antidepressant agents for the treatment of chronic pain and depression
TRANSCRIPT

Antidepressant Agents for the Treatment of Chronic Pain and Depression
Michael W. Jann, Pharm.D., and Julian H. Slade, Pharm.D.
Depression and painful somatic symptoms commonly occur together.Depression and chronic pain can have devastating effects on a patient’s health,productivity, and overall quality of life. When moderate-to-severe pain exists,it can impair patient function while making treatment more difficult orresistant, with increased severity in depressive symptoms and worseoutcomes. A variety of chronic pain syndromes exist, including diabeticneuropathy. A high prevalence of patients with chronic pain displaydepressive symptoms. Treatment for these conditions relies on pharmacologictherapy coupled with diligent, periodic assessments of changes in symptomseverity. The link between pain and depression lies in the central andperipheral nervous systems. The brain stem serves as an importantconnection between the higher brain centers and the spinal cord. In the brainstem, the neurotransmitters serotonin and norepinephrine modulate paintransmission through ascending and descending neural pathways. Bothserotonin and norepinephrine are also key neurotransmitters involved withthe pathophysiology of depression. Tricyclic antidepressants are effectivetreatments for pain and depression; selective serotonin reuptake inhibitorsprovide less benefit. Duloxetine and venlafaxine, which are serotonin andnorepinephrine reuptake inhibitors, were shown in clinical trials to alleviatepain and depressive symptoms. Diabetic neuropathy and other chronic painsyndromes were also shown to benefit from duloxetine and venlafaxine.Antidepressants remain fundamental therapeutic agents for depression andanxiety disorders. Their extended use into chronic pain, depression withphysical pain, physical pain with or without depression, and other potentialmedical conditions should be recognized.Key Words: depression, chronic pain, serotonin, norepinephrine, neuropathy,antidepressants, tricyclic antidepressants, TCA, duloxetine, paroxetine, venlafaxine.(Pharmacotherapy 2007;27(11):1571–1587)
OUTLINE
Biologic Link Between Chronic Pain and DepressionChronic Pain ModelDepression ModelInterrelationship Between Chronic Pain and
Depression
Role of Antidepressant AgentsAnimal Pain Models and In Vitro DataClinical Trials
Antidepressant Agents in the Treatment of DiabeticNeuropathyTricyclic Antidepressants and Selective Serotonin
Reuptake InhibitorsSerotonin and Norepinephrine Reuptake Inhibitors
Antidepressants in the Treatment of Other PainSyndromes
Nonpharmacologic Intervention with Vagal NerveStimulation
Conclusion
From the Department of Pharmacy Practice, MercerUniversity College of Pharmacy and Health Sciences,Atlanta, Georgia (both authors).
Address reprint requests to Michael W. Jann, Pharm.D.,Mercer University College of Pharmacy and Health Sciences,3001 Mercer University Drive, Atlanta, GA 30341; e-mail:[email protected].

PHARMACOTHERAPY Volume 27, Number 11, 2007
Chronic pain syndromes and depression aremajor medical problems facing our society.Approximately 80% of depressed outpatientswho completed self-rating questionnairesreported painful somatic symptoms that includedstomach pain, neck and back pain, headache, andnonspecific generalized pain.1 Among hospital-ized patients with depression, 92% reported atleast one painful symptom, and 76% reported thepresence of multiple painful symptoms.1 Chronicpain originates from a variety of medicalillnesses. Although the term “chronic” can beimprecise, we define it as a time period of at least3–6 months’ duration. Pain can be categorizedinto three groups: nociceptive (somatic andvisceral), neuropathic (central [e.g., stroke],peripheral [e.g., nerve compression by cancer,diabetic neuropathy], or mixed [e.g., postherpeticneuralgia]), and psychogenic. Nociceptive painoccurs when a tissue or organ is damaged byinjury or disease. Neuropathic pain is a result ofdirect damage to the nervous system or spinalcord. Psychogenic pain has no discerniblephysical source.
Pain is an unpleasant experience, and it isreasonable that its symptoms are closely linked todepression. In a population of patients withvarious sources of chronic pain, 28% reported atleast one depressive symptom and 43% fulfilledthe diagnosis for major depression.1 Thefrequency of clinical depression in patients withother diseases in which chronic pain is asignificant component is staggering and has beenreported to be 30–54%.2
Other symptoms that overlap both chronicpain and depression are found in anxietydisorders and would be more commonlyassociated with generalized anxiety disorder. Inpatients with generalized anxiety disorder, thepsychological symptoms of excessive anxiety,constant worries that are difficult to control,feeling on edge, and poor concentrationcombined with the physical symptoms of fatigue,muscle tension, restlessness, and sleepdisturbance can easily lead to depression.Whereas patients with chronic pain can havemany symptoms also found in generalizedanxiety, sleep disturbance may be one of the mostcommon indistinguishable symptoms among thethree categories of chronic pain, depression, andanxiety.3 It can be difficult for clinicians todiscern the origin of these overlapping symptomsthat lead to subsequent problems. We examinethe relationship between chronic pain anddepression and provide a pharmacologic rationale
for the efficacy of antidepressants for thetreatment of both debilitating conditions. Wealso discuss the implications of antidepressantsthat possess both norepinephrine and serotonergicproperties that have been shown to be effective intreating chronic pain and depression in clinicalstudies.
Biologic Link Between Chronic Pain andDepression
Chronic Pain Model
As previously stated, pain can be grouped intothree basic categories. It is beyond the scope ofthis article to review extensively its complexperipheral and central mechanisms. However,the basic pathophysiology of pain is provided toestablish a foundation for its treatment withantidepressant agents.
For pain to occur, an organic or environmentalstimulus must be converted into an electro-chemical signal, and then transmitted to higherbrain centers for interpretation. At that point, itis determined whether the signal is innocuous ornoxious in nature. Pain has been described as acomplex emotional experience involving not onlythe transduction of noxious stimuli, but alsocognitive and emotional processing by the brain.4
Pain is not homogeneous and involves multiplegenetic and biochemical mechanisms, nervoussystem pathways, and neuronal plasticity.4–7 Webriefly review only nociceptive and neuropathicpain mechanisms.
For nociceptive pain that originates from anoxious stimulus, the process is initiated at thenociceptor. The two main nociceptor classesinclude the lightly myelinated, medium-diameter,rapidly conducting Ad fibers, and theunmyelinated, small-diameter, more slowlyconducting C fibers.4 Thereby, Ad fibers mediaterapid, acute, sharp pain, and C fibers mediatedelayed, more diffuse, dull pain. A wide range ofstimuli triggers the pain sequence, which couldinvolve a rapid and/or delayed response. Eachstimulus has a corresponding receptor thattriggers the pain process (Table 1). For example,heat or thermal exposure elicits a rapid responsethrough activation of vanilloid receptor subtype 1and vanilloid-like receptor subtype 1 on Adfibers, launching the pain process. Tissuedamage from other mechanisms (e.g., medicaldisease) can release various biochemical stimuli(e.g., glutamate), which act through theircorresponding receptors to commence thedelayed pain process by way of the C fibers.4
1572

TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
Neuropathic pain is associated with disease orinjury of the peripheral or central nervous systemand presents difficult therapeutic paradigms forclinicians. Diabetes mellitus, immune disorders,cancer, and ischemic disorders are examples ofdisease processes that can lead to neuropathicpain. Classification of neuropathic pain can bebased on its location in the periphery, spinalcord, or brain (Table 2). Some disorders couldhave multiple locations (e.g., multiple sclerosis).8
An essential aspect of neuropathic pain is apartial or complete loss of afferent sensoryfunction and paradoxic hypersensitivities (i.e.,hyperalgesia and allodynia). Hyperalgesia is thelowering of the pain threshold and an increasedresponse to noxious stimuli, whereas allodynia isthe evocation of pain by nonnoxious stimuli.Mechanical hyperalgesia can be divided intothree groups: static, punctuate, and dynamic.These groups are easily distinguishable, as statichyperalgesia begins from gentle pressure,punctate starts with a pinprick, and dynamiccomes from a light brush that evokes a painfulsensation. Allodynia is characterized by sensationsor stimuli that are not considered painful, such astouch, warmth, cold, or simple movementseliciting a painful response. Like nociceptivepain, similar biochemical and molecularmechanisms occur in neuropathic pain with the
involvement of Ad and C fibers.4, 8
Both nociceptive and neuropathic pain stemfrom the primary sensory neurons and terminatein the dorsal horn of the spinal cord. The dorsalhorn is the first site of synaptic transfer to thebrain and can be influenced by neuronal plasticityor modulation integral to pain generation andpain hypersensitivity.5 Neural pathways from thespinal cord dorsal horn activate many brainstructures through an ascending pathway thatinvolves the autonomic, perceptual, and cognitivesystems, which elicit the pain response displayedclinically.4
Depression Model
Clinical symptoms of depression can begrouped into three basic categories: emotional(depressed mood, lack of motivation, disinterestin social activity, anxiety), cognitive (inability toconcentrate, poor memory), and physical(insomnia, headache, fatigue, and stomach, back,and neck pain). The physical pain aspects ofdepression are well recognized among clinicians.For example, the Hamilton Rating Scale forDepression (HAM-D), a clinical rating scaledeveloped in 1960 to assess depression, contains21 items, eight of which are questions that askpatients about their physical symptoms.
Theories about the biologic basis for depressionhave evolved over more than 25 years. Theprincipal biochemical basis for depression hasfocused on two neurotransmitters: serotonin andnorepinephrine.9, 10 These two neurotrans-mitters have also been implicated in theunderlying pathophysiology of chronic pain.4–7
Serotonergic and norepinephrine neurons overlapin the brain, and these two systems interactbiochemically and neuroanatomically. In patientswith depression, alterations or reductions ofthese two neurotransmitters and their respectivereceptors become dysfunctional over time,leading to a dysregulated system. The followingsix criteria for dysregulation have been proposed:
1573
Table 1. Stimuli and Their Associated Receptors ThatMediate Nociceptive Pain
Stimulus ReceptorNerve growth factor Tyrosine kinase A (TrkA)Bradykinin Bradykinin receptor subtype 2
(BK2)Serotonin Serotonergic receptor subtype 3
(5-HT3)Adenosine Adenosine 5′-triphosphate–gated5′-triphosphate ion channel with receptor (P2X3)
Heat Vanilloid receptor subtype 1 (VR1)and vanilloid-like receptorsubtype 1 (VRL-1)
Hydrogen Acid-sensing ion channel subtype3 (ASIC3) and VR1
Lipids Prostaglandin E2, cannabinoid 1,and VR1 (PGE2-CB1-VR1)
Pressure Degenerin and epithelial sodiumchannel (EDG/EnaC)
Glutamate a-Amino-3-hydroxy-5-methyl-4-isoxazolepropionateacid,N-methyl-D-aspartate, andglutamate (AMPA-NMDA-mGluR)
Substance P Neurokinin subtype 1 (NK1)Brain-derived Tyrosine kinase B (TrkB)neurotropic factor
Adapted from reference 4.
Table 2. Type of Neuropathic Pain and Anatomic Associations
Anatomic Location Type of Neuropathic PainBrain Stroke, multiple sclerosis, cancer,
syringomyeliaSpinal cord Multiple sclerosis, injury, cancer,
syringomyeliaPeriphery Neuropathies, herpes zoster, injury,
amputations, cancer, plexopathies,trigeminal neuralgia
Adapted from reference 8.

