anemias - judoctors · classification of anemia •according to morphology: •size: normo, micro,...
TRANSCRIPT

Anemias

Definition
• Reduction of total RBC MASS below average levels
• Reduction of oxygen carrying capacity of the blood
• Leads to tissue hypoxia
• Practically, measure by Hemoglobin concentration, and Hematocrit (ratio of packed RBCs to total blood volume). They correlate well with anemia when the plasma volume is normal

Classification of anemia according to cause
• Blood loss: acute, chronic
• Diminished RBC production
• Increased destruction (hemolytic anemia)
• Extrinsic factors (infection, antibody, mechanical)
• Intrinsic RBC abnormalities:
1) Hereditary (membrane, enzyme, Hg abnormalities)
2) Acquired (Paroxysmal nocturnal hematuria)

Classification of anemia
• According to morphology:
• Size: normo, micro, macrocytic
• Color: normo, hypochromic
• Shape: anisopoikelocytosis (spherocytes, sickle, schistiocytes)
• Hypochromic microcytic anemia usually reflects impaired Hg synthesis
• Macrocytic anemia reflects stem cell disease and maturation

RBC indices
• Hg concentration, Men: 13.8-18.0 g/dL, Women: 12-15
• Hematocrit: volume % of RBCs in blood, 45% men, 40% women
• Mean Cell Volume (MCV): average size in femtoliter, 10−15 (normal: 80-99 fL)
• Mean Cell Hg (MCH): average mass of Hg inside the RBC in picograms, 10−12, normal (27-31)
• Mean Cell Hg Concentration (MCHC): the average concentration of Hg in a given volume of packed red cells, expressed in grams per deciliter
• RBC count: number of cells/L Male: 4.7-6.1 million/ microliterFemale: 4.2-5.4
• Reticulocyte index (0.5-1.5%)
• Red cell distribution width (RDW): the coefficient of variation in red cell volume

Clinical features of anemia
• Dizziness
• Fatigue
• Pallor
• Headache
• Hypotension
• Tachycardia
• Dyspnea
• Special types: jaundice, bone and joint pain, growth retardation

Anemia of acute blood loss
• Symptoms are related to decreased intravascular volume, might cause cardiovascular shock and death
• Body responds by shifting fluid from interstitial to intravascular space, causing dilutional anemia and hypoxia
• Erythropoietin secretion is stimulated, activating BM erythropoiesis
• Mature RBCs as well as Reticulocytes appear in blood after 5 days
• In internal hemorrhage, iron is restored from extravasated RBCs and used again in erythropoiesis
• In external and GIT hemorrhage, iron is lost, which complicated anemia
• The anemia is normochromic normocytic, with reticulocytosis
• Leukocytosis (secondary to stress)
• Thrombocytosis (secondary to high erythropoietin)

Anemia of chronic blood loss
• Occurs when the rate of RBC loss exceeds regeneration
• Mostly associated with iron deficiency anemia

Hemolytic Anemia
• Normally, RBCs age is around 120 days, aged RBCs are engulfed by phagocytic cells in spleen, liver and BM
• In Hemolytic anemia; premature destruction of RBCs
• Accumulation of Hg degradation products
• Secondary increased erythropoiesis
• Extravascular hemolysis: increased phagocyticactivity
• Intravascular hemolysis: occurs inside blood vessels

Extravascular Hemolysis
• Generally caused when the RBC is less deformable or having abnormal shape
• Abnormal RBC shape prevents its normal movement in splenic sinusoids
• Prolonged time of RBCs passage attracts histiocytes to engulf them
• Free Hg from destructed RBCs binds Haptoglobin in serum
• Hg within phagocytes is converted to bilirubin• The triad of extravascular HA is: Anemia, splenomegaly
and jaundice

Intravascular Hemolysis
• Less common• Caused by mechanical damage, complement
fixation, microorganism, exogenous toxins• Due to large amount of free Hg, haptoglobin is
cleared from the serum• free Hg in serum is oxidized to Methemoglobin
(metHg),• Excess free Hg and met Hg are excreted in urine
(hemoglobinuria) causing dark urine• Renal hemosiderosis may occur

Hereditary Spherocytosis
• This inherited disorder is caused by intrinsic defects in the red cell membrane skeleton that render red cells spheroid, less deformable, and vulnerable to splenic sequestration and destruction
• The prevalence of HS is highest in northern Europe
• AD inheritance pattern, in 75% of cases
• The remaining patients have a more severe form of the disease that is usually caused by the inheritance of two different defects (a state known as compound heterozygosity)
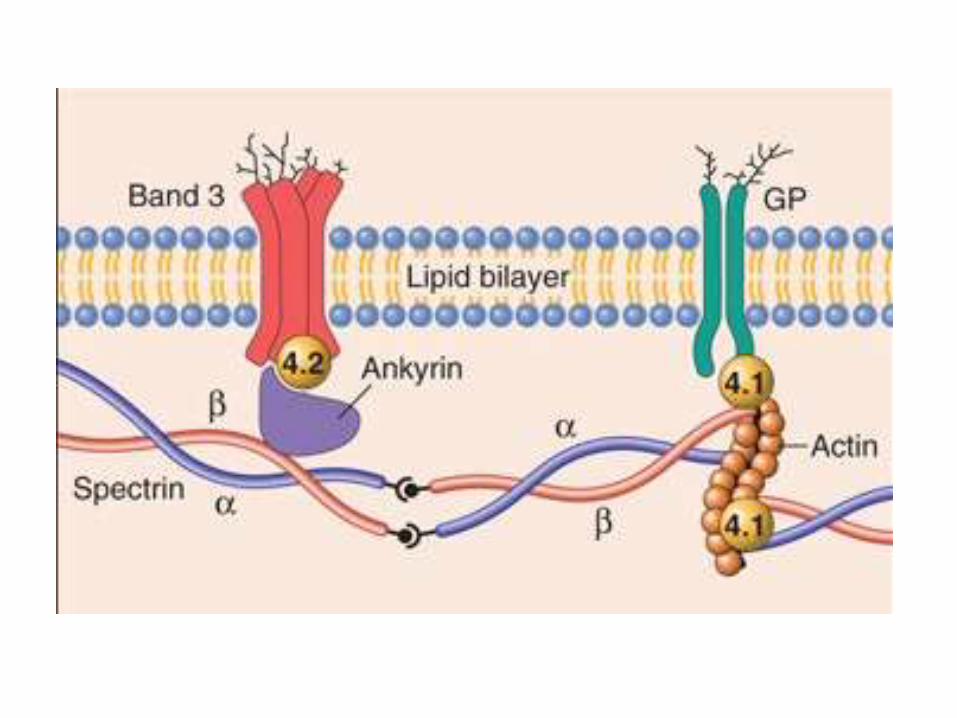

Pathophysiology
• Normal RBC is durable and elastic
• Spectrin is the major internal membrane protein, consists of two helical polypeptides; α, β
• The tail of Spectrin binds Actin
• Spectrin-Actin complex, is connected by Ankyrinand band 4.2 to band 3, a transmembraneprotein
• Protein 4.1 binds the tail of spectrin to Glycophorin A, a transmembrane protein
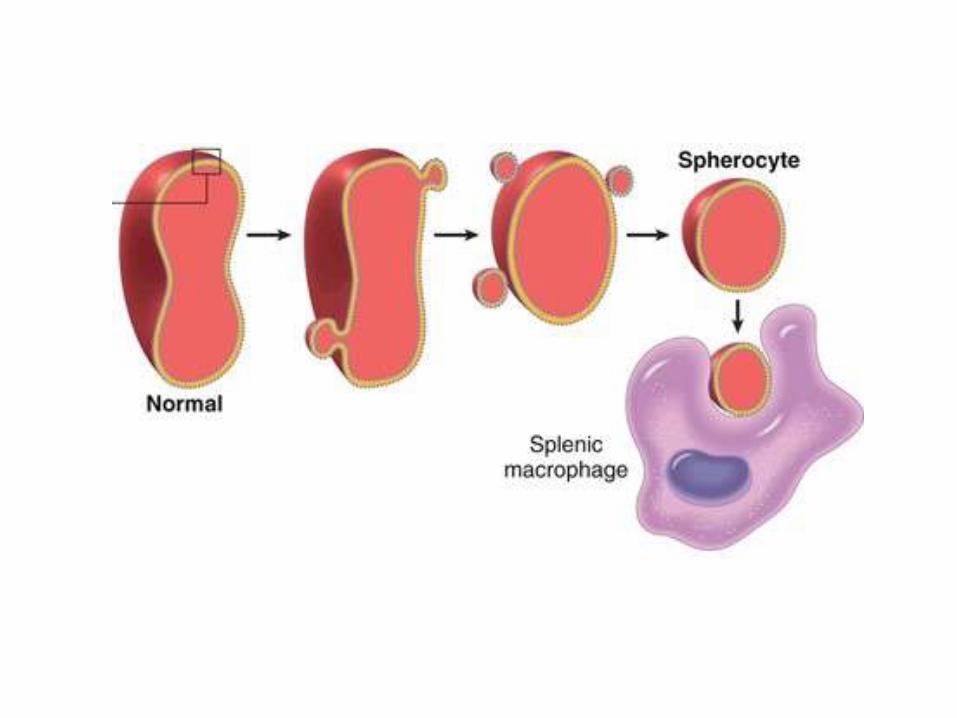

Pathophysiology
• Various mutations involving α-spectrin, β-spectrin, ankyrin, band 4.2, or band 3 that weaken the interactions between these proteins cause red cells to lose membrane fragments
• Most mutations are frame-shift, resulting in absent protein
• To accommodate the resultant change in the ratio of surface area to volume these cells adopt an irreversible spherical shape
• Spherocytic cells are less deformable than normal ones and therefore become trapped in the splenic cords, where they are phagocytosed by macrophages
• Na+ influx in spherocytes is twice than normal, while K+ efflux is the same, facilitating hemolysis in the microenvironment of the spleen
• RBC Life span is dropped to less than 20 days

Clinical features
• Congestion of RBCs in the spleen causes splenomegaly and anemia
• Jaundice, pigmented gall bladder stones
• Reticulocytosis, BM erythroid hyperplasia, hemosiderosis
• Family Hx of anemia or splenectomy
• Abnormal osmotic fragility test
• Increased MCHC in 50% of cases
• Treatment: splenectomy

Morphology
• Blood film: RBCs are round, small, hyperchromatic, no visible central pallor
• “Howell-Jolly” bodies are seen in post splenectomy. A fragment of chromosome which is detached and left in the cytoplasm after the extrusion of the nucleus, secondary to accelerated erythropoiesis. Appears as 1 or 2 eccentric dots
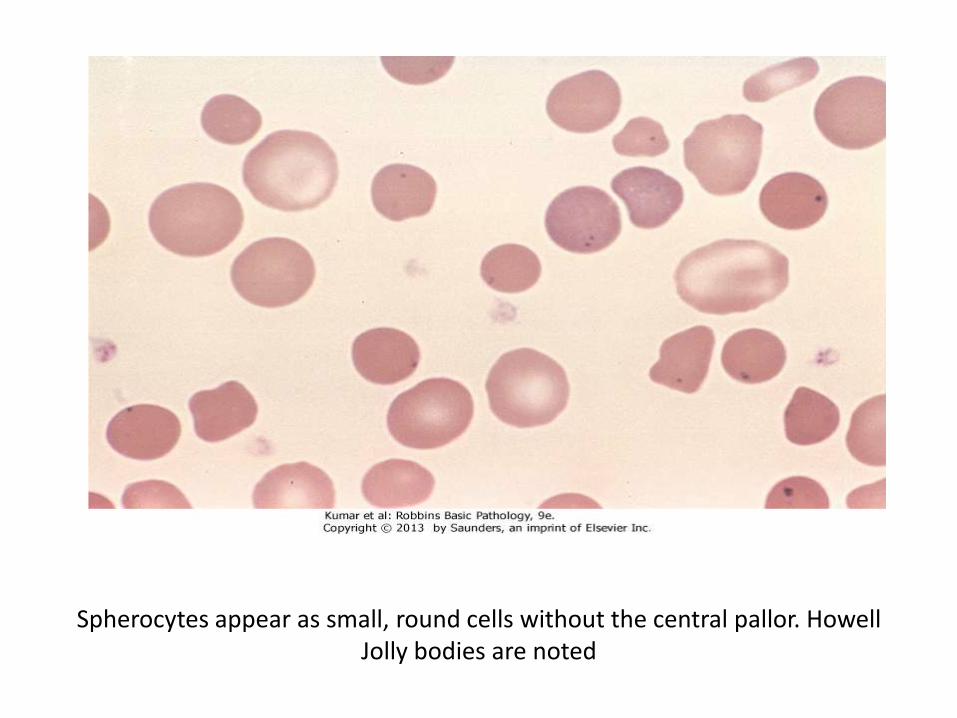
Spherocytes appear as small, round cells without the central pallor. Howell Jolly bodies are noted

Glucose-6-Phosphate Dehydrogenase Deficiency
• G6PD reduces nicotinamide adenine dinucleotidephosphate (NADP) to NADPH while oxidizing glucose-6-phosphate
• NADPH then provides reducing equivalents needed which protects against oxidant injury by catalyzing the breakdown of compounds such as H2O2
• G6PD deficiency is a recessive X-linked trait, placing males at higher risk for symptomatic disease.

Types of G6PD deficiency
• Several hundred G6PD genetic variants are known, but most are harmless
• The normal enzyme is G6PD-B
• Only two variants, designated G6PD-A and G6PD Mediterranean, cause most of the clinically significant hemolytic anemias
• G6PD-A is present in about 10% of American blacks; G6PD Mediterranean is prevalent in the Middle East

Pathophysiology
• The half-life of G6PD-A is moderately reduced, whereas that of G6PD Mediterranean is functionally abnormal
• Because mature red cells do not synthesize new proteins, G6PD-A or G6PD Mediterranean enzyme activities fall quickly to levels inadequate to protect against oxidant stress as red cells age. Thus, older red cells are much more prone to hemolysis than younger ones

Pathophysiology
• Oxidants cause both intravascular and extravascular hemolysis in G6PD-deficient individuals
• Exposure of G6PD-deficient red cells to high levels of oxidants causes the cross-linking of reactive sulfhydryl groups on globinchains, which become denatured and form membrane-bound precipitates known as Heinz bodies
• These are seen as dark inclusions within red cells stained with crystal violet. Heinz bodies can damage the membrane sufficiently to cause intravascular hemolysis
• Splenic macrophages identify Heinz bodies and pluck them out resulting in indentation. The remaining RBC is known as “bite cells”

Causes of hemolytic crisis
• Hemolysis happens upon exposure to oxidant stress• The most common triggers are infections, in which oxygen-derived
free radicals are produced by activated leukocytes • Many infections can trigger hemolysis; viral hepatitis, pneumonia,
and typhoid fever• The other important initiators are drugs and certain foods• Most important drugs are the antimalarials (e.g., primaquine and
chloroquine), sulfonamides, nitrofurantoins• Some drugs cause hemolysis only in individuals with the more
severe Mediterranean variant. • The most frequently cited food is the fava bean (Favism)• Uncommonly, G6PD deficiency presents as neonatal jaundice or a
chronic low-grade hemolytic anemia in the absence of infection or known environmental triggers

Clinical features
• Majority of patients are asymptomatic, anemia develops when the enzyme level drops below 20% of normal activity
• Hemolytic crisis appear 2-3 days after exposure to oxidant• Only old RBCs hemolize, HB level drops, RBCs appear
normochromic normocytic, patients have bone pain• Chronic hemolysis (splenomegaly and GB stones) are
absent• G6PD-A usually is self-limited• G6PD-Mediterranian has more severe crisis, might need
blood transfusion• Recovery is associated with reticulocytosis• Dx: enzyme assay (measure conversion to NAPDH)

Pyrovate kinase deficiency
• AR inheritance
• PK is an enzyme in the anaerobic glycolysis pathway (main pathway in RBCs)
• PK deficiency causes decreased ATP level which is essential for cell membrane pumps
• Intracellular Na accumulates, causing swelling of RBCs and rigidity
• Spleen clears abnormal shaped RBCs
• 2,3 diphosphoglycerate (DPG) level increases inside RBCs, facilitating O2 release, ameliorating the anemia

Clinical
• Degree of anemia varies according to type of mutation, ranging from neonatal jaundice to anemia presenting in adulthood with jaundice, GB stones and splenomegaly
• Anemia is exacerbated by stress
• Blood film shows NN anemia, variable reticulocytosis, anisopoikelocytosis
• Diagnosis: enzyme assay
• Treatment: splenectomy

Paroxysmal Nocturnal Hematuria
• Acquired disease
• Normally, proteins are anchored into the lipid bilayer in two ways. Most have a hydrophobic region that spans the cell membrane; these are called transmembrane proteins. The others are attached to the cell membrane through a covalent linkage to a specialized phospholipid called glycosylphosphatidylinositol (GPI)
• In PNH, there is a mutation in the phosphatidylinositol glycancomplementation group A gene (PIGA), which synthesizes GPI
• Thus, GPI and their normally anchored proteins are absent
• Because the causative mutations occur in a hematopoietic stem cell, all of its clonal progeny (red cells, white cells, and platelets) are deficient in GPI-linked proteins
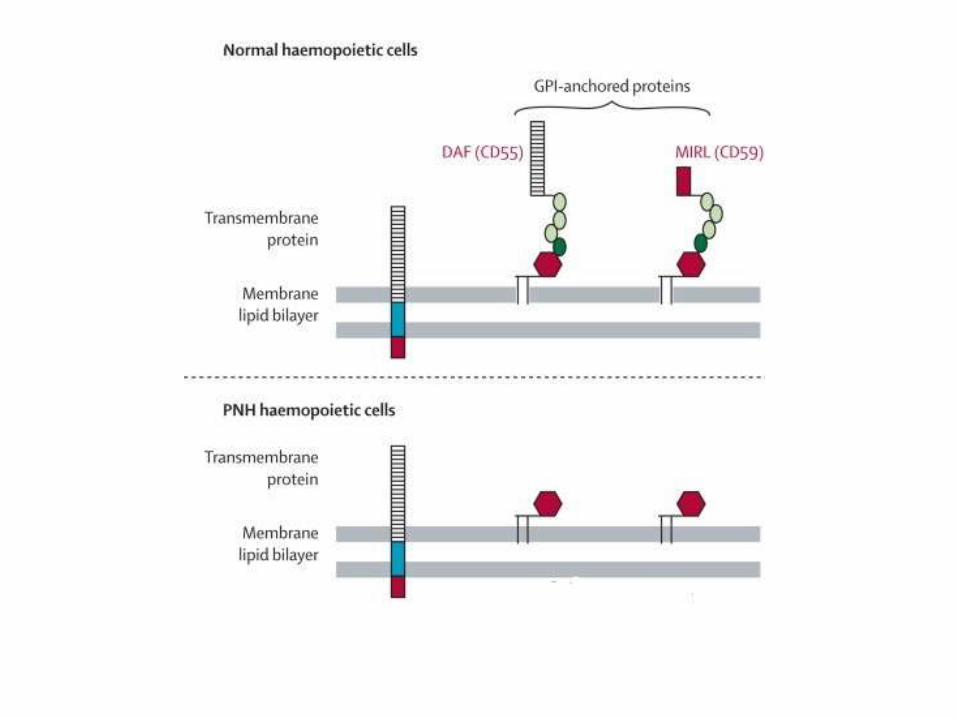

Patholophysiology
• Normal individuals harbor small numbers of bone marrow cells with PIGA mutations
• In PNH, autoimmune reaction occurs against normal clones resulting in predominance of GPI-deficient clone
• PNH blood cells are deficient in three GPI-linked proteins that regulate complement activity: (1) decay-accelerating factor, or CD55; (2) membrane inhibitor of reactive lysis, or CD59; and (3) C8 binding protein. Of these factors, the most important is CD59, a potent inhibitor of C3 convertase that prevents the spontaneous activation of the alternative complement pathway

Clinical features
• Red cells, platelets, and granulocytes deficient in these GPI-linked factors are abnormally susceptible to lysis or injury by complement. In red cells this manifests as intravascular hemolysis, which is caused by the C5b-C9 membrane attack complex
• The hemolysis is intravascular, paroxysmal and nocturnal in only 25% of cases; chronic hemolysis without dramatic hemoglobinuria is more typical. The tendency for red cells to lyse at night is explained by a slight decrease in blood pH during sleep, which increases the activity of complement
• Thrombosis is the leading cause of death in PNH. About 40% of patients suffer from venous thrombosis, often involving the hepatic, portal, or cerebral veins. Autolysis of some platelets causes aggregation of others secondary to released prothrombotic factors
• In severe cases, pancytopenia develops • About 5% to 10% of patients eventually develop acute myeloid leukemia
or a myelodysplastic syndrome, possibly because hematopoietic stem cells have suffered some type of genetic damage.

