anormalidades cromosómicas
TRANSCRIPT

ANORMALIDADES
CROMOSÓMICAS

Los trastornos genéticos son mucho más frecuentes de lo que se suele considerar.
Las enfermedades genéticas que se observan en la práctica médica representan sólo Ia punta del iceberg.
Menos errores genotípicos extremos que permiten el desarrollo embrionario y el nacimiento con vida.
Se estima que el 50% de los abortos espontáneos durante los primeros meses de gestación tienen una anomalía cromosómica demostrable
1% RNV anomalía cromosómica grave5% antes de los 25 años

25,000 2%99.9% 1% 3millones pb

TRES PRINCIPALES CATEGORÍAS DE TRASTORNOS GENÉTICOS
MonogénicosCromosómico
Complejos numéricos

Genéticaestudia uno o varios genes y sus efectos fenotípicos.
Genómica es el estudio de todos los genes del genoma y sus interacciones
Cariotipo anormal: Cariotipo que presenta anomalías cromosómicas o cromosomopatías

GEN Unidad de material hereditario.
Segmento de ADN (ácido desoxirribonucleico) que funciona como una clave para una proteína
determinada, con una estructura lineal y compuesto de muchos subelementos, los nucleótidos, del orden
de centenares (estos a su vez son unidades de recombinación y mutación).
El lugar físico que ocupa cada gen en el cromosoma se denomina locus.

GEN RECESIVO
Se expresa sólo cuando ambos cromosomas del par son portadores del alelo mutante.Para que este alelo se observe en el fenotipo el organismo debe poseer dos copias del mismo, provenientes uno de la madre y otro del padre.

GEN DOMINANTESe refiere al alelo que se manifiesta en un fenotipo, se
expresan cuando sólo un cromosoma de un par es portador del alelo mutante.
Los genes dominantes, son los que mas predominan, como los ojos de color café o negro que los verdes o azules

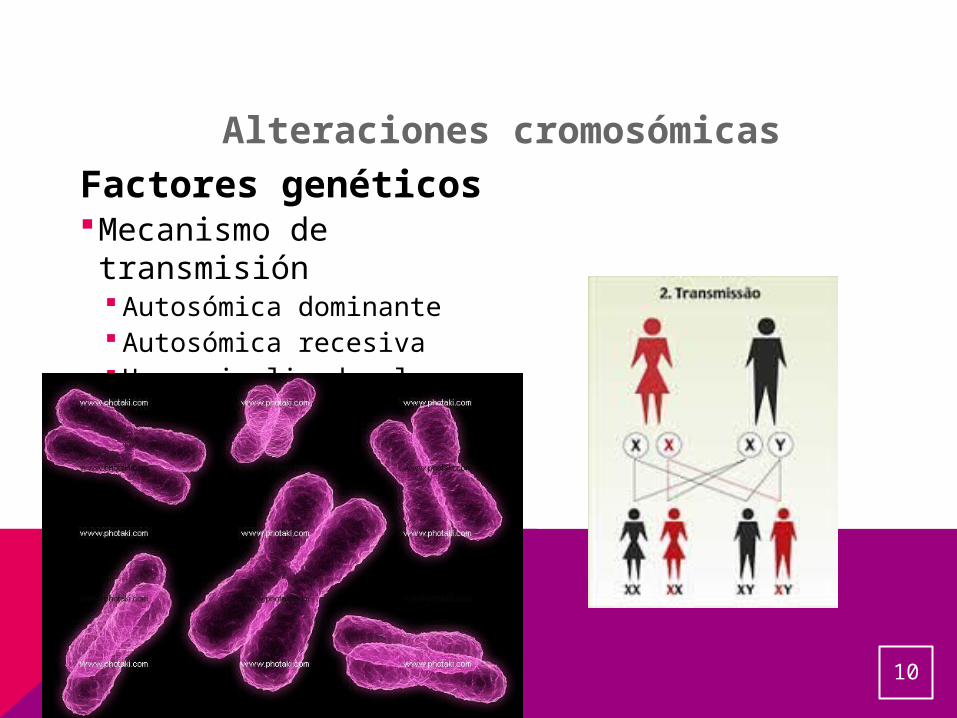
Factores genéticosMecanismo de transmisión Autosómica dominante Autosómica recesiva Herencia ligada al sexo
10
Alteraciones cromosómicas