PHARMACOTHERAPY Volume 27, Number 11, 2007
the system is impaired in one or more regulatoryor homeostatic mechanisms; basal output of thesystem is erratic; normal periods of functioningare disrupted; the system is less responsive toenvironmental stimuli; a slow return to basalactivity occurs after the disturbance; and effectiveagents restore or reregulate the system.11 Basically,norepinephrine and serotonin concentrations andoutput become erratic in patients with depres-sion, and antidepressants attempt to restore a“normal” firing rate in neuronal areas andneurotransmitter activity at the synaptic cleft.
Both the serotonin and norepinephrinepathways in the brain and their associatedsymptoms have been determined. Both pathwaysoriginate in the brain stem and project to variousbrain regions (Figure 1). Serotonin pathwaysoriginate at the raphe nucleus and project to thefrontal cortex, basal ganglia, hypothalamus, andlimbic areas. Norepinephrine pathways originatein the locus ceruleus and project to the frontalcortex, limbic areas, hypothalamus, andcerebellum. The clinical symptoms for mooddisturbance can be associated with the frontalcortex and limbic regions. Loss of appetite,weight loss or gain, and loss of pleasure can beconnected to the hypothalamus. Therefore,depressive symptoms originate from variousbrain areas that result in a complex set of clinicalpresentations to the health care professional.Each symptom can vary over time in intensityand duration, challenging the role of pharmaco-therapeutic interventions.
Interrelationship Between Chronic Pain andDepression
The link between the higher brain centersinvolved with depression and pain and theperipheral body regions occurs in the brain stem,with neurotransmission relayed through thespinal cord. Abnormal body activity andfunctions (e.g., musculoskeletal) are suppressedfrom the consciousness by the serotonin andnorepinephrine descending pathways in thespinal cord that originate in the brain stem.12, 13
This suppression is not always constant andfunctions as a homeostatic regulator. Forexample, these descending pathways suppressthe body’s input from minor discomforts such asaching muscles and joints.
As the descending serotonin and norepi-nephrine neurons arise from the brain stem, twoareas within the brain stem have been identifiedas the source of these neurons (Figure 1). Aspreviously mentioned, the dorsal raphe nucleusserves as a basis for serotonin neurons, and thelocus ceruleus serves as a foundation fornorepinephrine neurons.14 In fact, specificnorepinephrine cell groups A5 and A7 in thelocus ceruleus have been identified and provideanatomic evidence of neuronal projections fromthe brain stem to the spinal cord.15–17
A dysfunctional serotonin and norepinephrinesystem that promotes emotional and vegetativedepressive symptoms is likely to also havedysfunctional descending serotonin andnorepinephrine pathways, which provides theexplanation for depressed patients who alsocomplain of headache, abdominal pain, andmusculoskeletal pain in the lower back, joints,and neck, as well as fatigue and energy loss. Theserotonin and norepinephrine descendingneurons project from the brain stem into thespinal cord’s dorsal horn. Within this area, acomplex set of biochemical actions takes placeinvolving other types of neurotransmitters (e.g.,g-aminobutyric acid) and peptides (substance P)modulating the pain stimuli that originates in theperipheral neurons (Figure 2).18
There are two main primary afferent nociceptorpathways that lead into the spinal cord from theperiphery (Figure 2).19 One pathway ischaracterized by a host of peptides that includessubstance P, and the other pathway has bindingaction to the peptide IB4 lectin. Both pathwaysare composed of Ad and C fibers and terminate inthe superficial region of the dorsal horn.Another peripheral pathway that involves
1574
Figure 1. Origins and projections of the serotonin andnorepinephrine pathways, and interactions between thebrain stem and other higher brain areas, with their clinicalmanifestations.
Serotonin: raphe nucleus
Norepinephrine: locus ceruleus
Sleep centers
Cerebellum
Limbic regions
Hypothalamus
Frontal cortex
Depression
Cognition problems
Agitation
Psychomotor retardation
Agitation
Psychomotor retardation
Loss of appetiteLoss of pleasure Guilt
Suicidal ideationsWeight gain or loss
Brain stem
Spinal cord
Sleep disturbance

TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
norepinephrine neurons contains sympatheticpostganglionic neurons, as well as neuropeptideY. A painful stimulus from the periphery canaffect multiple pathways, eliciting the complexset of mechanisms leading to and found withinthe central nervous system.
As stated earlier, numerous receptor systemslocated on these two nociceptor pathways existthat form the starting point of pain and itsinteractions with depression. Glutamate, a majorexcitatory neurotransmitter, affects both path-ways and could serve as one common pharma-cologic model for the rapid action of the Adfibers and the slower actions of C fibers.5 Manyother receptor systems and their correspondingstimuli complete this discussion of the circuitryof pain modulation that incorporates depressivesymptomatology. This discussion focuses onlyon serotonin and norepinephrine pathways.Other models are certainly involved, mostnotably, enkephalin and opioid peptide links.18
Many questions remain, such as, what are thefunctional consequences of coexistent neuro-
transmitters and other neuromodulator receptorsystems in a single neuron? Are there presynapticinfluences on descending pathways? What typesof interactions occur between serotonin andnorepinephrine terminals in the spinal cord?Regardless of these many unanswered questions,depression and pain possess a physiologicallylinked basis with a discrete central nervoussystem network involving opioid-like peptides,biogenic amines, glutamate, and other transmitters.
Role of Antidepressant Drugs
For many years, antidepressants have beenused in different chronic pain syndromes with orwithout the presence of clinical depression.Numerous case reports and noncontrolled studieshave appeared in the literature. More than 40double-blind, placebo-controlled trials in whichantidepressants had been used to treat headache,facial pain, rheumatic pain, peripheral neuropathicpain, central pain, chronic pain of variousorigins, and cancer pain reported that theseagents benefit these patients.20 The studies alsoindicated that the antidepressant’s beneficialeffect on pain does not seem to be related to itseffect on mood. By incorporating these conceptswith the newer models of pain and depressiondiscussed in the previous sections, we unify theseconcepts together with the animal studies thathave evaluated the potential of antidepressantagents to improve pain symptoms.
Animal Pain Models and In Vitro Data
Based on the interrelationship between painand depression, the serotonin and norepinephrinereuptake inhibitors (SNRIs) may be preferredover other antidepressant pharmacologic classes(e.g., selective serotonin reuptake inhibitors [SSRIs])because of their dual action on noradrenergic(norepinephrine) and serotonergic (serotonin)activities in the central nervous system. Animalmodels have shown a dose-dependent responsewith SNRIs’ ability to decrease pain sensitivity,whereas SSRIs were ineffective.21 In a comparisonof tertiary tricyclic antidepressants (TCAs),SSRIs, and SNRIs in animal models of pain, TCAsand SNRIs had positive results of at least 80% inacute and chronic pain tests; the response rates toSSRIs were considerably lower for acute pain(44%) and chronic pain (33%).22 Mice deficientin serotonin and norepinephrine transportersshowed altered increased pain sensitivity toallodynia after nerve injury, and TCAs decreasedpain sensitivity, which could provide a basic
1575
Figure 2. Peripheral pathways and descending projectionsfrom the brain stem to the spinal cord. The serotonin andnorepinephrine descending neurons project from the brainstem into the spinal cord’s dorsal horn, where a complex setof biochemical actions takes place involving other types ofneurotransmitters (e.g., g-aminobutyric acid [GABA]) andpeptides (substance P), modulating the pain stimuli thatoriginate in the peripheral neurons. A host of peptides thatincludes substance P characterize one primary afferentnociceptor pathway, and the other pathway is identified asIB4 for its binding action to the peptide IB4 lectin. Anotherperipheral pathway that involves norepinephrine neuronscontains sympathetic postganglionic neurons (SPGN) andalso includes neuropeptide Y.
Norepinephrine: locus ceruleus
Serotonin: raphe nucleusBrain stem
Ascending pathway
Descending pathway
Receptors and primary afferent nociceptor pathways
SerotoninNorepinephrine
Norepinephrine or neuropeptide Y
Substance P(?)
Spinal cord
SPGN
IB4
Peptides
Pain stimulusGABA(?)

