antihypertensive therapy and its effects on potassium homeostasis
TRANSCRIPT

VOL. 8 NO. 1 JANUARY 2006 THE JOURNAL OF CLINICAL HYPERTENSION 67
The role of potassium in cardiovascular disease and the importance of preserving potassium balance have emerged as clinical hot points, particularly as they relate to cardioprotective and renoprotective therapies that secondarily promote potassium reten-tion. Antihypertensive medications that most com-monly influence serum potassium levels and/or total body potassium include β blockers and potassium-wasting and potassium-sparing diuretics as well as angiotensin-converting enzyme inhibitors and angio-tensin receptor blockers. Uncertainty exists as to the best way to monitor potassium levels when any of these drug therapies are used, particularly in the set-ting of chronic kidney disease and/or heart failure. Guidelines for the monitoring of serum potassium levels in the setting of antihypertensive therapy are at best makeshift and often drawn from the know-how of the treating physician. (J Clin Hypertens. 2006:8;67–73) ©2006 Le Jacq Ltd.
The measurement of serum potassium (K+), although easily accomplished, is seldom standardized.
Indeed, the normal range for K+ values is itself highly variable between laboratories; the lower limit fluctu-ates between 3.5 and 3.8 mmol/L and the upper limit between 5.0 and 5.5 mmol/L. Interpretation of a spe-cific serum K+ value initially requires an understanding of sampling conditions. For example, a serum K+ value derived from a serum sample (red-top tube) is typically 0.1 to 0.3 mmol/L higher than one obtained from a plasma sample (green- or purple-top tube).1
Blood samples obtained using poor technique can also falsely increase serum K+ values (pseudohy-perkalemia). Prolonged use of a tourniquet above a venipuncture site or extended fist clenching produces tissue hypoxia and promotes the escape of K+ from tissues into the plasma compartment.2 Serum K+ val-ues also show evidence of a circadian rhythm (aver-age peak-to-trough difference ≈0.60 mmol/L, with lowest values at night).3 In addition, serum K+ values transiently decrease after meals as the result of insulin stimulating an intracellular flux of K+. These consid-erations are necessary interpretive elements for serum K+ values in those receiving antihypertensive medica-tions known to adversely impact K+ homeostasis.
EXTERNAL AND INTERNAL FACTORS INFLUENCING SERUM K+
The economy of K+ in the body is divided into ele-ments of both internal and external balance with antihypertensive medications able to influence both components.
INTERNAL K+ BALANCE AND ANTIHYPERTENSIVE THERAPYInternal K+ balance in large measure relates to factors that encourage the intracellular migration of K+ such as insulin and β-adrenergic stimulation
Antihypertensive Therapy and Its Effects on Potassium Homeostasis
Domenic A. Sica, MD
From the Section of Clinical Pharmacology and Hypertension, Division of Nephrology, Medical College of Virginia of Virginia Commonwealth University, Richmond, VAAddress for correspondence: Domenic A. Sica, MD, Professor of Medicine and Pharmacology, Chairman, Section of Clinical Pharmacology and Hypertension, Division of Nephrology, Medical College of Virginia of Virginia Commonwealth University, Box 980160, MCV Station, Richmond, VA 23298-0160E-mail: [email protected]
C u r r e n t C o n c e p t s o f P h a r m a c o t h e r a p y i n H y p e r t e n s i o nD o m e n i c A . S i c a , M D , S e n i o r E d i t o r
www.lejacq.com ID: 5139
The Journal of Clinical Hypertension® (ISSN 1524-6175) is published monthly by Le Jacq Ltd., Three Parklands Drive, Darien, CT 06820-3652. Copyright ©2005 by Le Jacq Ltd., All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. The opinions and ideas expressed in this publication are those of the authors and do not necessarily reflect those of the Editors or Publisher. For copies in excess of 25 or for commercial purposes, please contact Sarah Howell at [email protected] or 203.656.1711 x106.