Autoimmune hemolytic anemia
• A group of anemias in which an abnormal immunoglobulin is attached to RBC membrane causing damage and lysis
• Direct Coombs antiglobulin test: the patient's RBCs are mixed with sera containing antibodies that are specific for human immunoglobulin. If either immunoglobulin is present on the surface of the red cells, the multivalent antibodies cause agglutination, which is easily appreciated visually as clumping
• Indirect Coombs antiglobulin test, the patient's serum is tested for its ability to agglutinate commercially available red cells bearing particular defined antigens

Warm Antibody Type
• 70% of immunohemolytic anemia.
• 50% are idiopathic (primary); the others are related to a predisposing condition or exposure to a drug.
• Most causative antibodies are of the IgG class; less commonly, IgA antibodies
• A common target is the Rh antigen on RBCs
• The red cell hemolysis is mostly extravascular. IgG-coated red cells bind to Fc receptors on phagocytes, which remove red cell membrane during "partial" phagocytosis. As in hereditary spherocytosis, the loss of membrane converts the red cells to spherocytes, which are sequestered and removed in the spleen. Moderate splenomegaly due to hyperplasia of splenic phagocytes is usually seen

Drug induced hemolytic anemia
• Antigenic drugs. In this setting hemolysis usually follows large, intravenous doses of the offending drug and occurs 1 to 2 weeks after therapy is initiated. These drugs, like penicillin and cephalosporins, bind to the red cell membrane and are recognized by anti-drug antibodies. Sometimes the antibodies bind only to the drug, as in penicillin-induced hemolysis. In other cases, such as in quinidine-induced hemolysis, the antibodies recognize a complex of the drug and a membrane protein. The responsible antibodies sometimes fix complement and cause intravascular hemolysis, but more often they act as opsonins that promote extravascular hemolysiswithin phagocytes
• Tolerance-breaking drugs. These drugs, of which the antihypertensive agent α-methyldopa is the prototype, induce in some unknown manner the production of antibodies against red cell antigens, particularly the Rhblood group antigens. About 10% of patients taking α-methyldopa develop autoantibodies, as assessed by the direct Coombs test, and roughly 1% develop clinically significant hemolysis

Cold Agglutinin Type
• This form of immunohemolytic anemia is caused by IgM antibodies that bind red cells avidly at low temperatures (0°-4°C). It is less common than warm antibody immunohemolytic anemia, accounting for 15% to 30% of cases
• Cold agglutinin antibodies sometimes appear transiently following certain infections, such as with Mycoplasma pneumoniae, Epstein-Barr virus, cytomegalovirus, influenza virus, and human immunodeficiency virus (HIV). In these settings the disorder is self-limited and the antibodies rarely induce clinically important hemolysis. Chronic cold agglutinin immunohemolytic anemia occurs in association with certain B-cell neoplasms or as an idiopathic condition.
• Clinical symptoms result from binding of IgM to red cells in vascular beds where the temperature may fall below 30°C, such as in exposed fingers, toes, and ears. IgM binding agglutinates red cells and fixes complement rapidly. As the blood recirculates and warms, IgM is released, usually before complement-mediated hemolysis can occur. However, the transient interaction with IgM is sufficient to deposit sublytic quantities of C3b, an excellent opsonin, which leads to the removal of affected red cells by phagocytes in the spleen, liver, and bone marrow. The hemolysis is of variable severity
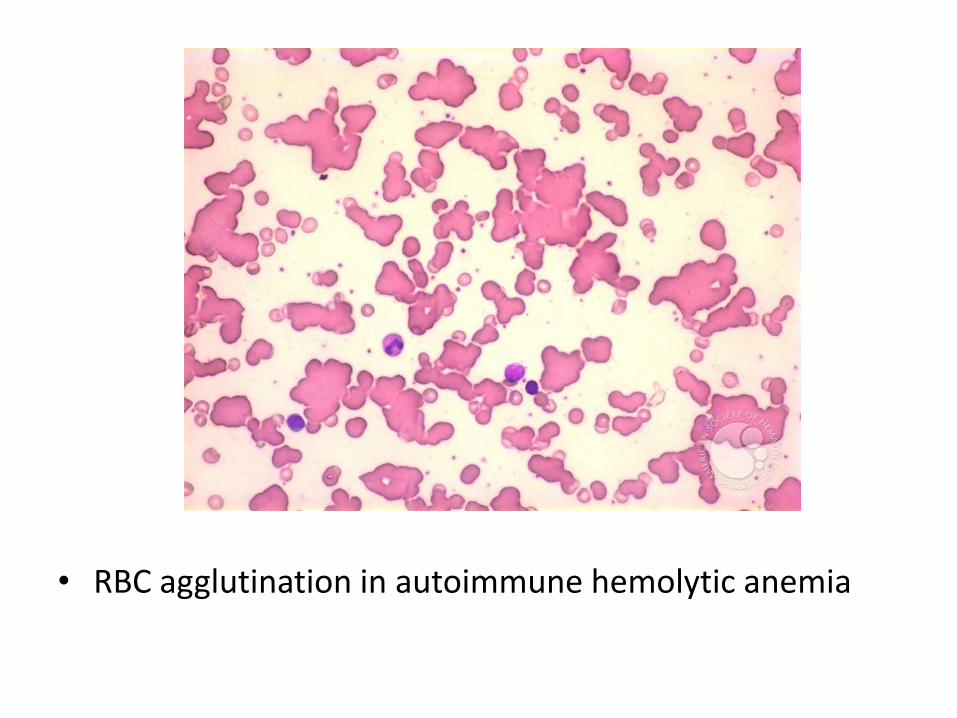
• RBC agglutination in autoimmune hemolytic anemia

Cold Hemolysin Type
• Cold hemolysins are autoantibodies responsible for an unusual entity known as paroxysmal cold hemoglobinuria
• This rare disorder causes substantial, sometimes fatal, intravascular hemolysis and hemoglobinuria
• The autoantibodies are IgGs that bind to the P blood group antigen on the red cell surface in cool, peripheral regions of the body. Complement-mediated lysis occurs when the cells recirculate to warm central regions, since the complement cascade functions more efficiently at 37°C
• Most cases are seen in children following viral infections; in this setting the disorder is transient, and most of those affected recover within 1 month.

Hemolytic Anemia Resulting from Trauma to Red Cells
• Physical damage to RBCs
• Cardiac valve prosthesis
• Microangiopathic disease (disseminated intravascular coagulation DIC, thrombotic thrombocytopenic purpura (TTP), hemolytic-uremic syndrome (HUS), malignant hypertension, systemic lupus erythematosus, and disseminated cancer): aggregates of fibrin and platelets causes damage to RBCs
• RBCs appear as fragments (schistocytes)

Schistocytes


Hemoglobinopathies
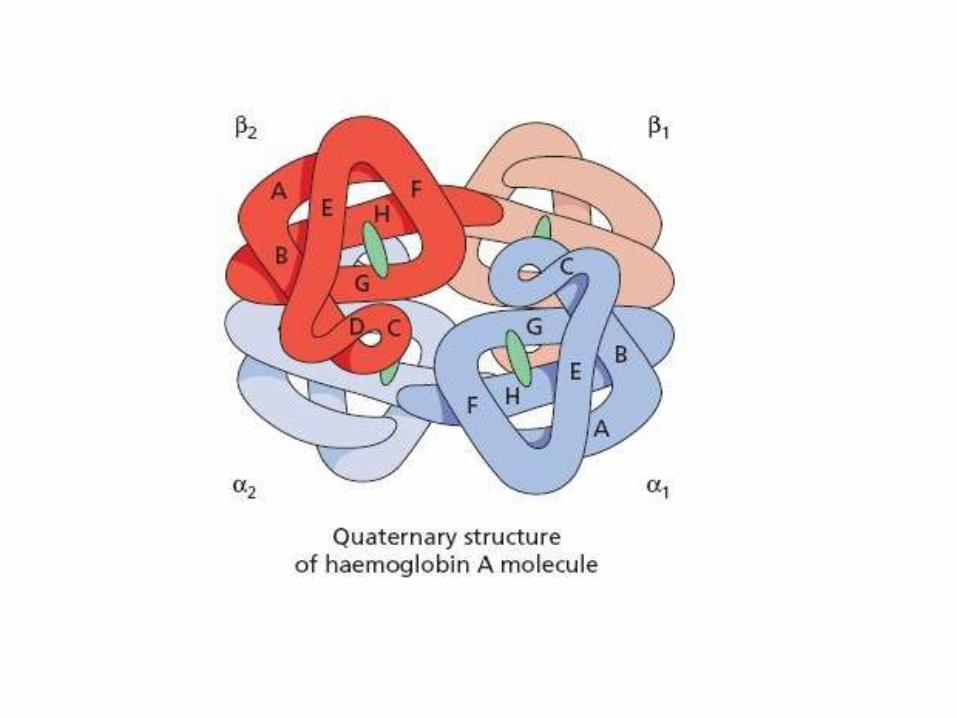

Normal Hemoglobin Structure
• Two pairs of globin chains with a haem group attached.
• 7 chains are synthesized in normal subjects: 4 embryonic: Hg Gower 1,2 Hg Portland 1,2. Hg F (fetal), Hg A, Hg A2
• The individual chains synthesized in postnatal life are designated α, β, γ, and δ
• Hg A has two α chains and two β chains (α2 β2)• Hg A2 has two α chains and two δ chains (α2 δ2),
1.5-3%• Hg F has two α chains and two γ chains (α2 γ2),
1%
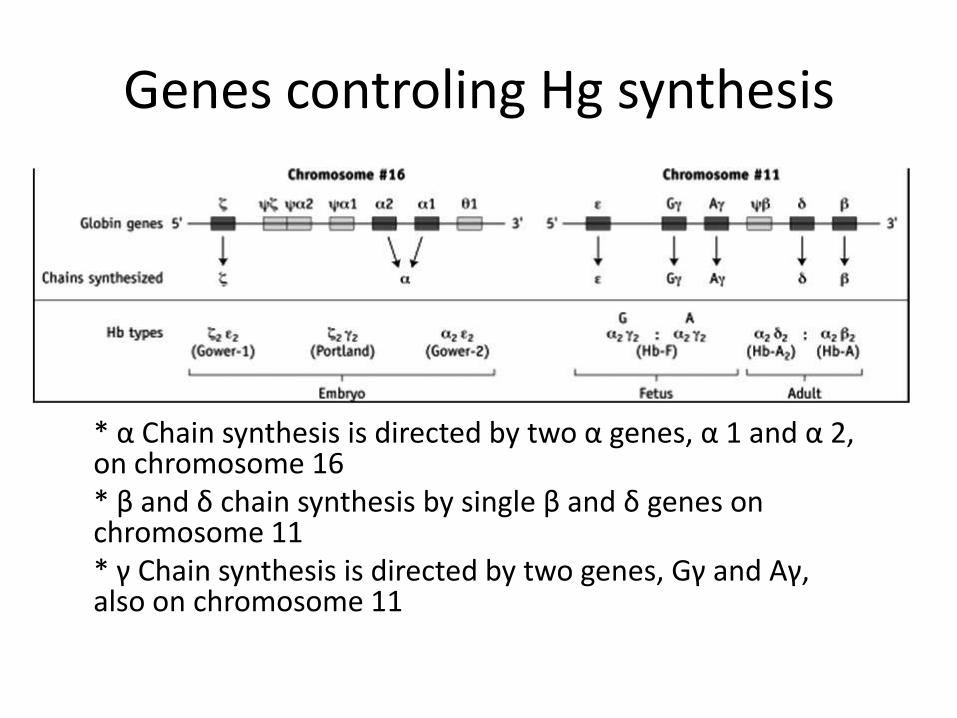
Genes controling Hg synthesis
* α Chain synthesis is directed by two α genes, α 1 and α 2, on chromosome 16 * β and δ chain synthesis by single β and δ genes on chromosome 11 * γ Chain synthesis is directed by two genes, Gγ and Aγ, also on chromosome 11

Thalassemia
• The thalassemia syndromes are a heterogeneous group of disorders caused by inherited mutations that decrease the synthesis of adult hemoglobin, HgA (α2β2)
• Endemic in Middle East, tropical Africa, India, Asia
• β-Thalassemia is caused by deficient synthesis of β chains, whereas α-thalassemia is caused by deficient synthesis of α chains
• The hematologic consequences of diminished synthesis of one globin chain stem not only from hemoglobin deficiency but also from a relative excess of the other globin chain, particularly in β-thalassemia

β-Thalassemias
• caused by mutations that diminish the synthesis of β-globin chains
• β0 mutations, associated with absent β-globin synthesis
• β+ mutations, characterized by reduced (but detectable) β-globin synthesis
• 100 different causative mutations, mostly consisting of point mutations
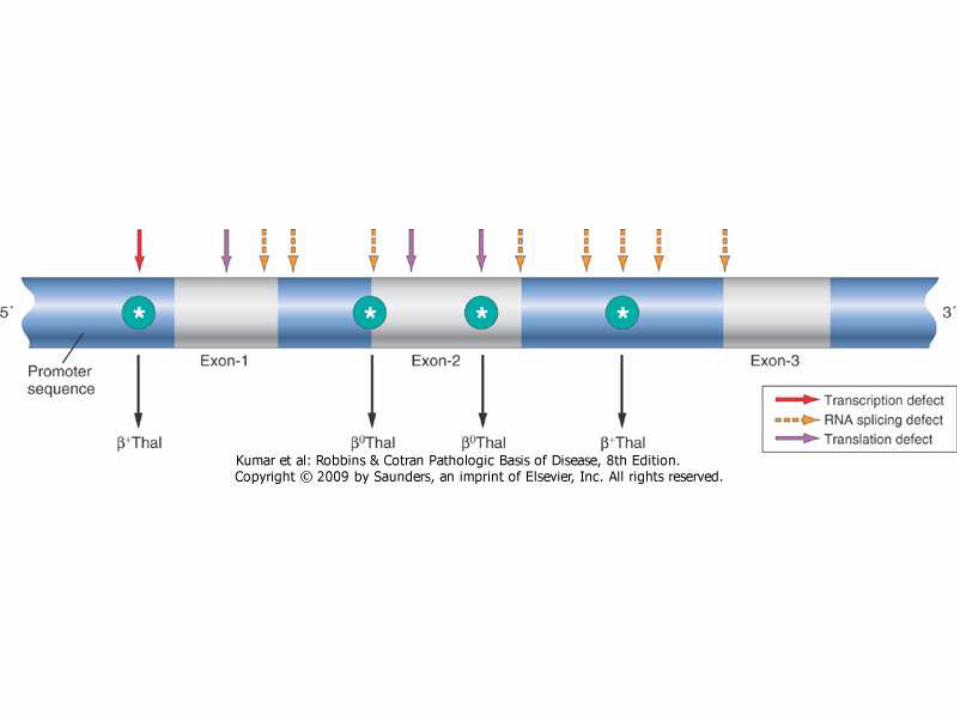

Types of mutations
• Splicing mutations: most common cause of β+-thalassemia. Most of these mutations lie within introns, while a few are located within exons. Some of these mutations destroy the normal RNA splice junctions and completely prevent the production of normal β-globin mRNA, resulting in β0-thalassemia
• Promoter region mutations. These mutations reduce transcription by 75% to 80%. Some normal β-globin is synthesized; thus, these mutations are associated with β+-thalassemia.
• Chain terminator mutations. These are the most common cause of β0-thalassemia. Two subtypes of mutations fall into this category. The most common type creates a new stop codon within an exon; the second introduces small insertions or deletions that shift the mRNA reading frames, Both block translation and prevent the synthesis of any functional β-globin

Pathophysiology
• The deficit in HgA synthesis produces "underhemoglobinized" hypochromic, microcytic red cells with subnormal oxygen transport capacity
• Diminished survival of red cells and their precursors, which results from the imbalance in α- and β-globin synthesis. Unpaired α chains precipitate within red cell precursors, forming insoluble inclusions, which damage cell membrane and results in cell death in RBC precursors (Ineffective erythropoiesis)
• Those red cells that are released from the marrow also bear inclusions and membrane damage and are prone to splenic sequestration and extravascular hemolysis

Pathophysiology
• In severe β-thalassemia, uncompensated anemia leads to massive erythroid hyperplasia in the marrow and extensive extramedullary hematopoiesis in spleen, liver and LNs
• The expanding mass of red cell precursors erodes the bony cortex, impairs bone growth, and produces skeletal abnormalities
• The metabolically active erythroid progenitors steal nutrients from other tissues that are already oxygen-starved, causing severe cachexia in untreated patients.
• Ineffective erythropoiesis suppresses the circulating levels of hepcidin, a critical negative regulator of iron absorption. Low levels of hepcidin and the iron load of repeated blood transfusions inevitably lead to severe iron overload
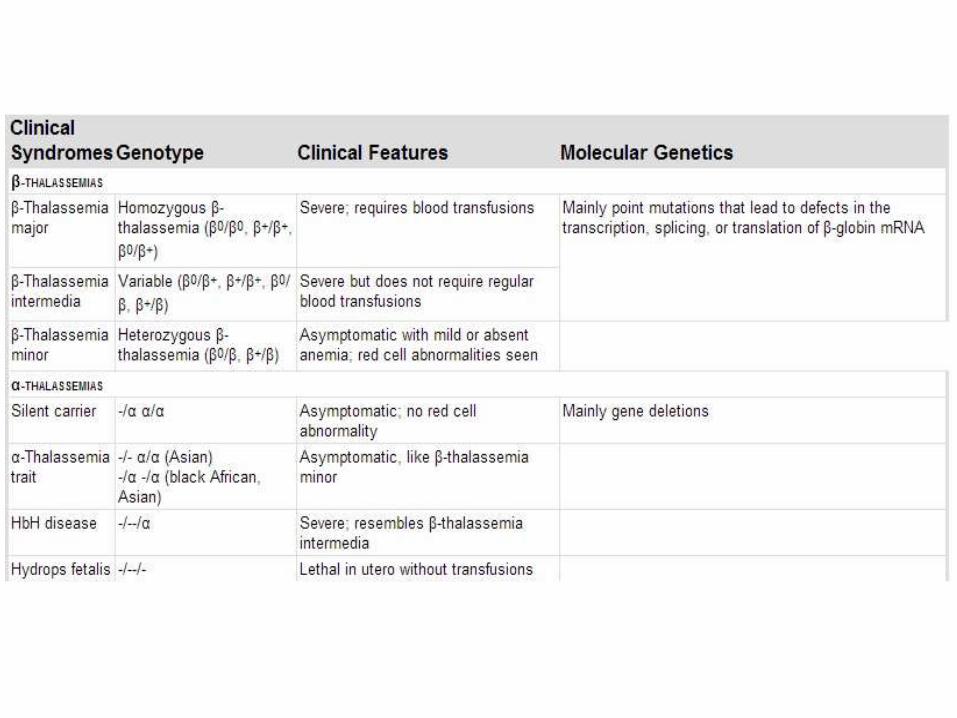

Clinical syndromes
• In general, individuals with two β-thalassemia alleles (β+/β+, β+/β0, or β0/β0) have a severe, transfusion-dependent anemia called β-thalassemia major
• Heterozygotes with one β-thalassemia gene and one normal gene (β+/β or β0/β) usually have a mild asymptomatic microcytic anemia. This condition is referred to as β-thalassemia minor or β-thalassemia trait
• A third genetically heterogeneous variant of moderate severity is called β-thalassemia intermedia. This category includes milder variants of β+/β+ or β+/β0-thalassemia and unusual forms of heterozygous β-thalassemia

α-thalassemia
• Normally, there are four α-globin genes, and the severity of α-thalassemia depends on how many α-globin genes are affected
• As in β-thalassemias, the anemia occurs both from inadequate hemoglobin synthesis and the effects of excess unpaired non-α chains (β, γ, and δ)
• In newborns with α-thalassemia, excess unpaired γ-globin chains form γ4 tetramers known as hemoglobin Barts, whereas in older children and adults excess β-globin chains form β4 tetramers known as HgH.
• Since free β and γ chains are more soluble than free α chains, hemolysis and ineffective erythropoiesis are less severe than in β-thalassemias.
• Gene deletion is the most common cause of reduced α-chain synthesis.

