets Theestimation of the predictive performance is therefore much moreconservative in the work of Frimurer et al while it may be too highfor some of the prediction tools where the split between training andtest data has been established just by random selection
Note that similarity to a neural network is somewhat different frommolecular similarity because the network can correlate all the vectorcomponents with each other in a nonlinear fashion Two very differ-ent compounds may appear very similar for a neural net in terms of
functionality while two quite similar compounds can be interpretedas having different properties A redundant data set will typically notconstrain the network weight structure as much as a nonredundantdata set Nonredundant data from complete and representative datasets will therefore in most cases lead to well-performing predictorseven if the weightstraining examples ratio is larger
METHODS FOR CLASSIFICATION OF DRUG-LIKESTRUCTURESSeveral methods have been developed to determine the suitability ofcompounds to be used as drugs (pharmaceuticals) based on know-ledge about the molecular structure and key physical propertiesReviews about a variety of methods have been published recently(Walters and Murcko 2002 Clark and Pickett 2000 Muegge 2003)
The task is to select compounds from combinatorial librariesin such a way that the probability of identifying a new drugthat will make it to the market is optimized To minimize theprobability of leaving out potential drugs in the HTS it is import-ant to ensure adequate diversity of the molecular structures used(Gillet et al 1999) A variety of methods for compound selec-tion have been proposed ranging from simple intuitive to advancedcomputational methods
Intuitive and graphical methodsMany researchers have developed simple counting methods and ahighly popular method is the lsquorule of fiversquo proposed by Lipinski et al(1997) Although the lsquorule of fiversquo originally addresses the possibil-ity or risk for poor absorption or permeation based on molecularweight (MW) the octanolndashwater partition coefficient (log P alsocalled lipophilicity) number of hydrogen bond donors (HBD) andacceptors (HBA) respectively it has been widely used to distinguishbetween drug-like and non-drug-like compounds A number of data-bases have included Lipinskisrsquo lsquorule of fiversquo data for each compoundusing calculated values for log P This is the case for SPECS MDLSCD ACD ACDLabs Physico-Chemical databases
Frimurer et al (2000) performed a detailed quantitative analysisassessment of the predictive value of Lipinskisrsquo lsquorule of fiversquo It wasfound that the correlation coefficient is close to zero in fact slightlynegative when evaluated on a nonredundant data set Oprea (2000)also discusses drug-related chemical databases and the performanceof the lsquorule of fiversquo He concludes that the lsquorule of fiversquo does not dis-tinguish between lsquodrugsrsquo and lsquonon-drugsrsquo because the distributionof the parameters used does not differ significantly between thesetwo groups of compounds The problem with the lsquorule of fiversquo isthat it represents conditions which are necessary but not sufficientfor a drug molecule Many (in fact most) of the molecules whichfulfill these conditions are not drug molecules The work of Lip-inski has had however a tremendous pioneering value concerningquantitative evaluation of lsquodrug-likenessrsquo of chemical structures
Muegge et al (2001) proposed a simple selection scheme fordrug-likeness based on the presence of certain functional groupspharmacophore points in the molecule This method and other meth-ods using functional group filters are significantly less accurate thanthe various machine learning methods discussed (Table 3)
Chemistry space or chemography methods are somewhat similarand are based on the hypothesis that drug-like molecules share certainproperties and thus cluster in graphical representations Analysis ofhow well molecular diversity is captured using a variety of descriptors
2151
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

Prediction methods and databases within chemoinformatics
Techniques have also been developed for redundancy reduction ofsequences containing functional sites such as signal peptide cleav-age sites in secreted proteins (Nielsen et al 1996) or translationinitiation sites in mRNA sequences (Pedersen and Nielsen 1997)
In this respect chemoinformatics is still in its infancy comparedto the thorough validation methods used in bioinformatics Forchemoinformatics applications redundancy within databases canaffect the learning capabilities of neural networks decision treesand other methods whereas redundancy between different databasesgenerally introduces noise and reduces the discriminating power ofthe model
Redundancy in this context means that the compounds in theirvectors component for component are similar Similarity of thechemical compounds within a data set can lead to over-fitting of amodel such that predictions for compounds similar to those used inthe training set are excellent but for different compounds the predic-tions are not accurate to the same level There are several examplesof compound clustering techniques where the Tanimoto coefficient(Patterson et al 1996) has been used as the metric quantifying thesimilarity between compounds normally with the goal of identifyingleads by similarity or for increasing the diversity of libraries used forscreening (Reynolds et al 1998 2001 Voigt et al 2001 Willett2003) The Tanimoto coefficient T compares binary fingerprintvectors and is defined as
T (x y) = Nxy(Nx + Ny minus Nxy) (2)
where Nxy is the number of 1 bits shared in the fingerprints ofmolecules x and y Nx the number of 1 bits in the fingerprintof molecule x and Ny the number of 1 bits in the fingerprint ofmolecule y A Tanimoto coefficient approaching zero indicates thatthe compounds being compared are very different while a Tanimotocoefficient approaching one indicates that they are very similar
In the drug-likeness predictor developed by Frimurer et al(2000) the concept of redundancy reduction normally used withinbioinformatics was transferred to compound data extracted fromMDDR and ACD In this case the compounds were representedby CONCORD atom types and the Tanimoto coefficient (Patter-son et al 1996) was used to calculate the similarity between anytwo compounds Using a given threshold for maximal similarity interms of a Tanimoto coefficient of say 085 the redundancy in a dataset can be reduced to a well-defined level This type of technique alsoallows for the creation of common benchmark principles and couldreplace frozen benchmark data sets which often become outdated asdatabases grow over time The performance of prediction techniquesdeveloped on different data sets can therefore be compared in a fairmanner despite the fact that the underlying data sets differ in size
In most of the chemoinformatics applications developed so far thiskind of data set cleaning has not been carried out Instead test setshave been selected randomly from large overall data sets therebyintroducing many cases where highly similar pairs of examplesare found both in the training and test parts of the data sets Theestimation of the predictive performance is therefore much moreconservative in the work of Frimurer et al while it may be too highfor some of the prediction tools where the split between training andtest data has been established just by random selection
Note that similarity to a neural network is somewhat different frommolecular similarity because the network can correlate all the vectorcomponents with each other in a nonlinear fashion Two very differ-ent compounds may appear very similar for a neural net in terms of
functionality while two quite similar compounds can be interpretedas having different properties A redundant data set will typically notconstrain the network weight structure as much as a nonredundantdata set Nonredundant data from complete and representative datasets will therefore in most cases lead to well-performing predictorseven if the weightstraining examples ratio is larger
METHODS FOR CLASSIFICATION OF DRUG-LIKESTRUCTURESSeveral methods have been developed to determine the suitability ofcompounds to be used as drugs (pharmaceuticals) based on know-ledge about the molecular structure and key physical propertiesReviews about a variety of methods have been published recently(Walters and Murcko 2002 Clark and Pickett 2000 Muegge 2003)
The task is to select compounds from combinatorial librariesin such a way that the probability of identifying a new drugthat will make it to the market is optimized To minimize theprobability of leaving out potential drugs in the HTS it is import-ant to ensure adequate diversity of the molecular structures used(Gillet et al 1999) A variety of methods for compound selec-tion have been proposed ranging from simple intuitive to advancedcomputational methods