ALTERACIONES MONOGENÉTICAS
Polimorfismo de un solo nucleótidos SNP
Exones - Intrones
1%
Mutaciones puntales: sustitución de una única base
Mutaciones repetitivas en secuencias no codificantes
Deleciones o inserciones
Mutación por repetición de trinucleótidos

CAC -CTA AC. GLUTAMICO VALINA
X FRAGIL CGG
FMR1 250-4000

DEFECTOS ENZIMÁTICOS
Galactosemia
Deficiencia de galactosa-1-fosfato uridintransfersa
Albinismo
Deficiencia de tirisinasa indispensable para la sintesis melanina- precusor de la tirosina



EFERMEDAD DE TAY-SACHS
La enfermedad de Tay-Sachs ocurre cuando el cuerpo carece de hexosaminidasa A, una proteína que ayuda a descomponer un químico que se encuentra en el tejido nervioso, llamado gangliósidos. Sin esta proteína, los gangliósidos, en particular, se acumulan en las neuronas en el cerebro.
Enfermedad de Tay-Sachs un gen defectuoso en el cromosoma 15.
. La mayoría infantil, en la cual el daño neurológico, generalmente bebés aún está dentro del útero y los síntomas por lo general aparecen cuando el niño tiene de 3 a 6 meses de edad.
La enfermedad tiende a empeorar muy rápidamente y el niño por lo general muere a la edad de 4 ó 5 años.
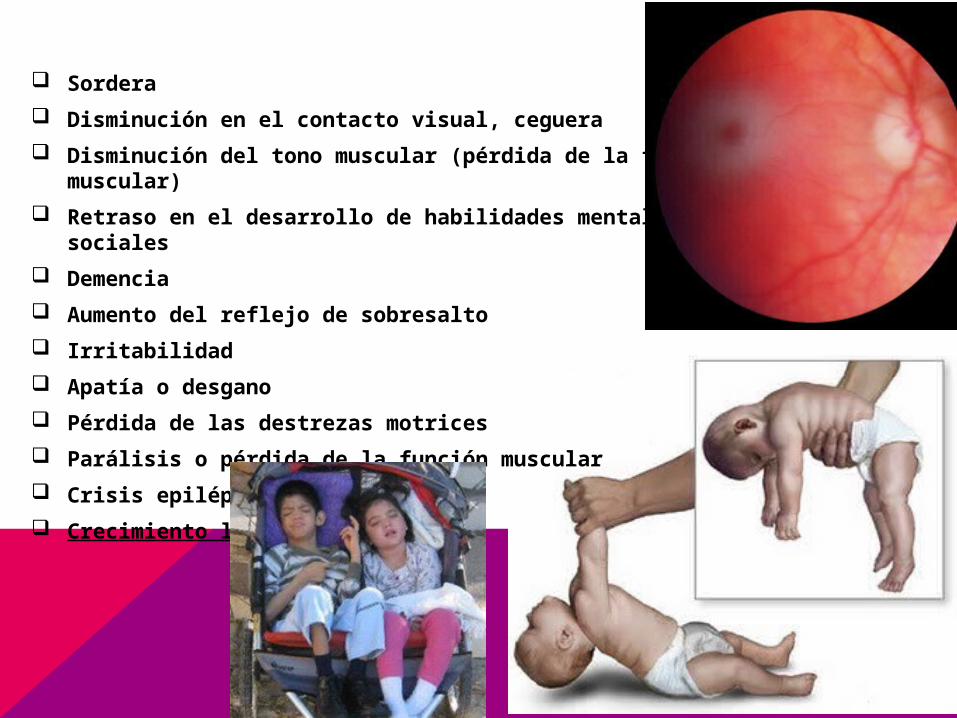
Sordera
Disminución en el contacto visual, ceguera
Disminución del tono muscular (pérdida de la fuerza muscular)
Retraso en el desarrollo de habilidades mentales y sociales
Demencia
Aumento del reflejo de sobresalto
Irritabilidad
Apatía o desgano
Pérdida de las destrezas motrices
Parálisis o pérdida de la función muscular
Crisis epiléptica
Crecimiento lento


ENFERMEDAD DE HUNTINGTON
Es un trastorno que se transmite de padres a hijos, en el cual las neuronas en ciertas partes del cerebro se desgastan o se degeneran.
Causas
La enfermedad de Huntington es causada por un defecto genético en el cromosoma N.° 4. El defecto hace que una parte del ADN, llamada repetición CAG, ocurra muchas más veces de lo que se supone que debe ser. Normalmente, esta sección del ADN se repite de 10 a 28 veces, pero en una persona con la enfermedad de Huntington, se repite de 36 a 120 veces.


