bfam project bf-s15t07 “efficient clustering algorithms for genome-wide expression analysis“...
Post on 18-Dec-2015
214 views
TRANSCRIPT

BFAM Project BF-S15T07“Efficient clustering algorithms
for genome-wide expressionanalysis“
BFAM Project BF-S15T08“Modeling and visualization of
biochemical networks“
Misc. projects inBioinformatics
Jens Ernst ([email protected]) Sebastian Wernicke ([email protected])Arno Buchner ([email protected])Jan Griebsch ([email protected])
Hanjo Täubig ([email protected])Moritz Maass ([email protected])
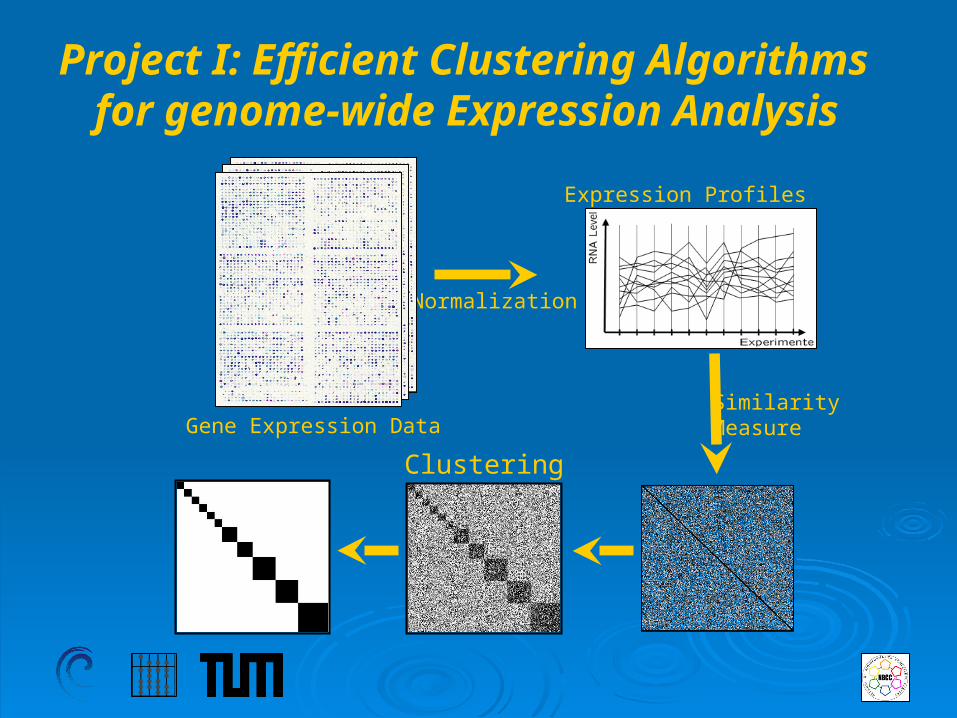
Project I: Efficient Clustering Algorithms for genome-wide Expression Analysis
Gene Expression Data
Expression Profiles
SimilarityMeasure
Normalization
Clustering

1. Retrospect: The SR-Algorithm
• Powerful algorithm for similarity-based clustering
• Based on methods of spectral graph theory,numerical linear algebra and randomization
• Applicable not only to gene expression profilesbut to any class of biological objects where pair-wise similarity is defined
• Thoroughly mathematically analyzed with respectto noise-robustness and running time
• Complexity: Θ(n2), and hence optimal
• New: Parallelized version and optimized version forsparse similarity matrices.

Output quality as a function of n and the amount of noise (false positive, false negative rate α). The number of clusters is specified to the algorithm.
500 – 2000 genes forming 4 clusters with 20%-49% false positives/negatives
αn0.45
1.0
0.45
n
α
2. Tests on Synthetic Data (1)

500 – 4000 genes forming 4 clusters with 20%-45% false positives/negatives
0.45
1.0
nα
n
α
0.45
Output quality as a function of n and the amount of noise (false positive, false negative rate α). The number of clusters is found by the algorithm.