Intuitive and graphical methodsMany researchers have developed simple counting methods and ahighly popular method is the lsquorule of fiversquo proposed by Lipinski et al(1997) Although the lsquorule of fiversquo originally addresses the possibil-ity or risk for poor absorption or permeation based on molecularweight (MW) the octanolndashwater partition coefficient (log P alsocalled lipophilicity) number of hydrogen bond donors (HBD) andacceptors (HBA) respectively it has been widely used to distinguishbetween drug-like and non-drug-like compounds A number of data-bases have included Lipinskisrsquo lsquorule of fiversquo data for each compoundusing calculated values for log P This is the case for SPECS MDLSCD ACD ACDLabs Physico-Chemical databases
Frimurer et al (2000) performed a detailed quantitative analysisassessment of the predictive value of Lipinskisrsquo lsquorule of fiversquo It wasfound that the correlation coefficient is close to zero in fact slightlynegative when evaluated on a nonredundant data set Oprea (2000)also discusses drug-related chemical databases and the performanceof the lsquorule of fiversquo He concludes that the lsquorule of fiversquo does not dis-tinguish between lsquodrugsrsquo and lsquonon-drugsrsquo because the distributionof the parameters used does not differ significantly between thesetwo groups of compounds The problem with the lsquorule of fiversquo isthat it represents conditions which are necessary but not sufficientfor a drug molecule Many (in fact most) of the molecules whichfulfill these conditions are not drug molecules The work of Lip-inski has had however a tremendous pioneering value concerningquantitative evaluation of lsquodrug-likenessrsquo of chemical structures
Muegge et al (2001) proposed a simple selection scheme fordrug-likeness based on the presence of certain functional groupspharmacophore points in the molecule This method and other meth-ods using functional group filters are significantly less accurate thanthe various machine learning methods discussed (Table 3)
Chemistry space or chemography methods are somewhat similarand are based on the hypothesis that drug-like molecules share certainproperties and thus cluster in graphical representations Analysis ofhow well molecular diversity is captured using a variety of descriptors
2151
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160
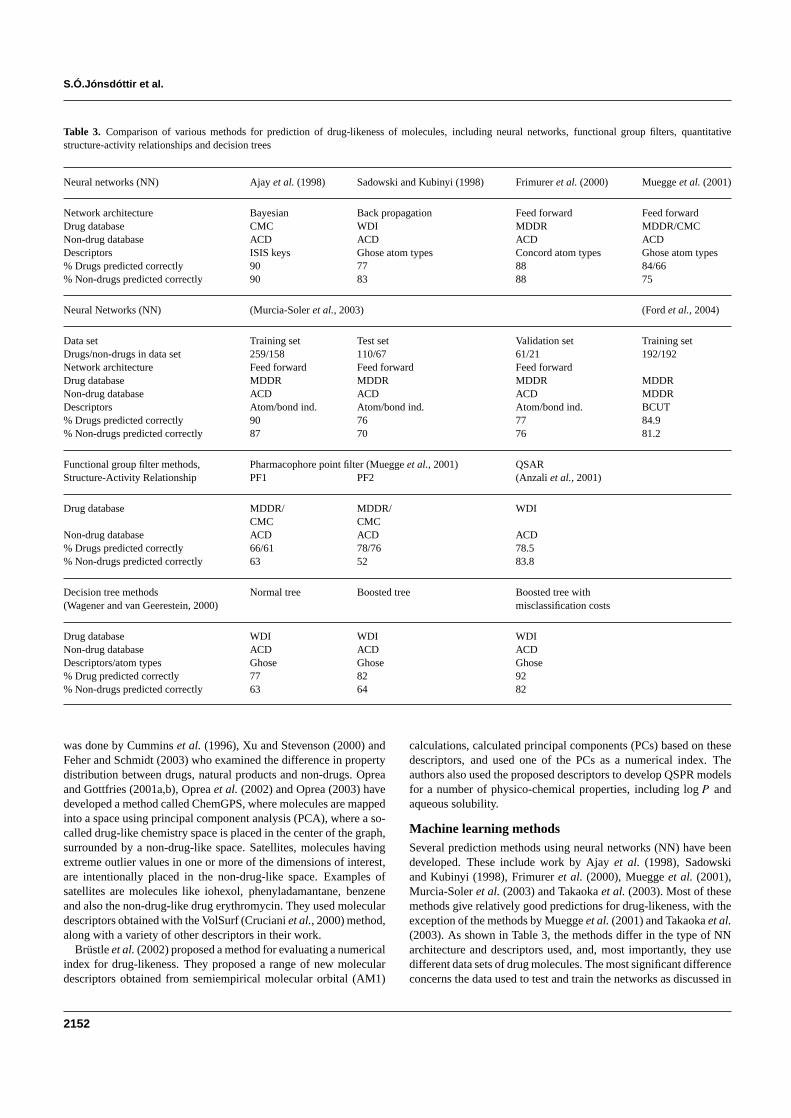
SOacuteJoacutensdoacutettir et al
Table 3 Comparison of various methods for prediction of drug-likeness of molecules including neural networks functional group filters quantitativestructure-activity relationships and decision trees
Neural networks (NN) Ajay et al (1998) Sadowski and Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)
Network architecture Bayesian Back propagation Feed forward Feed forwardDrug database CMC WDI MDDR MDDRCMCNon-drug database ACD ACD ACD ACDDescriptors ISIS keys Ghose atom types Concord atom types Ghose atom types Drugs predicted correctly 90 77 88 8466 Non-drugs predicted correctly 90 83 88 75
Neural Networks (NN) (Murcia-Soler et al 2003) (Ford et al 2004)
Data set Training set Test set Validation set Training setDrugsnon-drugs in data set 259158 11067 6121 192192Network architecture Feed forward Feed forward Feed forwardDrug database MDDR MDDR MDDR MDDRNon-drug database ACD ACD ACD MDDRDescriptors Atombond ind Atombond ind Atombond ind BCUT Drugs predicted correctly 90 76 77 849 Non-drugs predicted correctly 87 70 76 812
Functional group filter methods Pharmacophore point filter (Muegge et al 2001) QSARStructure-Activity Relationship PF1 PF2 (Anzali et al 2001)
Drug database MDDR MDDR WDICMC CMC
Non-drug database ACD ACD ACD Drugs predicted correctly 6661 7876 785 Non-drugs predicted correctly 63 52 838
Decision tree methods Normal tree Boosted tree Boosted tree with(Wagener and van Geerestein 2000) misclassification costs
Drug database WDI WDI WDINon-drug database ACD ACD ACDDescriptorsatom types Ghose Ghose Ghose Drug predicted correctly 77 82 92 Non-drugs predicted correctly 63 64 82
was done by Cummins et al (1996) Xu and Stevenson (2000) andFeher and Schmidt (2003) who examined the difference in propertydistribution between drugs natural products and non-drugs Opreaand Gottfries (2001ab) Oprea et al (2002) and Oprea (2003) havedeveloped a method called ChemGPS where molecules are mappedinto a space using principal component analysis (PCA) where a so-called drug-like chemistry space is placed in the center of the graphsurrounded by a non-drug-like space Satellites molecules havingextreme outlier values in one or more of the dimensions of interestare intentionally placed in the non-drug-like space Examples ofsatellites are molecules like iohexol phenyladamantane benzeneand also the non-drug-like drug erythromycin They used moleculardescriptors obtained with the VolSurf (Cruciani et al 2000) methodalong with a variety of other descriptors in their work
Bruumlstle et al (2002) proposed a method for evaluating a numericalindex for drug-likeness They proposed a range of new moleculardescriptors obtained from semiempirical molecular orbital (AM1)
calculations calculated principal components (PCs) based on thesedescriptors and used one of the PCs as a numerical index Theauthors also used the proposed descriptors to develop QSPR modelsfor a number of physico-chemical properties including log P andaqueous solubility
Machine learning methodsSeveral prediction methods using neural networks (NN) have beendeveloped These include work by Ajay et al (1998) Sadowskiand Kubinyi (1998) Frimurer et al (2000) Muegge et al (2001)Murcia-Soler et al (2003) and Takaoka et al (2003) Most of thesemethods give relatively good predictions for drug-likeness with theexception of the methods by Muegge et al (2001) and Takaoka et al(2003) As shown in Table 3 the methods differ in the type of NNarchitecture and descriptors used and most importantly they usedifferent data sets of drug molecules The most significant differenceconcerns the data used to test and train the networks as discussed in
2152
Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

Prediction methods and databases within chemoinformatics
the section about redundancy of data sets and to a lesser degree thechoice of descriptors for identifying the chemical structures Fordet al (2004) have developed an NN method for predicting if com-pounds are active as protein kinase ligands or not where they usedthe drug-likeness method of Ajay et al (1998) to remove unsuit-able compounds from the training set Like in the work by Frimureret al (2000) they use Tanimoto coefficient for establishing a diversetraining set
Ajay et al use the so-called ISIS keys and various other molecu-lar properties including those proposed in the lsquorule of fiversquo asdescriptors The descriptors used by Sadowski and Kubinyi arethe atom types of Ghose (Viswanadhan et al 1989) originallydeveloped for the prediction of log P Frimurer et al use the so-calledCONCORD atom types as descriptors In this work the descriptorselection procedure is also inverted in the sense that the weights inthe trained NNs are inspected after training was completed Therebyone may obtain a ranking of the importance of different descriptorsfor prediction tasks like drug-likeness Obviously such a rankingcannot reveal the correlations between descriptors which may behighly important for the classification performance The power ofthe NNs is that they can take such correlations into account but theycan be difficult to visualize or describe in detail
Murcia-Soler et al (2003) assign probabilities for the ability ofeach molecule to act as a drug in their method They use varioustopological indices as descriptors reflecting types of atoms andchemical bonds in the molecules Their paper contains a detailedand interesting analysis of the performance on training test and val-idation data sets (Table 3) Takaoka et al (2003) developed an NNmethod by assigning compound scores for drug-likeness and easyof synthesis based on chemistsrsquo intuition Their method predicts thedrug-likeness of drug molecules with 80 accuracy but the accuracyof the prediction of non-drug-likeness is hard to assess
Wagener and van Geerestein (2000) developed a quite successfulmethod using decision trees The first step in their method is similar tothe method by Muegge et al (2001) where the molecules are sortedbased on which key functional groups they contain Going throughseveral steps the connectivity of the atoms in each functional groupis determined and atom types are assigned using the atom types ofGhose This basic classification is then used as an input (leaf note) ina decision tree which is trained to determine if a compound is drug-like or non-drug-like To increase the accuracy a technique calledlsquoboostingrsquo can be used where weights are assigned to each data pointin the training set and optimized in such a way that they reflect theimportance of each data point By including misclassification costsin the training of the tree the predictions can be improved even more
Gillet et al (1998) proposed a method which uses a geneticalgorithm (GA) using weights of various properties obtained fromsubstructural analysis Using the properties from lsquorule of fiversquo andvarious topological indices as descriptors the molecules are sortedand ranked and from these results a score is calculated It is seen thatdrugs and non-drugs have different distributions of scores with someoverlap Also the distribution varies for different types of drugs
