antioxidant function of corneal aldh3a1 in cultured stromal fibroblasts
TRANSCRIPT

Free Radical Biology & Medicine 41 (2006) 1459–1469www.elsevier.com/locate/freeradbiomed
Original Contribution
Antioxidant function of corneal ALDH3A1 in cultured stromal fibroblasts
Natalie Lassen a,1, Aglaia Pappa a,1,2, William J. Black a, James V. Jester b, Brian J. Day c,Elysia Min c, Vasilis Vasiliou a,⁎
a Molecular Toxicology and Environmental Health Sciences Program, Department of Pharmaceutical Sciences,University of Colorado Health Sciences Center, 4200 East Ninth Avenue, Denver, CO 80262, USA
b Department of Ophthalmology, University of California at Irvine, Irvine, CA 92697, USAc Department of Medicine, National Jewish Medical and Research Center, Denver, CO, USA
Received 3 March 2006; revised 29 June 2006; accepted 8 August 2006Available online 12 August 2006
Abstract
Aldehyde dehydrogenase 3A1 (ALDH3A1) is highly expressed in epithelial cells and stromal keratocytes of mammalian cornea and isbelieved to play an important role in cellular defense. To explore a potential protective role against oxidative damage, a rabbit corneal fibroblasticcell line (TRK43) was stably transfected with the human ALDH3A1 and subjected to oxidative stress induced by H2O2, mitomycin C (MMC), oretoposide (VP-16). ALDH3A1-transfected cells were more resistant to H2O2, MMC, and VP-16 compared to the vector-transfected cells. Alltreatments induced apoptosis only in vector-transfected cells, which was associated with increased levels of 4-hydroxy-2-nonenal (4-HNE)-adducted proteins. Treatment with H2O2 resulted in a rise in reduced glutathione (GSH) levels in all groups but was more pronounced in theALDH3A1-expressing cells. Treatment with the DNA-damaging agents led to GSH depletion in control groups, although the depletion wassignificantly less in ALDH3A1-expressing cells. Increased carbonylation of ALDH3A1 but not significant decline in enzymatic activity wasobserved after all treatments. In conclusion, our results suggest that ALDH3A1 may act to protect corneal cells against cellular oxidative damageby metabolizing toxic lipid peroxidation products (e.g., 4-HNE), maintaining cellular GSH levels and redox balance, and operating as anantioxidant.© 2006 Elsevier Inc. All rights reserved.
Keywords: ALDH3A1; Apoptosis; Cornea; GSH; Fibroblasts; Lipid peroxidation
The mammalian cornea, an avascular tissue at the anteriorsurface of the eye, is structurally and functionally organizedto serve as a protective barrier between the externalenvironment and the internal ocular tissues. There are three
Abbreviations: ALDH, aldehyde dehydrogenase; ALDH3A1, aldehydedehydrogenase 3A1; UV, ultraviolet; 4-HNE, 4-hydroxy-2-nonenal; LPO, lipidperoxidation; MMC, mitomycin C; VP-16, etoposide; ROS, reactive oxygenspecies; GSSG, oxidized glutathione; GSH, reduced glutathione; TKT,transketolase; SV40, simian virus 40; DNP, 2,4-dinitrophenylhydrazone;TRK43, rabbit corneal fibroblastic cell line; ΔpCEP4, mammalian expressionvector; mock-TRK43, rabbit corneal epithelial cell line stably transfected withthe vectorΔpCEP4 alone; ALDH3A1-TRK43, rabbit corneal epithelial cell linestably transfected with human ALDH3A1 cDNA.⁎ Corresponding author. Fax: +1 303 315 6281.E-mail address: [email protected] (V. Vasiliou).
1 These authors contributed equally to this work.2 Current address: Department of Molecular Biology and Genetics, Democri-
tus University of Thrace, Dimitras 19, 68100 Alexandroupolis, Greece.
0891-5849/$ - see front matter © 2006 Elsevier Inc. All rights reserved.doi:10.1016/j.freeradbiomed.2006.08.009
distinguished components of the cornea: the epithelium, thestroma, and the endothelium [1]. The epithelium, the mostsuperficial surface of the eye, consists of several cell layersthat are constantly replaced by proliferation and differentia-tion of the corneal limbal stem cells. The stroma lies beneaththe epithelium and is the largest layer of the cornea,constituting approximately 90% of corneal thickness. It ismade up of a highly organized extracellular matrix, producedlargely by stromal keratocytes, that is responsible for theremarkable strength and light transparency of the cornea. Theinner cellular layer, the endothelium, comprises a single layerof cells that separates the stroma and the aqueous humor andis responsible for regulating corneal osmotic balance [2].Notable unique features of the cornea are its opticaltransparency, light refraction, and resistance to oxidativedamage and neoplasia [1].
The cornea is constantly exposed to light and harmfulenvironmental factors (such as ultraviolet (UV) radiation),

1460 N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
conditions that are known to induce cellular damage through theformation of reactive oxygen species (ROS). ROS can initiatelipid peroxidation (LPO) and cause protein modifications andDNA damage, leading eventually to cell death by apoptotic andnecrotic mechanisms [3,4]. However, the corneal tissue seemsto be well protected against oxidative damage. This uniqueproperty of the cornea is likely to be reflected in the geneexpression pattern of this tissue. The cornea expresses highlevels of certain proteins, named crystallins [5], some of whichare known to have protective effects against oxidative stress [6].The first recognized corneal crystallin was aldehyde dehydro-genase 3A1 (ALDH3A1) in the bovine cornea [7]. Othercorneal crystallins that have been identified include transketo-lase (TKT) in murine, rabbit, and human corneas; ALDH1A1 inthe rabbit cornea; α-enolase in the human, murine, and chickencorneas; isocitrate dehydrogenase in the bovine cornea;peptidylprolyl cis–trans-isomerase and argininosuccinatelyase in the chicken cornea; glutathione S-transferase-relatedprotein in the squid cornea; and gelsolin and actin in the fishcornea [8,9].
ALDH3A1 is a member of the aldehyde dehydrogenasesuperfamily [10] and catalyzes the NAD(P)+-dependent oxida-tion of aromatic and medium chain aldehydes, includingaldehydic products of LPO such as 4-HNE [11]. Immunohisto-chemical studies in the human cornea revealed that ALDH3A1 isexpressed in both epithelial cells and stromal keratocytes, but notin endothelial cells [11]. The ability of ALDH3A1 to protectcorneal tissue against UV-induced oxidative damage seems to becentral to its role as corneal crystallin. SWR/J mice, a strainprone to corneal hazing after UVB irradiation [12], lackALDH3A1 expression [13], presumably due to mutationsdetected in the structural Aldh3a1 gene [14]. In addition, theability of ALDH3A1 to prevent apoptosis induced by 4-HNEand UV radiation has been well documented [6,15]. Themechanism of such protection involves 4-HNE detoxificationby ALDH3A1, thereby preventing accumulation of 4-HNE-adducted proteins and activation of caspase-3 [6]. In addition tometabolism of the aldehydic products of LPO, several otherhypotheses have been proposed regarding the protective role ofALDH3A1 against oxidative stress. These include directabsorption of UV-light [7,16], direct scavenging of ROS [17],and production of the antioxidant NAD(P)H (NADP andNADPH) during metabolism [18]. NADPH is an essentialreducing equivalent for the regeneration of GSH from itsoxidized form (GSSG) via the glutathione reductase/peroxidasesystem and for the activity of the NADPH-dependent thior-edoxin system [19]. Accordingly, it acts as an indirectantioxidant by maintaining the reducing power of glutathioneand thioredoxin. NAD(P)H may also act as a direct antioxidantby contributing to the reduction of glutathiyl radicals or tyrosylradicals [20]. Finally, a chaperone-like activity [21,22] and anovel function as a cell cycle regulator [23] have also beenproposed for the ALDH3A1 protein.
