atkins & de paula: elements of physical chemistry: 5e chapter 11: chemical kinetics: accounting...
TRANSCRIPT

Atkins & de Paula: Elements of Physical Chemistry:
5e
Chapter 11: Chemical Kinetics:
Accounting for the Rate Laws

End of chapter 11 assignments
Discussion questions:• 2, 5
Exercises:• 5, 13, 14, 18, 25
Use Excel if data needs to be graphed

Time for me to “nag” Time for me to “nag” you!you!
• Did you:– Read the chapter?– Work through the example problems?– Connect to the publisher’s website &
access the “Living Graphs”?– Examine the “Checklist of Key Ideas”?– Work assigned end-of-chapter
exercises?• Review terms & concepts that
you should remember from previous chapters or courses

Reaction schemesReaction schemes
• All chemical rxns proceed toward a state of equilibrium
• The reverse rxn becomes increasingly important as the rxn proceeds
• Many rxns proceed through a series of intermediates
• These intermediates may be important

Rate LawsRate Laws
• Rate laws are always determined experimentally.
• Reaction order is always defined in terms of reactant (not product) concentrations.
14.214.2

Rate LawsRate Laws
• The order of a reactant is not related to the stoichiometric coefficient of the reactant in the balanced chemical equation.
• The stoichiometric coefficient of the reactant in the balanced chemical equation is the exponent in the K expression.
14.2
Kc = [C]c[D]d
[A]a[B]baA + bB cC + dD
F2 (g) + 2ClO2 (g) 2FClO2 (g)
rate = k [F2][ClO2]1

Digression: Rate law and KDigression: Rate law and Keqeq
• Keq is concerned with the initial and final conditions of the rxn (overall)
• Rate law, order, “kinetics” are concerned with everything in between
• Let me show you what IUPAC says about the rate law and Keq

Rate law and KRate law and Keqeq
The equilibrium law or the equilibrium constant are in the first instance derived from the thermodynamics of the reaction, the value of the equilibrium constant Kc depending entirely upon the characteristics of the reactants and these of the reaction products.
--continued

Rate law and KRate law and Keqeq
Rate of reaction and reaction mechanism are kinetic aspects of the reaction. They have nothing to do with the expression for Kc or with the value of Kc. It would be simplistic and imprudent to assume all sorts of unrealistic reaction equations to obtain an expression for Kc.

Rate law and KRate law and Keqeq
• Quoted from IUPAC’s web site• http://www.iupac.org/• Specifically: http://www.iupac.org/didac/Didac
%20Eng/Didac02/Content/E06%20-%20E07%20-%20E08.htm

Order & molecularityOrder & molecularity
The overall reaction order is the sum of the powers of the concentrations of all of the substances appearing in the experimental rate law for the reaction; hence, it is the sum of the individual orders (exponents) associated with a given reactant (or product). Reaction order is an experimentally determined, not theoretical, quantity, although theory may attempt to predict it.
Quoted from another book by Atkins…

Order & molecularityOrder & molecularity
Molecularity is the number of reactant molecules partici-pating in an elementary reaction. This concept has meaning only for an elementary reaction, but reaction order applies to any reaction. In general, reaction order bears no necessary relation to the stoichiometry of the reaction, with the exception of elementary reactions, where the order of the reaction corresponds to the number of molecules participating in the reaction; that is, to its molecularity. Thus for an elementary reaction, overall order and molecularity are the same and are determined by the stoichiometry.
Quoted from another book by Atkins…
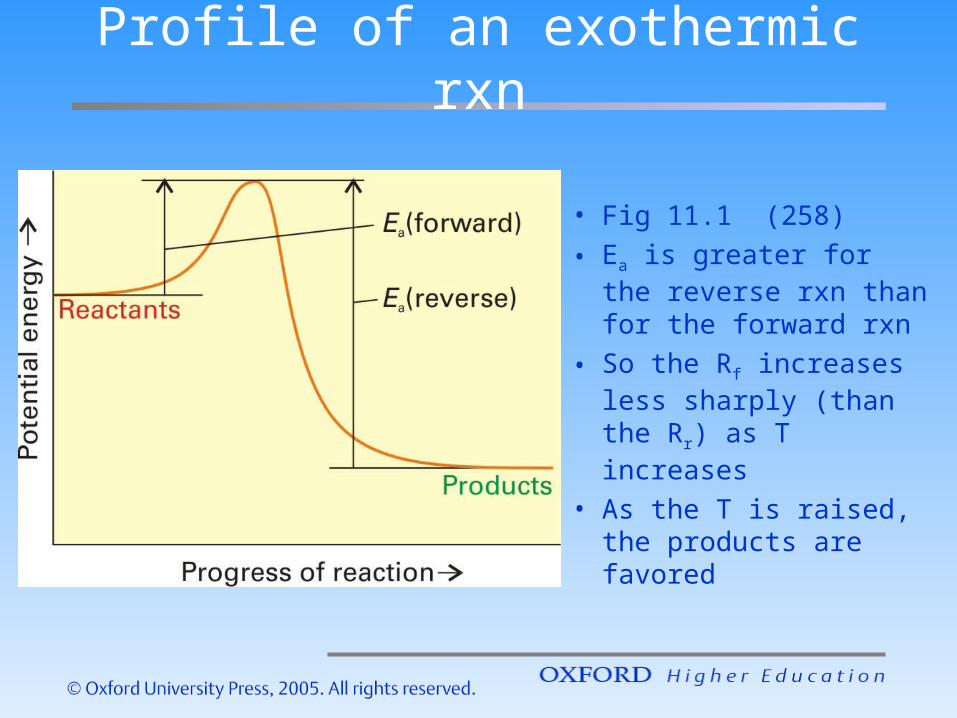
Profile of an exothermic rxn
• Fig 11.1 (258)
• Ea is greater for the reverse rxn than for the forward rxn
• So the Rf increases less sharply (than the Rr) as T increases
• As the T is raised, the products are favored

