b promotes pi3k-akt signaling and prostate cancer cell ... · akt, we further examined whether...
TRANSCRIPT

SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
B IOCHEM ISTRY
1Ludwig Institute for Cancer Research and Science for Life Laboratory, UppsalaUniversity, Uppsala SE 751 24, Sweden. 2Unit of Pathology, Department of MedicalBiosciences, Umeå University, Umeå SE 901 85, Sweden. 3Laboratory of Biochemistry,Showa Pharmaceutical University, Tokyo 194-8543, Japan.*These authors contributed equally to this work.†Corresponding author. Email: [email protected] (M.L.); [email protected] (C.-H.H.)
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
Copyright © 2017
The Authors, some
rights reserved;
exclusive licensee
American Association
for the Advancement
of Science. No claim
to original U.S.
Government Works
Dow
nloaded from
TGF-b promotes PI3K-AKT signaling and prostate cancercell migration through the TRAF6-mediatedubiquitylation of p85aAnahita Hamidi,1 Jie Song,2 Noopur Thakur,1 Susumu Itoh,3 Anders Marcusson,1 Anders Bergh,2
Carl-Henrik Heldin,1*† Maréne Landström1,2*†
Transforming growth factor–b (TGF-b) is a pluripotent cytokine that regulates cell fate and plasticity in normaltissues and tumors. The multifunctional cellular responses evoked by TGF-b are mediated by the canonicalSMAD pathway and by noncanonical pathways, including mitogen-activated protein kinase (MAPK) pathwaysand the phosphatidylinositol 3′-kinase (PI3K)–protein kinase B (AKT) pathway. We found that TGF-b activatedPI3K in a manner dependent on the activity of the E3 ubiquitin ligase tumor necrosis factor receptor–associatedfactor 6 (TRAF6). TRAF6 polyubiquitylated the PI3K regulatory subunit p85a and promoted the formation of acomplex between the TGF-b type I receptor (TbRI) and p85a, which led to the activation of PI3K and AKT. Lys63-linked polyubiquitylation of p85a on Lys513 and Lys519 in the iSH2 (inter–Src homology 2) domain was requiredfor TGF-b–induced activation of PI3K-AKT signaling and cell motility in prostate cancer cells and activatedmacrophages. Unlike the activation of SMAD pathways, the TRAF6-mediated activation of PI3K and AKT wasnot dependent on the kinase activity of TbRI. In situ proximity ligation assays revealed that polyubiquitylationof p85a was evident in aggressive prostate cancer tissues. Thus, our data reveal a molecular mechanism bywhich TGF-b activates the PI3K-AKT pathway to drive cell migration.
h
on February 14, 2020ttp://stke.sciencem
ag.org/
INTRODUCTIONTransforming growth factor–b (TGF-b) family members are importantregulators of normal epithelial cell differentiation, cytostasis, andapoptosis but can also promote tumorigenesis (1, 2). TGF-b binds totype II and type I serine and threonine kinase receptors (TbRII andTbRI, respectively) and induces the formation of a heterotetramericcomplex of two TbRIs and two TbRIIs. In the receptor complex, theconstitutively active TbRII phosphorylates TbRI resulting in activationof the TbRI kinase, leading to phosphorylation andnuclear translocationof receptor-associated SMADs (R-SMADs),where they, in complexwithSMAD4, regulate the expression of certain target genes (3, 4).
TGF-b also signals through non-SMADpathways, such as the extra-cellular signal–regulated kinase 1 and 2 (ERK1/2), c-Jun N-terminal ki-nase (JNK), p38 mitogen-activated protein kinase (MAPK), andphosphatidylinositol 3′-kinase (PI3K)–protein kinase B (AKT) path-ways (5, 6). Both TbRI and TbRII are required for TGF-b–induced ac-tivation of class IA PI3K (7). The PI3K family is divided into threeclasses based on their substrate specificities and structures. Class IAmembers (hereafter referred to as PI3K) are heterodimers of a regula-tory subunit (p85a, p85b, p55a, p55g, or p50a) and a catalytic subunit(p110a, p110b, or p110g) (8). PI3K generates phosphatidylinositol3,4,5-trisphosphate (PIP3) from phosphatidylinositol 4,5-bisphosphate(PIP2) (9). PIP3 interacts with and thereby translocates AKT to the plasmamembrane, where AKT becomes activated by phosphorylation at Thr308
by phosphoinositide-dependent kinase 1 (PDK1) and at Ser473 bymTORC2 (mammalian target of rapamycin complex 2) and other kinases(10). There are three isoformsofAKT(AKT1,AKT2, andAKT3); primar-ily, AKT1 has been linked to oncogenesis (11).
An important mechanism of the regulation of signaling pathwaysinvolves posttranscriptional modifications via ubiquitin chains, whichare bound covalently to an acceptor lysine of the target protein (12).Whereas Lys48-linked polyubiquitylation of proteins targets its sub-strates for proteasomal degradation (13), Lys63-linked polyubiquityla-tion often regulates the function and/or localization of proteins. TheRING E3 ligase tumor necrosis factor receptor–associated factor 6(TRAF6), which is known to induce Lys63-linked polyubiquitylationof its substrates, interacts with a consensus motif present in TbRI.TGF-b induces autoubiquitylation and activation of TRAF6 andLys63-linked polyubiquitylation and activation of TGF-b–associatedkinase 1 (TAK1), which lead to the activation of downstream p38MAPK (14). TRAF6 also promotes proteolytic cleavage of TbRI, re-leasing the intracellular domain of TbRI, which, after translocationto the nucleus, induces a transcriptional program (15).
Here, we aimed to elucidate the mechanisms by which TGF-b in-duces the activation of PI3K and AKT. Our results showed that TRAF6is required for TGF-b–induced Lys63-linked ubiquitylation of the p85asubunit of PI3K, which leads to PI3K activation, promoting the activa-tion of AKT and its recruitment to the plasma membrane. Moreover,we found that TGF-b–induced cellmigration is dependent on PI3K andTRAF6. Notably, by analyzing tissue sections by an in situ proximityligation assay (PLA), we observed a correlation between polyubiquity-lated p85a and tumor aggressiveness in patients with prostate cancer.The current study provides evidence for the molecular mechanismswhereby TGF-b promotes activation of the PI3K-AKT pathway anddemonstrates its correlationwith aggressive disease for prostate cancerpatients.
RESULTSAKT interacts with TbRI in a TRAF6-dependent mannerAKT is activated upon stimulation by TGF-b (16). To elucidate themolecular mechanism of the TGF-b–induced activation of AKT, we
1 of 15

SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
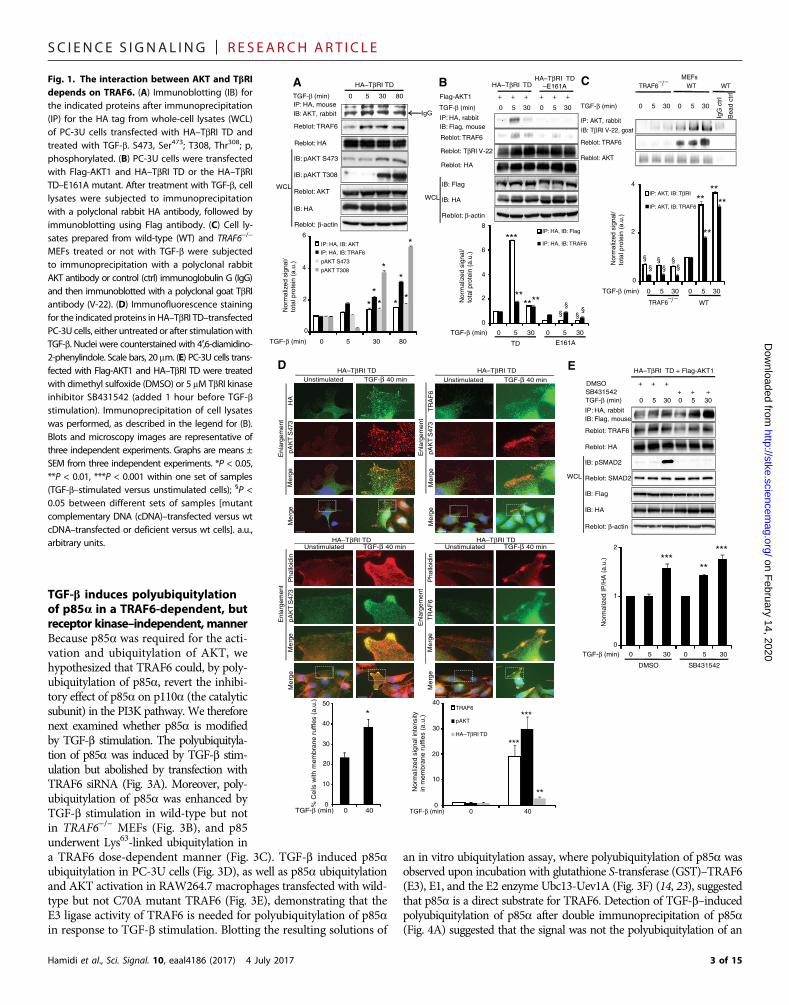
performed coimmunoprecipitation and coimmunostaining analy-ses to determine whether a complex containing AKT and TbRI isformed. In transfected human prostate cancer (PC-3U) cells, weobserved an interaction between hemagglutinin (HA)–taggedTbRI TD [a TbRI mutant that is partly constitutively active (14)]and endogenous AKT that was increased by TGF-b stimulation(Fig. 1A). TRAF6, which induces Lys63-linked polyubiquitylationand activation of AKT upon stimulation by insulin-like growth fac-tor 1 (IGF-1), interleukin-1b (IL-1b), and lipopolysaccharide (17),was also pulled down by the TbRI antibody, as reported previously(14), and AKT activation correlated with its interaction with TbRI(Fig. 1A). To explore whether TRAF6 is necessary for the interactionbetween HA–TbRI TD and AKT, cells were transiently transfectedwith HA–TbRI TD or the E161A mutant HA–TbRI TD, in whichthe TRAF6 binding motif is mutated (14). The interaction of AKTwith HA–TbRI TD–E161A was decreased compared to HA–TbRI TD(Fig. 1B). Furthermore, an interaction between endogenous TbRI andAKT in wild-type mouse embryonic fibroblasts (MEFs) that wasenhanced upon TGF-b stimulation was significantly reduced inTRAF6−/−MEFs (Fig. 1C). Consistent with the coimmunoprecipitationresults, a coimmunostaining analysis showed that activated AKT,TRAF6, and TbRI colocalized in cell membrane ruffles upon TGF-bstimulation (Fig. 1D).
Because TRAF6 is required for the interaction between TbRI andAKT, we further examined whether TRAF6 interacts with AKTupon TGF-b stimulation by transfecting human embryonic kidney(HEK) 293T cells with HA-tagged AKT and Flag-tagged wild-typeTRAF6. Immunoprecipitation of cell lysates with an HA antiserum,followed by immunoblotting with a Flag antiserum, revealed a bandwith the size of TRAF6; the density of that band was increased incells stimulated with TGF-b. In contrast, no coimmunoprecipitationwas seen in lysates from cells transfected with the enzymaticallyinactive C70A mutant TRAF6 (fig. S1A). Activation of AKT, asmonitored by phosphorylation of Thr308 and Ser473, correlated withits interaction with TRAF6 (fig. S1A). Immunofluorescence imagingof endogenous TRAF6 and phospho-AKT (pAKT) (Ser473) in PC-3Ucells showed that these proteins colocalized in membrane ruffles.Both the colocalization and the number of membrane ruffles wereenhanced in cells stimulated with TGF-b (fig. S1B). In cells depletedof TRAF6 by small interfering RNA (siRNA), no Ser473-pAKT wasobserved at the cell membrane in response to TGF-b stimulation(fig. S1B).
The fact that the C70A mutant TRAF6 did not coimmunoprecipi-tate with HA-AKT suggests that Cys70 mediated the interaction be-tween TRAF6 and its substrate, in line with the report from Yin et al.(18), or that ubiquitin chains mediated the interaction between TRAF6and AKT. Therefore, we performed in vivo ubiquitylation assays (14)using PC-3U cells. After preparing lysates from TGF-b–stimulated ornontreated cells, endogenous AKT was immunoprecipitated andsubjected to immunoblotting with antibodies against polyubiquitin.TGF-b induced robust polyubiquitylation of AKT, which correlatedin time with the phosphorylation and activation of AKT (fig. S2A).Double immunoprecipitation of AKT was performed, followed by im-munoblotting with antibodies against Lys63-linked polyubiquitin (fig.S2B), and suggested that the polyubiquitin signal was not from anotherinteracting protein.
To examine the nature of the polyubiquitin chains, we trans-fected PC-3U cells with HA-tagged wild-type ubiquitin (wild-type3×HA-Ub) or mutant ubiquitins in which all lysine residues, except
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
for Lys48 (Lys48-only 3×HA-Ub) or Lys63 (Lys63-only 3×HA-Ub),were mutated to arginine residues. TGF-b–induced ubiquityla-tion of AKT was seen when wild-type or Lys63-only Ub plasmids,but not Lys48-only Ub plasmid, were expressed, indicating thatTGF-b induced Lys63-linked polyubiquitylation of AKT (fig. S2C).We further examined whether phosphorylated AKT undergoesLys63-linked polyubiquitylation by immunoprecipitating Thr308-pAKTand immunoblotting for Lys63-linked polyubiquitin. The same AKTmolecules that underwent activation by phosphorylation also dis-played Lys63-linked polyubiquitylation upon TGF-b stimulation(fig. S2D).
Because the E3 ligase activity of TRAF6 was required for AKTbinding to TRAF6 (fig. S1A) and AKT underwent Lys63-linkedpolyubiquitylation upon TGF-b stimulation (fig. S2, A to D), we ex-plored whether TRAF6 is responsible for AKT ubiquitylation. TGF-b–induced phosphorylation (fig. S3A) and ubiquitylation (fig. S3B) ofAKT were seen in wild-type but not in TRAF6−/−MEFs. Moreover, thephosphorylation of AKT increased with increasing amounts of trans-fected TRAF6 (fig. S3C), and both phosphorylation (fig. S3D) and ubi-quitylation (fig. S3E) of AKT were seen after transfection of wild-typebut not that of C70A mutant TRAF6. Thus, our observations suggestthat TGF-b causes Lys63-linked polyubiquitylation and activation ofAKT in a TRAF6-dependent manner.
The interaction between TbRI and AKT was not dependent on thekinase activity of TbRI because it also occurred in the presence ofTbRI kinase inhibitor SB431542, evident by immunoblotting forpSMAD2 as a control (Fig. 1E). Furthermore, neither the TbRI kinaseinhibitors (SB431542 and SB505124; fig. S4, A to C) nor the TbRI andTbRII kinase inhibitor LY2109761 (fig. S4, B and D) inhibited AKTphosphorylation. In addition, the activation of AKT by TGF-b did notdepend on the kinase activity of TAK1, which is downstream ofTRAF6, because the TAK1 inhibitor chloro-radicicol A (19) did notabolish AKT phosphorylation (fig. S5A). Moreover, AKT activation byTGF-b did not involve activation of the platelet-derived growth factorreceptor (PDGFR); PDGFR efficiently activates AKT (20) and inter-acts with TGF-b receptors (21), but the PDGFR kinase inhibitorAG1296 did not inhibit TGF-b–induced AKT phosphorylation(fig. S5B). We have previously reported that inhibitory SMAD7,which binds to TbRI where it competes with R-SMADs and preventstheir activation (4), is required for the TGF-b–induced activation ofAKT (22). Consistent with this finding, we found that AKT phospho-rylation and Lys63-linked ubiquitylation of AKT and the PI3K regula-tory subunit p85a were increased by TGF-b stimulation in wild-typebut not in Smad7−/− MEFs (fig. S6).