Clinical features
• Silent carrier: a single gene deletion, patients have microcytosis but no anemia, asymptomatic
• α-Thalassemia Trait: deletion of two genes, clinically identical to β-thalassemia minor: microcytosis, minimal or no anemia, and no abnormal physical signs
• Hemoglobin H Disease: deletion of 3 genes, common in Asia, clinically resembles β-thalassemia intermedia, HgH has very high affinity to oxygen, leading to tissue hypoxia. It also precipitates within the RBC which results in extravascular hemolysis
• Hydrops fetalis: deletion of 4 genes. Patients die in utero unless transfused. Babies have severe pallor, hepatosplenomegaly and edema. Treatment: life-long transfusion or BM transplant

Diagnosis
• Blood film: hypochromic microcytic anemia, target cells, basophilic stippling
• Hg electrophoresis: different globin chains have different electrical charges. Hg is separated on gel and an electrical current is applied. Each type of Hg migrate a specific distance and hence can be recognized
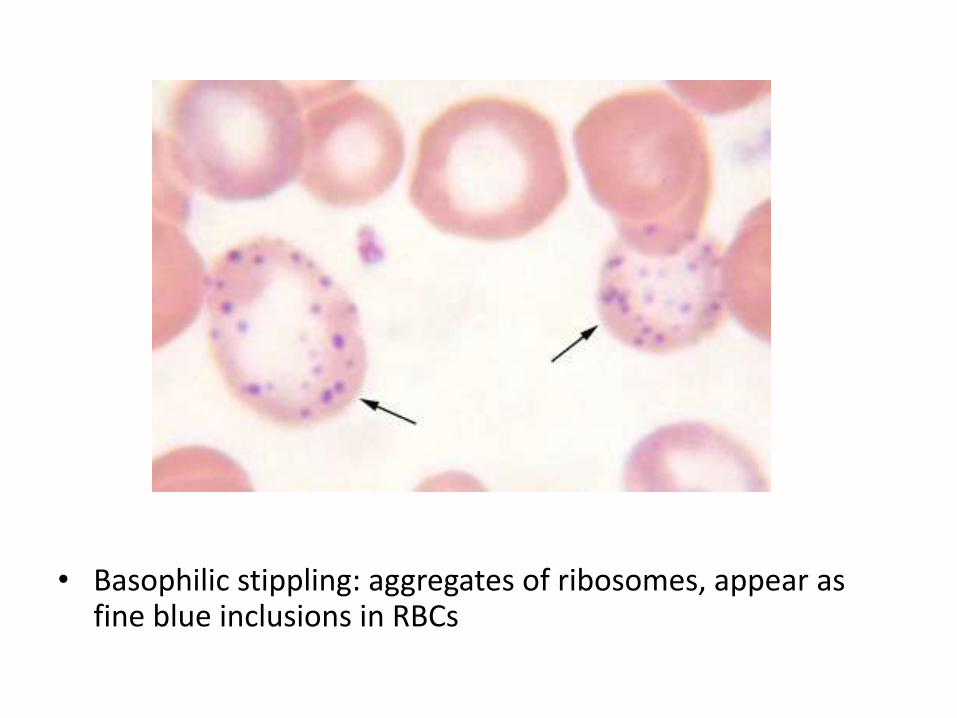
• Basophilic stippling: aggregates of ribosomes, appear as fine blue inclusions in RBCs

Sickle Cell Anemia

Sickle Cell Disease/ AnemiaSickle Cell Disease/ Anemia
• Hereditary hemoglobinopathy that occurs primarily in individuals of African descent
• Sickle cell disease is caused by a point mutation in the sixth codon of β-globin that leads to the replacement of a glutamate residue with a valine residue
• The abnormal physiochemical properties of the resulting sickle hemoglobin (HbS) are responsible for the disease

• Sickle Cell Trait: heterozygosity of HgS, carriers are largely asymptomatic, HgS ≈ 40%
• Sickle Cell Disease: homozygosity of HbS, symptomatic, HgS ≈ 80%
• Both types of Hg are protective against Malaria falciparum infection

Pathophysiology
• HbS molecules undergo polymerization when deoxygenated. Initially the red cell cytosol converts from a freely flowing liquid to a viscous gel as HbS aggregates form. With continued deoxygenation aggregated HbS molecules assemble into long needle-like fibers within red cells, producing a distorted sickle shape
• This causes damage to cell membrane. Ca+2 enters the cell and causes protein cross-linking. K+ and H2O moves out of the cell from damaged membrane
• With repeated sickling, more damage happens until the cell shape is irreversibly changed even if oxygenated again
• Sickle cells are fragile, leading to intravascular hemolysis• Sickle cells are removed by macrophages, leading to extravascular
hemolysis too

Pathophysiology
• The presence of HbS underlies the major pathologic manifestations:
(1) chronic hemolysis
(2) microvascular occlusions
(3) tissue damage

Interaction of HgS with the other types of hemoglobin
• In heterozygotes with sickle cell trait: HgA interferes with HbS polymerization. As a result, red cells in heterozygous individuals do not sickle except under conditions of profound hypoxia
• HgF inhibits the polymerization of HbS even more than HgA; hence, infants do not become symptomatic until they reach 5 or 6 months of age, when the level of HgF normally falls

Mean cell hemoglobin concentration
• Higher HgS concentrations increase the probability that aggregation and polymerization will occur during any given period of deoxygenation. Thus, intracellular dehydration, which increases the MCHC, facilitates sickling
• HgC is a variant of Beta chain. It tends to cause dehydration in RBC. Thus, if it was combined with HgS (HgSC disease), sickling takes place
• Conversely, conditions that decrease the MCHC reduce the disease severity. This occurs when the individual is homozygous for HbS but also has coexistent α-thalassemia, which reduces Hb synthesis and leads to milder disease.

Intracellular PH
• A decrease in pH reduces the oxygen affinity of hemoglobin, thereby increasing the fraction of deoxygenated HbS at any given oxygen tension and augmenting the tendency for sickling
• Acidosis and hypoxia occurs in inflammation

Transit time of red cells through microvascular beds
• Transit times in most normal microvascular beds are too short for significant aggregation of deoxygenated HbS to occur
• Sickling is confined to microvascular beds with slow transit times (Spleen, BM and inflamed tissue)

Blood morphology in sickle cell crisis
• peripheral blood demonstrates variable numbers of irreversibly sickled cells, reticulocytosis, and target cells, which result from red cell dehydration
• The bone marrow is hyperplastic as a result of a compensatory erythroid hyperplasia
• Expansion of the marrow leads to bone resorption and secondary new bone formation, resulting in prominent cheekbones and changes in the skull that resemble a crew-cut in x-rays
• Extramedullary hematopoiesis can also appear • The increased breakdown of hemoglobin can cause
pigment gallstones and hyperbilirubinemia

Splenic changes
• In early childhood, the spleen is enlarged up to 500 gm by red pulp congestion, which is caused by the trapping of sickled red cells in the cords and sinuses
• With time, however, the chronic erythrostasis leads to splenic infarction, fibrosis, and progressive shrinkage, so that by adolescence or early adulthood only a small residual splenic tissue is left; this process is called autosplenectomy
• Howell-Jolly bodies (small nuclear remnants) are also present in some red cells due to the asplenia
• Patients are at increased risk of bacterial infections (Pneumococcus pneumoniae and Haemophilus influenzae septicemia and meningitis)

Vaso-occlusive crisis
• Also called pain crises, are episodes of hypoxic injury and infarction that cause severe pain in the affected region
• Although infection, dehydration, and acidosis (all of which favor sickling) can act as triggers, in most instances no predisposing cause is identified
• The most commonly involved sites are the bones, lungs, liver, brain, spleen, penis (priapism), skin (leg ulcers), pulmonary vessels (cor pulmonale)
• In children, painful bone crises are extremely common and often difficult to distinguish from acute osteomyelitis. These frequently manifest as the hand-foot syndrome or dactylitis of the bones of the hands or feet, or both (growth retardation)
• Acute chest syndrome is a particularly dangerous type of vaso-occlusive crisis involving the lungs, which typically presents with fever, cough, chest pain, and pulmonary infiltrates

Sequestration crises
• Occurs in children with intact spleens
• Massive entrapment of sickle red cells leads to rapid splenic enlargement, hypovolemia, and sometimes shock
• These complications may be fatal in several cases
• Survival from sequestration crises and the acute chest syndrome requires treatment with exchange transfusions.

Aplastic crises
• Acute event
• Infection of red cell progenitors by parvovirus B19, which causes a transient cessation of erythropoiesis and a sudden worsening of the anemia

Diagnosis
• Blood film
• Sickling test: application of oxygen-consuming reagent to blood
• Hemoglobin electrophoresis is also used to demonstrate the presence of HbS

Treatment
• Hydroxyurea: (1) an increase in red cell HbF levels, which occurs by unknown mechanisms; and (2) an anti-inflammatory effect, which stems from an inhibition of white cell production
• Blood exchange

Sickle cells and malaria
• Sickle cell trait provides protection against malaria infection, especially for Plasmodium Falciparum
• Endemic areas of malaria overlaps geographically with areas of sickle cell disease
• Mechanism of protection is not completely known• Suggested explanation: organism causes oxidative stress
inside RBC, which causes hypoxia and sickling, then these RBCs are removed from circulation by phagocytes
• The environment in infected sickle cells produces more oxygen radicals, harming the organism itself
• Duffy antigen is the receptor for P. Vivax, this antigen is absent in many people with sickle cell trait
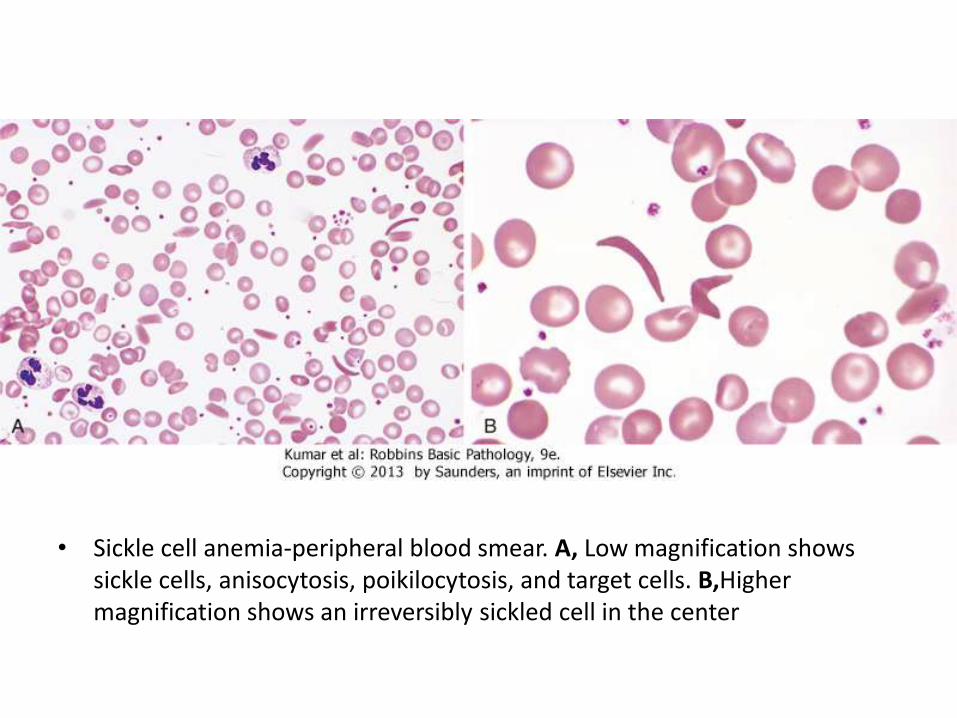
• Sickle cell anemia-peripheral blood smear. A, Low magnification shows sickle cells, anisocytosis, poikilocytosis, and target cells. B,Highermagnification shows an irreversibly sickled cell in the center


ANEMIAS OF DIMINISHED ERYTHROPOIESIS

ANEMIAS OF DIMINISHED ERYTHROPOIESIS
• Anemias secondary to inadequate RBC production
• Nutritional
• Renal failure
• Chronic inflammation
• Bone marrow failure

Iron deficiency anemia
• The most common anemia worldwide
• The total body iron is about 2 gm in women and 6 gm in men
• Ideal diet constitutes 10-20 mg of iron, only 10% is absorped
• Most dietary iron occurs in meat products
• Iron is present in human body as functional (80%) and stored (20%)
• Functional iron is found in hemoglobin, myoglobin, catalaseand cytochromes
• Stored iron is found is hemosiderin and ferritin
• Free iron is highly toxic
• Iron absorption occurs in the proximal duodenum

Mechanism of iron homeostasis
Absorption• Regulated• intestine may increase rate
of absorption by 10x• Responsive to:• Iron status• Erythropoietic demand• Hypoxia• Inflammation (↓)
Loss• Not regulated• Insensible losses:• Physiologic exfoliation
(enterocytes, keratinocytes, endometrium), 1-2 mg/day
• Bleeding• Reproduction
Body Iron status is regulated mainly at the level of absorption
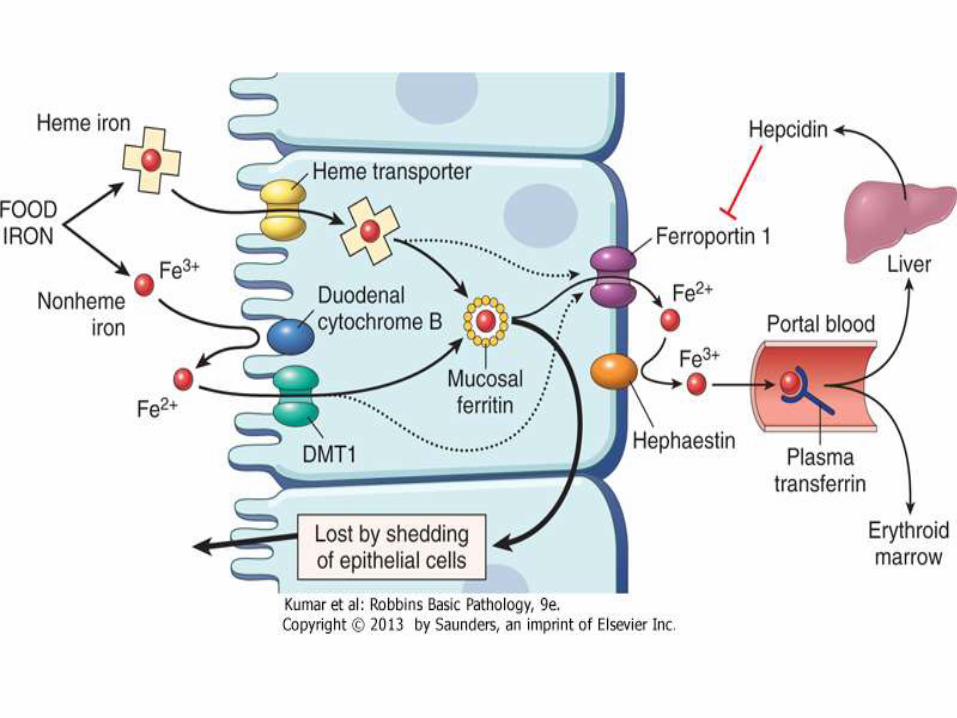

Iron absorption and transport
• Dietary non-heme iron is in ferric status (Fe3+)
• Iron crosses cell membrane in ferrous status (Fe2+)
• Reduced by cytochrome
• Oxidized by Hephaestin
• Stored in ferritin in ferric status
• Transported by transferrin in ferrous status
• Presents as ferrous in non-oxygenated heme
• Oxidized to ferrous when O2 is bound to Hg
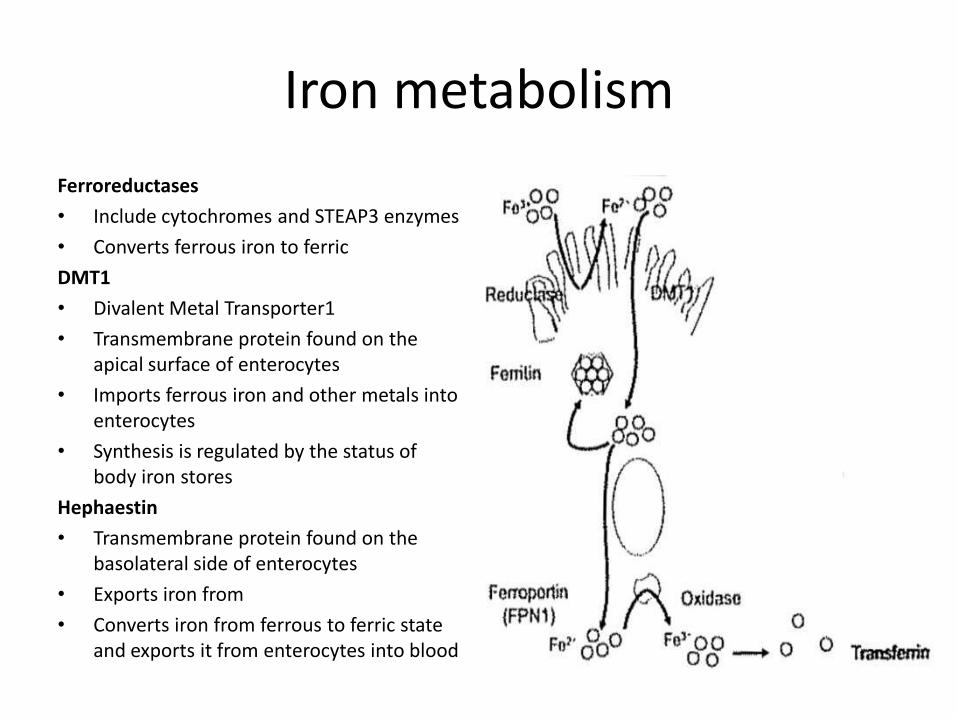
Iron metabolism
Ferroreductases
• Include cytochromes and STEAP3 enzymes
• Converts ferrous iron to ferric
DMT1
• Divalent Metal Transporter1
• Transmembrane protein found on the apical surface of enterocytes
• Imports ferrous iron and other metals into enterocytes
• Synthesis is regulated by the status of body iron stores
Hephaestin
• Transmembrane protein found on the basolateral side of enterocytes
• Exports iron from
• Converts iron from ferrous to ferric state and exports it from enterocytes into blood