Anzali et al (2001) developed a QSAR type method for predictingbiological activities with the computer system PASS (prediction ofactivity spectra for substances) Both data for drugs and non-drugswere used in the training of the model and subsequently used fordiscriminating between drugs and non-drugs
The NN decision trees GA and QSAR methods are classifiedas machine learning whereas lsquorule of fiversquo and functional group
Fig 4 Chemoinformatics integrates information on ADMET properties inthe relationship between chemistry space biological targets and diseasesThis figure can be viewed in colour on Bioinformatics online
filters are simple intuitive methods A short overview of the reportedpredictive capability of the various methods is given in Table 3 Itis seen that the NN methods and the decision tree methods give thebest results As discussed previously it is difficult to compare thedifferent NN methods in quantitative terms due to differences in sizeand redundancy of the data sets used to train and test the methods
Lead-likenessAnother possibility is to determine whether a molecule is lead-like rather than drug-like As discussed by Hann et al (2001) itis often not feasible to optimize the drug-like molecules by addingfunctional groups as the molecules then become too large and over-functionalized Instead one could target new leads with computerizedmethods and in such a way give additional flexibility in targetingnew potential drugs
According to Oprea et al (2001a) not much information aboutmolecular structures of leads is available in the literature In theirpaper several lead structures are given and an analysis of the dif-ference between leads and drugs is presented According to theiranalysis drugs have higher molecular weight higher lipophilicityadditional rotational bonds only slightly higher number of hydrogen-bond acceptors and the same number of hydrogen-bond donorscompared to leads As pointed out by the authors this analysis con-tains too few molecules to be statistically significant but it indicatesuseful trends Similar trends are also observed by Hann et al (2001)The design of lead-like combinatorial libraries is also discussed byTeague et al (1999) and Oprea conducted studies of chemical spacenavigation of lead molecules (Oprea 2002a) and their properties(Oprea 2002b) Various methods for ranking molecules in lead-discovery programs are discussed by Wilton et al (2003) includingtrend vectors substructural analysis bioactive profiles and binarykernel discrimination
PREDICTION OF PROPERTIES
ADMET propertiesADMET properties stand for absorption distribution metabolismexcretion and toxicity of drugs in the human organism In recent yearsincreased attention has been given to modeling these properties butstill there is a lack of reliable models One of the first attempts toaddress the modeling of one these properties the intestinal absorp-tion was the lsquorule of fiversquo proposed by Lipinski et al (1997) Anumber a review articles on ADMET properties have been pub-lished recently (Beresford et al 2002 Ekins et al 2002 van de
2153
SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

SOacuteJoacutensdoacutettir et al
Waterbeemd and Gifford 2003 Livingstone 2003) Boobis et al(2002) made an expert report on the state of models for predictionof ADMET properties Boobis et al (2002) and van de Waterbeemdand Gifford (2003) give a fairly detailed overview of methods andcomputer programs for prediction of ADMET properties
One of the major problems concerning modeling of ADMET prop-erties is the lack of reliable experimental data for training the modelsFor metabolism and toxicity databases are available but for theother properties experimental information is scarce A number ofprograms for modeling of ADMET properties have been developedrecently and ADMET modules have been included in some examplesof molecular modeling software Only models for absorption are dis-cussed in greater detail here whereas modeling of the other ADMETproperties is only discussed briefly with references to recent reviewsFigure 4 shows how ADMET properties relate to targets and diseasesfrom a chemoinformatics perspective
Absorption According to Beresford et al (2002) reasonable modelsexist for intestinal absorption and bloodndashbrain-barrier (BBB) penet-ration It is however important to bear in mind that the reason for loworal bioavailability is often not due to poor overall absorption butdue to limited first pass through the gut wall membrane Recentlyvan de Waterbeemd et al (2001) published a property guide foroptimization of drug absorption and pharmacokinetics
At least three commercially available computer programs havebeen developed for prediction of the intestinal absorption IDEATMGastroPlusTM and OraSpotterTM IDEATM uses an absorption modelproposed by Grass (1997) and later improved by Norris et al(2000) and GastroPlusTM uses the ACAT (advanced compartmentalabsorption and transit) model proposed by Agoram et al (2001)In the ACAT model the GI tract is divided into nine compartmentsone for the stomach seven for the small intestine and one for thecolon and the absorption process is described using a set of over80 differential equations The ACAT model uses compound specificparameters like permeability solubility and dose as well as physio-logical information such as species GI transit time food status etcA detailed overview of these two methods is given by Parrott andLaveacute (2002) and the link between drug absorption and the permeab-ility and the solubility is discussed by Pade and Stavhansky (1998)The OraSpotterTM program uses information about the molecularstructure only which are turned into SMILES The descriptors arethen evaluated from the SMILES
Parrott and Laveacute (2002) developed models for the intestinalabsorption using IDEATM 20 and GastroPlusTM 310 They usedthree approaches in their work (1) Prediction of absorption classusing chemical structure data only (2) Predictions based upon meas-ured solubility and predicted permeability (3) Predictions basedupon measured solubility and measured CACO-2 permeability TheRMS deviations were in the range of 19ndash24 for the modelsdeveloped and the best model was obtained with the IDEA pro-gram using both measured solubility and measured permeabilityHowever the IDEA and GastroPlus programs gave fairly similarresults in this study
Wessel et al (1998) developed a QSPR type model using non-linear GANN techniques for the prediction of human intestinalabsorption based on Caco-2 measurements of permeability The dataset used contains measured values for 86 drug and drug-like com-pounds selected from various sources The descriptors used weregenerated with the ADAPT (automated data analysis and pattern
recognition toolkit) software the MOPAC program using the AM1semi-empirical quantum chemical method derived from 2D struc-tures using graph theory and by using substructure fragments Afairly good model with RMS deviation of 16 was obtained
Palm et al (1998) evaluated the relationship between variousmolecular descriptors and transport of drugs across the intestinalepithelium also using measurements for Caco-2 cells and Clark(1999) developed a method using the polar molecular surface areaof the molecules as a descriptor A minimalistic model based on thelsquorule of fiversquo was proposed by Oprea and Gottfries (1999) Zamoraet al (2001) used chemometrics methods for predicting drug per-meability and new QSAR models were proposed by Kulkarni et al(2002) Egan and Lauri (2002) published a review on methods forprediction of passive intestinal permeability
BBB penetration The BBB separates the brain from the systemicblood circulation Passage through the BBB is a necessity for orallyadministrated drugs targeting receptors and enzymes in the centralnervous system (CNS) whereas generally it is an unwanted propertyfor peripherally acting drugs BBB penetration is usually expressedas log BB = log(CbrainCblood) ie the logarithm to the ratio of theconcentration of the drug in the brain to that in the blood Experi-mental log BB values range between minus20 and +10 and compoundswith log BB gt 03 are characterized as BBB penetrating whereascompounds with log BB lt minus10 are poorly distributed to the brain
One of the first attempts to predict BBB penetration dates backto 1988 when Young et al (1988) obtained a correlation betweenlog BB and log P or derivatives of log P for a small series of histam-ine H2 antagonists In the following years a number of models forBBB penetration were published and these models were discussed inrecent reviews (Ekins et al 2000 Norinder and Haeberlein 2002) Inthe following we will focus on more recently reported models Nearlyall models developed until 2000 were based on a relatively smallnumber of compounds (57ndash65 compounds) with a limited structuraldiversity including several compounds being far from drug-likeKelder et al (1999) expanded the number of available compounds byreporting log BB for an additional 45 drug-like compounds In addi-tion to developing models for BBB penetration they also showedthat CNS active drugs could be distinguished from non-CNS activedrugs based on their polar surface area