The approach to equilibriumThe approach to equilibrium
• Derivation 11.1 (259)• You should work through this
derivation
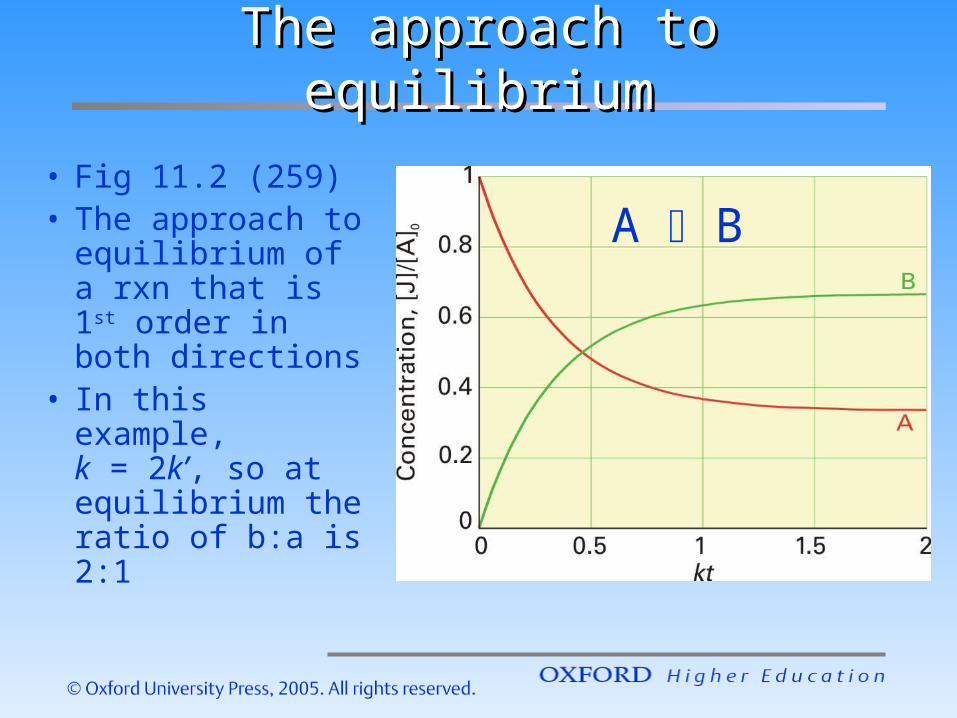
The approach to equilibriumThe approach to equilibrium
• Fig 11.2 (259)• The approach
to equilibrium of a rxn that is 1st order in both directions
• In this example, k = 2k’, so at equilibrium the ratio of b:a is 2:1
A B

Relaxation methodsRelaxation methods
• Some externally applied influence shifts the rxn equilibrium abruptly– Temperature jump– Pressure jump
• The adjustment back to equilibrium is called “relaxation”
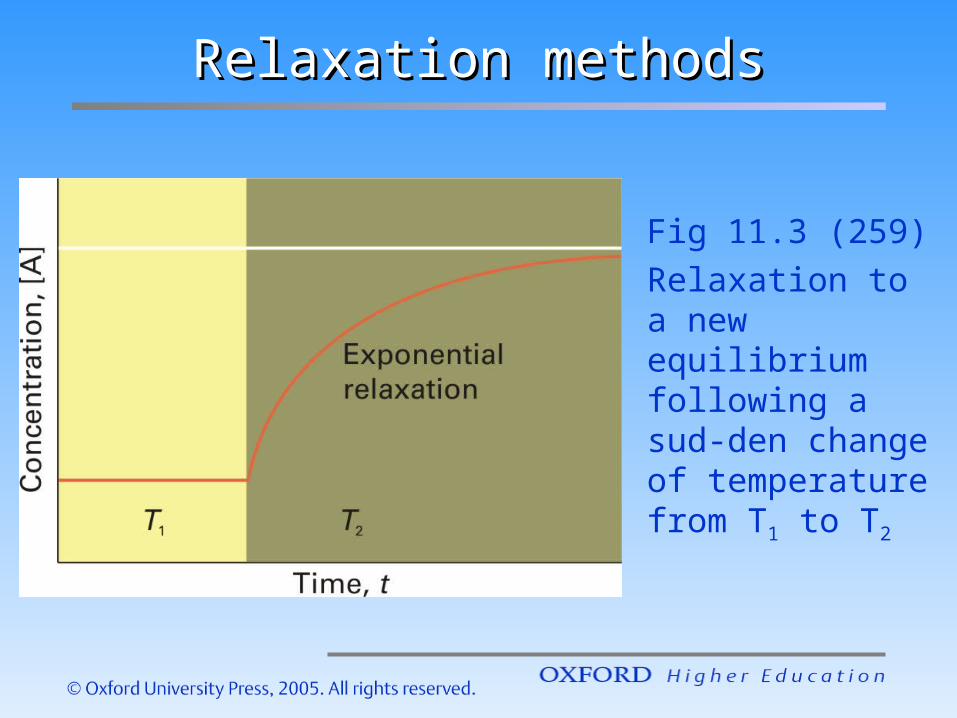
Relaxation methodsRelaxation methods
Fig 11.3 (259)Relaxation to a new equilibrium following a sud-den change of temperature from T1 to T2

Relaxation methodsRelaxation methods
• When a sudden temperature jump shifts the equilibrium of a 1st order rxn:
• x = x0e–t/ and = ka + kb
• x is the shift in equilibrium at the new t• x0 is the shift in equilibrium
immediately after the temperature change
is the relaxation time• t is the time (s)
1

Consecutive reactionsConsecutive reactions
Common examples are radioactive decay chains and biochemical rxn sequences (e.g., gylcoysis, Krebs cycle, etc)

The U
raniu
m D
eca
y S
eri
es
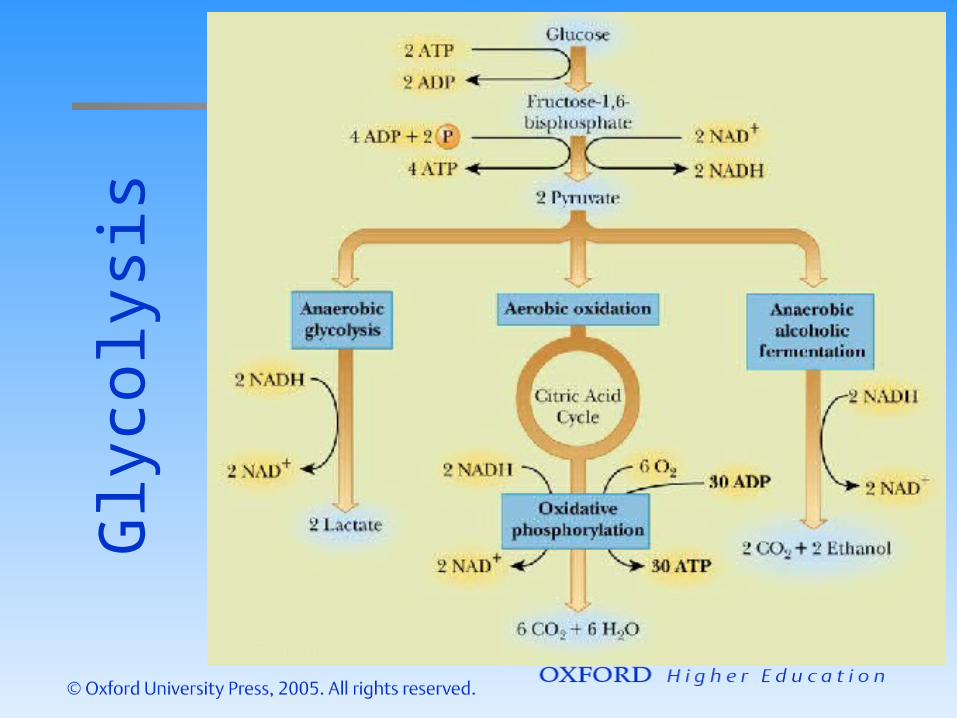
Gly
coly
sis
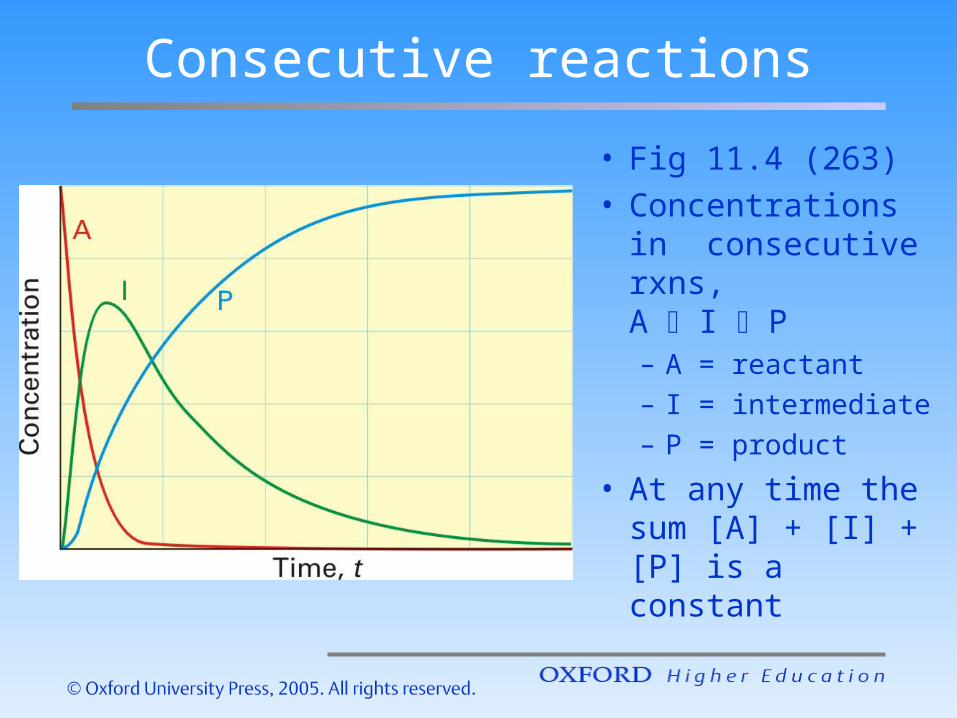
Consecutive reactions
• Fig 11.4 (263)• Concentrations
in consecutive rxns, A I P– A = reactant– I = intermediate– P = product
• At any time the sum [A] + [I] + [P] is a constant