The aim of the present study was to extend our investiga-tions into the protective role of ALDH3A1 in cornealkeratocytes. Recent studies have shown that ALDH3A1 isabundantly expressed in human and mouse corneal keratocytes
[9,11], but is replaced by ALDH1A1 in rabbit cornealkeratocytes [24]. Further, culture of keratocytes in serum-containing medium markedly down regulates expression ofALDH in keratocytes [9]. Therefore, we have used animmortalized rabbit corneal fibroblast cell line [25], whichdoes not express ALDH3A1 and shows markedly reduced orabsent expression of ALDH1A1. This study shows for the firsttime that rabbit corneal fibroblasts stably transfected tooverexpress ALDH3A1 are more resistant to oxidative damageinduced by H2O2 or the DNA-damaging agent MMC or VP-16than are vector-transfected cells. In addition, this studyprovides evidence regarding the mechanisms by whichALDH3A1 may protect against cellular damage.
Materials and methods
Reagents
Unless otherwise specified, all tissue culture media, supple-ments, growth factors, assay reagents, protein inhibitors, andbuffers were from Gibco BRL (Gaithersburg, MD, USA) orSigma–Aldrich Co (St. Louis, MO, USA). The LipofectaminePlus reagent and hygromycin were purchased from Invitrogen(Carlsbad, CA, USA). The bicinchoninic acid kit was purchasedfrom Pierce Chemical Co. (Rockford, IL, USA). The poly-vinylidene difluoride (PVDF) membranes were obtained fromImmobilon-P Millipore (Bedford, MA, USA). Protein A/G–agarose beads were supplied by Calbiochem (La Jolla, CA,USA). 4-HNE was from Cayman Chemical Co. (Ann Arbor,MI, USA) and the monoclonal anti-4-HNE antibody waspurchased from Oxis International Inc. (Portland, OR, USA).The monoclonal antibody against human ALDH3A1 hasrecently been developed by us [11]. Horseradish peroxidase-conjugated secondary antibody was obtained from The JacksonLaboratory (West Grove, PA, USA). The chemiluminescencekit was purchased from NEN Life Science Products (Boston,MA, USA). The Oxyblot detection kit was purchased fromChemicon International (Temecula, CA, USA).
Cell culture
The development and characterization of the rabbit cornealfibroblastic cell line (TRK43) from SV40 transformation ofnormal rabbit corneal fibroblasts has been described elsewhere[25]. These cells express markedly reduced levels of ALDH1A1and TKT and lack expression of ALDH3A1. TRK43 cells weregrown in Dulbecco's modified Eagle's medium supplementedwith 10% fetal bovine serum and 100 U/ml penicillin–100 μg/ml streptomycin solution and maintained at 37°C in ahumidified 5% CO2 incubator. Cells in exponential growth(about 70% confluent) and at passages between 10 and 20 wereused.
Generation of ALDH3A1-transfected cell lines
The mammalian expression vector (ΔpCEP4) and theALDH3A1 expression construct (ΔpCEP4-ALDH3A1) have

1461N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
been described before [26]. Stable transfection of TRK43 cellswith either ΔpCEP4 or ΔpCEP4-ALDH3A1 was performedusing the Lipofectamine Plus reagent according to themanufacturer's protocol. Stably transfected cells were selectedin culture medium containing 0.4 mg/ml hygromycin for 4–6 weeks before individual colonies were obtained. Hygromycin-resistant clones were isolated from individual colonies andfurther expanded and characterized for ALDH3A1 expressionby enzyme activity assays and Western blot analyses.
Preparation of cell lysates, corneal extracts, and proteindetermination
Cells were washed twice with ice-cold PBS, lysed in celllysis buffer (25 mM Tris, 0.25 M sucrose (pH 7.4), 0.5 μg/mlleupeptin, 0.5 μg/ml aprotinin, 1 μg/ml pepstatin, 100 μg/mlphenylmethylsulfonyl fluoride, 0.1% Triton X-100), andsonicated for 30 s (using a Branson sonifier, Model 250;VWR Scientific, Willard, OH, USA) at an intermediate setting(output∼3). The lysates were cooled on ice for 3–5 min and thesonicating–cooling cycle was repeated three more times.Finally, cell lysates were centrifuged at 18,000 g for 30 minat 4°C. Eyes of 8-week-old C57BL/6J mice were enucleatedafter sacrifice by CO2 narcosis. Whole corneas were firstexcised, rinsed in PBS, and homogenized in lysis buffer and thelysates were processed as described above. All animals weretreated in full compliance with the Association for Research inVision and Ophthalmology resolution on usage and treatment ofanimals in research. Protein concentrations in cell lysates weremeasured using the bicinchoninic acid method, according to themanufacturer's details.
ALDH3A1 enzyme activity assay
Determination of ALDH activity was performed spectropho-tometrically (Beckman Model DU-640; Fullerton, CA, USA)using 5.0 mM benzaldehyde (dissolved in 20% methanol) as asubstrate and 2.5 mMNADP+ as a coenzyme, by monitoring theproduction of NADPH at 340 nm and 25°C, as describedpreviously [14]. Enzyme-specific activities were expressed asnmol NADPH/min/mg protein.
Western blot analysis
Levels of ALDH3A1, presence of 4-HNE-adducted proteins,and detection of protein carbonylation were determined byWestern blot analysis. Thirty micrograms of cellular protein(18,000 g supernatant) was separated by SDS–polyacrylamidegel electrophoresis (SDS–PAGE) using 10% polyacrylamidegels. Proteins were then transferred onto PVDF membranes orprocessed for Coomassie blue staining. The membranes wereblocked overnight with 5% nonfat milk in 50 mM Tris/150 mMNaCl (pH 7.5), containing 0.1% Tween 20, and subsequentlyprobed with mouse monoclonal anti-human ALDH3A1 (1:1) ormonoclonal anti-4-HNE antibody (1:1000) for 2 h at roomtemperature. Horseradish peroxidase-conjugated secondaryantibody goat anti-mouse IgG was used at a dilution of 1:5000
(30 min, room temperature). Labeled protein bands weredetected by enhanced chemiluminescence. Densitometry datawere normalized to β-actin and analyzed using the Scion Imagesoftware (http://www.scioncorp.com).
Cytotoxicity assays
Cells (3 × 104) were plated in 96-well culture plates, allowedto attach overnight, and then treated in triplicate with H2O2 (0–1000 μM), MMC (0–200 μg/ml), or VP-16 (0–1000 μM) incomplete medium for 16 h. After treatment, cells were allowedto recover by replacement of the culture medium with freshcomplete medium and further incubation for 2 h. After therecovery period, cell viability was determined by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide]assay [27], which measures cell viability based on the abilityof mitochondrial succinyl dehydrogenase in living cells toconvert the yellow substrate MTT into a dark blue formazanproduct, or the sulforhodamine B (SRB) assay, which detectsthe binding of SRB to the basic amino acids of cellular proteins[28]. In the case of the MTTassay, culture medium was replacedwith 1 mg/ml MTT in serum-free medium and the cells wereincubated for 2 h. The MTT solution was then gently removedand the formazan crystals were solubilized by the addition of100 μl DMSO. The optical density of the resultant solution wasmeasured on a microplate reader (Thermomax, MolecularDevices Corp., Sunnyvale, CA, USA) at a wavelength of550 nm. In the case of the SRB assay, cells were fixed by theaddition of 25 μl of cold 50% (w/v) trichloroacetic acid. After1 h incubation at 4°C, wells were washed five times with waterand stained with 0.4% (w/v) SRB in 1% (v/v) acetic acid for30 min at room temperature. Wells were washed five times with1% acetic acid to rinse out the unbound dye and allowed to air-dry. Absorbance was measured at 540 nm after solubilization ofthe dye in 10 mM Tris base. The acute EC50 values for H2O2,MMC, and VP-16 were calculated from the cytotoxicity data byregression analysis using the equation of a four-parameterlogistic curve (SigmaPlot software, version 7.0, 2001).