Joslashrgensen et al (2001) presented a model based on all availableBBB penetration data at that time (105 compounds) They basedtheir model on an atom-weighted surface area approach wherethe contribution of each atom to the BBB penetration dependedon the atom type and its solvent accessibility Rose et al (2002)developed a model for the same set of compounds using electrotopo-logical state descriptors whereas Kaznessis et al (2001) generated anumber of physically significant descriptors from Monte Carlo sim-ulations in water and used those in a model for BBB penetrationHou and Xu (2002) developed a model using GA and Feher et al(2000) proposed a very simple model based on only three descriptorslog P number of hydrogen bonds and polar surface areas It appearsthat these descriptors or related descriptors describe the structuralrequirements for BBB penetration most effectively (Norinder andHaeberlein 2002)
It is interesting that the quantitative models for the prediction ofBBB penetration although based on very different methods yieldedcomparable results This probably reflects the relatively small num-ber of compounds included in the modeling the lack of structural
2154
Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

Prediction methods and databases within chemoinformatics
diversity as well as uncertainties associated with the experimentallog BB values Several drug molecules are actively transported acrossmembranes including the BBB by various transporters which mayinclude active transport to the CNS as well as efflux transporters(Bodor and Buchwald 1999 Kusuhara and Sugiyama 2002 Sunet al 2002)
A number of models for the classification of compounds as CNSactive or CNS inactive have been published An NN based on 2DUnity fingerprints was developed by Keserucirc et al (2000) for identific-ation of CNS active compounds in virtual HTS Engkvist et al (2003)showed that a model based on substructure analysis performed as wellas a more complex NN based model Doniger et al (2002) developedand compared an NN and a support vector machine approach Crivoriet al (2000) published a method for the classification of compoundsas CNS active or CNS inactive The model used descriptors from 3Dmolecular interaction fields generated by VolSurf and multivariatestatistical methods for the subsequent data analysis Based on a testset of 110 molecules the model predicted log BB correctly with a 90accuracy for an external set of 120 compounds In a recent paper byWolohan and Clark (2003) the combination of descriptors developedfrom interaction fields and subsequently analyzed by multivariatestatistics were further developed and applied not only for the pre-diction of BBB penetration but also for addressing the more generalproblem of oral bioavailability
Other ADMET properties Metabolism of drugs in the gut wall andin the liver is a major issue where reactions due to cytochrome P450liver enzymes are of particular importance (Lewis 1996) Metabol-ism of xenobiotics is very complicated and thus difficult to modeladequately Nicholson and Wilson (2003) recently published a reviewarticle on xenobiotic metabolism and de Groot and Ekins (2002)and Lewis and Dickins (2002) published a review on metabolismthrough cytochrome P450 dependent reactions Langowski and Long(2002) studied various enzyme modeling systems databases andexpert systems for the prediction of xenobiotic metabolism Theydiscussed three expert systems META (Klopman et al 1994Talafous et al 1994 Klopman et al 1997) MetabolExpert andMETEOR (Greene et al 1997 Buttom et al 2003) and twoenzyme modeling systems Enzymatic reactions are primarily stud-ied using various molecular modeling methods and as it is nota central chemoinformatics problem it is outside the scope ofthis paper
Toxicity of drugs is another extremely important problem leadingto a significant number of drug failures Greene (2002) publisheda review paper about a number of commercial prediction systemsincluding DEREK (Greene et al 1997 Judson et al 2003) Onco-Logic (Dearden et al 1997) HazardExpert COMPACT (Parkeet al 1990) CASEMulti-CASE (Klopman 1992) and TOPKATKatritzky et al (2001) Espinosa et al (2002) and Tong et al (2002)have developed QSAR models for the prediction of toxicity
Up to now not much effort has been put into in silico modeling ofdistribution of drugs within the human organism and almost noneon excretion Experimental quantities which measure distribution ofdrugs in the human body adequately are not readily available butaccording to Boobis et al (2002) log P octanolndashwater distributioncoefficients (log D) and in vivo pharmacokinetic data can be used asa measure of distribution Transporters plasma-protein binding andother aspects of distribution are discussed by van de Waterbeemdand Gifford (2003)
Physico-chemical propertiesAs mentioned above physico-chemical properties of interest todrug discovery are mainly acidndashbase dissociation constants (pKa)aqueous solubilities octanolndashwater partition coefficients (log P) andoctanolndashwater distribution coefficients (log D) These properties aregenerally relatively well studied and many predictive methods areavailable in the literature Recently van de Waterbeemd (2003)and Livingstone (2003) published reviews on physico-chemicalproperties relevant to the drug discovery process Many relativelygood models are available for log P log D and pKa of drugs(van de Waterbeemd 2003) and a large number of in silico modelshave been developed for the aqueous solubility of drugs and drug-like compounds as discussed in recent reviews by Blake (2000)Husskonen (2001) and Jorgensen and Duffy (2002) There is how-ever continued interest in improved models for the solubility ofdrugs due to the importance of this property It is important to men-tion that the solubility relates to the solid phase and is significantlymore difficult to model than the other physico-chemical propertiesDifficulties in modeling properties relating to the solid phase arediscussed by Dyekjaeligr and Joacutensdoacutettir (2003)
Some of the available models for aqueous solubility of organiccompounds are based on structure information only using NN(Huuskonen et al 1998 Huuskonen 2000 McElroy and Jurs 2001Yan and Gasteiger 2003 Taskinen and Yliruusi 2003 Wegnerand Zell 2003) and QSPR (Katritzky et al 1998 Yaffe et al2001 Zhong and Hu 2003) methods Other models use experi-mental quantities like log P melting points heats of fusion andvapor pressures as descriptors (Ran et al 2001 Sangvhi et al2003) sometimes together with calculated structural descriptors(McFarland et al 2001 Thompson et al 2003) and group con-tribution methods have also been developed (Kuumlhne et al 1995Klopmann and Zhu 2001) Traditionally solubility is calculatedusing equilibrium thermodynamics (Prausnitz et al 1986) Veryaccurate predictions can be obtained but the physical parametersneeded are only seldom available for drugs and drug-like moleculesJoacutensdoacutettir et al (2002) have developed an in silico method usingmolecular mechanics calculations
To date the research efforts have mainly been focused on theaqueous solubility of drugs and very little attention has been devotedto their solubility in buffered solutions and the pH dependence ofsolubility (Avdeef et al 2000 Sangvhi et al 2003) For understand-ing dissolution of drugs in the human organism it is crucial to focusincreasingly on solubility in a more realistic environment and toacquire larger amounts of experimental data for the pH dependenceof solubility
ROLE OF DATABASES AND CHEMOINFORMATICSIN FUTURE DRUG DISCOVERYAs documented in this review chemoinformatics is a rapidlygrowing field which has become extremely important in phar-maceutical research in the last couple of years The generationstorage retrieval and utilization of data related to chemicalstructures has merged established disciplines and catalyzed thedevelopment of new techniques Thus chemoinformatics bridgesestablished areas like molecular modeling computational chem-istry statistics and bioinformatics and is closely related toother emerging fields as chemogenomics metabonomics andpharmacogenomics
2155
SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

SOacuteJoacutensdoacutettir et al
Chemoinformatics methods are already used extensively in thedrug discovery and development process by the pharmaceuticalindustry and many powerful methods have been proposed The pre-dictive methods available are of various qualities and complexitiesranging from simple rules-of-thumb to sophisticated 3D methodsinvolving simulation of ensembles of molecules containing thou-sands of atoms In the years to come improved in silico methodsfor the prediction of properties based on structural informationwill merge and be used to assist in identifying more suitable hitsand leads