THE JOURNAL OF CLINICAL HYPERTENSION VOL. 8 NO. 1 JANUARY 200668
(mainly a β2-adrenergic effect).4 Conversely, a lack of insulin and β-adrenergic blockade can be expected to have the opposite effect on the cel-lular translocation of K+. In this regard, the total body K+ content of a 70-kg adult is approximately 4000 mmol. The large majority of body K+ resides intracellularly, with about 60 mmol (<2%) of total body K+ located extracellularly. As such, since the quantity of K+ located outside cells is so small, slight shifts one way or the other can result in sig-nificant changes in serum K+ values.5
Reports of small increases in serum K+ subse-quent to β-blocker therapy for essential hyper-tension were widespread by the mid-1970s. This process was seen with nonselective and cardiose-lective β blockade as well as with combined α-β blockade.6 The changes in serum K+ concentration that accompany the administration of β blockers arise from internal K+ shifts—a mechanism sup-ported by the finding that urinary excretion of K+ is not reduced when β blockade increases serum K+ values.7
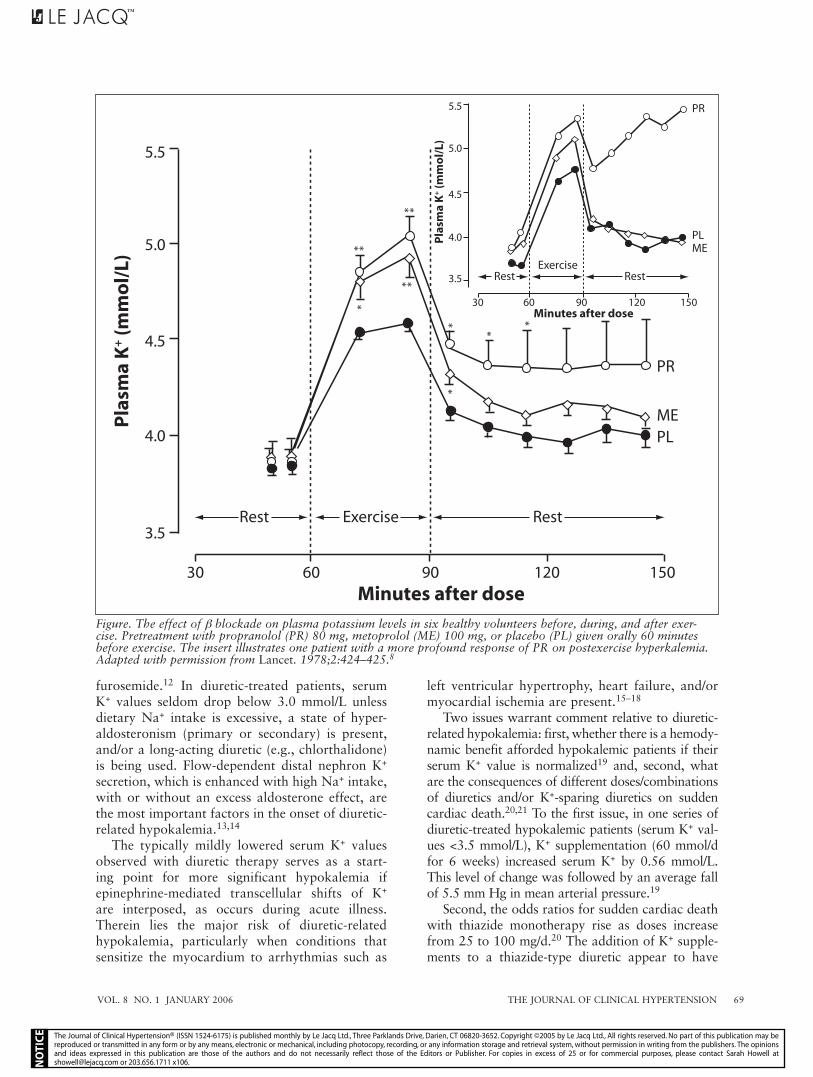
The increment in serum K+ from β-blocker ther-apy is of minor importance, except in patients with significantly reduced renal function. For example, β-blocker therapy can increase serum K+ values by 1 mmol/L or more in end-stage renal disease patients. Vigorous physical exercise is normally accompanied by transient hyperkalemia with β blockade, further increasing serum K+ concentrations both during and after sustained exercise (Figure).8
Although calcium channel blockers increase the cellular uptake of K+, drugs in this class incon-sistently effect serum K+ concentrations at usual doses.9,10 Intentional ingestion of large amounts of verapamil, however, can significantly reduce serum K+ concentrations. In one case, profound hypoka-lemia (2.8 mmol/L) was seen within 40 minutes of ingesting 4.16 g of immediate-release verapamil.11
TREATMENT CONSIDERATIONSAll forms of redistributional hyperkalemia, includ-ing those that are medication-related, evolve inde-pendent of the underlying state of total body K+ balance. Therefore, laboratory values obtained during such situations cannot be used to accurately gauge whether a true total body excess of K+ exists or, in the instance of a mixed picture—a patient with a known basis for K+ excess and the presence of factors associated with redistribution—to estab-lish the true level of the excess. In such a setting, usual acute care measures for hyperkalemia should be instituted, since the time course for recovery can be unpredictable. The duration of β blocker-related
redistributional hyperkalemia can be fairly pro-longed. This is particularly so when renally cleared β blockers, such as atenolol, are administered to patients with an inherent inability to eliminate K+, as in the case of advanced chronic kidney disease.