DNA fragmentation assay
Cells (5 × 105) were treatedwithMMC (25 and 50μg/ml), VP-16 (50 and 100 μM), or H2O2 (400 and 800 μM) for 16 h. Aftertreatment, cells were collected, washed with PBS, resuspended inlysis buffer (10 mMTris–HCl, pH 7.4, containing 10 mMEDTA,10 mM NaCl, 0.5% SDS, and 0.1 mg/ml proteinase K), andincubated overnight at 50°C. DNAwas extracted using phenol–chloroform and subsequently incubated with RNase A(0.1 mg/ml) for 1 h at 37°C. DNA samples were analyzedusing conventional electrophoresis in a 1.5% (w/v) agarose geland visualized under UV illumination (Fluor-S MultiImager;Bio-Rad Laboratories, Richmond, CA, USA).
Immunoprecipitation and detection of ALDH3A1 carbonylation
For immunoprecipitation experiments, equal numbers ofexponentially growing ALDH3A1-TRK43 cells (5 × 105) were

Table 1ALDH3A1 enzyme activity in TRK43 transfected cell lines
Cell line ALDH3A1 activity a
TRK43 (parental cells) UndetectableMock-TRK43 UndetectableALDH3A1-5 19.3 ± 0.3ALDH3A1-6 3951.3 ± 2.4ALDH3A1-10 4532.6 ± 1.8ALDH3A1-11 20.7 ± 1.1ALDH3A1-12 13.9 ± 1.7ALDH3A1-15 863.6 ± 2.8ALDH3A1-16 80.4 ± 2.7ALDH3A1-17 3923.7 ± 4.4
Data are expressed as means ± SE from triplicates in two separate experiments.a Specific activities are expressed as nmol NADPH/min/mg protein and were
assayed using benzaldehyde and NADP+ as substrate and coenzyme,respectively (as described under Materials and methods).
1462 N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
treated with H2O2 (500 μM), MMC (50 μg/ml), or VP-16(100 μM) for 16 h, harvested by trypsinization, washed in PBS,and then lysed in freshly prepared lysis buffer with proteaseinhibitors (as described above). Equal aliquots of supernatant(6 mg) were added to tubes containing anti-ALDH3A1 mono-clonal antibody and allowed to incubate overnight at 4°C undergentle agitation. Protein A/G–agarose beads (35 μl) were thenadded, and the incubations were continued for an additional90 min at 4°C. Protein A/G–agarose beads were collected bycentrifugation and washed four times with 50 mM Tris–HCl, pH8.0, containing 150 mM NaCl, 1% (v/v) Nonidet P-40 and thenwashed one time with 50 mM Tris–HCl, pH 8.0. The protein A/G–agarose beads were processed for detection of carbonylgroups in oxidatively modified immunoprecipitated proteins byan antibody-mediated process using the Oxyblot detection kitaccording to the manufacturer's instructions. Briefly, theimmunoprecipitated proteins were treated with 2,4-dinitrophe-nylhydrazine to derivatize the carbonyl groups to 2,4-dinitro-phenylhydrazone (DNP). They were then separated on 10%polyacrylamide gels, transferred to PVDF membranes, andprobed using an antibody specific to the DNP-derivatizedresidues on oxidatively damaged proteins. Blots were strippedand subsequently reprobed for ALDH3A1 to assess oxidation ofALDH3A1.
Measurement of intracellular GSH levels by high-performanceliquid chromatography (HPLC)
Cells (4 × 105) were seeded onto 30-mm culture plates andexposed to complete medium containing H2O2 (500 μM), MMC(50 μg/ml), or VP-16 (100 μM). After 16 h treatment, cells wereharvested by trypsinization and resuspended in 2 ml of PBS. Analiquot (50 μl) was counted manually using a hemocytometer andtrypan blue staining for normalizing the GSH results. Theremaining cells were collected by centrifugation and lysed (asdescribed above). Cellular proteins were precipitated by adding10% metaphosphoric acid (150 μl/ml). The samples were cooledon ice and centrifuged at 18,000 g for 20 min at 4°C. Super-natants were then analyzed for GSH content by HPLC withelectrochemical detection as previously described [29]. Samplecontents of GSH were expressed as nmol GSH per 106 cells.
Statistical analysis
All experiments were performed in triplicate and repeated atleast three times. Data are presented as means ± SE. Allcomparisons of mean values between groups were performedusing one-way ANOVA. A level of p < 0.05 was consideredstatistically significant.
Results
Expression of ALDH3A1 in transfected rabbit cornealfibroblasts
To study whether ALDH3A1 has a protective role againstcellular oxidative damage in rabbit corneal fibroblasts, we
stably transfected TRK43 cells with the human ALDH3A1cDNA being under control of a viral gene promoter. Naive(parental) TRK43 cells do not express ALDH3A1 endoge-nously (Table 1). Several ALDH3A1-expressing TRK43 celllines were isolated and enzyme activity assays and Westernblot analyses were conducted to confirm expression of thehuman ALDH3A1. Enzymatic activity profiles revealed threetypes of ALDH3A1-expressing cell lines (Table 1). Cell linesALDH3A1-5, -11, -12, and -16 were characterized by lowactivity (<100 nmol NADPH/min/mg); the ALDH3A1-15 cellline showed moderate activity (∼860 nmol NADPH/min/mg);and ALDH3A1-6, -10, and -17 cell lines demonstrated thehighest activities (>3000 nmol NADPH/min/mg) (Table 1). Noenzymatic activity was detected in control cells transfected withthe vector alone (mock-TRK43; Table 1). Western blot analysisusing a specific antibody against human ALDH3A1 detectedthe expression of a single protein band of 54 kDa only in thosecell lines transfected with the ALDH3A1 expression construct.The intensity of the immunoreactive bands confirmed thecorrelation between the amount of ALDH3A1 detected byimmunodetection (Fig. 1A) and the levels of enzymatic activity(Table 1).
ALDH3A1 protects corneal fibrolasts against H2O2−, MMC-,or VP-16-induced cytotoxicity and apoptosis
Earlier studies have shown that exposure of mammalian cellsto H2O2, MMC, or VP-16 induces cell death and apoptosis [30–34]. The ability of these agents to induce cell death in mock- andALDH3A1-TRK43 cell lines was assessed. Mock- andALDH3A1-TRK43 cell lines expressing low (ALDH3A1-11),moderate (ALDH3A1-15), and high (ALDH3A1-10,ALDH3A1-17) transgene levels were exposed to variousconcentrations of H2O2, MMC, or VP-16 for 16 h. At first,MTT assays were chosen to assess the toxicity caused by theseoxidants. MTT assay measures the ability of mitochondrialsuccinyl dehydrogenase in living cells to reduce the tetrazoliumsalts to formazan end-product, thus providing an indication ofcell growth and viability. Representative dose–response cellviability curves obtained with mock- and ALDH3A1-10 (high

Fig. 1. (A) Western blot analysis of ALDH3A1 in transfected TRK43 cell lines.Cytosolic proteins (30 μg) from mock- or ALDH3A1-transfected TRK43 celllines (ALDH3A1-5, ALDH3A1-6, ALDH3A1-10, ALDH3A1-11, ALDH3A1-12, ALDH3A1-15, ALDH3A1-16, ALDH3A1-17) were extracted and assayedfor ALDH3A1 expression byWestern blot analysis, using specific antibodies (asdescribed under Materials and methods). The same membrane was reprobed forβ-actin (bottom) for loading correction. (B) Coomassie blue staining of proteins(30 μg) extracted from mock- and ALDH3A1-TRK43 cell lines and analyzed bySDS–PAGE (as described under Materials and methods). Position of the proteinband corresponding to ALDH3A1 is indicated by the purified humanALDH3A1 (4.5 μg). Extracted proteins (30 μg) from mouse cornea were usedto compare physiological expression of ALDH3A1 with induced expression ofALDH3A1 in the ALDH3A1-transfected clones. M, molecular weight markers.