Structural and property databases provide the foundation ofchemoinformatics and a variety of databases containing structur-ally derived data for organic compounds are available Although thenumber of entries in a given database is often considered the mostimportant measure of the quality or usability of a database there isincreased awareness concerning the quality of the data rather thanon the number of entries
The recent literature presents a number of methods for the clas-sification of the drug-likeness of compounds based on subsets ofmolecules from databases representing drug-like and non-drug-likemolecules Classification methods which predict the lead-likenessof molecules or their affinity toward specific targets would beeven more useful and thus the generation of databases of lead-like molecules etc is highly desired All such methods are limitedto likely drugs but we could eventually gather information aboutnon-drug-like drugs and find common features in those as well
To ensure good predictive power of such models the redundancyof the data sets used for training and testing the model is crucial Itis thus very important to balance the data in such a way that certainstructural features are not over-represented Researchers using manyother databases eg protein structure databases have encountered asimilar problem They realized that the use of all available 3D proteinstructures in PDB for analyzing and extracting loops motifs side-chains etc could be problematic due to the over-representation ofsome proteins or protein families relative to others Accordingly anumber of data sets of non-homologous protein structures have beendeveloped
Also much focus has been devoted to in silico modeling ofADMET and physico-chemical properties of drugs and drug can-didates A variety of different methods for the prediction of relevantphysico-chemical properties like log P pKa and aqueous solubilityof organic compounds are available and in general these methodsperform satisfactorily considering the data available In the caseof the aqueous solubility a large number of methods have beendeveloped and there is continued interest in this property The effectof the pH value of the solution on solubility needs to be studied inmuch greater detail
Concerning ADMET properties a number of in silico models havebeen proposed for intestinal absorption and BBB penetration somemodels for metabolism and toxicity and only very few models fordistribution and excretion The total amount of data available for acertain end point often limits the possibility of developing improvedpredictive models for ADMET properties Permeability of the BBBis such a case where measured values are only available for a littlemore than 100 compounds The lack of structural diversity withinthese compounds is another limiting factor
Properties like metabolism oral bioavailability etc of drugswithin the human organism involve several individual processesand thus sub-processes and bottlenecks governing these processes
need to be identified Due to lack of reliable data combined with avery complex mode of action of the processes involved the presentmethods often fail In some cases where a method yields promisingresults for a set of compounds the transferability of the method toanother series or class of compounds may be questionable Withinthis area there is much need for developing better and more robustpredictive methods but there is also a need for determining andcollecting larger amount of experimental data for the individualprocesses
Oral bioavailability is a particularly difficult property to model asit involves a huge number of processes within the human organismand depends on all the ADMET and physico-chemical propertiesdiscussed above The molecule has to dissolve be adsorbed into thebloodstream transported to the target and not metabolized on itsway Thus for an orally administrated drug to reach its final des-tination a whole range of properties need to be within acceptablelimits and thus a model which defines such limits would be veryuseful Dynamic modeling of processes within a living cell withsystems biology methods is growing rapidly and is expected to havea huge impact on future drug design in particular on the model-ing of the oral bioavailability As discussed by Parsons et al (2004)chemicalndashgenetic and genetic interaction profiles can potentially beintegrated to provide information about the pathways and targetsaffected by bioactive compounds Such methods could thus be veryuseful for identifying the mechanism of action and cellular targetsof bioactive compounds Improved understanding of how differentdrugs affect one another within the human organism is also of greatimportance and thus much interest is presently devoted to studiesof drugndashdrug interactions An extremely exciting perspective is ofcourse also to use pharmacogenomic methods for examining howindividual patients respond to specific drugs and how that dependson their genetic makeup
Although more data better data and an improved understand-ing of the interplay between the different processes in the humanorganism are required the present level of available data has alreadymade chemoinformatics an effective tool in the drug discovery anddevelopment process
DATABASES AND COMPUTER PROGRAMSThe Accelrysrsquo Biotransformation database is marketed by Accelrys
San Diego USA at URL httpwwwaccelryscom
The Accelrysrsquo Metabolism database is marketed by AccelrysSan Diego USA at URL httpwwwaccelryscom
ACDLabs Physico-Chemical Property databases are marketed byAdvanced Chemical Development Inc Toronto Canada at URLhttpwwwacdlabscom
The AQUASOL dATAbASE of Aqueous Solubility sixth edition ismarketed by the University of Arizona Tucson Arizona USA atURL httpwwwpharmacy arizonaeduoutreachaquasol
The Available Chemical Directory (ACD) is marketed by MDLInformation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The BIOSTER database is marketed by Accelrys San Diego USAat URL http wwwaccelryscom
The Cambridge Structural Database (CSD) is maintained and distrib-uted by the Cambridge Crystallographic Data Centre CambridgeUK at URL httpwww ccdccamacuk
2156
Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

Prediction methods and databases within chemoinformatics
CASE and MultiCASE MultiCASE Inc Beachwood OH USAURL httpwww multicasecom
The Chapman amp HallCRC Dictionary of Drugs produced byChapman amp HallCRC London UK is a part of CombinedChemical Dictionary (CCD) and is available online at URLhttpwwwchemnetbasecom
The Chemicals Available for Purchase (CAP) database is mar-keted by Accelrys San Diego USA at URL httpwwwaccelryscom
The Comprehensive Medical Chemistry (CMC) database is marketedby MDL Information Systems Ltd San Lenandro USA at URLhttpwwwmdlcom
CrossFire Beilstein is marketed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom
DEREK LHASA limited Department of Chemistry University ofLeeds Leeds UK URL httpwwwchemleedsacukluk
The Derwent World Drug Index (WDI) and Derwent Drug Fileare published by Thomson Derwent London UK at URLhttpwwwderwentcomproductslrwdi
The Design Institute of Physical Properties database (DIPPR)is maintained by Michigan Technology University HoughtonUSA in collaboration with AIChE Information is foundat URLs httpdipprchemmtuedu and httpwwwaicheorgdippr
GastroPlusTM version 40 Simulations Plus Inc Lancaster CaUSA URL http wwwsimulations-pluscom
GRID version 21 Molecular Discovery Ldt Ponte San Giovanni -PG Italy URL httpwwwmoldiscoverycom
HazardExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
IDEATM version 22 and IDEA pkEXPRESSTM LION bioscienceAG Heidelberg Germany URL httpwwwlionbiosciencecom
MDL Drug Data Report (MDDR) is marketed by MDL Inform-ation Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Metabolite database is marketed by MDL Informa-tion Systems Ltd San Lenandro USA at URL httpwwwmdlcom
The MDL Screening Compounds Directory (formerly ACD-SC)ismarketed by MDL Information Systems Ltd San Lenandro USAat URL httpwwwmdlcom
The MDL Toxicity database is marketed by MDL InformationSystems Ltd San Lenandro USA at URL httpwwwmdlcom
The MedChem Bio-loom database is published by BioByte CorpClaremont USA at URL httpwwwbiobytecom and also dis-tributed by Daylight Chemical Informations Systems Inc MissionViejo USA at URL httpwwwdaylightcom
MetabolExpert CompuDrug International Inc Sedone AZ USAURL http wwwcompudrugcom
METEOR LHASA limited Department of Chemistry Universityof Leeds Leeds UK URL httpwwwchemleedsacukluk
The National Cancer Institute database (NCI) is a publicly avail-able database distributed by MDL Information Systems LtdSan Lenandro USA at URL httpwwwmdlcom and DaylightChemical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
OraSpotterTM version 30 ZyxBioLLC Cleveland OH USA URLhttpwwwzyxbiocom
The Physical Properties Database (PHYSPROP) is marketed bySyracuse Research Corporation (SRC) North Syracuse USA atURL httpwwwsyrrescomesc
The Properties of Organic Compounds database is produced byChapman amp HallCRC London UK available online at URLhttpwwwchemnetbasecom