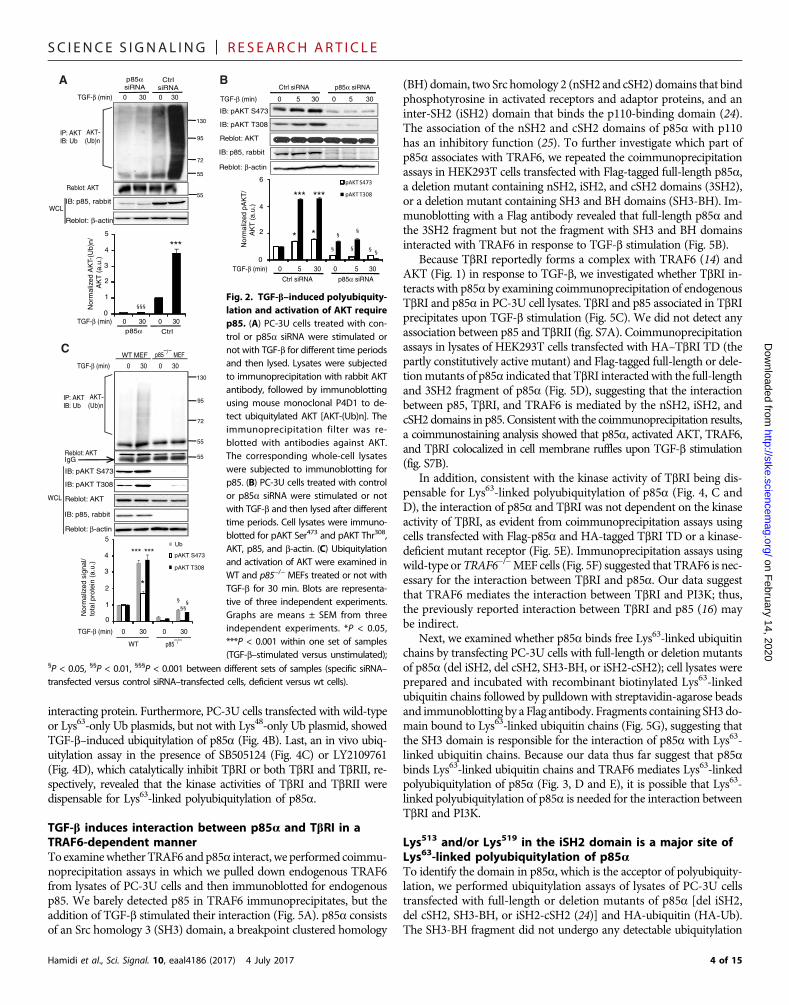
TGF-b–induced polyubiquitylation and activation of AKTrequire PI3KThe phosphorylation of AKT upon TGF-b stimulation involved thePI3K pathway because it was inhibited by the PI3K inhibitorsLY294002 and wortmannin (fig. S4A). Given that the p85a regulatorysubunit of PI3K is acting upstream of PI3K, we hypothesized that p85awould be required for the activation of AKT and in the TGF-b signalingpathway. We thus examined whether the p85a regulatory subunit ofPI3K is needed for AKT ubiquitylation and activation. Ubiquitylation(Fig. 2A) and phosphorylation (Fig. 2B) of AKT did not occur afterknocking down p85a, confirming that p85a was required for the acti-vation of AKT. In addition, in wild-type MEFs, TGF-b induced poly-ubiquitylation and phosphorylation of AKT, neither of which was seenin p85−/− MEFs (Fig. 2C).
2 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
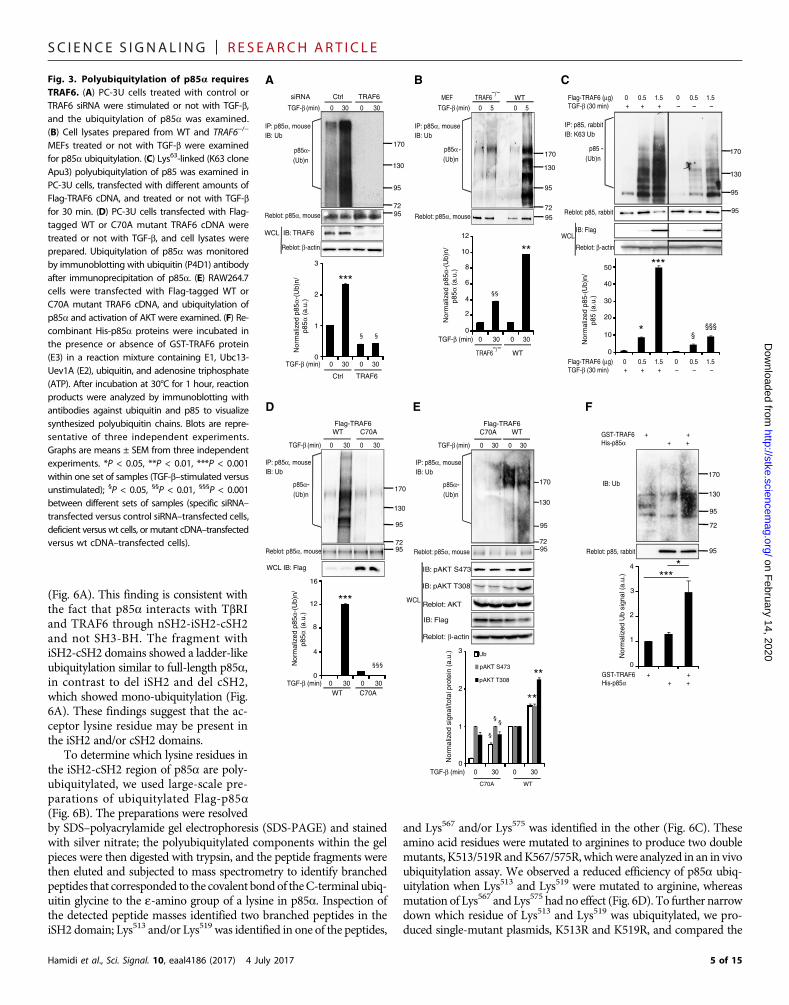
TGF-b induces polyubiquitylationof p85a in a TRAF6-dependent, butreceptor kinase–independent, mannerBecause p85a was required for the acti-vation and ubiquitylation of AKT, wehypothesized that TRAF6 could, by poly-ubiquitylation of p85a, revert the inhibi-tory effect of p85a on p110a (the catalyticsubunit) in the PI3K pathway. We thereforenext examined whether p85a is modifiedby TGF-b stimulation. The polyubiquityla-tion of p85a was induced by TGF-b stim-ulation but abolished by transfection withTRAF6 siRNA (Fig. 3A). Moreover, poly-ubiquitylation of p85a was enhanced byTGF-b stimulation in wild-type but notin TRAF6−/− MEFs (Fig. 3B), and p85underwent Lys63-linked ubiquitylation in
a TRAF6 dose-dependent manner (Fig. 3C). TGF-b induced p85aubiquitylation in PC-3U cells (Fig. 3D), as well as p85a ubiquitylationand AKT activation in RAW264.7 macrophages transfected with wild-type but not C70A mutant TRAF6 (Fig. 3E), demonstrating that theE3 ligase activity of TRAF6 is needed for polyubiquitylation of p85ain response to TGF-b stimulation. Blotting the resulting solutions ofHamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
an in vitro ubiquitylation assay, where polyubiquitylation of p85a wasobserved upon incubation with glutathione S-transferase (GST)–TRAF6(E3), E1, and the E2 enzyme Ubc13-Uev1A (Fig. 3F) (14, 23), suggestedthat p85a is a direct substrate for TRAF6. Detection of TGF-b–inducedpolyubiquitylation of p85a after double immunoprecipitation of p85a(Fig. 4A) suggested that the signal was not the polyubiquitylation of an
IP: HA, rabbitIB: Flag, mouse
TGF-β (min) 0 5 30 0 5 30
Flag-AKT1 + + + + + +
IB: HA
Reblot: β-actin
HA–TβRI TDHA–TβRI TD
–E161A
IB: Flag
WCL
Reblot: TβRI V-22
Reblot: HA
IgG IP: HA, mouseIB: AKT, rabbit
TGF-β (min) 0 5 30 80
HA–TβRI TD
Reblot: TRAF6
Reblot: HA
IB: pAKT S473
WCL
IB: pAKT T308
Reblot: AKT
Reblot: β-actin
IB: HA
A B
IP: AKT, rabbit
IB: TβRI V-22, goat
Reblot: TRAF6
Reblot: AKT
WTTRAF6 –/–
MEFsC
IP: HA, rabbitIB: Flag, mouse
Reblot: TRAF6
DMSO + + + SB431542 + + +
IB: HA
Reblot: β-actin
HA–TβRI TD + Flag-AKT1
IB: Flag
WCL
Reblot: HA
E
pAK
T S
473
HA
HA–TβRI TD HA–TβRI TDUnstimulated TGF-β 40 min
Me
rge
pAK
T S
473
TR
AF
6M
erg
e
pAK
T S
473
Pha
lloid
inM
erg
eM
erg
e
Me
rge
Me
rge
Unstimulated TGF-β 40 min
TR
AF
6P
hallo
idin
Me
rge
Me
rge
Enl
arge
men
t
Enl
arge
men
t
Enl
arge
men
t
Enl
arge
men
t
D
WT
IgG
ctr
l
Bea
d ct
rl
IP: HA, IB: AKT
IP: HA, IB: TRAF6
pAKT S473
pAKT T308
TGF-β (min) 0 5 30 80
Nor
mal
ized
sig
nal/
tota
l pro
tein
(a.
u.)
0
2
4
6
*
*
TGF-β (min) 0 5 30 0 5 30
0
2
4
6
8IP: HA, IB: Flag
IP: HA, IB: TRAF6
TD E161A
***
**
IP: AKT, IB: TβRI
IP: AKT, IB: TRAF6
TGF-β (min) 0 5 30 0 5 30
0
2
4
****
TRAF6 WT
TGF-β (min) 0 5 30 0 5 30
DMSO SB431542
0
1
2
*****
***
Nor
mal
ized
IP/H
A (
a.u.
)
Nor
mal
ized
sig
nal/
tota
l pro
tein
(a.
u.)
Nor
mal
ized
sig
nal/
tota
l pro
tein
(a.
u.)
TGF-β (min) 0 400
10
20
30
40TRAF6
pAKT
HA–TβRI TD
Nor
mal
ized
sig
nal i
nten
sity
in m
embr
ane
ruffl
es (
a.u.
)
***
***
**
TGF-β (min) 0 40
% C
ells
with
mem
bran
e ru
ffles
(a.
u.)
0
10
20
30
40
50
*
**
*
* *
*
** ** §§ §
§
**
**
§§
§§
§§
IB: pSMAD2
Reblot: SMAD2
Reblot: TRAF6
TGF-β (min) 0 5 30 0 5 30
TGF-β (min) 0 5 30 0 5 30
HA–TβRI TD HA–TβRI TDUnstimulated TGF-β 40 min Unstimulated TGF-β 40 min
–/–
Fig. 1. The interaction between AKT and TbRIdepends on TRAF6. (A) Immunoblotting (IB) forthe indicated proteins after immunoprecipitation(IP) for the HA tag from whole-cell lysates (WCL)of PC-3U cells transfected with HA–TbRI TD andtreated with TGF-b. S473, Ser473; T308, Thr308; p,phosphorylated. (B) PC-3U cells were transfectedwith Flag-AKT1 and HA–TbRI TD or the HA–TbRITD–E161A mutant. After treatment with TGF-b, celllysates were subjected to immunoprecipitationwith a polyclonal rabbit HA antibody, followed byimmunoblotting using Flag antibody. (C) Cell ly-sates prepared from wild-type (WT) and TRAF6−/−
MEFs treated or not with TGF-b were subjectedto immunoprecipitation with a polyclonal rabbitAKT antibody or control (ctrl) immunoglobulin G (IgG)and then immunoblotted with a polyclonal goat TbRIantibody (V-22). (D) Immunofluorescence stainingfor the indicated proteins in HA–TbRI TD–transfectedPC-3U cells, either untreated or after stimulationwithTGF-b. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole. Scale bars, 20 mm. (E) PC-3U cells trans-fected with Flag-AKT1 and HA–TbRI TD were treatedwith dimethyl sulfoxide (DMSO) or 5 mM TbRI kinaseinhibitor SB431542 (added 1 hour before TGF-bstimulation). Immunoprecipitation of cell lysateswas performed, as described in the legend for (B).Blots and microscopy images are representative ofthree independent experiments. Graphs are means ±SEM from three independent experiments. *P < 0.05,**P < 0.01, ***P < 0.001 within one set of samples(TGF-b–stimulated versus unstimulated cells); §P <0.05 between different sets of samples [mutantcomplementary DNA (cDNA)–transfected versus wtcDNA–transfected or deficient versus wt cells]. a.u.,arbitrary units.
3 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
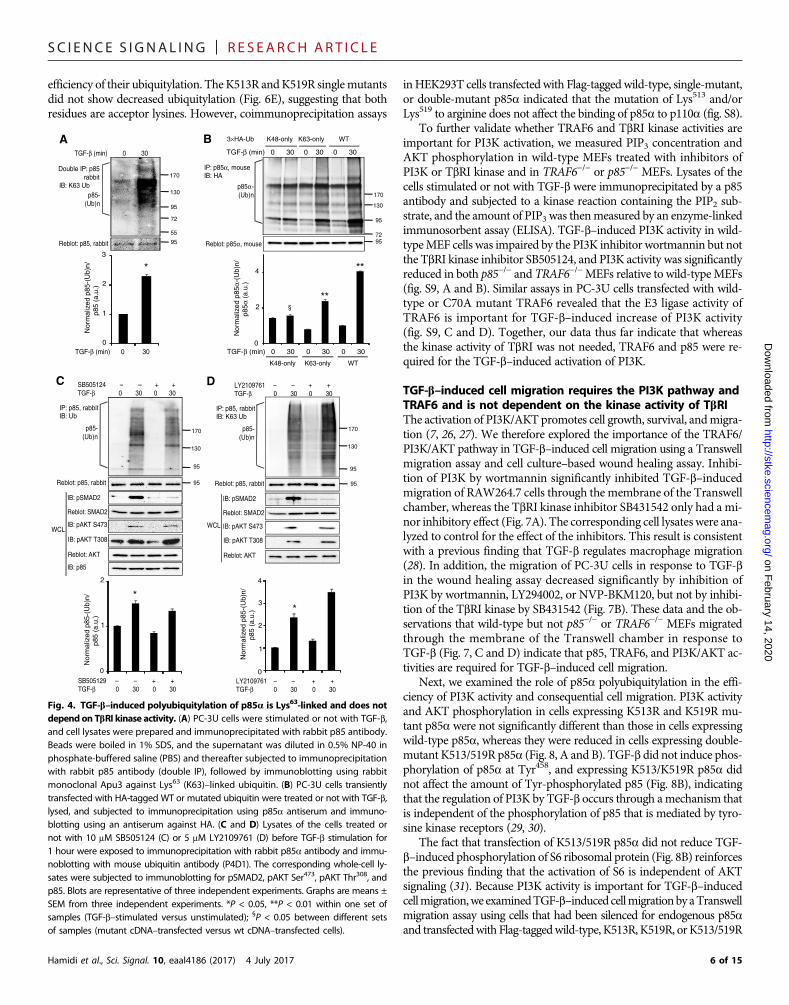
interacting protein. Furthermore, PC-3U cells transfected with wild-typeor Lys63-only Ub plasmids, but not with Lys48-only Ub plasmid, showedTGF-b–induced ubiquitylation of p85a (Fig. 4B). Last, an in vivo ubiq-uitylation assay in the presence of SB505124 (Fig. 4C) or LY2109761(Fig. 4D), which catalytically inhibit TbRI or both TbRI and TbRII, re-spectively, revealed that the kinase activities of TbRI and TbRII weredispensable for Lys63-linked polyubiquitylation of p85a.