Elementary reactionsElementary reactions
• Many reactions occur as a series of steps called “elementary reactions”
• Molecularity is the number of molecules coming together to react– Unimolecular– Bimolecular – What’s next?
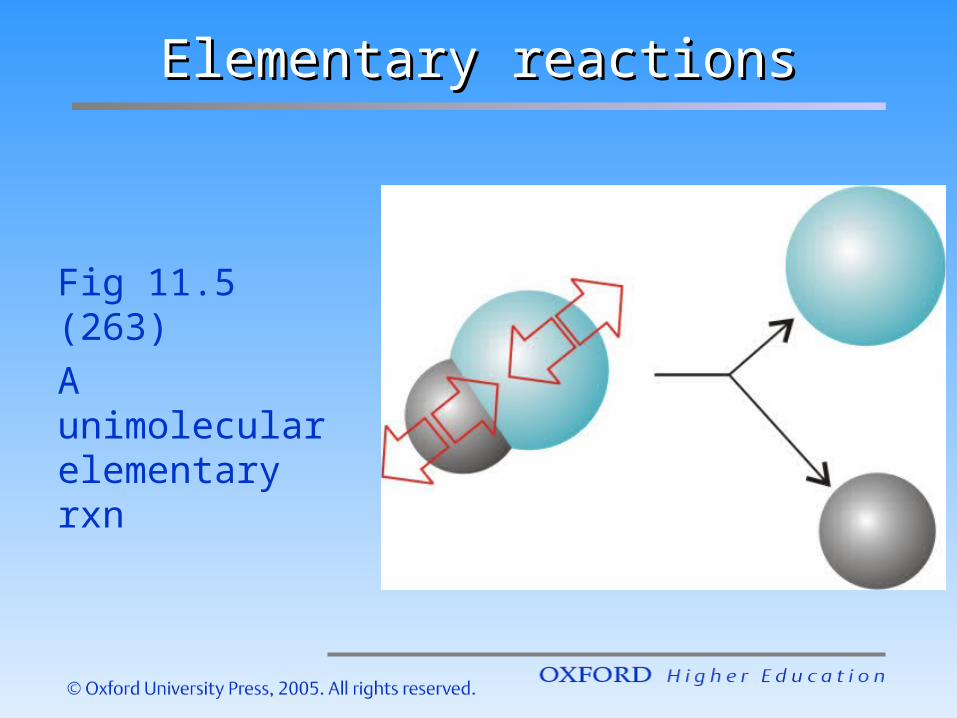
Elementary reactionsElementary reactions
Fig 11.5 (263)A unimolecular elementary rxn
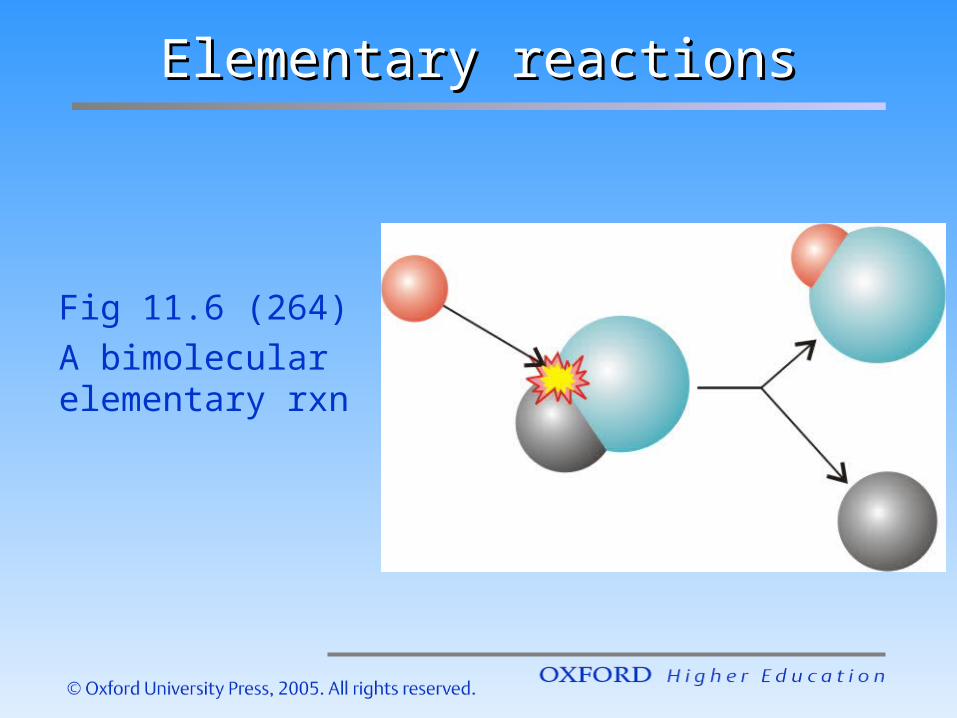
Elementary reactionsElementary reactions
Fig 11.6 (264)A bimolecular elementary rxn

The formulation of rate lawsThe formulation of rate laws
• Assuming that a chemical rxn is a series of elementary rxns…
• One of these elementary rxns is the rate determining step
• An acceptable rate law for an overall rxn is expressed only in terms of the concentrations of the species in the overall rxn, not in terms of an intermediate
(264)

The formulation of rate lawsThe formulation of rate laws
• Consider this rxn:
• 2 NO(g) + O2(g) d 2 NO2(g)
• Experimentally: rate = k[NO]2[O2] (which is 3rd order overall)
• How can we explain this? Is it a termolecular rxn (2 NOs and 1 O2)?
• Let’s see…
(264)

Proposed mechanism: 2 NO(g) + OProposed mechanism: 2 NO(g) + O22(g) (g) dd 2 2 NONO22
Step 1. NO + NO d N2O2
Rate of formation of N2O2 = ka[NO]2
Step 2. N2O2 d NO + NO
Rate of decomposition of N2O2 = k’a[N2O2]
Step 3. N2O2 + O2 d NO2 + NO2
Rate of consumption of N2O2 = 2kb[N2O2][O2]
Thus the net rate of formation of N2O2 is =
ka[NO]2 – k’a[N2O2] – 2kb[N2O2][O2]Does text have an error in this last eqn?

