aryl hydrocarbon receptor agonists induce microrna...
TRANSCRIPT

Therapeutic Discovery
Aryl Hydrocarbon Receptor Agonists Induce MicroRNA-335Expression and Inhibit LungMetastasis of EstrogenReceptorNegative Breast Cancer Cells
Shu Zhang1, KyoungHyun Kim1, Un Ho Jin1, Catherine Pfent2, Huojun Cao1, Brad Amendt1, Xinyi Liu1,Heather Wilson-Robles3, and Stephen Safe1,4
AbstractThe aryl hydrocarbon receptor (AHR) was initially identified as a receptor that bound 2,3,7,8-tetrachlor-
odibenzo-p-dioxin (TCDD) and related environmental toxicants; however, there is increasing evidence that the
AHR is an important new drug target for treating multiple diseases including breast cancer. Treatment of
estrogen receptor (ER)-negative MDA-MB-231 and BT474 breast cancer cells with TCDD or the selective AHR
modulator 6-methyl-1,3,-trichlorodibenzofuran (MCDF) inhibited breast cancer cell invasion in a Boyden
chamber assay. These results were similar to those previously reported for the antimetastic microRNA-335
(miR-335). Both TCDD and MCDF induced miR-335 in MDA-MB-231 and BT474 cells and this was accom-
panied by downregulation of SOX4, a miR-335-regulated (inhibited) gene. The effects of TCDD andMCDF on
miR-335 and SOX4 expression and interactions of miR-335 with the 30-UTR target sequence in the SOX4 gene
were all inhibited in cells transfected with an oligonucleotide (iAHR) that knocks down the AHR, thus
confirming AHR-miR-335 interactions. MCDF (40 mg/kg/d) also inhibited lung metastasis of MDA-MB-231
cells in a tail vein injectionmodel, showing that the AHR is a potential new target for treating patientswith ER-
negative breast cancer, a disease where treatment options and their effectiveness are limited.Mol Cancer Ther;
11(1); 108–18. �2011 AACR.
Introduction
The aryl hydrocarbon receptor (AHR) was initiallyidentified as an intracellular protein that boundwith highaffinity to the environmental toxicant 2,3,7,8-tetrachloro-dibenzo-p-dioxin (TCDD; ref. 1). It was also shown thattheAHRboundother structurally related aromatic hydro-carbons and several carcinogenic polycyclic aromatichydrocarbons. Studies with AHR�/� mice confirmed arole for this receptor in mediating the toxic and carcino-genic effects of these compounds (2–4). Subsequent stud-ies showed that the AHR binds structurally diverse com-pounds including aromatic compounds, flavonoids, andmany other phytochemicals, various drugs includingsulindac and omeprazole, and endogenous biochemicals
such as bilirubin, arachadonic acid derivatives, and indi-goids (5–12).
The identification of AHR ligands with well-describedhealth-promoting or beneficial pharmaceutical effects hasalso spurred research on development of drugs that targetthe AHR for treatment of specific tumors, immune dis-orders, and inflammatory disease and also for enhancedproduction of hematopoietic stem cells (13–21). 6-Methyl-1,3,8-trichlorodibenzofuran (MCDF) was initially charac-terized as anAHRantagonist that blockedTCDD-inducedtoxicities in rodent models (22–25); however, MCDF didnot inhibit TCDD-induced antiestrogenic activity inthe rodent uterus, human breast cancer cells, or tumors(26–29). MCDF exhibited AHR agonist activity for theseresponses and, in carcinogen-induced female Sprague–Dawley rats, MCDF alone and in combination withtamoxifen was a potent inhibitor of mammary tumorgrowth but did not inhibit tamoxifen-induced bonelengthening (29). In subsequent studies, we showed thatMCDF, TCDD, and related halogenated aromatics inhib-ited growth of several estrogen receptor (ER)-negativebreast cancer cell lines (30), suggesting a potential role forselective AHR modulators (SAhRMS) such as MCDF fortreatment of patients with ER-negative tumors for whichthe prognosis is poor and treatment options are limitedprimarily to cytotoxic drug therapy (31).
AHR agonists also inhibit breast cancer cell invasive-ness and colony formation and promote breast cancer cell
Authors' Affiliations: 1Institute of Biosciences and Technology, TexasA&M Health Science Center, Houston; Departments of 2Veterinary Patho-biology, 3Small Animal Clinical Sciences, and 4Veterinary Physiology andPharmacology, Texas A&M University, College Station, Texas
Note: Supplementary data for this article are available at Molecular CancerTherapeutics Online (http://mct.aacrjournals.org/).
Corresponding Author: Stephen Safe, Department of Veterinary Physi-ology and Pharmacology, Texas A&M University, 4466 TAMU, Vet. Res.Bldg. 410,CollegeStation, TX77843.Phone: 979-845-5988; Fax: 979-862-4929; E-mail: [email protected]
doi: 10.1158/1535-7163.MCT-11-0548
�2011 American Association for Cancer Research.
MolecularCancer
Therapeutics
Mol Cancer Ther; 11(1) January 2012108
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548

differentiation (32), andwe hypothesized that thismay bedue, in part, to AHR-microRNA (miR) interactions. Usinga miR array as a screening tool, we identified microRNA-335 (miR-335) as anAh-responsivemiR inducedbyMCDFand TCDD in ER-negative breast cancer cells. MiR-335 isan antimetasticmiR, and overexpression ofmiR-335 in theMDA-MB-231 LM2 cell variant inhibits breast cancer cellinvasion and metastasis in vivo (33). This study reportsthat AHR agonistsMCDF and TCDD inducemiR-335 anddecrease SOX-4 expression in breast cancer cells, andMCDF inhibits lung metastasis of these cells in an in vivotail vein injection model. These results show the potentialclinical importance of the AHR as a drug target forinhibiting breast cancer metastasis and indicate a role forAHR-miR-335 interactions in mediating inhibition oftumor metastasis.
Materials and Methods
Cell lines and cell cultureTheMDA-MB-231 and BT474 cell were purchased from
the American Type Culture Collection (ATCC) and wereauthenticated as indicated below.
MDA-MB-231 from ATCC:
http://www.atcc.org/ATCCAdvancedCatalogSearch/ProductDetails/tabid/452/Default.aspx?ATCCNum¼HTB-26&Template¼cellBiology
BT474 from ATCC:
http://www.atcc.org/ATCCAdvancedCatalogSearch/ProductDetails/tabid/452/Default.aspx?ATCCNum¼HTB-20&Template¼cellBiology
Cells were initially grown and multiple aliquots werefrozen and stored at �80�C for future use. Cells werepurchased more than 6 months ago and were not furthertested or authenticated by the authors. MDA-MB-231 andBT474 cells were maintained in Dulbecco’s ModifiedEagle’sMedium supplementedwith 10% FBS and 10ml/L100� antibiotic antimycotic solution (Sigma-Aldrich).Cells were maintained at 37�C in the presence of 5%CO2 and the solvent [dimethyl sulfoxide (DMSO)] usedin the experiments was 0.1% or more.
Antibodies and reagentsCYP1A1 and b-actin antibodies were purchased from
Santa Cruz Biotechnology. All the short interfering RNAswere prepared by Dharmacon Research. Reporter lysisbuffer and luciferase reagent for luciferase studies werepurchased fromPromega.b-Galactosidase (b-Gal) reagentwas obtained from Tropix. Lipofectamine reagents weresupplied by Invitrogen.Western Lightning chemilumine-scence reagents were from Perkin-Elmer Life Sciences.SOX4 30-UTR reporter constructwas generously providedby Dr. J. Massagu�e (Memorial Sloan-Kettering CancerCenter, New York, NY). MCDF and TCDD were synthe-
sized in this laboratory to more than 98% purity as deter-mined by gas chromatographic analysis.
Scratch assay and Boyden chamber assayCellswere seeded in 6-well plates and allowed to attach
for 16 hours. The medium was then changed to DMEM/F-12 medium containing 2.5% charcoal-stripped FBS, andthen treated with either vehicle (DMSO) or the com-pounds for 24 hours before the scratch was made. Ascratch through the central axis of the plate was gentlymade using a sterile pipette tip. Cells were 70% confluentwhen the scratch was made. Cells were then washed andtreated with either vehicle (DMSO) or the compounds.Migration of the cells into the scratch was observed at 9preselected points (3 points per well) at 0, 8, and 16 hours.Results of this study were obtained at a 16-hour timepoint. A 48-well micro-chemotaxis chamber (CorningIncorporated) was used for Boyden chamber assay.To evaluate invasion, the 8-mm pore filter separating the2 wells was coated with Matrigel (Becton-DickinsonLabware; 50 mg/filter). Either vehicle (DMSO) or thecompounds were dissolved in culture medium andplaced in both the upper and lower well, whereas5 � 103 cells were added to each upper well. After 24hours, the filter was removed and fixed with methanol.Cell invasion was evaluated by counting spread cellsadhering to the lower filter surface.