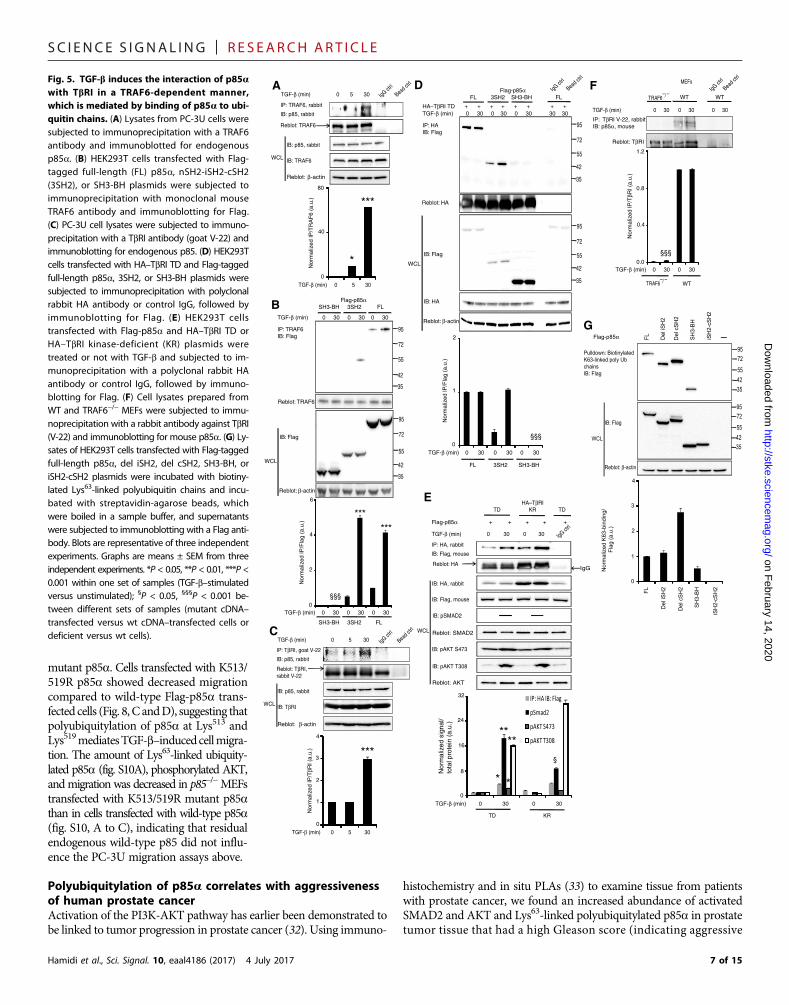
TGF-b induces interaction between p85a and TbRI in aTRAF6-dependent mannerTo examinewhetherTRAF6 andp85a interact, we performed coimmu-noprecipitation assays in which we pulled down endogenous TRAF6from lysates of PC-3U cells and then immunoblotted for endogenousp85. We barely detected p85 in TRAF6 immunoprecipitates, but theaddition of TGF-b stimulated their interaction (Fig. 5A). p85a consistsof an Src homology 3 (SH3) domain, a breakpoint clustered homology
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
(BH) domain, two Src homology 2 (nSH2 and cSH2) domains that bindphosphotyrosine in activated receptors and adaptor proteins, and aninter-SH2 (iSH2) domain that binds the p110-binding domain (24).The association of the nSH2 and cSH2 domains of p85a with p110has an inhibitory function (25). To further investigate which part ofp85a associates with TRAF6, we repeated the coimmunoprecipitationassays in HEK293T cells transfected with Flag-tagged full-length p85a,a deletion mutant containing nSH2, iSH2, and cSH2 domains (3SH2),or a deletion mutant containing SH3 and BH domains (SH3-BH). Im-munoblotting with a Flag antibody revealed that full-length p85a andthe 3SH2 fragment but not the fragment with SH3 and BH domainsinteracted with TRAF6 in response to TGF-b stimulation (Fig. 5B).
Because TbRI reportedly forms a complex with TRAF6 (14) andAKT (Fig. 1) in response to TGF-b, we investigated whether TbRI in-teracts with p85a by examining coimmunoprecipitation of endogenousTbRI and p85a in PC-3U cell lysates. TbRI and p85 associated in TbRIprecipitates upon TGF-b stimulation (Fig. 5C). We did not detect anyassociation between p85 and TbRII (fig. S7A). Coimmunoprecipitationassays in lysates of HEK293T cells transfected with HA–TbRI TD (thepartly constitutively active mutant) and Flag-tagged full-length or dele-tionmutants of p85a indicated that TbRI interactedwith the full-lengthand 3SH2 fragment of p85a (Fig. 5D), suggesting that the interactionbetween p85, TbRI, and TRAF6 is mediated by the nSH2, iSH2, andcSH2 domains in p85. Consistent with the coimmunoprecipitation results,a coimmunostaining analysis showed that p85a, activated AKT, TRAF6,and TbRI colocalized in cell membrane ruffles upon TGF-b stimulation(fig. S7B).
In addition, consistent with the kinase activity of TbRI being dis-pensable for Lys63-linked polyubiquitylation of p85a (Fig. 4, C andD), the interaction of p85a and TbRI was not dependent on the kinaseactivity of TbRI, as evident from coimmunoprecipitation assays usingcells transfected with Flag-p85a and HA-tagged TbRI TD or a kinase-deficient mutant receptor (Fig. 5E). Immunoprecipitation assays usingwild-type orTRAF6−/−MEF cells (Fig. 5F) suggested that TRAF6 is nec-essary for the interaction between TbRI and p85a. Our data suggestthat TRAF6 mediates the interaction between TbRI and PI3K; thus,the previously reported interaction between TbRI and p85 (16) maybe indirect.
Next, we examined whether p85a binds free Lys63-linked ubiquitinchains by transfecting PC-3U cells with full-length or deletion mutantsof p85a (del iSH2, del cSH2, SH3-BH, or iSH2-cSH2); cell lysates wereprepared and incubated with recombinant biotinylated Lys63-linkedubiquitin chains followed by pulldown with streptavidin-agarose beadsand immunoblotting by a Flag antibody. Fragments containing SH3do-main bound to Lys63-linked ubiquitin chains (Fig. 5G), suggesting thatthe SH3 domain is responsible for the interaction of p85a with Lys63-linked ubiquitin chains. Because our data thus far suggest that p85abinds Lys63-linked ubiquitin chains and TRAF6 mediates Lys63-linkedpolyubiquitylation of p85a (Fig. 3, D and E), it is possible that Lys63-linked polyubiquitylation of p85a is needed for the interaction betweenTbRI and PI3K.
Lys513 and/or Lys519 in the iSH2 domain is a major site ofLys63-linked polyubiquitylation of p85aTo identify the domain in p85a, which is the acceptor of polyubiquity-lation, we performed ubiquitylation assays of lysates of PC-3U cellstransfected with full-length or deletion mutants of p85a [del iSH2,del cSH2, SH3-BH, or iSH2-cSH2 (24)] and HA-ubiquitin (HA-Ub).The SH3-BH fragment did not undergo any detectable ubiquitylation
-
p85α Ctrl
IB: p85, rabbit
Reblot: AKT
Reblot: β-actin
TGF-β (min) 0 5 30 0 5 30
IB: pAKT T308
IB: pAKT S473
72
95
130
55
A
AKT(Ub)n
Reblot: AKT
IP: AKTIB: Ub
TGF-β (min) 0 30 0 30Ctrl siRNA p85α siRNA
IB: p85, rabbit55
B
72
95
130
55
TGF-β (min) 0 30 0 30
C
***
p85αsiRNA
CtrlsiRNA
Nor
mal
ized
AK
T-(U
b)n/
A
KT
(a.
u.)
TGF-β (min) 0 30 0 30
TGF-β (min) 0 5 30 0 5 30
Nor
mal
ized
pA
KT
/
A
KT
(a.
u.)
0
2
4
6
0
2
4
5
3
1
Ctrl siRNA p85α siRNA
*** ***
TGF-β (min) 0 30 0 30
3
Reblot: AKT
IB: pAKT S473
IB: pAKT T308
Reblot: β-actin
WCL
IB: p85, rabbit
Nor
mal
ized
sig
nal/
tota
l pro
tein
(a.
u.)
WT
§§§
* * §
§ §
§
§§
-AKT(Ub)n
IP: AKTIB: Ub
Reblot: AKT55
WT MEF–/– MEFp85
4
5Ub
pAKT S473
pAKT T308
*** ***
IgG
Reblot: β-actin
WCL
2
1
0
*§ §
§§
–/–p85
Fig. 2. TGF-b–induced polyubiquity-lation and activation of AKT requirep85. (A) PC-3U cells treated with con-trol or p85a siRNA were stimulated ornot with TGF-b for different time periodsand then lysed. Lysates were subjectedto immunoprecipitation with rabbit AKTantibody, followed by immunoblottingusing mouse monoclonal P4D1 to de-tect ubiquitylated AKT [AKT-(Ub)n]. Theimmunoprecipitation filter was re-blotted with antibodies against AKT.The corresponding whole-cell lysateswere subjected to immunoblotting forp85. (B) PC-3U cells treated with controlor p85a siRNA were stimulated or notwith TGF-b and then lysed after differenttime periods. Cell lysates were immuno-blotted for pAKT Ser473 and pAKT Thr308,AKT, p85, and b-actin. (C) Ubiquitylationand activation of AKT were examined inWT and p85−/− MEFs treated or not withTGF-b for 30 min. Blots are representa-tive of three independent experiments.Graphs are means ± SEM from threeindependent experiments. *P < 0.05,***P < 0.001 within one set of samples(TGF-b–stimulated versus unstimulated);
§P < 0.05, §§P < 0.01, §§§P < 0.001 between different sets of samples (specific siRNA–transfected versus control siRNA–transfected cells, deficient versus wt cells).
4 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
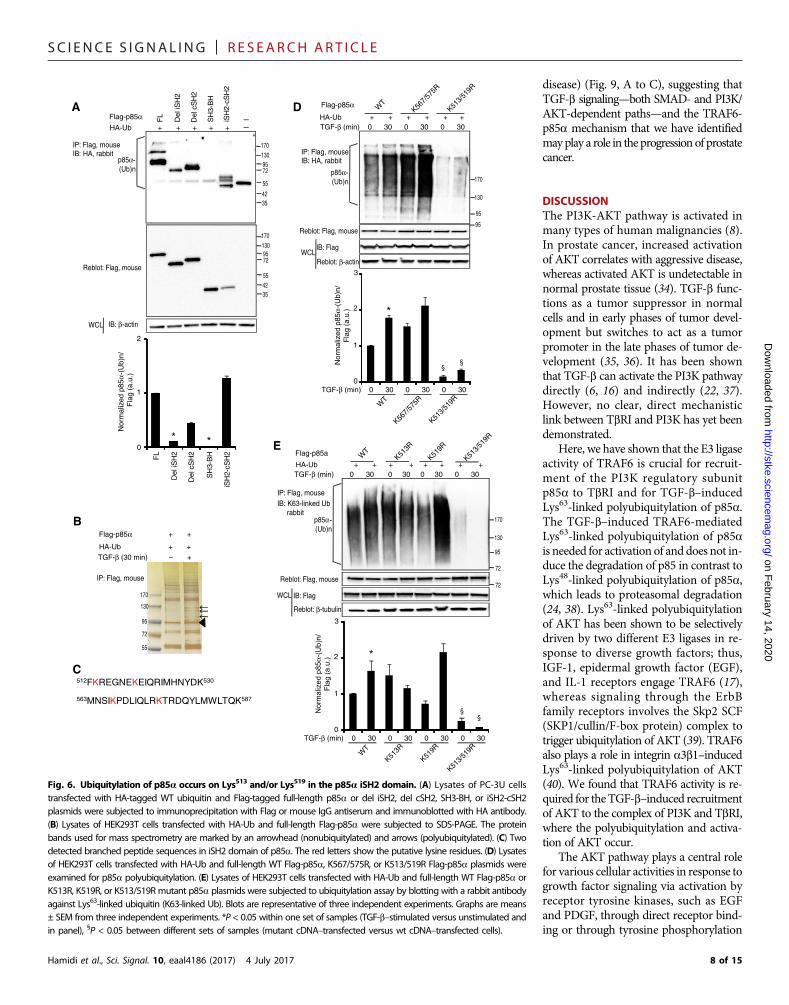
(Fig. 6A). This finding is consistent withthe fact that p85a interacts with TbRIand TRAF6 through nSH2-iSH2-cSH2and not SH3-BH. The fragment withiSH2-cSH2 domains showed a ladder-likeubiquitylation similar to full-length p85a,in contrast to del iSH2 and del cSH2,which showed mono-ubiquitylation (Fig.6A). These findings suggest that the ac-ceptor lysine residue may be present inthe iSH2 and/or cSH2 domains.
To determine which lysine residues inthe iSH2-cSH2 region of p85a are poly-ubiquitylated, we used large-scale pre-parations of ubiquitylated Flag-p85a(Fig. 6B). The preparations were resolved
by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and stainedwith silver nitrate; the polyubiquitylated components within the gelpieces were then digested with trypsin, and the peptide fragments werethen eluted and subjected to mass spectrometry to identify branchedpeptides that corresponded to the covalent bond of theC-terminal ubiq-uitin glycine to the e-amino group of a lysine in p85a. Inspection ofthe detected peptide masses identified two branched peptides in theiSH2 domain; Lys513 and/or Lys519 was identified in one of the peptides,Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
and Lys567 and/or Lys575 was identified in the other (Fig. 6C). Theseamino acid residues were mutated to arginines to produce two doublemutants, K513/519R andK567/575R,whichwere analyzed in an in vivoubiquitylation assay. We observed a reduced efficiency of p85a ubiq-uitylation when Lys513 and Lys519 were mutated to arginine, whereasmutation of Lys567 and Lys575 had no effect (Fig. 6D). To further narrowdown which residue of Lys513 and Lys519 was ubiquitylated, we pro-duced single-mutant plasmids, K513R and K519R, and compared the
A
D
siRNA Ctrl TRAF6
TGF-β (min) 0 30 0 30
p85α-(Ub)n
Reblot: p85α, mouse
WCL IB: TRAF6
IP: p85α, mouseIB: Ub
TGF-β (min) 0 5 0 5
p85α -(Ub)n
Reblot: p85α, mouse
WTTRAF6 –/–MEF
72
95
130
170
B
72
95
130
170
TGF-β (min) 0 30 0 30
p85α-(Ub)n
Flag-TRAF6 WT C70A
WCL IB: Flag
E
95 95
95
72
95
130
170
IP: p85α, mouseIB: Ub
IP: p85α, mouseIB: Ub
Reblot: p85α, mouse
***
TGF-β (min) 0 30 0 30
Nor
mal
ized
p85
α-(U
b)n/
p
85α
(a.u
.)
0
2
3
1
TGF-β (min) 0 30 0 30
WT
0
2
4
6
8
10
12
**
TGF-β (min) 0 30 0 30
Ctrl TRAF6
WT C70A
0
4
12
16
Nor
mal
ized
p85
α-(U
b)n/
p
85α
(a.u
.)
***
TGF-β (min) 0 30 0 30
p85α-(Ub)n
Flag-TRAF6C70A WT
IP: p85α, mouseIB: Ub
Reblot: p85α, mouse
Reblot: AKT
IB: pAKT T308
IB: pAKT S473
IB: Flag
Reblot: β-actin
WCL
TGF-β (min) 0 30 0 30
C70A WT
Nor
mal
ized
sig
nal/t
otal
pro
tein
(a.
u.)
0
1
2
3 Ub
pAKT T308
pAKT S473
**
170
130
95
7295
§ §
§§
§§§
**
§§
§
Flag-TRAF6 (μg) 0 0.5 1.5 0 0.5 1.5TGF-β (30 min) + + + – – –
Reblot: p85, rabbit
IP: p85, rabbitIB: K63 Ub
p85 -(Ub)n
IB: Flag
Reblot: β-actin
WCL
95
130
170
C
0
20
30
10
Nor
mal
ized
p85
-(U
b)n/
p
85 (
a.u.
)
40
50
Flag-TRAF6 (μg) 0 0.5 1.5 0 0.5 1.5TGF-β (30 min) + + + – – –
*
***
§§§§
95
F
72
95
130
170
GST-TRAF6 + +His-p85α + +
IB: Ub
Reblot: p85, rabbit
0
2
3
1
4
GST-TRAF6 + +His-p85α + +
Nor
mal
ized
Ub
sign
al (
a.u.