Comportamientos antisociales
Alucinaciones
Irritabilidad
Malhumor
Inquietud o impaciencia
Paranoia
Psicosis
Los movimientos anormales e inusuales abarcan:
Movimientos faciales, incluyendo muecas
Girar la cabeza para cambiar la posición de los ojos
Movimientos espasmódicos rápidos y súbitos de los brazos, las piernas, la cara y otras partes del cuerpo
Movimientos lentos e incontrolables
Marcha inestable
Demencia que empeora lentamente, incluyendo:
Desorientación o confusión
Pérdida de la capacidad de discernimiento
Pérdida de la memoria
Cambios de personalidad
Cambios en el lenguaje

DEFECTOS DE LOS RECEPTORES Y DE LA ESTRUCTURA, FUNCIÓN O CANTIDAD DE PROTEÍNAS
Hipercolesterolemia familiar
Falta de receptores LDL-acumula
Talasemía
Cantidad reducida de globulinas


Mutación en los genes de proteínas estructurales
SX DE MARFAN
Trastorno de los tejidos conjuntivos cambio en esqueleto, ojos y aparato cardiovascular
1-5000 Autosómica dominante
Defecto en la glocoproteína Fribilina 1
FBN1 Y FBN2 15q21.1 y 5q 23.3


Anom
alía
sNuméricas
Estructurales

Euploide
Aneuploidía
Mosaicismo
Técnica de FISH

Estructurales
Deleción Translocación Duplicación Isocromoso
maCromosoma
en anillo

DELECIONES
INTERSTICIAL TERMINAL

Paracéntricas Pericéntricas
INVERSIONES

TIPOS DE TRANSLCACIÓN

DUPLICACIONES

Numéricas
Trisomias Monosomias
Nulosomias


SÍNDROME DE CRI-DU-CHAT
LARINGEAS
RETROGNATÍA
MICROCEFALÍA
EPICANTO
MÁS EN MUJERES
TELECANTO
CARA DE LA LUNA LLENA
CARIOTIPO: 46 XX Ó XY 5 ,del(q)

SÍNDROME DE CRI-DU-CHAT

SÍNDROME DE ANGELMAN
BRAQUICEFALÍAPERIODOS DE RISAMOVIMIENTOS ATACTICOSMICROCEFALÍACONVULSIONESDIFICULTAD PARA HABLARDELECION HEREDADA DEL CROMOSOMA
DE LA MADRE
CARIOTIPO: 46 XX Ó XY ( 15q- )

SÍNDROME DE ANGELMAN

SÍNDROME DE PRADER WILLI
OBESIDAD
HIPOPLASÍA DE TESTICULOS
HIPERFAGÍA
HIPOTONÍA
DELECIÓN HEREDADA DEL PADRE
CARIOTIPO:46 XX Ó XY ( 15q- )

SÍNDROME DE PRADER-WILLI

NUMÉRICAS

TRISOMIA 21
Capacidad mental limitada
Retraso en el crecimiento
Pliegue epicántico
Hipotonía muscular
Protrusión de la lengua por hipoplasia maxilar y del paladar,
Defectos cardiacos
Clinodactilia del quinto dedo