Iron metabolism
Transferrin (TF)• 90 kD serum glycoprotein• 10% of non-albumin protein in
serum• Binds 2 iron atoms in Ferric state
(Fe3+)• Carries iron throughout the
circulation• Delivers it to cell transferrin
receptors on erthroid precursors• Normally, only 1/3 is saturated
with iron
• All molecules solublize iron in aqueous environments and minimize its reactivity
Ferritin
• Stores iron within cells
• Found mainly in RES, intracellular
• Stored in hepatocytes in the liver (from serum TF), and in macrophages in BM and spleen (mainly from hemecatabolism)
• Holds up to 4500 iron atoms
• Oxido-reductase reactivity
• Iron can be mobilized when needed
Hemosiderin
• Aggregates of partially degraded ferritin
• Found normally in trace amounts in RES
• Stained blue with Prussian stain

Hepcidin
• Synthesized in the liver
• Hepcidin level in the serum correlates positively with the iron stores in the body
• Hepcidin inhibits Ferroportin, causing retention of iron inside enterocytes which shed outside the body
• Hepcidin production is increased in anemia of chronic disease secondary to IL-6

Iron metabolism
• Normal serum iron level is 120 μg/dL in men and 100 μg/dL in women
• Serum iron level does not reflect the actual status of total body iron
• Serum ferritin is derived from the storage pool of body iron, hence, it correlated well with body iron
• Exception: in stress conditions, ferritin increases with other acute phase reactants
• Recycling is a major source of iron available for erythropoesis. Macrophages is RES engulf old RBCs, degrade heme and the retrieved iron is either stored in ferritin or delivered to erythroid cells

Causes of iron deficiency
• Decreased dietary intake (vegetarians)
• Impaired absorption (GI disease)
• Increased demand (pregnancy)
• Chronic blood loss (GI bleeding, menorrhagea)
People at increased risk of anemia are: infants, elderly, teenagers, low socioeconomic class

Iron deficiency
• Iron deficiency develops insidiously
• Iron stores are depleted first, marked by a decline in serum ferritin and the absence of stainable iron in the bone marrow
• Then a decrease in serum iron and a rise in the serum transferrin
• Ultimately, the capacity to synthesize hemoglobin, myoglobin, and other iron-containing proteins is diminished, leading to microcytic anemia, impaired work and cognitive performance, and even reduced immunocompetence

Clinical features
• RBCs appear as microcytic and hypochromic, Target cells
• Low serum ferritin and iron levels, low transferrinsaturation, increased total iron-binding capacity, and
• Response to iron therapy • Erythropoietin levels are increased, but the
marrow response is blunted by the iron deficiency; thus, marrow cellularity usually is only slightly increased
• Thrombocytosis
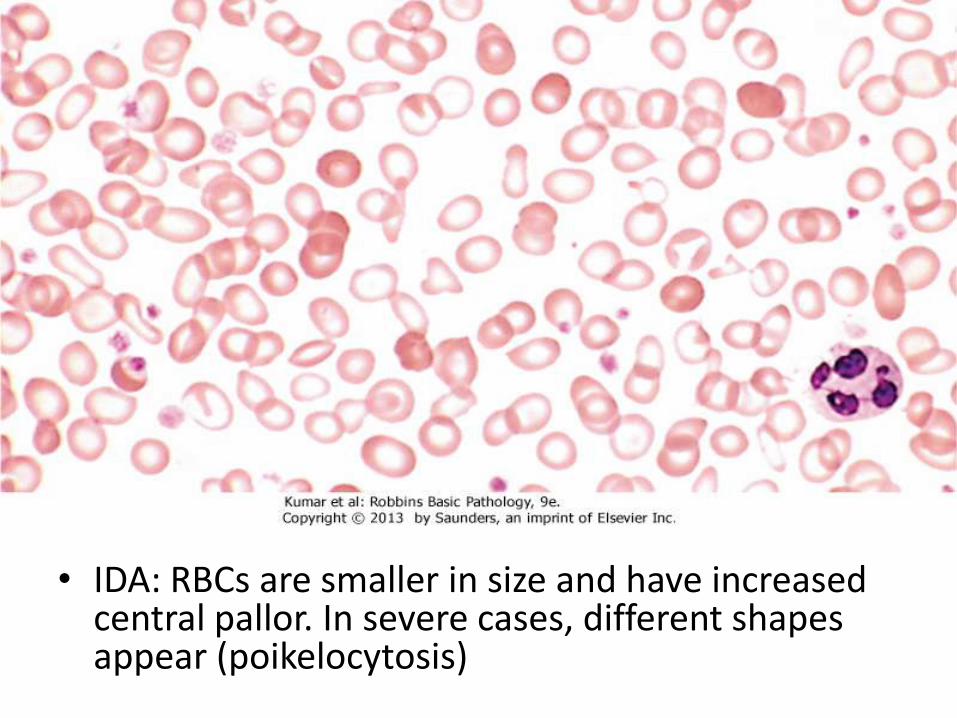
• IDA: RBCs are smaller in size and have increased central pallor. In severe cases, different shapes appear (poikelocytosis)
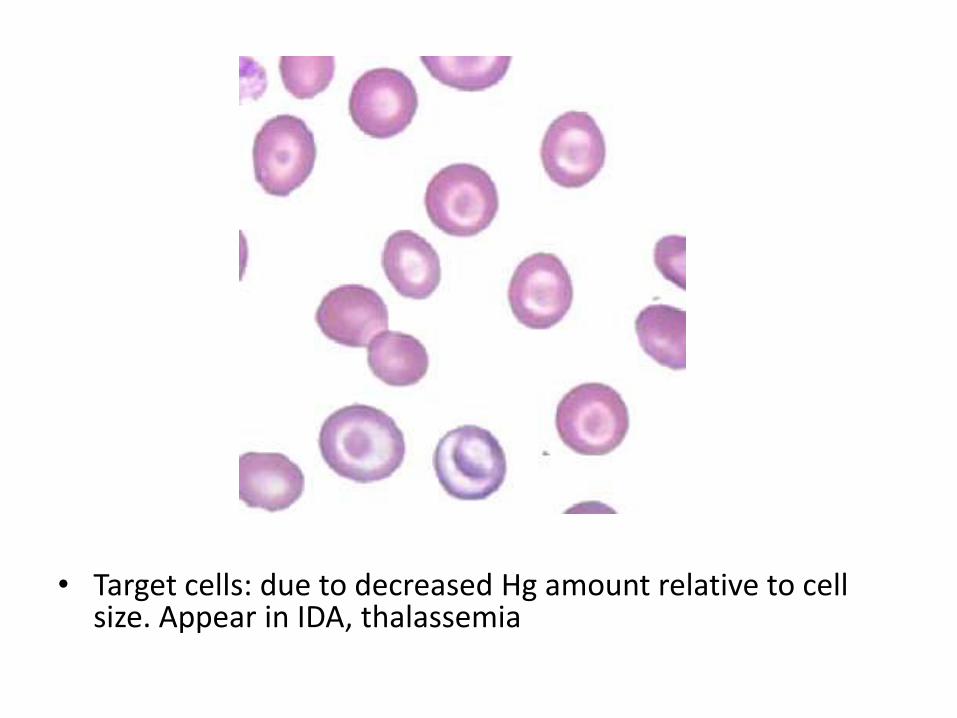
• Target cells: due to decreased Hg amount relative to cell size. Appear in IDA, thalassemia

Megaloblastic Anemia
• Anemia associated with impairment in DNA synthesis in hematopoietic cells special morphologic features (large immature erythroid precursors)
• Two types: Vitamin B12 and folate deficiency
• Vitamin B12 and folate are coenzymes required for synthesis of thymidine

Causes of Vit B12 deficiency
• Low intake (vegans)
• Impaired GI absorption (intrinsic factor deficiency, malabsorption disease, gastrectomy)
• Bacterial overgrowth, parasitic infection, fish tapeworm infestation

Causes of folate deficiency
• Low intake (inadequate diet, infancy)
• Impaired absorption (malabsorption, chronic alcoholism, anti-convulsants, oral contraceptives)
• Increased loss (dialysis)
• Impaired utilization (methotrexate, Vit B12 deficiency)

Pernicious Anemia
• Abnormal autoreactive T-cell response initiates direct gastric mucosal injury, also triggers formation of autoantibodies
• Type 1 antibody: blocks Vit B12 from binding to intrinsic receptors
• Type 2 antibody: blocks Vit B12-intrinsic factor complex to its ileal receptor
• Type 3 antibody: blocks Proton pumps on parietal cells (not specific)
• With time, anemia develops, gastric glands become atrophic
• Neurologic symptoms develop secondary to spinal cord demyelination

Morphology
• PB: RBCs are large and oval and no central pallor, with anisopoikelocytosis. Reticulocytes are low. Neutrophilsare large and have hypersegmented nuclear lobes (5 or more)
• BM: hypercellular. Megaloblastic changes in erythroidprecursors (large size and immature nucleus despite cytoplasmic maturation)
• Granulocytic precursors and megakaryocytes are also large with multilobation
• Increased erythropoietic level as well as impaired DNA synthesis leads to increased apoptosis in nucleated RBCs and hemolysis
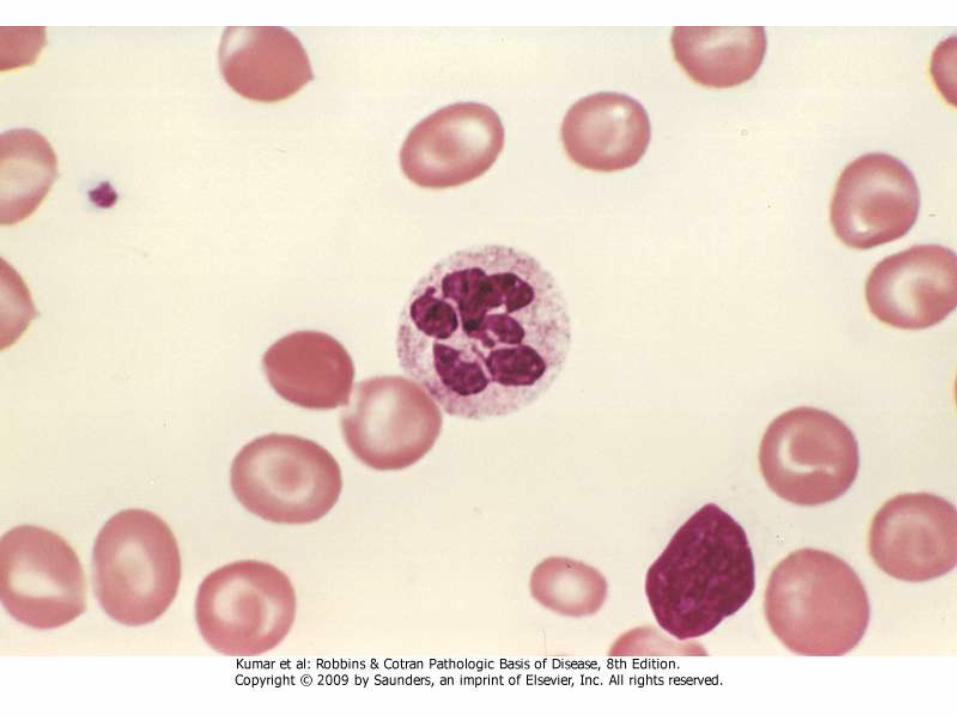
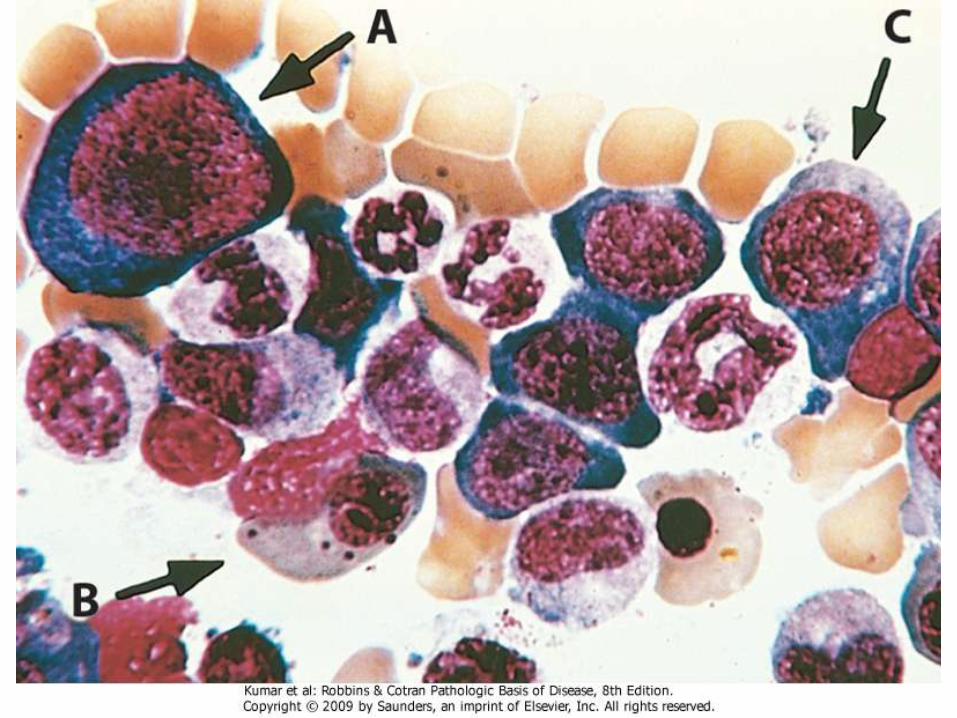

Anemia of Chronic Disease
• Most common anemia in hospitalized people• It is associated with a reduction in the proliferation of
erythroid progenitors and impaired iron utilization • The chronic illnesses associated with this form of
anemia can be grouped into three categories:• Chronic microbial infections, such as osteomyelitis,
bacterial endocarditis, and lung abscess• Chronic immune disorders, such as rheumatoid
arthritis and regional enteritis• Neoplasms, such as carcinomas of the lung and breast,
and Hodgkin lymphoma

Pathophysiology
• The anemia of chronic disease occurs in the setting of persistent systemic inflammation and is associated with low serum iron, reduced total iron-binding capacity, and abundant stored iron in tissue macrophages
• Certain inflammatory mediators, particularly interleukin-6 (IL-6), stimulate an increase in the hepatic production of hepcidin
• Hepcidin inhibits ferriportin function in macrophages and reduces the transfer of iron from the storage pool to developing erythroid precursors in the bone marrow
• Hepcidin suppresses erythropoietin production, hence, erythroid cells proliferation is not compensating well

Pathologic findings
• The red cells can be normocytic and normochromic, or hypochromic and microcytic, as in anemia of iron deficiency
• There are increased storage iron in marrow macrophages, high serum ferritin level, and reduced total iron-binding capacity
• Only successful treatment of the underlying condition reliably corrects the anemia
• However, some patients, particularly those with cancer, benefit from administration of erythropoietin.

Aplastic Anemia
• A syndrome of chronic primary hematopoietic failure and attendant pancytopenia
• In the majority of patients autoimmune mechanisms are suspected
• Both inherited or acquired abnormalities of hematopoietic stem cells also occur

Causes
• Most cases of "known" etiology follow exposure to chemicals and drugs.
• Certain drugs and agents (including many cancer chemotherapy drugs and the organic solvent benzene) cause marrow suppression that is dose related and reversible
• In other instances, aplastic anemia arises in an unpredictable, idiosyncratic fashion following exposure to drugs that normally cause little or no marrow suppression. The implicated drugs include chloramphenicol and gold salts
• Persistent marrow aplasia can also appear after a variety of viral infections, most commonly viral hepatitis of the non-A, non-B, non-C, non-G type, which is associated with 5% to 10% of cases
• Irradiation causes BM suppression • In the majority of cases, no known cause can be identified

Fanconi anemia
• Inherited form of aplastic anemia
• AR, defects in a multiprotein complex that is required for DNA repair
• Marrow hypofunction becomes evident early in life and is often accompanied by multiple congenital anomalies, such as hypoplasia of the kidney and spleen and bone anomalies

Pathophysiology
• Two major etiologies have been invoked: an extrinsic, immune-mediated suppression of marrow progenitors; and an intrinsic abnormality of stem cells
• Stem cells first be antigenically altered by exposure to drugs, infectious agents, or other unidentified environmental insults. Activated TH1 cells produce cytokines such as interferon-γ (IFNγ) and TNF that suppress and kill hematopoietic progenitors
• Immunosuppressive drugs such as cyclosporine produce responses in 60% of patients
• The antigens recognized by the autoreactive T cells are not well defined. In some instances GPI-linked proteins may be the targets, possibly explaining the previously noted association of aplasticanemia and PNH

Pathophysiology
• In the second group, aplastic anemia results from cytogenetic abnormality
• Abnormally short tolemers are seen in 10% of cases
• In minority of cases, acute leukemia complicates aplastic anemia
• In some genetically altered stem cells, the abnormal antigen expression activates T-cells which kills them

Pathologic findings
• The markedly hypocellular bone marrow is largely devoid of hematopoietic cells; often only fat cells, fibrous stroma
• Marrow aspirates often yield little material (a "dry tap")
• If the anemia necessitates multiple transfusions, systemic hemosiderosis can appear
• Reticulocytopenia
• No splenomegaly

Pure Red Cell Aplasia
• Only erythroid progenitors are suppressed • It may occur in association with neoplasms, particularly thymoma, drug exposures,
autoimmune disorders• A special form of red cell aplasia occurs in individuals infected with parvovirus B19,
which preferentially infects and destroys red cell progenitors. • Normal individuals clear parvovirus infections within 1 to 2 weeks; as a result, the
aplasia is transient and clinically unimportant. However, in persons with moderate to severe hemolytic anemias, even a brief cessation of erythropoiesis results in rapid worsening of the anemia, producing an aplastic crisis.
• In those who are severely immunosuppressed (such as persons with advanced HIV infection), an ineffective immune response sometimes permits the infection to persist, leading to chronic red cell aplasia and a moderate to severe anemia
• Blackfan-Diamond anemia: a congenital form of pure red cell aplasia than appears in infancy

Myelophthisic anemia
• Bone marrow failure secondary to physical occupation of the marrow spaces by a pathologic process
• Examples: metastatic cancer, granulomatousdisease, fibrosis

Chronic Renal Failure
• Anemia tends to be roughly proportional to the severity of the uremia
• Caused by:• 1) diminished synthesis of erythropoietin by the
damaged kidneys, which leads to inadequate red cell production
• 2) iron deficiency due to platelet dysfunction and increased bleeding, which is often encountered in uremia
• Uremia causes a change in RBC membrane shape, known as echinocytes

Liver disease
• whether toxic, infectious, or cirrhotic is associated with anemia attributed to decreased marrow function
• Folate and iron deficiencies caused by poor nutrition and excessive bleeding often exacerbate anemia in this setting
• Erythroid progenitors are preferentially affected depression of the white cell count and platelets is less common but also occurs.
• The anemia is often slightly macrocytic due to lipid abnormalities associated with liver failure, which cause red cell membranes to acquire phospholipid and cholesterol as they circulate in the peripheral blood, a characteristic shape of acanthocytes

Hypothyroidism
• Causes decrease in cell metabolism
• Results in mild normochromic normocyticanemia

POLYCYTHEMIA
• Erythrocytosis: increase in red cells per unit volume of peripheral blood, usually in association with an increase in hemoglobin concentration.
• May be absolute (defined as an increase in total red cell mass) or relative
• Relative polycythemia results from dehydration, such as occurs with water deprivation, prolonged vomiting, diarrhea, or the excessive use of diuretics.
• Absolute polycythemia is described as primary when the increased red cell mass results from an autonomous proliferation of erythroid progenitors, and secondary when the excessive proliferation stems from elevated levels of erythropoietin

Absolute polycythemia
• Primary polycythemia (polycythemia vera) is a clonal, neoplastic myeloproliferative disorder
• Secondary polycythemia occurs as an Adaptive process (lung disease, high-altitude living, cyanotic heart disease), Paraneoplastic: erythropoietin-secreting tumors (e.g., renal cell carcinoma, hepatomacellularcarcinoma, cerebellar hemangioblastoma)or Surreptitious: endurance athletes