)
****
95
Reblot: β-actin
Nor
mal
ized
p85
α-(U
b)n/
p
85α
(a.u
.)
8
TRAF6 –/–
Fig. 3. Polyubiquitylation of p85a requiresTRAF6. (A) PC-3U cells treated with control orTRAF6 siRNA were stimulated or not with TGF-b,and the ubiquitylation of p85a was examined.(B) Cell lysates prepared from WT and TRAF6−/−
MEFs treated or not with TGF-b were examinedfor p85a ubiquitylation. (C) Lys63-linked (K63 cloneApu3) polyubiquitylation of p85 was examined inPC-3U cells, transfected with different amounts ofFlag-TRAF6 cDNA, and treated or not with TGF-bfor 30 min. (D) PC-3U cells transfected with Flag-tagged WT or C70A mutant TRAF6 cDNA weretreated or not with TGF-b, and cell lysates wereprepared. Ubiquitylation of p85a was monitoredby immunoblotting with ubiquitin (P4D1) antibodyafter immunoprecipitation of p85a. (E) RAW264.7cells were transfected with Flag-tagged WT orC70A mutant TRAF6 cDNA, and ubiquitylation ofp85a and activation of AKT were examined. (F) Re-combinant His-p85a proteins were incubated inthe presence or absence of GST-TRAF6 protein(E3) in a reaction mixture containing E1, Ubc13-Uev1A (E2), ubiquitin, and adenosine triphosphate(ATP). After incubation at 30°C for 1 hour, reactionproducts were analyzed by immunoblotting withantibodies against ubiquitin and p85 to visualizesynthesized polyubiquitin chains. Blots are repre-sentative of three independent experiments.Graphs are means ± SEM from three independentexperiments. *P < 0.05, **P < 0.01, ***P < 0.001within one set of samples (TGF-b–stimulated versusunstimulated); §P < 0.05, §§P < 0.01, §§§P < 0.001between different sets of samples (specific siRNA–transfected versus control siRNA–transfected cells,deficient versus wt cells, or mutant cDNA–transfectedversus wt cDNA–transfected cells).
5 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
efficiency of their ubiquitylation. TheK513R andK519R singlemutantsdid not show decreased ubiquitylation (Fig. 6E), suggesting that bothresidues are acceptor lysines. However, coimmunoprecipitation assays
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
inHEK293T cells transfected with Flag-taggedwild-type, single-mutant,or double-mutant p85a indicated that the mutation of Lys513 and/orLys519 to arginine does not affect the binding of p85a to p110a (fig. S8).
To further validate whether TRAF6 and TbRI kinase activities areimportant for PI3K activation, we measured PIP3 concentration andAKT phosphorylation in wild-type MEFs treated with inhibitors ofPI3K or TbRI kinase and in TRAF6−/− or p85−/− MEFs. Lysates of thecells stimulated or not with TGF-b were immunoprecipitated by a p85antibody and subjected to a kinase reaction containing the PIP2 sub-strate, and the amount of PIP3 was thenmeasured by an enzyme-linkedimmunosorbent assay (ELISA). TGF-b–induced PI3K activity in wild-typeMEF cells was impaired by the PI3K inhibitor wortmannin but notthe TbRI kinase inhibitor SB505124, and PI3K activity was significantlyreduced in both p85−/− and TRAF6−/−MEFs relative to wild-typeMEFs(fig. S9, A and B). Similar assays in PC-3U cells transfected with wild-type or C70A mutant TRAF6 revealed that the E3 ligase activity ofTRAF6 is important for TGF-b–induced increase of PI3K activity(fig. S9, C and D). Together, our data thus far indicate that whereasthe kinase activity of TbRI was not needed, TRAF6 and p85 were re-quired for the TGF-b–induced activation of PI3K.
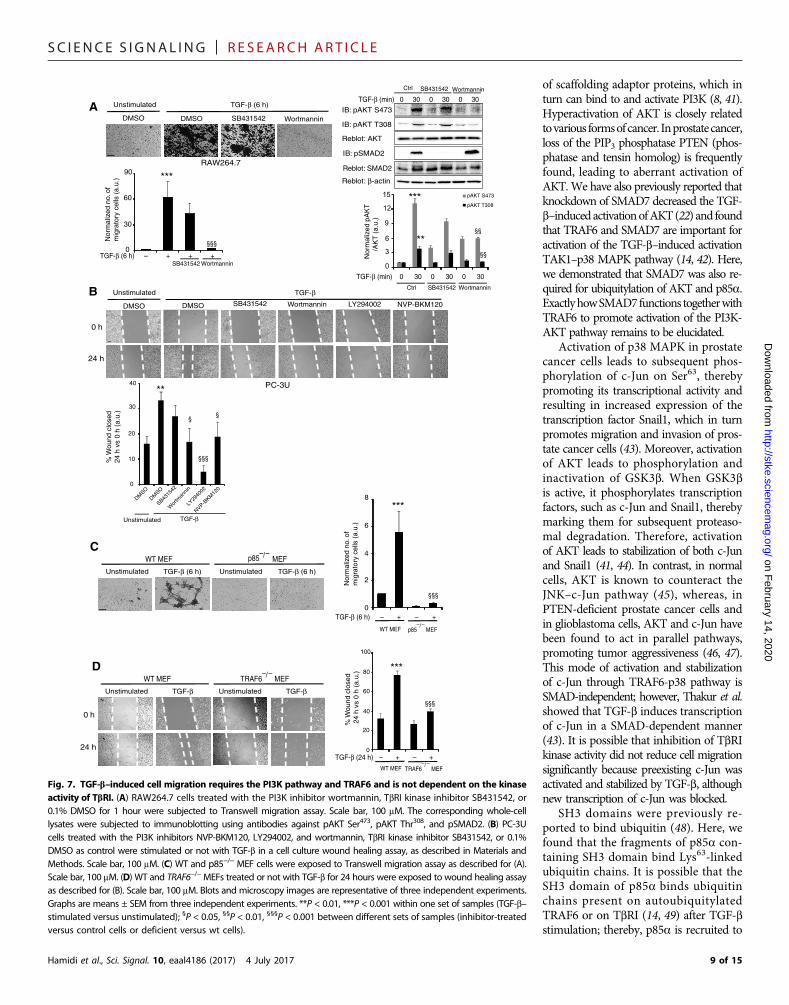
TGF-b–induced cell migration requires the PI3K pathway andTRAF6 and is not dependent on the kinase activity of TbRIThe activation of PI3K/AKT promotes cell growth, survival, andmigra-tion (7, 26, 27). We therefore explored the importance of the TRAF6/PI3K/AKT pathway in TGF-b–induced cell migration using a Transwellmigration assay and cell culture–based wound healing assay. Inhibi-tion of PI3K by wortmannin significantly inhibited TGF-b–inducedmigration of RAW264.7 cells through the membrane of the Transwellchamber, whereas the TbRI kinase inhibitor SB431542 only had a mi-nor inhibitory effect (Fig. 7A). The corresponding cell lysateswere ana-lyzed to control for the effect of the inhibitors. This result is consistentwith a previous finding that TGF-b regulates macrophage migration(28). In addition, the migration of PC-3U cells in response to TGF-bin the wound healing assay decreased significantly by inhibition ofPI3K by wortmannin, LY294002, or NVP-BKM120, but not by inhibi-tion of the TbRI kinase by SB431542 (Fig. 7B). These data and the ob-servations that wild-type but not p85−/− or TRAF6−/− MEFs migratedthrough the membrane of the Transwell chamber in response toTGF-b (Fig. 7, C and D) indicate that p85, TRAF6, and PI3K/AKT ac-tivities are required for TGF-b–induced cell migration.
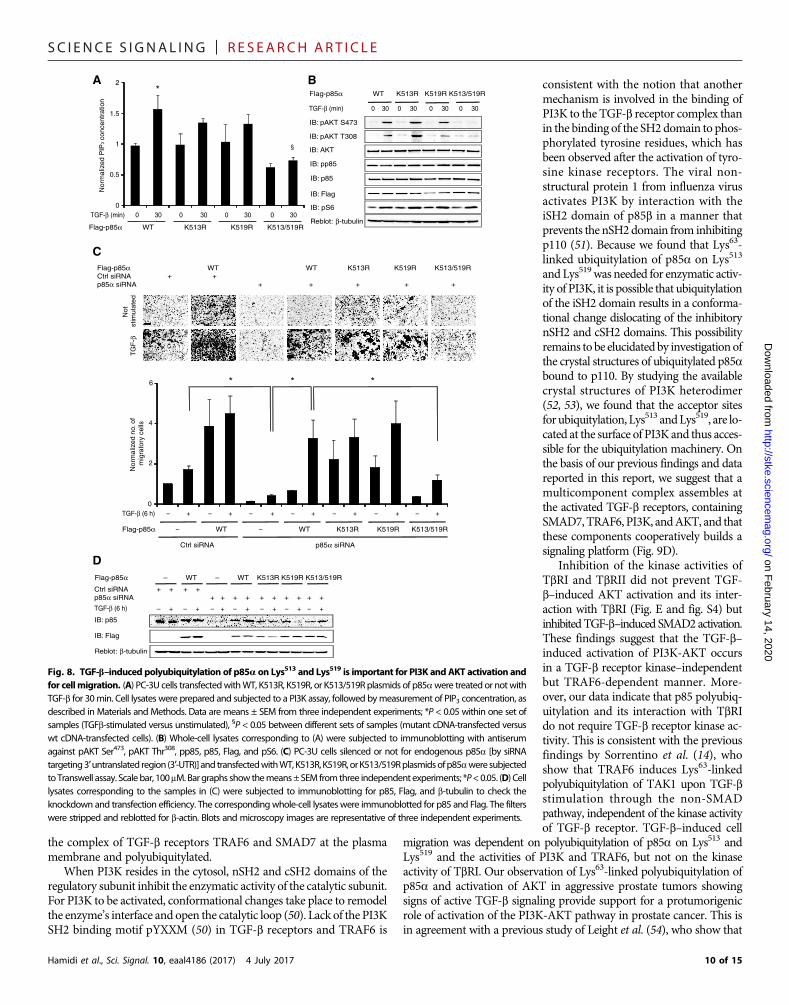
Next, we examined the role of p85a polyubiquitylation in the effi-ciency of PI3K activity and consequential cell migration. PI3K activityand AKT phosphorylation in cells expressing K513R and K519R mu-tant p85a were not significantly different than those in cells expressingwild-type p85a, whereas they were reduced in cells expressing double-mutant K513/519R p85a (Fig. 8, A and B). TGF-b did not induce phos-phorylation of p85a at Tyr458, and expressing K513/K519R p85a didnot affect the amount of Tyr-phosphorylated p85 (Fig. 8B), indicatingthat the regulation of PI3K by TGF-b occurs through amechanism thatis independent of the phosphorylation of p85 that is mediated by tyro-sine kinase receptors (29, 30).
The fact that transfection of K513/519R p85a did not reduce TGF-b–induced phosphorylation of S6 ribosomal protein (Fig. 8B) reinforcesthe previous finding that the activation of S6 is independent of AKTsignaling (31). Because PI3K activity is important for TGF-b–inducedcellmigration,we examinedTGF-b–induced cellmigrationbyaTranswellmigration assay using cells that had been silenced for endogenous p85aand transfectedwith Flag-taggedwild-type, K513R, K519R, orK513/519R
72
95
130
170p85α-(Ub)n
Reblot: p85α, mouse
IP: p85α, mouseIB: HA
3×HA-Ub K48-only K63-only WT
K48-only K63-only WT
0
2
4
Nor
mal
ized
p85
α-(U
b)n/
p
85α
(a.u
.)
**
**
TGF-β (min) 0 30 0 30 0 30
TGF-β (min) 0 30 0 30 0 30
B
§
D
TGF-β (min) 0 30
Reblot: p85, rabbit
Double IP: p85 rabbitIB: K63 Ub
p85-(Ub)n
72
95
130
55
170
SB505124 – – + +TGF-β 0 30 0 30
Reblot: p85, rabbit
IP: p85, rabbitIB: Ub
p85-(Ub)n
IB: pSMAD2
Reblot: SMAD2
WCLIB: pAKT S473
IB: pAKT T308
Reblot: AKT
IB: p85
TGF-β (min) 0 30 0
2
3
1
Nor
mal
ized
p85
-(U
b)n/
p8
5 (a
.u.)
*
A
C
0
2
Nor
mal
ized
p85
-(U
b)n/
p
85 (
a.u.
)
1
SB505129 – – + +TGF-β 0 30 0 30
*
95
130
170
95
130
170
LY2109761 – – + +TGF-β 0 30 0 30
IP: p85, rabbitIB: K63 Ub
p85-(Ub)n
95 95
95 95
0
2
Nor
mal
ized
p85
-(U
b)n/
p
85 (
a.u.
)
1
LY2109761 – – + +TGF-β 0 30 0 30
3
4
*
Reblot: p85, rabbit
IB: pSMAD2
Reblot: SMAD2
WCL IB: pAKT S473
IB: pAKT T308
Reblot: AKT
Fig. 4. TGF-b–induced polyubiquitylation of p85a is Lys63-linked and does notdependon TbRI kinase activity. (A) PC-3U cells were stimulated or not with TGF-b,and cell lysates were prepared and immunoprecipitated with rabbit p85 antibody.Beads were boiled in 1% SDS, and the supernatant was diluted in 0.5% NP-40 inphosphate-buffered saline (PBS) and thereafter subjected to immunoprecipitationwith rabbit p85 antibody (double IP), followed by immunoblotting using rabbitmonoclonal Apu3 against Lys63 (K63)–linked ubiquitin. (B) PC-3U cells transientlytransfected with HA-tagged WT or mutated ubiquitin were treated or not with TGF-b,lysed, and subjected to immunoprecipitation using p85a antiserum and immuno-blotting using an antiserum against HA. (C and D) Lysates of the cells treated ornot with 10 mM SB505124 (C) or 5 mM LY2109761 (D) before TGF-b stimulation for1 hour were exposed to immunoprecipitation with rabbit p85a antibody and immu-noblotting with mouse ubiquitin antibody (P4D1). The corresponding whole-cell ly-sates were subjected to immunoblotting for pSMAD2, pAKT Ser473, pAKT Thr308, andp85. Blots are representative of three independent experiments. Graphs are means ±SEM from three independent experiments. *P < 0.05, **P < 0.01 within one set ofsamples (TGF-b–stimulated versus unstimulated); §P < 0.05 between different setsof samples (mutant cDNA–transfected versus wt cDNA–transfected cells).
6 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
mutant p85a. Cells transfected with K513/519R p85a showed decreased migrationcompared to wild-type Flag-p85a trans-fected cells (Fig. 8, C andD), suggesting thatpolyubiquitylation of p85a at Lys513 andLys519mediatesTGF-b–inducedcellmigra-tion. The amount of Lys63-linked ubiquity-lated p85a (fig. S10A), phosphorylated AKT,andmigration was decreased in p85−/−MEFstransfected with K513/519R mutant p85athan in cells transfected with wild-type p85a(fig. S10, A to C), indicating that residualendogenous wild-type p85 did not influ-ence the PC-3U migration assays above.