The Protein Data Bank (PDB) is operated by Rutgers The StateUniversity of New Jersey the San Diego Supercomputer Centerat the University of California San Diego and the Centerfor Advanced Research in Biotechnology of the National Insti-tute of Standards and Technology and funded by NSF NIHand the Department of Energy It is found at URL httpwwwrcsborgpdb
Proteinndashprotein interaction databases List of databases with linksto each database at URLs httpwwwcbiomskccorgprlindexphp httpproteomewayneeduPIDBLhtml and httpwwwimb-jenadejcbppi
SPECSnet database is provided by Specs Riswijk The Netherlandsat URL http wwwspecsnet
The Spresi database is marketed by Infochem GmbH MuumlnchenGermany at URL httpwwwinfochemde An older versionof the database Spresi95 is available from Daylight Chem-ical Information Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
TOPKAT (TOxicity Predictions by Komputer Assisted Tech-nology) Accelrys San Diego USA URL httpwwwaccelryscom
The ToxExpress Reference Database and ToxExpress Solutionsare marketed by GeneLogic Inc Gaithersburg USA at URLhttpwwwgenelogiccom
Toxicological Data Network (TOXNET) is a collection of databasespublished by various services and made accessible free of chargeover the internet by National Library of Medicine Bethesda USAat URL httptoxnetnlmnihgov
The TSCA93 database is published by the US EnvironmentalProtection Agency and is distributed by Daylight Chem-ical Informations Systems Inc Mission Viejo USA at URLhttpwwwdaylightcom
VolSurf version 3011 Molecular Discovery Ldt Ponte San Gio-vanni - PG Italy URL httpwwwmoldiscoverycom
The WOrld of Molecular BioActivities database (WOMBAT) is pub-lished by Sunset Molecular Discovery LLC Santa Fe USA atURL httpwwwsunsetmolecularcom and distributed by Day-light Chemical Informations Systems Inc Mission Viejo USA atURL httpwwwdaylightcom
ACKNOWLEDGEMENTSThe information about the databases are mostly found on the inter-net pages provided by the suppliers We contacted Molecular DesignLimited (MDL) Accelrys Infochem and Derwent for additionalinformations which they kindly gave to us We should also liketo thank Tudor Oprea for information about the WOMBAT databaseand Olga Rigina for helping with information about proteinndashproteininteraction databases
2157
SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

SOacuteJoacutensdoacutettir et al
REFERENCESAgoramB et al (2001) Predicting the impact of physiological and biochemical
processes on oral drug bioavailability Adv Drug Delivery Rev 50 S41ndashS67Ajay et al (1998) Can we learn to distinguish between lsquodrug-likersquo and lsquonondrug-likersquo
molecules J Med Chem 41 3314ndash3324AnzaliS et al (2001) Discrimination between drugs and nondrugs by prediction of
activity spectra for substances (PASS) J Med Chem 44 2432ndash2437AvdeefA et al (2000) pH-metric solubility 2 Correlation between the acid-base
titration and the saturation shake-flask solubility pH methods Pharm Res 1785ndash89
BaderGD et al (2003) BIND the biomolecular interaction network database NucleicAcids Res 31 248ndash250
BajorathJ (2002) Chemoinformatics methods for systematic comparison of moleculesfrom natural and synthetic sources and design of hybrid libraries J Comput-AidedMol Des 16 431ndash439
BajorathJ (2004) Chemoinformatics Concepts Methods and Applications (Methodsin Molecular Biology) Humana Press Totowa
BaldiP and BrunakS (2001) Bioinformatics The Machine Learning ApproachAdaptive Computation and Machine Learning 2nd edn The MIT Press Cambridge
BeresfordAP et al (2002) The emerging importance of predictive ADME simulationin drug discovery Drug Discov Today 7 109ndash116
BlakeJF (2000) Chemoinformaticsndashpredicting the physicochemical properties oflsquodrug-likersquo molecules Curr Opin Biotech 11 104ndash107
BodorN and BuchwaldP (1999) Recent advances in the brain targeting of neuro-pharmaceuticals by chemical delivery systems Adv Drug Delivery Rev 36229ndash254
BoumlhmH-J and SchneiderG (2000) Virtual Screening for Bioactive MoleculesWiley-VCH Weinheim
BoobisA et al (2002) Congress Report In silico prediction of ADME and parmaco-kinetics Report of an expert meeting organised by COST B15 Eur J Pharm Sci17 183ndash193
Browne LJ et al (2002) Chemogenomics - pharmacology with genomics toolsTargets 1 59
BruumlstleM et al (2002) Descriptors physical properties and drug-likeness J MedChem 45 3345ndash3355
BurkertU and AllingerNL (1982) Molecular Mechanics ACS Monograph Vol 177American Chemical Society Washington DC
ButtomWG et al (2003) Using absolute and relative reasoning in the prediction of thepotential metabolism of xenobiotics J Chem Inf Comput Sci 43 1371ndash1377
ClarkDE (1999) Rapid calculation of polar molecular surface area and its applica-tion to the prediction of transport phenomena 1 Prediction of intestinal absorptionJ Pharm Sci 88 807ndash814
ClarkDE and PickettSD (2000) Computational methods for the prediction of lsquodrug-likenessrsquo Drug Discovery Today 5 49ndash58
CostanzoMC et al (2000) The Yeast Proteome Database (YPD) and Caeohabditiselegans proteome database (WormPD) comprehensive resources for the organizationand comparison of model organism protein information Nucleic Acids Res 2873ndash76
CramerCJ (2002) Essentials of Computational Chemistry Theories and ModelsJohn Wiley and Sons New York
CramerRD III et al (1988) Comparative molecular field analysis (CoMFA) 1 Effect ofshape on binding of steroids to carrier proteins J Am Chem Soc 110 5959ndash5967
CrivoriP et al (2000) Predicting blood-brain barrier permeation from three-dimensionalmolecular structure J Med Chem 43 2204ndash2216
CrucianiG et al (2000) Molecular fields in quantitative structure-permeation relation-ships the VolSurf approach J Mol Struct (Theochem) 503 17ndash30
CumminsDJ et al (1996) Molecular diversity in chemical databases comparisonof medicinal chemistry knowledge bases and database of commercially availablecompounds J Chem Inf Comput Sci 36 750ndash763
de Groot MJ and EkinsS (2002) Pharmacophore modeling of cytochromes P450Adv Drug Delivery Rev 54 367ndash383
DeardenJC et al (1997) The development and validation of expert systems forpredicting toxicity ATLA 25 223ndash252
DonigerS et al (2002) Predicting CNS permeability of drug molecules comparison ofneural network and support vector machine algorithms J Comp Bio 9 849ndash864
DurantJL et al (2002) Reoptimization of MDL keys for use in drug discoveryJ Chem Inf Comput Sci 42 1273ndash1280
DurbinR et al (1998) Biological Sequence Analysis Probabilistic Models of Proteinsand Nucleic Acids Cambridge University Press Cambridge
DyekjaeligrJD and Joacutensdoacutettir SOacute (2003) QSPR models based on molecularmechanics and quantum chemical calculations 2 Thermodynamic properties
of alkanes alcohols polyols and ethers Ind Eng Chem Res 424241ndash4259
DyekjaeligrJD et al (2002) QSPR models based on molecular mechanics and quantumchemical calculations 1 Construction of Boltzmann-averaged descriptors foralkanes alcohols diols ethers and cyclic compounds J Mol Model 8 277ndash289
EganWJ and LauriG (2002) Prediction of intestinal permeability Adv Drug DeliveryRev 54 273ndash289
EkinsS et al (2000) Progress in predicting human ADME parameters in silicoJ Pharm Tox Methods 44 251ndash272
EkinsS et al (2002) Toward a new age of virtual ADMETOX and multidimensionaldrug discovery J Comput-Aided Mol Des 16 381ndash401
EngkvistO et al (2003) Prediction of CNS activity of compound libraries usingsubstructure analysis J Chem Inf Comput Sci 43 155ndash160
EspinosaG et al (2002) An integrated SOM-Fuzzy ARTMAP neural system for theevaluation of toxicity J Chem Inf Comput Sci 42 343ndash359
FeherM and SchmidtJM (2003) Property distributions differences between drugsnatural products and molecules from combinatorial chemistry J Chem Inf ComputSci 43 218ndash227
FeherM et al (2000) A simple model for the prediction of blood-brain partitioningInt J Pharm 201 239ndash247
FordMG et al (2004) Selecting compounds for focused screening using lineardiscriminant analysis and artificial neural networks J Mol Graph Model 22467ndash472
FrimurerTM et al (2000) Improving the odds in discriminating lsquodrug-likersquo from lsquonondrug-likersquo compounds J Chem Inf Comput Sci 40 1315ndash1324
FujitaT et al (1964) J Am Chem Soc 86 5175ndash5180GallopMA et al (1994) Application of combinatorial technologies to drug dis-
covery 1 Background and peptide combinatorial libraries J Med Chem 371233ndash1251
GasteigerJ (2003) Handbook of Chemoinformatics From Data to Knowledge vols1ndash4 Wiley-VCH Weinheim
GasteigerJ and EngelT (2003) Chemoinformatics A Textbook Wiley-VCHWeinheim
GeladiP and KowalskiBR (1986a) An example of 2-block predictive partial least-squares regression with simulated data Anal Chim Acta 185 19ndash32
GeladiP and KowalskiBR (1986b) Partial least-squares regression a tutorial AnalChim Acta 185 1ndash17
GilletVJ et al (1998) Identification of biological activity profiles using substructuralanalysis and genetic algorithms J Chem Inf Comput Sci 38 165ndash179
GilletVJ et al (1999) Selecting combinatorial libraries to optimize diversity andphysical properties J Chem Inf Comput Sci 39 169ndash177
GilletVJ et al (2003) Similarity searching using reduced graph J Chem Inf ComputSci 43 338ndash345