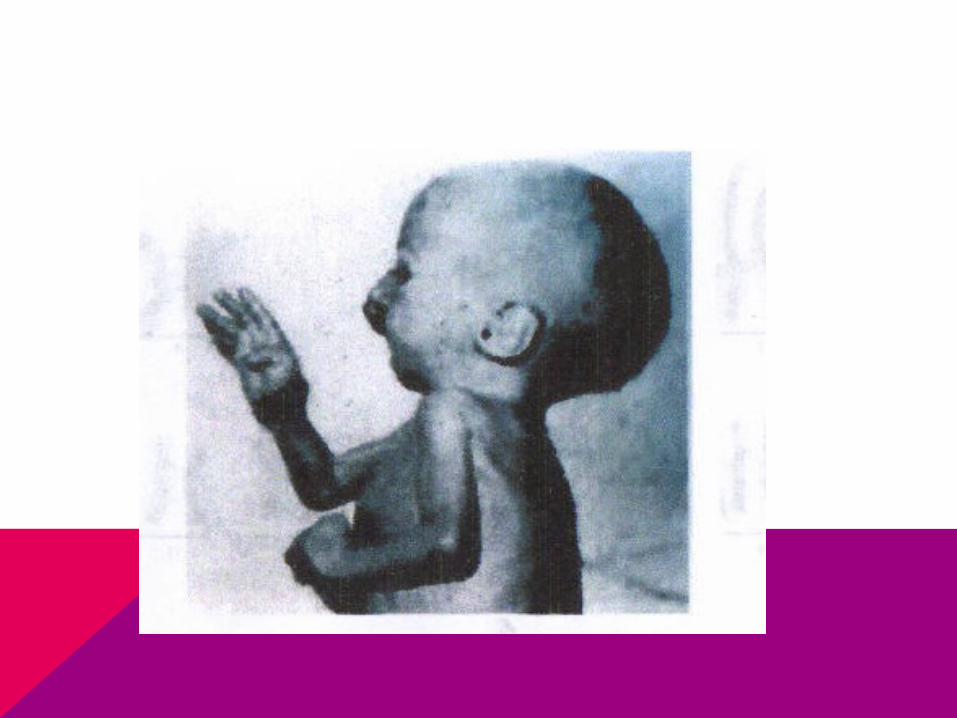
TRISOMIA 18
Los lactantes son pequeños al nacer
Sindactilia
Malformaciones del pabellón de la oreja y oreja de implantación baja
Manos en puño
Occipucio prominente
Defectos en el septo ventricular.
Muerte en el primer bimestre de vida
Pie Zambo
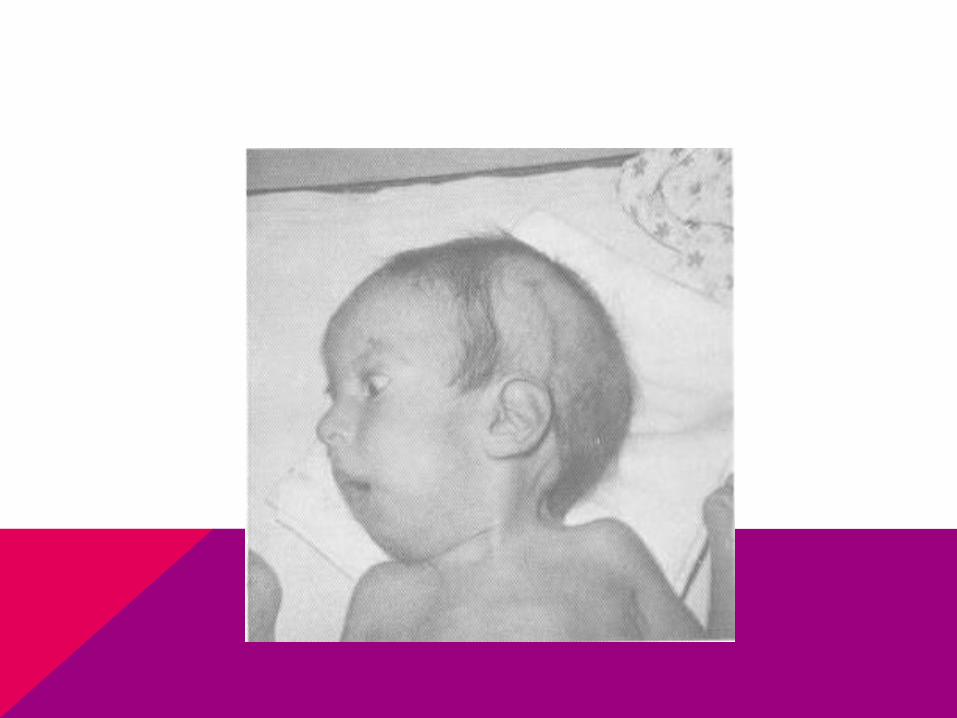