Polyubiquitylation of p85a correlates with aggressivenessof human prostate cancerActivation of the PI3K-AKT pathway has earlier been demonstrated tobe linked to tumor progression in prostate cancer (32). Using immuno-
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
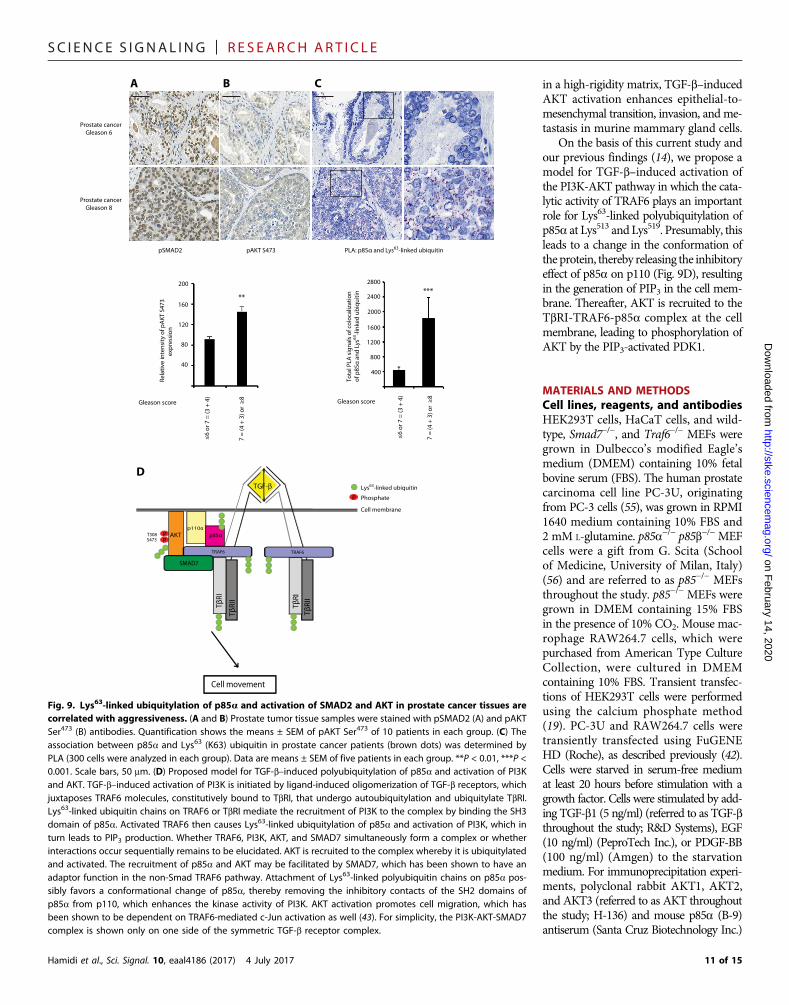
histochemistry and in situ PLAs (33) to examine tissue from patientswith prostate cancer, we found an increased abundance of activatedSMAD2 and AKT and Lys63-linked polyubiquitylated p85a in prostatetumor tissue that had a high Gleason score (indicating aggressive
IP: TRAF6, rabbit
IB: p85, rabbit
Reblot: TRAF6
IB: p85, rabbit
IB: TRAF6WCL
TGF-β (min) 0 5 30
Reblot: β-actin
IgG ct
rl
Bead c
trl
TGF-β (min) 0 5 30 IgG ct
rl
Bead c
trl
IP: TβRI, goat V-22
IB: p85, rabbit
Reblot: TβRI, rabbit V-22
IB: p85, rabbit
IB: TβRIWCL
Reblot: β-actin
C
TGF-β (min) 0 5 30
0
40
80
*
***
Nor
mal
ized
IP/T
RA
F6
(a.u
.)
TGF-β (min) 0 5 30
Nor
mal
ized
IP/T
βRI (
a.u.
)
0
2
3
1
4
***
A
BTGF-β (min) 0 30 0 30 0 30
Flag-p85α SH3-BH 3SH2 FL
IP: TRAF6IB: Flag
Reblot: TRAF6
WCL
IB: Flag
Reblot: β-actin
55
72
95
42
35
55
72
95
42
35
TGF-β (min) 0 30 0 30 0 30 0
2
4
6
Nor
mal
ized
IP/F
lag
(a.u
.)
SH3-BH 3SH2 FL
******
IP: TβRI V-22, rabbitIB: p85α, mouse
Reblot: TβRI
WTTRAF6 –/ –
MEFs
WT
TGF-β (min) 0 30 0 300.0
0.4
0.8
1.2
Nor
mal
ized
IP/T
βRI (
a.u.
)
WT
TGF-β (min) 0 30 0 30 0 30
F
TGF-β (min) 0 30 0 30 0 30 30 30
Flag-p85α FL 3SH2 SH3-BH FL
IP: HAIB: Flag
Reblot: HA
WCL
IB: Flag
Reblot: β-actin
HA–TβRI TD + + + + + + + +
55
72
95
42
35
IB: HA
TGF-β (min) 0 30 0 30 0 30
FL 3SH2 SH3-BH
0
1
2
Nor
mal
ized
IP/F
lag
(a.u
.)
D
§§§
§§§
§§§
IP: HA, rabbit
IB: Flag, mouse
Reblot: HA
IB: HA, rabbit
IB: Flag, mouse
WCL
0 30 0 30TGF-β (min)
Reblot: SMAD2
IgG ct
rl
IgG
IB: pSMAD2
IB: pAKT T308
IB: pAKT S473
Reblot: AKT
Flag-p85α + + + + +
HA–TβRITD KR TD
IgG ct
rl
Bead c
trl
TGF-β (min) 0 30 0 30
0
8
32
Nor
mal
ized
sig
nal/
tota
l pro
tein
(a.
u.)
16
24
TD KR
*
****
*
§
E
55
72
95
42
35
Flag-p85α FL Del
iSH
2
Del
cSH
2
SH3-
BH
iSH
2-cS
H2
–Pulldown: BiotinylatedK63-linked poly UbchainsIB: Flag
WCL
IB: Flag
Reblot: β-actin
55
7295
4235
55
7295
4235
G
0
1
2
3
4
Nor
mal
ized
K63
-bin
ding
/
Fl
ag (a
.u.)
FL
Del
iSH
2
Del
cSH
2
SH3-
BH
iSH
2-cS
H2
IgG ct
rl
Bead c
trl
TRAF6 –/ –
Fig. 5. TGF-b induces the interaction of p85awith TbRI in a TRAF6-dependent manner,which is mediated by binding of p85a to ubi-quitin chains. (A) Lysates from PC-3U cells weresubjected to immunoprecipitation with a TRAF6antibody and immunoblotted for endogenousp85a. (B) HEK293T cells transfected with Flag-tagged full-length (FL) p85a, nSH2-iSH2-cSH2(3SH2), or SH3-BH plasmids were subjected toimmunoprecipitation with monoclonal mouseTRAF6 antibody and immunoblotting for Flag.(C) PC-3U cell lysates were subjected to immuno-precipitation with a TbRI antibody (goat V-22) andimmunoblotting for endogenous p85. (D) HEK293Tcells transfected with HA–TbRI TD and Flag-taggedfull-length p85a, 3SH2, or SH3-BH plasmids weresubjected to immunoprecipitation with polyclonalrabbit HA antibody or control IgG, followed byimmunoblotting for Flag. (E) HEK293T cellstransfected with Flag-p85a and HA–TbRI TD orHA–TbRI kinase-deficient (KR) plasmids weretreated or not with TGF-b and subjected to im-munoprecipitation with a polyclonal rabbit HAantibody or control IgG, followed by immuno-blotting for Flag. (F) Cell lysates prepared fromWT and TRAF6−/− MEFs were subjected to immu-noprecipitation with a rabbit antibody against TbRI(V-22) and immunoblotting for mouse p85a. (G) Ly-sates of HEK293T cells transfected with Flag-taggedfull-length p85a, del iSH2, del cSH2, SH3-BH, oriSH2-cSH2 plasmids were incubated with biotiny-lated Lys63-linked polyubiquitin chains and incu-bated with streptavidin-agarose beads, whichwere boiled in a sample buffer, and supernatantswere subjected to immunoblotting with a Flag anti-body. Blots are representative of three independentexperiments. Graphs are means ± SEM from threeindependent experiments. *P < 0.05, **P < 0.01, ***P <0.001 within one set of samples (TGF-b–stimulatedversus unstimulated); §P < 0.05, §§§P < 0.001 be-tween different sets of samples (mutant cDNA–transfected versus wt cDNA–transfected cells ordeficient versus wt cells).
7 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
disease) (Fig. 9, A to C), suggesting thatTGF-b signaling—both SMAD- and PI3K/AKT-dependent paths—and the TRAF6-p85a mechanism that we have identifiedmayplay a role in the progression of prostatecancer.
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
DISCUSSIONThe PI3K-AKT pathway is activated inmany types of human malignancies (8).In prostate cancer, increased activationof AKT correlates with aggressive disease,whereas activated AKT is undetectable innormal prostate tissue (34). TGF-b func-tions as a tumor suppressor in normalcells and in early phases of tumor devel-opment but switches to act as a tumorpromoter in the late phases of tumor de-velopment (35, 36). It has been shownthat TGF-b can activate the PI3K pathwaydirectly (6, 16) and indirectly (22, 37).However, no clear, direct mechanisticlink between TbRI and PI3K has yet beendemonstrated.
Here, we have shown that the E3 ligaseactivity of TRAF6 is crucial for recruit-ment of the PI3K regulatory subunitp85a to TbRI and for TGF-b–inducedLys63-linked polyubiquitylation of p85a.The TGF-b–induced TRAF6-mediatedLys63-linked polyubiquitylation of p85ais needed for activation of and does not in-duce the degradation of p85 in contrast toLys48-linked polyubiquitylation of p85a,which leads to proteasomal degradation(24, 38). Lys63-linked polyubiquitylationof AKT has been shown to be selectivelydriven by two different E3 ligases in re-sponse to diverse growth factors; thus,IGF-1, epidermal growth factor (EGF),and IL-1 receptors engage TRAF6 (17),whereas signaling through the ErbBfamily receptors involves the Skp2 SCF(SKP1/cullin/F-box protein) complex totrigger ubiquitylation of AKT (39). TRAF6also plays a role in integrin a3b1–inducedLys63-linked polyubiquitylation of AKT(40). We found that TRAF6 activity is re-quired for theTGF-b–induced recruitmentof AKT to the complex of PI3K and TbRI,where the polyubiquitylation and activa-tion of AKT occur.
The AKT pathway plays a central rolefor various cellular activities in response togrowth factor signaling via activation byreceptor tyrosine kinases, such as EGFand PDGF, through direct receptor bind-ing or through tyrosine phosphorylation
Flag-p85α WT
K513/
519R
K567/
575R
p85α-(Ub)n
Reblot: Flag, mouse
IP: Flag, mouseIB: HA, rabbit
WCLIB: Flag
HA-Ub + + + + + +
130
170
95
B
55
7295
4235
130170
Flag-p85α FL
Del
iSH
2
Del
cS
H2
SH
3-B
H
iSH
2-cS
H2
–HA-Ub + + + + + –
A
p85α-(Ub)n
IP: Flag, mouseIB: HA, rabbit
55
7295
4235
130170
Reblot: Flag, mouse
TGF-β (min) 0 30 0 30 0 30
Flag-p85a WT
K513R
K519R
K513/
519R
p85α-(Ub)n
Reblot: Flag, mouse
IP: Flag, mouse
WCL IB: Flag
HA-Ub + + + + + + + +
130
170
95
IB: K63-linked Ub rabbit
TGF-β (min) 0 30 0 30 0 30 0 30
55
72
95
130
170
IP: Flag, mouse
Flag-p85α + +
HA-Ub + +TGF-β (30 min) – +
C512FKREGNEKEIQRIMHNYDK530
563MNSIKPDLIQLRKTRDQYLMWLTQK587
Reblot: β-actin
D
E
Reblot: β-tubulin
WCL IB: β-actin
0
1
2
FL
Del
iSH
2
Del
cS
H2
SH
3-B
H
iSH
2-cS
H2
Nor
mal
ized
p85
α-(U
b)n/
Fla
g (a
.u.)
* *
0
1
2
Nor
mal
ized
p85
α-(U
b)n/
F
lag
(a.u
.)
3
TGF-β (min) 0 30 0 30 0 30
*
§§
0
1
2
Nor
mal
ized
p85
α-(U
b)n/
F
lag
(a.u
.)
3
TGF-β (min) 0 30 0 30 0 30 0 30
*
§§
WT
K513/
519R
K567/
575R
WT
K513R
K519R
K513/
519R
95
72
72
Fig. 6. Ubiquitylation of p85a occurs on Lys513 and/or Lys519 in the p85a iSH2 domain. (A) Lysates of PC-3U cellstransfected with HA-tagged WT ubiquitin and Flag-tagged full-length p85a or del iSH2, del cSH2, SH3-BH, or iSH2-cSH2plasmids were subjected to immunoprecipitation with Flag or mouse IgG antiserum and immunoblotted with HA antibody.(B) Lysates of HEK293T cells transfected with HA-Ub and full-length Flag-p85a were subjected to SDS-PAGE. The proteinbands used for mass spectrometry are marked by an arrowhead (nonubiquitylated) and arrows (polyubiquitylated). (C) Twodetected branched peptide sequences in iSH2 domain of p85a. The red letters show the putative lysine residues. (D) Lysatesof HEK293T cells transfected with HA-Ub and full-length WT Flag-p85a, K567/575R, or K513/519R Flag-p85a plasmids wereexamined for p85a polyubiquitylation. (E) Lysates of HEK293T cells transfected with HA-Ub and full-length WT Flag-p85a orK513R, K519R, or K513/519R mutant p85a plasmids were subjected to ubiquitylation assay by blotting with a rabbit antibodyagainst Lys63-linked ubiquitin (K63-linked Ub). Blots are representative of three independent experiments. Graphs are means± SEM from three independent experiments. *P < 0.05 within one set of samples (TGF-b–stimulated versus unstimulated andin panel), §P < 0.05 between different sets of samples (mutant cDNA–transfected versus wt cDNA–transfected cells).
8 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
of scaffolding adaptor proteins, which inturn can bind to and activate PI3K (8, 41).Hyperactivation of AKT is closely relatedtovarious formsofcancer. Inprostatecancer,loss of the PIP3 phosphatase PTEN (phos-phatase and tensin homolog) is frequentlyfound, leading to aberrant activation ofAKT.We have also previously reported thatknockdown of SMAD7 decreased the TGF-b–inducedactivationofAKT(22) and foundthat TRAF6 and SMAD7 are important foractivation of the TGF-b–induced activationTAK1–p38 MAPK pathway (14, 42). Here,we demonstrated that SMAD7 was also re-quired for ubiquitylation of AKT and p85a.ExactlyhowSMAD7functions togetherwithTRAF6 to promote activation of the PI3K-AKT pathway remains to be elucidated.
Activation of p38 MAPK in prostatecancer cells leads to subsequent phos-phorylation of c-Jun on Ser63, therebypromoting its transcriptional activity andresulting in increased expression of thetranscription factor Snail1, which in turnpromotes migration and invasion of pros-tate cancer cells (43). Moreover, activationof AKT leads to phosphorylation andinactivation of GSK3b. When GSK3bis active, it phosphorylates transcriptionfactors, such as c-Jun and Snail1, therebymarking them for subsequent proteaso-mal degradation. Therefore, activationof AKT leads to stabilization of both c-Junand Snail1 (41, 44). In contrast, in normalcells, AKT is known to counteract theJNK–c-Jun pathway (45), whereas, inPTEN-deficient prostate cancer cells andin glioblastoma cells, AKT and c-Jun havebeen found to act in parallel pathways,promoting tumor aggressiveness (46, 47).This mode of activation and stabilizationof c-Jun through TRAF6-p38 pathway isSMAD-independent; however, Thakur et al.showed that TGF-b induces transcriptionof c-Jun in a SMAD-dependent manner(43). It is possible that inhibition of TbRIkinase activity did not reduce cell migrationsignificantly because preexisting c-Jun wasactivated and stabilized by TGF-b, althoughnew transcription of c-Jun was blocked.
SH3 domains were previously re-ported to bind ubiquitin (48). Here, wefound that the fragments of p85a con-taining SH3 domain bind Lys63-linkedubiquitin chains. It is possible that theSH3 domain of p85a binds ubiquitinchains present on autoubiquitylatedTRAF6 or on TbRI (14, 49) after TGF-bstimulation; thereby, p85a is recruited to
A
C
Unstimulated
0 h
24 h
TGF-βSB431542 Wortmannin LY294002 NVP-BKM120
PC-3U
WT MEF TRAF6 –/– MEF
% W
ound
clo
sed
24 h
vs
0 h
(a.u
.)