1463N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
expression) TRK43 cells after treatment with H2O2, MMC, orVP-16 and their EC50 values for each of the correspondingtreatments are shown in Fig. 2A. MTT assays performed onTRK43 cells expressing low (ALDH3A1-11) and high(ALDH3A1-17 and ALDH3A1-10) transgene levels showedthat stable expression of ALDH3A1 resulted in a dose-dependent protection against oxidant-induced cell death, asdemonstrated by the relative fold increase in EC50 values of theALDH3A1-expressing cell lines compared to control (mock)(Fig. 2B). Because the MTT assay depends on metabolicallyactive cells, and to exclude the possibility that treatments hadpossibly influenced the cellular metabolic conditions, weapplied another assay with a different endpoint, the SRB proteinbinding assay. The SRB assay is independent of metabolicconditions and detects the binding of the anionic dye SRB to thebasic amino acids of cellular proteins and, thus, assesses cellularprotein synthesis, providing a quantitative measurement of cellnumber. The protective effects of ALDH3A1 expressionobtained by MTT assays were further confirmed by SRB assays(Fig. 2C). TRK43 cells expressing low (ALDH3A1-11),medium (ALDH3A1-15), and high ALDH3A1 (ALDH3A1-17and ALDH3A1-10) levels demonstrated a dose-dependentprotection against all treatments as demonstrated by the relative
fold increase in EC50 values of the ALDH3A1-expressing celllines compared to control (mock) (Fig. 2C). Although bothMTTand SRB assays detected the gene-dosage-dependent protectionof ALDH3A1, differences in the sensitivity between the assaysfor certain oxidants (e.g., MMC) were observed, which could beexplained by differences in the endpoints of the assays used(Figs. 2B and C). It is noteworthy that two independent TRK43clones with high expression levels of ALDH3A1 (ALDH3A1-10 and ALDH3A1-17; Table 1) demonstrated almost equalprotection against oxidant treatment and that was confirmed inboth assays used (Figs. 2B and C). Based on the above results,the biological effects of ALDH3A1 stable transfection arespecific and not a consequence of a heterologous protein over-expression. Because the highest levels of ALDH3A1were foundin the ALDH3A1-10 cell line (although not as much as theprotein levels physiologically found in the mouse cornea; Fig.1B), this cell line (referred to as ALDH3A1-TRK43 in the rest ofthe text) was designated to study the role of ALDH3A1 insubsequent experiments.
The ability of H2O2, MMC, or VP-16 to induce apoptosis incorneal fibroblasts and the antiapoptotic role of ALDH3A1wereinvestigated by DNA fragmentation assays and changes in cellmorphology. DNA fragmentation, characterized by extensivecleavage of cellular DNA into oligonucleosome-sized frag-ments, is a cellular parameter considered to be a marker of theapoptotic process [35]. We found that treatment with H2O2 (400and 800 μM), MMC (25 and 50 μg/ml), or VP-16 (50 and100 μM) induced DNA fragmentation in mock-TRK43 cells, butnot in ALDH3A1-TRK43 cells (Figs. 3A–C). ALDH3A1-TRK43 cell lines expressing low levels of ALDH3A1 were notprotected against H2O2− (400 and 800 μM), MMC- (25 and50 μg/ml), or VP-16- (50 and 100 μM) induced DNA frag-mentation, in contrast to the cells expressing moderate and hightransgene levels (data not shown). Consistent with and com-plementary to the DNA fragmentation results were the morpho-logical observations made in treated mock- and ALDH3A1-TRK43 cells. Mock-TRK43 cells were characterized by morepronounced cell shrinkage than the ALDH3A1-TRK43 cellsobserved after 8 and 16 h treatment with H2O2 (500 μM), MMC(50 μg/ml), or VP-16 (100 μM) (data not shown).
ALDH3A1 inhibits the formation of 4-HNE-adducted proteinsinduced by H2O2, MMC, and VP-16
All three agents used in this study are known as beingcapable of inducing LPO in mammalian cells [36–38]. It is wellestablished that 4-HNE is one of the most reactive products ofLPO that interact with proteins and nucleic acids, therebyaltering their cellular functions [39]. To examine whethertreatment with these agents induces LPO in rabbit cornealfibroblasts, we determined the formation of 4-HNE throughdetection of 4-HNE-adducted proteins. Mock- and ALDH3A1-TRK43 cells were incubated with MMC (25 μg/ml), VP-16(100 μM), or H2O2 (500 μM) for 16 h and cell lysates weresubjected to SDS–PAGE and Western blotting using anantibody specific for detecting the products of reaction between4-HNE and proteins. As shown in Fig. 4, treatment with any of

Fig. 2. Gene-dosage-dependent protection of ALDH3A1 against oxidant cytotoxicity. (A) Representative viability curves of ALDH3A1-10 (filled circles) and mock-TRK43 (open circles) cells after treatment with H2O2 (top), MMC (middle), or VP-16 (bottom) obtained by MTT assay. The estimated EC50 values of mock-TRK43and ALDH3A1-10 cells for each treatment were determined from the viability curves and are indicated. (B) Gene-dosage-dependent protection of ALDH3A1 againstoxidant cytotoxicity determined by MTT assay. (C) Gene-dosage-dependent protection of ALDH3A1 against oxidant cytotoxicity determined by SRB assay. Mock-and ALDH3A1-TRK43 cells with low (ALDH3A1-11), moderate (ALDH3A1-15), or high (ALDH3A1-10; ALDH3A1-17) transgene expression were plated in 96-well plates and treated with various concentrations of H2O2, MMC, and VP-16 for 16 h. EC50 values were determined from cell viability curves as described underMaterials and methods. Results are expressed as the fold increase in EC50 values of ALDH3A1-TRK43 clones relative to control (mock). Data are presented asmeans ± SE from triplicates in three separate experiments. *p<0.05, one-way ANOVA, compared to mock cells under the same treatment conditions.
1464 N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
the agents resulted in increased 4-HNE-protein adduct forma-tion in mock-TRK43 cells. In contrast, there was very little orno accumulation of 4-HNE-protein adducts in ALDH3A1-TRK43 cells induced by these treatments. These data indicatethat treatment of rabbit corneal fibroblasts with H2O2, MMC, orVP-16 induces 4-HNE formation and that ALDH3A1 canprotect these cells by decreasing the protein modificationsassociated with 4-HNE-adduct formation.
Modification of ALDH3A1 in ALDH3A1-TRK43 cells aftertreatment with H2O2, MMC, and VP-16
To determine whether ALDH3A1 expression and/or enzy-matic activity was altered after treatment with H2O2, MMC, orVP-16, we examined the protein expression levels and enzymaticactivity as well as oxidation of the ALDH3A1 protein after 16 hexposure to H2O2 (500 μM), MMC (50 μg/ml), or VP-16
(100 μM) in ALDH3A1-TRK43 cells. As shown in Fig. 5A,ALDH3A1 enzyme activities were unchanged 8 and 16 h aftertreatment with these agents. No changes were detected inALDH3A1 protein levels, as determined byWestern blot analysisin ALDH3A1-TRK43 cells under the same conditions (Fig. 5B).However, increased carbonylation of ALDH3A1 protein wasinduced by treatment with H2O2, MMC, or VP-16 (Fig. 6).
