amyloid-targeted therapeutics in alzheimer's disease: use ... · alzheimer’s disease...
TRANSCRIPT

by Mercè Boada, Pilar Ortiz, FernandoAnaya, Isabel Hernández, Joan Muñoz,Laura Núñez, Javier Olazarán, IsabelRoca, Gemma Cuberas, Lluís Tárraga,Mar Buendia, Ramón P. Pla, Isidre Ferrerand Antonio Páez
CURRENT CONCEPTS ABOUTALZHEIMER’S DISEASE
Symptoms, epidemiology and diagnosis Alzheimer’s disease (AD) is a chronic, pro-gressive and ultimately fatal neurodegener-ative disorder in which normal thinking andmemory appear to be disrupted, probablydue to impaired or blocked transmission ofcomplex messages between brain cells.Symptoms that characterize AD can begrouped by cognitive dysfunction symptoms(memory loss, language difficulties,impaired intellectual and coordinationskills), psychiatric symptoms (depression,hallucinations, delusions, agitation) and aseries of symptoms associated with difficul-ties in performing daily life activities such asshopping, driving and, in severe cases,dressing and eating unaided.
AD primarily affects the elderly. About 6% ofpeople aged over 65 are affected1 and AD isthe most common cause of dementia in thispopulation (50–70%), followed by vascular
dementia (30–40%), and mixed dementia(15–20%). It is estimated that 24.3 millionpeople have dementia today worldwide,with 4.6 million new cases of dementiaevery year.1 The prevalence of dementiaincreases exponentially from approximately1% at 60–65 years of age to more than30–35% in people older than 80 years. Thedirect and indirect costs of AD and otherdementias are enormous, with the world-wide average annual cost per person withdementia estimated to be USD 10,700 in2005.2
Late-onset AD is considered a complex dis-order in which multiple genetic and non-genetic factors must work together to pro-duce the clinical phenotype. APOEε4 alleleis the only well-established major geneticrisk factor involved in late-onset AD.3
Carriers of one APOEε4 copy have a 2- to 4-fold risk of developing AD as compared withnoncarriers, and APOEε4 homozygotesmultiply their AD risk by 154. In addition toAPOE gene, there may be other loci thatcould be associated with an increased ordecreased risk; however, the true effects ofthese loci remain controversial as manyreported associations are not subsequentlyconfirmed in other studies. Reduced samplesizes, different recruitment sources andstrategies or diverse inclusion or exclusioncriteria are methodological problems inher-
Drug News Perspect 22(6), July/August 2009
325Copyright © 2009 Prous Science, S.A.U. or its licensors. All rights reserved. CCC: 0214-0934/2009. DOI: 10.1358/dnp.2009.22.6.1395256
LOOKING AHEAD
AMYLOID-TARGETEDTHERAPEUTICS IN ALZHEIMER'SDISEASE: USE OF HUMANALBUMIN IN PLASMAEXCHANGE AS A NOVELAPPROACH FOR AβMOBILIZATION
The fact that 90% ofcirculating Aβ is boundto albumin led to thehypothesis that ifendogenous albuminwere replaced through aplasma exchangeschedule, the existingdynamic equilibrium setbetween the CSF andplasma Aβ may bealtered.
SUMMARY
A clinical investigation program wascarried out to replace endogenousalbumin of patients with mild to mod-erate Alzheimer’s disease (AD) with 5%Human Albumin Grifols® through aplasma exchange (PE) schedule, inorder to alter the dynamic equilibriumbetween albumin-bound Aβ in plasmaand Aβ in cerebrospinal fluid. In a pilotproof-of-concept study, 7 patientsunderwent 6 PE in 3 weeks and 1 yearof follow-up. Plasma Aβ determina-tions demonstrated a variation patternin levels in relation with the PEs.Cognitive status scores (MMSE andADAS-Cog) were more stable thanexpected. In a phase II clinical trial, 29patients were randomized into PE-treated and control groups with 1 yearfollow-up. Interim results point towardthe occurrence of Aβ40 mobilization inthe PE-treated patients, who scoredbetter in cognitive tests (differences at9 months: 2.5 in MMSE and 5.5 inADAS-cog). These results suggest thata PE program with 5% Human AlbuminGrifols may have a promising role in thetreatment of mild to moderate AD.