Tests on Synthetic Data (2)
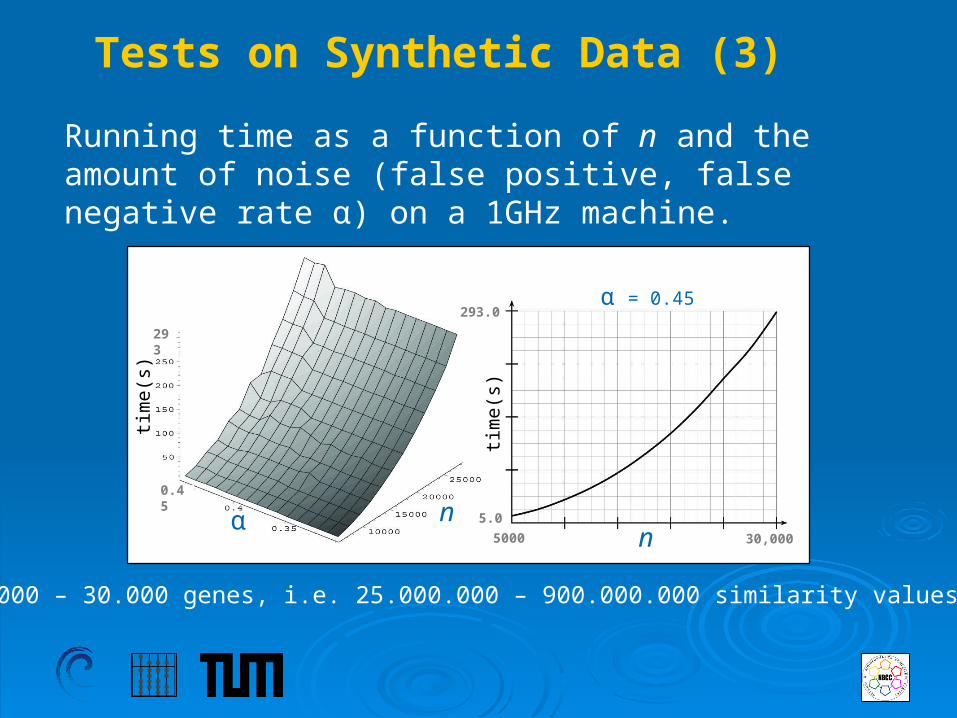
Running time as a function of n and the amount of noise (false positive, false negative rate α) on a 1GHz machine.
Tests on Synthetic Data (3)
5.000 – 30.000 genes, i.e. 25.000.000 – 900.000.000 similarity values
nα5000 30,000
5.0
293.0
tim
e(s)
tim
e(s)
0.45
293
n
α = 0.45

4. Clustering Protein Interaction Networks
• Experiments with a network from the STRING system provided by the Bork group at EMBL.
• Data: Escherichia coli, orthologous group-based
• Edge scores: Interaction intensities defined by
score=1-(1-neighborhood score)x(1-fusion score)x (1-co-occurence score)
[ Courtesy of C. von Mering, Nucleic Acids Res. 2003 Jan 1;31(1):258-61 ]
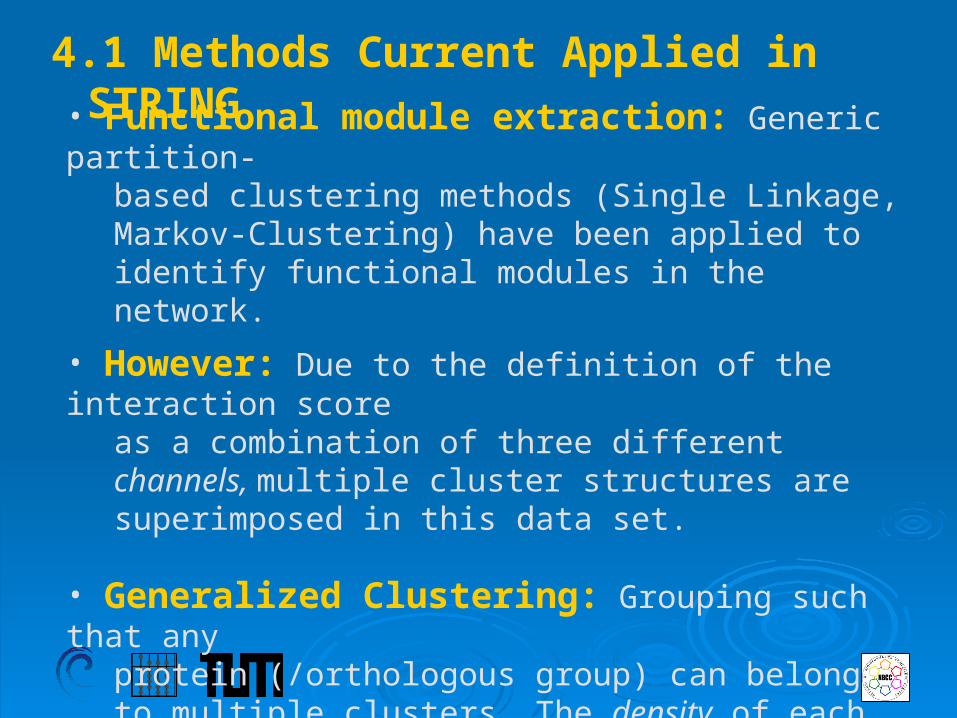
• Functional module extraction: Generic partition-based clustering methods (Single Linkage, Markov-Clustering) have been applied to identify functional modules in the network.