PHARMACOTHERAPY Volume 27, Number 11, 2007
pharmacologic model for antidepressant efficacyin pain.22
An in vitro comparison was made between thenewer SNRIs duloxetine and venlafaxine inassessing the inhibition of monoamine uptakeand transporter binding.23 Duloxetine wasshown to have more potent tritiated hydrogenbinding affinity than that of paroxetine(serotonin) and nisoxetine (norepinephrine) onboth human cell lines and rat brain area andsynaptosomes. Venlafaxine also displayed theseproperties, but at a lower potency. In another invitro study, duloxetine, venlafaxine, desipramine,and other antidepressants were evaluated fortheir inhibition of monoamine transporterbinding.24 Duloxetine showed potent inhibition(mean concentration of which the inhibitorelicits 50% of maximal inhibition [Ki]) of bothnorepinephrine and serotonin transporterscompared with the other agents (norepinephrine:duloxetine Ki 7.5 nmol/L vs venlafaxine Ki 2483nmol/L and desipramine Ki 3.8 nmol/L;serotonin: duloxetine Ki 0.8 nmol/L vsvenlafaxine Ki 82 nmol/L and desipramine Ki 179nmol/L). Duloxetine was equally potent assertraline for serotonin inhibition (0.9 nmol/L)and much more potent than sertraline in norepi-nephrine inhibition (715 nmol/L), demonstratingthat duloxetine is a potent norepinephrine andserotonin inhibitor that differs from otherantidepressants.
Duloxetine was compared with other anti-depressants in a variety of animal pain models.25
Duloxetine 3–15 mg/kg in a dose-dependentmanner was reported to significantly attenuate(p<0.05) late-phase paw-licking behavior,whereas paroxetine lacked any effect. In aformalin model, duloxetine was reported to bemore potent than venlafaxine and amitriptylineand also did not produce any motor coordinationproblems with use of the rotorod (a rotating-roddevice used in rodent studies) test. Duloxetinewas also shown to be effective in reversingmechanical allodynia behavior in the L5–6 spinalnerve ligation model of neuropathic pain, butwas only minimally effective for the tail-flickmodel of acute nociceptive pain.
In another study, duloxetine was comparedwith gabapentin, morphine, and ibuprofen inacute nociceptive pain models and inflammatoryor persistent pain models.26 Duloxetine did notproduce a significant effect on response latencyin the mouse tail-flick model, but had a modestincrease in the mouse hot-plate test. Morphineproduced dose-dependent analgesic effects in
both of these tests. In models of inflammatory orpersistent pain, duloxetine, gabapentin, andibuprofen reversed thermal hyperalgesia andmechanically induced allodynia in a dose-dependent manner. Duloxetine and gabapentindid not have a substantial effect on the rotorodtest compared with morphine. Ibuprofen only atdoses of 1000 mg/kg produced modest effects onthe rotorod test, and doses less than 300 mg/kghad no effect.
Venlafaxine’s in vitro profile somewhat resemblesthat of duloxetine for both norepinephrine andserotonin activities, and venlafaxine was reportedto be effective in several animal models of pain.27
For example, venlafaxine showed a dose-dependent improvement in reducing pain in ratswith the vincristine model of neuropathy.28
Venlafaxine 10-, 20-, 40-, and 80-mg/kg doseswere evaluated. At 40 and 80 mg/kg, venlafaxineproduced maximal effects on C-reflex inhibitionwith a median effective dose calculated at 27.2mg/kg. Although some benefit was observedwith the venlafaxine 10- and 20-mg/kg doses, itwas not significant over a 2-hour time courseduring the study. These results may indicate thathigher venlafaxine doses could be needed fortreating chronic pain symptoms. The need forhigher venlafaxine doses could be due to itsweaker potency for norepinephrine activity.
In summary, both duloxetine and venlafaxinewere shown to possess significant dose-dependenteffects in alleviating pain in a variety of animalmodels. Both of these agents were comparedwith other antidepressants including TCAs andSSRIs. The SSRIs did not produce any consistentimprovement in the pain models. The tertiaryTCAs had improvements in the animal painmodels, but also had predictable adverse effects(e.g., motor coordination problems). The animalmodels support the long-established clinicalobservations of TCAs being effective and wellrecognized in various chronic pain syndromes,but also show that these agents have predictableadverse effects that can limit their use. Thesefindings in animal models provide a pharmacologicbasis to evaluate the efficacy of duloxetine andvenlafaxine in clinical studies of various painsyndromes in patients with or without depression.
Clinical Trials
Depression with Physical Pain Symptoms
Although many double-blind, placebo-controlled trials of antidepressants in patientswith depression and pain have been conducted,
1576

TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
these studies were usually completed in a smallnumber of patients (< 100).20 We present newerinformation with these agents from largemulticenter trials. Table 3 presents a summary ofthe clinical trials that analyzed the efficacy of
antidepressants in patients with depression andsymptoms of physical pain. Duloxetine was themost studied agent in clinical trials that focusedon these two paradigms of depression andphysical pain. These clinical studies were
1577
Table 3. Summary of Clinical Trials of Antidepressants for the Treatment of Depression and Pain
Study No. ofDesign Patients Duration Treatment Rating Scales CommentsDB, PC29 353 9 wks Duloxetine 40, 60, or HAM-D-17, VAS, Duloxetine 80 mg showed significant
80 mg/day SSI, CGIS, PGI-I, improvement in overall pain scoresParoxetine 20 mg/day QoL-D (p<0.01), reduced awake pain (p<0.05),Placebo and shoulder pain (p<0.05); paroxetine,
duloxetine 40 mg, and placebo showedno significant improvements in painscores; data for duloxetine 60 mg werenot reported
DB, PC30 245 9 wks Duloxetine 60 mg/day HAM-D-17, VAS, Duloxetine > placebo in decreasingPlacebo CGIS, PGI-I, HAM-D-17 total scores (p<0.001) at
QoL-D 2 wks; overall pain scores were lowerfor duloxetine at 9 wks (p=0.055) withthe most profound effect in back pain(p<0.001)
DB, PC31 512 9 wks Duloxetine 60 mg/day HAM-D-17, CGIS, Duloxetine > placebo in overall painPlacebo PGI-I, QoL-D, (p=0.016), back pain (p=0.002), and
VAS, SSI shoulder pain (p=0.021); VAS scoreswere significantly lower in remittersthan nonremitters (p<0.001)
DB, PC32 282 7 wks Duloxetine 60 mg/day HAM-D-17, BPI, Duloxetine showed a strong trend over Placebo VAS, CGIS, placebo in BPI scores (p=0.066) at end
PGI-I point but had significant differences atweeks 1, 2, and 5; in patients with ≥ 1depressive episode, duloxetine > placebo,as seen in previous studies
DB, PC33 207 12 wks Duloxetine 120 mg/day FIQ, CGIS, PGI-I, Duloxetine > placebo in FIQ total scorePlacebo BPI, SF-36, (p=0.027), but not in pain score
QoL-D, SDS, (p=0.130); significant improvementsBDI-II, BAI noted in CGIS (p=0.048), BPI (p=0.004),
and PGI-I (p=0.033); 38% of patientshad fibromyalgia + major depression:duloxetine improved both conditions
Open 186 1 yr Venlafaxine HAM-D-21, VAS 73 patients with chronic pain andlabel34 ≥ 150 mg/day QoL-12 depression had improved HAM-D
(p<0.001) and VAS (p<0.001) scores;QoL scores improved in depressedpatients with or without pain (p<0.001)
Open 573 9 mo Paroxetine 20 mg/day HSCL-20, PHQ-9, Physical pain symptoms improved onlylabel35 Fluoxetine 20 mg/day PHQ-15, SF-36, during first month, then plateaued with
Sertraline 50 mg/day WLQ, CDS minimal resolution; depressioncontinually and gradually improved over9 mo; remitters (HSCL-20 score < 6) andpartial responders (≥ 50% decrease)improved more than nonresponders;predictors of nonresponse were advancedage, poor physical function, low energylevel, and presence of depression
DB = double-blind; PC = placebo-controlled; HAM-D-17or -21 = Hamilton Rating Scale for Depression 17 or 21 items; VAS = Visual AnalogScale; BPI = Brief Pain Inventory; SSI = Somatic Symptom Inventory; CGIS = Clinical Global Impression of Severity; PGI-I = Patient GlobalImpression of Improvement; QoL-D = Quality of Life in Depression Scale; HSCL-20 = Hopkins Symptoms Checklist 20 items; FIQ =Fibromyalgia Impact Questionnaire; PHQ-9 or -15 = Patient Health Questionnaire 9 items or 15 items (somatic symptoms); SDS = SheehanDisability Scale; BDI = Beck Depression Inventory–II; BAI = Beck Anxiety Inventory; SF-36 = Short Form–36 General Health Survey; WLQ =Work Limitations Questionnaire; CDS = Chronic Disease Score; and QoL-12 = Quality of Life–12 items.