Correspondence: M. Boada, [email protected]

ent to case-control genetic analyses thatmay explain part of the controversies.4
Early diagnosis of AD has become increas-ingly important as disease-modifyingapproaches to treatment are being devel-oped. Although the only definitive diagnosisof AD can be made via brain biopsy orautopsy, there currently are diagnostic crite-ria allowing standardization of the diagnos-tic process for physicians. Such criteriainclude clinical observation of symptoms,neurologic examination, and results fromdiagnostic tests such as memory screeningand psychometric tests, neuroimaging, andcerebrospinal and other fluid markersassessments.5-7
Pathophysiology and molecularmechanismsNeuropathological characteristics of ADinclude the presence of extracellular neurit-ic plaques and intraneuronal neurofibrillarytangles in areas of the brain parenchymainvolved in memory and/or in brain vessels,predominantly in the amygdala, hippocam-pus and neocortex.8 β-Amyloid peptide (Aβ)is the proteinaceous component of the amy-loid fibrillar deposits that are usually pres-ent in neuritic plaques. Neurofibrillary tan-gles are composed of paired helical fila-ments of which hyperphosphorylated tauproteins form the primary component.These lesions are found in nerve cell bodiesand in apical dendrites provoking cytoskele-tal changes in AD-affected neurons.Although there is an inter-relation and asynergetic effect between Aβ aggregationand the propagation of tau pathology,9
observations from autopsied AD brains indi-cate that plaques precede tangles. It is cur-rently accepted that Aβ production anddeposition are central to the pathogenesisof AD,10 although whether Aβ is the ultimatecause is still under debate.
The presence of amyloid plaques is associ-ated with neurotoxic events, oxidative stressand neuroinflammatory reactions.11,12
However, it is still unclear whether Aβ neu-rotoxicity is a preliminary cause or rather alate event in the pathophysiology of AD.Affected neurons become dysfunctional,show synaptic and dendritic desarboriza-tion, have reduced levels of neurotransmit-ters, and finally undergo neuronal apopto-sis.13,14
The amyloid cascade: Aβ aggregationand neuritic plaquesThe amyloid cascade hypothesis is one ofthe several hypotheses that nowadays try toexplain the pathogenesis observed in AD.10
According to this hypothesis, pathologicmetabolism of β-amyloid precursor protein(Aβ PP or APP), the originator of the Aβ pep-tide,15 is the initiating event, subsequentlyleading to the aggregation of Aβ to formneuritic plaques, which would favor the for-mation of neurofibrillary tangles, loss ofsynaptic connections, death of tangle-bear-ing neurons and dementia.
APP is a type-I integral membrane glyco-protein containing the Aβ region (4 kD) thatis synthesized in the neuronal rough endo-plasmic reticulum.16 Secretory vesicles con-taining full-length APP are transferredthrough the Golgi apparatus to the trans-Golgi network and are then axonally trans-ported to the presynaptic outer plasmamembrane. APP is processed by several dif-ferent proteases called secretases, following
either a nonamyloidogenic pathway or apathogenic amyloidogenic pathway (Fig. 1).
In the nonamyloidogenic pathway, mem-brane-bound APP is constitutively cleavedby α-secretase within the Aβ sequence,therefore preventing the formation of amy-loidogenic peptides. APP cleavage by α-secretase gives rise to the release of a largesoluble N-terminal fragment named sAPP-α and a 10-kD membrane-bound 83-residue COOH-terminal fragment (C83) intothe extracellular space. Subsequently, γ-secretase cleaves C83 to produce a non-pathogenic soluble peptide (p3) and a resid-ual APP intracellular domain (AICD). ThesAPP-α fragment has been described aspossessing neurotrophic and neuroprotec-tive properties.17
In the amyloidogenic pathway, APP iscleaved by β-secretase (BACE1) releasing asoluble N-terminal fragment (sAPP-β) anda membrane-bound 12-kD C-terminal frag-ment (C99), which is cleaved by γ-secretase
LOOKING AHEAD Drug News Perspect 22(6), July/August 2009
326 M. Boada et al. pp. 325-339
Figure 1. The amyloid cascade hypothesis and Aβ clearance. In the nonamyloidogenic pathway, mem-brane-bound β-amyloid precursor protein (APP) is constitutively cleaved by α-secretase (α-Sec), givingrise to the release of a soluble N-terminal fragment (α-sAPP) and a membrane-bound fragment (C83),which is subsequently cleaved by γ-secretase (γ-Sec) to produce a nonpathogenic soluble peptide (p3)and a residual APP intracellular domain (AICD). In the amyloidogenic pathway, APP is cleaved by β-sec-retase releasing a soluble N-terminal fragment (β-sAPP) and a membrane-bound fragment (C99),which is cleaved by γ-secretase to produce AICD and a heterogeneous generation of Aβ40 and Aβ42,whose hydrophobic properties facilitate the formation of amyloid plaque in the cerebrospinal fluid(CSF). Aβ can be cleared from the CSF by transcytosis through the blood–brain barrier (BBB) mediatedby low-density lipoprotein receptor-related protein-1 (LRP1), while the receptor for glycation end prod-ucts (RAGE) mediates Aβ influx into the brain across the BBB.