Westernblotanalysis andquantitative real-timePCRCells (2 � 105) were plated in 6-well plates in DMEM/
F-12 (Sigma-Aldrich) media containing 2.5% charcoal-stripped FBS for 16 hours and then treated with eithervehicle (DMSO) or the compounds. Cellular lysates andtheir subsequent separation by electrophoresis was car-ried out as previously described using b-actin as a loadingcontrol (30). Total RNAwas extracted, real-time PCRwascarried out as previously described and normalized toTBP (34). ThePCRprofilewas as follows: one cycle of 95�Cfor 10 minutes, then 40 cycles of 95�C for 15 seconds and60�C for 1 minute. The comparative CTmethod was usedfor relative quantitation of samples. The following pri-mers for CYP1A1, SOX4, TBP, and PTPRN2 were synthe-sized by Integrated DNA Technologies: CYP1A1, for-ward, 50-CTT CCG ACA CTC TTC CTT CG-30; CYP1A1,reverse, 50-GGT TGA TCT GCC ACG GTT T-30; SOX4,forward, 50-CAA ACC AAC AAT GCC GAG AAC-30;SOX4, reverse, 50-CTC TTT TTC TGC GCC GGT TTG-30; TBP, forward, 50-TGC ACA GGA GCC AAG AGTGAA-30; TBP, reverse, 50-CAC ATC ACA GCT CCC CACCA-30; PTPRN2, forward, 50-TCT GGC CTC ATC TACTGC CT-30; TBP, reverse, 50-CTT CAG GTG GTC CTCCAT GT-30.
Chromatin immunoprecipitation assayThe chromatin immunoprecipitation (ChIP) assay was
done using ChiP-IT Express Magnetic Chromatin Immu-noprecipitation Kit (Active Motif) according to manufac-turer’sprotocol.BT474orMDA-MB-231cells (5�106 cells)
AHR-Dependent Inhibition of Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Ther; 11(1) January 2012 109
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548

were treatedwith DMSO, TCDD, orMCDF for 1 hour andafter several steps including reversing DNA cross-links,DNAwasprepared byproteinaseKdigestion followedbyPCR amplification. The 335-DRE14 primers were 50-CAGGAG TGG GAC TAG CCC TCC TTG G-30 (sense), and50-GTG TTC TCT ACG ACC CCG AGG TGC-30 (anti-sense). The 335-DRE56primerswere 50-AGTCATTCCGT-TAGCTGGCTCCAC-30 (sense), and 50-TGG ACC TGGACC GAC ACC TGC AG-30 (antisense). The 335-DRE7primerswere 50-TTCCCTACGATGAAATTCTCTTGC-30 (sense), and 50-CGAAGGGTGGTC TTGAATGATG-30 (antisense). The positive control primers were 50-TCAGGG CTG GGG TCG CAG CGC TTC T-30 (sense), and50-GCT ACA GCC TAC CAG GAC TCG GCA G-30 (anti-sense), and they amplified a 122-bp region of humanCyp1A1 promoter (35).
Cell transfection and RNA interferenceCells were seeded in 6- or 12-well plates in phenol red-
free DMEM/F12medium (Sigma-Aldrich) supplementedwith 2.5% dextran/charcoal-stripped FBS. After 16 to 20hours when cells were 50% confluent, appropriateamounts of plasmids and short interfering RNAduplexeswere transfected using Lipofectamine 2000 reagent(Invitrogen) according to the recommendations of themanufacturer and as previously described (30). Afterincubation, cells were collected for Western blot analysisand quantitative real-time PCR assay.
Tail vein injection of cells for metastasis inathymic mice
Mice were purchased from Harlan, MDA-MB-231cancer cells (106 cells/animal) were introduced throughtail-vein injection. After 6 days, mice were gavaged dailyfor 21 days with MCDF (40 mg/kg/d; 7 mice) or cornoil (vehicle; 7 mice), then euthanized and lungs wereanalyzed formetastatic tumors. RNAwasharvestedusingthe mirVana miRNA Isolation Kit (Ambion) and the ABmicroRNA Assay Kit (Applied Biosystems) was used toreverse transcribe the miR335 in total RNA samples. ThePromega kit was used to reverse transcribe the b2MmRNA. Three mice not injected with cancer cells werealso used as controls.