White blood cells disorders
Non-neoplastic

Leukopenia
• Leukopenia results most commonly from a decrease in blood granulocytes (neutrophils)
• Lymphopenia is much less common; it is associated with rare congenital immunodeficiency diseases, advanced human immunodeficiency virus (HIV) infection, and treatment with high doses of corticosteroids

Neutropenia
• A reduction in the number of granulocytes in blood is known as neutropenia or, when severe, agranulocytosis
• Neutropenic persons are susceptible to bacterial and fungal infections, which can be fatal. The risk of infection rises sharply as the neutrophil count falls below 500 cells/μL

Causes
Decreased production
• Part of aplastic, myelophthisic, megaloblasticanemias
• Drugs: chemotherapy, antiepileptic, antithyroid
• Isolated: congenital disorders (Schwachman-Diamond syndrome, Kostmann disease), acquired (some T-cell lymphoma)

Causes
Increased destruction
• Overwhelming infection
• Cyclic neutropenia
• Splenomegaly

Clinical features
• Patients are very susceptible to infections
• Fever, malaise, fatigue
• Infections usually begin in the oral cavity
• Treatment: treat the underlying cause (stop the offending drug), Granulocytic-Colony Stimulating Factor

Reactive Leukocytosis
• An increase in the number of white cells in the blood is common in a variety of inflammatory states caused by microbial and nonmicrobialstimuli. Leukocytoses are relatively nonspecific and are classified according to the particular white cell series that is affected

Neutrophilia
• Infection (bacterial)
• Burn
• Tissue necrosis (myocardial infarction)

Eosinophilia
• Allergic reactions
• Parasitic infections
• Drug reactions
• Rheumatologic diseases
• Some malignancies (Hodgkin lymphoma)

Monocytosis
• Chronic infections
• Inflammatory bowel disease
• Rheumatologic diseases

Lymphocytosis
• Viral infections
• Tuberculosis

Reactive Lymphadenitis
• Infections and nonmicrobial inflammatory stimuli often activate immune cells residing in lymph nodes, which act as defensive barriers
• Any immune response against foreign antigens can lead to lymph node enlargement (lymphadenopathy). The infections causing lymphadenitis are varied and numerous, and may be acute or chronic
• In most instances the histologic appearance of the lymph node reaction is nonspecific

Reactive Lymphadenitis
• Acute lymphadenitis: neutrophilic infiltration
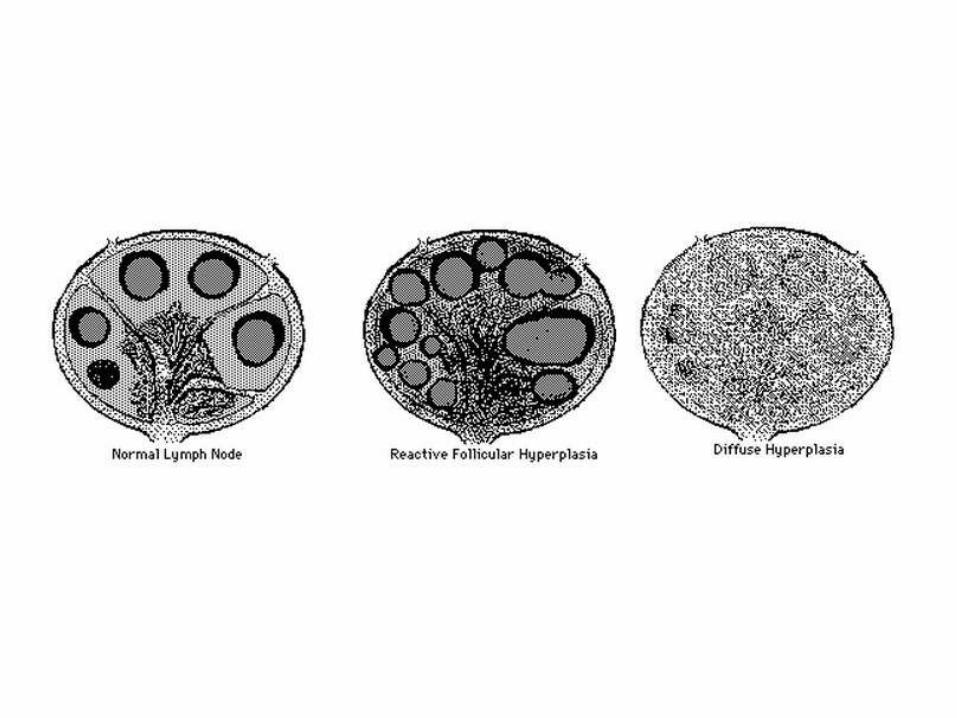
• Chronic lymphadenitis:
• Follicular hyperplasia: proliferation of germinal center B-cells resulting in enlarged follicles, occur in HIV, Toxoplasmosis, Rheumatologic diseases
• Paracortical (diffuse) hyperplasia: proliferation of T-cells in the interfollicular areas, caused by viral infection, drug reaction, post vaccination
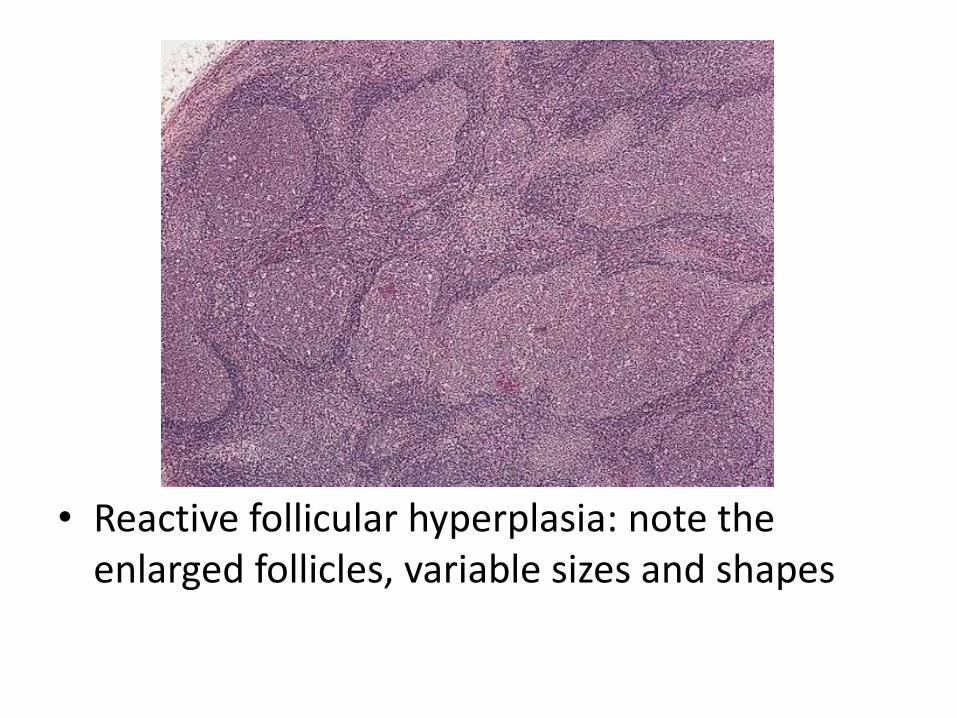
• Reactive follicular hyperplasia: note the enlarged follicles, variable sizes and shapes

White Blood Cells
Neoplastic disorders

NEOPLASTIC PROLIFERATIONS OF WHITE CELLS
• Lymphoid neoplasms, which include lymphomas and plasma cell neoplasms. In many instances tumors are composed of cells resembling some normal stage of lymphocyte differentiation, a feature that serves as one of the bases for their classification.
• Myeloid neoplasms arise from progenitor cells that give rise to the formed elements of the blood: granulocytes, red cells, and platelets. The myeloid neoplasms fall into three fairly distinct subcategories: acute myeloid leukemias, in which immature progenitor cells accumulate in the bone marrow; myeloproliferativedisorders, in which an inappropriate increase in the production of formed blood elements leads to elevated blood cell counts; and myelodysplastic syndromes, which are characteristically associated with ineffective hematopoiesis and cytopenias
• Histiocytic neoplasms include proliferative lesions of macrophages and dendritic cells

Lymphoid neoplasms

Lymphoma
• Many types, classified based on cell of origin (B or T), cell size and stage of maturation
• They vary widely in their clinical presentation and behavior• Most lymphomas arise in lymph nodes, however, they can arise
from any organ• Some characteristically manifest as leukemias, with involvement of
the bone marrow and the peripheral blood• Generally classified as Hodgkin and non-Hodgkin lymphomas• Non-Hodgkin lymphomas are also generally classified as low or
high-grade lymphoma • Plasma cell tumors usually arise within the bones and manifest as
discrete masses, causing systemic symptoms related to the production of monoclonal immunoglobulin.
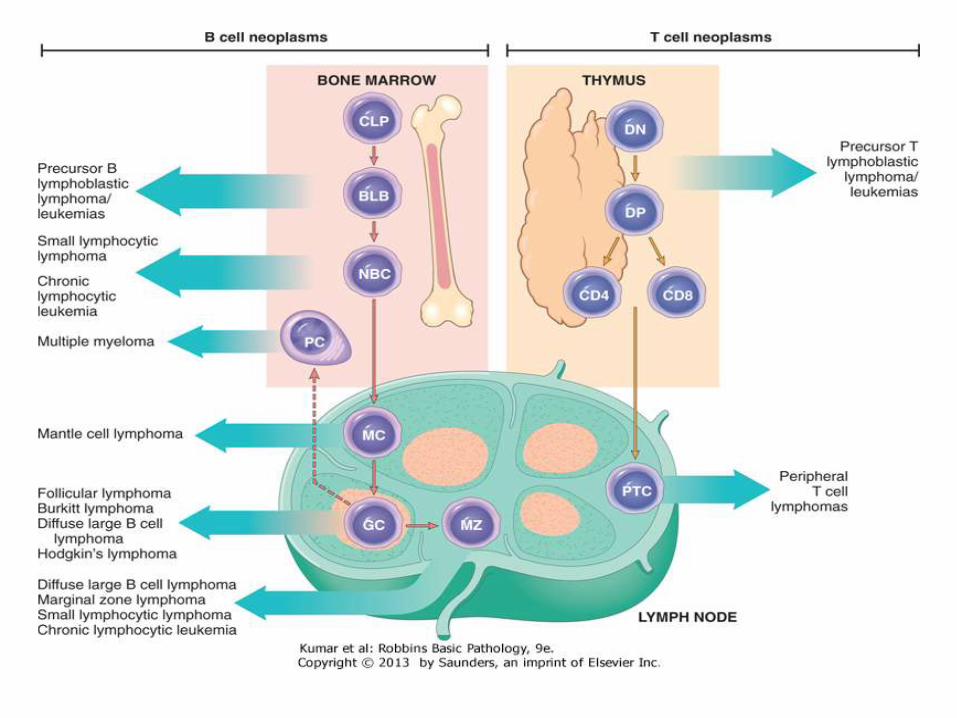

Diagnosis of lymphoma
• In lymphomas, there is proliferation of lymphocytes which came from an original cell, thus, they are monoclonal
• There is effacement of the lymph node normal architecture
• Normal lymphocytes have specific immunophenotype based on the degree of differentiation
• Neoplastic lymphocytes show aberrant immunophenotype
• Cytogenetic abnormality is very common and sometimes is defining the type of lymphoma
• Lymphoma is usually associated with disturbed immune system

Acute Lymphoblastic Leukemia/ Lymphoma
• An aggressive, high-grade type of lymphoma
• Arises from precursor lymphoid cells (lymphoblasts), B or T
• B-ALL is the most common cancer is children, arises from BM, affecting blood, and sometimes LNs
• T-ALL occurs mainly in male adolescents, arises from thymus, then affecting blood, BM and other tissues
• Lymphoblasts develop mutations in transcription genes which regulate both lymphocyte differentiation and proliferation

Clinical features
• Abrupt, stormy onset of symptoms• Clinical signs and symptoms related to suppressed marrow
function, including fatigue (due to anemia), fever (reflecting infections resulting from neutropenia), and bleeding (petechiae, ecchymoses, epistaxis, gum bleeding) secondary to thrombocytopenia
• Bone pain and tenderness, resulting from marrow expansion and infiltration of the subperiosteum
• Generalized lymphadenopathy, splenomegaly, and hepatomegaly due to dissemination of the leukemic cells. These are more pronounced in ALL than in AML
• Central nervous system manifestations, including headache, vomiting, and nerve palsies resulting from meningeal spread
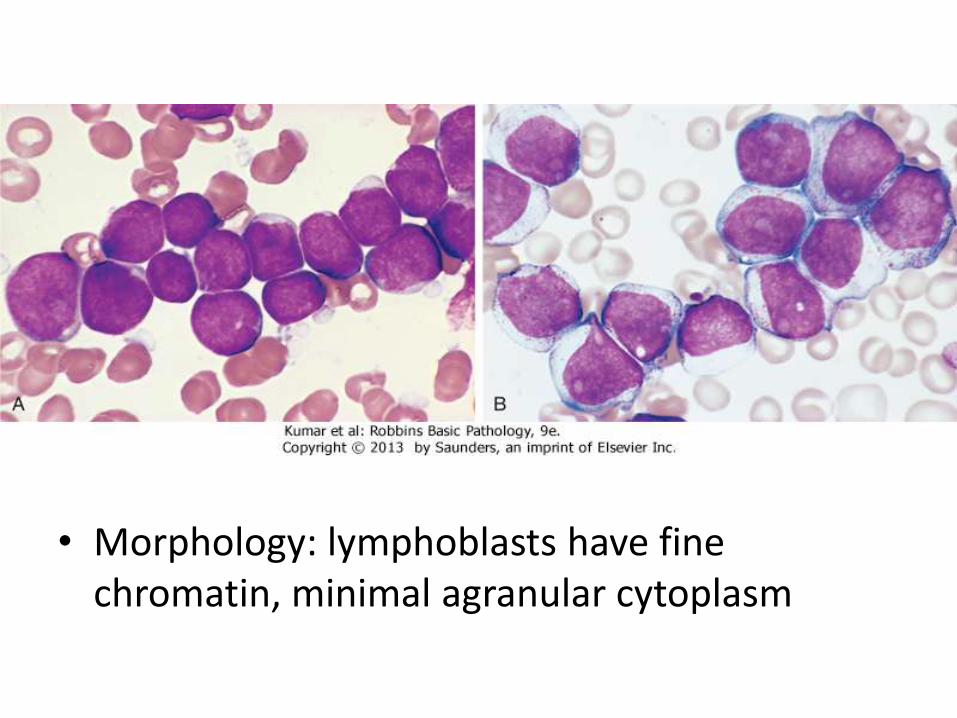
• Morphology: lymphoblasts have fine chromatin, minimal agranular cytoplasm

Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma
• Low grade B-cell lymphoma• Cells are small, round, mature looking similar to
normal lymphocytes• Affects BM and blood (CLL), or LN (SLL)• Bcl2 (anti-apoptotic protein) is up-regulated• The most common leukemia in elderly• Causes derangement in immune system
(hypogammaglobulinemia), or hemolytic anemia• Indolent course, stays stable for years• 10% transforms into high-grade lymphoma
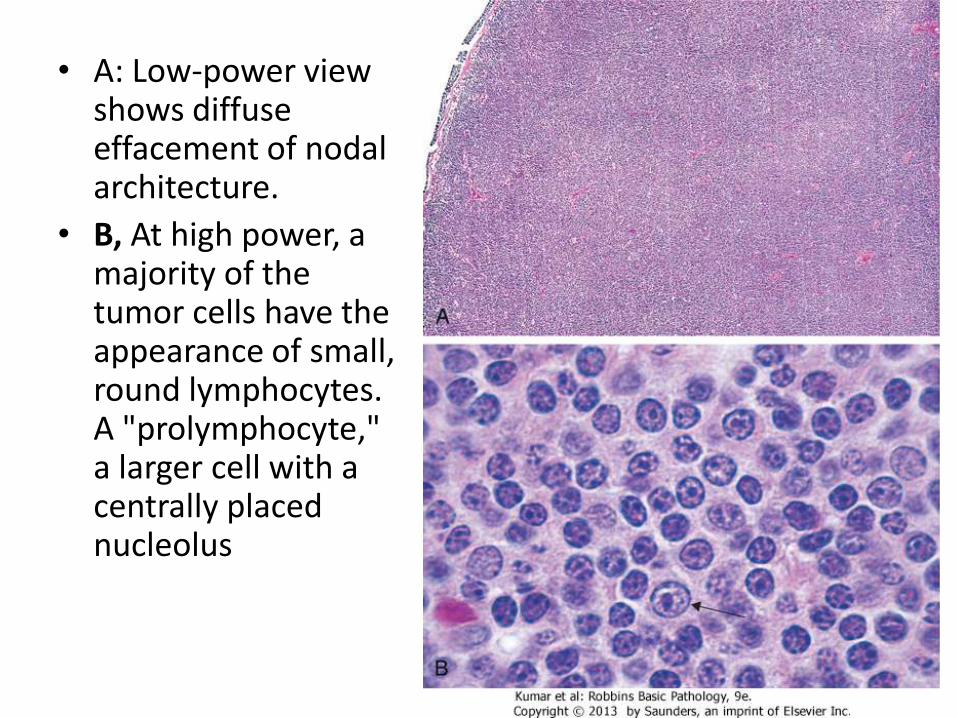
• A: Low-power view shows diffuse effacement of nodal architecture.
• B, At high power, a majority of the tumor cells have the appearance of small, round lymphocytes. A "prolymphocyte," a larger cell with a centrally placed nucleolus
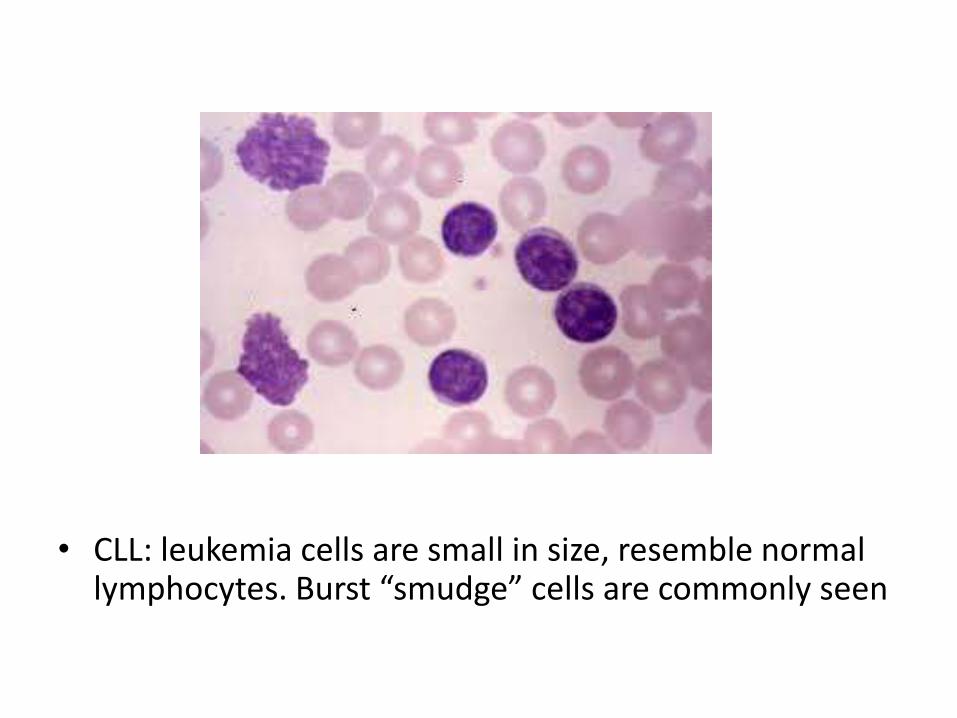
• CLL: leukemia cells are small in size, resemble normal lymphocytes. Burst “smudge” cells are commonly seen

Follicular Lymphoma
• Common, low-grade B-cell lymphoma• Affects elderly• Arises from germinal center B-cell• Lymphoma cells have specific translocation t(14:18), in
which Bcl2 gene on chr18 fuses with IgH gene on chr14, causing overexpression of Bcl2
• Patients has generalized lymphadenopathy• Lymphoma cells proliferate to form abnormal, large,
crowded follicles• Patients have indolent course, transforms into high
grade lymphoma in 40% of cases
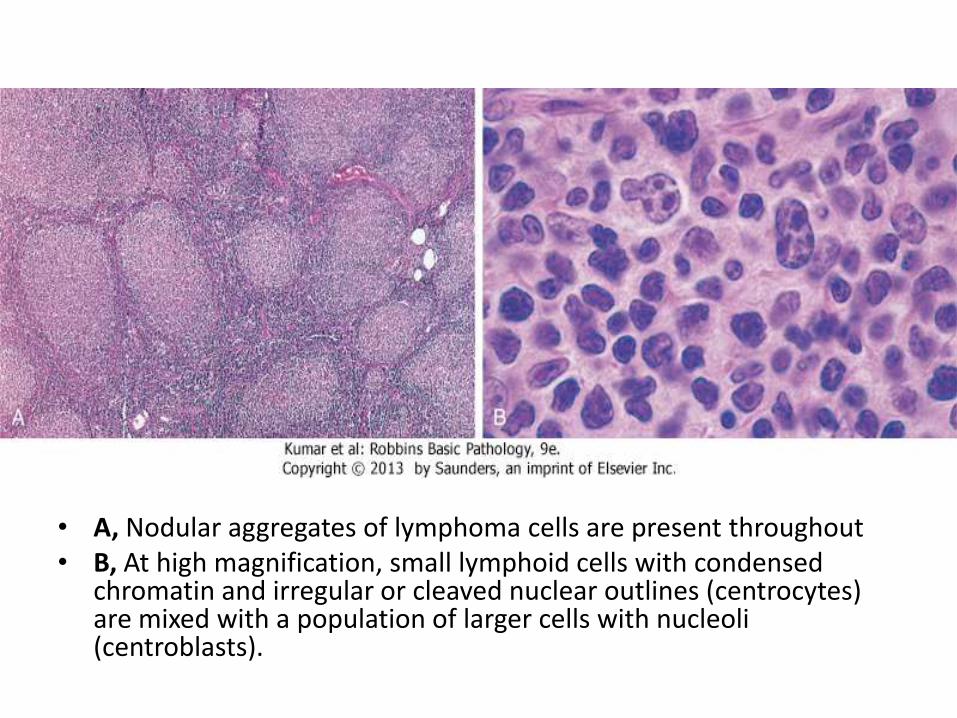
• A, Nodular aggregates of lymphoma cells are present throughout• B, At high magnification, small lymphoid cells with condensed
chromatin and irregular or cleaved nuclear outlines (centrocytes) are mixed with a population of larger cells with nucleoli (centroblasts).