PHARMACOTHERAPY Volume 27, Number 11, 2007
conducted in adult populations with a minimumage of 18 years.
A clinical study compared duloxetine with theSSRI paroxetine.29 Subjects were randomlyassigned to receive duloxetine 40, 60, or 80mg/day, paroxetine 20 mg/day, or placebo for 9weeks. The HAM-D (17 items) was used as theprimary indicator of efficacy. Secondarymeasures included the 100-mm Visual AnalogScale (VAS), Somatic Symptom Inventory (SSI),Clinical Global Impression of Severity scale(CGIS), and the Patient Global Impression ofImprovement scale (PGI-I); these werecompleted at every visit. The Quality of Life inDepression scale (QoL-D) was conducted atbaseline and after 9 weeks of treatment.Significant improvement in VAS overall painseverity was found only with duloxetine 80mg/day (p<0.01) at weeks 6 and 8. Althoughimprovement in VAS scores were observed withduloxetine 80 mg/day at week 1, it was notstatistically significant. Duloxetine 40 mg/day,paroxetine 20 mg/day, and placebo showedminimal improvement, which also lackedstatistical significance. On analysis of the HAM-D subscale item 13, which specifically assessedpainful physical symptoms (backaches,headaches, and muscle aches), significantimprovement was reported with duloxetine 80mg/day (p<0.05) but not with the other treat-ments. The SSI scores did not show improve-ment in any treatment group. This preliminarystudy does indicate that duloxetine 80 mg/daycould be effective in the treatment of physicalpainful symptoms, whereas a lower 40-mg dosewas ineffective. Lack of efficacy was also foundwith paroxetine, which supports the theory thatan antidepressant with a dual pharmacologicmechanism of action is needed to achievetherapeutic benefit for both depression and pain.
Another double-blind, placebo-controlledclinical study evaluated duloxetine 60 mg/dayversus placebo and was conducted with anassigned randomization of 1:1.30 Unlike otherstudies, this was the only study, to our knowledge,to screen for depressed patients who also hadlevels of self-reported pain before the start oftreatment. (In the other studies, patients werenot required to meet a minimum threshold forpain, and the studies were not powered for painoutcomes.) Study drug consisted of threeduloxetine 20-mg capsules or three identicalplacebo capsules taken once/day for 9 weeks.The dose could be decreased to two capsules fortolerability reasons (only once during the study)
but had to be increased back to three capsulesafter 3 weeks and remain at that level for theremainder of the study. Prescription pain drugswere not allowed. Antihypertensive drugs wereallowed only if the subject had been taking astable dose for at least 3 months. The primaryefficacy evaluation was by the HAM-D, and thesecondary measures included the CGIS, VAS forpain assessment, and PGI-I, which were completedat every visit. The QoL-D was conducted atbaseline and after 9 weeks of treatment.
Study results indicated that duloxetine showedsignificant improvement in total HAM-D scoresat 2 weeks (p<0.001) with sustained improvementat 9 weeks based on the consistent reduction intotal HAM-D scores versus placebo. The meanchange between baseline and study end-pointscores for duloxetine and placebo at 9 weeks was-10.91 and -6.05 points, respectively (p<0.001).The analysis of the HAM-D subscale item 13reported significant improvement withduloxetine compared with placebo, whencomparing baseline with study end-point scores(duloxetine -0.78 vs placebo -0.49, p<0.013). Allother secondary measures also showed significantimprovement with duloxetine versus placebo(p<0.001). The most commonly reportedadverse events were nausea, dry mouth, andsomnolence. This study indicated that duloxetinecould be an effective agent for both depressionand painful physical symptoms and established aminimum dose-response threshold.
Results of two subsequent multicenter clinicaltrials with duloxetine substantiated the efficacyof the 60-mg/day dosage versus placebo.31, 32 Thestudy methodology was similar to the previousstudy30 with duloxetine 60 mg/day in whichrandomization was in a 1:1 duloxetine:placeboratio. The clinical assessments were alsoidentical and included the HAM-D 17 items,CGIS, VAS, PGI-I, QoL-D, and SSI, whichfocused on symptomatic pain. The VAS wasexpanded to include subscales of overall pain,headaches, back pain, interference with dailyactivities, and time in pain while awake. One ofthe studies32 also used the Brief Pain Inventory(BPI) as its primary efficacy instrument for painassessment. The other study31 was designed tohave 80% power to detect a 2.73-point differencein the total HAM-D score but was not poweredfor pain outcomes. The former study32 waspowered at 80% to detect a treatment effect sizeof 0.36 with an a of 0.05 and a 15% increase insample size to factor for early subjectterminations. These parameters yielded a
1578

TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
treatment group of 141 patients/group with anestimated group difference of VAS scores of 20 orgreater (scale 0–100 mm). Both studies used thelikelihood mixed-effects model repeatedmeasures to assess the mean changes frombaseline for all efficacy outcomes. The primaryanalysis addressed the association betweenpainful physical symptoms and depressionremission (defined as HAM-D score ≤ 7) andresponse (defined as ≥ 50% reduction in totalHAM-D score).
In the first study,31 the VAS scores significantlyimproved for all subscales except for headache.Duloxetine was superior to placebo in inter-ference with daily activities and pain while awake(p<0.05) and with shoulder pain, back pain, andoverall pain (p<0.05) when comparing baselinewith study end point. The VAS overall painseverity showed significant improvement in theduloxetine group at 2 weeks versus the placebogroup (p<0.005) and continued to be sustained atweek 9 (p<0.05). Weak correlations betweenVAS overall pain scores and HAM-D total scoreswere found throughout the study (e.g., week 3, r= 0.226). Of interest, improvement was foundwith total HAM-D scores. Therefore, althoughimprovement in depression and pain scores werefound, the time of improvement and themagnitude of improvement differed betweenthese two paradigms. In the duloxetine group,remission rates were 38.8% versus 24.8% (p=0.027)for pain responders versus nonresponders. In theplacebo group, the remission rates were 32.6%versus 12.3% (p<0.001) in the pain respondersversus nonresponders. These findings indicatedthat pain response is needed for remission tooccur in a significant number of subjects. Thelikelihood of pain nonresponse could contributetoward diminished efficacy with duloxetine.Patients with early pain response noted at 2weeks had a significantly higher probability ofdepression response compared with patients notshowing early pain response (35.4% vs 20.9%,p=0.009). All other secondary scales used in thetrial demonstrated significant improvementsfrom baseline to study end point. Significantcorrelations between QoL-D and VAS pain scoreswere found (p<0.001). Treatment-emergentadverse events were not reported in this study.
Results from the second study32 showed thatthe mean change in BPI score in the duloxetinegroup had significant improvement by week 1(p<0.005) with noted improvements at weeks 2and 5 (p<0.05). Improved BPI scores wereobserved at other time periods, but these scores
were not statistically significant compared withplacebo. Mean changes from baseline to endpoint in VAS pain measures, CGIS, and PGI-I didnot differ significantly between groups. Whenpooled BPI assessments from all the visits wereanalyzed together, duloxetine was shown to besignificantly greater than placebo for meanimprovements in pain severity, worst pain, leastpain, average pain, and pain right now (p<0.05).Improvement ranged from 33.5–49.6% for theduloxetine group compared with 19.0–39.3% forthe placebo group. Mean change in total HAM-Dscores did not differ significantly between thetwo groups (duloxetine -10.85 vs placebo -10.27,p=0.544). The data were then analyzed bycomparing patients with no previous depressiveepisode with patients with one or more previousepisodes. Significant improvements were foundwith duloxetine in the mean change in averageBPI scores (p=0.012) and BPI walkinginterference scores (p<0.001) in patients withone or more previous episodes. These resultsshowed that patients with previous depressiveepisodes and physical pain symptoms respondedto duloxetine compared with placebo. Nausea,dry mouth, and fatigue were the most commonlyreported adverse events with duloxetine.
Physical Pain With or Without Depression
A multicenter, double-blind, placebo-controlledtrial was conducted to investigate the potentialbenefits of duloxetine in patients with primaryfibromyalgia with or without depression.33 Thepathophysiology of fibromyalgia is unknown, butevidence for dysfunctional serotonin andnorepinephrine neurotransmission might play arole in its pain modulation. Only patients withprimary fibromyalgia were allowed into the studyand were required to score 4 or higher on thepain intensity item (scale 0–10) of theFibromyalgia Impact Questionnaire (FIQ). Afterscreening, patients had a 1-week, single-blind,placebo lead-in phase and then were randomlyassigned in a 1:1 ratio to receive duloxetine orplacebo. Duloxetine dosage was a forced titrationmethod from 20 mg/day to 60 mg twice/day forthe first 2 weeks in the following manner: 20mg/day for 5 days, 20 mg twice/day for 5 days, 40mg twice/day for at least 2 days, and then 60 mgtwice/day by week 2. The dosage of 60 mgtwice/day remained fixed throughout theremainder of the study.
The coprimary efficacy measures were the FIQpain severity score and the total FIQ score.
1579