Proposed mechanism: 2 NO(g) + OProposed mechanism: 2 NO(g) + O22(g) (g) dd 2 2 NONO22
Net rate of formation of N2O2 =
ka[NO]2 – k’a[N2O2] – 2kb[N2O2][O2]
• Solving this eqn involves solving a very difficult differential eqn and gives a complex expression
• This drives us to consider…“The Steady-State Approximation”
(265)

The steady-state The steady-state approximationapproximation
• Our assumption: the concentrations of all intermediates remain constant and small throughout the rxn (except at the very beginning and at the very end)
• So, the net rate of formation of N2O2 = 0
• So, ka[NO]2 – k’a[N2O2] – 2kb[N2O2][O2] = 0
• Solving for [N2O2]
• Solving for [NO2] gives 11.10, which accounts for overall 3rd order rxn (265)

The rate-determining stepThe rate-determining step
• If step 3 is much faster than steps 1 and 2, (this is the case if the [O2] is high) then k’a can be ignored
• Now the rate law simplifies to 11.12
• [NO2] = = 2ka[NO]2
• Now the rxn is 2nd order and [O2] does not appear in the rate law
kb [NO]2[O2]
2kakb[NO]2[O2]

The rate-determining stepThe rate-determining step
The rate determining step (RDS) must have two characteristics:
1. It must be the slowest step, and2. It must be a crucial part of the
path from reactants to products
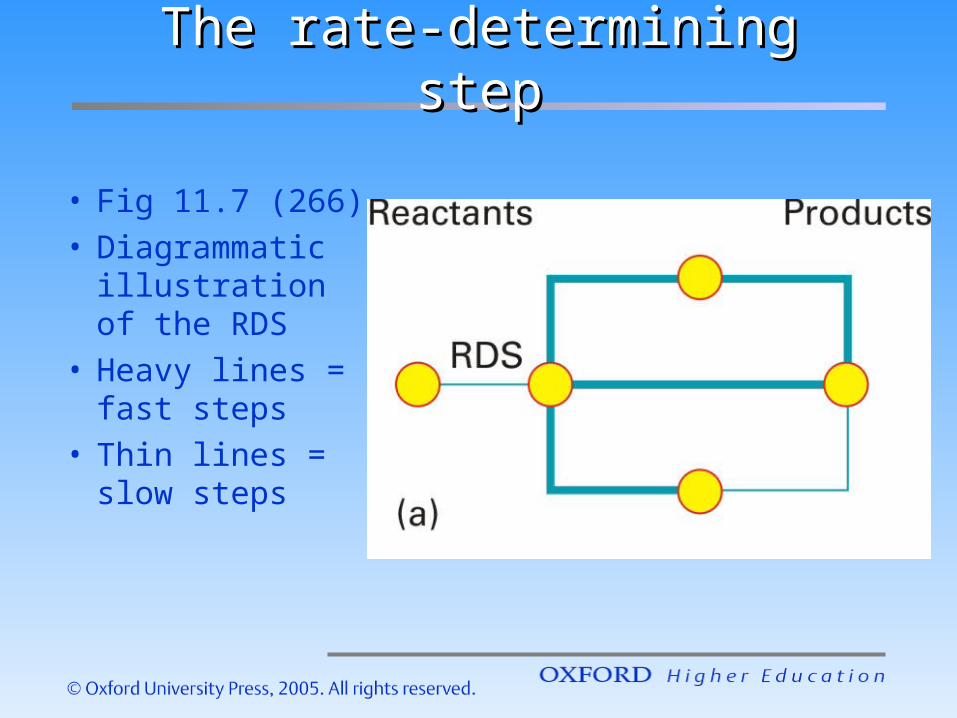
The rate-determining stepThe rate-determining step
• Fig 11.7 (266)• Diagrammati
c illustration of the RDS
• Heavy lines = fast steps
• Thin lines = slow steps
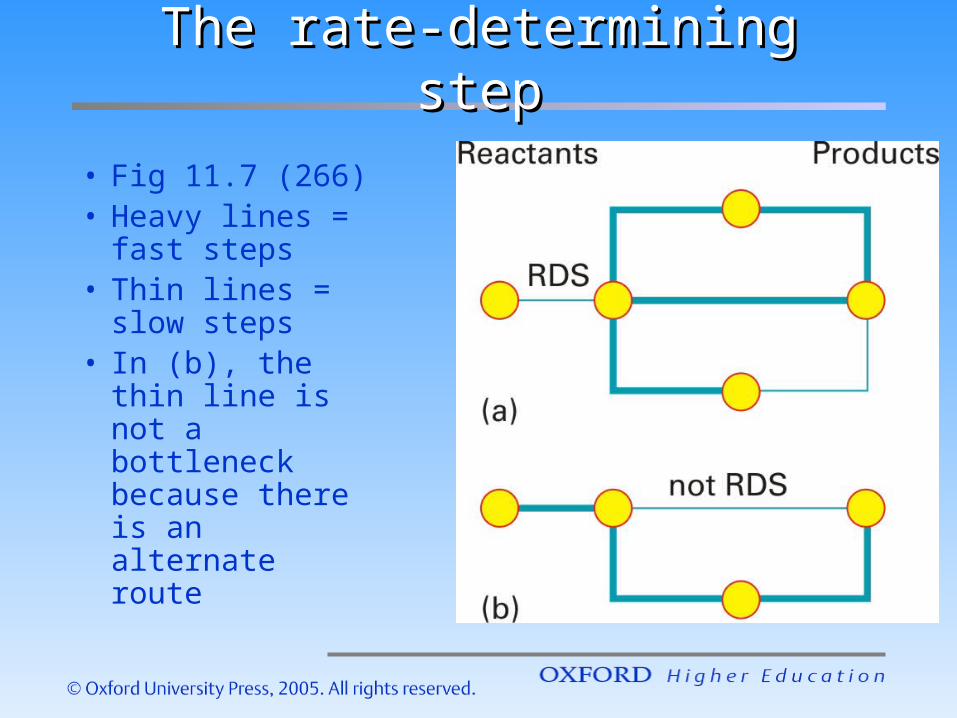
The rate-determining stepThe rate-determining step
• Fig 11.7 (266)• Heavy lines =
fast steps• Thin lines =
slow steps• In (b), the thin
line is not a bottleneck because there is an alternate route