Diffuse Large B Cell Lymphoma
• most common type of lymphoma in adults, accounting for approximately 50% of adult NHLs, also arises in children
• Arises de novo, as a transformation from low grade B-cell lymphoma, in the setting of chronic immune stimulation
• High-grade lymphoma, progressive and fatal if not treated
• 80% of patients achieve complete remission after treatment with chemotherapy
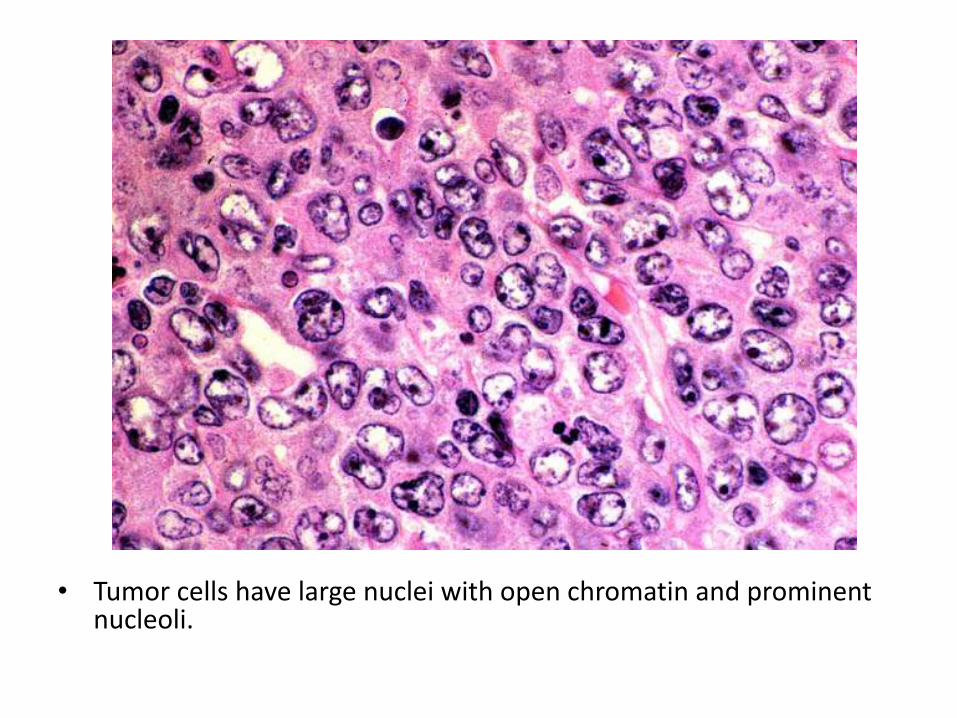
• Tumor cells have large nuclei with open chromatin and prominent nucleoli.

Burkitt lymphoma
• High-grade B-cell lymphoma
• Endemic in Africa, sporadic worldwide
• High association with EBV
• t(8:14), myc gene fuses with IgH gene, causing overexpression of myc, which activates other transcription factors and causes continuous cell proliferation
• Lymphoma commonly arises in extranodal sites (jaw, ileum)
• Lymphoma is rapidly growing and fatal if not treated
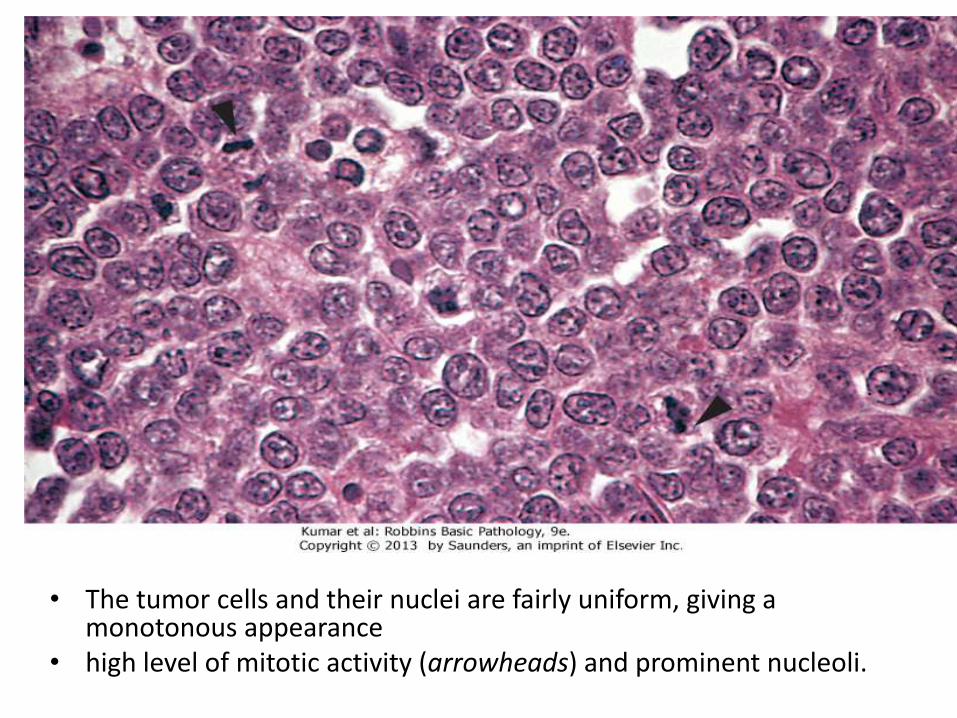
• The tumor cells and their nuclei are fairly uniform, giving a monotonous appearance
• high level of mitotic activity (arrowheads) and prominent nucleoli.
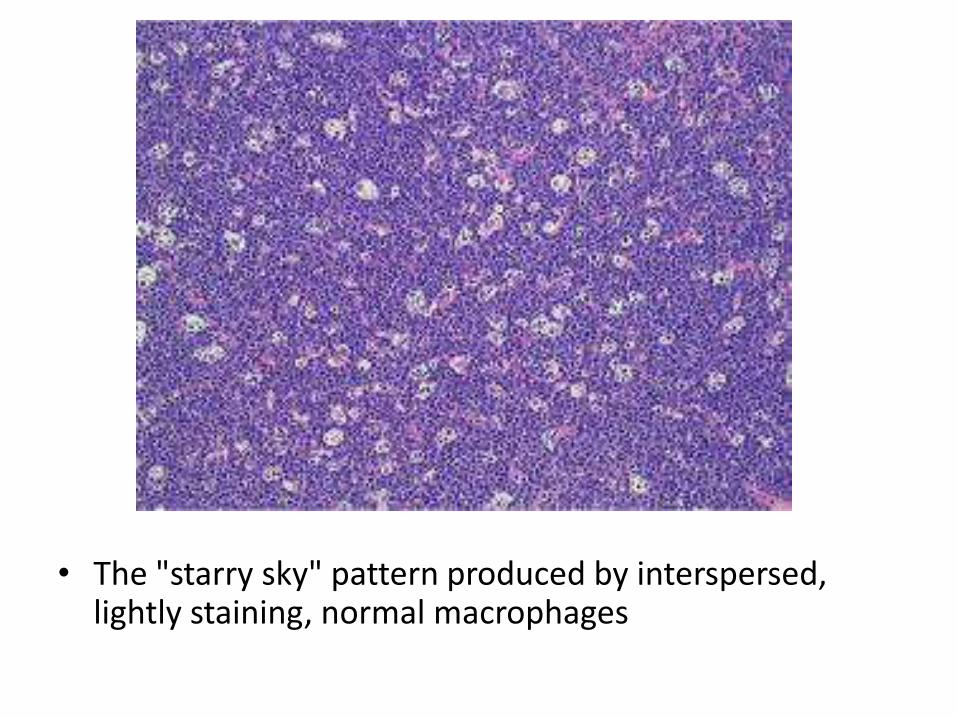
• The "starry sky" pattern produced by interspersed, lightly staining, normal macrophages

Hodgkin Lymphoma
• a group of lymphoid neoplasms that differ from NHL in several respects
• More often localized to a single axial group of nodes (cervical, mediastinal, para-aortic)
• Orderly spread by contiguity
• Mesenteric nodes and Waldeyer ring rarely involved
• Extra-nodal presentation rare

Hodgkin Lymphoma
• It is characterized by the presence of neoplasticgiant cells called Reed-Sternberg cells
• RS cells constitute only a minority of tumor size, the rest is composed of reactive lymphocytes, histiocytes and granulocytes
• neoplastic RS cells are derived from crippled, germinal center or post-germinal center, B cells
• Immunophenotype is very different from normal B-cells
• EBV plays a role in the evolution of disease

Clinical features
• Common in children and young adults
• Presents as painless lymphadenopathy
• Constitutional symptoms (B-symptoms), such as fever, night sweats, and weight loss are common
• Spread: nodal disease first, then splenic disease, hepatic disease, and finally involvement of the marrow and other tissues
• Radiation therapy is effective in early phases, then chemotherapy
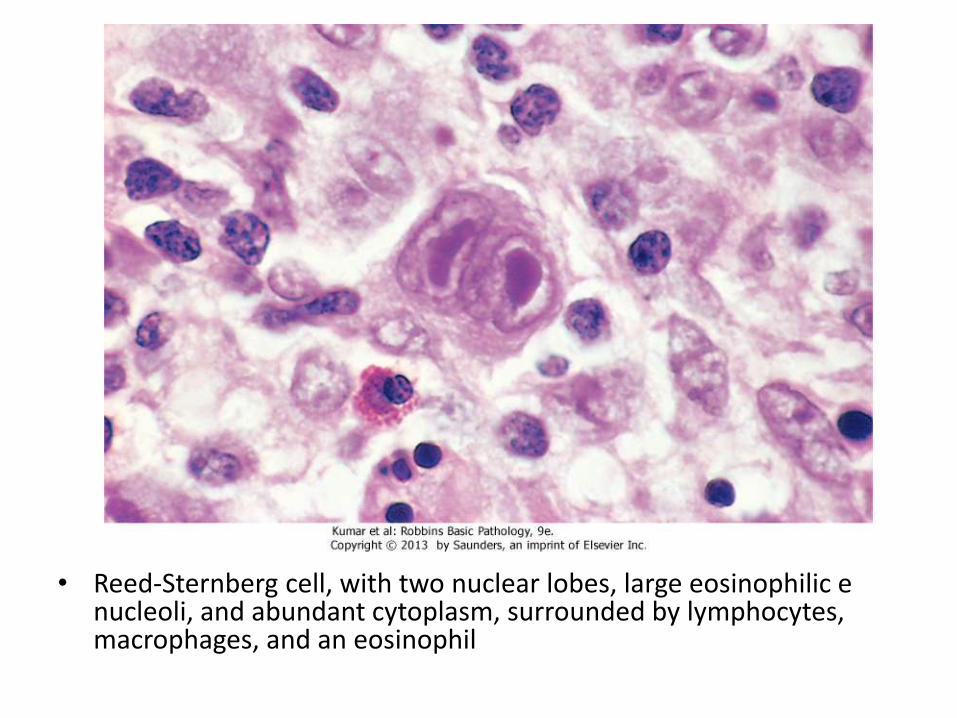
• Reed-Sternberg cell, with two nuclear lobes, large eosinophilic e nucleoli, and abundant cytoplasm, surrounded by lymphocytes, macrophages, and an eosinophil

Plasma cell myeloma
• Neoplasm of plasma cells that secrets monoclonal Immunoglobulin (M-protein)or part of it
• Aggressive tumor, difficult to control
• Affects elderly
• Commonly associated with renal failure, hypercalcemia, osteosclerosis, BM failure
• Clinically known as multiple myeloma

M-protein
• unlike normal plasma cells, in which the production and coupling of heavy and light chains are tightly balanced, neoplastic plasma cells often synthesize excess light or heavy chains along with complete Igs
• Occasionally only light chains or heavy chains are produced • The most common Ig is IgG, then IgA• The free light chains are small enough to be excreted in the
urine, where they are called Bence-Jones proteins• M-protein causes RBCs to stick together, appearing as a
stack of coins (Rouleuax formation)• Sometimes, light chain structures are deposited in tissues
as “amyloid”
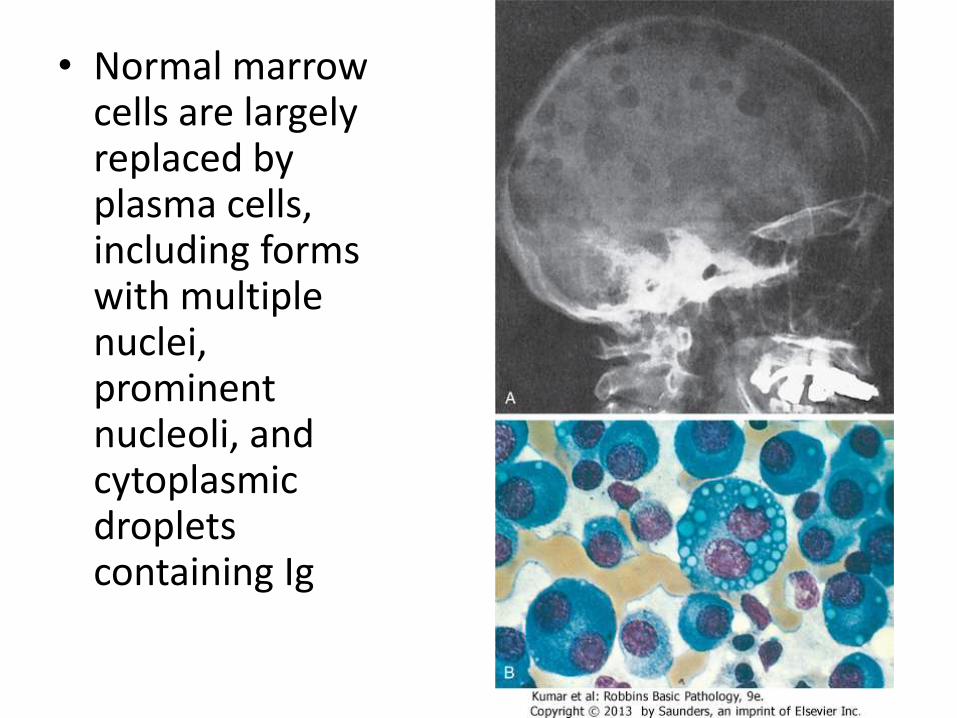
• Normal marrow cells are largely replaced by plasma cells, including forms with multiple nuclei, prominent nucleoli, and cytoplasmicdroplets containing Ig

Myeloid neoplasms

Myeloid neoplasms
• Arise from hematopoietic progenitor cells, capable of differentiation to granulocytic, erythrocytic or megakaryocytic lineages
• These diseases primarily involve the marrow and to a lesser degree the secondary hematopoietic organs (the spleen, liver, and lymph nodes), and usually present with symptoms related to altered hematopoiesis
• 3 types: acute myeloid leukemia, myelodysplasticsyndrome, myeloproliferative neoplasm

Acute myeloid leukemia
• caused by acquired oncogenic mutations that impede differentiation, and increases proliferation, leading to the accumulation of immature myeloid blasts in the marrow
• Accumulated blasts leads to marrow failure and complications related to anemia, thrombocytopenia, and neutropenia.
• AML occurs at all ages, but the incidence rises throughout life

Classifications
• WHO Classification
1) AML-recurrent cytogenetic abnormality
2) AML-Myelodysplasia related changes (complicates MDS)
3) Therapy-related myeloid neoplasm
4) AML- not otherwise specified

Older Classification (FAB)
• M0: AML with minimal differentiation
• M1: AML without maturation
• M2: AML with maturation
• M3: Acute promyelocytic leukemia
• M4: Acute myelomonocytic leukemia
• M5: Acute monocytic leukemia
• M6: Acute erythrocytic leukemia
• M7: Acute megakaryocytic leukemia

AML-recurrent cytogenetic abnormality
• AML with t(8;21); Full range of myelocyticmaturation (M2); Auer rods easily found; abnormal cytoplasmic granules, favorable Px
• AML with inv(16); Myelocytic and monocyticdifferentiation; abnormal eosinophilicprecursors with abnormal basophilic granules (M4eos), favorable Px
• AML with t(15;17); promyelocytic proliferation (M3), intermediate Px

Morphology
• BM shows 20% of more blasts Myeloblasts have delicate nuclear chromatin, two to four nucleoli, and more voluminous cytoplasm than lymphoblasts
• Auer rods: distinctive needle-like azurophilicgranules, sometimes seen
• Blasts commonly appear in peripheral blood
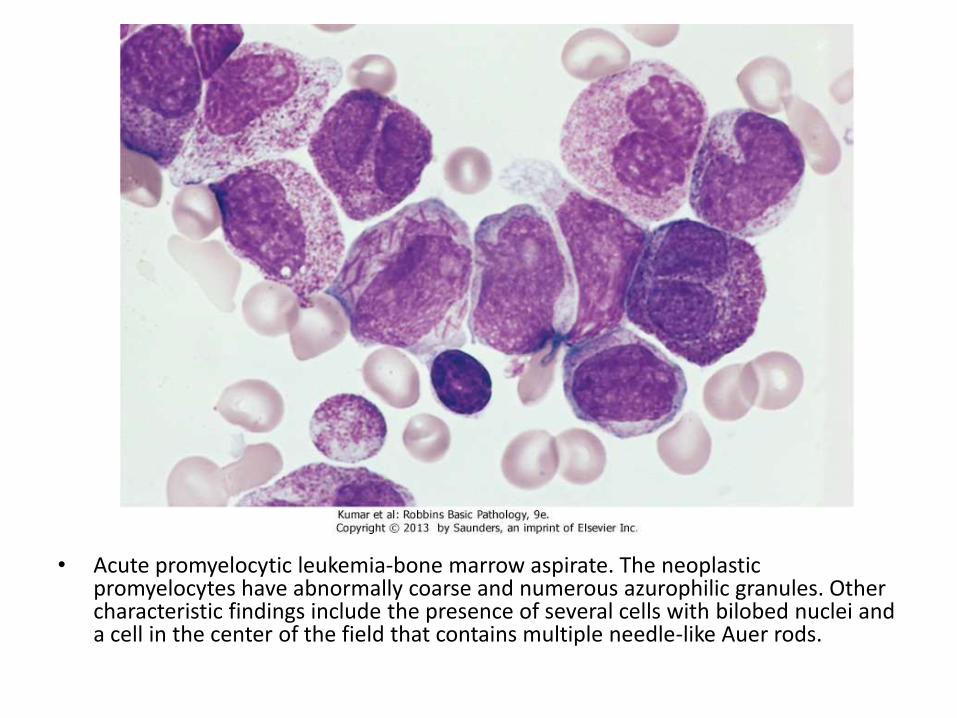
• Acute promyelocytic leukemia-bone marrow aspirate. The neoplasticpromyelocytes have abnormally coarse and numerous azurophilic granules. Other characteristic findings include the presence of several cells with bilobed nuclei and a cell in the center of the field that contains multiple needle-like Auer rods.