to produce AICD and a heterogeneous gen-eration of Aβ40 and Aβ42, whose hydropho-bic properties facilitate the formation ofamyloid plaque in the brain cerebrospinalfluid (CSF).18,19 Aβ accumulates not only inthe core of neuritic plaques but also on thevessel walls (amyloid angiopathy). Thespread of AD pathology can be mediated bysoluble extracellular Aβ that induces neuro-toxicity and tau hyperphosphorylation insurrounding cells.
Aβ can be cleared off the CSF by transcyto-sis through the blood–brain barrier (BBB)mediated by low-density lipoprotein recep-tor-related protein-1 (LRP1), while the recep-tor for glycation end products (RAGE) medi-ates Aβ influx into the brain across the BBB(Fig. 1).20
Tau hyperphosphorylation andneurofibrillar tangles Tau proteins are found in all cell types andare major components of neurons wherethey are predominantly associated with axonmicrotubules. The main function of the tauprotein is to modulate microtubule forma-tion dynamics by site-specific phosphoryla-tion. Normal microtubule assembly occurs intwo separate phases: nucleation and elon-gation. Nucleation occurs when tubulindimers polymerize to form protofilamentswhich posteriorly arrange in groups throughlateral contacts to form a hollow cylinder;subsequently these microtubules elongateby the continuation of this process. Tau pro-teins dynamically stabilize microtubules bybinding to several tubulin molecules simul-taneously. When decreased axonal transportis required, certain motifs within the micro-tubule binding repeats of tau are phosphory-lated by affinity-regulating kinases, thusreducing the binding of tau to microtubulesand favoring disassembly.21,22 In pathologicalconditions, tau can be hyperphosphorylatedat additional sites, thus increasing thepropensity of tau to oligomerize and accu-mulate as intracellular paired helical fila-ments that can eventually form insolubleaggregates as neurofibrillary tangles.Disruption of normal phosphorylation eventsresults in the deregulation of neurite out-growth and impaired axonal transport.23
Apoptosis and neuronal lossThe concept that the accumulation of largeamounts of Aβ in brain amyloid plaques
inducing neuronal death is the hallmark ofAD still remains controversial. Neuronal lossis particularly difficult to assess, and oppo-site views have been expressed concerningits course as well as its relation with Aβ andseverity in AD. Albeit some studies suggestAβ is neurotoxic to cells,13,24 some authorshave identified a weak correlation betweendementia and neuritic plaques.25,26
Increasing evidence indicates that neuronaldeath in AD is the result of apoptotic mech-anisms, with Aβ playing a key role.27,28 Aβmay exert its neurotoxic effects in a varietyof ways, including disruption of mitochondr-ial function,29 induction of apoptoticgenes,30,31 formation of ion channels32 trig-gering loss of calcium homeostasis,24 stimu-lation of the JNK pathway,33 or activation ofmicroglia cells leading to the expression ofproinflammatory genes12,34 and an increasein reactive oxygen species.11,35 For someauthors, caspases might play a dual role inAD influencing the proteolytic processing ofAPP and increasing Aβ formation, ultimate-ly regulating the apoptotic death of neu-rons.36
THERAPEUTICS
Current management: preventing declineof cognitive mechanisms Despite years of intensive research, a safeand effective treatment has not yet beenencountered. The currently approved thera-pies are only available for the symptomatictreatment of AD, show no long-term effica-cy, and do not prevent disease progression.Current therapeutic agents includecholinesterase inhibitors and N-methyl-D-aspartate (NMDA) receptor antagonists.