PHARMACOTHERAPY Volume 27, Number 11, 2007
Secondary measures included other FIQsubscales such as fatigue, morning tiredness, andstiffness. Other secondary assessments includedthe BPI, PGI-I, Short Form–36 General HealthSurvey (SF-36), QoL-D, CGIS, and SheehanDisability Scale. The severity of depression andanxiety was evaluated by the Beck DepressionInventory-II (BDI-II) and Beck Anxiety Inventory(BAI). The FIQ and BPI were completed at eachweekly visit, whereas the remaining assessmentswere conducted at weeks 4, 8, and 12.
The study results reported that duloxetineshowed significant improvement in total FIQscores versus placebo at week 4 (p=0.005) andconsistent improvement to week 12 (-5.53points, p=0.027). Early improvement was notedat weeks 1 and 2, but the change in total FIQscore was not significant. However, improve-ment in the pain severity FIQ score was found tobe significant at weeks 1 and 2 (p=0.004 andp<0.001, respectively). Maximum pain relief wasnoted to occur at week 4 (p<0.001). A strongtrend was found in the response rate (defined asat least 50% reduction in total FIQ score) in theduloxetine group compared with the placebogroup (27.7% vs 16.7%, p=0.06). Significantdifferences (p<0.05) between duloxetine andplacebo were found for the BPI, PGI-I, and CGISscores, but not with the BDI-II and BAI scores.These findings demonstrate that duloxetine canbe effective for the physical pain symptomsregardless of the presence of depression oranxiety in patients with fibromyalgia. Efficacywas reported to be greater in female patientscompared with male patients; however, morethan 88% of the study patients were women.This study’s results could be biased toward thefemale population. Another possibility is thatmale patients may not recognize this disease and,therefore, would not seek treatment.
A 1-year, open-label, single-center studyevaluated the efficacy of venlafaxine extendedrelease (XR) for the treatment of chronic painassociated with depression.34 Patients with avariety of chronic pain syndromes for at least 3months entered the study. Patients wererecruited by referral from their primary carephysicians who were treating the patients fortheir depression. These pain syndromes includedchronic back pain, migraine, and chronicregional pain syndromes. Venlafaxine-XR wasstarted after at least a 10-day washout fromprevious antidepressants except for fluoxetine,which had a 30-day washout. Venlafaxineimmediate-release was started at 12.5 mg
twice/day for 3 days, and then increased to 37.5mg twice/day for an additional 3 days.Venlafaxine-XR was then started at 37.5 mg/dayand the dose increased every 3 days to 75 mg/dayand then to 150 mg/day or higher. The meanstudy dose was 225 mg/day. Only nonsteroidalantiinflammatory drugs as needed werepermitted for intermittent pain. The HAM-D andVAS were used to evaluate depression and pain;QoL-12 was used to assess quality of life. Patientswere evaluated in two groups: depression onlyand depression with pain. After 1 year, HAM-Dscores were significantly lower in both groupscompared with baseline scores (depression-onlygroup 8.9 vs 17.6, p<0.001; depression and paingroup 8.9 vs 16.8, p<0.001). The VAS scores forthe depression and pain group also showedsignificant improvement from baseline to thestudy end point (8.4 vs 3.6, p<0.001). Baselineand study end-point QoL-12 scores were similarfor both patient groups, which showedsignificant improvement (p<0.001). Only 11patients discontinued treatment because ofadverse events, which were nausea, anxiousness,agitation, and sexual side effects.
In another open-label study, depressed patientswere evaluated on the outcome of physicalsymptoms during 9 months of antidepressanttreatment.35 Patients were included in this studyif the primary care physician thought that anti-depressant therapy was warranted based on theirclinical evaluation. Patients were randomlyassigned to receive one of three SSRIs—paroxetine 20 mg/day, fluoxetine 20 mg/day, orsertraline 50 mg/day. Depression outcomes wereassessed by the Hopkins Symptoms Checklist–20items (HSCL-20) and the 9-item Patient HealthQuestionnaire (PHQ). The HSCL includes a 13-item depression subscale and 7 items that allowfor the assessment of the Diagnostic andStatistical Manual of Mental Disorders, FourthEdition (DSM-IV). The PHQ-9 is a self-administered test that includes nine DSM-IVdepressive symptoms and is a validated measureof depression severity. The PHQ-15, which alsowas used, is a different assessment that evaluatesphysical symptoms. Quality-of-life evaluationsincluded the SF-36 and three scales from theWork Limitations Questionnaire (outputdemand, time management, interpersonalrelationships). Medical comorbidity wascalculated for each patient with the ChronicDisease Score. Evaluations were conducted atbaseline and at 1, 3, 6, and 9 months afterenrollment. Major depression was noted in 74%
1580

TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
of the patients, dysthymia in 18%, and minordepression in 8%. At least 33–50% of thepatients had physical complaints, with 10–20%having severe symptoms (e.g., fatigue, sleepdisturbance, pain, and headaches).
The SSRI treatments resulted in a substantialimprovement in physical pain symptoms asshown by PHQ-15 scores for only the firstmonth, then the scores plateaued with minimalimprovement during the remainder of the study.Depressed patients without physical painsymptoms had the most benefit when PHQ-9scores were compared with those of depressedpatients with physical pain symptoms. Similar tothe PHQ-15 scores, improvement in PHQ-9scores was found to occur in the first month oftreatment with consistent benefits. Remittersand partial responders to antidepressant therapy(defined as ≥ 50% improvement in HSCL-20scores after 3 mo) had significant improvement(p<0.001) in both pain and nonpain physicalsymptoms compared with nonresponders.Significant differences were not found betweenremitters and partial responders. A logisticregression analysis revealed that significantpredictors of nonresponse included advancedage, presence of depression, poor physicalfunctioning, and low energy level (p<0.10).
This study showed that physical symptoms indepressed patients only initially improved withSSRI treatment, and unlike depression, minimalresolution occurred afterward. The fixed-dosedesign with the SSRIs could have been a limitingfactor. Whether further improvements couldtake place with increased drug doses, especiallywith sertraline, was not investigated. However,this was an open-label study, which lends supportthat SSRIs may not be the best drug choice fordepressed patients with physical painfulsymptoms; a double-blind, placebo-controlledtrial would strengthen these findings.
Summary
Most of the above-mentioned clinical trialswere conducted mainly with duloxetine andother agents in a double-blind, placebo-controlledenvironment. These studies demonstrate thatantidepressants are safe and effective treatmentsof physical painful symptoms whether or notcomorbid depression and/or anxiety are presentfor a relatively short time period of up to 12weeks. An early response was noted during thefirst few weeks, with continued benefitsthroughout the study. The trials with a double-
blind, placebo-controlled design typically were9–12 weeks in duration compared with the muchlonger duration of 9–12 months for the open-label studies. The shorter study duration for thedouble-blind, placebo-controlled trials is directlyrelated to their costs, subject compliance, andretention. Nevertheless, whether the clinicaltrials used double-blind, placebo-controlled oropen-label methodologies, they showed aconsistent pattern of clinical efficacy for drugresponse.
Antidepressant Agents in the Treatment ofDiabetic Neuropathy
A focus on the treatment of diabetic neuropathywith antidepressants was selected because of therecent approval by the United States Food andDrug Administration (FDA) of duloxetine forthis specific disease state. Although numerousstudies have been published with TCAs indifferent pain syndromes including diabeticneuropathy,20 only a relatively small number ofstudies that have compared TCAs with SSRIshave explored the requirement of a noradrenergiceffect in an agent for analgesia to occur inpatients with diabetic neuropathy. Studies withSNRIs in this population of patients have alsobeen conducted. In addition, combinationtherapy for diabetic neuropathy (e.g., pregabalin)can be prescribed where different pharmacologicapproaches are used to maximize therapeuticefficacy in reducing these painful symptoms.
Tricyclic Antidepressants and Selective SerotoninReuptake Inhibitors
Imipramine was compared with paroxetine in20 patients with diabetic neuropathy.36 After 1week of taking placebo, subjects were randomlyassigned into a three-way crossover study toreceive imipramine, paroxetine, or placebo for 2weeks and then cross over to another treatmentgroup. The treatment group duration was 2weeks, and each subject participated in all threetreatment groups. There was no washout periodbetween treatments except for in three patientswho were identified as poor metabolizers ofcytochrome P450 (CYP) 2D6 and in whom a2–3-week washout was allowed. Subjects weregiven a fixed imipramine daily dose of either 50or 75 mg to achieve a plasma concentration(imipramine + desipramine) of 400–600 nmol/L,and paroxetine was administered as 40 mg/day.Clinical assessments were conducted by patient
1581