HOLOPROSENCEFALÍACICLOPÍAMICROCEFALÍAMICROFTALMÍAPOLIDACTILÍAPIE EQUINOVAROONFALOCELELABIO LEPORINO Y PALADAR HENDIDO
CARIOTIPO: 47 XX Ó XY + 13

TRISOMIA 13

SINDROME DOBLE Y
IMPULSIVOS
CONVULSIONES
AGRESIVIDAD CON OTROS YY
DEPRESIÓN LEVE
CARIOTIPO: 47 XYY

SÍNDROME METAHEMBRA
ESCASA MESTRUACIÓN
UÑAS HIPERCONVEXAS
ATLETICAS
VOZ GRUESA
HORMONAS FEMENINAS
CARIOTIPO: 47XXX

SÍNDROME DE KLINEFELTER
TESTICULOS PEQUEÑOSESTERILIDADCRIPTORQUIDÍAGINECOMASTIAMIEMBROS LARGOSALTOSATROFÍA TESTICULAR
CARIOTIPO: 47 XXY

SÍNDROME DE KLINEFELTER

EJERCIC
IO


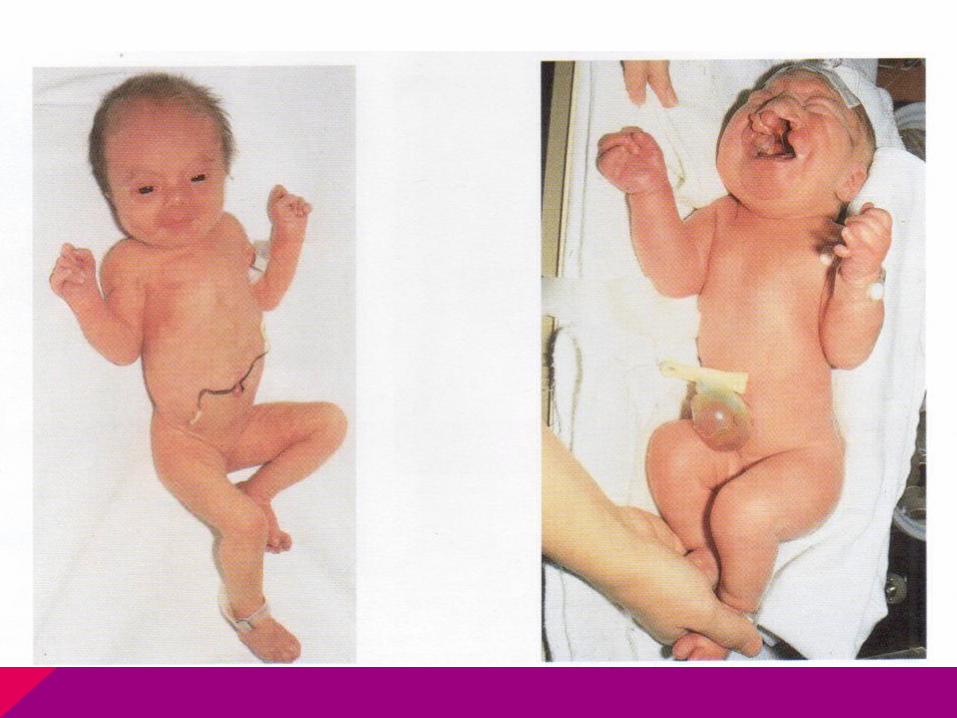
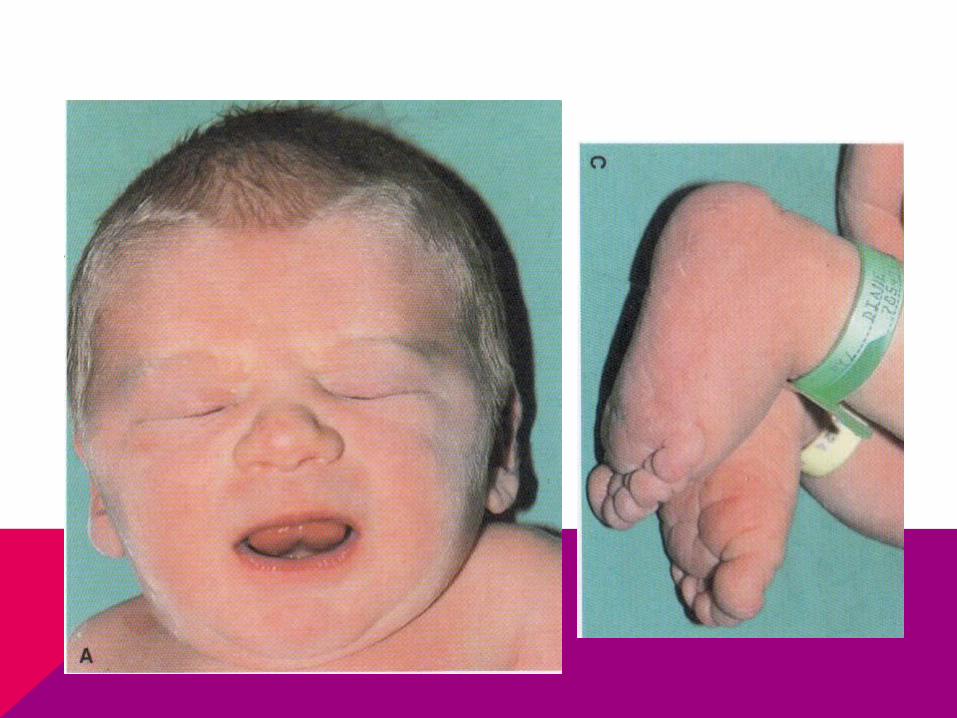
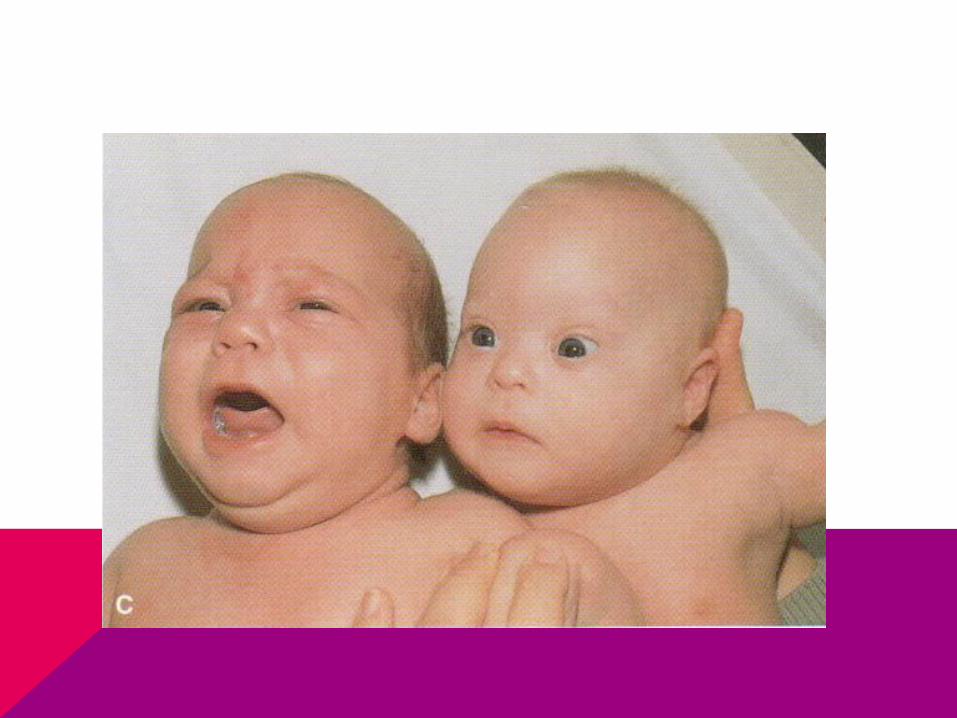
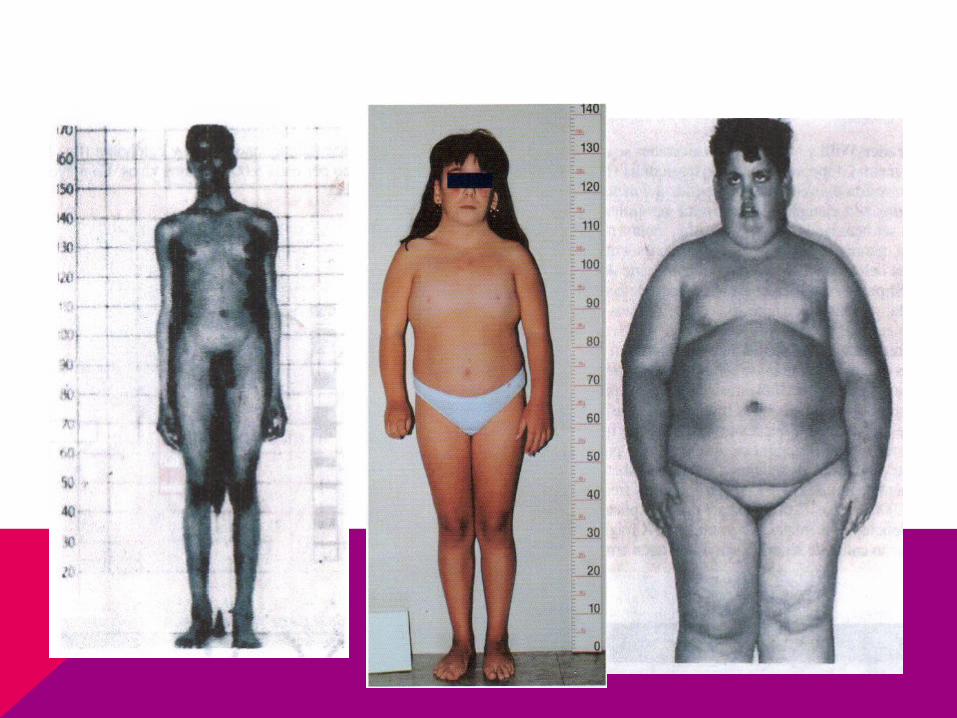