0
10
20
30
40
Unstimulated
DMSO
SB4315
42
Wor
tman
nin
LY29
4002
NVP-BKM
120
**
§
§§§
§
SB431542 Wortmannin
***
0
30
60
90
Nor
mal
ized
no.
of
mig
rato
ry c
ells
(a.
u.)
B
TGF-β (6 h)
SB431542 Wortmannin
§§§
Reblot: AKT
IB: pAKT S473
IB: pAKT T308
Reblot: β-actin
IB: pSMAD2
Reblot: SMAD2
TGF-β (min) 0 30 0 30 0 30
0
3
6
9
12
15
Nor
mal
ized
pA
KT
/A
KT
(a.
u.)
SB431542 Wortmannin
pAKT S473
pAKT T308***
**§§
§§
TGF-β (min) 0 30 0 30 0 30
Ctrl
Ctrl SB431542 Wortmannin
0 h
24 h
Unstimulated TGF-β Unstimulated TGF-β
TGF-β (24 h) – + – +
WT MEF TRAF6 –/ –
MEF
0
20
40
60
80
100
***
§§§
% W
ound
clo
sed
24
h vs
0 h
(a.
u.)D
WT MEF p85 –/– MEF
Unstimulated TGF-β (6 h) Unstimulated TGF-β (6 h)
TGF-β (6 h) – + – +0
2
4
Nor
mal
ized
no.
of
mig
rato
ry c
ells
(a.
u.) 6
8
WT MEF p85 –/–
MEF
***
§§§
DMSO
RAW264.7
DMSO DMSO
Unstimulated
DMSO
TGF-β (6 h) – + + +
DMSO
TGF-β
Fig. 7. TGF-b–induced cell migration requires the PI3K pathway and TRAF6 and is not dependent on the kinaseactivity of TbRI. (A) RAW264.7 cells treated with the PI3K inhibitor wortmannin, TbRI kinase inhibitor SB431542, or
0.1% DMSO for 1 hour were subjected to Transwell migration assay. Scale bar, 100 mM. The corresponding whole-celllysates were subjected to immunoblotting using antibodies against pAKT Ser473, pAKT Thr308, and pSMAD2. (B) PC-3Ucells treated with the PI3K inhibitors NVP-BKM120, LY294002, and wortmannin, TbRI kinase inhibitor SB431542, or 0.1%DMSO as control were stimulated or not with TGF-b in a cell culture wound healing assay, as described in Materials andMethods. Scale bar, 100 mM. (C) WT and p85−/− MEF cells were exposed to Transwell migration assay as described for (A).Scale bar, 100 mM. (D) WT and TRAF6−/−MEFs treated or not with TGF-b for 24 hours were exposed to wound healing assayas described for (B). Scale bar, 100 mM. Blots and microscopy images are representative of three independent experiments.Graphs are means ± SEM from three independent experiments. **P < 0.01, ***P < 0.001 within one set of samples (TGF-b–stimulated versus unstimulated); §P < 0.05, §§P < 0.01, §§§P < 0.001 between different sets of samples (inhibitor-treatedversus control cells or deficient versus wt cells).9 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
the complex of TGF-b receptors TRAF6 and SMAD7 at the plasmamembrane and polyubiquitylated.
When PI3K resides in the cytosol, nSH2 and cSH2 domains of theregulatory subunit inhibit the enzymatic activity of the catalytic subunit.For PI3K to be activated, conformational changes take place to remodelthe enzyme’s interface and open the catalytic loop (50). Lack of the PI3KSH2 binding motif pYXXM (50) in TGF-b receptors and TRAF6 is
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
consistent with the notion that anothermechanism is involved in the binding ofPI3K to the TGF-b receptor complex thanin the binding of the SH2 domain to phos-phorylated tyrosine residues, which hasbeen observed after the activation of tyro-sine kinase receptors. The viral non-structural protein 1 from influenza virusactivates PI3K by interaction with theiSH2 domain of p85b in a manner thatprevents the nSH2domain from inhibitingp110 (51). Because we found that Lys63-linked ubiquitylation of p85a on Lys513
and Lys519was needed for enzymatic activ-ity of PI3K, it is possible that ubiquitylationof the iSH2 domain results in a conforma-tional change dislocating of the inhibitorynSH2 and cSH2 domains. This possibilityremains to be elucidatedby investigationofthe crystal structures of ubiquitylated p85abound to p110. By studying the availablecrystal structures of PI3K heterodimer(52, 53), we found that the acceptor sitesfor ubiquitylation, Lys513 andLys519, are lo-cated at the surface of PI3K and thus acces-sible for the ubiquitylation machinery. Onthe basis of our previous findings and datareported in this report, we suggest that amulticomponent complex assembles atthe activated TGF-b receptors, containingSMAD7,TRAF6, PI3K, andAKT, and thatthese components cooperatively builds asignaling platform (Fig. 9D).
Inhibition of the kinase activities ofTbRI and TbRII did not prevent TGF-b–induced AKT activation and its inter-action with TbRI (Fig. E and fig. S4) butinhibitedTGF-b–induced SMAD2activation.These findings suggest that the TGF-b–induced activation of PI3K-AKT occursin a TGF-b receptor kinase–independentbut TRAF6-dependent manner. More-over, our data indicate that p85 polyubiq-uitylation and its interaction with TbRIdo not require TGF-b receptor kinase ac-tivity. This is consistent with the previousfindings by Sorrentino et al. (14), whoshow that TRAF6 induces Lys63-linkedpolyubiquitylation of TAK1 upon TGF-bstimulation through the non-SMADpathway, independent of the kinase activityof TGF-b receptor. TGF-b–induced cell
migration was dependent on polyubiquitylation of p85a on Lys513 andLys519 and the activities of PI3K and TRAF6, but not on the kinaseactivity of TbRI. Our observation of Lys63-linked polyubiquitylation ofp85a and activation of AKT in aggressive prostate tumors showingsigns of active TGF-b signaling provide support for a protumorigenicrole of activation of the PI3K-AKT pathway in prostate cancer. This isin agreement with a previous study of Leight et al. (54), who show that
A
0
0.5
1
1.5
2
TGF-β (min) 0 30 0 30 0 30 0 30
Nor
mal
ized
PIP
3 co
ncen
trat
ion
Flag-p85α WT K513R K519R K513/519R
*
§ IB: AKT
IB: pAKT S473
IB: pAKT T308
Reblot: β-tubulin
IB: Flag
IB: pS6
IB: pp85
IB: p85
0 30 0 30 0 30 0 30TGF-β (min)
BFlag-p85α WT K513R K519R K513/519R
C
No
tst
imu
late
dT
GF
-β
Flag-p85α WT WT K513R K519R K513/519RCtrl siRNA + +p85α siRNA + + + + +
0
2
4
6
Flag-p85α – WT – WT K513R K519R K513/519R
TGF-β (6 h) – + – + – + – + – + – + – +
Ctrl siRNA p85α siRNA
* * *
Nor
mal
ized
no.
of
m
igra
tory
cel
ls
TGF-β (6 h) – + – + – + – + – + – + – +
Flag-p85α – WT – WT K513R K519R K513/519R
Reblot: β-tubulin
IB: Flag
IB: p85
p85α siRNA + + + + + + + + + +Ctrl siRNA + + + +
D
Fig. 8. TGF-b–induced polyubiquitylation of p85a on Lys513 and Lys519 is important for PI3K and AKT activation andfor cellmigration. (A) PC-3U cells transfectedwithWT, K513R, K519R, or K513/519R plasmids of p85awere treated or notwithTGF-b for 30min. Cell lysates were prepared and subjected to a PI3K assay, followed bymeasurement of PIP3 concentration, asdescribed in Materials and Methods. Data are means ± SEM from three independent experiments; *P < 0.05 within one set ofsamples (TGFb-stimulated versus unstimulated), §P < 0.05 between different sets of samples (mutant cDNA-transfected versuswt cDNA-transfected cells). (B) Whole-cell lysates corresponding to (A) were subjected to immunoblotting with antiserumagainst pAKT Ser473, pAKT Thr308, pp85, p85, Flag, and pS6. (C) PC-3U cells silenced or not for endogenous p85a [by siRNAtargeting3′untranslated region (3′-UTR)] and transfectedwithWT,K513R,K519R, orK513/519Rplasmidsofp85aweresubjectedto Transwell assay. Scalebar, 100mM.Bargraphs showthemeans±SEM fromthree independent experiments; *P<0.05. (D) Celllysates corresponding to the samples in (C) were subjected to immunoblotting for p85, Flag, and b-tubulin to check theknockdown and transfection efficiency. The correspondingwhole-cell lysates were immunoblotted for p85 and Flag. The filterswere stripped and reblotted for b-actin. Blots and microscopy images are representative of three independent experiments.
10 of 15
SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
Dow
in a high-rigidity matrix, TGF-b–inducedAKT activation enhances epithelial-to-mesenchymal transition, invasion, and me-tastasis in murine mammary gland cells.
On the basis of this current study andour previous findings (14), we propose amodel for TGF-b–induced activation ofthe PI3K-AKT pathway in which the cata-lytic activity of TRAF6 plays an importantrole for Lys63-linked polyubiquitylation ofp85a at Lys513 and Lys519. Presumably, thisleads to a change in the conformation ofthe protein, thereby releasing the inhibitoryeffect of p85a on p110 (Fig. 9D), resultingin the generation of PIP3 in the cell mem-brane. Thereafter, AKT is recruited to theTbRI-TRAF6-p85a complex at the cellmembrane, leading to phosphorylation ofAKT by the PIP3-activated PDK1.
on February 14, 2020
http://stke.sciencemag.org/
nloaded from
MATERIALS AND METHODSCell lines, reagents, and antibodiesHEK293T cells, HaCaT cells, and wild-type, Smad7−/−, and Traf6−/− MEFs weregrown in Dulbecco’s modified Eagle’smedium (DMEM) containing 10% fetalbovine serum (FBS). The human prostatecarcinoma cell line PC-3U, originatingfrom PC-3 cells (55), was grown in RPMI1640 medium containing 10% FBS and2 mM L-glutamine. p85a−/− p85b−/− MEFcells were a gift from G. Scita (Schoolof Medicine, University of Milan, Italy)(56) and are referred to as p85−/− MEFsthroughout the study. p85−/− MEFs weregrown in DMEM containing 15% FBSin the presence of 10% CO2. Mouse mac-rophage RAW264.7 cells, which werepurchased from American Type CultureCollection, were cultured in DMEMcontaining 10% FBS. Transient transfec-tions of HEK293T cells were performedusing the calcium phosphate method(19). PC-3U and RAW264.7 cells weretransiently transfected using FuGENEHD (Roche), as described previously (42).Cells were starved in serum-free mediumat least 20 hours before stimulation with agrowth factor. Cells were stimulated by add-ing TGF-b1 (5 ng/ml) (referred to as TGF-bthroughout the study; R&D Systems), EGF(10 ng/ml) (PeproTech Inc.), or PDGF-BB(100 ng/ml) (Amgen) to the starvationmedium. For immunoprecipitation experi-ments, polyclonal rabbit AKT1, AKT2,and AKT3 (referred to as AKT throughoutthe study; H-136) and mouse p85a (B-9)antiserum (Santa Cruz Biotechnology Inc.)
Prostate cancer
Gleason 6
Prostate cancer
Gleason 8
pSMAD2
***
To
tal P
LA s
ign
als
of
colo
caliz
atio
no
f p
85
α a
nd
Lys
63-l
inke
d u
biq
uit
in
Gleason score≤
6 o
r 7
= (
3 +
4)
7 =
(4
+ 3
) o
r ≥
8
**
Gleason score
≤6
or
7 =
(3
+ 4
)
7 =
(4
+ 3
) o
r ≥
8
Re
lati
ve in
ten
sity
of
pA
KT
S4
73
exp
ress
ion
pAKT S473
400
800
1200
1600
2000
2400
2800
40
80
120
160
200
A B C
PLA: p85α and Lys63-linked ubiquitin
Cell movement
P
Lys63-linked ubiquitin
Phosphate
Cell membrane
p110α
PP
S473T308
TβR
I
Tβ
RII
AKT
TRAF6
p85α
TβR
I
TGF-β
Tβ
RII
SMAD7
TRAF6
D
Fig. 9. Lys63-linked ubiquitylation of p85a and activation of SMAD2 and AKT in prostate cancer tissues arecorrelated with aggressiveness. (A and B) Prostate tumor tissue samples were stained with pSMAD2 (A) and pAKTSer473 (B) antibodies. Quantification shows the means ± SEM of pAKT Ser473 of 10 patients in each group. (C) Theassociation between p85a and Lys63 (K63) ubiquitin in prostate cancer patients (brown dots) was determined byPLA (300 cells were analyzed in each group). Data are means ± SEM of five patients in each group. **P < 0.01, ***P <0.001. Scale bars, 50 mm. (D) Proposed model for TGF-b–induced polyubiquitylation of p85a and activation of PI3Kand AKT. TGF-b–induced activation of PI3K is initiated by ligand-induced oligomerization of TGF-b receptors, whichjuxtaposes TRAF6 molecules, constitutively bound to TbRI, that undergo autoubiquitylation and ubiquitylate TbRI.Lys63-linked ubiquitin chains on TRAF6 or TbRI mediate the recruitment of PI3K to the complex by binding the SH3domain of p85a. Activated TRAF6 then causes Lys63-linked ubiquitylation of p85a and activation of PI3K, which inturn leads to PIP3 production. Whether TRAF6, PI3K, AKT, and SMAD7 simultaneously form a complex or whetherinteractions occur sequentially remains to be elucidated. AKT is recruited to the complex whereby it is ubiquitylatedand activated. The recruitment of p85a and AKT may be facilitated by SMAD7, which has been shown to have anadaptor function in the non-Smad TRAF6 pathway. Attachment of Lys63-linked polyubiquitin chains on p85a pos-sibly favors a conformational change of p85a, thereby removing the inhibitory contacts of the SH2 domains ofp85a from p110, which enhances the kinase activity of PI3K. AKT activation promotes cell migration, which hasbeen shown to be dependent on TRAF6-mediated c-Jun activation as well (43). For simplicity, the PI3K-AKT-SMAD7complex is shown only on one side of the symmetric TGF-b receptor complex.
11 of 15

SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
were used. Polyclonal rabbit HA (Y-11), monoclonal mouse HA (F-7),monoclonal mouse ubiquitin (P4D1), polyclonal rabbit and goat TbRI(V-22), and monoclonal mouse TRAF6 antibodies (used for immuno-fluorescence) were purchased from Santa Cruz Biotechnology Inc.Mouse monoclonal Lys63-linked polyubiquitin antibodies (HWA4C4and Apu3) were purchased from Enzo Life Sciences Inc. andMillipore, respectively. Monoclonal rabbit pAKT (Ser473 and Thr308),rabbit phospho-PI3K p85 (Tyr458)/p55 (Tyr199), monoclonal rabbitp85 (recognizing both p85a and p85b; 19H8), monoclonal rabbitp110a, and rabbit pS6 ribosomal protein (Ser235/236) antibodies werefrom Cell Signaling. Mouse monoclonal Penta·His antibody was fromQiagen. Monoclonal mouse b-actin and monoclonal rabbit TRAF6(used for immunoblotting) were obtained from Abcam. Tetramethylrhodamine isothiocyanate–labeled phalloidin and monoclonal mouseFLAG (M2) antibodies were obtained from Sigma. Rabbit Alexa Fluor488 and Alexa Fluor 555 and mouse Alexa Fluor 488 antibodies raisedin donkey were purchased from Invitrogen. Light chain–specific per-oxidase IgG, fraction monoclonal rabbit IgG, and light chain–specificperoxidase AffiniPure mouse IgG were purchased from Jackson Im-munoresearch Laboratories Inc. Biotinylated poly-Ub wild-type chains(2 to 7) (K63-linked) were purchased from Boston Biochem.
InhibitorsThe PI3K inhibitors LY294002, wortmannin (KY12420), and NVP-BKM120were purchased fromCalbiochem. The TbRI kinase inhibitorsSB431542 and SB505124 were from Tocris and Sigma, respectively.LY2109761, which inhibits the kinases of both TbRI and TbRII, waspurchased from Cayman Chemical. The PDGFR kinase inhibitorsimatinib and AG1296 were obtained from Novartis Pharma AGand Sigma, respectively.