EXTERNAL K+ BALANCE AND ANTIHYPERTENSIVE THERAPYThe gastrointestinal and renal systems are the major determinants of external K+ balance, with a normally functioning gut characteristically con-serving or eliminating K+ based on total body stores. The gastrointestinal abnormalities most relevant to external K+ balance include diarrhea and/or vomiting. Of note, magnesium (Mg2+) losses can be substantial with sustained diarrhea and may go unrecognized in that serum Mg2+ val-ues poorly reflect total body stores of this cation. Total body Mg2+ deficiency, with or without sig-nificantly reduced Mg2+ values, can compromise the body’s ability to conserve administered K+ and is therefore something to be considered in the case of “refractory hypokalemia.”1 Antihypertensive therapy rarely effects changes in serum K+ values as a consequence of adverse gastrointestinal effects; however, the occasional patient may experience diarrhea with an antihypertensive medication and in the process develop hypokalemia. This is typi-cally a self-limiting event that corrects with medi-cation discontinuation.
Renal factors influencing K+ balance include uri-nary flow rate, extracellular fluid volume, sodium (Na+) intake, acid-base balance, mineralocorticoid excess, renal tubular diseases, Mg2+ depletion, and level of renal function.5 Alterations in systemic K+ balance attributable to antihypertensive medica-tion most typically occur on a renal basis. This is the case for several drug classes, including K+-wast-ing and K+-sparing diuretics as well as angiotensin-converting enzyme (ACE) inhibitors and angioten-sin receptor blockers (ARBs).
K+-WASTING DIURETICSA serum K+ value below 3.5 mmol/L, the most common definitional criterion for a diagnosis of hypokalemia, is not an uncommon finding in patients treated with loop or high-dose thiazide-type diuretics.12 During the first several days of thiazide diuretic therapy, plasma K+ falls by an average of 0.6 mmol/L (in a dose-dependent man-ner) in subjects not taking K+ supplements and on an unrestricted Na+ diet. In comparison, the aver-age fall (≈0.3 mmol/L) with a loop diuretic is less, particularly with a short-acting compound such as
The Journal of Clinical Hypertension® (ISSN 1524-6175) is published monthly by Le Jacq Ltd., Three Parklands Drive, Darien, CT 06820-3652. Copyright ©2005 by Le Jacq Ltd., All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. The opinions and ideas expressed in this publication are those of the authors and do not necessarily reflect those of the Editors or Publisher. For copies in excess of 25 or for commercial purposes, please contact Sarah Howell at [email protected] or 203.656.1711 x106.
VOL. 8 NO. 1 JANUARY 2006 THE JOURNAL OF CLINICAL HYPERTENSION 69
furosemide.12 In diuretic-treated patients, serum K+ values seldom drop below 3.0 mmol/L unless dietary Na+ intake is excessive, a state of hyper-aldosteronism (primary or secondary) is present, and/or a long-acting diuretic (e.g., chlorthalidone) is being used. Flow-dependent distal nephron K+ secretion, which is enhanced with high Na+ intake, with or without an excess aldosterone effect, are the most important factors in the onset of diuretic-related hypokalemia.13,14
The typically mildly lowered serum K+ values observed with diuretic therapy serves as a start-ing point for more significant hypokalemia if epinephrine-mediated transcellular shifts of K+ are interposed, as occurs during acute illness. Therein lies the major risk of diuretic-related hypokalemia, particularly when conditions that sensitize the myocardium to arrhythmias such as
left ventricular hypertrophy, heart failure, and/or myocardial ischemia are present.15–18
Two issues warrant comment relative to diuretic-related hypokalemia: first, whether there is a hemody-namic benefit afforded hypokalemic patients if their serum K+ value is normalized19 and, second, what are the consequences of different doses/combinations of diuretics and/or K+-sparing diuretics on sudden cardiac death.20,21 To the first issue, in one series of diuretic-treated hypokalemic patients (serum K+ val-ues <3.5 mmol/L), K+ supplementation (60 mmol/d for 6 weeks) increased serum K+ by 0.56 mmol/L. This level of change was followed by an average fall of 5.5 mm Hg in mean arterial pressure.19