PHARMACOTHERAPY Volume 27, Number 11, 2007
self-ratings with use of the VAS and by a singlephysician rating the neuropathy with use of a 6-item observation scale.
Both imipramine and paroxetine were reportedto be significantly better than placebo in reducingmedian VAS scores. The improvement in medianVAS scores from baseline was placebo 141.5,paroxetine 81.5, and imipramine 37.0 (p<0.001).Of interest, imipramine was shown to haveslightly greater effect than paroxetine. In theneuropathy findings, both agents showedsignificant improvement compared with placeboin five of six items (not hypesthesia). Improve-ment with imipramine was significant comparedwith paroxetine in four of six items: pain,dysesthesia, nightly aggravation, and sleepdisturbance (p<0.05). Although imipramine’sadverse-effect profile could partially explain theimprovements in nightly aggravation and sleepdisturbance, benefits in pain efficacy were clearlynoted. Significantly more adverse effects werereported with imipramine than paroxetine andincluded dry mouth, sweating, fatigue, andpalpitations that resulted in five patientsdropping out of the study. Four patients reportedwithdrawal symptoms after imipramine discon-tinuation. This study showed that imipramineand paroxetine were effective agents in reducingdiabetic neuropathy pain in this short time span,with imipramine being slightly more effectivethan paroxetine, but with more adverse effects.
Amitriptyline, desipramine, and fluoxetinewere evaluated in a double-blind, crossoverstudy.37 Fifty-seven subjects were randomlyassigned to one of two treatment groups: onegroup compared amitriptyline with desipramineand the other compared fluoxetine with placebo.After a 1-week baseline period, subjects weretreated with antidepressants for 6 weeks, had awashout period for the next 2 weeks, and thencrossed over to the other agent for the next 6weeks. The placebo selected for the fluoxetinecomparison was benztropine to mimic the drymouth adverse effect of desipramine andamitriptyline. Patients rated their pain daily bychoosing from a scale of 13 words describingdifferent magnitudes of pain intensity thatshowed internal consistency, reliability, andobjectivity. At the end of each study period, eachpatient made a global rating of pain relief in sixincrements that ranged from complete relief topain worsening. The mean daily doses wereamitriptyline 105 mg, desipramine 111 mg,fluoxetine 40 mg, and placebo (benztropine 1.3mg).
For the desipramine versus amitriptyline arm(29 patients), both drugs were reported to beeffective in decreasing weekly pain from baselineto study point without significant differencesbetween each agent. Seventy-four percent and61% of patients receiving amitriptyline anddesipramine, respectively, reported with theglobal descriptors of moderate or greater painrelief. In the fluoxetine versus placebo arm,fluoxetine did not differ significantly fromplacebo in the weekly pain relief scores (p=0.34).Only 48% of the fluoxetine group reportedmoderate or greater pain relief in the globalassessment compared with 41% in the placebogroup (p=NS). A few (16) patients had depressionin the fluoxetine versus placebo arm, and inthose patients fluoxetine was reported todecrease their pain to a similar extent as that inthe amitriptyline versus desipramine arm. Thepain improvement in the amitriptyline versusdesipramine arm occurred with the samemagnitude in depressed and nondepressedpatients. The most commonly reported adverseeffects were dry mouth (amitriptyline,desipramine, and placebo) and headache(fluoxetine). This study showed that modestdoses of amitriptyline and desipramine wereeffective in pain relief, and despite moderatefluoxetine doses, pain relief occurred only indepressed patients. These studies indicate thatTCAs should be the first-line agents to treatpatients with diabetic neuropathy, and SSRIsshould be reserved for those patients who havecoexistent depression. Again, efficacy from TCAssupport the hypothesis that a noradrenergicpharmacologic profile is needed, and since theseagents also possess a serotonergic property, adual-action antidepressant can be moreefficacious compared with a compound with onlya single pharmacologic specificity (e.g., SSRIs).
Serotonin and Norepinephrine ReuptakeInhibitors
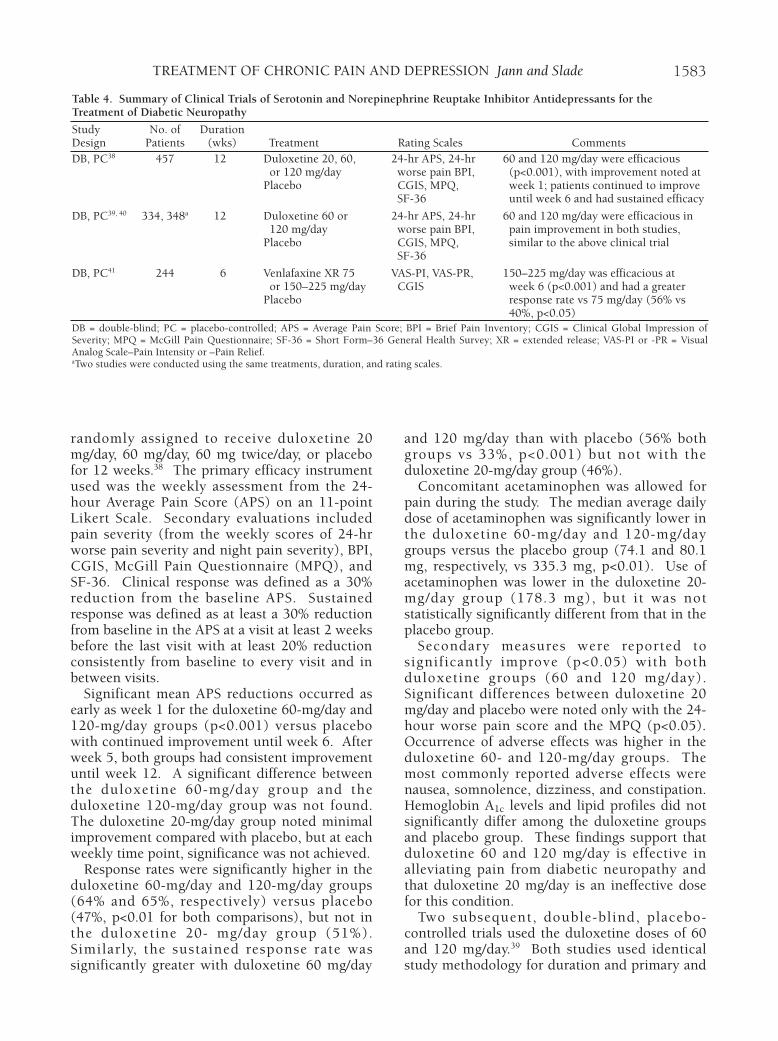
Several double-blind, placebo-controlled trialswith the SNRIs duloxetine and venlafaxine wereconducted in patients with diabetic mellitus type1 or 2 who experienced pain due to bilateralperipheral neuropathy per the MichiganNeuropathy Screening Instrument (Table 4).38–40
These studies enrolled an adult population with aminimum age of 18 years.
Duloxetine
In one study with duloxetine, subjects were
1582
TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
randomly assigned to receive duloxetine 20mg/day, 60 mg/day, 60 mg twice/day, or placebofor 12 weeks.38 The primary efficacy instrumentused was the weekly assessment from the 24-hour Average Pain Score (APS) on an 11-pointLikert Scale. Secondary evaluations includedpain severity (from the weekly scores of 24-hrworse pain severity and night pain severity), BPI,CGIS, McGill Pain Questionnaire (MPQ), andSF-36. Clinical response was defined as a 30%reduction from the baseline APS. Sustainedresponse was defined as at least a 30% reductionfrom baseline in the APS at a visit at least 2 weeksbefore the last visit with at least 20% reductionconsistently from baseline to every visit and inbetween visits.
Significant mean APS reductions occurred asearly as week 1 for the duloxetine 60-mg/day and120-mg/day groups (p<0.001) versus placebowith continued improvement until week 6. Afterweek 5, both groups had consistent improvementuntil week 12. A significant difference betweenthe duloxetine 60-mg/day group and theduloxetine 120-mg/day group was not found.The duloxetine 20-mg/day group noted minimalimprovement compared with placebo, but at eachweekly time point, significance was not achieved.
Response rates were significantly higher in theduloxetine 60-mg/day and 120-mg/day groups(64% and 65%, respectively) versus placebo(47%, p<0.01 for both comparisons), but not inthe duloxetine 20- mg/day group (51%).Similarly, the sustained response rate wassignificantly greater with duloxetine 60 mg/day
and 120 mg/day than with placebo (56% bothgroups vs 33%, p<0.001) but not with theduloxetine 20-mg/day group (46%).
Concomitant acetaminophen was allowed forpain during the study. The median average dailydose of acetaminophen was significantly lower inthe duloxetine 60-mg/day and 120-mg/daygroups versus the placebo group (74.1 and 80.1mg, respectively, vs 335.3 mg, p<0.01). Use ofacetaminophen was lower in the duloxetine 20-mg/day group (178.3 mg), but it was notstatistically significantly different from that in theplacebo group.
Secondary measures were reported tosignificantly improve (p<0.05) with bothduloxetine groups (60 and 120 mg/day).Significant differences between duloxetine 20mg/day and placebo were noted only with the 24-hour worse pain score and the MPQ (p<0.05).Occurrence of adverse effects was higher in theduloxetine 60- and 120-mg/day groups. Themost commonly reported adverse effects werenausea, somnolence, dizziness, and constipation.Hemoglobin A1c levels and lipid profiles did notsignificantly differ among the duloxetine groupsand placebo group. These findings support thatduloxetine 60 and 120 mg/day is effective inalleviating pain from diabetic neuropathy andthat duloxetine 20 mg/day is an ineffective dosefor this condition.
Two subsequent, double-blind, placebo-controlled trials used the duloxetine doses of 60and 120 mg/day.39 Both studies used identicalstudy methodology for duration and primary and
1583
Table 4. Summary of Clinical Trials of Serotonin and Norepinephrine Reuptake Inhibitor Antidepressants for theTreatment of Diabetic Neuropathy
Study No. of DurationDesign Patients (wks) Treatment Rating Scales CommentsDB, PC38 457 12 Duloxetine 20, 60, 24-hr APS, 24-hr 60 and 120 mg/day were efficacious
or 120 mg/day worse pain BPI, (p<0.001), with improvement noted atPlacebo CGIS, MPQ, week 1; patients continued to improve
SF-36 until week 6 and had sustained efficacy
DB, PC39, 40 334, 348a 12 Duloxetine 60 or 24-hr APS, 24-hr 60 and 120 mg/day were efficacious in120 mg/day worse pain BPI, pain improvement in both studies,
Placebo CGIS, MPQ, similar to the above clinical trialSF-36
DB, PC41 244 6 Venlafaxine XR 75 VAS-PI, VAS-PR, 150–225 mg/day was efficacious ator 150–225 mg/day CGIS week 6 (p<0.001) and had a greater
Placebo response rate vs 75 mg/day (56% vs40%, p<0.05)
DB = double-blind; PC = placebo-controlled; APS = Average Pain Score; BPI = Brief Pain Inventory; CGIS = Clinical Global Impression ofSeverity; MPQ = McGill Pain Questionnaire; SF-36 = Short Form–36 General Health Survey; XR = extended release; VAS-PI or -PR = VisualAnalog Scale–Pain Intensity or –Pain Relief.aTwo studies were conducted using the same treatments, duration, and rating scales.