Clinical features
• Patients present shortly after developing fever, malaise, fatigue, bleeding
• Procoagulants and fibrinolytic factors released by leukemic cells, especially in AML with the t(15;17), exacerbate the bleeding tendency
• Infection is common
• AML is a difficult disease to treat. About 60% of patients achieve complete remission with chemotherapy, but only 15% to 30% remain free of disease for 5 years

Myelodysplastic Syndromes
• Group of clonal stem cell disorders characterized by maturation defects that are associated with ineffective hematopoiesis and a high risk of transformation to AML
• The clone retains the capacity to differentiate but does so in an ineffective and disordered fashion
• These abnormal cells stay within the bone marrow and hence the patients have peripheral blood cytopenias
• The hallmark of MDS is persistent peripheral cytopenia and BM morphologic dysplasia

MDS
• MDS may be either primary (idiopathic) or secondary to previous genotoxic drug or radiation therapy (t-MDS)
• t-MDS usually appears from 2 to 8 years after the genotoxic exposure
• All forms of MDS can transform to AML, but transformation occurs with highest frequency and most rapidly in t-MDS
• Cytogenetic analysis commonly reveals chromosomal aberrations

Types of MDS
• 1) Refractory cytopenia with unilineage dysplasia• 2) Refractory cytopenia with multilineage
dysplasiaThere is persistant cytopenia in 1 or more lines,
accompanies by BM dysplasia in the same or more lines (e.g anemia, with dysplaia in both erythroid and myeloid precursors). The number of blasts in BM is <5% of all cells
Anemia is the most common form of cytopenia, while isolated neutropenia or thrombocytopenia are rare
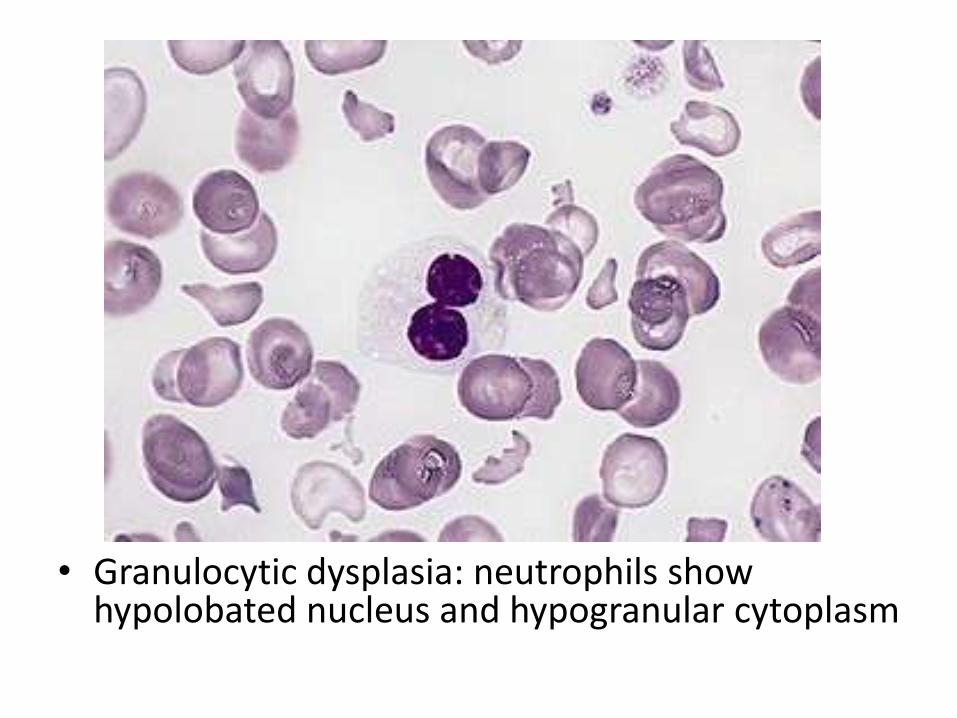
• Granulocytic dysplasia: neutrophils show hypolobated nucleus and hypogranular cytoplasm
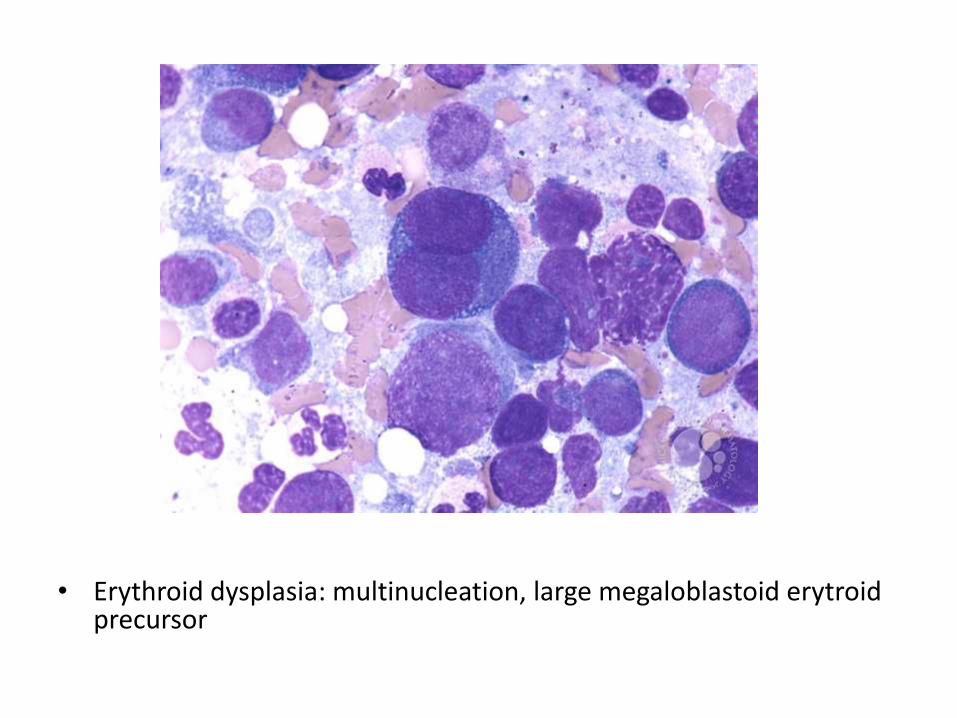
• Erythroid dysplasia: multinucleation, large megaloblastoid erytroidprecursor
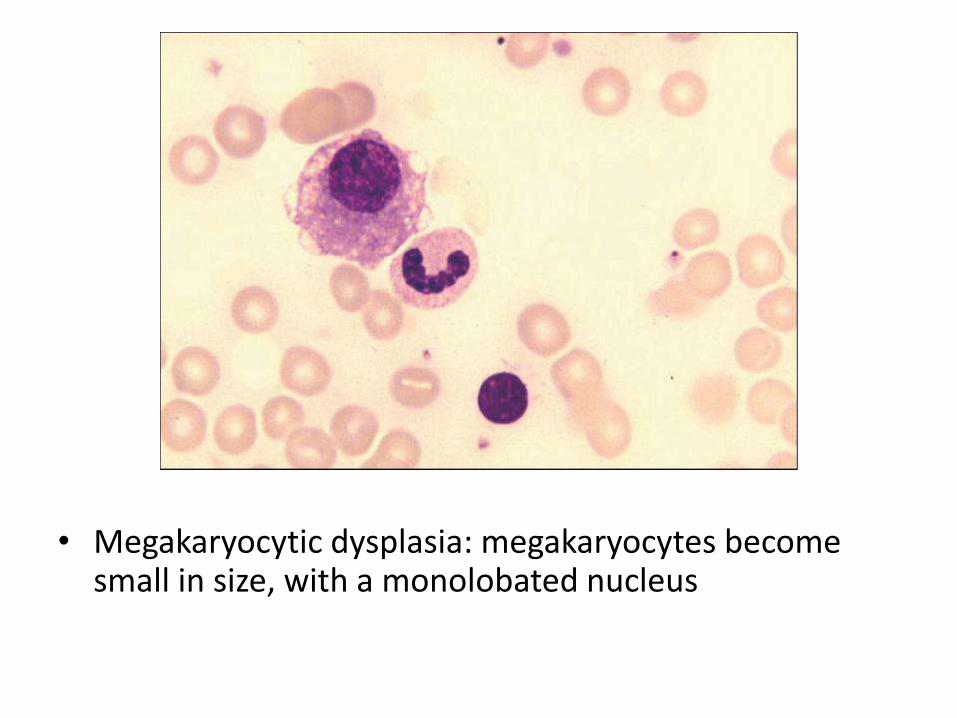
• Megakaryocytic dysplasia: megakaryocytes become small in size, with a monolobated nucleus

Types of MDS
• 3) Refractory anemia with ring sideroblasts
• There is persistent isolated anemia. Erythroidprecursors show a ring of iron around the nucleus. Results from abnormal iron accumulation in the mitochondria
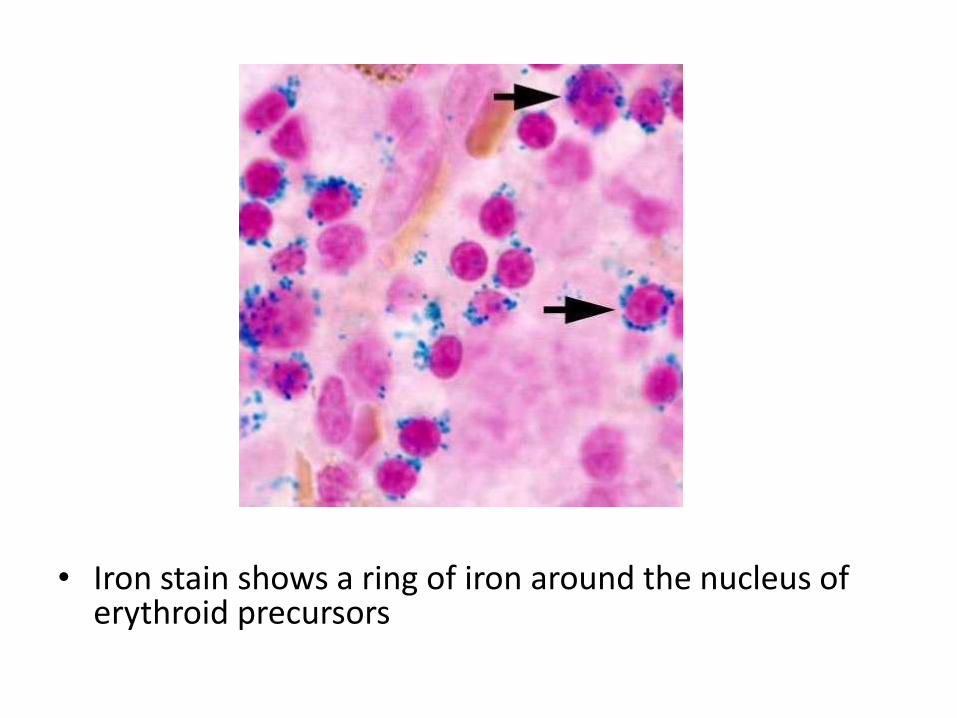
• Iron stain shows a ring of iron around the nucleus of erythroid precursors

Types of MDS
• Refractory Anemia with Excess Blasts
• Any type of MDS, with increased BM blasts between 5-20% of all cells
• Heralds progression to AML

Myeloproliferative neoplasms
• Chronic myeloproliferative disorders are marked by the hyperproliferation of neoplastic myeloid progenitors that retain the capacity for terminal differentiation
• There is a persistent increase in one or more formed elements of the peripheral blood
• The neoplastic progenitors tend to seed secondary hematopoietic organs (the spleen, liver, and lymph nodes), resulting in hepatosplenomegaly (caused by neoplasticextramedullary hematopoiesis)

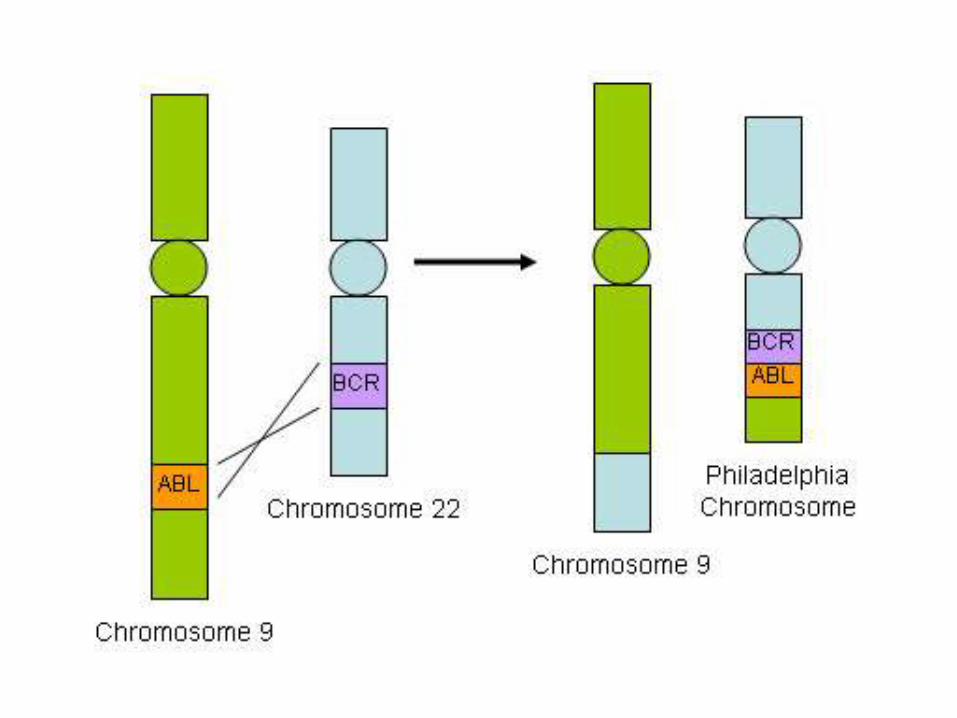
Chronic Myelogenous Leukemia
• a balanced (9;22) translocation that moves ABL from chromosome 9 to a position on chromosome 22 adjacent to BCR
• The new chr22 is known as Pheladelphiachromosome
• The BCR-ABL fusion gene has a tyrosine kinaseactivity, stimulating the proliferation and prolonged survival of granulocytic and megakaryocytic cells

morphology
• Peripheral blood shows markedly increased WBC count, sometimes exceeding 100,000 cell/uL
• Most of the cells are neutrophils, metamyelocytes and myelocytes
• Basophils and eosinophils are also increased• The bone marrow is hypercellular owing to
increased numbers of granulocytic and megakaryocytic precursors
• Spleen is enlarged with extramedullaryhematopoiesis
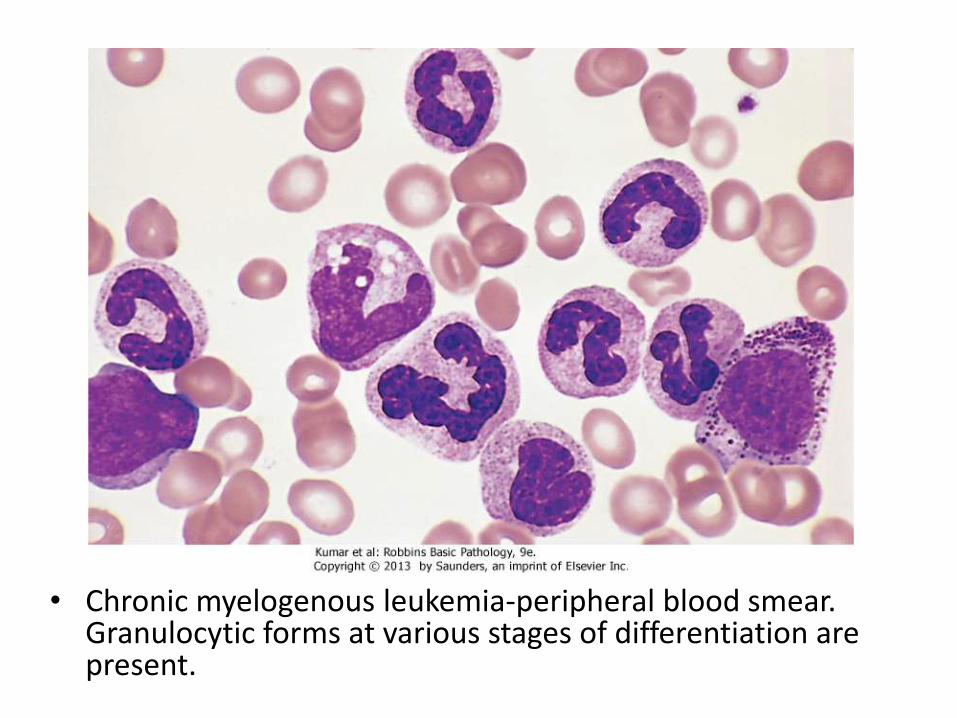
• Chronic myelogenous leukemia-peripheral blood smear. Granulocytic forms at various stages of differentiation are present.