Second, the odds ratios for sudden cardiac death with thiazide monotherapy rise as doses increase from 25 to 100 mg/d.20 The addition of K+ supple-ments to a thiazide-type diuretic appear to have
Rest
30
3.5
4.0
4.5
5.0
5.5
60 90 120 150
Exercise Rest
Minutes after dose
Minutes after dose
Pla
sma
K+ (m
mo
l/L)
Pla
sma
K+ (m
mo
l/L)
PLME
PR
RestRest
30
3.5
4.0
4.5
5.0
5.5
60 90 120 150
Exercise
MEPL
PR
*
**
*
*
* **
*
**
Figure. The effect of β blockade on plasma potassium levels in six healthy volunteers before, during, and after exer-cise. Pretreatment with propranolol (PR) 80 mg, metoprolol (ME) 100 mg, or placebo (PL) given orally 60 minutes before exercise. The insert illustrates one patient with a more profound response of PR on postexercise hyperkalemia. Adapted with permission from Lancet. 1978;2:424–425.8
The Journal of Clinical Hypertension® (ISSN 1524-6175) is published monthly by Le Jacq Ltd., Three Parklands Drive, Darien, CT 06820-3652. Copyright ©2005 by Le Jacq Ltd., All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. The opinions and ideas expressed in this publication are those of the authors and do not necessarily reflect those of the Editors or Publisher. For copies in excess of 25 or for commercial purposes, please contact Sarah Howell at [email protected] or 203.656.1711 x106.

THE JOURNAL OF CLINICAL HYPERTENSION VOL. 8 NO. 1 JANUARY 200670
little effect on the risk of sudden cardiac death. Alternatively, the risk of sudden cardiac death was substantially reduced in patients receiving a thiazide and K+-sparing diuretic in combination. Differences in serum K+ concentrations did not explain the dif-ferences between thiazide/K+ supplements and thia-zides/K+-sparing diuretics. It is tempting to speculate that these between-group differences derived from
the favorable effect of K+-sparing diuretics on Mg2+ balance; however, these studies were not designed to test this hypothesis.20
K+-SPARING DIURETICSAll aldosterone receptor antagonists (ARAs) increase serum K+ levels in a dose-dependent man-ner.22,23 There are several variables that govern the incidence of ARA-related hyperkalemia (the definition of hyperkalemia can be highly variable), with a reduced glomerular filtration rate (GFR) being the single most important variable (Table I). Thus, patients with a primary underlying renal disease (and, in particular, diabetic renal disease) or conditions set apart by a high frequency of accompanying renal failure—as is the case with heart failure—will be most prone to an increase in serum K+ values with ARA therapy.24–27
Although there are dose-ranging studies with ARAs in the setting of a normal GFR, there is little such information regarding these compounds when used in reduced GFR states.27 Heart failure, how-ever, can provide a starting point for understanding the hyperkalemia risk with ARA therapy in chronic kidney disease in that the GFR is often reduced in this disease state. For example, the Randomized Aldactone Evaluation Study (RALES)22 was pre-ceded by a short, dose-finding study (dose range, 12.5–75.0 mg/d) in which hyperkalemia (serum K+ ≥5.5 mEq/L) occurred in 13%, 20%, and 24% of patients treated with spironolactone 25, 50, and 75 mg/d, respectively.
It should be noted that spironolactone use has been linked to a reduction in GFR in patients with resistant hypertension—a process that can independently influence the tendency to develop hyperkalemia. In a study by Nishizaka et al.,27 five subjects (three of whom had diabetes) treated with low-dose spironolactone (12.5–50 mg/d) experi-enced a fall in GFR in tandem with a substantial reduction in BP. With lowering of the spironolac-tone dose and stabilization of blood pressure, the GFR returned to baseline in three of these subjects. In the remaining subjects, the GFR normalized with discontinuation of the spironolactone. This experience suggests that the acute drop in GFR in these subjects may have related more so to the sub-acute drop in BP reduction rather than to a direct nephrotoxic effect of spironolactone.