• However: Due to the definition of the interaction scoreas a combination of three different channels, multiple cluster structures are superimposed in this data set.
• Generalized Clustering: Grouping such that any protein (/orthologous group) can belong to multiple clusters. The density of each cluster should be as high as possible, whereas the inter-cluster connectivity (excluding overlaps) should be minimized.
4.1 Methods Current Applied in STRING
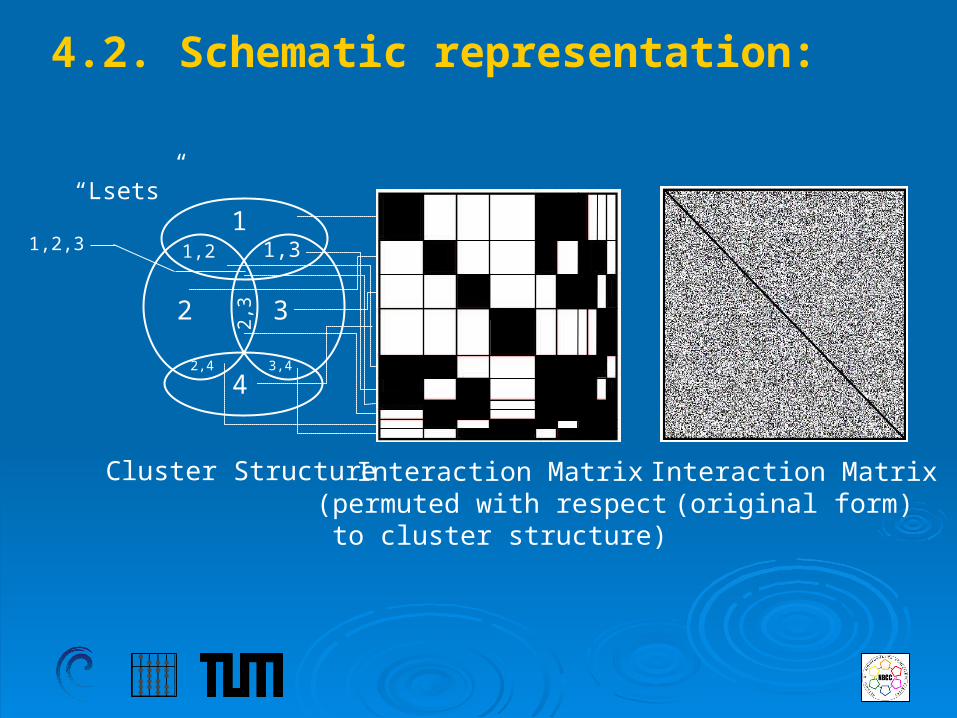
4.2. Schematic representation:
1
2 3
4
1,2 1,32,
3
2,4 3,4
1,2,3
“Lsets”
Cluster Structure Interaction Matrix(permuted with respect
to cluster structure)
Interaction Matrix(original form)

1. Construction of elementary sets by SR-techniques
Result: A partition of the protein set into a fixed number k of elementary sets. The value of k may safely be overestimated.