There is evidence that biological dysfunctionor imbalance in neurotransmission, particu-larly cholinergic and glutamatergic, isinvolved in the etiology of AD. The neuro-transmitter acetylcholine is essential forprocessing memory and learning. Deficits inboth concentration and function of acetyl-choline have been found in patients with AD,caused by either a loss of cholinergic neu-rons or decreased acetylcholinesteraseactivity.37 Cholinesterase inhibitors have amoderate but worthwhile effect in stabiliz-ing symptoms. The current drugs(donezepil, rivastigmine and galantamine)are adequate for mild and moderate AD.38-41
On the other hand, overactivation of NMDAreceptors, which are pivotal in learning and
memory, by the neurotransmitter glutamatehas been linked to neuronal damage thatmay result in cognitive decline in patientswith AD.37 Memantine is a partial NMDAreceptor antagonist that appears to beeffective in slowing down cognition declinein moderate to severe AD patients.42
However, the search for effective treatmentstrategies for AD continues and specialattention is being paid to potential targetsfor drug and therapeutics development,such as the enzymes and moleculesinvolved in the mechanisms that can lead tothe development of the disease. A numberof products targeting not only Aβ formationand aggregation but also tau pathology,oxidative stress, inflammation, excitotoxicityand neurodegeneration are currently underactive investigation.43-45
New perspectives: targeting neuriticplaqueAmong the many novel therapeuticapproaches under investigation for AD,strategies oriented towards reducing theproduction of cytotoxic Aβ in order to preventthe accumulation of amyloid deposits or toreduce the existing neuritic plaque seemparticularly appealing.46 Pharmacologic tar-gets as detailed in the following sections andpoints of action of putative therapeuticagents can be seen in Figure 1.
Reduction of APP Modulation of APP production is the topupstream targeting of Aβ. Intracellular traf-ficking of APP may be regulated by multiplefactors such as signal transduction enzymesor hormone stimulation. Interfering withthese factors may affect intracellular levelsof APP and thus the proteolytic processingof APP, thereby reducing the overall levels ofAβ. Compounds such as phenserine, anacetylcholinesterase inhibitor, and deferox-amine, a Fe3+ chelator, have been alsodescribed as possessing the capacity tolower the rate of APP messenger RNA syn-thesis, resulting in a substantial reduction ofAβ levels.47,48 In a phase III clinical study,however, phenserine failed to demonstrateefficacy compared to placebo in cognitiontests.
Activation of α-secretase Favoring APP processing through the neu-roprotective, nonamyloidogenic pathway
Drug News Perspect 22(6), July/August 2009 LOOKING AHEAD
327M. Boada et al. pp. 325-339

seems to be a logical alternative strategy toreduce the burden of cytotoxic Aβ. Thisprocess should involve the pharmacologicalactivation or upregulation of α-secretase.Multiple enzymes have been identified aspossessing α-secretase-like activity. Fourmembers of the ADAM (a disintegrin andmetalloproteinase) family, ADAM 9, ADAM10, ADAM 17 (TACE) and more recentlyADAM 19, have been proposed as α-secre-tases.49,50 In particular, ADAM 10 has beenpostulated to exert a predominant role invivo as the physiologically relevant constitu-tive α-secretase.51 Competition between α-secretase and β-secretase for the substrateAPP has been demonstrated in vivo, andevidence suggested that overexpression ofADAM 10 inhibited the production of Aβ,prevented plaque formation and alleviatedthe associated neurological effects.52
Several mechanisms for α-secretase upreg-ulation have been described, includingADAM10 gene expression enhancement andstimulation of molecular signaling.51
Moreover, low cholesterol levels have beenassociated with higher levels of α-secretaseADAM 10 activity.53 Statins (e.g., batimastat,marimastat, simvastatin, atorvastatin) arewell-known cholesterol-lowering drugs thathave been suggested to regulate α-secre-tase resulting in anti-AD efficacy.54,55
Inhibition of ββ-secretase As one of the major players involved in theneurotoxic Aβ-generating amyloidogenicpathway, β-secretase may be a key thera-peutic target against AD. β-Secretase is anintegral membrane aspartyl protease prima-rily expressed in the brain and often termedBACE1 for β-site APP-cleaving enzyme 1.56
While recent reports indicate that BACE1expression is tightly regulated, proposedphysiological roles include participation in awide range of processes such as axonalgrowth, brain development and myelination,although many of these functions within thecentral nervous system are not completelyunderstood.57 Overexpression of BACE1 isassociated with neurodegeneration andBACE1 is upregulated in at least some ADbrains.58 Development of effective BACE1inhibitors has proven challenging, mainlydue to difficulties found in successful BBBcrossing and delivery to the brain.59 CurrentBACE1 inhibition agents under investigationinclude OM-99-1, OM-99-2, ATG-Z1 andCTS-21166.55,60