Histologic image analysisLungs from each mouse were fixed in 10% neutral-
buffered formalin, embedded in paraffin, and sectionedto 5 mm in thickness for hematoxylin and eosin (H&E)staining. H&E-stained slides were scanned to create ahigh-quality TIF image using the Nikon Super COOL-SCAN 5000. All nonlung tissue was edited from theimage using ImageJ software (NIH; ref. 36). The imagewas converted into an 8-bit binary image and the num-ber of pixels was calculated. Next, all normal lung tissuewas edited from the images so that only tumor metas-tases remained and the process was repeated to calcu-late pixels. To determine the percentage of tumor tototal lung, the number of pixels from the tumors was
divided by the number of pixels from the total lungtissue.
Statistical analysisStatistical significancewas determined byANOVAand
Scheffe’s test and the levels of probability are noted. Theresults are expressed as means � SEM for at least 3separate experiments for each treatment group.
Results
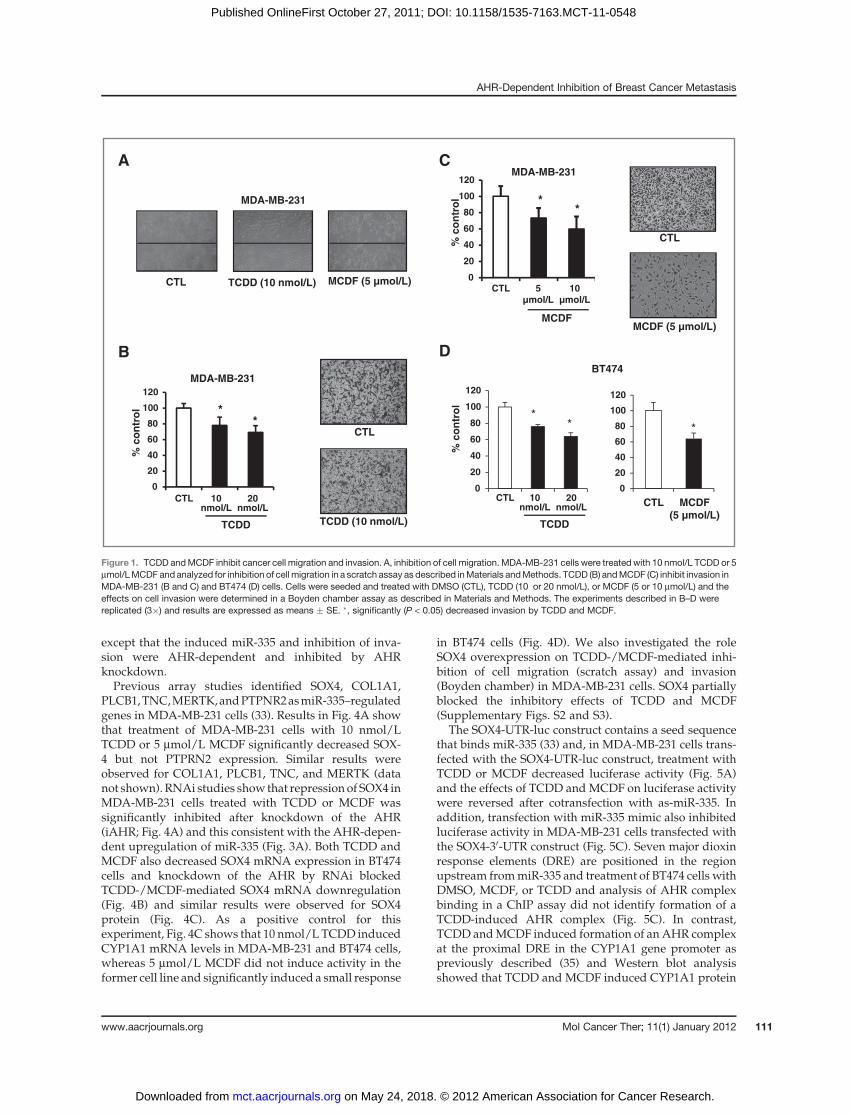
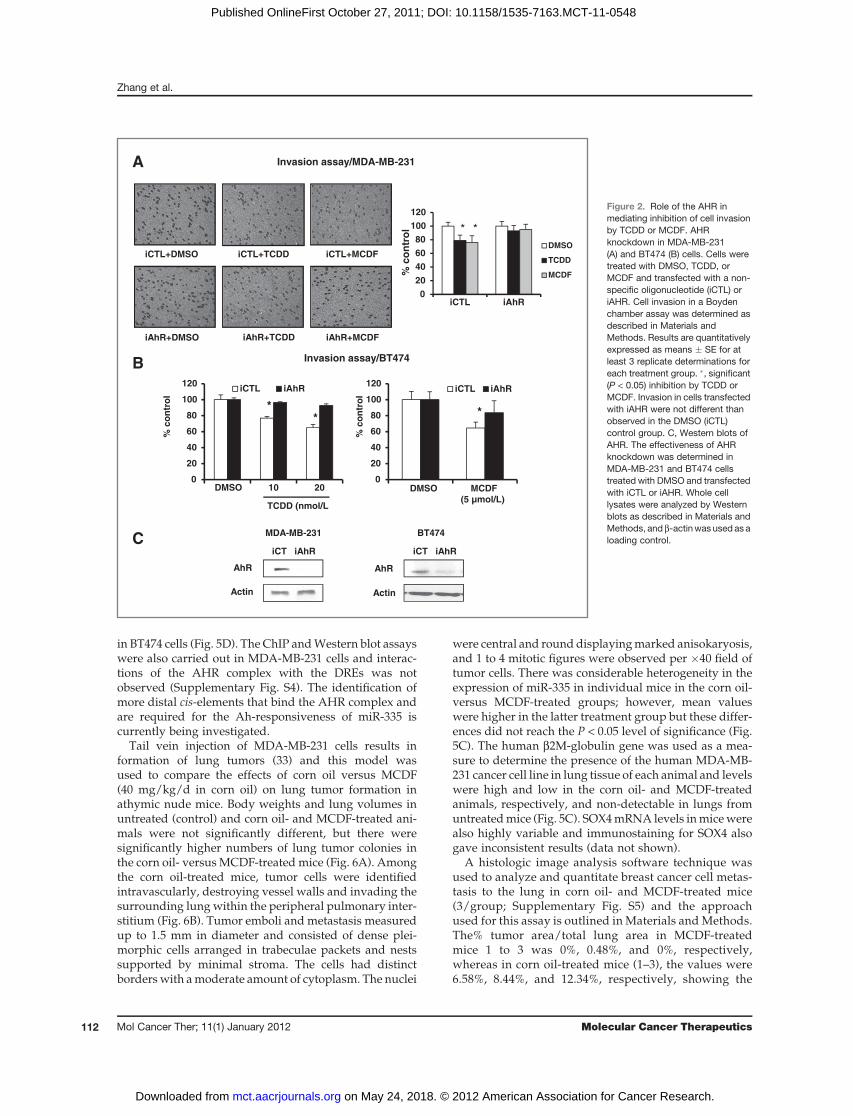
Figure 1A shows that 10 nmol/L TCDD and 5 mmol/LMCDF inhibited MDA-MB-231 cell migration in ascratch assay, whereas no significant migration wasobserved in control or treated BT474 cells (data notshown). Supplementary Fig. S1 shows that MCDF (butnot TCDD) inhibited proliferation of BT474 and MDA-MB-231 cells using the MTT assay, and this effect maycontribute to inhibition ofMDA-MB-231 cell migration byMCDF. Results in Fig. 1B and C show that 10 and 20nmol/L TCDD and 5 and 10 mmol/L MCDF significantlydecreased MDA-MB-231 cell invasion in a Boyden cham-ber assay, and Fig. 1D confirms that 10 and 20 nmol/LTCDDand5mmol/LMCDFalso inhibitedBT474 invasionin theBoyden chamber assay. Results illustrated inFig. 2Ashow that inhibition of AHR expression by RNA inter-ference (RNAi) significantly blocked TCDD- and MCDF-mediated inhibition of MDA-MB-231 cell invasion andsimilar results were observed in BT474 cells (Fig. 2B),showing a role for the AhR in mediating this response.The effectiveness of AHR knockdown is illustrated inFig. 2C, which shows that iAHR significantly decreasedAHRprotein levels in bothMDA-MB-231 andBT474 cells.
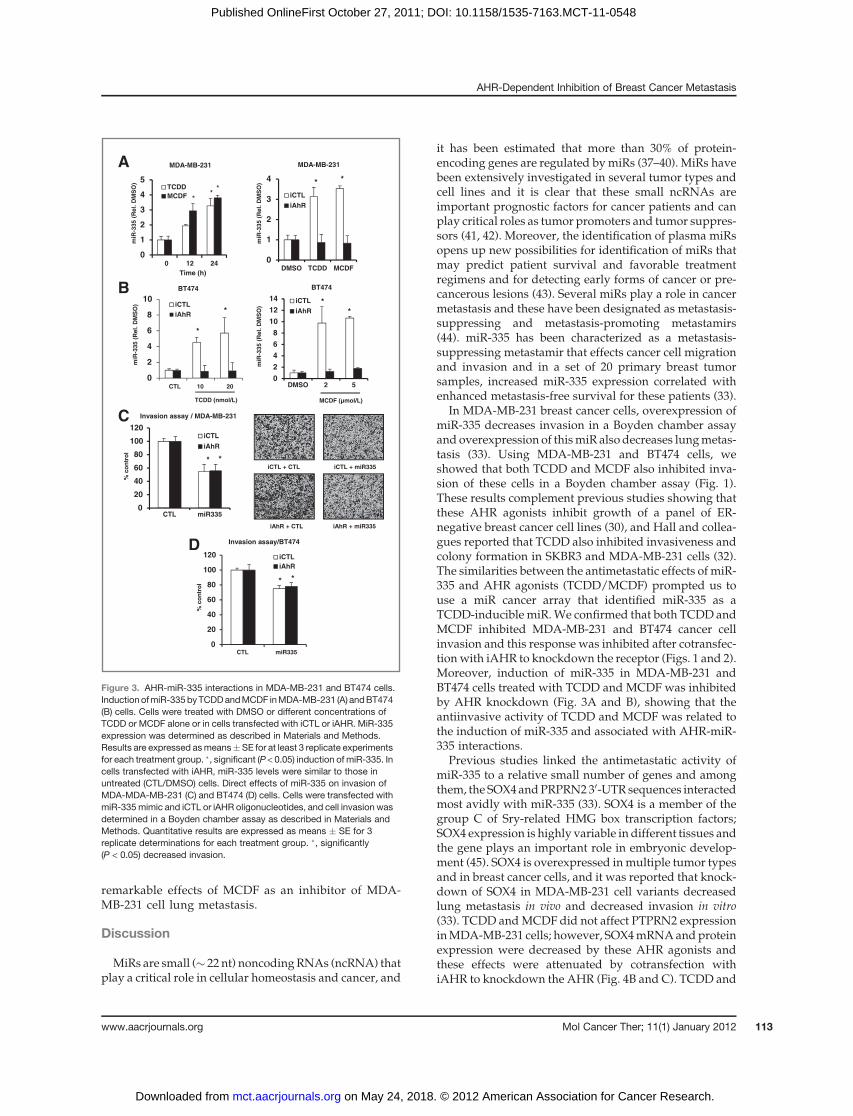
Wehypothesized that theAHR-dependent inhibition ofcell migration may be due, in part, to regulation of miRsand, the effects of TCDD and MCDF were initially inves-tigated using a cancer microRNA-PCR array (from SABiosciences). On the basis of published results (33), wealso examined the effects of these compounds onmiR-335expression. Figure 3A confirms that both 10 nmol/LTCDD and 5 mmol/L MCDF significantly induced miR-335 after treatment for 12 and 24 hours. Moreover, theinduction of miR-335 in MDA-MB-231 cells treated withTCDD and MCDF for 24 hours was decreased afterknockdown of the AHR by RNAi (Fig. 3A). Resultsin Fig. 3B also show that TCDD and MCDF also inducedmiR-335 expression in BT474 cells and this response wasalso abrogated after knockdown of AHR by RNAi con-firming that induction ofmiR-335 by TCDD andMCDF inboth cell lines wasAHR-dependent. Figure 3C shows thatoverexpression of miR-335 in MDA-MB-231 cells inhib-ited cell invasion in a Boyden chamber assay and similarresults were observed in BT474 cells (Fig. 3D). The effectsof the miR-335 mimic were direct (AHR-independent)and were observed in cells transfected with a nonspecificiCTL oligonucleotide or iAHR. These results confirm thatmiR-335 directly decreases cancer cell invasion and werecomparable with that observed for TCDD and MCDF
Zhang et al.