Intra- and inter-Lset edge densities:
k = 150;
Mean intra-Lset density: 0.309
Inter-Lset connectivity: 0.024
Lsets belonging to the same cluster
4.2. Construction of Intersecting Clusters:
Frequency distribution of edge densities withinand between Lsets
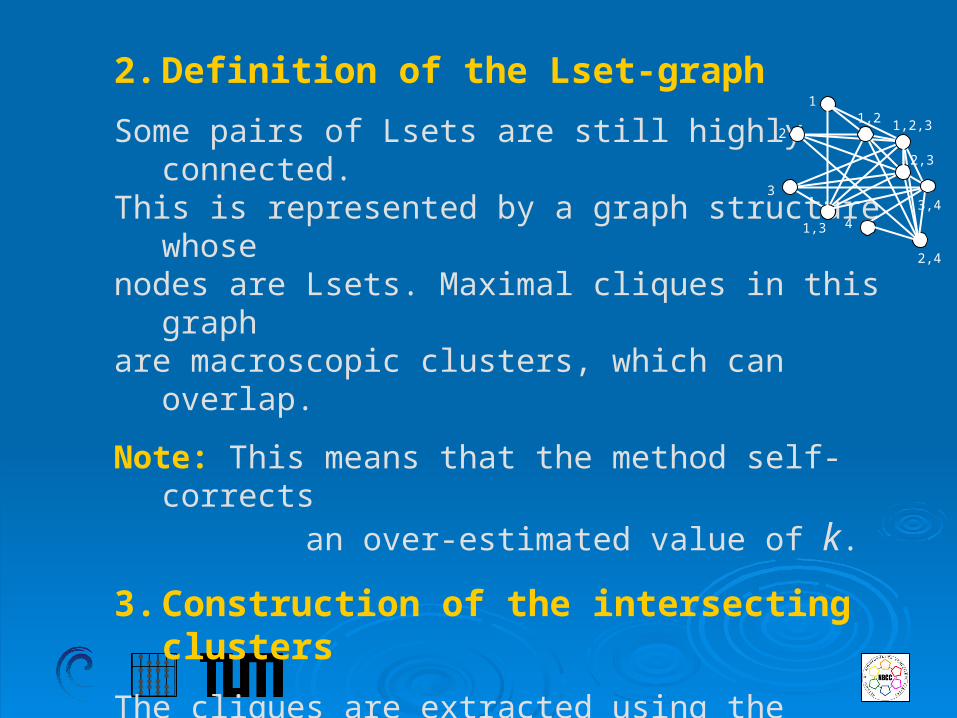
2. Definition of the Lset-graph
Some pairs of Lsets are still highly connected.This is represented by a graph structure whosenodes are Lsets. Maximal cliques in this graph are macroscopic clusters, which can overlap.
Note: This means that the method self-corrects an over-estimated value of k.
3. Construction of the intersecting clusters
The cliques are extracted using the Tsukiyama-algorithm.
Result: 144 clusters
Intra-cluster density: 0.269
Inter-cluster connectivity: 0.020 (excl. overlaps)
1
2,3
2,4
3
1,3 43,4
1,2,31,2
2

Quality assessment based on biological expert knowledge: currently pending
The clusters are being compared with a known set of protein-to-pathway assignments.
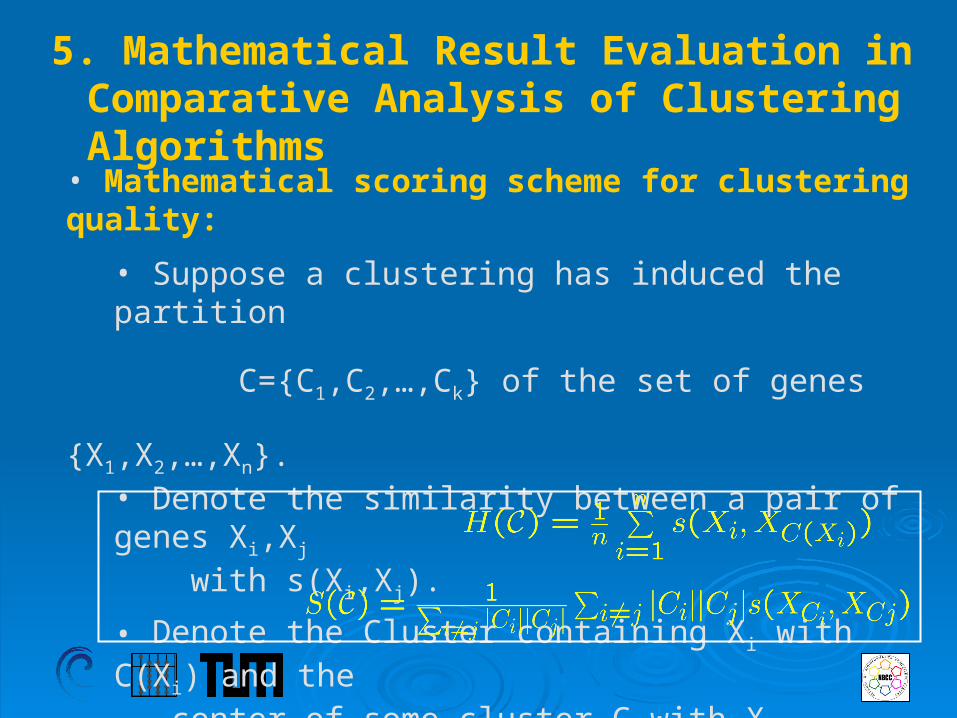
5. Mathematical Result Evaluation in Comparative Analysis of Clustering Algorithms
• Mathematical scoring scheme for clustering quality:
• Suppose a clustering has induced the partition
C={C1,C2,…,Ck} of the set of genes {X1,X2,…,Xn}.