ALDH3A1 maintains GSH levels
The catalytic activity of ALDH3A1 generates NAD(P)H,which is linked to the regeneration of reduced GSH fromoxidized disulfide GSSG via the glutathione reductase/perox-idase system. Due to the role of GSH in maintaining the cellularredox balance [40], we examined the levels of reduced GSH inboth mock- and ALDH3A1-TRK43 cell lines after 16 htreatment with H2O2 (500 μM), MMC (50 μg/ml), or VP-16
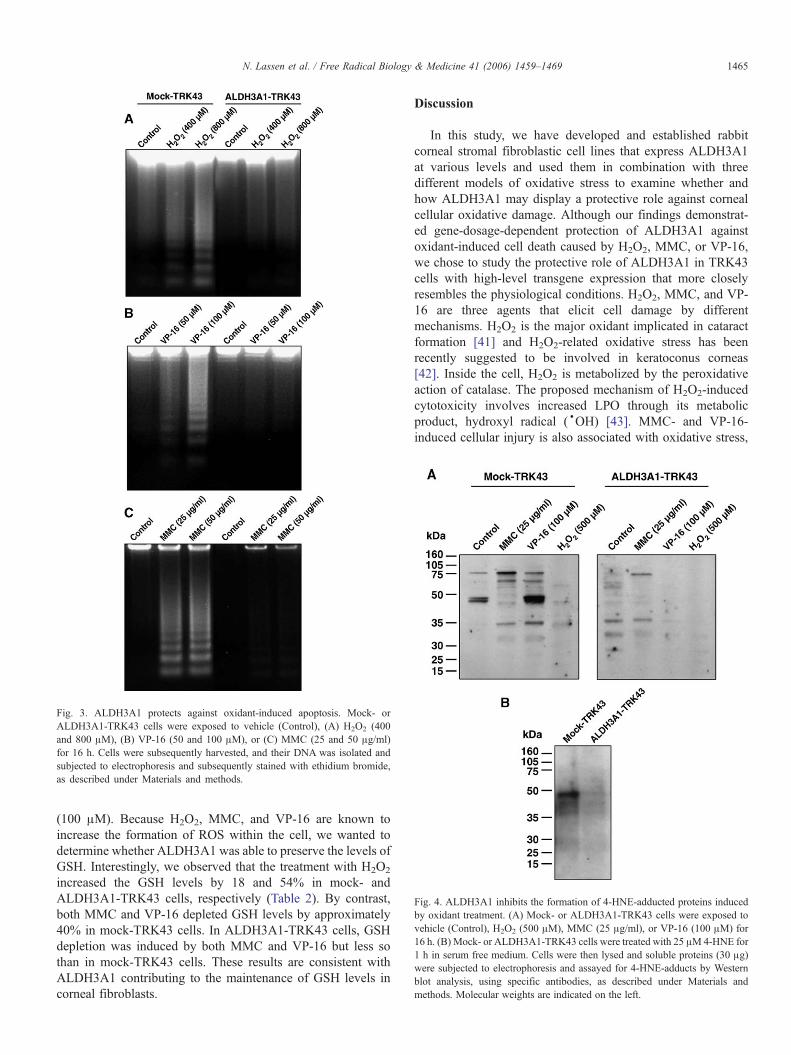
Fig. 4. ALDH3A1 inhibits the formation of 4-HNE-adducted proteins inducedby oxidant treatment. (A) Mock- or ALDH3A1-TRK43 cells were exposed tovehicle (Control), H2O2 (500 μM), MMC (25 μg/ml), or VP-16 (100 μM) for16 h. (B) Mock- or ALDH3A1-TRK43 cells were treated with 25 μM4-HNE for1 h in serum free medium. Cells were then lysed and soluble proteins (30 μg)were subjected to electrophoresis and assayed for 4-HNE-adducts by Westernblot analysis, using specific antibodies, as described under Materials andmethods. Molecular weights are indicated on the left.
Fig. 3. ALDH3A1 protects against oxidant-induced apoptosis. Mock- orALDH3A1-TRK43 cells were exposed to vehicle (Control), (A) H2O2 (400and 800 μM), (B) VP-16 (50 and 100 μM), or (C) MMC (25 and 50 μg/ml)for 16 h. Cells were subsequently harvested, and their DNA was isolated andsubjected to electrophoresis and subsequently stained with ethidium bromide,as described under Materials and methods.
1465N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
(100 μM). Because H2O2, MMC, and VP-16 are known toincrease the formation of ROS within the cell, we wanted todetermine whether ALDH3A1 was able to preserve the levels ofGSH. Interestingly, we observed that the treatment with H2O2
increased the GSH levels by 18 and 54% in mock- andALDH3A1-TRK43 cells, respectively (Table 2). By contrast,both MMC and VP-16 depleted GSH levels by approximately40% in mock-TRK43 cells. In ALDH3A1-TRK43 cells, GSHdepletion was induced by both MMC and VP-16 but less sothan in mock-TRK43 cells. These results are consistent withALDH3A1 contributing to the maintenance of GSH levels incorneal fibroblasts.
Discussion
In this study, we have developed and established rabbitcorneal stromal fibroblastic cell lines that express ALDH3A1at various levels and used them in combination with threedifferent models of oxidative stress to examine whether andhow ALDH3A1 may display a protective role against cornealcellular oxidative damage. Although our findings demonstrat-ed gene-dosage-dependent protection of ALDH3A1 againstoxidant-induced cell death caused by H2O2, MMC, or VP-16,we chose to study the protective role of ALDH3A1 in TRK43cells with high-level transgene expression that more closelyresembles the physiological conditions. H2O2, MMC, and VP-16 are three agents that elicit cell damage by differentmechanisms. H2O2 is the major oxidant implicated in cataractformation [41] and H2O2-related oxidative stress has beenrecently suggested to be involved in keratoconus corneas[42]. Inside the cell, H2O2 is metabolized by the peroxidativeaction of catalase. The proposed mechanism of H2O2-inducedcytotoxicity involves increased LPO through its metabolicproduct, hydroxyl radical (·OH) [43]. MMC- and VP-16-induced cellular injury is also associated with oxidative stress,

Fig. 6. Treatment with oxidative agents increases carbonylation of ALDH3A1protein. (A) ALDH3A1-TRK43 cells were treated with vehicle (Control, C),H2O2 (500 μM), MMC (50 μg/ml), or VP-16 (100 μM) and harvested after 16 h.Total lysates were immunoprecipitated using ALDH3A1 monoclonal antibody.DNP-derivatized (+) and not derivatized (−) proteins were separated by SDS–PAGE and transferred to PVDF membrane. Oxidized proteins were recognizedby an antibody specific to the DNP-derivatized residues and detected byenhanced chemiluminescent reagents. Molecular weights are indicated on theleft. (B) The same PVDF membrane was stripped and reprobed withALDH3A1 monoclonal antibody. Bands labeled by the ALDH3A1 antibodyare superimposed on the bands of the respective molecular weight seen withthe anti-DNP antibody, as indicated by the arrow on the right. (C) Quantitativedensitometric analysis of oxidized ALDH3A1 using the Scion Image software.Relative increases in band optical densities (arbitrary units) are normalized forthe total ALDH3A1 in each lane (B). Standard error was less than 10% in allcases. *p < 0.05, one-way ANOVA, compared to respective vehicle-treated(control) cells.
Fig. 5. Treatment with oxidative agents does not alter ALDH3A1 expressionor function. ALDH3A1-TRK43 cells were treated with vehicle (control, C),H2O2 (500 μM), MMC (50 μg/ml), or VP-16 (100 μM) for 8 or 16 h andALDH3A1 (A) enzymatic activities and (B) protein levels were determined asdescribed under Materials and methods. Specific enzymatic activities werenormalized as a percentage of the activity in control cells, whereasALDH3A1 protein bands were normalized relative to their respective β-actin expression. Data were obtained from triplicates in two separateexperiments. Standard error was less than 10% in all cases, and there wasno statistical difference in all the treatments compared to respective vehicle-treated (control) cells (p < 0.05, one-way ANOVA).