Kinetic controlKinetic control
Consider these competing rxns:
• A + B d P1
Rate of formation of P1 = k1[A][B]
• A + B d P2
Rate of formation of P2 = k2[A][B]
=[P2]
[P1]
k2
k1
Represents kinetic control

Kinetic controlKinetic control
If a rxn is allowed to reach equilibrium, then thermodynamics (rather than kinetic considerations) determines the proportions of products, and the ratio of concentrations is controlled by standard Gibbs energies of reactants and products
(267)
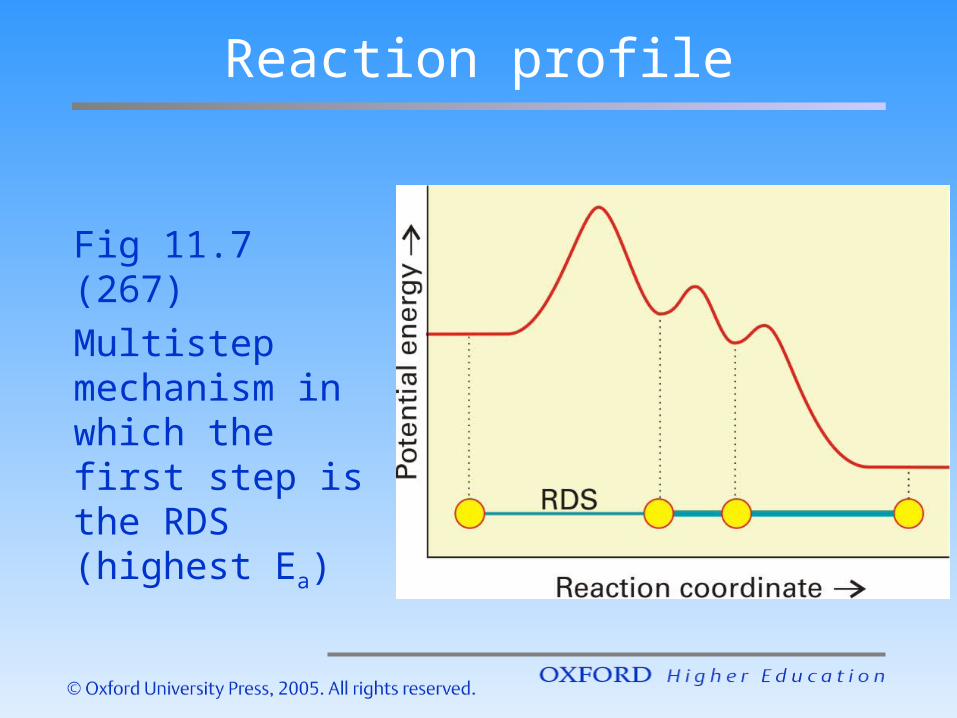
Reaction profile
Fig 11.7 (267)Multistep mechanism in which the first step is the RDS (highest Ea)

Unimolecular reactionsUnimolecular reactions
• Many gas-phase rxns proceed by two gas molecules colliding but are 1st order
• These collisions are bimolecular events and the rxn should be 2nd order
• Explain! • The Lindemann mechanism posits that
the RDS is unimolecular
(267f)

Activation control & diffusion Activation control & diffusion controlcontrol
Now we consider reactions in
solution

Activation control & diffusion Activation control & diffusion controlcontrol
• kd is the rate constant (of diffusion)
• Diffusion-controlled limit—the rate of the rxn is controlled by the rate at which reactants diffuse (kd)
• Activation-controlled limit—the rate of the rxn depends on the rate at which energy accumulates in the “encounter pair” (ka)

Activation control & diffusion Activation control & diffusion controlcontrol
• Now read next-to-last paragraph, p.269
• “A lesson to learn from this analysis is the concept of the rate-determining step is rather subtle. Thus, in the…”

Activation control & diffusion Activation control & diffusion controlcontrol
• Vis-a-vis the rate of diffusion in a liquid, the rate constant (kd) is related to the coefficient of viscosity,
• How are kd and related?
kd =
is the Greek lowercase letter eta
8RT3

DiffusionDiffusion
This section relates directly to the lab experiment you’ve worked on this semester
(270)

DiffusionDiffusion
• Terms: diffusion, random walk, flux (J)• Rate of diffusion Concentration gradient
• J =
• J = – D Concentration gradient (11.18b)
• D is the diffusion coefficient (area/time)
• This is Fick’s first law of diffusion
Number of particles passing through a window
Area of window Time interval
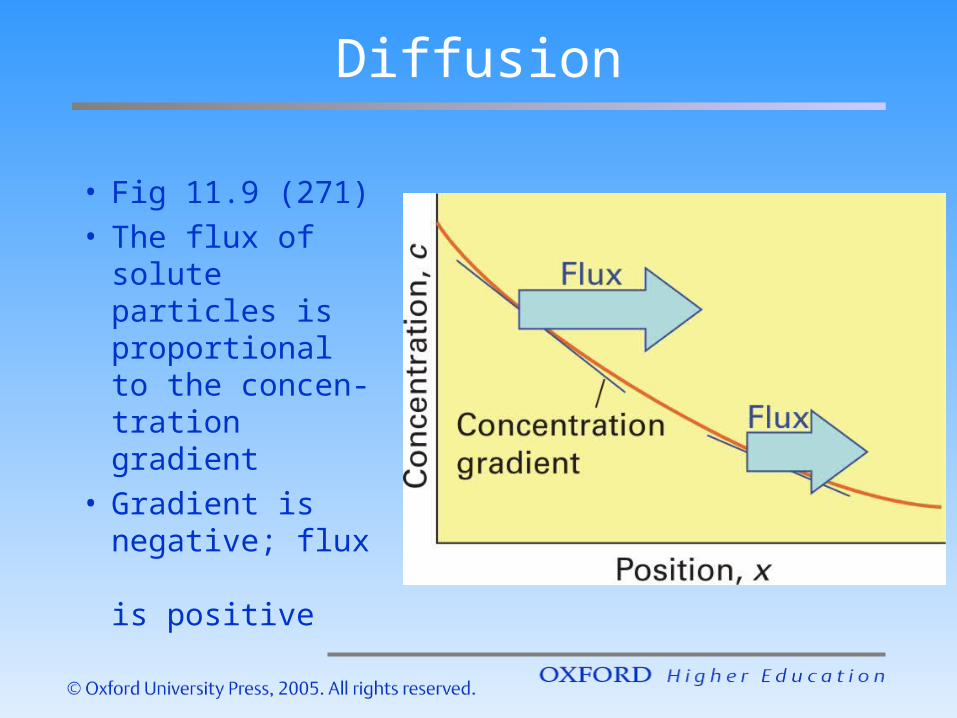
Diffusion
• Fig 11.9 (271)• The flux of
solute particles is proportional to the concen-tration gradient
• Gradient is negative; flux is positive
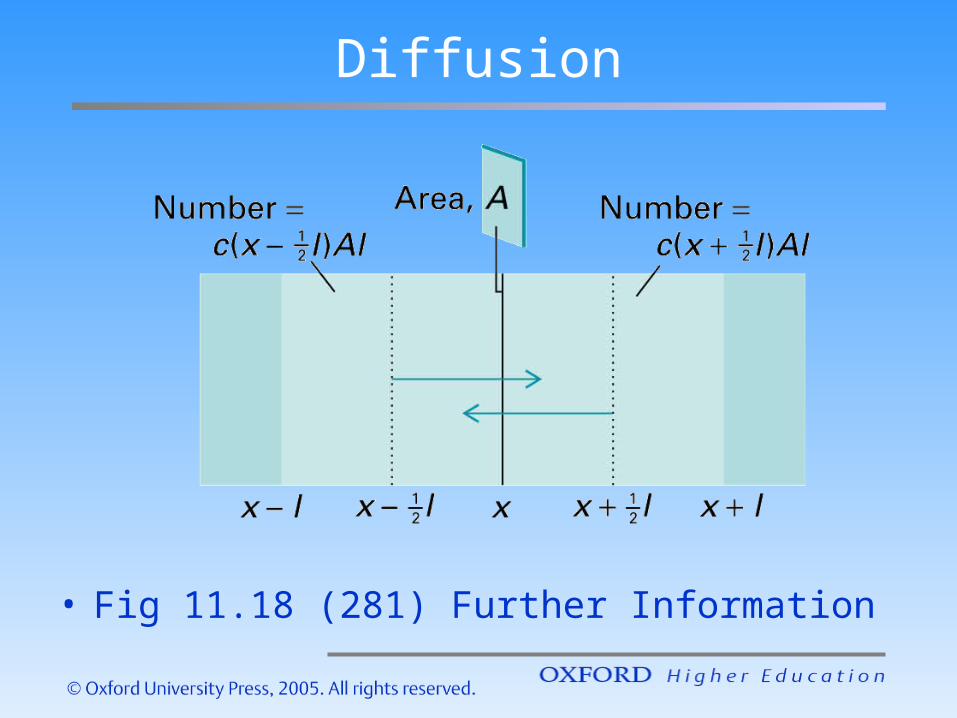
Diffusion
• Fig 11.18 (281) Further Information