Mol Cancer Ther; 11(1) January 2012 Molecular Cancer Therapeutics110
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548
except that the induced miR-335 and inhibition of inva-sion were AHR-dependent and inhibited by AHRknockdown.Previous array studies identified SOX4, COL1A1,
PLCB1,TNC,MERTK,andPTPNR2asmiR-335–regulatedgenes in MDA-MB-231 cells (33). Results in Fig. 4A showthat treatment of MDA-MB-231 cells with 10 nmol/LTCDD or 5 mmol/L MCDF significantly decreased SOX-4 but not PTPRN2 expression. Similar results wereobserved for COL1A1, PLCB1, TNC, and MERTK (datanot shown). RNAi studies show that repression of SOX4 inMDA-MB-231 cells treated with TCDD or MCDF wassignificantly inhibited after knockdown of the AHR(iAHR; Fig. 4A) and this consistent with the AHR-depen-dent upregulation of miR-335 (Fig. 3A). Both TCDD andMCDF also decreased SOX4 mRNA expression in BT474cells and knockdown of the AHR by RNAi blockedTCDD-/MCDF-mediated SOX4 mRNA downregulation(Fig. 4B) and similar results were observed for SOX4protein (Fig. 4C). As a positive control for thisexperiment, Fig. 4C shows that 10 nmol/L TCDD inducedCYP1A1 mRNA levels in MDA-MB-231 and BT474 cells,whereas 5 mmol/L MCDF did not induce activity in theformer cell line and significantly induced a small response
in BT474 cells (Fig. 4D). We also investigated the roleSOX4 overexpression on TCDD-/MCDF-mediated inhi-bition of cell migration (scratch assay) and invasion(Boyden chamber) in MDA-MB-231 cells. SOX4 partiallyblocked the inhibitory effects of TCDD and MCDF(Supplementary Figs. S2 and S3).
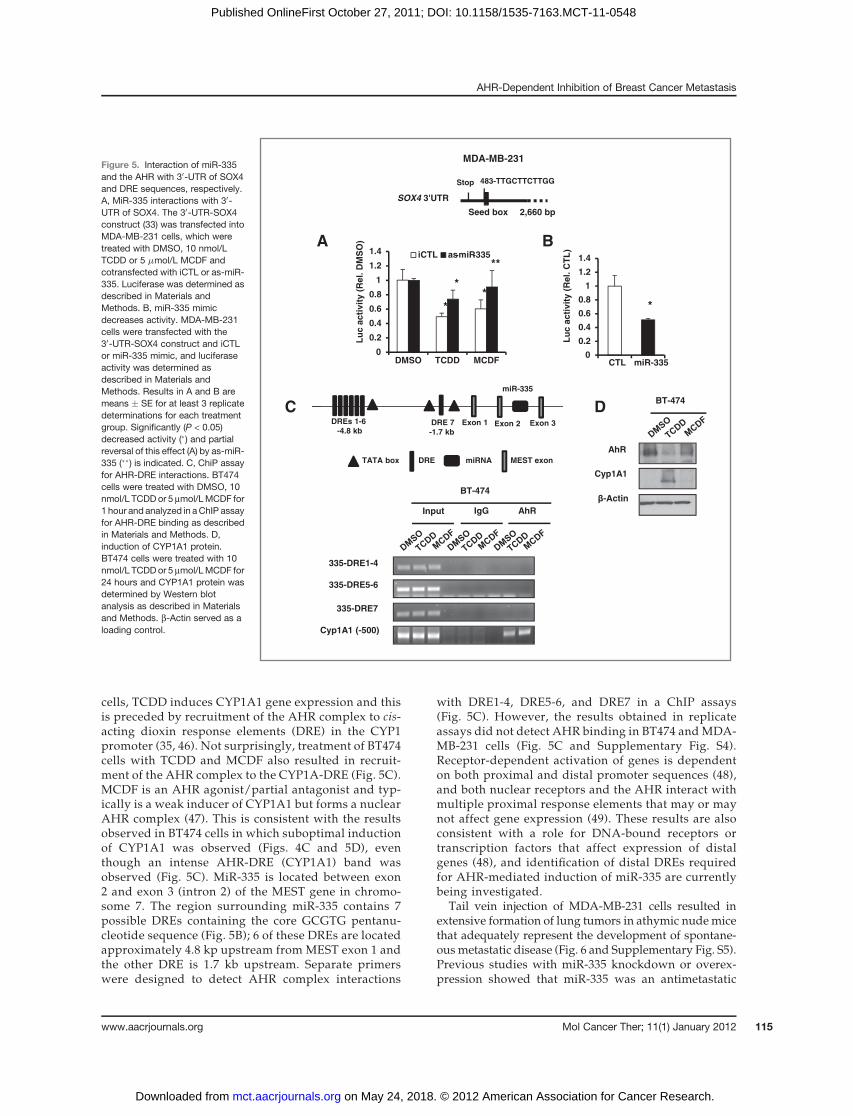
The SOX4-UTR-luc construct contains a seed sequencethat binds miR-335 (33) and, in MDA-MB-231 cells trans-fected with the SOX4-UTR-luc construct, treatment withTCDD or MCDF decreased luciferase activity (Fig. 5A)and the effects of TCDD and MCDF on luciferase activitywere reversed after cotransfection with as-miR-335. Inaddition, transfection with miR-335 mimic also inhibitedluciferase activity in MDA-MB-231 cells transfected withthe SOX4-30-UTR construct (Fig. 5C). Seven major dioxinresponse elements (DRE) are positioned in the regionupstream frommiR-335 and treatment of BT474 cells withDMSO, MCDF, or TCDD and analysis of AHR complexbinding in a ChIP assay did not identify formation of aTCDD-induced AHR complex (Fig. 5C). In contrast,TCDD andMCDF induced formation of anAHR complexat the proximal DRE in the CYP1A1 gene promoter aspreviously described (35) and Western blot analysisshowed that TCDD and MCDF induced CYP1A1 protein
0
20
40
60
80
100
120
A
MDA-MB-231
TCDD (10 nmol/L)CTL MCDF (5 µmol/L)
B
MDA-MB-231
0
20
40
60
80
100
120
% c
on
tro
l
CTL
TCDD (10 nmol/L)
**
C
MCDFMCDF (5 µmol/L)
CTL
% c
on
tro
l
**
DBT474
0
20
40
60
80
100
120
1 2 3CTL
TCDD
20nmol/L
10nmol/L1 2 3CTL
TCDD
20nmol/L
10nmol/L
CTL 10
µmol/L
5
µmol/L
% c
on
tro
l
0
20
40
60
80
100
120
1 2MCDF
(5 µmol/L)
CTL
**
*
MDA-MB-231
Figure 1. TCDD andMCDF inhibit cancer cell migration and invasion. A, inhibition of cell migration. MDA-MB-231 cells were treated with 10 nmol/L TCDD or 5mmol/LMCDF and analyzed for inhibition of cell migration in a scratch assay as described inMaterials andMethods. TCDD (B) andMCDF (C) inhibit invasion inMDA-MB-231 (B and C) and BT474 (D) cells. Cells were seeded and treated with DMSO (CTL), TCDD (10 or 20 nmol/L), or MCDF (5 or 10 mmol/L) and theeffects on cell invasion were determined in a Boyden chamber assay as described in Materials and Methods. The experiments described in B–D werereplicated (3�) and results are expressed as means � SE. �, significantly (P < 0.05) decreased invasion by TCDD and MCDF.
AHR-Dependent Inhibition of Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Ther; 11(1) January 2012 111
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548
in BT474 cells (Fig. 5D). The ChIP andWestern blot assayswere also carried out in MDA-MB-231 cells and interac-tions of the AHR complex with the DREs was notobserved (Supplementary Fig. S4). The identification ofmore distal cis-elements that bind the AHR complex andare required for the Ah-responsiveness of miR-335 iscurrently being investigated.