Immunoblotting and immunostainingHEK293 cell lines and PC-3U cells were grown in 10-cm dishes.Forty-eight hours after transfection, cells were starved for 20 hours,stimulated with TGF-b, PDGF-BB, or EGF for the indicated timeperiods, washed once with ice-cold PBS, and lysed in ice-cold lysisbuffer [150 mM NaCl, 50 mM tris (pH 8), 1% Triton X-100, 10% (v/v)glycerol, 1% (v/v) NP-40, 0.5% sodium deoxycholate, 1 mM aprotinin,1 mM Pefabloc, and 1 mM sodium orthovanadate]. After centrifuga-tion, supernatants were collected, and protein concentrations weremeasured by using the BCA (bicinchoninic acid) protein assay kit(Thermo Fisher Scientific). Equal amounts of proteins were used forimmunoprecipitations. Samples were subjected to SDS-PAGE in 8 or10% polyacrylamide gels, followed by blotting onto polyvinylidene di-fluoride membranes and immunoblotting, as described previously (22).
For analysis by immunostaining, PC-3U cells were seeded in six-wellplates and grown on coverslips 24 hours before transfection. Immuno-stainingwas performed, as described previously (42). Photomicrographswere obtained using an Axioplan 2 microscope (Carl Zeiss MicroImagingInc.) with a digital camera (RETIGAEXi) using a PlanApochromat ×63/1.4oil DIC objective lens (Carl Zeiss MicroImaging Inc.). Photographswere taken at room temperature. Primary images were acquired usingtheZEN2011 program. Imagememory contentwas reduced, andbright-ness contrast was adjusted using Photoshop 6.0 (Adobe). Pictures shownare representative images from three different experiments.
In vivo ubiquitylation assaysPC-3U andMEF cells were washed once in PBS, scraped in 1ml of PBS,and centrifuged at 400g for 5 min. Noncovalent protein interactions
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
were dissociated in 1% SDS, and the solution was boiled for 10 min.Samples were diluted 1:10 in PBS, containing 0.5%NP-40, 1 mM apro-tinin, and 1mMPefabloc. The sampleswere cleared by centrifugation at12,000g for 10 min and subjected to immunoprecipitation, followed byimmunoblotting.
In vitro ubiquitylation assaysRecombinant GST-TRAF6 (E3) and His-p85a (SignalChem; about0.1 mg at maximum concentration) were incubated in 20 mM tris(pH 7.4), 50 mM NaCl, 10 mM MgCl2, 10 mM dithiothreitol, 10 mMATP, ubiquitin (0.5 mg/ml) (Sigma), 2 mM ubiquitin aldehyde (Biomol),100 mM MG132 (Sigma), 0.1 mg of E1 (human recombinant fromBiomol), and 0.2 mg of E2 Ubc13-Uev1A (Biomol) at 30°C for 1 hourand then subjected to SDS-PAGE.
Scratch wound healing assayPC-3U andMEF cells were cultured in six-well plates. Forty-eight hourslater, the cells were treated or not with different inhibitors. TGF-b wasadded to the cells after 1 hour, and “wounds”were made using a 200-mlpipette tip. Pictures of thewoundswere taken immediately and 24hourslater using a Zeiss Axiovert 40CFL microscope. Primary images wereacquired using the AxioVision program and analyzed by the TScratchprogram. The percentage of the open wound was calculated by dividingthe area of the gap after 24 hours by that at 0 hour. Then, the percentageof the wound closure was calculated by subtracting the open woundpercentage from 100. Pictures shown are representative images fromthree different experiments.
Transwell migration assayMigration assays were performed in Transwell cell culture chamberswith polycarbonate filters (24-well, 8-mm pore, Corning Costar). Thefilterswere coatedwith fibrinogen (1mg/ml) for 6 hours and then dried.Cells pretreated or not with the inhibitors for 1 hour were washed andresuspended at a density of 200,000 cells in 200 ml of the starvation me-dium without or with only TGF-b or TGF-b and the inhibitor andloaded to the upper chamber. Starvation medium (500 ml) without orwith only TGF-b or TGF-b and the inhibitor was poured to the lowerreservoir. After 6 hours ofmigration, cells on the uppermembranewerescraped off, and those that migrated to the lower membrane were fixedby 4% formaldehyde, stained by the Giemsa staining method, andphotographed using the Zeiss Axiovert 40CFL microscope. Primaryimages were acquired using the Zen program. Quantification of thenumber of the migratory cells was done by measuring the number ofpixels using Photoshop 6.0 (Adobe). Pictures shown are representativeimages from three different experiments.
PlasmidsExpression vectors for wild-type 3×HA-Ub, Lys48-only 3×HA-Ub,and Lys63-only 3×HA-Ub were gifts from V. M. Dixit (Genentech).FLAG-tagged TRAF6-C70A was a gift from Z. J. Chen (University ofTexas Southwestern Medical Center, Dallas, TX). Flag-AKT1was a gift from W. C. Sessa (Yale University School of Medicine,New Haven, CT). HA-tagged AKT1 was purchased from Addgene.HA–TbRI TD (also called ALK5 TD) was a gift from P. ten Dijke(University of Leiden, Netherlands). The HA–TbRI TD–E161Aplasmid with HA fused to the C terminus of the receptor was de-scribed previously (14). Flag-tagged p85a constructs were gifts fromJ. Y. Ahn (School of Medicine, Sungkyunkwan University, Suwon,Korea).
12 of 15

SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
siRNA transfectionSMARTpool siRNA for TRAF6 and p85a and Individual siGENOMEPIK3R1 siRNA that targets 3′-UTR were synthesized by DharmaconResearch. The SMARTpool TRAF6 and p85a siRNA were transfectedwith DharmaFECT (Dharmacon Research) according to the manufac-turer’s protocol. The Individual siGENOME PIK3R1 siRNAwas trans-fected with Lipofectamine 3000 (Invitrogen).
Site-directed mutagenesisSite-directed mutagenesis was done using a QuikChange LightningSite-Directed Mutagenesis kit (Agilent Technologies). The followingprimers were synthesized by Sigma: K567R, ggataaggtctggtctaatgctgtt-catacgtttgtcaatttc and gaaattgacaaacgtatgaacagcattagaccagaccttatcc; K575R,ggtctctcgtccttctcagctggataaggtctgg and ccagaccttatccagctgagaaggacgagagacc;K513R, gccttcacgtctaaacttttctatgtattctttgctgtaccg and cggtacagcaaagaatacata-gaaaagtttagacgtgaaggc; andK519R, gcataatcctttgtatttctctctcattgccttcacgtttaaacand gtttaaacgtgaaggcaatgagagagaaatacaaaggattatgc.
PI3K activity detected by ELISAAn ELISA assay (96-well) for detection of PIP3 was purchased fromEchelon Biosciences Inc. MEFs or transfected PC-3U cells werelysed, and PI3K reactions were set up according to the manufacturer’sprocedure. The PIP3 product of the kinase reactions was detectedby ELISA.
Mass spectrometric analysis of polyubiquitylated p85aHEK293T cells in five 10-cm dishes were transfected, and ubiqui-tylation assays were performed, as described above. p85 was immu-noprecipitated and then subjected to SDS-PAGE and silver staining(57). Bands corresponding to nonubiquitylated and polyubiquity-lated p85a were cut out and trypsin-digested in the gel (58). In brief,the protein bands were destained, washed, and treated with ammoni-um bicarbonate. After complete drying under nitrogen, trypsin (por-cine; modified sequence grade from Promega) was allowed to absorbinto the gel piece. After overnight digestion, the peptides were extractedand then desalted and concentrated on a handmade microcolumn,POROS R2 20, packed in a gel loader tip (59). The peptides were elutedwith acetonitrile-containing matrix (4-hydroxy-a-cyanocinnamic acid)directly onto the target, and spectra were taken using a Bruker Biflex IIImatrix-assisted laser desorption/ionization–time-of-flight mass spec-trometer from Bruker Daltonics. The search for ubiquitylated peptideswas accomplished by GPMAW (Lighthouse Data) by scanning for theexpected addition of the most C-terminal tryptic peptide of humanubiquitin [GG (114.05 Da)] onto a lysine residue in the tryptic digestof human p85a. Spectra and associated data are provided in fig. S12.
In situ PLAThe prostate cancer tissue sections were pretreated with deparaffiniza-tion, antigen retrieval, and permeabilization. PLAwas performedwith aDuolink Detection kit for Brightfield (Sigma) according to the manu-facturer’s instructions. Images were acquired with Pannoramic 250Flash. Signals were quantified usingDuolink ImageTool. p85a antibodywas from Novus Biologicals. pSMAD2 and pAKT Ser473 antibodieswere from Cell Signaling.
Malignant prostate cancer tissues were collected from men under-going prostatectomy. The tumors had Gleason scores ranging from6 (3 + 3) to 9 (4 + 5) and were staged as pT2 or pT3. All experi-ments were performed in agreement with the relevant guidelinesand regulations for handling human tissues. The use of patient tis-
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
sues was approved by the Regional Ethical Review Board (permitno. 03-482).
ImmunohistochemistryImmunohistochemistry was performed as previously described(33). In brief, the tissue slides were deparaffinized in xylene, rehydratedthrough graded alcohols, incubated for antigen retrieval in the Retriever2100 (ProteoGenix). Endogenous peroxidase activity was blocked with3% hydrogen peroxidase in methanol. After incubation with theprimary antibody at 4°C overnight, the sections were incubated withREAL EnVision detection system (DAKO). The reaction was visua-lized by REAL DAB+ chromogen (DAKO) under microscopic control.After stopping the reaction with tap water, the sections were treatedwith hematoxylin, dehydrated, and mounted. Negative controls areshown in fig. S11.
Statistical analysisStatistical analyses between all groups were performed using one-wayanalysis of variance (ANOVA).F test was used to deriveP values.Quan-tification of immunoblots was done by measuring the density of thebands using the AIDA 2D densitometry program. Data are means ±SEM of three independent experiments (n = 3), except for figs, S6, S8,and S10 (B and C), which were repeated twice (n = 2). P values of <0.05were considered statistically significant (*P < 0.05, **P < 0.01, ***P <0.001 within one set of samples; §P < 0.05, §§P < 0.01, §§§P < 0.001 be-tween different sets of samples).
SUPPLEMENTARY MATERIALSwww.sciencesignaling.org/cgi/content/full/10/486/eaal4186/DC1Fig. S1. AKT interacts with TRAF6 in a TGF-b–dependent manner.Fig. S2. TGF-b induces Lys63-linked polyubiquitylation and activation of AKT.Fig. S3. TGF-b–induced polyubiquitylation and activation of AKT require the E3 ligase activityof TRAF6.Fig. S4. TGF-b–induced activation of AKT depends on the PI3K pathway and is not dependenton the kinase activities of TbRI or TbRII.Fig. S5. TGF-b–induced activation of AKT is not dependent on the kinase activity of TAK1 or thePDGFR.Fig. S6. SMAD7 is required for Lys63-linked polyubiquitylation of p85a and AKT.Fig. S7. TRAF6, pAKT Ser473, TbRI, and p85a colocalize at the cell membrane.Fig. S8. Mutation of Lys513 and Lys519 to arginine does not affect binding of p85a to p110a.Fig. S9. PI3K activity is increased by TGF-b stimulation and requires the E3 ligase activity ofTRAF6 but not the kinase activity of TbRI.Fig. S10. TGF-b–induced polyubiquitylation of p85a on Lys513 and Lys519 is important for AKTactivation and migration of p85−/− MEFs.Fig. S11. Negative control for immunohistochemistry and in situ PLA.Fig. S12. Mass spectrometry analysis.
REFERENCES AND NOTES1. J. Massagué, TGFb in cancer. Cell 134, 215–230 (2008).2. C.-H. Heldin, M. Landström, A. Moustakas, Mechanism of TGF-b signaling to growth arrest,
apoptosis, and epithelial–mesenchymal transition. Curr. Opin. Cell Biol. 21, 166–176(2009).
3. C.-H. Heldin, K. Miyazono, P. ten Dijke, TGF-b signalling from cell membrane to nucleusthrough SMAD proteins. Nature 390, 465–471 (1997).
4. Y. Mu, S. K. Gudey, M. Landström, Non-Smad signaling pathways. Cell Tissue Res. 347,11–20 (2012).
5. Y. E. Zhang, Non-Smad pathways in TGF-b signaling. Cell Res. 19, 128–139 (2009).6. S. Lamouille, R. Derynck, Cell size and invasion in TGF-b–induced epithelial to
mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178,437–451 (2007).
7. L. Zhang, F. Zhou, P. ten Dijke, Signaling interplay between transforming growth factor-breceptor and PI3K/AKT pathways in cancer. Trends Biochem. Sci. 38, 612–620 (2013).
8. L. M. Thorpe, H. Yuzugullu, J. J. Zhao, PI3K in cancer: Divergent roles of isoforms, modesof activation and therapeutic targeting. Nat. Rev. Cancer 15, 7–24 (2015).
13 of 15

SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from
9. D. Stokoe, L. R. Stephens, T. Copeland, P. R. J. Gaffney, C. B. Reese, G. F. Painter,A. B. Holmes, F. McCormick, P. T. Hawkins, Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B. Science 277, 567–570 (1997).
10. D. D. Sarbassov, D. A. Guertin, S. M. Ali, D. M. Sabatini, Phosphorylation and regulation ofAkt/PKB by the rictor-mTOR complex. Science 307, 1098–1101 (2005).
11. A. Carnero, The PKB/AKT pathway in cancer. Curr. Pharm. Des. 16, 34–44 (2010).12. A. Hershko, A. Ciechanover, The ubiquitin system. Annu. Rev. Biochem. 67, 425–479
(1998).13. F. Ikeda, N. Crosetto, I. Dikic, What determines the specificity and outcomes of ubiquitin
signaling? Cell 143, 677–681 (2010).14. A. Sorrentino, N. Thakur, S. Grimsby, A. Marcusson, V. von Bulow, N. Schuster, S. Zhang,
C.-H. Heldin, M. Landström, The type I TGF-b receptor engages TRAF6 to activate TAK1 in areceptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207 (2008).
15. Y. Mu, R. Sundar, N. Thakur, M. Ekman, S. K. Gudey, M. Yakymovych, A. Hermansson,H. Dimitriou, M. T. Bengoechea-Alonso, J. Ericsson, C.-H. Heldin, M. Landström, TRAF6ubiquitinates TGFb type I receptor to promote its cleavage and nuclear translocation incancer. Nat. Commun. 2, 330 (2011).
16. J. Y. Yi, I. Shin, C. L. Arteaga, Type I transforming growth factor b receptor binds to andactivates phosphatidylinositol 3-kinase. J. Biol. Chem. 280, 10870–10876 (2005).
17. W.-L. Yang, J. Wang, C.-H. Chan, S.-W. Lee, A. D. Campos, B. Lamothe, L. Hur, B. C. Grabiner,X. Lin, B. G. Darnay, H.-K. Lin, The E3 ligase TRAF6 regulates Akt ubiquitination andactivation. Science 325, 1134–1138 (2009).
18. Q. Yin, S.-C. Lin, B. Lamothe, M. Lu, Y.-C. Lo, G. Hura, L. Zheng, R. L. Rich, A. D. Campos,D. G. Myszka, M. J. Lenardo, B. G. Darnay, H. Wu, E2 interaction and dimerizationin the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658–666 (2009).
19. A. Hamidi, V. von Bulow, R. Hamidi, N. Winssinger, S. Barluenga, C.-H. Heldin,M. Landström, Polyubiquitination of transforming growth factor b (TGFb)-associatedkinase 1 mediates nuclear factor-kB activation in response to different inflammatorystimuli. J. Biol. Chem. 287, 123–133 (2012).
20. J. A. Romashkova, S. S. Makarov, NF-kB is a target of AKT in anti-apoptotic PDGFsignalling. Nature 401, 86–90 (1999).