DiffusionDiffusion
• Diffusion is often aided by convection • The diffusion eqn below lets us predict
the rate at which the concentration of a solute changes in a “non-uniform” solution
• Fick’s second law:Rate of change of concentration in a region =
D curvature of the concentration in the region (11.19)
• So, what’s a “wrinkle”? 271
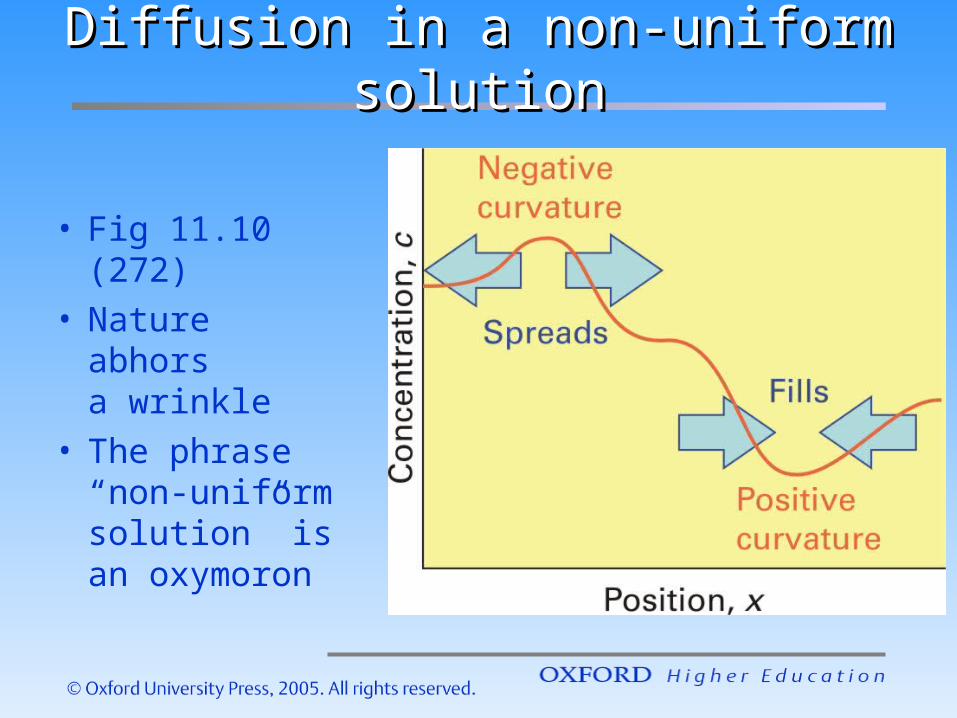
Diffusion in a non-uniform solutionDiffusion in a non-uniform solution
• Fig 11.10 (272)
• Nature abhors
a wrinkle• The phrase
“non-uniform solution” is an oxymoron

Table 11.1 Diffusion coefficients at 25°C, D/(10-9 m2 s-1)
(270)
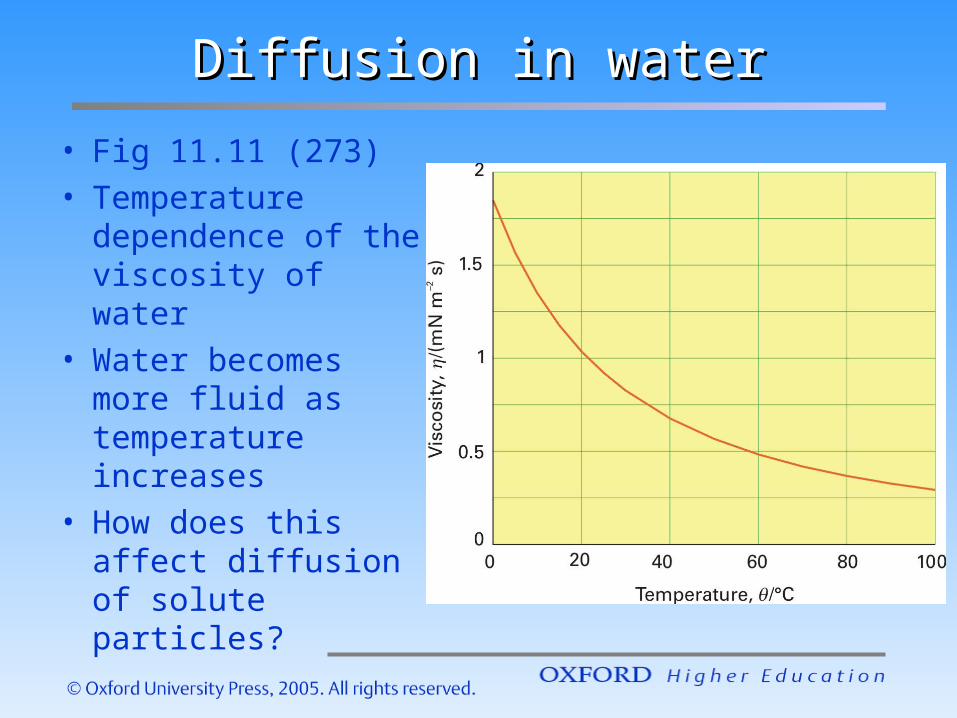
Diffusion in waterDiffusion in water
• Fig 11.11 (273)• Temperature
dependence of the viscosity of water
• Water becomes more fluid as temperature increases
• How does this affect diffusion of solute particles?