ACE INHIBITORS AND ARBsHyperkalemia is an ACE inhibitor- and ARB-asso-ciated side effect that has a clear physiologic basis, as is the case for ARAs.25,28–31 ACE inhibitors and
Table I. Risk Factors for Hyperkalemia* With Aldosterone Receptor Antagonist TherapyInappropriate dosing**
Spironolactone >25 mg/dEplerenone >50 mg/d
Pretherapy serum K+ >4.5 mEq/LHigh potassium diet, potassium supplements, or salt
substitutesDiabetes mellitusAdvanced stages of heart failure with accompanying
reductions in renal function (typically Clcreat <50 mL/min)†
Volume depletionLoop and/or thiazide diuretic-relatedIntercurrent illnesses (typically gastrointestinal)
Deterioration in renal function with spironolactone or eplerenoneRelated to blood pressure/volume status consideration in
the setting of concomitant renin-angiotensin-aldosterone system blockade
Older ageRelated to level of renal function
Caucasian raceDrugs commonly used to modify potassium homeostasis
β BlockersNonsteroidal anti-inflammatory drugs or cyclooxygenase
inhibitorsHeparin Regular and low-molecular weight
Digoxin intoxicationTrimethoprim Calcineurin inhibitorsCyclosporineTacrolimus
*Hyperkalemia is typically defined as a serum K+ (potassium) value >6.0 mEq/L; **the dose of spironolactone and eplere-none designated as inappropriate is one in excess of that used in the pivotal heart failure clinical trials. Doses higher than those employed in the clinical trials can be considered as appropriate if the aldosterone receptor antagonist is being given for reasons other than cardioprotection, such as for K+ sparing in a persistently hypokalemic patient or when being used for resistant hypertension; †hyperkalemia with an aldosterone receptor antagonist can still occur at Clcreat (cre-atine clearance) values >50 mL/min in those with risk factors other than reduced renal function, such as diabetes
The Journal of Clinical Hypertension® (ISSN 1524-6175) is published monthly by Le Jacq Ltd., Three Parklands Drive, Darien, CT 06820-3652. Copyright ©2005 by Le Jacq Ltd., All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. The opinions and ideas expressed in this publication are those of the authors and do not necessarily reflect those of the Editors or Publisher. For copies in excess of 25 or for commercial purposes, please contact Sarah Howell at [email protected] or 203.656.1711 x106.

VOL. 8 NO. 1 JANUARY 2006 THE JOURNAL OF CLINICAL HYPERTENSION 71
ARBs will increase the serum K+ value in virtu-ally all treated subjects, but only to a degree (0.1–0.2 mEq/L) that is barely discernible clinically. Hyperkalemia, when it occurs with either of these drug classes, remains highly definitional in nature. To register as an incident case of hyperkalemia relating to ACE inhibitor or ARB therapy, a spe-cific threshold value (generally >5.5–6.0 mmol/L) needs to be reached. This definitional approach results in many patients with a significant numeric change in serum K+ values (but not reaching a value of ≥5.5 mEq/L) going unrecognized.
The frequency with which serum K+ values should be monitored in an ACE inhibitor-treated patient should be based on pretherapy K+ values, the level of renal function, the presence of diabetes, whether concomitant medications are being given that might influence systemic K+ balance, and past occurrences of hyperkalemia (Table I).25,32 Potassium supplements, K+-sparing diuretics, and salt substitutes (≈60 mmol/tsp of potassium chloride) increase the probability of developing hyperkalemia if combined with an ACE inhibitor or an ARB.25 Nonsteroidal anti-inflammato-ry drugs (NSAIDs) and cyclooxygenase inhibitors also can exaggerate the rise in serum K+ seen with either an ACE inhibitor or an ARB by reducing aldosterone concentrations and thereby decreasing K+ excretion. It is uncommon even under the most extreme cir-cumstances to see more than a 2.0-mmol/L increase in serum K+ values with use of an ACE inhibitor or an ARB. When changes of this magnitude occur, it is generally in conjunction with a sudden fall in GFR, prompted by either the ACE inhibitor or ARB (as is the case with renal artery stenosis or due to a volume-depleting intercurrent illness).