PHARMACOTHERAPY Volume 27, Number 11, 2007
secondary efficacy assessments as that of theabove-mentioned study.38 Clinical response ratesand sustained response rates were not reported.Each study showed that both duloxetine doseshad significant improvement in 24-hour APSscores by week 1 (p<0.001) with continuedimprovement at week 2. In one study (334patients), improvement plateaued after week 2and remained consistent until week 12. Afterweek 5, the duloxetine 120-mg/day group had aslight improvement, greater than that in theduloxetine 60-mg/day group, but the differencewas not significant. For the second study (348patients), improvement gradually continued untilweek 5, then plateaued, and improvementremained consistent throughout the time points.No significant differences between bothduloxetine groups were found throughout thestudy in mean 24-hour APS scores. All secondarymeasures of improvement with both duloxetinegroups were found to be significant comparedwith placebo (p<0.05).
Pooled safety data from all three of thesestudies were presented, and nausea, somnolence,dizziness, and fatigue were the most frequentlyreported.38–40 A slightly higher rate of theseadverse effects was found with the duloxetine120-mg/day group, except for nausea for whichthe frequency was identical to that in the 60-mg/day group. A slightly greater change infasting blood glucose level from baseline wasfound to be significant for the duloxetine 120-mg/day group versus placebo (+9.9 vs -2.47mg/dl, p<0.01) but not for the duloxetine 60-mg/day group (+8.1 mg/dl). No significantchange in hemoglobin A1c level was foundbetween duloxetine 60 mg/day and 120 mg/daycompared with placebo.
In summary, these three pivotal studies38–40
demonstrate that duloxetine 60 mg/day is thebest effective dose for treatment of diabeticneuropathy. If the patient does not adequatelyrespond to that dose, a further dose increase maybe warranted depending on tolerability.Duloxetine 20 mg/day was not shown to benefitpatients, but a 40-mg/day dose was not evaluated.As duloxetine is partially metabolized byCYP2D6, polymorphism status may play a role inthe dose-response relationship for this agent, andfuture studies should be conducted to elucidatethis potential correlation.
Venlafaxine Extended-Release
Venlafaxine XR was evaluated in patients with
painful diabetic neuropathy.41 Patients wererandomly assigned to three groups: placebo,venlafaxine XR 75 mg/day, or venlafaxine XR150–225 mg/day for 6 weeks. During the first 3weeks, patients were in a dose-titration phase;they started at 37.5 mg/day for 1 week and thenincreased to 75 mg/day at week 2. In the higherdose group, the dose was increased to 150mg/day at week 3. By week 4, the number ofcapsules could be individually adjusted to themaximum dose of 225 mg/day within this group.Primary efficacy was assessed with the 100-mmVAS for pain intensity (VAS-PI) and pain relief(VAS-PR). Secondary measurements includedthe CGIS and percentage of patients achieving a50% reduction in pain intensity.
The venlafaxine XR 150–225-mg/day groupshowed a statistically significant difference versusthe placebo group only at week 6 (p<0.001) forVAS-PI and VAS-PR scores. Although improve-ment was found during the other study weeks,the difference was not statistically significant.Minimal improvement was found in thevenlafaxine XR 75-mg/day group, but it did notachieve statistical significance at any time point.Similar findings with the CGIS scores were alsoreported. The response rate was significantlyhigher for the venlafaxine XR 150–225-mg/daygroup versus placebo (56% vs 34%, p<0.01) andalso for the venlafaxine XR 75-mg/day group(40%, p<0.05). Nausea and somnolence were themost commonly reported adverse effects for bothvenlafaxine XR groups. A higher number ofpatients in the venlafaxine XR 75-mg/day group(20%) had a postural decrease in systolic bloodpressure exceeding 25 mm Hg compared withvenlafaxine XR 150–225 mg/day (12%) andplacebo (13%). Seven venlafaxine XR–treatedpatients were reported to have clinicallysignificant electrocardiographic changes duringtreatment, but only four of these patientswithdrew from the study. Overall, venlafaxineXR was suggested to be efficacious and safe inthis short-term study.
This study was of shorter duration comparedwith the duloxetine clinical trials. Althoughdifferent clinical assessments were used, itappears that venlafaxine XR does provide somebenefit in patients with pain due to diabeticneuropathy.
Treatment of Other Pain Syndromes
Antidepressants, especially TCAs, have beenused for a variety of chronic pain syndromes.20
1584

TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
Updated information is presented only for theSNRIs. Venlafaxine has been evaluated in a smallseries of pain syndromes associated with differentdiseases.27 A few double-blind, placebo-controlled studies and useful case reports arepresented in this section.
Venlafaxine was compared with imipramine in32 patients with painful polyneuropathy for atleast 6 months in whom the diagnosis wasconfirmed by nerve conduction studies.42
Patients aged 20–70 years were eligible, and mostof the patients had pain due to diabeticneuropathy (15 patients) or nondiabetic pain (17patients). Patients were randomly assigned in athree-way crossover study to receive venlafaxine,imipramine, or placebo for 4 weeks with a 1-week washout period between treatments.Venlafaxine was dosed as 37.5 mg twice/day forweek 1, then 75 mg twice/day for week 2, andthen 112.5 mg twice/day for the remaining 2weeks. Imipramine was dosed 25 mg twice/dayfor week 1, 50 mg twice/day for week 2, and then75 mg twice/day for the final 2 weeks. Painratings were collected by using a 0–10-point scalefor constant pain (burning or pressing), painparoxysms, touch-evoked pain, and weekly painpressure, but only baseline and week-4 scoreswere evaluated. Patients self-rated their globalpain relief as complete, good, moderate, slight, ornone, or worse pain. Serum venlafaxine andimipramine concentrations were obtained atweek 4 of each study period.
Both venlafaxine and imipramine werereported to be effective in improving all of thepain scores (p<0.05). No significant differencebetween venlafaxine and imipramine was found.In the patient’s global self-ratings, only 7 patientsreceiving placebo reported benefit (21 patientshad no relief or worse pain) compared withvenlafaxine (13 had benefit, 17 had no relief orworse pain) and imipramine (17 had benefit, 12had no relief or worse pain). From the globalevaluation, venlafaxine had only a strong trendtoward efficacy compared with placebo(p=0.073), whereas imipramine was reported tobe significant versus placebo (p=0.003). Again,no significant differences between venlafaxineand imipramine were found (p=0.16).
The most commonly reported adverse eventsfor imipramine were dry mouth and sweating,whereas only tiredness was reported forvenlafaxine. Significantly higher mean serumvenlafaxine and total drug (venlafaxine +metabolite) concentrations were found for theresponders versus nonresponders (p=0.02 and
p=0.006, respectively) in patients assigned toreceive venlafaxine. No correlations were foundbetween imipramine and imipramine plusdesipramine serum concentrations and response.Venlafaxine was reported to be a usefulalternative to TCAs for painful polyneuropathy.The duration of this study was much shorter thanthat of previous studies, and full benefit of thedrugs may not have been realized. The adverse-effect profile was not unexpected for these twodrugs. Since higher drug concentrations andimprovement was found with venlafaxine, thisfinding should be further explored.
In a case report, venlafaxine was noted tobenefit a patient with cancer who had neuropathyfrom oxaliplatin.43 After the 10th cycle withoxaliplatin, therapy was stopped because ofdevelopment of hypesthesias of the fingers(patient was unable to grip utensils). Venlafaxinewas started at 37.5 mg/day, and the patientreported a quick recovery of the sensation in thefingers. Three days later, the venlafaxine dosagewas increased to 37.5 mg twice/day, which wasmaintained for the next 6 months without anydisruption in functional ability.
In another case report, oxaliplatin was stoppedafter nine cycles because of painful neuropathy inboth legs. Venlafaxine 37.5 mg twice/day wasstarted, and after 2 weeks of treatment, noimprovement was noted; venlafaxine wasdiscontinued. Topiramate 50 mg/day was startedand increased to twice/day for 1 week. Thepatient noticed pain relief and was able to walk;this effect was sustained during the month offollow-up.
Only one of the case reports showed thatvenlafaxine could benefit some difficult cases ofpatients with neuropathy from other conditions.Although the second case report did not reportany improvement with venlafaxine, the dosagemay have been too low and the time period tooshort to fully evaluate its potential use. Thispatient’s condition was very serious, however,and topiramate was beneficial.
Nonpharmacologic Intervention with VagalNerve Stimulation
Nonpharmacologic interventions could also beuseful in treating patients with chronic pain anddepression. For example, vagal nerve stimulation(VNS) was recently approved by the FDA fortreatment of refractory depression.44, 45 Continuedantidepressant treatment is recommended alongwith VNS. The vagal nerve afferents have been
1585