• Denote the similarity between a pair of genes Xi,Xj
with s(Xi,Xj).
• Denote the Cluster containing Xi with C(Xi) and the center of some cluster C with XC.
Cluster Homogeneity:
Separation:
• Remarks:
1. The cluster analysis was conducted in the form of a blind test. Use of expert knowledge or supervised learning techniques was not intended for.
2. No prior selection of genes was asked for.
3. Normalization/standardization of expression data or the similarity-/distance measure were not explicitly required.
• Choice of similarity measure s for the evaluation:
Pearson Correlation Coefficient (due to invariance under scaling and translation of expression profiles, which was used by some participants).
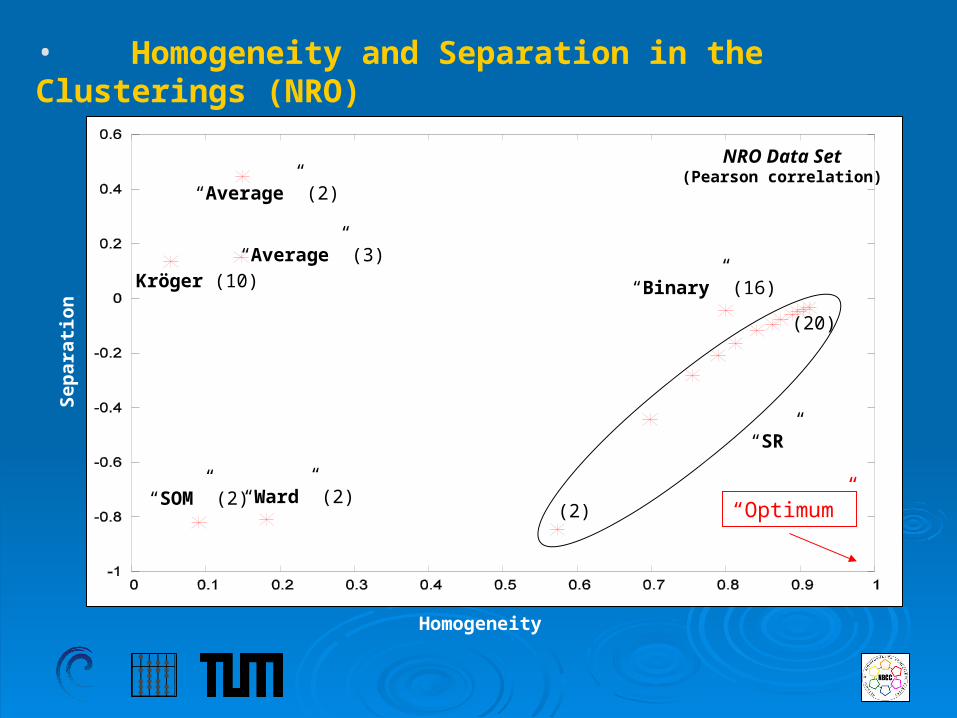
• Homogeneity and Separation in the Clusterings (NRO)
Homogeneity
“SOM” (2) “Ward” (2)
Kröger (10)“Average” (3)
“Average” (2)
“Binary” (16)