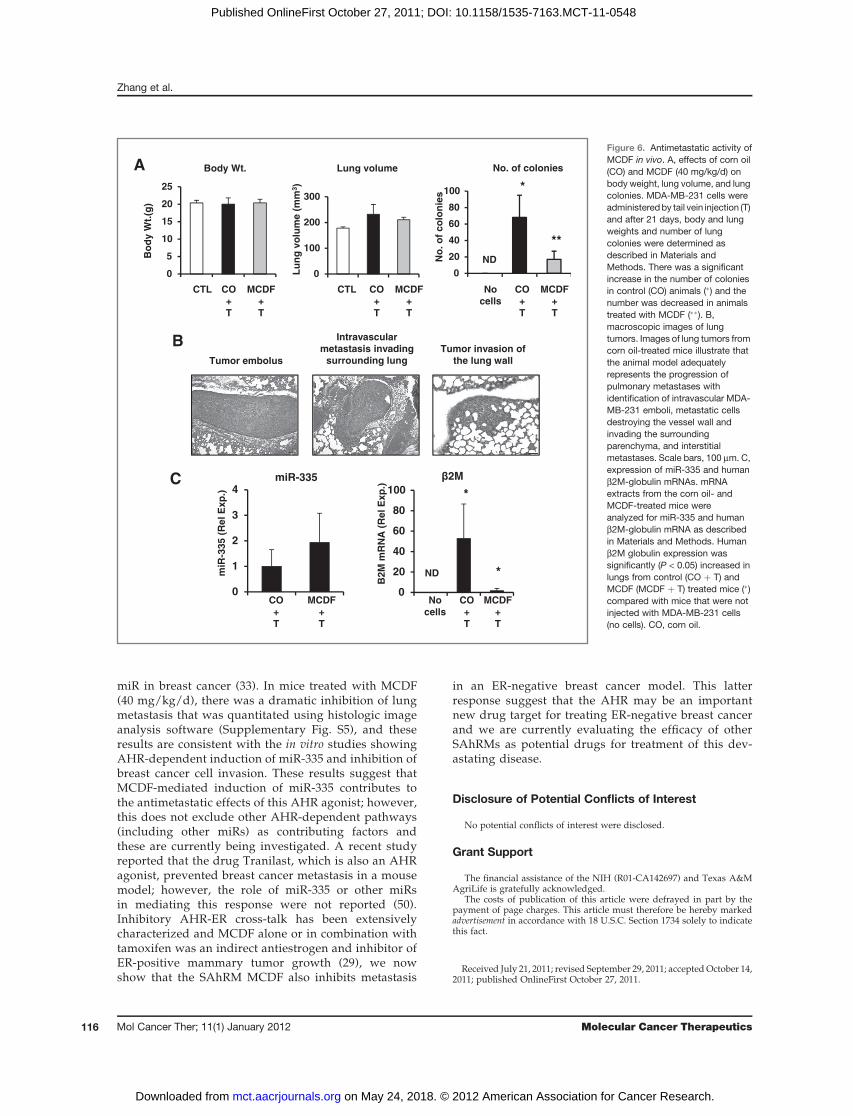
Tail vein injection of MDA-MB-231 cells results information of lung tumors (33) and this model wasused to compare the effects of corn oil versus MCDF(40 mg/kg/d in corn oil) on lung tumor formation inathymic nude mice. Body weights and lung volumes inuntreated (control) and corn oil- and MCDF-treated ani-mals were not significantly different, but there weresignificantly higher numbers of lung tumor colonies inthe corn oil- versusMCDF-treated mice (Fig. 6A). Amongthe corn oil-treated mice, tumor cells were identifiedintravascularly, destroying vessel walls and invading thesurrounding lung within the peripheral pulmonary inter-stitium (Fig. 6B). Tumor emboli and metastasis measuredup to 1.5 mm in diameter and consisted of dense plei-morphic cells arranged in trabeculae packets and nestssupported by minimal stroma. The cells had distinctborderswith amoderate amount of cytoplasm. The nuclei
were central and round displayingmarked anisokaryosis,and 1 to 4 mitotic figures were observed per �40 field oftumor cells. There was considerable heterogeneity in theexpression of miR-335 in individual mice in the corn oil-versus MCDF-treated groups; however, mean valueswere higher in the latter treatment group but these differ-ences did not reach the P < 0.05 level of significance (Fig.5C). The human b2M-globulin gene was used as a mea-sure to determine the presence of the human MDA-MB-231 cancer cell line in lung tissue of each animal and levelswere high and low in the corn oil- and MCDF-treatedanimals, respectively, and non-detectable in lungs fromuntreatedmice (Fig. 5C). SOX4mRNA levels inmicewerealso highly variable and immunostaining for SOX4 alsogave inconsistent results (data not shown).
A histologic image analysis software technique wasused to analyze and quantitate breast cancer cell metas-tasis to the lung in corn oil- and MCDF-treated mice(3/group; Supplementary Fig. S5) and the approachused for this assay is outlined inMaterials andMethods.The% tumor area/total lung area in MCDF-treatedmice 1 to 3 was 0%, 0.48%, and 0%, respectively,whereas in corn oil-treated mice (1–3), the values were6.58%, 8.44%, and 12.34%, respectively, showing the
A Invasion assay/MDA-MB-231
iCTL+DMSO iCTL+TCDD iCTL+MCDF
iAhR+DMSO iAhR+TCDD iAhR+MCDF
0
20
40
60
80
100
120
iCTL iAhR
DMSO
TCDD
MCDF% c
on
tro
l * *
B
% c
on
tro
l
TCDD (nmol/L
0
20
40
60
80
100
120iCTL iAhR
0
20
40
60
80
100
120iCTL iAhR
Invasion assay/BT474
**
*
% c
on
tro
l
DMSO 10 20 DMSO
AhR
Actin
iCT iAhRC
MDA-MB-231
AhR
Actin
iCT iAhR
BT474
MCDF
(5 µmol/L)
Figure 2. Role of the AHR inmediating inhibition of cell invasionby TCDD or MCDF. AHRknockdown in MDA-MB-231(A) and BT474 (B) cells. Cells weretreated with DMSO, TCDD, orMCDF and transfected with a non-specific oligonucleotide (iCTL) oriAHR. Cell invasion in a Boydenchamber assay was determined asdescribed in Materials andMethods. Results are quantitativelyexpressed as means � SE for atleast 3 replicate determinations foreach treatment group. �, significant(P < 0.05) inhibition by TCDD orMCDF. Invasion in cells transfectedwith iAHR were not different thanobserved in the DMSO (iCTL)control group. C, Western blots ofAHR. The effectiveness of AHRknockdown was determined inMDA-MB-231 and BT474 cellstreated with DMSO and transfectedwith iCTL or iAHR. Whole celllysates were analyzed by Westernblots as described in Materials andMethods, and b-actinwas used as aloading control.
Zhang et al.
Mol Cancer Ther; 11(1) January 2012 Molecular Cancer Therapeutics112
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548
remarkable effects of MCDF as an inhibitor of MDA-MB-231 cell lung metastasis.
Discussion
MiRs are small (� 22 nt) noncodingRNAs (ncRNA) thatplay a critical role in cellular homeostasis and cancer, and
it has been estimated that more than 30% of protein-encoding genes are regulated bymiRs (37–40). MiRs havebeen extensively investigated in several tumor types andcell lines and it is clear that these small ncRNAs areimportant prognostic factors for cancer patients and canplay critical roles as tumor promoters and tumor suppres-sors (41, 42). Moreover, the identification of plasma miRsopens up new possibilities for identification of miRs thatmay predict patient survival and favorable treatmentregimens and for detecting early forms of cancer or pre-cancerous lesions (43). Several miRs play a role in cancermetastasis and these have been designated as metastasis-suppressing and metastasis-promoting metastamirs(44). miR-335 has been characterized as a metastasis-suppressing metastamir that effects cancer cell migrationand invasion and in a set of 20 primary breast tumorsamples, increased miR-335 expression correlated withenhanced metastasis-free survival for these patients (33).
In MDA-MB-231 breast cancer cells, overexpression ofmiR-335 decreases invasion in a Boyden chamber assayand overexpression of thismiR also decreases lungmetas-tasis (33). Using MDA-MB-231 and BT474 cells, weshowed that both TCDD and MCDF also inhibited inva-sion of these cells in a Boyden chamber assay (Fig. 1).These results complement previous studies showing thatthese AHR agonists inhibit growth of a panel of ER-negative breast cancer cell lines (30), and Hall and collea-gues reported that TCDD also inhibited invasiveness andcolony formation in SKBR3 and MDA-MB-231 cells (32).The similarities between the antimetastatic effects of miR-335 and AHR agonists (TCDD/MCDF) prompted us touse a miR cancer array that identified miR-335 as aTCDD-inducible miR.We confirmed that both TCDD andMCDF inhibited MDA-MB-231 and BT474 cancer cellinvasion and this response was inhibited after cotransfec-tionwith iAHR to knockdown the receptor (Figs. 1 and 2).Moreover, induction of miR-335 in MDA-MB-231 andBT474 cells treated with TCDD and MCDF was inhibitedby AHR knockdown (Fig. 3A and B), showing that theantiinvasive activity of TCDD and MCDF was related tothe induction of miR-335 and associated with AHR-miR-335 interactions.