PHARMACOTHERAPY Volume 27, Number 11, 2007
implicated to influence nociception and pain.45
Several case reports have shown that VNSreduces pain in patients with migraine andcluster headaches and those with epilepsy.46, 47
Perhaps, VNS combined with antidepressantscould be useful in patients with chronic painsyndromes and depression. Other nonpharmaco-logic interventions such as transcranial magnetictherapy also used for depression may be useful inpatients with chronic pain.
Conclusion
Chronic pain and depression pose significantclinical issues to the health care system, withlong-term consequences for patients. Coexistenceof both diseases often negatively affects thepatient’s performance at work. Some patients canbecome disabled and unable to continue in theiremployment, thereby increasing health careutilization and the economic impact to society.The biologic process for chronic pain anddepression appears to share similar mechanismswhere serotonin and norepinephrine are involvedin the dorsal raphe nucleus and locus ceruleus.Antidepressants with serotonin and norepi-nephrine activity were found to be effective inthe treatment of depression associated withphysical pain symptoms and chronic painsyndromes. The SSRIs were clearly shown tohave marginal benefit compared with the SNRIs.Tricyclic antidepressants have been used forchronic pain syndromes but possess adverseeffects that could limit their long-term use. TheSNRIs like venlafaxine and duloxetine have beenstudied extensively in double-blind, placebo-controlled trials and in open-label studies, whichhave demonstrated the efficacy of these drugs fordepression with physical pain symptoms,physical pain with or without depression,diabetic neuropathy, and other associated painsyndromes.
To date, only one antidepressant agent,duloxetine, has been FDA approved for diabeticneuropathy. The SNRIs, which have a dualpharmacologic profile of serotonin and norepi-nephrine action, have been consistently shown tobe efficacious for pain symptoms. Combinationtherapy that uses different pharmacologicapproaches also could be prescribed.
Antidepressants remain the standard of care fortreating depression as well as generalized andother anxiety disorders. Their use extendsbeyond these areas, however, and it is wellaccepted that the antidepressants are efficacious
in treating chronic pain syndromes. Numerousclinical studies supported these findings.Clinicians should be knowledgeable about thevarious uses of antidepressants in the treatmentof these debilitating medical conditions. Futuredevelopment of antidepressants may involvemultiple neurotransmitters beyond serotonin andnorepinephrine that could include dopaminergicpathways, neuropeptides, corticotropin-releasingfactor, and other neuropharmacologic systemsthat account for these wide therapeuticapplications.
References1. Lepine JP, Briley M. The epidemiology of pain and depression.
Hum Psychopharmacol Clin Exp 2004;19:S3–7.2. Banks SM, Kerns RD. Explaining the high rates of depression
in chronic pain: a diathesis-stress framework. Psychol Bull1996;119:95–110.
3. Burt VK. Plotting the course to remission: the search for betteroutcomes in the treatment of depression. J Clin Psychiatry2004;65(suppl 12):20–5.
4. Julius D, Basbaum AI. Molecular mechanisms of nociception.Nature 2001;413:203–10.
5. Woolf CJ, Salter MW. Neuronal plasticity: increasing the gainin pain. Science 2000;288:1765–8.
6. Costigan M, Woolf CJ. Pain: molecular mechanisms. J Pain2000;1(suppl 1):35–44.
7. Mannion RJ, Woolf CJ. Pain mechanisms and management: acultural perspective. Clin J Pain 2000;16:S144–56.
8. Jensen TS, Gottrup H, Sindrup SH, Bach FW. The clinicalpicture of neuropathic pain. Eur J Pharmacol 2001;429:1–11.
9. Wong DT, Bymaster FP. Dual serotonin and noradrenalineuptake inhibitor class of antidepressants: potential for greaterefficacy or just hype? Prog Drug Res 2002;58:171–222.
10. Blier P, Abbott FV . Putative mechanisms of action ofantidepressant drugs in affective and anxiety disorders andpain. J Psychiatry Neurosci 2001;26:37–43.
11. Siever LJ, Davis KL. Overview: toward a dysregulationhypothesis of depression. Am J Psychiatry 1985;142:1017–31.
12. Stahl SM. The psychopharmacology of painful physicalsymptoms in depression. J Clin Psychiatry 2002;63:382–3.
13. Stahl SM, Briley M. Understanding pain in depression. HumPsychopharmacol Clin Exp 2004;19:S9–13.
14. Kwiat GC, Basbaum AI. The origin of brainstem noradrenergicand serotonergic projections to the spinal cord dorsal horn inthe rat. Somatosens Mot Res 1992;9:157–73.
15. Proudfit HK, Clark FM. The projections of locus coeruleusneurons to the spinal cord. In: Barnes CD, Pompeiano O, eds.Progress in brain research. New York: Elsevier SciencePublications, 1991:123–41.
16. Clark FM, Proudfit HK. The projections of noradrenergicneurons in the A5 catecholamine cell group to the spinal cordin the rat: anatomical evidence that A5 neurons modulatenociception. Brain Res 1993;616:200–10.
17. Clark FM, Proudfit HK. The projections of the noradrenergicneurons A7 catecholamine cell group to the spinal cord in therat demonstrated by anterograde tracing combined withimmunochemistry. Brain Res 1991;547:279–88.
18. Basbaum AI, Fields HL. Endogenous pain control systems:brainstem spinal pathways and endorphin circuitry. Ann RevNeurosci 1984;7:309–38.
19. Mogil JS, Yu L, Basbaum AI. Pain genes? Natural variation andtransgenic mutants. Ann Rev Neurosci 2000;23:777–811.
20. Magni G. The use of antidepressants in the treatment ofchronic pain: a review of the current evidence. Drugs1991;42:730–48.
1586

TREATMENT OF CHRONIC PAIN AND DEPRESSION Jann and Slade
21. Mochizucki D . Serotonin and noradrenaline reuptakeinhibitors in animal models of pain. Hum PsychopharmacolClin Exp 2004;19:S15–19.
22. Mico JA, Ardid D, Berrocoso E, Eschalier A. Antidepressantsand pain. Trends Pharmacol Sci 2006;27:348–54.
23. Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG, et al.Comparative affinity of duloxetine and venlafaxine forserotonin and norepinephrine transporters in vitro and in vivo,human serotonin receptor subtypes and other neuronalreceptors. Neuropsychopharmacology 2001;25:871–80.
24. Koch S, Hemrick-Lemke SK, Thompson LK, et al. Comparisonof effects of dual transporter inhibitors on monoaminetransporters and extracellular levels in rats. Neuro-pharmacology 2003;45:935–44.
25. Iyengar S, Webster AA, Hemrick-Luecke SK, Xu JY, SimmonsRMA. Efficacy of duloxetine, a potent and balanced serotonin-norepinephrine reuptake inhibitor in persistent pain models inrats. J Pharmacol Exp Ther 2004;311:576–84.
26. Jones CK, Peters SC, Shannon HE. Efficacy of duloxetine, apotent and balanced serotonin and noradrenergic reuptakeinhibitor, in inflammatory and acute pain models in rodents. JPharmacol Exp Ther 2005;312:726–32.
27. Grothe DR, Scheckner B, Albano D . Treatment of painsyndromes with venlafaxine. Pharmacotherapy 2004;24:621–9.
28. Marchand F, Alloui A, Pelissier T, et al. Evidence for ahypertensive effect of venlafaxine in vincristine-inducedneuropathy in rat. Brain Res 2003;980:117–20.
29. Goldstein DJ, Lu Y, Detke MJ, Wiltse C, Mallinckrodt C,Demitrack MA. Duloxetine in the treatment of depression: adouble-blind placebo-controlled comparison with paroxetine. JClin Psychopharmacol 2004;24:389–99.
30. Detke MJ, Lu Y, Goldstein DJ, Hayes JR, Demitrack MA.Duloxetine, 60 mg once daily, for major depressive disorder: arandomized double-blind placebo-controlled trial. J ClinPsychiatry 2002;63:308–15.
31. Fava M, Mallinckrodt CH, Detke MJ, Watkin JG, WohlreichMM. The effect of duloxetine on painful physical symptoms indepressed patients: do improvements in these symptoms resultin higher remission rates? J Clin Psychiatry 2004;65:521–30.
32. Brannan SK, Mallinckrodt CH, Brown EB, Wholreich MM,Watkin JG, Schatzberg AF. Duloxetine 60 mg once-daily in thetreatment of painful physical symptoms in patients with majordepressive disorder. J Psychiatr Res 2005;39:43–53.
33. Arnold LM, Lu Y, Crofford LJ, et al . A double-blind,multicenter trial comparing duloxetine with placebo in thetreatment of fibromyalgia patients with or without majordepressive disorder. Arthritis Rheum 2004;50:2974–84.
34. Bradley RH, Barkin RL, Jerome J, et al. Efficacy of venlafaxine
for the long term treatment of chronic pain with associatedmajor depressive disorder. Am J Ther 2003;10:318–23.
35. Greco T, Eckert G, Kroenke K. The outcome of physicalsymptoms with treatment of depression. J Gen Intern Med2004;19:813–18.
36. Sindrup SH, Jensen TS. Efficacy of pharmacological treatmentsof neuropathic pain: an update and effect related to mechanismof drug action. Pain 1999;83:389–400.
37. Max MB, Lynch SA, Muir J, et al. Effects of desipramine,amitriptyline, and fluoxetine on pain in diabetic neuropathy. NEngl J Med 1992;326:1250–6.
38. Goldstein DJ, Lu Y, Detke MJ, Lee TC, Iyengar S. Duloxetinevs placebo in patients with painful diabetic neuropathy. Pain2005;116:109–18.
39. Raskin J, Pritchett YL, Bailey RK, et al. Duloxetine in thetreatment of diabetic peripheral neuropathic pain—results from3 clinical trials. Presented at the American academy of nursepractitioners, Fort Lauderdale, FL, June 14–17, 2005.
40. Raskin J, Pritchett YL, Wang F, et al. A double-blind,randomized multicenter trial comparing duloxetine withplacebo in the management of diabetic peripheral neuropathicpain. Pain Med 2005;6:346–56.
41. Rowbotham MC, Goli V, Kunz NR, Lei D. Venlafaxineextended release in the treatment of painful diabeticneuropathy: a double-blind, placebo-controlled study. Pain2004;110:697–706.
42. Sindrup SH, Bach FW, Madsen C, Gram LF, Jensen TS.Venlafaxine versus imipramine in painful polyneuropathy.Neurology 2003;60:1284–9.
43. Durand JP, Alexandre J, Guillevin L, Goldwasser F. Clinicalactivity of venlafaxine and topiramate against oxaliplatin-induced disabling permanent neuropathy. Anti Cancer Drugs2005;16:587–91.
44. Rush AJ, Marangell BM, Sackeim HA, et al. Vagus nervestimulation for the treatment-resistant depression: arandomized controlled acute phase trial. Biol Psychiatr2005;58:347–54.
45. George MS, Rush AJ, Marangell BM, et al. A one-yearcomparison of vagus nerve stimulation with treatment as usualfor treatment-resistant depression. Biol Psychiatr2005;58:364–73.
46. Janig W, Khasar SG, Levine JD, Maio FJP. The role of vagalvisceral afferents in the control of nociception. In: Mayer EA,Saper CB, eds. Progress in brain research. New York: ElsevierScience Publications, 2000:273–87.
47. Multon S, Schoenen J. Pain control by vagus nerve stimulation:from animal to man…and back. Acta Neurol Belg2005;105:62–7.
1587