ACE inhibitor-/ARB-related hyperkalemia is class- and not compound-specific. There is scant experi-mental evidence to suggest that one ACE inhibitor or ARB carries a lesser risk of hyperkalemia. If dif-ferences in the incidence of hyperkalemia truly exist amongst ACE inhibitors (or ARBs)—as has been described for fosinopril—it is a phenomenon prob-ably coupled to the absence of drug accumulation in chronic kidney disease for certain ACE inhibitors.33,34 A final consideration is whether ARB therapy is asso-ciated with a lesser rate of hyperkalemia than is the case for ACE inhibitors. In this regard, the absolute change in serum K+ with an ARB is somewhat less than that observed with an ACE inhibitor.31
TREATMENT CONSIDERATIONSACE inhibitors, ARBs, β blockers, and ARAs all have well established outcomes benefits; therefore, even in patients likely to develop hyperkalemia,
every effort should be made to implement and/or continue the use of these compounds in high-risk
Table II. Approach With ARA and/or ACE/ARB Therapy to the Patient Prone to Hyperkalemia Estimate the GFR to define the specific risk of
hyperkalemia.*Measure serum K+ 1–2 weeks after starting therapy and with
a dose increase. Remind the patient that any intercurrent illness
accompanied by volume loss requires physician notification since the risk of hyperkalemia increases under these circumstances.
Provide a low-potassium diet; determine whether salt substitutes are being used.**
Decrease the dose of any K+ supplement in tandem with beginning therapy.
Prescribe a loop and/or thiazide diuretic in sufficient doses to maintain a consistent urine flow rate.Excessive diuresis can have an opposing effect on serum K+
values if GFR falls in the process.ARA therapy can increase the diuretic response to loop
and/or thiazide diuretic therapy and can decrease GFR.Whenever possible, discontinue drugs that interfere with K+
homeostasis:NSAIDs or COXIBs decrease K+ excretion;Heparin decreases aldosterone production.
If serum K+ increases >5.0 mEq/L, consider discontinuation or dosage reduction of an ACE inhibitor, ARB, and/or the ARA (often the ARA should be the discontinued drug).
Utilize a reduced dose of an ACE inhibitor or one such as trandolapril or fosinopril that does not accumulate in the setting of renal failure. ARBs also do not accumulate in renal failure states.
Empirically reduce the ARA dose or convert to every-other-day therapy.†
If spironolactone therapy is the basis for hyperkalemia, then empirically switch to eplerenone.††
ARA=aldosterone receptor antagonist; ACE=angiotensin-converting enzyme inhibitor; ARB=angiotensin receptor blocker; GFR=glomerular filtration rate; K+=potassium; NSAIDs=nonsteroidal anti-inflammatory drugs; COXIBs=cyclooxygenase inhibitors; *change in serum creati-nine poorly gauges level of renal function. A predictive equation based on serum creatinine, age, gender, race, and body size should be used to estimate GFR. See http://www.nkdep.nih.gov/healthprofessionals/tools/index; **salt substitutes typically have 60 mEq/tsp of potassium chloride; †there is little informa-tion available that addresses the survival benefits of spirono-lactone or eplerenone at doses <25 and 50 mg/d, respectively. Moreover, it is unclear in the patient prone to hyperkalemia whether simple dosage reduction is adequate to correct this ten-dency; ††this relates to the shorter half-life and absence of active metabolites for eplerenone; however, head-to-head studies that compare equivalent doses of eplerenone and spironolactone for hyperkalemia risk have yet to be undertaken.
The Journal of Clinical Hypertension® (ISSN 1524-6175) is published monthly by Le Jacq Ltd., Three Parklands Drive, Darien, CT 06820-3652. Copyright ©2005 by Le Jacq Ltd., All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. The opinions and ideas expressed in this publication are those of the authors and do not necessarily reflect those of the Editors or Publisher. For copies in excess of 25 or for commercial purposes, please contact Sarah Howell at [email protected] or 203.656.1711 x106.

THE JOURNAL OF CLINICAL HYPERTENSION VOL. 8 NO. 1 JANUARY 200672
patients. ACE inhibitor (or ARB) therapy alone (with or without a diuretic) is not a common cause of hyperkalemia. It is only when an ARA, such as spironolactone or eplerenone, is added to an ACE inhibitor or an ARB that the risk of hyperkalemia truly materializes. When an ARA is added to an ACE inhibitor/ARB (with or without a β blocker), dietary K+ intake should be preemptively restrict-ed. This maneuver alone may suffice to forestall the onset of hyperkalemia.
Limiting the use of K+ supplements in anticipa-tion of a rise in K+ values with these therapies is an important consideration. Weekly or biweekly determinations of serum K+ values are advisable until a patient is stabilized on a regimen comprised of an ACE inhibitor/ARB and an ARA. Once stabilized, serum K+ values can be obtained less frequently, but still need to be obtained regularly. Typically, it takes 2–4 weeks after starting an ARA to arrive at a steady state for K+ balance. Serum K+ sampling in the setting of ACE inhibitor or ARB therapy alone typically is less intense since the risk of hyperkalemia is less so than when either of these drug classes are combined with an ARA.