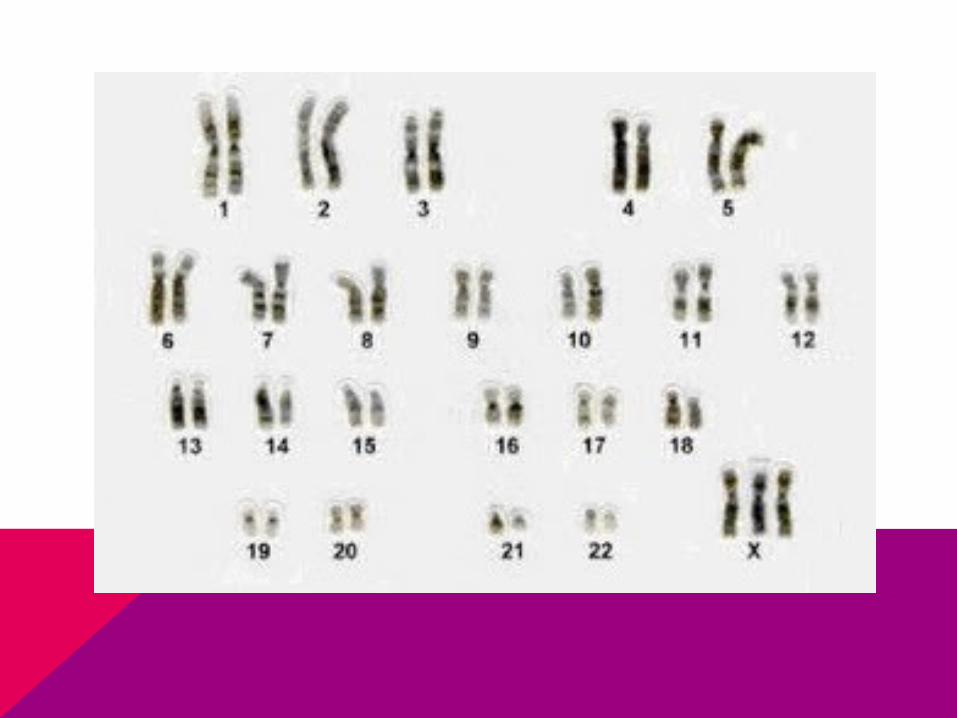



CASO CLÍNICO
Llega niña a consulta por retraso de la menarca, de estatura baja y no tiene desarrollados los caracteres sexuales secundarios