GoldenJ (2003) Towards a tractable genome Knowledge management in drugdiscovery Curr Drug Discovery Feb 17ndash20
GoodfordPJ (1985) A computational procedure for determining energetically favor-able binding-sites on biologically important macromolecules J Med Chem 28849ndash857
GordonEM et al (1994) Application of combinatorial technologies to drug discovery2 Combinatorial organic synthesis library screeing strategies and future directionsJ Med Chem 37 1385ndash1401
GotoS et al (1998) LIGAND chemical database for enzyme reactions Bioinformatics14 591ndash599
GotoS et al (2002) LIGAND database of chemical compounds and reaction inbiological pathways Nucleic Acids Res 30 402ndash404
GrassGM (1997) Simulation models to predict oral drug absorption from in vitro dataAdv Drug Delivery Rev 23 199ndash219
GreeneN (2002) Computer systems for the prediction of toxicity an update Adv DrugDelivery Rev 54 417ndash431
GreeneN et al (1997) Knowledge-based expert system for toxicity and metabol-ism prediction DEREK StAR and METEOR SAR QSAR Environ Res 10299ndash313
GuumlntherJ et al (2003) Utilizing structural knowledge in drug design strategiesapplications using relibase J Mol Biol 326 621ndash636
HannMM et al (2001) Molecular complexicity and its impact on the probability offinding leads for drug discovery J Chem Inf Comput Sci 41 856ndash864
HanschC and FujitaT (1964) J Am Chem Soc 86 1616ndash1626HendlichM et al (2003) Relibase design and development of a database for
comprehensive analysis of protein ndash ligand interactions J Mol Biol 326 607ndash620HermjakobH et al (2004) IntActmdashan open source molecular interaction database
Nucleic Acids Res 32 D452ndashD455
2158
Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

Prediction methods and databases within chemoinformatics
HobohmU et al (1992) Selection of representative protein data sets Protein Sci 1409ndash417
HodgesPE et al (1999) The yeast proteome database (YPD) a model for the organ-ization and presentation of genome-wide functional data Nucleic Acids Res 2769ndash73
HopfingerAJ and Duca JS (2000) Extraction of pharmacophore information formHigh-throughput Screens Curr Opin Biotech 11 97ndash103
HopkinsAL and Groom CR (2002) The drugable genome Nat Rev Drug Discovery1 727ndash730
HouT and XuX (2002) ADME evaluation in drug discovery 1 Applications of geneticalgorithms to the prediction of bloodndashbrain partitioning of a large set of drugsJ Mol Model 8 337ndash349
HuuskonenJ (2000) Estimation of aqueous solubility for a diverse set of organiccompounds based on molecular topology J Chem Inf Comput Sci 40773ndash777
HuuskonenJ (2001) Estimation of aqueous solubility in drug design Comb ChemHigh T Scr 4 311ndash316
HuuskonenJ et al (1998) Aqueous solubility prediction of drugs based on moleculartopology and neural network modeling J Chem Inf Comput Sci 38 450ndash456
JensenF (1999) Introduction to Computational Chemistry John Wiley and SonsNew York
JiZL et al (2003) Internet resources for proteins associated with drug therapeuticeffect adverse reactions and ADME Drug Discov Today 12 526ndash529
JoacutensdoacutettirSOacute et al (2002) Modeling and measurements of solidndashliquid and vaporndashliquid equilibria of polyols and carbohydrates in aqueous solution CarbohydrateRes 337 1563ndash1571
JoslashrgensenFS et al (2001) Prediction of bloodndashbrain barrier penetration In HoumlltjeH-Dand Sippl W (eds) Rational Approaches to Drug Design Prous Science PressBarcelona pp 281ndash285
JorgensenWL and DuffyEM (2002) Prediction of drug solubility from structureAdv Drug Delivery Rev 54 355ndash366
JudsonPN et al (2003) Using argumentation for absolute reasoning about the potentialtoxicity of chemicals J Chem Inf Comput Sci 43 1364ndash1370
KarelsonM (2000) Molecular Descriptors in QSARQSPR John Wiley amp Sons IncKatritzkyAR et al (1998) QSPR studies of vapor pressure aqueous solubility and the
prediction of waterndashair partition coefficients J Chem Inf Comput Sci 38 720ndash725KatritzkyAR et al (2001) Theoretical descriptors for the correlation of aquatice toxicity
of environmental pollutant by quantitative structurendashtoxicity relationships J ChemInf Comput Sci 41 1162ndash1176
KaznessisYN et al (2001) Prediction of bloodndashbrain partitioning using Monte Carlosimulations of molecules in water J Comput-Aided Mol Des 15 697ndash708
KelderJ et al (1999) Polar molecular surface as a dominating determinant for oralabsorption and brain penetration of drugs Pharm Res 16 1514ndash1519
KeserucircGM et al (2000) A neural network based virtual high throughput screening testfor the prediction of CNS activity Comb Chem High T Scr 3 535ndash540
KlopmanG (1992) MULTI-CASE 1 A hierarchical computer automated structureevaluation program Quant Struct-Act Relat 11 176ndash184
KlopmanG et al (1994) META 1 A program for the evaluation of metabolictransformations of chemicals J Chem Inf Comput Sci 34 1320ndash1325
KlopmanG et al (1997) META 3 A genetic algorithm for metabolic transform prioritiesoptimization J Chem Inf Comput Sci 37 329ndash334
KlopmannF and ZhuH (2001) Estimation of the aqueous solubility of organicmolecules by group contribution approach J Chem Inf Comput Sci 41439ndash445
KubinyiH (2003) Drug research myths hype and reality Nat Rev Drug Discovery2 665ndash668
KuumlhneR et al (1995) Group contribution methods to estimate water solubility of organiccompounds Chemosphere 30 2061ndash2077
KulkarniA et al (2002) Predicting Caco-2 permeation coefficients of organic moleculesusing membrane-interaction QSAR analysis J Chem Inf Comput Sci 42 331ndash342
KusuharaH and Sugiyama Y (2002) Role of transporters in the tissue-selective distri-bution and elimination of drugs transporters in the liver small intestine brain andkidney J Cont Release 78 43ndash54
LangowskiJ and LongA (2002) Computer systems for the prediction of xenobioticmetabolism Adv Drug Delivery Rev 54 407ndash415
LeachAR and GilletVJ (2003) An Introduction to Chemoinformatics KluwerAcademics Publishers Dordrecht
LengauerT (2002) BioinformaticsmdashFrom Genomes to Drugs Vol 14 Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim
LewisDFV (1996) The Cytochromes P450 Structure Function and MechanismTaylor and Francis London
LewisDFV and Dickins M (2002) Substrate SARs in human P450s Drug DiscovToday 7 918ndash925
LideDR (2003) CRC Handbook of Chemistry and Physics 84th edn CRC Press BocaRaton FL
LipinskiCA et al (1997) Experimental and computational approaches to estimatesolubility and permeability in drug discovery and development settings Adv DrugDelivery Rev 23 3ndash25
LivingstoneDJ (2003) Theoretical property prediction Curr Top Med Chem 31171ndash1192
MacchiaruloA et al (2004) Ligand selectivity and competition between enzymesin silico Nat Biotechnol 22 1039ndash1045
McElroyNR and JursPC (2001) Prediction of aqueous solubility of heteroatom-containing organics compounds from molecular structure J Chem Inf ComputSci 41 1237ndash1247
McFarlandJW et al (2001) Estimating the water solubilities of crystalline compoundsfrom their chemical structures alone J Chem Inf Comput Sci 41 1355ndash1359
MewesHW et al (2002) MIPS a database for genome and protein sequences NucleicAcids Res 30 31ndash34
MiledZB et al (2003) An efficient implementation of a drug candidate databaseJ Chem Inf Comput Sci 43 25ndash35
MillerMA (2002) Chemical databases techniques in drug discovery Nat Rev DrugDiscovery 1 220ndash227
MountDW (2001) Bioinformatics Sequence and Genome Analysis Cold SpringHarbor Laboratory Press Cold Spring Harbor
MueggeI (2003) Selection criteria for drug-like compounds Med Res Rev 23302ndash321
MueggeI et al (2001) Simple selection criteria for drug-like chemical matter J MedChem 44 1841ndash1846
Murcia-SolerM et al (2003) Drugs and nondrugs an effective discrimination withtopological methods and artificial neural networks J Chem Inf Comput Sci 431688ndash1702
Murray-RustP and RzespaHS (1999) Chemical markup Language and XML Part IBasic principles J Chem Inf Comput Sci 39 928
NicholsonJK and WilsonID (2003) Understanding lsquoglobalrsquo systems biology meta-bolism and the continuum of metabolism Nat Rev Drug Discov 2 668ndash677
NielsenH et al (1996) Defining a similarity threshold for a functional protein sequencepattern The signal peptide cleavage site Proteins 24 165ndash177
NorinderU and HaeberleinM (2002) Computational approaches to the prediction ofthe blood-brain distribution Adv Drug Delivery Rev 54 291ndash313
NorrisDA et al (2000) Development of predictive pharmacokinetic simulation modelsfor drug discovery J Contr Release 65 55ndash62
OlssonT and OpreaTI (2001) Chemoinformatics a tool for decision-marker in drugdiscovery Curr Opin Drug Disc Dev 4 308ndash313
OpreaTI (2000) Property distribution of drug-related chemical databases J ComputAided Mol Des 14 251ndash264
OpreaTI (2002a) Chemical space navigation in lead discovery Curr Opin ChemBiol 6 384ndash389
OpreaTI (2002b) Current trends in lead discovery are we looking for the appropriateproperties J Comput-Aided Mol Des 16 325ndash334
OpreaTI (2003) Chemoinformatics and the quest for leads in drug discovery InGasteigerJ (ed) Handbook of ChemoinformaticsmdashFrom Data to KnowledgeWiley-VCH Weinheim Vol 4 pp 1509ndash1531