Previous studies linked the antimetastatic activity ofmiR-335 to a relative small number of genes and amongthem, the SOX4andPRPRN230-UTR sequences interactedmost avidly with miR-335 (33). SOX4 is a member of thegroup C of Sry-related HMG box transcription factors;SOX4 expression is highly variable in different tissues andthe gene plays an important role in embryonic develop-ment (45). SOX4 is overexpressed inmultiple tumor typesand in breast cancer cells, and it was reported that knock-down of SOX4 in MDA-MB-231 cell variants decreasedlung metastasis in vivo and decreased invasion in vitro(33). TCDD andMCDF did not affect PTPRN2 expressioninMDA-MB-231 cells; however, SOX4mRNAandproteinexpression were decreased by these AHR agonists andthese effects were attenuated by cotransfection withiAHR to knockdown the AHR (Fig. 4B and C). TCDD and
A
0
1
2
3
4
5TCDD
MCDF
MDA-MB-231
0 12 24
Time (h)
miR
-335 (
Rel. D
MS
O)
*
**
*
0
1
2
3
4
DMSO TCDD MCDF
iCTL
iAhR
MDA-MB-231
* *
miR
-335 (
Rel. D
MS
O)
B
0
2
4
6
8
10
12
14
DMSO 2 5
iCTL
iAhR
MCDF (µmol/L)
0
2
4
6
8
10iCTL
iAhR
CTL 10 20
TCDD (nmol/L)
BT474 BT474
miR
-335 (
Rel. D
MS
O)
*
**
*
miR
-335 (
Rel. D
MS
O)
C
0
20
40
60
80
100
120
CTL miR335
iCTL
iAhR
iCTL + CTL iCTL + miR335
iAhR + miR335iAhR + CTL
% c
on
tro
l
Invasion assay / MDA-MB-231
* *
D
0
20
40
60
80
100
120 iCTL
iAhR
% c
on
tro
l
CTL miR335
Invasion assay/BT474
* *
Figure 3. AHR-miR-335 interactions in MDA-MB-231 and BT474 cells.Induction ofmiR-335byTCDDandMCDF inMDA-MB-231 (A) andBT474(B) cells. Cells were treated with DMSO or different concentrations ofTCDD or MCDF alone or in cells transfected with iCTL or iAHR. MiR-335expression was determined as described in Materials and Methods.Results are expressed asmeans�SE for at least 3 replicate experimentsfor each treatment group. �, significant (P < 0.05) induction of miR-335. Incells transfected with iAHR, miR-335 levels were similar to those inuntreated (CTL/DMSO) cells. Direct effects of miR-335 on invasion ofMDA-MDA-MB-231 (C) and BT474 (D) cells. Cells were transfected withmiR-335mimic and iCTL or iAHR oligonucleotides, and cell invasion wasdetermined in a Boyden chamber assay as described in Materials andMethods. Quantitative results are expressed as means � SE for 3replicate determinations for each treatment group. �, significantly(P < 0.05) decreased invasion.
AHR-Dependent Inhibition of Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Ther; 11(1) January 2012 113
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548

MCDF decreased luciferase activity in MDA-MB-231cells transfected with p0-UTR-SOX4-luc, which containedthe miR-335–binding site (Ref. 33; Fig. 5A), and theseresults were consistent with AHR-mediated regulationof miR-335-SOX4 (UTR) interactions. Moreover, SOX4overexpression partially blocks TCDD- andMCDF-medi-
ated inhibition of MDA-MB-231 cell migration and inva-sion (Supplementary Figs. S2 and S3) and this confirmsprevious studies showing that SOX4 was a prometastaticfactor (33).
The AHR is a ligand-activated nuclear receptor andin most cell lines including ER-negative breast cancer
SOX4
MDA-MB-231
DMSO
TCDD (10 nmol/L)
MCDF (5µmol/L
iAhR + + +
+ +
+ +
+ +
- - -- - - -
- - - -- - - -
β-Actin
C
A
0
0.5
1
1.5
2
DMSO TCDD MCDF
SOX4
PTPRN2
MDA-MB-231
0
0.5
1
1.5
iCTL iAhR
DMSO
MCDF
MDA-MB-231 / SOX4
**
*
0
0.5
1
1.5
2DMSO
TCDD
iCTL iAhR
*
mR
NA
(R
el.
DM
SO
)
SO
X4
mR
NA
(R
el.
DM
SO
)
**
**
B BT474
0
0.2
0.4
0.6
0.8
1
1.2
1.4
DMSO 1 10
iCTL
iAhR
TCDD (nmol/L)
*
*
MCDF (µmol/L)
0
0.2
0.4
0.6
0.8
1
1.2
1.4 iCTL
iAhR
BT474
DMSO 2 5
**
SO
X4
mR
NA
(R
el.
DM
SO
)
SO
X4
mR
NA
(R
el.
DM
SO
)**
**
****
D
0
1
2
3
4
5
DMSO TCDD MCDF
MDA-MB-231
*
CY
P1A
1 m
RN
A
(Re
l. D
MS
O)
0
50
100
150
200
BT474
CY
P1A
1 m
RN
A
(Re
l. D
MS
O)
*
*
Figure 4. AHR-mediatedsuppression of SOX4. Suppressionof SOX4 in MDA-MB-231 (A) orBT474 (B) cells by TCDD or MCDF.Cells were treated with DMSO,TCDD or MCDF alone ortransfected with iCTL or iAHR. After24 hours, mRNA levels weredetermined by real-time PCR asdescribed in Materials andMethods. Results in A and B aregiven as means � SE for at least 3replicate determinations for eachtreatment group. Significant(P < 0.05) inhibition of SOX4expression by TCDD or MCDF (�)and reversal of these effects byiAHR (��) are indicated. C,suppression of SOX4 protein. Cellswere treated with DMSO, TCDD, orMCDF and transfected with iAHR oriCTL oligonucleotides, and wholecell lysates were analyzed byWestern blots as described inMaterials and Methods. D,induction of CYP1A1 by TCDD orMCDF. Cells were treated withDMSO, 10 nmol/L TCDDor 5 mmol/L MCDF for 24 hours,and CYP1A1 mRNA levels weredetermined by real-time PCR asdescribed in Materials andMethods. Results are given asmeans � SE for at least 3 replicatedeterminations. �, significant(P < 0.05) induction of CYP1A1mRNA.
Zhang et al.
Mol Cancer Ther; 11(1) January 2012 Molecular Cancer Therapeutics114
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548
cells, TCDD induces CYP1A1 gene expression and thisis preceded by recruitment of the AHR complex to cis-acting dioxin response elements (DRE) in the CYP1promoter (35, 46). Not surprisingly, treatment of BT474cells with TCDD and MCDF also resulted in recruit-ment of the AHR complex to the CYP1A-DRE (Fig. 5C).MCDF is an AHR agonist/partial antagonist and typ-ically is a weak inducer of CYP1A1 but forms a nuclearAHR complex (47). This is consistent with the resultsobserved in BT474 cells in which suboptimal inductionof CYP1A1 was observed (Figs. 4C and 5D), eventhough an intense AHR-DRE (CYP1A1) band wasobserved (Fig. 5C). MiR-335 is located between exon2 and exon 3 (intron 2) of the MEST gene in chromo-some 7. The region surrounding miR-335 contains 7possible DREs containing the core GCGTG pentanu-cleotide sequence (Fig. 5B); 6 of these DREs are locatedapproximately 4.8 kp upstream from MEST exon 1 andthe other DRE is 1.7 kb upstream. Separate primerswere designed to detect AHR complex interactions
with DRE1-4, DRE5-6, and DRE7 in a ChIP assays(Fig. 5C). However, the results obtained in replicateassays did not detect AHR binding in BT474 andMDA-MB-231 cells (Fig. 5C and Supplementary Fig. S4).Receptor-dependent activation of genes is dependenton both proximal and distal promoter sequences (48),and both nuclear receptors and the AHR interact withmultiple proximal response elements that may or maynot affect gene expression (49). These results are alsoconsistent with a role for DNA-bound receptors ortranscription factors that affect expression of distalgenes (48), and identification of distal DREs requiredfor AHR-mediated induction of miR-335 are currentlybeing investigated.
Tail vein injection of MDA-MB-231 cells resulted inextensive formation of lung tumors in athymic nudemicethat adequately represent the development of spontane-ousmetastatic disease (Fig. 6 and Supplementary Fig. S5).Previous studies with miR-335 knockdown or overex-pression showed that miR-335 was an antimetastatic
Figure 5. Interaction of miR-335and the AHR with 30-UTR of SOX4and DRE sequences, respectively.A, MiR-335 interactions with 30-UTR of SOX4. The 30-UTR-SOX4construct (33) was transfected intoMDA-MB-231 cells, which weretreated with DMSO, 10 nmol/LTCDD or 5 mmol/L MCDF andcotransfected with iCTL or as-miR-335. Luciferase was determined asdescribed in Materials andMethods. B, miR-335 mimicdecreases activity. MDA-MB-231cells were transfected with the30-UTR-SOX4 construct and iCTLor miR-335 mimic, and luciferaseactivity was determined asdescribed in Materials andMethods. Results in A and B aremeans � SE for at least 3 replicatedeterminations for each treatmentgroup. Significantly (P < 0.05)decreased activity (�) and partialreversal of this effect (A) by as-miR-335 (��) is indicated. C, ChiP assayfor AHR-DRE interactions. BT474cells were treated with DMSO, 10nmol/L TCDDor 5 mmol/LMCDF for1 hour and analyzed in aChIP assayfor AHR-DRE binding as describedin Materials and Methods. D,induction of CYP1A1 protein.BT474 cells were treated with 10nmol/L TCDDor 5 mmol/LMCDF for24 hours and CYP1A1 protein wasdetermined by Western blotanalysis as described in Materialsand Methods. b-Actin served as aloading control.