The circumstances that most commonly dis-rupt K+ homeostasis after a steady state has been reached for ARA effect (with or without an ACE inhibitor or ARB) are those marked by rapid volume loss, such as diarrheal or upper gastroin-testinal illnesses (Tables I and II). Loop and thia-zide-type diuretics should also be carefully used in the setting of antihypertensive therapy-related hyperkalemia; whereas an increase in urine flow rate may facilitate urinary K+ excretion, an exces-sive diuretic response can prove detrimental. Under such circumstances, patients should be advised to discontinue ARA treatment (and ACE inhibitor or ARB therapy) until the intercurrent illness has resolved (or volume contraction is corrected) and their K+ status is reevaluated. Once an episode of hyperkalemia has resolved, ACE inhibitor, ARB, and/or ARA therapy can be cautiously reintro-duced if the benefit sought from such therapy exceeds the risk of recurrent hyperkalemia.
CONCLUSIONSHyperkalemia, as defined by a serum K+ value of >5.5–6.0 mmol/L, can occur with several common-ly used antihypertensive therapies. An important management step for antihypertensive medication-related hyperkalemia is to discontinue the offending medication at the time of the incident. Thereafter, the continued treatment with one or the other of these agents becomes discretionary. Hard and fast
rules cannot be put forward for the patient with clinically significant hyperkalemia, and each patient should be managed on an individualized basis.
REFERENCES 1 Sica DA, Struthers AD, Cushman WC, et al. Importance
of potassium in cardiovascular disease. J Clin Hypertens (Greenwich). 2002;4:198–206.
2 Don BR, Sebastian A, Cheitlin M, et al. Pseudohyperkalemia caused by fist clenching during phlebotomy. N Engl J Med. 1990;322:1290–1292.
3 Solomon R, Weinberg MS, Dubey A. The diurnal rhythm of plasma potassium: relationship to diuretic therapy. J Cardiovasc Pharmacol. 1991;17:854–859.
4 Moratinos J, Reverte M. Effects of catecholamines on plasma potassium: the role of alpha- and beta-adrenocep-tors. Fundam Clin Pharmacol. 1993;7:143–153.
5 Halperin ML, Kamel KS. Potassium. Lancet. 1998;352:135–140. 6 Nowicki M, Szewczyk-Seifert G, Klimek D, et al. Carvedilol
does not modulate moderate exercise-induced hyperkalemia in hemodialysis patients. Clin Nephrol. 2002;57:352–358.
7 Traub YM, Rabinov M, Rosenfeld JB, et al. Elevation of serum potassium during beta blockade: absence of relation-ship to the renin-aldosterone system. Clin Pharmacol Ther. 1980;28:765–768.
8 Carlsson E, Fellenius E, Lundborg P, et al. β-adrenocep-tor blockers, plasma-potassium, and exercise. Lancet. 1978;2:424–425.
9 Solomon R, Dubey A. Diltiazem enhances potassium dis-posal in subjects with end-stage renal disease. Am J Kidney Dis. 1992;19:420–426.
10 Mimran A, Ribstein J, Sissmann J. Effects of calcium antagonists on adrenaline-induced hypokalaemia. Drugs. 1993;46(suppl 2):103–107.
11 Minella RA, Schulman DS. Fatal verapamil toxicity and hypokalemia. Am Heart J. 1991;121:1810–1812.
12 Morgan DB, Davidson C. Hypokalaemia and diuretics: an analysis of publications. BMJ. 1980;280:905–908.
13 Khuri RN, Strieder WN, Giebisch G. Effects of flow rate and potassium intake on distal tubular potassium transfer. Am J Physiol. 1975;228:1249–1261.
14 Velazquez H, Wright FS. Control by drugs of renal potassium handling. Annu Rev Pharmacol Toxicol. 1986;26:293–309.
15 MacMahon S, Collins G, Rautaharju P, et al. Electro-cardiographic left ventricular hypertrophy and effects of antihypertensive drug therapy in hypertensive patients in the Multiple Risk Factor Intervention Trial. Am J Cardiol. 1989;63:202–210.
16 Macdonald JE, Struthers AD. What is the optimal serum potassium level in cardiovascular patients? J Am Coll Cardiol. 2004;43:155–161.
17 Kafka H, Langevin L, Armstrong P. Serum magnesium and potassium in acute myocardial infarction: influences on ven-tricular arrhythmia. Arch Intern Med. 1987;147:465–469.