OpreaTI and GottfriesJ (1999) Toward minimalistic modeling of oral drug absorptionJ Mol Graph Model 17 261ndash274
OpreaTI and GottfriesJ (2001a) CHEMGPS a chemical space navigation tool InHoumlltjeH-D and SipplW (eds) Rational Approaches to Drug Design Prous SciencePress Barcelona pp 437ndash446
OpreaTI and GottfriesJ (2001b) Chemography the art of navigating in chemicalspace J Comb Chem 3 157ndash166
OpreaTI et al (2001a) Is there a difference between leads and drugs A historicalperspective J Chem Inf Comput Sci 41 1308ndash1315
OpreaTI et al (2001b) Quo vadis scoring functions Toward an integratedpharmacokinetic and binding affinity prediction framework In GhoseAK andViswanadhanVN (eds) Combinatorial Library Design and Evaluation for DrugDesign Marcel Dekker Inc New York pp 233ndash266
OpreaTI et al (2002) Pharmacokinetically based mapping device for chemical spacenavigation J Comb Chem 4 258ndash266
OrengoCA et al (2002) Bioinformatics Genes Proteins and Computers AdvancedTexts BIOS Scientific Publishers Abingdon
PadeV and StavhanskyS (1998) Link between drug absorption solubility and permeab-ility measurements in Caco-2 Cells J Pharm Sci 87 1604ndash1607
2159
SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160

SOacuteJoacutensdoacutettir et al
PalmK et al (1998) Evaluation of dynamic polar surface area as predictor of drugabsorption comparison with other computational and experimental predictorsJ Med Chem 41 5382ndash5392
ParkeDV et al (1990) The safety evaluation of drugs and chemical by the use of com-puter optimized molecular parametric analysis of chemical toxicity (COMPACT)ATLA 18 91ndash102
ParrottN and LaveacuteT (2002) Prediction of intestinal absorption comparative assess-ment of GASTROPLUSTM and IDEATM Eur J Pharm Sci 17 51ndash61
ParsonsAB et al (2004) Integration of chemical-genetic and genetic interactiondata links bioactive compounds to cellular target pathways Nat Biotechnol 2262ndash69
PattersonDE et al (1996) Neighborhood behavior a useful concept for validation ofmolecular diversity descriptors J Med Chem 39 3049ndash3059
PearlmanRS (2000) CONCORD Userrsquos Manual Tripos Inc St Louis MOPedersenAG and NielsenH (1997) Neural network prediction of translation initi-
ation sites in eukaryotes perspectives for EST and Genome analysis ISMB 5226ndash233
PeriS et al (2003) Development of human protein reference database as an ini-tial platform for approaching systems biology in humans Genome Res 132363ndash2371
PrausnitzJM et al (1986) Molecular Thermodynamics and Fluid Phase Equilibria2nd edn Prentice-Hall NJ
RanY et al (2001) Prediction of aqueous solubility of organic compounds by generalsolubility equation (GSE) J Chem Inf Comput Sci 41 1208ndash1217
ReynoldsCH et al (1998) Lead discovery using stochastic cluster analysis (SCA) anew method for clustering structurally similar compounds J Chem Inf Comp Sci38 305ndash312
ReynoldsCH et al (2001) Diversity and coverage of structural sublibraries selectedusing the SAGE and SCA algorithms J Chem Inf Comp Sci 41 1470ndash1477
RichardAM and Williams CR (2002) Distributed structure-searchable toxicity(DSSTox) puclic database network a proposal Mutat Res 499 27ndash52
RoseK et al (2002) Modeling bloodndashbrain barrier partitioning using the electrotopo-logical state J Chem Inf Comput Sci 42 651ndash666
SadowskiJ and KubinyiH (1998) A scoring scheme for discriminating between drugsand nondrugs J Med Chem 41 3325ndash3329
SanderC and Schneider R (1991) Database of homology-derived protein structuresand the structural meaning of sequence alignment Proteins 9 56ndash68
SangvhiT et al (2003) Estimation of aqueous solubility by the General SolubilityEquation (GSE) the easy way QSAR Comb Sci 22 258ndash262
SelloG (1998) Similarity measures is it possible to compare dissimilar structuresJ Chem Inf Comput Sci 38 691ndash701
StahuraFL and BajorathJ (2002) Bio- and chemo-informatics beyond data man-agement crucial challenges and future opportunities Drug Discov Today 7S41ndashS47
SunH et al (2003) Drug efflux transporters in the CNSrsquo Adv Drug Delivery Rev 5583ndash105
TakaokaY et al (2003) Development of a method for evaluating drug-likeness andease of synthesis using a data set in which compounds are assigned scores based onchemistsrsquo intuition J Chem Inf Comput Sci 43 1269ndash1275
TalafousJ et al (1994) META 2 A dictionary model of mammalian xenobioticmetabolism J Chem Inf Comput Sci 34 1326ndash1333
TaskinenJ and YliruusiJ (2003) Prediction of physcochemical properties based onneural networks modelling Adv Drug Delivery Rev 55 1163ndash1183
TeagueSJ et al (1999) The design of leadlike combinatorial libraries Angew ChemInt Ed 38 3743ndash3748
ThompsonJD et al (2003) Predicting aqueous solubilities from aqueous free ener-gies of solvation and experimental or calculated vapor pressure of pure substancesJ Chem Phys 119 1661ndash1670
TodeschiniR and ConsonniV (2000) Handbook of Molecular Descriptors Vol 11Methods and Principles in Medicinal Chemistry Wiley-VCH Weinheim
TongW et al (2002) Development of quantitative structure-activity relationships(QSPRs) and their use for priority setting in the testing strategy of endocrinedisruptors Reg Res Persp J May 1ndash21
van de Waterbeemd H (2003) Physico-chemical approaches to drug absorption In vande Waterbeemd H LennernaumlsH and ArthurssonP (eds) Drug BioavailabilityEstimation of Solubility Permeability Absorption and Bioavailability Methods andPrinciples in Medicinal Chemistry Wiley-VCH Weinheim Vol 18 pp 3ndash20
van de Waterbeemd H and GiffordE (2003) ADMET in silico modelling Towardsprediction paradise Nat Rev Drug Discovery 2 192ndash204
van de Waterbeemd H et al (2001) Property-based design optimization of drugabsorption and pharmacokinetics J Med Chem 44 1313ndash1333
van de Waterbeemd H et al (2003) Drug Bioavailability Estimation of SolubilityPermeability Absorption and Bioavailability Vol 18 of Methods and Principles inMedicinal Chemistry Wiley-VCH Weinheim
ViswanadhanVN et al (1989) Atomic physicochemical parameters for three dimen-sional structure directed quantitative structurendashactivity relationships 4 Additionalparameters for hydrophopic and dispersive interactions and their application foran automated superposition of certain naturally occurring nucleoside antibioticsJ Chem Inf Comput Sci 29 163ndash172
VoigtJH et al (2001) Comparison of the NCI open database with seven large chemicalstructural databases J Chem Inf Comp Sci 41 702ndash712
WagenerM and van Geerestein VJ (2000) Potential drugs and nondrugs predictionand identification of important structural features J Chem Inf Comput Sci 40280ndash292
WaltersWP and Murcko MA (2002) Prediction of lsquodrug-likenessrsquo Adv Drug DeliveryRev 54 255ndash271
WaltersWP et al (1998) Virtual screeningmdashan overview Drug Discovery Today 3160ndash178
WegnerJK and ZellA (2003) Prediction of aqueous solubility and partition coefficientoptimized by a genetic algorithm based descriptor selection method J Chem InfComput Sci 43 1077ndash1084
WeiningerD (1988) SMILES a chemical language and information system 1 Intro-duction to methodology and encoding rules J Chem Inf Comput Sci 2831ndash36
WesselMD et al (1998) Prediction of human intestinal absorption of drug compoundsfrom molecular structure J Chem Inf Comput Sci 38 726ndash735
WillettP (2000) Chemoinformaticsmdashsimilarity and diversity in chemical librariesCurr Opin Biotech 11 85ndash88
WillettP (2003) Similarity-based approaches to virtual screening Biochem Soc Trans31 603ndash606
WiltonD et al (2003) Comparison of ranking methods for virtual screening in lead-discovery programs J Chem Inf Comput Sci 43 469ndash474
WolohanPRN and Clark RD (2003) Predicting drug pharmacokinetic proper-ties using molecular interaction fields and SIMCA J Comput-Aided Mol Des17 65ndash76
XeneariosI et al (2002) DIP the database of interacting proteins a researchtool for studying cellular networks of protein interactions Nucl Acids Res 30303ndash305
XuJ and StevensonJ (2000) Drug-like index a new approach to measure drug-likecompounds and their diversity J Chem Inf Comput Sci 40 1177ndash1187
YaffeD et al (2001) A fuzzy ARTMAP based on quantitative structurendashproperty rela-tionships (QSPRs) for predicting aqueous solubility of organic compounds J ChemInf Comput Sci 41 1177ndash1207
YanA and GasteigerJ (2003) Prediction of aqueous solubility of organic compoundsbased on a 3d structure representation J Chem Inf Comput Sci 43 429ndash434
YoungRC et al (1988) Development of a new physicochemical model for brain pen-etration and its application to the design of centrally acting H2 receptor histamineantagonists J Med Chem 31 656ndash671
ZamoraI (2001) Prediction of oral drug permeability In HoumlltjeH and SipplW(eds) Rational Approaches to Drug Design Prous Science Press Barcelonapp 271ndash280
ZanzoniA et al (2002) MINT a molecular interaction database FEBS Lett 513135ndash140
ZhongCG and HuQH (2003) Estimation of the aqueous solubility oforganic compounds using molecular connectivity indices J Pharm Sci 922284ndash2294
2160