A
0
0.2
0.4
0.6
0.8
1
1.2
1.4
DMSO TCDD MCDF
iCTL as-miR-335
MDA-MB-231
**
0
0.2
0.4
0.6
0.8
1
1.2
1.4
*
CTL miR-335
SOX4 3'UTR
Stop
Seed box
483-TTGCTTCTTGG
2,660 bp
Lu
c a
cti
vit
y (
Rel. D
MS
O)
Lu
c a
cti
vit
y (
Rel. C
TL
)
B
Input IgG AhR
BT-474
335-DRE1-4
335-DRE5-6
335-DRE7
Cyp1A1 (-500)
AhR
Cyp1A1
β-Actin
BT-474
DRE 7
-1.7 kb
DREs 1-6
-4.8 kb
miR-335
TATA box DRE miRNA
Exon 1 Exon 2 Exon 3
MEST exon
D C
**
*
AHR-Dependent Inhibition of Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Ther; 11(1) January 2012 115
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548
miR in breast cancer (33). In mice treated with MCDF(40 mg/kg/d), there was a dramatic inhibition of lungmetastasis that was quantitated using histologic imageanalysis software (Supplementary Fig. S5), and theseresults are consistent with the in vitro studies showingAHR-dependent induction of miR-335 and inhibition ofbreast cancer cell invasion. These results suggest thatMCDF-mediated induction of miR-335 contributes tothe antimetastatic effects of this AHR agonist; however,this does not exclude other AHR-dependent pathways(including other miRs) as contributing factors andthese are currently being investigated. A recent studyreported that the drug Tranilast, which is also an AHRagonist, prevented breast cancer metastasis in a mousemodel; however, the role of miR-335 or other miRsin mediating this response were not reported (50).Inhibitory AHR-ER cross-talk has been extensivelycharacterized and MCDF alone or in combination withtamoxifen was an indirect antiestrogen and inhibitor ofER-positive mammary tumor growth (29), we nowshow that the SAhRM MCDF also inhibits metastasis
in an ER-negative breast cancer model. This latterresponse suggest that the AHR may be an importantnew drug target for treating ER-negative breast cancerand we are currently evaluating the efficacy of otherSAhRMs as potential drugs for treatment of this dev-astating disease.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Grant Support
The financial assistance of the NIH (R01-CA142697) and Texas A&MAgriLife is gratefully acknowledged.
The costs of publication of this article were defrayed in part by thepayment of page charges. This article must therefore be hereby markedadvertisement in accordance with 18 U.S.C. Section 1734 solely to indicatethis fact.
Received July 21, 2011; revised September 29, 2011; acceptedOctober 14,2011; published OnlineFirst October 27, 2011.
B Intravascular
metastasis invading
surrounding lung
Tumor invasion of
the lung wallTumor embolus
0
5
10
15
20
25
Body Wt.
Bo
dy W
t.(g
)
0
100
200
300
Lung volume
Lu
ng
vo
lum
e (
mm
3)
0
20
40
60
80
100
No
. o
f c
olo
nie
s
A
ND
CTL CO
+
T
MCDF
+
T
CTL CO
+
T
MCDF
+
T
No
cells
CO
+
T
MCDF
+
T
No. of colonies
C
miR
-335 (
Rel
Exp
.)
0
1
2
3
4miR-335
0
20
40
60
80
100
ND
β2M
Β2
M m
RN
A (
Re
lE
xp
.)
CO
+
T
MCDF
+
T
No
cells
CO
+
T
MCDF
+
T
*
*
*
**
Figure 6. Antimetastatic activity ofMCDF in vivo. A, effects of corn oil(CO) and MCDF (40 mg/kg/d) onbody weight, lung volume, and lungcolonies. MDA-MB-231 cells wereadministeredby tail vein injection (T)and after 21 days, body and lungweights and number of lungcolonies were determined asdescribed in Materials andMethods. There was a significantincrease in the number of coloniesin control (CO) animals (�) and thenumber was decreased in animalstreated with MCDF (��). B,macroscopic images of lungtumors. Images of lung tumors fromcorn oil-treated mice illustrate thatthe animal model adequatelyrepresents the progression ofpulmonary metastases withidentification of intravascular MDA-MB-231 emboli, metastatic cellsdestroying the vessel wall andinvading the surroundingparenchyma, and interstitialmetastases. Scale bars, 100 mm. C,expression of miR-335 and humanb2M-globulin mRNAs. mRNAextracts from the corn oil- andMCDF-treated mice wereanalyzed for miR-335 and humanb2M-globulin mRNA as describedin Materials and Methods. Humanb2M globulin expression wassignificantly (P < 0.05) increased inlungs from control (CO þ T) andMCDF (MCDF þ T) treated mice (�)compared with mice that were notinjected with MDA-MB-231 cells(no cells). CO, corn oil.
Zhang et al.
Mol Cancer Ther; 11(1) January 2012 Molecular Cancer Therapeutics116
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548

References1. Poland A, Glover E, Kende AS. Stereospecific, high affinity binding of
2,3,7,8- tetrachlorodibenzo-p-dioxin by hepatic cytosol: evidence thatthe binding species is receptor for induction of aryl hydrocarbonhydroxylase. J Biol Chem 1976;251:4936–46.
2. Schmidt JV, Su GH, Reddy JK, Simon MC, Bradfield CA. Character-ization of a murine Ahr null allele: involvement of the Ah receptor inhepatic growth and development. Proc Natl Acad Sci U S A 1996;93:6731–6.
3. Peters JM, Narotsky MG, Elizondo G, Fernandez-Salguero PM,Gonzalez FJ, Abbott BD. Amelioration of TCDD-induced teratogenesisin aryl hydrocarbon receptor (AhR)-null mice. Toxicol Sci 1999;47:86–92.
4. Ciatto S, Bonardi R. Is breast cancer ever cured? Follow-up study of5623 breast cancer patients. Tumori 1991;77:465–7.
5. Denison MS, Nagy SR. Activation of the aryl hydrocarbon receptor bystructurally diverse exogenous and endogenous chemicals. Annu RevPharmacol Toxicol 2003;43:309–34.
6. AshidaH, Fukuda I, Yamashita T, Kanazawa K. Flavones and flavonolsat dietary levels inhibit a transformation of aryl hydrocarbon receptorinduced by dioxin. FEBS Lett 2000;476:213–7.
7. Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA.Aromatic hydrocarbon responsiveness-receptor agonists generatedfrom indole-3-carbinol in vitro and in vivo - comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin.ProcNatlAcadSciUSA1991;88:9543–7.
8. Ciolino HP, MacDonald CJ, Memon OS, Bass SE, Yeh GC. Sulindacregulates the aryl hydrocarbon receptor-mediated expression ofPhase 1 metabolic enzymes in vivo and in vitro. Carcinogenesis2006;27:1586–92.
9. DzeletovicN,McGuire J,DaujatM,Tholander J, EmaM,Fujii-KuriyamaY, et al. Regulation of dioxin receptor function by omeprazole. J BiolChem 1997;272:12705–13.
10. Sinal CJ, Bend JR. Aryl hydrocarbon receptor-dependent inductionof Cyp1a1 by bilirubin in mouse hepatoma Hepa 1c1c7 cells. MolPharmacol 1997;52:590–9.
11. DiNatale BC, Murray IA, Schroeder JC, Flaveny CA, Lahoti TS,Laurenzana EM, et al. Kynurenic acid is a potent endogenous arylhydrocarbon receptor ligand that synergistically induces interleukin-6in thepresenceof inflammatory signaling. Toxicol Sci 2010;115:89–97.
12. Adachi J, Mori Y, Matsui S, Takigami H, Fujino J, Kitagawa H, et al.Indirubin and indigo are potent aryl hydrocarbon receptor ligandspresent in human urine. J Biol Chem 2001;276:31475–8.
13. Safe S,QinC,McDougal A. Development of selective aryl hydrocarbonreceptor modulators (SARMs) for treatment of breast cancer. ExpertOpin Investig Drugs 1999;8:1385–96.
14. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al.Control of Treg and TH17 cell differentiation by the aryl hydrocarbonreceptor. Nature 2008;453:65–71.
15. Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, RenauldJC, et al. The aryl hydrocarbon receptor links TH17-cell-mediatedautoimmunity to environmental toxins. Nature 2008;453:106–9.
16. Kerkvliet NI, Steppan LB, VorachekW,OdaS, Farrer D,WongCP, et al.Activation of aryl hydrocarbon receptor by TCDD prevents diabetes inNOD mice and increases Foxp3þ T cells in pancreatic lymph nodes.Immunotherapy 2009;1:539–47.
17. DiNatale BC, Schroeder JC, Francey LJ, Kusnadi A, Perdew GH.Mechanistic insights into the events that lead to synergistic inductionof interleukin 6 transcription upon activation of the aryl hydrocarbonreceptor and inflammatory signaling. JBiol Chem2010;285:24388–97.