“SR”
(2)
(20)
Sep
arat
ion
“Optimum”
NRO Data Set(Pearson correlation)
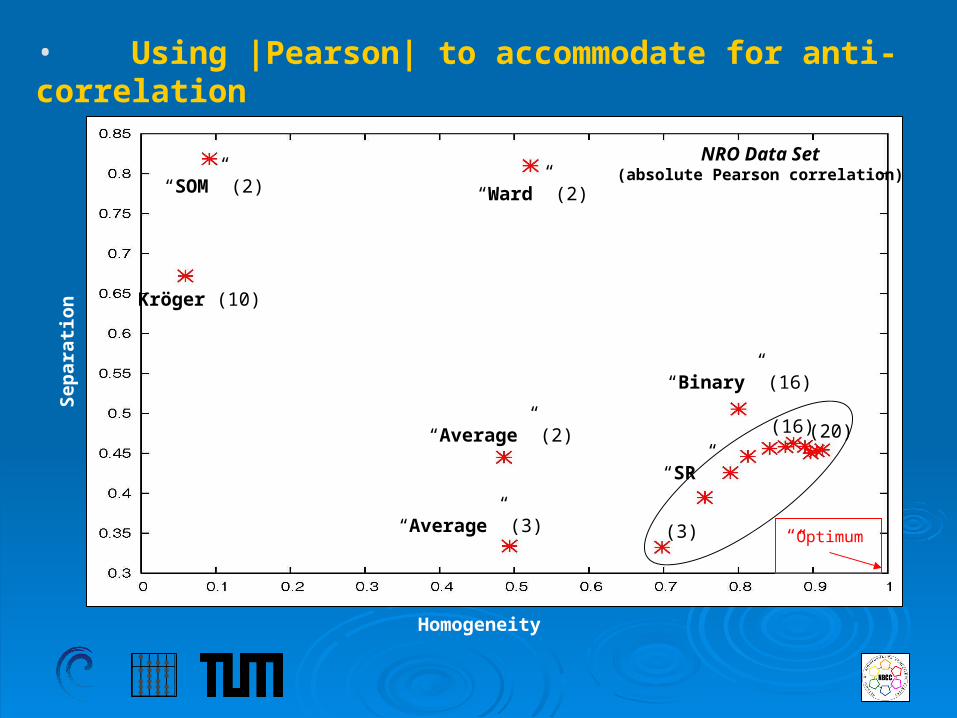
• Using |Pearson| to accommodate for anti-correlation
Homogeneity
“SOM” (2) “Ward” (2)
Kröger (10)
“Average” (3)
“Average” (2)
“Binary” (16)
“SR”
(3)
(20)
Sep
arat
ion
“Optimum”
NRO Data Set(absolute Pearson correlation)
(16)
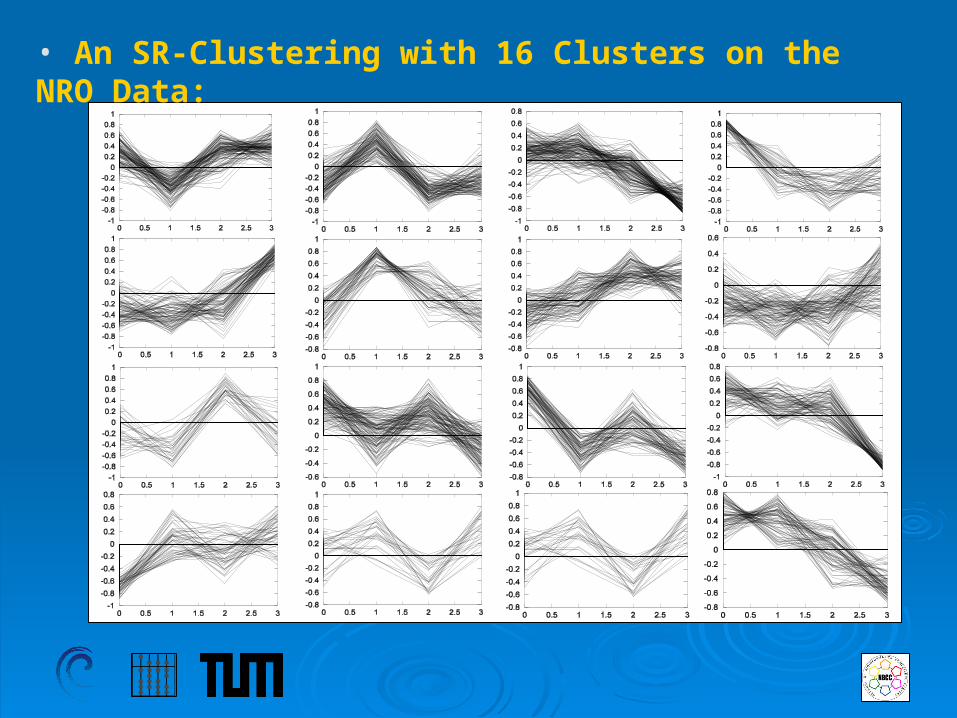
• An SR-Clustering with 16 Clusters on the NRO Data:
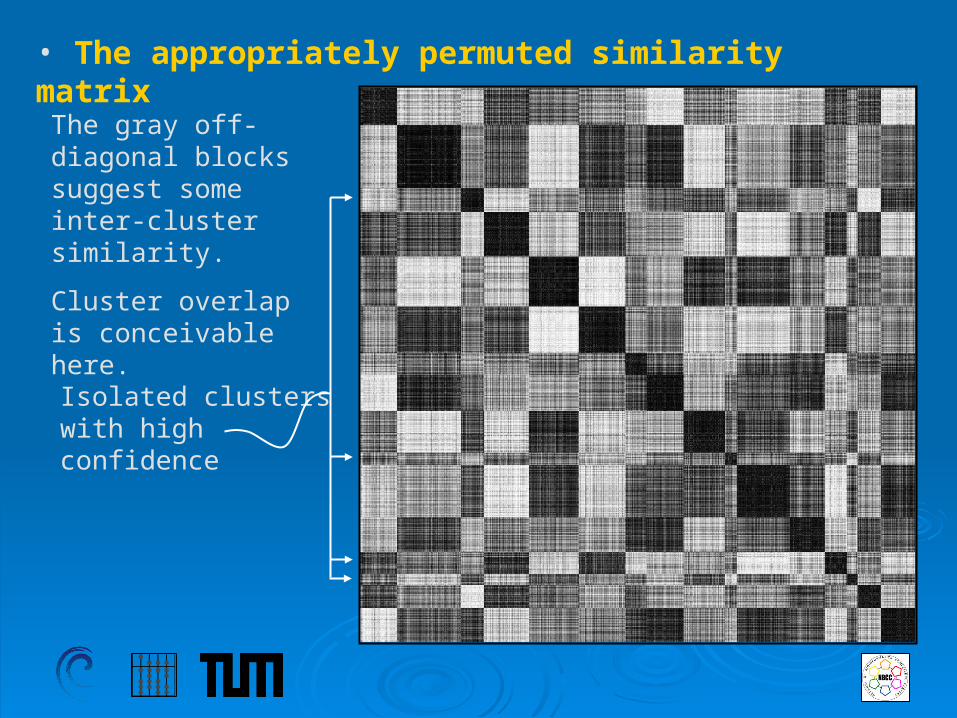
• The appropriately permuted similarity matrix
The gray off-diagonal blocks suggest some inter-cluster similarity.
Cluster overlap is conceivable here.
Isolated clusters with highconfidence

6. Cooperation within the BFAM Network:
1. Cooperation with Genomatix Software GmbH:• Extension of cluster analysis by integration of information
from biological databases and expert knowledge
2. Cooperation with Genomatix Software GmbH, BiomaxInformatics GmbH, the group of Prof. Lasser and the group of Prof. Kriegel:• Comparative analysis of clustering algorithms
3. Publications:[1] „Similarity-Based Clustering Algorithms for Gene Expression Profiles“,
J. Ernst, Dissertation, Technische Universität München, 2002
[2] „Generalized Clustering of Gene Expression Profiles – A Spectral Approach“,
J. Ernst, Proc. of the Int. Conference on Bioinformatics, Bangkok, 2002
[3] „The Complexity of Detecting Fixed-Density Clusters“, H. Täubig et. al.,
Proc. of the 5th Italian Conference on Algorithms and Complexity, 2003

Chair for Efficient Algorithms
Graph Theory
Combinatorial Optimization
Randomized Algorithms
Computer Algebra
Petri Nets
Scheduling
Complexity Theory
Algorithms for Bioinformatics
Algorithm Visualization
Project“Clustering“
Project“Biological Networks“
Misc.Bioinformatics
Projects