Polycythemia Vera
• P vera is characterized by an excessive proliferation of erythroid, granulocytic, and megakaryocytic elements (panmyelosis), but most clinical signs and symptoms are related to an absolute increase in red cell mass
• P vera must be distinguished from relative polycythemia, which results from hemoconcentration
• Unlike reactive forms of absolute polycythemia, p verais associated with low levels of serum erythropoietin, which is a reflection of the growth factor-independent growth of the neoplastic clone

Pathophysiology
• Caused by a mutation in JAK2 gene, which sharply lowers the dependence of hematopoietic cells on growth factors for growth and survival, erythroidprecursors are more sensitive than other cells
• Marked erythrocytosis results in increased viscosity and vascular stasis, thromboses and infarctions
• Spleen and liver are enlarged• There is plethora and cyanosis in every tissue• Bone marrow is hypercellular• Disease progression occurs late, which transforms into
AML

Primary Myelofibrosis
• Brief period of granulopoiesis and megakaryopoiesis, rapidly followed my BM fibrosis and elemenation of hematopoietic elements
• The fibroblast proliferation is stimulated by platelet-derived growth factor and transforming growth factor β released from neoplasticmegakaryocytes
• Hematopoiesis takes place in spleen and liver• RBC’s escaping the fibotic stroma in BM are
deformed and take the shape of “tear-drops”

Morphology
• BM is initially hypercellular with increased megakaryocytes
• Later in disease, become fibrotic and hypocellular
• Peripheral blood shows thrombocytosis, nucleated RBCs, immature granulocytes and tear-drop cells
• Spleen shows marked extramedullaryhematopoiesis
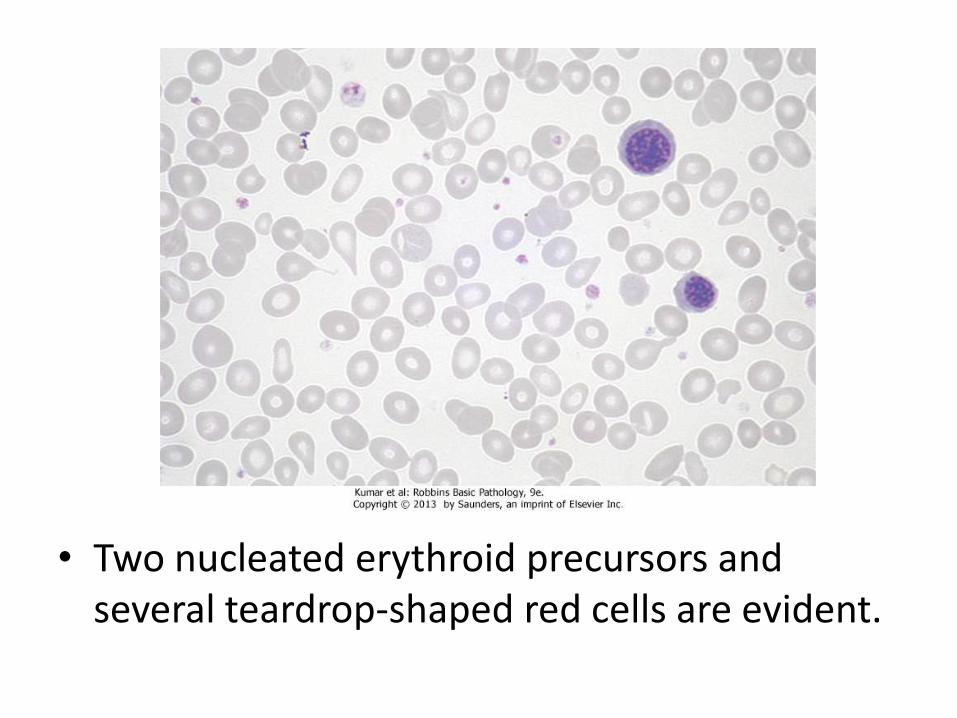
• Two nucleated erythroid precursors and several teardrop-shaped red cells are evident.


Coagulation disorders

Types
• Congenital or acquired
• Acquired deficiencies are most common and often involve several factors simultaneously.
• Vitamin K is required for the synthesis of prothrombin and clotting factors VII, IX, and X, and its deficiency causes a severe coagulation defect
• The liver synthesizes several coagulation factors and also removes many activated coagulation factors from the circulation; thus, hepatic parenchymaldiseases are common causes of complex hemorrhagic diatheses
• DIC also may lead to multiple concomitant factor deficiencies
• Rarely, autoantibodies may cause acquired deficiencies limited to a single factor
• Hereditary deficiencies of each of the coagulation factors have been identified
• Hemophilia A (a deficiency of factor VIII) and hemophilia B (Christmas disease, a deficiency of factor IX) are X-linked traits, whereas most deficiencies are autosomal recessive disorders
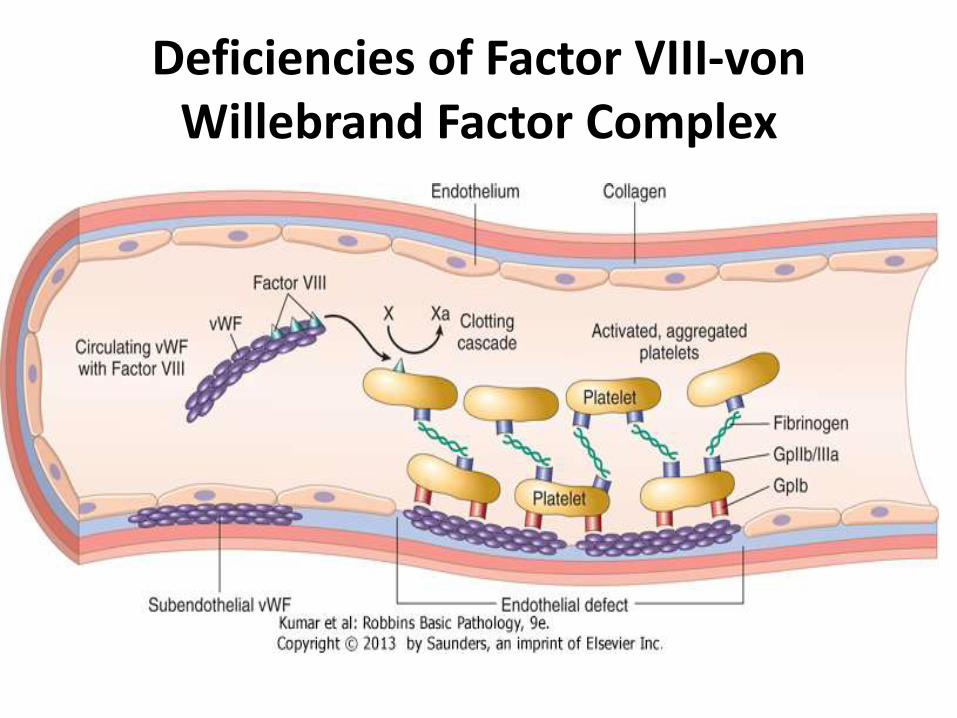
Deficiencies of Factor VIII-von Willebrand Factor Complex

• factor VIII is an essential cofactor for factor IX, which activates factor X in the intrinsic coagulation pathway
• Circulating factor VIII binds noncovalently to vWF
• Endothelial cells are the major source of plasma vWF, whereas most factor VIII is synthesized in the liver
• vWF is found in the plasma (in association with factor VIII), in platelet granules, in endothelial cells within cytoplasmic vesicles called Weibel-Palade bodies, and in the subendothelium, where it binds to collagen

• When endothelial cells are stripped away by trauma or injury, subendothelial vWF is exposed and binds to platelets, mainly through glycoprotein Ib and to a lesser degree through glycoprotein IIb/IIIa
• The most important function of vWF is to facilitate the adhesion of platelets to damaged blood vessel walls, a crucial early event in the formation of a hemostatic plug
• Inadequate platelet adhesion is believed to underlie the bleeding tendency in von Willebrand disease
• In addition to its role in platelet adhesion, vWF also stabilizes factor VIII; thus, vWF deficiency leads to a secondary deficiency of factor VIII.

von Willebrand Disease
• Autosomal dominant disorder • It usually presents as spontaneous bleeding from mucous membranes, excessive bleeding from
wounds, and menorrhagia• the most common inherited bleeding disorder, usually mild disease• People with von Willebrand disease have compound defects in platelet function and coagulation,
but in most cases only the platelet defect produces clinical findings • The classic and most common variant of von Willebrand disease (type I) is an autosomal dominant
disorder in which the quantity of circulating vWF is reduced but clinically insignificant • Type II is characterized by the selective loss of high-molecular-weight multimers of vWF resulting in
functional deficiency of vWF. In type IIA, the high-molecular-weight multimers are not synthesized, leading to a true deficiency. In type IIB, abnormal "hyperfunctional" high-molecular-weight multimers are synthesized that are rapidly removed from the circulation
• These high-molecular-weight multimers cause spontaneous platelet aggregation (a situation reminiscent of the very-high-molecular-weight multimer aggregates seen in TTP); indeed, some people with type IIB von Willebrand disease have mild chronic thrombocytopenia, presumably due to platelet consumption.

Hemophilia A-Factor VIII Deficiency
• X-linked recessive disorder • Approximately 30% of cases are caused by new mutations;
in the remainder, there is a positive family history • Severe hemophilia A is observed in people with marked
deficiencies of factor VIII (activity levels less than 1% of normal)
• Milder deficiencies may only become apparent when other predisposing conditions, such as trauma, are also present
• In about 10% of patients, the factor VIII concentration is normal by immunoassay, but the coagulant activity is low because of a mutation in factor VIII that causes a loss of function.

Clinical features
• In symptomatic cases there is a tendency toward easy bruising and massive hemorrhage after trauma or operative procedures
• In addition, "spontaneous" hemorrhages frequently are encountered in tissues that normally are subject to mechanical stress, particularly the joints, where recurrent bleeds (hemarthroses) lead to progressive deformities that can be crippling
• Petechiae are characteristically absent
• Typically, patients with hemophilia A have a prolonged PTT that is corrected by mixing the patient's plasma with normal plasma
• Specific factor assays are then used to confirm the deficiency of factor VIII
• In approximately 15% of those with severe hemophilia A replacement therapy is complicated by the development of neutralizing antibodies against factor VIII, probably because factor VIII is seen by the immune system as a "foreign" antigen. In these persons, the PTT fails to correct in mixing studies

Hemophilia B-Factor IX Deficiency
• X-linked disorder that is indistinguishable clinically from hemophilia A but much less common
• The PTT is prolonged. The diagnosis is made using specific assays of factor IX. It is treated by infusion of recombinant factor IX.

DISSEMINATED INTRAVASCULAR COAGULATION
• Systemic activation of coagulation and results in the formation of thrombi throughout the microcirculation
• As a consequence, platelets and coagulation factors are consumed and, secondarily, fibrinolysis is activated
• Thus, DIC can give rise to either tissue hypoxia and microinfarcts caused by myriad microthrombi or to a bleeding disorder related to pathologic activation of fibrinolysis and the depletion of the elements required for hemostasis (hence the term consumptive coagulopathy)

Pathophysiology
• Clotting is initiated by either the extrinsic pathway, which is triggered by the release of tissue factor (tissue thromboplastin); or the intrinsic pathway, which involves the activation of factor XII by surface contact, collagen, or other negatively charged substances
• Both pathways lead to the generation of thrombin Clotting normally is limited by the rapid clearance of activated clotting factors by the macrophages and the liver, endogenous anticoagulants (e.g., protein C), and the concomitant activation of fibrinolysis

Pathophysiology
• DIC usually is triggered by either (1) the release of tissue factor or thromboplastic substances into the circulation or (2) widespread endothelial cell damage
• Thromboplastic substances can be released into the circulation from a variety of sources-for example, the placenta in obstetric complications or certain types of cancer cells, particularly those of acute promyelocytic leukemia and adenocarcinomas
• In gram-negative and gram-positive sepsis (important causes of DIC), endotoxins or exotoxins stimulate the release of tissue factor from monocytes
• The net result of these alterations is the enhanced generation of thrombin and the blunting of inhibitory pathways that limit coagulation

Pathophysiology
• Severe endothelial cell injury can initiate DIC by causing the release of tissue factor and by exposing subendothelial collagen and von Willebrandfactor (vWF)
• Widespread endothelial injury can be produced by the deposition of antigen-antibody complexes (e.g., in systemic lupus erythematosus), by temperature extremes (e.g., after heat stroke or burn injury), or by infections (e.g., due to meningococci or rickettsiae)
• DIC is most often associated with sepsis, obstetric complications, malignancy, and major trauma (especially trauma to the brain)
• The initiating events in these conditions are multiple and complex. For example, in obstetric conditions, tissue factor derived from the placenta, retained dead fetus, or amniotic fluid enters the circulation; however, shock, hypoxia, and acidosis often coexist and can lead to widespread endothelial injury
• Trauma to the brain releases fat and phospholipids, which act as contact factors and thereby activate the intrinsic pathway

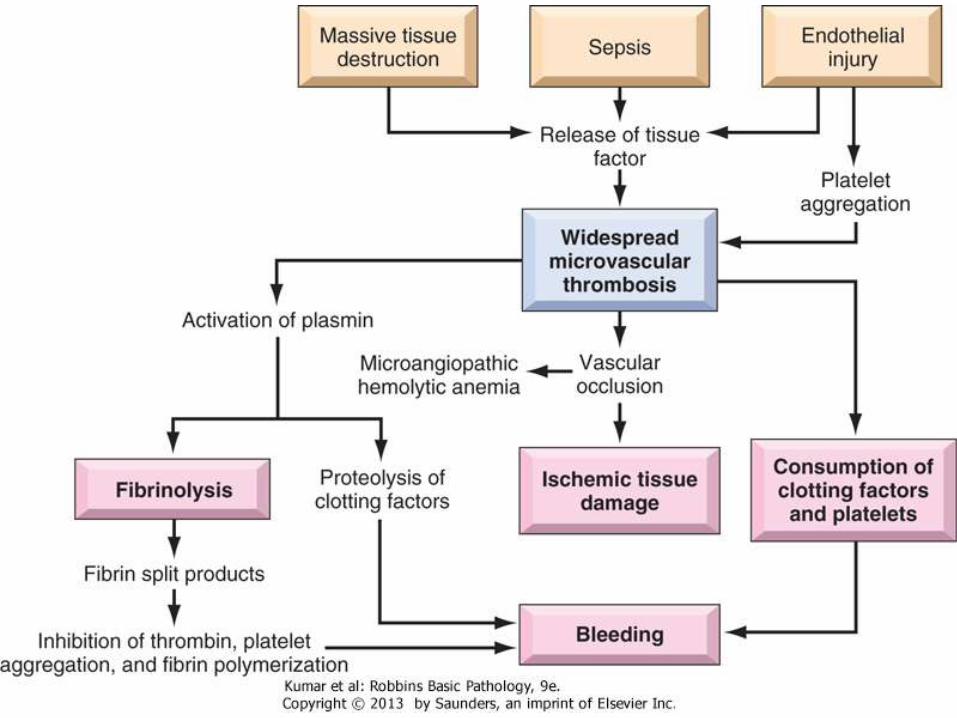
Pathophysiology
• Whatever the pathogenetic mechanism, DIC has two consequences. First, there is widespread fibrin deposition within the microcirculation, causing obstruction and ischemia
• Red cells are traumatized while passing through vessels narrowed by fibrin thrombi (microangiopathic hemolytic anemia)
• Second, a bleeding diathesis results from the depletion of platelets and clotting factors and the secondary release of plasminogenactivators
• Plasmin cleaves not only fibrin (fibrinolysis) but also factors V and VIII, thereby reducing their concentration further
• In addition, fibrinolysis creates fibrin degradation products. These inhibit platelet aggregation, have antithrombin activity, and impair fibrin polymerization, all of which contribute to the hemostaticfailure


Morphology
• In DIC microthrombi are most often found in the arterioles and capillaries of the kidneys, adrenals, brain, and heart, but no organ is spared
• The glomeruli contain small fibrin thrombi. These may be associated with only a subtle, reactive swelling of the endothelial cells or varying degrees of focal glomerulitis
• The microvascular occlusions give rise to small infarcts in the renal cortex. In severe cases the ischemia can destroy the entire cortex and cause bilateral renal cortical necrosis
• Microinfarcts also are commonly encountered in the brain and are often surrounded by microscopic or gross foci of hemorrhage. These can give rise to bizarre neurologic signs

Clinical features
• As might be imagined, depending on the balance between clotting and bleeding tendencies, the range of possible clinical manifestations is enormous
• In general, acute DIC (e.g., that associated with obstetric complications) is dominated by a bleeding diathesis, whereas chronic DIC (e.g., as occurs in those with cancer) tends to manifest with signs and symptoms related to thrombosis
• The abnormal clotting usually is confined to the microcirculation, but large vessels are involved on occasion
• The manifestations may be minimal, or there may be shock, with acute renal failure, dyspnea, cyanosis, convulsions, and coma
• Most often, attention is called to the presence of DIC by prolonged and copious postpartum bleeding or by the presence of petechiae and ecchymoses on the skin
• Laboratory evaluation reveals thrombocytopenia and prolongation of the PT and the PTT
• Fibrin split products are increased in the plasma
• The prognosis varies widely depending on the nature of the underlying disorder and the severity of the intravascular clotting and fibrinolysis
• Acute DIC can be life-threatening and must be treated aggressively fresh frozen plasma. Conversely, chronic DIC is sometimes identified unexpectedly by laboratory testing. In either circumstance, definitive treatment must be directed at the underlying cause.

THROMBOCYTOPENIA
• Isolated thrombocytopenia is associated with a bleeding tendency and normal coagulation tests
• A count less than 150,000 platelets/μL generally is considered to constitute thrombocytopenia. However, only when platelet counts fall to 20,000 to 50,000 platelets/μL is there an increased risk of post-traumatic bleeding
• Most bleeding occurs from small, superficial blood vessels and produces petechiae or large ecchymoses in the skin, the mucous membranes of the gastrointestinal and urinary tracts, and other sites. Larger hemorrhages into the central nervous system are a major hazard in those with markedly depressed platelet counts
• Results from either decreased production or increased consumption/destruction

Immune Thrombocytopenic Purpura
• Immune thrombocytopenic purpura (ITP) has two clinical subtypes. Chronic ITP is a relatively common disorder that tends to affect women between the ages of 20 and 40 years. Acute ITP is a self-limited form seen mostly in children after viral infections
• Antibodies directed against platelet membrane glycoproteins IIb/IIIa or Ib/IX complexes can be detected in roughly 80% of cases of chronic ITP
• The spleen is an important site of antiplatelet antibody production and the major site of destruction of the IgG-coated platelets
• Although splenomegaly is not a feature of uncomplicated chronic ITP, the importance of the spleen in the premature destruction of platelets is proved by the benefits of splenectomy, which normalizes the platelet count and induces a complete remission in more than two thirds of patients
• The bone marrow usually contains increased numbers of megakaryocytes, a finding common to all forms of thrombocytopenia caused by accelerated platelet destruction
• The onset of chronic ITP is insidious. Common findings include petechiae, easy bruising, epistaxis, gum bleeding, and hemorrhages after minor trauma. Fortunately, more serious intracerebral or subarachnoid hemorrhages are uncommon
• The diagnosis rests on the clinical features, the presence of thrombocytopenia, examination of the marrow, and the exclusion of secondary ITP

Thrombotic Thrombocytopenic Purpura
• TTP is associated with the pentad of fever, thrombocytopenia, microangiopathic hemolytic anemia, transient neurologic deficits, and renal failure
• A similar disease, Hemolytic Uremic Syndrome, has similar manifestations but is distinguished from TTP by the absence of neurologic symptoms, the dominance of acute renal failure, and frequent occurrence in children after infection with E.Coli
• In both, there is a widespread formation of platelet-rich thrombi in the microcirculation. The consumption of platelets leads to thrombocytopenia, and the narrowing of blood vessels by the platelet-rich thrombi results in a microangiopathic hemolytic anemia

Pathophysiology
• Deficiency in ADAMTS 13, a metalloprotease enzyme that degrades very-high-molecular-weight multimers of von Willebrand factor (vWF)
• Abnormal large vWF multimers accumulate in plasma
• Under some circumstances, these giant vWF multimers promote the formation of platelet microaggregates throughout the circulation
• The superimposition of an endothelial cell injury (caused by some other condition) can further promote microaggregateformation, thus initiating or exacerbating clinically evident TTP
• ADAMTS 13 deficiency can be inherited or acquired, the latter by way of autoantibodies that bind and inhibit the metalloprotease

Pathophysiology
• Although DIC, TTP and HUS share features such as microvascular occlusion and microangiopathichemolytic anemia, they are pathogenically distinct
• Unlike in DIC, in TTP and HUS activation of the coagulation cascade is not of primary importance, so results of laboratory tests of coagulation (such as the PT and the PTT) usually are normal
• TTP must be considered in any patient with unexplained thrombocytopenia and microangiopathichemolytic anemia, as any delay in diagnosis can be fatal