18 Packer M. Potential role of potassium as a determinant of morbidity and mortality in patients with systemic hypertension and congestive heart failure. Am J Cardiol. 1990;65:45E–51E.
19 Kaplan NM, Carnegie A, Raskin P, et al. Potassium supple-mentation in hypertensive patients with diuretic-induced hypokalemia. N Engl J Med. 1985;312:746–749.
20 Siscovick DS, Raghunathan TE, Psaty BM, et al. Diuretic therapy and the risk of primary cardiac arrest. N Engl J Med. 1994;330:1852–1857.
21 Cooper HA, Dries DL, Davis CE, et al. Diuretics and risk of arrhythmic death in patients with left ventricular dysfunc-tion. Circulation. 1999;100:1311–1315.
22 The RALES Investigators. Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure
The Journal of Clinical Hypertension® (ISSN 1524-6175) is published monthly by Le Jacq Ltd., Three Parklands Drive, Darien, CT 06820-3652. Copyright ©2005 by Le Jacq Ltd., All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. The opinions and ideas expressed in this publication are those of the authors and do not necessarily reflect those of the Editors or Publisher. For copies in excess of 25 or for commercial purposes, please contact Sarah Howell at [email protected] or 203.656.1711 x106.

VOL. 8 NO. 1 JANUARY 2006 THE JOURNAL OF CLINICAL HYPERTENSION 73
(The Randomized Aldactone Evaluation Study [RALES]). Am J Cardiol. 1996;78:902–907.
23 Sica DA. Eplerenone and serum potassium change—relationship to renal function. Am J Hypertens. 2003;16(suppl 1):A100.
24 Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyper-kalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med. 2004;351:543–551.
25 Sica DA, Hess M. Pharmacotherapy in congestive heart fail-ure: aldosterone receptor antagonism: interface with hyperka-lemia in heart failure. Congest Heart Fail. 2004;10:259–264.
26 Tamirisa KP, Aaronson KD, Koelling TM. Spironolactone-induced renal insufficiency and hyperkalemia in patients with heart failure. Am Heart J. 2004;148:971–978.
27 Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens. 2003;16:925–930.
28 Textor SC, Bravo EL, Fouad FM, et al. Hyperkalemia in azotemic patients during angiotensin-converting enzyme inhibition and aldosterone reduction with captopril. Am J Med. 1982;73:719–725.
29 Juurlink DN, Mamdani M, Kopp A, et al. Drug-drug interactions among elderly patients hospitalized for drug toxicity. JAMA. 2003;289:1652–1658.
30 Cruz CS, Cruz AA, Marcilio de Souza CA. Hyperkalaemia in congestive heart failure patients using ACE inhibitors and spi-ronolactone. Nephrol Dial Transplant. 2003;18:1814–1819.
31 Bakris GL, Siomos M, Richardson D, et al. ACE inhibition or angiotensin receptor blockade: impact on potassium in renal failure. VAL-K Study Group. Kidney Int. 2000;58:2084–2092.
32 Sica DA, Gehr TWB, Frishman WH. The renin-angiotensin axis: angiotensin-converting enzyme inhibitors and angio-tensin-receptor blockers. In: Frishman W, Sonnenblick S, Sica DA, eds. Cardiovascular Pharmacotherapeutics. 2nd ed. New York, NY: McGraw-Hill; 2003:131–156.
33 Keilani T, Schleuter W, Molteni A, et al. Converting enzyme inhibition with fosinopril does not suppress aldosterone and may not cause hyperkalemia despite moderate renal impairment. J Am Soc Nephrol. 1991;2:281.
34 Schoolwerth A, Sica DA, Ballermann BJ, et al. Renal considerations in angiotensin converting enzyme inhibitor therapy: a statement for healthcare professionals from the Council on the Kidney in Cardiovascular Disease and the Council for High Blood Pressure Research of the American Heart Association. Circulation. 2001;104:1985–1991.
The Journal of Clinical Hypertension® (ISSN 1524-6175) is published monthly by Le Jacq Ltd., Three Parklands Drive, Darien, CT 06820-3652. Copyright ©2005 by Le Jacq Ltd., All rights reserved. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopy, recording, or any information storage and retrieval system, without permission in writing from the publishers. The opinions and ideas expressed in this publication are those of the authors and do not necessarily reflect those of the Editors or Publisher. For copies in excess of 25 or for commercial purposes, please contact Sarah Howell at [email protected] or 203.656.1711 x106.