CatalysisCatalysis
• Homogeneous catalysis, catalyst and the rxn mixture are in the same phases (e.g., gas, solution, etc.)
• Heterogeneous catalysis, catalyst and the rxn mixture are in different phases
• (e.g., hydrogenation on a Pt surface) 273273
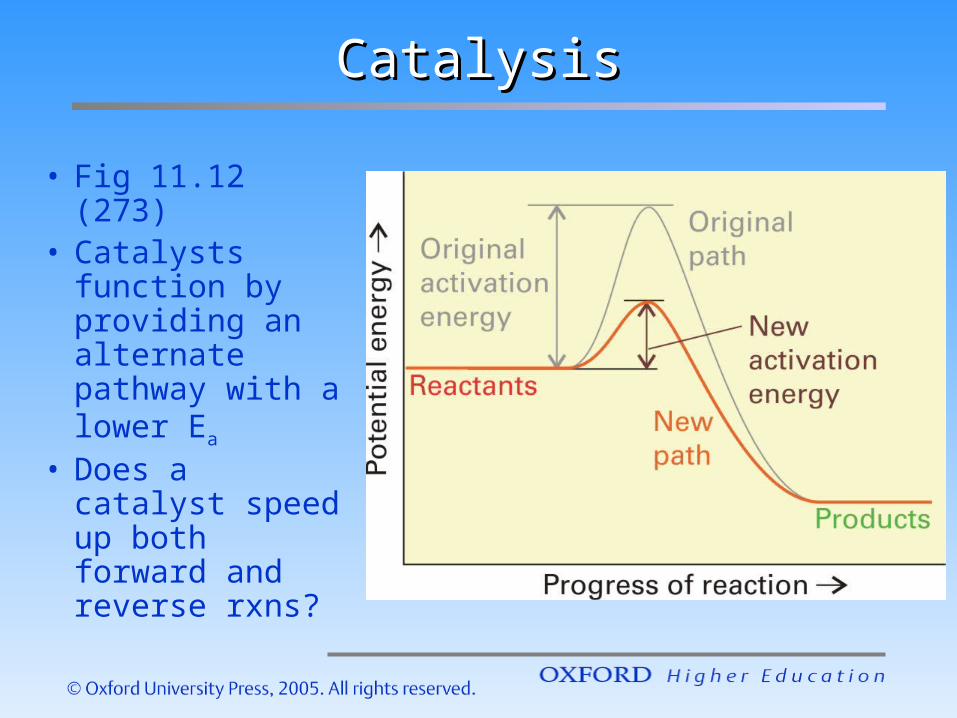
CatalysisCatalysis
• Fig 11.12 (273)
• Catalysts function by providing an alternate pathway with a lower Ea
• Does a catalyst speed up both forward and reverse rxns?
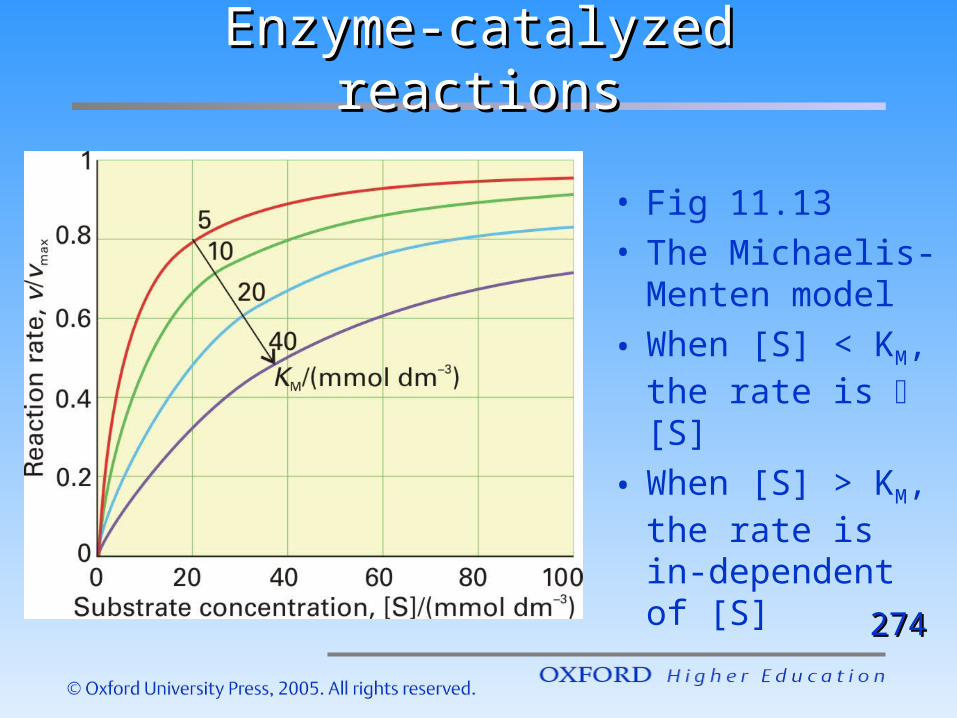
Enzyme-catalyzed reactionsEnzyme-catalyzed reactions
• Fig 11.13• The Michaelis-
Menten model
• When [S] < KM, the rate is [S]
• When [S] > KM, the rate is in-dependent of [S]
274274
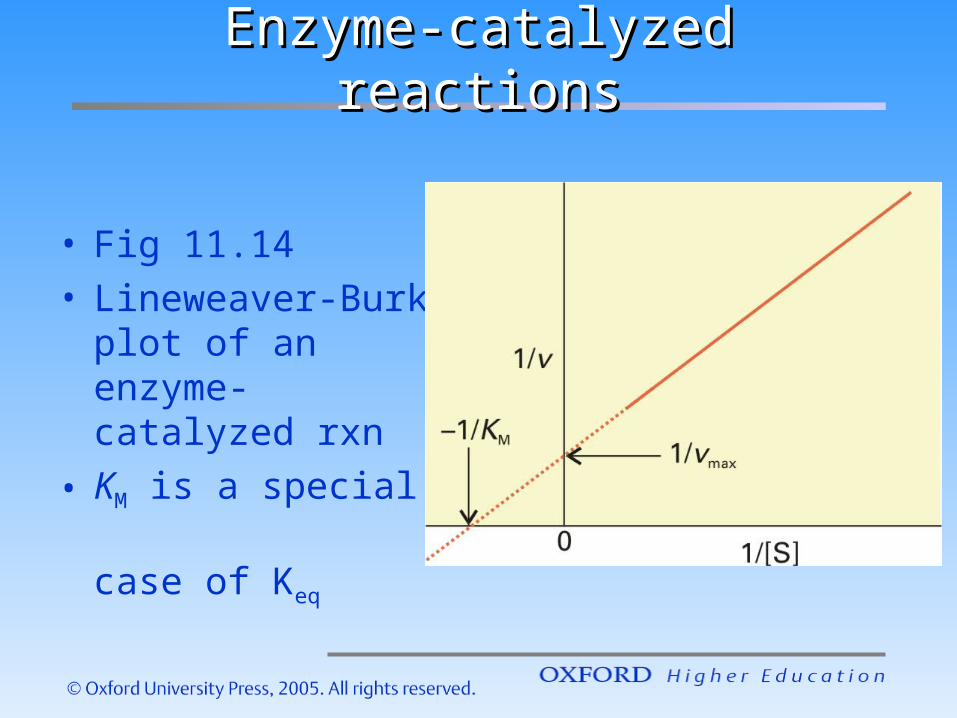
Enzyme-catalyzed reactionsEnzyme-catalyzed reactions
• Fig 11.14• Lineweaver-Burk
plot of an enzyme-catalyzed rxn
• KM is a special case of Keq
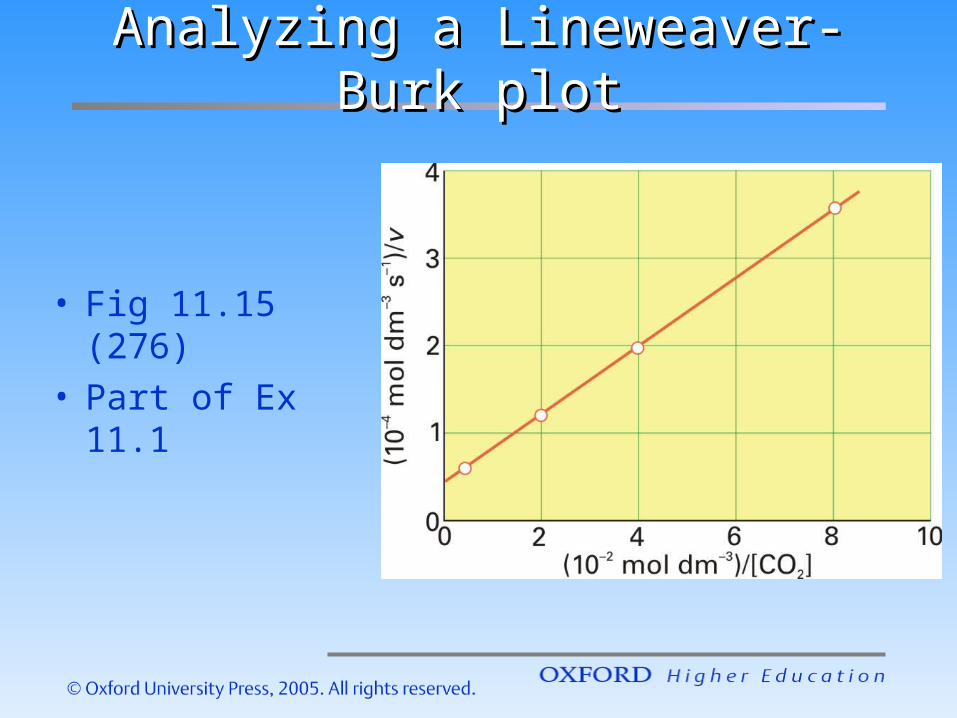
Analyzing a Lineweaver-Burk Analyzing a Lineweaver-Burk plotplot
• Fig 11.15 (276)
• Part of Ex 11.1