1466 N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
although through different mechanisms. MMC is an antitumoragent that alkylates and cross-links DNA. These effects aredependent on reductive bioactivation of MMC that occursthrough a one-electron pathway producing a semiquinone freeradical intermediate or a two-electron pathway producing ahydroquinone intermediate. The one-electron reduction ismainly catalyzed by NADPH-cytochrome P450 reductase[44], and the two-electron reduction is mainly mediated byNAD(P)H:quinone oxidoreductase [45]. In the presence ofoxygen, the semiquinone enters a redox cycle that forms aseries of ROS, such as superoxide and ·OH radicals, whichare involved in oxidative damage of cell components [46,47].Alternatively, the hydroquinone intermediate leads to theformation of stable MMC metabolites which, in turn, interactwith DNA to cause alkylation and cross-linking [44].Etoposide (VP-16), a semisynthetic derivative of podophyllo-toxin, is an antitumor agent currently used to treat a variety ofhematopoietic and solid tumors. VP-16, a DNA topoisome-rase II poison, contains a hindered phenolic ring that canparticipate in intracellular redox signaling reactions. Oxidativeactivation of VP-16 catalyzed by enzymes capable of one-electron transfer reactions (e.g., peroxidases, tyrosinase) leadsto the generation of etoposide phenoxyl radicals that promoteselective oxidation of protein thiols and consequent induction
of ROS generation that seems to contribute to apoptotic celldeath [38].
To the best of our knowledge, this is the first reportsuggesting the involvement of ALDH3A1 in the protection ofcorneal fibroblasts against oxidative stress. There are at leastthree possible mechanisms by which ALDH3A1 protectsagainst apoptosis induced by the three oxidative agents usedin this study. These include: (a) enzymatic detoxification ofaldehyde metabolites produced during LPO, (b) antioxidantfunction, and (c) generation of NADPH, which maintains theantioxidative power of glutathione and may also act as a directantioxidant. As mentioned above, it is well established thatH2O2, MMC, and VP-16 create oxidant conditions that lead toinduction of LPO. Among the 200 aldehydes generated duringLPO, 4-HNE represents 5% of the total aldehydes and is themost active one [39]. 4-HNE is a potent electrophile thatinteracts with proteins, phospholipids, and nucleic acids andcontributes significantly to the pathophysiologic impact ofoxidative stress [39,48]. We and other investigators have shownthat 4-HNE causes apoptotic cell death and that ALDH3A1inhibits 4-HNE-induced apoptosis and protein adduct formationin various cell lines [6,15]. We now present evidence that

Table 2Effects of oxidant exposure on levels of reduced glutathione (GSH) in mock-and ALDH3A1-TRK43 cell lines
Treatment GSH (nmol/106 cells ± SE) a GSH (% control ± SE) b
Mock-TRK43Control 48.8 ± 1.20 100.0 ± 2.4H2O2 57.6 ± 0.05 118.2 ± 0.1⁎
MMC 29.2 ± 0.02 59.8 ± 0.1⁎
VP-16 30.5 ± 0.22 62.5 ± 0.5⁎
ALDH3A1-TRK43Control 47.7 ± 0.86 100.0 ± 1.8H2O2 73.5 ± 0.21 154.1 ± 0.4⁎,⁎⁎
MMC 38.2 ± 0.84 80.1 ± 1.8⁎,⁎⁎
VP-16 41.4 ± 0.58 86.8 ± 1.2⁎,⁎⁎
Data are presented as the means±SE from three independent experiments.a GSH content was determined in mock- and ALDH3A1-TRK43 cell lines
after 16 h treatment with vehicle (control), H2O2 (500 μM), MMC (50 μg/ml),or VP-16 (100 μM) as described under Materials and methods. Results fromthree independent experiments are shown (n=3).b Cellular GSH content was normalized to the GSH content of vehicle-treated
cells (control).⁎ p < 0.05, one-way ANOVA, compared to respective vehicle-treated(control) cells.⁎⁎ p < 0.05, one-way ANOVA, compared to same treatment in mock-TRK43cells.
1467N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
treatment of rabbit corneal fibroblasts with H2O2, MMC, or VP-16 enhances LPO, as indicated by the increased levels of 4-HNE–protein adducts, and leads to apoptotic cell death. In thecase of ALDH3A1-expressing cells, reduced levels of 4-HNE-adducted proteins were observed. Based on the capacity ofALDH3A1 to metabolize 4-HNE [6,11], we can assume thatALDH3A1 protects rabbit fibroblasts against the apoptoticeffect of the three agents by metabolizing 4-HNE. Thesefindings are in agreement with our previous studies, whichdemonstrated that ALDH3A1 prevents apoptosis in humancorneal epithelial cells induced by VP-16 and MMC [23].
The second possible mechanism by which ALDH3A1protects corneal rabbit fibroblasts against apoptosis includesan antioxidant function of this protein. It is known that ROSgenerated after treatment with oxidants can readily oxidizeproteins [49]. In addition to the fragmentation of polypeptidechains, oxidation of proteins leads to modifications of aminoacid side chains, such as generation of methionine sulfoxides,cysteine sulfonic acids, cysteine disulfide bonds, or tyrosinecross-links [50]. Another alteration is the introduction ofcarbonyl groups into proteins at lysine, arginine, proline, orthreonine residues; protein carbonylation is the most generaland well-used marker of severe oxidative damage [51].Oxidative modifications of proteins, if not degraded by thecellular proteolytic systems [48], could impair a wide range ofbiochemical functions, resulting in alterations or even disrup-tion of metabolic pathways [51]. It is well known thataccumulation of oxidized proteins occurs during aging and inpathologies such as cataract, diabetes, atherosclerosis, andneurodegenerative diseases [49]. Because ALDH3A1 is presentin the cornea of most mammalian species in amounts thatexceed any likely catalytic role [52], several other functionshave been postulated for this corneal crystallin, including a
direct antioxidant function. Our results support this notion inthat we observed a two- to threefold increase in carbonylation ofALDH3A1 after treatment with oxidants without any noticeabledecrease in enzymatic activity or ALDH3A1 protein levels.That no significant changes were detected in ALDH3A1enzymatic activity was somewhat unexpected given that veryoften protein oxidation is associated with loss of function [51].However, it is conceivable that oxidized residues in theALDH3A1 protein do not affect its enzymatic activity. Thefact that ALDH3A1 is expressed in such high levels in thecornea is also consistent with its function as antioxidant. Such isthe case for albumin, another abundant corneal protein, whichacts as a native antioxidant by scavenging hydrogen peroxide[53]. ALDH3A1, due to its abundance in the cornea, maybecome preferably oxidized over other vital proteins underconditions of oxidative stress, thus preventing more extensivecellular damage and leading to cell survival.
The third possible mechanism by which ALDH3A1 mayprotect corneal rabbit fibroblasts against oxidative stress-inducedcell death is by supplying NADPH through its metabolicfunction, thus enhancing the cellular antioxidant capacity.NADPH is an essential reducing equivalent for both theregeneration of GSH and the normal function of the NADPH-dependent thioredoxin system. In addition to its indirectantioxidant functions, NAD(P)H acts also as a directly operatingantioxidant [20]. In mammalian cells, major sources of NADPHfor thiol-dependent antioxidant functions include the cytosolicand mitochondrial NADP+-dependent isocitrate dehydrogenase[54,55], the cytosolic glucose-6 phosphate dehydrogenase [56],and the mitochondrial malic enzyme [57]. Because ALDH3A1 isa major protein in the cornea, and it uses both NADP+ and NAD+
as coenzymes [58], it may significantly contribute to theproduction of reducing power in the form of NAD(P)H in thistissue. This is supported by our previous study in which wereported an increase in the cellular concentration of NADPH afterexposure to 4-HNE in ALDH3A1-transfected corneal epithelialcells [6]. We now provide evidence that ALDH3A1-transfectedcells respond to H2O2-induced oxidative stress by increasingcellular GSH. In agreement with these data, ALDH3A1prevented glutathione depletion caused by MMC and VP-16.The differences in cellular responses (increase or prevention ofGSH depletion) depend upon the nature of the oxidant and mayreflect the different mechanisms by which these agents induceoxidative damage. In all cases, ALDH3A1 seems to maintaincellular GSH levels, possibly through the production of NADPHduring the metabolism of LPO aldehydes generated by oxidant-induced ROS production. Our demonstration of less cytotoxiceffects of VP-16 and MMC in ALDH3A1-expressing cellscompared to naive cells is consistent with previous findings onthe protection of acute myeloblastic leukemia cells against VP-16-induced apoptosis by high glutathione levels [59] andenhanced toxicity of MMC on small lung carcinoma cellscaused by depletion of intracellular GSH [60]. Because thecellular sensitivity to these agents is highly dependent on theredox status of the cell, it is also possible that ALDH3A1 andGSH act together compensatively to defend against oxidativedamage.