21. H. Porsch, M. Mehić, B. Olofsson, P. Heldin, C.-H. Heldin, Platelet-derived growth factorb-receptor, transforming growth factor b type I receptor, and CD44 protein modulateeach other’s signaling and stability. J. Biol. Chem. 289, 19747–19757 (2014).
22. S. Edlund, S. Y. Lee, S. Grimsby, S. Zhang, P. Aspenström, C.-H. Heldin, M. Landström,Interaction between Smad7 and b-catenin: Importance for transforming growth factorb-induced apoptosis. Mol. Cell. Biol. 25, 1475–1488 (2005).
23. L. Deng, C. Wang, E. Spencer, L. Yang, A. Braun, J. You, C. Slaughter, C. Pickart,Z. J. Chen, Activation of the IkB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 103, 351–361(2000).
24. H. R. Ko, C. K. Kim, S. B. Lee, J. Song, K.-H. Lee, K. K. Kim, K. W. Park, S.-W. Cho, J.-Y. Ahn,P42 Ebp1 regulates the proteasomal degradation of the p85 regulatory subunit ofPI3K by recruiting a chaperone-E3 ligase complex HSP70/CHIP. Cell Death Dis. 5,e1131 (2014).
25. J. Yu, Y. Zhang, J. McIlroy, T. Rordorf-Nikolic, G. A. Orr, J. M. Backer, Regulation of the p85/p110 phosphatidylinositol 3′-kinase: Stabilization and inhibition of the p110a catalyticsubunit by the p85 regulatory subunit. Mol. Cell. Biol. 18, 1379–1387 (1998).
26. M. Ekman, Y. Mu, S. Y. Lee, S. Edlund, T. Kozakai, N. Thakur, H. Tran, J. Qian,J. Groeden, C.-H. Heldin, M. Landström, APC and Smad7 link TGFb type I receptors tothe microtubule system to promote cell migration. Mol. Biol. Cell 23, 2109–2121(2012).
27. C. Porta, C. Paglino, A. Mosca, Targeting PI3K/Akt/mTOR signaling in cancer. Front. Oncol.4, 64 (2014).
28. J.-S. Kim, J.-G. Kim, M.-Y. Moon, C.-Y. Jeon, H.-Y. Won, H.-J. Kim, Y.-J. Jeon, J.-Y. Seo,J.-I. Kim, J. Kim, J.-Y. Lee, P.-H. Kim, J.-B. Park, Transforming growth factor–b1 regulatesmacrophage migration via RhoA. Blood 108, 1821–1829 (2006).
29. C. F. de la Cruz-Herrera, M. Baz-Martínez, V. Lang, A. El Motiam, J. Barbazán,R. Couceiro, M. Abal, A. Vidal, M. Esteban, C. Muñoz-Fontela, A. Nieto, M. S. Rodríguez,M. Collado, C. Rivas, Conjugation of SUMO to p85 leads to a novel mechanism of PI3Kregulation. Oncogene 35, 2873–2880 (2016).
30. E. Y. Skolnik, B. Margolis, M. Mohammadi, E. Lowenstein, R. Fischer, A. Drepps,A. Ullrich, J. Schlessinger, Cloning of PI3 kinase–associated p85 utilizing a novelmethod for expression/cloning of target proteins for receptor tyrosine kinases. Cell65, 83–90 (1991).
31. M. Razmara, C.-H. Heldin, J. Lennartsson, Platelet-derived growth factor-induced Aktphosphorylation requires mTOR/Rictor and phospholipase C-g1, whereas S6phosphorylation depends on mTOR/Raptor and phospholipase D. Cell Commun. Signal.11, 3 (2013).
32. S. J. Assinder, Q. Dong, Z. Kovacevic, D. R. Richardson, The TGF-b, PI3K/Akt and PTENpathways: Established and proposed biochemical integration in prostate cancer.Biochem. J. 417, 411–421 (2009).
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
33. J. Song, Y. Mu, C. Li, A. Bergh, M. Miaczynska, C.-H. Heldin, M. Landström, APPL proteinspromote TGFb-induced nuclear transport of the TGFb type I receptor intracellulardomain. Oncotarget 7, 279–292 (2016).
34. B. Wegiel, A. Bjartell, Z. Culig, J. L. Persson, Interleukin-6 activates PI3K/Akt pathwayand regulates cyclin A1 to promote prostate cancer cell survival. Int. J. Cancer 122,1521–1529 (2008).
35. C.-H. Heldin, M. Vanlandewijck, A. Moustakas, Regulation of EMT by TGFb in cancer.FEBS Lett. 586, 1959–1970 (2012).
36. Y. Katsuno, S. Lamouille, R. Derynck, TGF-b signaling and epithelial-mesenchymaltransition in cancer progression. Curr. Opin. Oncol. 25, 76–84 (2013).
37. H. Xia, L. L. P. J. Ooi, K. M. Hui, MicroRNA-216a/217-induced epithelial-mesenchymaltransition targets PTEN and SMAD7 to promote drug resistance and recurrence of livercancer. Hepatology 58, 629–641 (2013).
38. S. Kuchay, S. Duan, E. Schenkein, A. Peschiaroli, A. Saraf, L. Florens, M. P. Washburn,M. Pagano, FBXL2- and PTPL1-mediated degradation of p110-free p85b regulatorysubunit controls the PI(3)K signalling cascade. Nat. Cell Biol. 15, 472–480 (2013).
39. C.-H. Chan, C.-F. Li, W.-L. Yang, Y. Gao, S.-W. Lee, Z. Feng, H.-Y. Huang, K. K. C. Tsai,L. G. Flores, Y. Shao, J. D. Hazle, D. Yu, W. Wei, D. Sarbassov, M.-C. Hung, K. I. Nakayama,H.-K. Lin, The Skp2-SCF E3 ligase regulates Akt ubiquitination, glycolysis, herceptinsensitivity, and tumorigenesis. Cell 149, 1098–1111 (2012).
40. E. M. Yazlovitskaya, H.-Y. Tseng, O. Viquez, T. Tu, G. Mernaugh, K. K. McKee, K. Riggins,V. Quaranta, A. Pathak, B. D. Carter, P. Yurchenco, A. Sonnenberg, R. T. Böttcher,A. Pozzi, R. Zent, Integrin a3b1 regulates kidney collecting duct development viaTRAF6-dependent K63-linked polyubiquitination of Akt. Mol. Biol. Cell 26, 1857–1874(2015).
41. B. D. Manning, L. C. Cantley, AKT/PKB signaling: Navigating downstream. Cell 129,1261–1274 (2007).
42. S. Edlund, S. Bu, N. Schuster, P. Aspenström, R. Heuchel, N.-E. Heldin, P. ten Dijke,C.-H. Heldin, M. Landström, Transforming growth factor-b1 (TGF-b)–induced apoptosisof prostate cancer cells involves Smad7-dependent activation of p38 by TGF-b-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 14,529–544 (2003).
43. N. Thakur, S. K. Gudey, A. Marcusson, J. Y. Fu, A. Bergh, C.-H. Heldin, M. Landström,TGFb-induced invasion of prostate cancer cells is promoted by c-Jun-dependenttranscriptional activation of Snail1. Cell Cycle 13, 2400–2414 (2014).
44. B. P. Zhou, J. Deng, W. Xia, J. Xu, Y. M. Li, M. Gunduz, M. C. Hung, Dual regulation of Snailby GSK-3b-mediated phosphorylation in control of epithelial-mesenchymal transition.Nat. Cell Biol. 6, 931–940 (2004).
45. H.-F. Zhao, J. Wang, S.-S. Tony To, The phosphatidylinositol 3-kinase/Akt and c-JunN-terminal kinase signaling in cancer: Alliance or contradiction? (Review).Int. J. Oncol. 47, 429–436 (2015).
46. J. Cui, S.-Y. Han, C. Wang, W. Su, L. Harshyne, M. Holgado-Madruga, A. J. Wong, c-JunNH2-terminal kinase 2a2 promotes the tumorigenicity of human glioblastoma cells.Cancer Res. 66, 10024–10031 (2006).
47. I. Vivanco, N. Palaskas, C. Tran, S. P. Finn, G. Getz, N. J. Kennedy, J. Jiao, J. Rose, W. Xie,M. Loda, T. Golub, I. K. Mellinghoff, R. J. Davis, H. Wu, C. L. Sawyers, Identification ofthe JNK signaling pathway as a functional target of the tumor suppressor PTEN.Cancer Cell 11, 555–569 (2007).
48. S. D. Stamenova, M. E. French, Y. He, S. A. Francis, Z. B. Kramer, L. Hicke, Ubiquitin binds toand regulates a subset of SH3 domains. Mol. Cell 25, 273–284 (2007).
49. R. Sundar, S. K. Gudey, C.-H. Heldin, M. Landström, TRAF6 promotes TGFb-inducedinvasion and cell-cycle regulation via Lys63-linked polyubiquitination of Lys178 in TGFbtype I receptor. Cell Cycle 14, 554–565 (2015).
50. O. Vadas, J. E. Burke, X. Zhang, A. Berndt, R. L. Williams, Structural basis for activation andinhibition of class I phosphoinositide 3-kinases. Sci. Signal. 4, re2 (2011).
51. B. G. Hale, P. S. Kerry, D. Jackson, B. L. Precious, A. Gray, M. J. Killip, R. E. Randall,R. J. Russell, Structural insights into phosphoinositide 3-kinase activation by the influenzaA virus NS1 protein. Proc. Natl. Acad. Sci. U.S.A. 107, 1954–1959 (2010).
52. N. Miled, Y. Yan, W.-C. Hon, O. Perisic, M. Zvelebil, Y. Inbar, D. Schneidman-Duhovny,H. J. Wolfson, J. M. Backer, R. L. Williams, Mechanism of two classes of cancermutations in the phosphoinositide 3-kinase catalytic subunit. Science 317, 239–242(2007).
53. D. Mandelker, S. B. Gabelli, O. Schmidt-Kittler, J. Zhu, I. Cheong, C.-H. Huang, K. W. Kinzler,B. Vogelstein, L. M. Amzel, A frequent kinase domain mutation that changes theinteraction between PI3Ka and the membrane. Proc. Natl. Acad. Sci. U.S.A. 106,16996–17001 (2009).
54. J. L. Leight, M. A. Wozniak, S. Chen, M. L. Lynch, C. S. Chen, Matrix rigidity regulates aswitch between TGF-b1–induced apoptosis and epithelial–mesenchymal transition.Mol. Biol. Cell 23, 781–791 (2012).
55. P. Franzén, H. Ichijo, K. Miyazono, Different signals mediate transforming growthfactor-b1-induced growth inhibition and extracellular matrix production in prostaticcarcinoma cells. Exp. Cell Res. 207, 1–7 (1993).
14 of 15

SC I ENCE S I GNAL ING | R E S EARCH ART I C L E
56. M. Innocenti, E. Frittoli, I. Ponzanelli, J. R. Falck, S. M. Brachmann, P. P. Di Fiore, G. Scita,Phosphoinositide 3-kinase activates Rac by entering in a complex with Eps8, Abi1,and Sos-1. J. Cell Biol. 160, 17–23 (2003).
57. A. Shevchenko, M. Wilm, O. Vorm, M. Mann, Mass spectrometric sequencing of proteinssilver-stained polyacrylamide gels. Anal. Chem. 68, 850–858 (1996).
58. F. Gharahdaghi, C. R. Weinberg, D. A. Meagher, B. S. Imai, S. M. Mische, Massspectrometric identification of proteins from silver-stained polyacrylamide gel: Amethod for the removal of silver ions to enhance sensitivity. Electrophoresis 20,601–605 (1999).
59. J. Gobom, E. Nordhoff, E. Mirgorodskaya, R. Ekman, P. Roepstorff, Sample purification andpreparation technique based on nano-scale reversed-phase columns for the sensitiveanalysis of complex peptide mixtures by matrix-assisted laser desorption/ionization massspectrometry. J. Mass Spectrom. 34, 105–116 (1999).
Acknowledgments: We thank A. Moustakas and our colleagues at the Ludwig Institute forCancer Research, Uppsala Branch and the Department of Medical Biosciences, UmeåUniversity, Sweden for valuable discussions. We also thank S. K. Gudey for technicalassistance and U. Engström and Å. Engström for mass spectrometry analysis. Funding: This
Hamidi et al., Sci. Signal. 10, eaal4186 (2017) 4 July 2017
work was partly supported by grants from the Knut and Alice Wallenberg Foundation(KAW 2012.0090), Swedish Cancer Society (CAN 2014/674), and Swedish Medical ResearchCouncil (K2013-66X-15284-04-4 and 2015-02757). Author contributions: A.H., N.T., A.M.,and J.S. performed the experiments. A.B. provided the human samples. S.I. generatedthe Smad7−/− MEFs. C.-H.H. and M.L. planned the project. A.H., A.B., C.-H.H., and M.L.prepared the manuscript. Competing interests: The authors declare that they have nocompeting interests.
Submitted 9 June 2015Resubmitted 16 November 2016Accepted 14 June 2017Published 4 July 201710.1126/scisignal.aal4186
Citation: A. Hamidi, J. Song, N. Thakur, S. Itoh, A. Marcusson, A. Bergh, C.-H. Heldin,M. Landström, TGF-b promotes PI3K-AKT signaling and prostate cancer cell migrationthrough the TRAF6-mediated ubiquitylation of p85a. Sci. Signal. 10, eaal4186 (2017).
15 of 15
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from

αTRAF6-mediated ubiquitylation of p85 promotes PI3K-AKT signaling and prostate cancer cell migration through theβTGF-
LandströmAnahita Hamidi, Jie Song, Noopur Thakur, Susumu Itoh, Anders Marcusson, Anders Bergh, Carl-Henrik Heldin and Maréne
DOI: 10.1126/scisignal.aal4186 (486), eaal4186.10Sci. Signal.
signaling pathways are not as insulated from each other as was once thought.βthe TGF-TRAF6 mediates activation of the other ''SMAD-independent'' (JNK) pathway. These data suggest that, although distinct,
correlated with migration in cultured cells and prostate tumor grade in patient samples.αabundance of ubiquitylated p85, thus enabling phosphorylation of the catalytic PI3K subunit p110, but only in the presence of SMAD7. Theαsubunit p85
receptor triggered the recruitment of PI3K and the ubiquitin ligase TRAF6, which polyubiquitylated the regulatory PI3K to itsβRI. The binding of TGF-βmediated mechanism and a SMAD protein but is independent of the kinase function of T
−induced activation of PI3K depends on another ubiquitin ligase−β. found that the TGF-et alreceptor. However, Hamidi type IβSMAD-independent pathways (PI3K-AKT) is mediated by a direct interaction between PI3K and the TGF-
pathways are generally thought to be either dependent or independent of transcription factors called SMADs. One of the signaling stimulates various intracellular pathways that can promote migration in tumor cells. TheseβTGF-
PI3K connection−βThe TGF-
ARTICLE TOOLS http://stke.sciencemag.org/content/10/486/eaal4186
MATERIALSSUPPLEMENTARY http://stke.sciencemag.org/content/suppl/2017/06/28/10.486.eaal4186.DC1
CONTENTRELATED
http://stke.sciencemag.org/content/sigtrans/10/502/eaam7464.fullhttp://stke.sciencemag.org/content/sigtrans/8/358/ra1.fullhttp://stke.sciencemag.org/content/sigtrans/9/415/ra19.fullhttp://stke.sciencemag.org/content/sigtrans/9/418/ra27.fullhttp://stke.sciencemag.org/content/sigtrans/8/396/ra96.full
REFERENCES
http://stke.sciencemag.org/content/10/486/eaal4186#BIBLThis article cites 59 articles, 21 of which you can access for free
PERMISSIONS http://www.sciencemag.org/help/reprints-and-permissions
Terms of ServiceUse of this article is subject to the
is a registered trademark of AAAS.Science SignalingYork Avenue NW, Washington, DC 20005. The title (ISSN 1937-9145) is published by the American Association for the Advancement of Science, 1200 NewScience Signaling
Science. No claim to original U.S. Government Works.Copyright © 2017 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of
on February 14, 2020
http://stke.sciencemag.org/
Dow
nloaded from