EnzymesEnzymes
• Enzymes are biological catalysts
• Virtually all enzymes are proteins (polyamino acids)
• A few are RNAs• Since you are not biology
majors…

EnzymesEnzymes
• Fig 11.16• In
competitive inhibition, the substrate and inhibitor compete for the active site.

EnzymesEnzymes
Fig 11.17In one type of non-competitive inhibition, the inhibitor attaches to the enzyme, but not at the active site; this changes the 3D shape of the enzyme, rendering it inactive

The structure of chain reactionsThe structure of chain reactions
• An intermediate in one step generates a reactive intermediate in a later step, which generates another reactive intermediate in a still later step, which generates….
• These are called “chain carriers”

The structure of chain reactionsThe structure of chain reactions
• Initiation step• Propagation step• Branching step• Retardation step• Inhibition step• Termination step

The rate laws of chain reactionsThe rate laws of chain reactions
• Often free radical rxns• Often have complicated rate laws

Key Key ConceptConcept
ss

Key Key ConceptConcept
ss

Key Key ConceptConcept
ss

The EndThe End…of this chapter…”
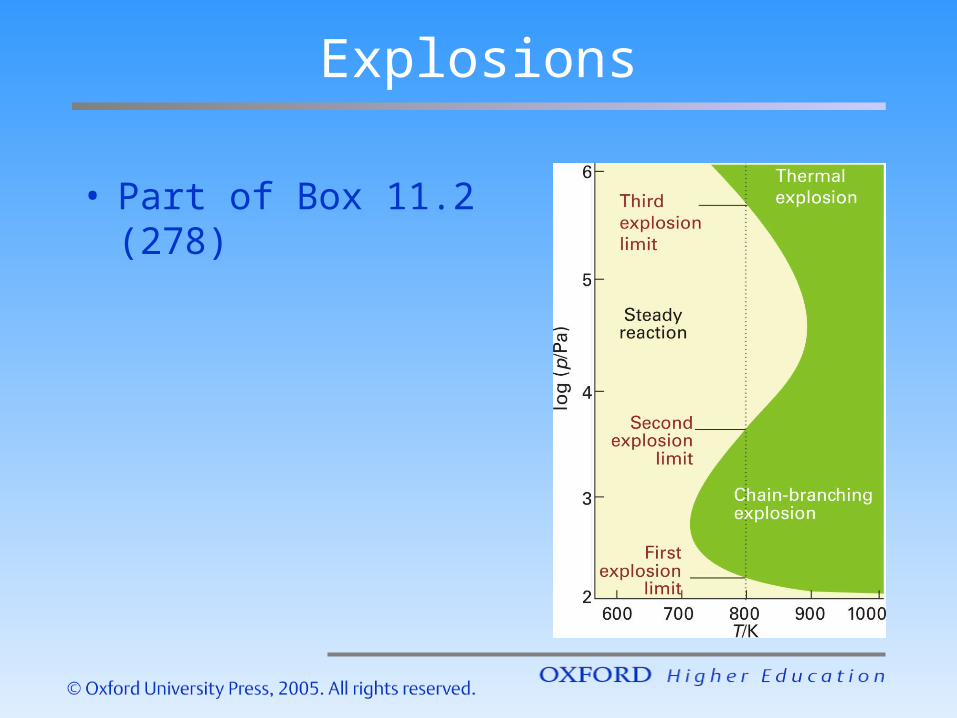
Explosions
• Part of Box 11.2 (278)

aA + bB cC + dD
Kc = [C]c[D]d
[A]a[B]b
Law of Mass Action
15.1
ratef = kf [A]a[B]b rater = kr [C]c[D]d
Let’s derive the equilibrium constant from first principles
Now define equilibrium: at equilibrium, ratef = rater
If ratef = rater then kf [A]a[B]b = kr [C]c[D]d
Collect constants on the left and variables on the right:
kf
kr
[C]c[D]d
[A]a[B]b= OR
ratef [A]a[B]b rater [C]c[D]d