1468 N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
To conclude, we have shown that ALDH3A1, an enzymeabundantly expressed in the cornea, can act as a multifunc-tional protein protecting against cellular oxidative damage bypreventing LPO-induced protein adduction, operating as anantioxidant, and maintaining intracellular GSH levels andredox balance, potentially through regenerating NADPH.Understanding the interplay between free radical biochemistryand ALDH3A1 will further elucidate the molecular mechan-isms by which this enzyme exerts its protective functions in thecornea.
Acknowledgments
We thank our colleagues, especially Dr. David Thompson,for valuable discussions and critical reading of the manuscript.This work was supported by NIH Grant EY11490 (V.V.).
References
[1] Klyce, S. D.; Beuerman, R. W. The Cornea. Churchill Livingstone, NewYork; 1988.
[2] Fischbarg, J.; Maurice, D. M. An update on corneal hydration control. Exp.Eye Res. 78:537–541; 2004.
[3] Hightower, K. R.; McCready, J. P.; Borchman, D. Membrane damage inUV-irradiated lenses. Photochem. Photobiol. 59:485–490; 1994.
[4] Shinohara, T.; Singh, D. P.; Chylack, L. T. Jr. Age-related cataract:immunity and lens epithelium-derived growth factor (LEDGF). J. Ocul.Pharmacol. Ther. 16:181–191; 2000.
[5] Piatigorsky, J. Enigma of the abundant water-soluble cytoplasmic proteinsof the cornea: the “refraction” hypothesis. Cornea 20:853–858; 2001.
[6] Pappa, A.; Chen, C.; Koutalos, Y.; Townsend, A. J.; Vasiliou, V. Aldh3a1protects human corneal epithelial cells from ultraviolet- and 4-hydroxy-2-nonenal-induced oxidative damage. Free Radical Biol. Med. 34:1178–1189; 2003.
[7] Abedinia, M.; Pain, T.; Algar, E. M.; Holmes, R. S. Bovine cornealaldehyde dehydrogenase: the major soluble corneal protein with a possibledual protective role for the eye. Exp. Eye Res. 51:419–426; 1990.
[8] Piatigorsky, J. Crystallin genes: specialization by changes in gene regulationmay precede gene duplication. J. Struct. Funct. Genom. 3:131–137; 2003.
[9] Jester, J. V.; Budge, A.; Fisher, S.; Huang, J. Corneal keratocytes:phenotypic and species differences in abundant protein expression and invitro light-scattering. Invest Ophthalmol. Visual Sci. 46:2369–2378; 2005.
[10] Vasiliou, V.; Bairoch, A.; Tipton, K. F.; Nebert, D. W. Eukaryotic aldehydedehydrogenase (ALDH) genes: human polymorphisms, and recommendednomenclature based on divergent evolution and chromosomal mapping.Pharmacogenetics 9:421–434; 1999.
[11] Pappa, A.; Estey, T.; Manzer, R.; Brown, D.; Vasiliou, V. Humanal1dehyde dehydrogenase 3A1 (ALDH3A1): biochemical characterizationand immunohistochemical localization in the cornea. Biochem. J. 376:615–623; 2003.
[12] Downes, J. E.; Swann, P. G.; Holmes, R. S. Differential corneal sensitivity toultraviolet light among inbred strains of mice: correlation of ultraviolet Bsensitivity with aldehyde dehydrogenase deficiency.Cornea 13:67–72; 1994.
[13] Pappa, A.; Sophos, N. A.; Vasiliou, V. Corneal and stomach expression ofaldehyde dehydrogenases: from fish to mammals. Chem. Biol. Interact.130–132:181–191; 2001.
[14] Shiao,T.;Tran,P.;Siegel,D.;Lee,J.;Vasiliou,V.Fouraminoacidchangesareassociated with the Aldh3a1 locus polymorphism in mice which may beresponsible for corneal sensitivity to ultraviolet light. Pharmacogenetics9:145–153; 1999.
[15] Haynes, R. L.; Szweda, L.; Pickin, K.; Welker, M. E.; Townsend, A. J.Structure–activity relationships for growth inhibition and induction ofapoptosis by 4-hydroxy-2-nonenal in RAW 264.7 cells. Mol. Pharmacol.58:788–794; 2000.
[16] Mitchell, J.; Cenedella, R. J. Quantitation of ultraviolet light-absorbingfractions of the cornea. Cornea 14:266–272; 1995.
[17] Uma, L.; Hariharan, J.; Sharma, Y.; Balasubramanian, D. Corneal aldehydedehydrogenase displays antioxidant properties. Exp. Eye Res. 63:117–120; 1996.
[18] Atherton, S. J.; Lambert, C.; Schultz, J.; Williams, N.; Zigman, S.Fluorescence studies of lens epithelial cells and their constituents. Photo-chem. Photobiol. 70:823–828; 1999.
[19] Chae, H. Z.; Chung, S. J.; Rhee, S. G. Thioredoxin-dependent peroxidereductase from yeast. J. Biol. Chem. 269:27670–27678; 1994.
[20] Kirsch, M.; De, G. H. NAD(P)H, a directly operating antioxidant? FASEBJ. 15:1569–1574; 2001.
[21] Uma, L.; Hariharan, J.; Sharma, Y.; Balasubramanian, D. Effect of UVBradiation on corneal aldehyde dehydrogenase. Curr. Eye Res. 15:685–690;1996.
[22] Manzer, R.; Pappa, A.; Estey, T.; Sladek, N.; Carpenter, J. F.; Vasiliou, V.Ultraviolet radiation decreases expression and induces aggregation ofcorneal ALDH3A1. Chem. Biol. Interact. 143–144:45–53; 2003.
[23] Pappa, A.; Brown, D.; Koutalos, Y.; DeGregori, J.; White, C.; Vasiliou, V.Human aldehyde dehydrogenase 3A1 inhibits proliferation and promotessurvival of human corneal epithelial cells. J. Biol. Chem. 280:27998–28006; 2005.
[24] Jester, J. V.; Moller-Pedersen, T.; Huang, J.; Sax, C. M.; Kays, W. T.;Cavangh, H. D.; Petroll, W. M.; Piatigorsky, J. The cellular basis of cornealtransparency: evidence for ‘corneal crystallins’. J. Cell Sci. 112 (Pt. 5):613–622; 1999.
[25] Barry-Lane, P. A.; Wilson, S. E.; Cavanagh, H. D.; Petroll, W. M.; Jester,J. V. Characterization of SV40-transfected cell strains from rabbit kerato-cytes. Cornea 16:72–78; 1997.
[26] Bunting, K. D.; Townsend, A. J. Protection by transfected rat or humanclass 3 aldehyde dehydrogenase against the cytotoxic effects of oxazapho-sphorine alkylating agents in hamster V79 cell lines: demonstration ofaldophosphamide metabolism by the human cytosolic class 3 isozyme.J. Biol. Chem. 271:11891–11896; 1996.
[27] Hansen, M. B.; Nielsen, S. E.; Berg, K. Re-examination and furtherdevelopment of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 119:203–210; 1989.
[28] Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica,D.; Warren, J. T.; Bokesch, H.; Kenney, S.; Boyd, M. R. New colorimetriccytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst.82:1107–1112; 1990.
[29] Day, B. J.; van Heeckeren, A. M.; Min, E.; Velsor, L. W. Role for cysticfibrosis transmembrane conductance regulator protein in a glutathioneresponse to bronchopulmonary Pseudomonas infection. Infect. Immun.72:2045–2051; 2004.