18. Murray IA, KrishnegowdaG, DiNatale BC, FlavenyC, Chiaro C, Lin JM,et al. Development of a selective modulator of aryl hydrocarbon (Ah)receptor activity that exhibits anti-inflammatory properties. Chem ResToxicol 2010;23:955–66.
19. Benson JM, Shepherd DM. Aryl hydrocarbon receptor activation byTCDD reduces inflammation associated with Crohn's disease. ToxicolSci 2011;120:68–78.
20. Murray IA, Morales JL, Flaveny CA, DiNatale BC, Chiaro C, GowdahalliK, et al. Evidence for ligand-mediated selective modulation of arylhydrocarbon receptor activity. Mol Pharmacol 2010;77:247–54.
21. Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE,et al. Aryl hydrocarbon receptor antagonists promote the expansion ofhuman hematopoietic stem cells. Science 2010;329:1345–8.
22. Astroff B, Zacharewski T, Safe S, Arlotto MP, Parkinson A, Thomas P,et al. 6-Methyl-1,3,8-trichlorodibenzofuran as a 2,3,7,8-tetrachlorodi-benzo-p-dioxin antagonist: inhibition of the induction of rat cyto-chrome P-450 isozymes and related monooxygenase activities. MolPharmacol 1988;33:231–6.
23. Harris M, Zacharewski T, Astroff B, Safe S. Partial antagonism of2,3,7,8-tetrachlorodibenzo-p-dioxin-mediated induction of aryl hydro-carbon hydroxylase by 6-methyl-1,3,8-trichlorodibenzofuran: mech-anistic studies. Mol Pharmacol 1989;35:729–35.
24. Bannister R, Biegel L, Davis D, Astroff B, Safe S. 6-Methyl-1,3,8-trichlorodibenzofuran (MCDF) as a 2,3,7,8- tetrachlorodibenzo-p-dioxin antagonist in C57BL/6 mice. Toxicology 1989;54:139–50.
25. YaoC, Safe S. 2,3,7,8-Tetrachlorodibenzo-p-dioxin-induced porphyr-ia in genetically inbred mice: partial antagonism and mechanisticstudies. Toxicol Appl Pharmacol 1989;100:208–16.
26. Astroff B, Safe S. 6-Substituted-1,3,8-trichlorodibenzofurans as2,3,7,8-tetrachlorodibenzo-p-dioxin antagonists in the rat: structure-activity relationships. Toxicology 1989;59:285–96.
27. Astroff B, Safe S. 6-Alkyl-1,3,8-trichlorodibenzofurans as antiestro-gens in female Sprague-Dawley rats. Toxicology 1991;69:187–97.
28. Zacharewski T, Harris M, Biegel L, Morrison V, Merchant M, Safe S. 6-Methyl-1,3,8-trichlorodibenzofuran (MCDF) as an antiestrogen inhuman and rodent cancer cell lines: evidence for the role of the Ahreceptor. Toxicol Appl Pharmacol 1992;13:311–8.
29. McDougal A, Wormke M, Calvin J, Safe S. Tamoxifen-induced anti-tumorigenic/antiestrogenic action synergized by a selective Ah recep-tor modulator. Cancer Res 2001;61:3901–7.
30. Zhang S, Lei P, Liu X, Li X, Walker K, Kotha L, et al. The arylhydrocarbon receptor as a target for estrogen receptor-negativebreast cancer chemotherapy. Endocr Relat Cancer 2009;16:835–44.
31. Moulder S, Hortobagyi GN. Advances in the treatment of breastcancer. Clin Pharmacol Ther 2008;83:26–36.
32. Hall JM, Barhoover MA, Kazmin D, McDonnell DP, Greenlee WF,Thomas RS. Activation of the aryl-hydrocarbon receptor inhibits inva-sive and metastatic features of human breast cancer cells and pro-motes breast cancer cell differentiation. Mol Endocrinol 2010;24:359–69.
33. Tavazoie SF, AlarconC, Oskarsson T, Padua D,WangQ, Bos PD, et al.Endogenous human microRNAs that suppress breast cancer metas-tasis. Nature 2008;451:147–52.
34. Li X, Mertens-Talcott SU, Zhang S, Kim KH, Ball J, Safe S. MicroRNA-27a indirectly regulates estrogen receptor a expression and hormoneresponsiveness in MCF-7 breast cancer cells. Endocrinology 2010;151:2462–73.
35. Okino ST, Quattrochi LC, Pookot D, Iwahashi M, Dahiya R. A dioxin-responsive enhancer 30 of the human CYP1A2 gene. Mol Pharmacol2007;72:1457–65.
36. Abramoff MD,Magalhaes PJ, RamSJ. Image processingwith ImageJ.Biophotonics International 2004;11:36–42.
37. Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribeshuman microRNAs. Nat Struct Mol Biol 2006;13:1097–101.
38. Filipowicz W, Jaskiewicz L, Kolb FA, Pillai RS. Post-transcriptionalgene silencing by siRNAs and miRNAs. Curr Opin Struct Biol2005;15:331–41.
39. Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, Epstein EJ, et al.Combinatorial microRNA target predictions. Nat Genet 2005;37:495–500.
40. Patel DJ, Ma JB, Yuan YR, Ye K, Pei Y, Kuryavyi V, et al. Structuralbiology of RNA silencing and its functional implications. Cold SpringHarb Symp Quant Biol 2006;71:81–93.
41. Lu J, Getz G, Miska EA, varez-Saavedra E, Lamb J, Peck D, et al.MicroRNA expression profiles classify human cancers. Nature 2005;435:834–8.
42. Farazi TA, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. JPathol 2011;223:102–15.
AHR-Dependent Inhibition of Breast Cancer Metastasis
www.aacrjournals.org Mol Cancer Ther; 11(1) January 2012 117
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548

43. Huang Z, Huang D, Ni S, Peng Z, Sheng W, Du X. Plasma microRNAsare promising novel biomarkers for early detection of colorectal can-cer. Int J Cancer 2010;127:118–26.
44. Hurst DR, Edmonds MD, Welch DR. Metastamir: the field of metas-tasis-regulatory microRNA is spreading. Cancer Res 2009;69:7495–8.
45. Penzo-Mendez AI. Critical roles for SoxC transcription factors indevelopment and cancer. Int J Biochem Cell Biol 2010;42:425–8.
46. AbdelrahimM, Ariazi E, Kim K, Khan S, Barhoumi R, Burghardt R, et al.3-Methylcholanthrene and other aryl hydrocarbon receptor agonistsdirectly activate estrogen receptor a. Cancer Res 2006;66:2459–67.
47. Piskorska-Pliszczynska J, Astroff B, Zacharewski T, HarrisM,Roseng-ren R, Morrison V, et al. Mechanism of action of 2,3,7,8-tetrachlor-odibenzo-p-dioxin antagonists: characterization of [125I]-6-methyl-8-
iodo-1,3- dichlorodibenzofuran-Ah receptor complexes. ArchBiochem Biophys 1991;284:193–200.
48. Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, et al.Chromosome-wide mapping of estrogen receptor binding revealslong-range regulation requiring the forkhead protein FoxA1. Cell2005;122:33–43.
49. Ahmed S, Valen E, Sandelin A, Matthews J. Dioxin increases theinteraction between aryl hydrocarbon receptor and estrogenreceptor alpha at human promoters. Toxicol Sci 2009;111:254–66.
50. Prud'hommeGJ, Glinka Y, Toulina A, Ace O, Subramaniam V, Jothy S.Breast cancer stem-like cells are inhibited by a non-toxic aryl hydro-carbon receptor agonist. PLoS One 2010;5:e13831.
Zhang et al.
Mol Cancer Ther; 11(1) January 2012 Molecular Cancer Therapeutics118
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548

2012;11:108-118. Published OnlineFirst October 27, 2011.Mol Cancer Ther Shu Zhang, KyoungHyun Kim, Un Ho Jin, et al. Negative Breast Cancer CellsExpression and Inhibit Lung Metastasis of Estrogen Receptor Aryl Hydrocarbon Receptor Agonists Induce MicroRNA-335
Updated version
10.1158/1535-7163.MCT-11-0548doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2011/10/21/1535-7163.MCT-11-0548.DC1
Access the most recent supplemental material at:
Cited articles
http://mct.aacrjournals.org/content/11/1/108.full#ref-list-1
This article cites 50 articles, 16 of which you can access for free at:
Citing articles
http://mct.aacrjournals.org/content/11/1/108.full#related-urls
This article has been cited by 4 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://mct.aacrjournals.org/content/11/1/108To request permission to re-use all or part of this article, use this link
on May 24, 2018. © 2012 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Published OnlineFirst October 27, 2011; DOI: 10.1158/1535-7163.MCT-11-0548