[30] Goswami, S.; Sheets, N. L.; Zavadil, J.; Chauhan, B. K.; Bottinger, E. P.;Reddy, V. N.; Kantorow, M.; Cvekl, A. Spectrum and range of oxidativestress responses of human lens epithelial cells to H2O2 insult. Invest.Ophthalmol. Visual Sci. 44:2084–2093; 2003.
[31] Kim, T. I.; Tchah, H.; Lee, S. A.; Sung, K.; Cho, B. J.; Kook, M. S.Apoptosis in keratocytes caused by mitomycin C. Invest. Ophthalmol.Visual Sci. 44:1912–1917; 2003.
[32] Chiarini, L. B.; Leal-Ferreira, M. L.; de Freitas, F. G.; Linden, R. Changingsensitivity to cell death during development of retinal photoreceptors.J. Neurosci. Res. 74:875–883; 2003.
[33] Zuliani, T.; Denis, V.; Noblesse, E.; Schnebert, S.; Andre, P.; Dumas, M.;Ratinaud, M. H. Hydrogen peroxide-induced cell death in normal humankeratinocytes is differentiation dependent. Free Radical Biol. Med.38:307–316; 2005.
[34] Clarke, A. A.; Philpott, N. J.; Gordon-Smith, E. C.; Rutherford, T. R. Thesensitivity of Fanconi anaemia group C cells to apoptosis induced bymitomycin C is due to oxygen radical generation, not DNA crosslinking.Br. J. Haematol. 96:240–247; 1997.
[35] Fesus, L.; Davies, P. J.; Piacentini, M. Apoptosis: molecular mechanismsin programmed cell death. Eur. J. Cell Biol. 56:170–177; 1991.
[36] Usatyuk, P. V.; Natarajan, V. Role of mitogen-activated protein kinasesin 4-hydroxy-2-nonenal-induced actin remodeling and barrier functionin endothelial cells. J. Biol. Chem. 279:11789–11797; 2004.

1469N. Lassen et al. / Free Radical Biology & Medicine 41 (2006) 1459–1469
[37] Premkumar, K.; Abraham, S. K.; Santhiya, S. T.; Ramesh, A. Protectiveeffects of saffron (Crocus sativus Linn.) on genotoxins-induced oxidativestress in Swiss albino mice. Phytother. Res. 17:614–617; 2003.
[38] Kagan, V. E.; Kuzmenko, A. I.; Tyurina, Y. Y.; Shvedova, A. A.; Matsura,T.; Yalowich, J. C. Pro-oxidant and antioxidant mechanisms of etoposidein HL-60 cells: role of myeloperoxidase. Cancer Res. 61:7777–7784;2001.
[39] Esterbauer, H.; Schaur, R. J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal malondialdehyde and related aldehydes. Free RadicalBiol. Med. 11:81–128; 1991.
[40] Dalton, T. P.; Chen, Y.; Schneider, S. N.; Nebert, D. W.; Shertzer, H. G.Genetically altered mice to evaluate glutathione homeostasis in health anddisease. Free Radical Biol. Med. 37:1511–1526; 2004.
[41] Spector, A. Oxidative stress-induced cataract: mechanism of action. FA-SEB J. 9:1173–1182; 1995.
[42] Kenney, M. C.; Chwa, M.; Atilano, S. R.; Tran, A.; Carballo, M.;Saghizadeh, M.; Vasiliou, V.; Adachi, W.; Brown, D. J. Increased levels ofcatalase and cathepsin V/L2 but decreased TIMP-1 in keratoconus corneas:evidence that oxidative stress plays a role in this disorder. Invest.Ophthalmol. Visual Sci. 46:823–832; 2005.
[43] Lim, P.; Wuenschell, G. E.; Holland, V.; Lee, D. H.; Pfeifer, G. P.;Rodriguez, H.; Termini, J. Peroxyl radical mediated oxidative DNA basedamage: implications for lipid peroxidation induced mutagenesis.Biochemistry 43:15339–15348; 2004.
[44] Tomasz, M.; Lipman, R. Reductive metabolism and alkylating activity ofmitomycin C induced by rat liver microsomes. Biochemistry 20:5056–5061; 1981.
[45] Siegel, D.; Gibson, N. W.; Preusch, P. C.; Ross, D. Metabolism ofmitomycin C by DT-diaphorase: role in mitomycin C-induced DNAdamage and cytotoxicity in human colon carcinoma cells. Cancer Res.50:7483–7489; 1990.
[46] Nakano, H.; Sugioka, K.; Nakano, M.; Mizukami, M.; Kimura, H.;Tero-Kubota, S.; Ikegami, Y. Importance of Fe2+-ADP and the relativeunimportance of OH in the mechanism of mitomycin C-induced lipidperoxidation. Biochim. Biophys. Acta 796:285–293; 1984.
[47] Pritsos, C. A.; Sartorelli, A. C. Generation of reactive oxygen radicalsthrough bioactivation of mitomycin antibiotics. Cancer Res. 46:3528–3532; 1986.
[48] Poli, G.; Schaur, R. J. 4-Hydroxynonenal in the pathomechanisms ofoxidative stress. IUBMB Life 50:315–321; 2000.
[49] Stadtman, E. R. Protein oxidation in aging and age-related diseases. Ann.N. Y. Acad. Sci. 928:22–38; 2001.
[50] le-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Proteincarbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta329:23–38; 2003.
[51] Stadtman, E. R.; Oliver, C. N.; Starke-Reed, P. E.; Rhee, S. G. Age-relatedoxidation reaction in proteins. Toxicol. Ind. Health 9:187–196; 1993.
[52] Piatigorsky, J. A case for corneal crystallins. J. Ocul. Pharmacol. Ther.16:173–180; 2000.
[53] Zhu, L.; Crouch, R. K. Albumin in the cornea is oxidized by hydrogenperoxide. Cornea 11:567–572; 1992.
[54] Lee, S. M.; Koh, H. J.; Park, D. C.; Song, B. J.; Huh, T. L.; Park, J. W.Cytosolic NADP(+)-dependent isocitrate dehydrogenase status modu-lates oxidative damage to cells. Free Radical Biol. Med. 32: 1185–1196;2002.
[55] Kim, H. J.; Kang, B. S.; Park, J. W. Cellular defense against heat shock-induced oxidative damage by mitochondrial NADP+-dependent isocitratedehydrogenase. Free Radical Res. 39:441–448; 2005.
[56] Salvemini, F.; Franze, A.; Iervolino, A.; Filosa, S.; Salzano, S.; Ursini,M. V. Enhanced glutathione levels and oxidoresistance mediated byincreased glucose-6-phosphate dehydrogenase expression. J. Biol. Chem.274:2750–2757; 1999.
[57] Hanukoglu, I.; Rapoport, R. Routes and regulation of NADPH productionin steroidogenic mitochondria. Endocr. Res. 21:231–241; 1995.
[58] Marselos, M.; Lindahl, R. Substrate preference of a cytosolic aldehydedehydrogenase inducible in rat liver by treatment with 3-methylcholan-threne. Toxicol. Appl. Pharmacol. 95:339–345; 1988.
[59] Siitonen, T.; Alaruikka, P.; Mantymaa, P.; Savolainen, E. R.; Kavanagh,T. J.; Krejsa, C. M.; Franklin, C. C.; Kinnula, V.; Koistinen, P. Protectionof acute myeloblastic leukemia cells against apoptotic cell death by highglutathione and gamma-glutamylcysteine synthetase levels during etopo-side-induced oxidative stress. Ann. Oncol. 10:1361–1367; 1999.
[60] Lee, C. S.; Park, S. Y.; Ko, H. H.; Han, E. S. Effect of change in cellularGSH levels on mitochondrial damage and cell viability loss due tomitomycin C in small cell lung cancer cells. Biochem. Pharmacol.68:1857–1867; 2004.