Inhibition/modulation of γ-secretase γ-Secretase is a high-molecular-weightcomplex composed of four major mem-brane proteins: presenilin 1 (PS1), nicastrin(NTC), presenilin enhancer 2 (PEN-2) andanterior pharynx defective 1 (Aph-1). γ-Secretase is ubiquitously expressed and cancleave a number of different membrane pro-teins besides C99. Notch 1 receptor is a par-ticularly relevant substrate of γ-secretase.61
Notch signaling regulates the capacity ofneurons to extend and elaborate neuritesbut it is also involved in embryogenesis aswell as cell differentiation and maturationevents in adulthood. For these reasons, γ-secretase inhibitors (e.g., begacestat, MK-0752 and flurizan, although the latter failedin a phase III clinical study) can interferewith vital physiological processes causingtoxicity.62 Research for alternatives to γ-sec-retase inhibitors is focused on the develop-ment of selective C99 proteolysis blockers(e.g., imatinib, LDDN-9918) and γ-secretasemodulators capable of reducing the forma-tion of pathogenic Aβ40 and Aβ42 (e.g.,ibuprofen, indomethacin), allowing γ-secre-tase to generate shorter, less fibrillogenicAβ peptides.63
Interfering with Aββ aggregationAD neurotoxicity is thought to result fromthe aggregation of Aβ into growing amyloidfibrils that form neuritic plaques.64 As a con-sequence, downstream strategies targetingAβwith the intention to inhibit this aggrega-tion or to disrupt the already formed amy-loid plaque in brain tissue are currentlyunder investigation (e.g., immunotherapy,small-molecule pharmacotherapy, metalchelation).65
Immunotherapy with Aβ-specific antibodies,which includes active (vaccination) or pas-sive immunization, is thought to act throughseveral mechanisms of action. Antibodiesagainst Aβ can prevent the formation ofplaques in some animal models and inhumans,66 although these treatments areassociated with deleterious immune reac-tions.67,68 Antibodies can bind Aβ in fibrilsand plaque, thus favoring disaggregation,producing soluble forms of Aβ that can beeliminated from the body.69 However,plaque-directed antibodies are required tocross the BBB. It is thought that antibodiescan enter the brain by passive diffusion atsites deficient in BBB.70 Moreover, Aβ-anti-body complexes may be cleared by FcRn
receptor-mediated transcytosis across theBBB.71
Small-molecule inhibitors of Aβ aggrega-tion under active development includeColostrinin, AZC-103, SEN-606, and evennatural products derived from Gingko bilo-ba, curcumin and nicotine. The Aβ aggrega-tion inhibitor tramiprosate (Alzhemed®)failed to show significant differences versusplacebo in AD patients. Amyloid plaquedegradation enhancers include small mole-cules such as aleplasinin (PAZ-417) as wellas short synthetic peptides that could beactive in disrupting the stability of the βsheet.
There is evidence that certain metal ions(Cu2+, Fe3+ and Zn2+) play a role in the pre-cipitation of cytotoxic Aβ. In this sense, thepossible capacity of metal chelators such asiodochlorohydroxyquin and PBT-2 (theproduct that replaced the withdrawn clio-quinol) to reverse amyloid-β plaque deposi-tion is under investigation.72,73
Aββ clearanceIn addition to antibody- or drug-mediatedAβ degradation in brain, extracellularmonomeric Aβ can be cleared from thebrain to the periphery, where it can then bedegraded or removed. The concentration ofAβ in brain interstitial fluid is tightly regulat-ed through transport across the BBB (Fig. 1).LRP1 is the major cell surface transporterprotein involved in Aβ clearance throughtranscytosis from the brain to the blood,74
while the RAGE mediates soluble Aβ influxinto the brain across the BBB.75 RAGE is apotential target for therapies aimed at low-ering the Aβ load in brain. Inhibitors ofRAGE–Aβ binding currently in the pipelinefor mild and moderate AD include PF-04494700 (phase II development).
There is growing evidence that Aβ levels inAD are increased in plasma and decreasedin CSF.76 This observation has led to thedesign of novel therapeutic strategies pro-posed to clear Aβ from the brain throughthe induction of an unbalance of Aβ trans-port dynamics across the BBB. Thus, thesequestration of Aβ in plasma may bothincrease the transport of free Aβ from CSFto plasma and reduce Aβ transport into thebrain in order to restore the intrinsic equilib-rium between brain and blood Aβ levels.77
Immunotherapy with antibodies binding toand clearing plasma Aβ has the advantage
LOOKING AHEAD Drug News Perspect 22(6), July/August 2009
328 M. Boada et al. pp. 325-339

of not having to undergo BBB crossing andis proven capable of reducing brain amyloidburden in mouse models.78
A novel approach: plasma exchange withalbumin replacementPlasma exchange is a process used to elim-inate patient’s plasma and replacing it withanother solution in order to maintain nor-mal volemia and osmotic balance. Toachieve this effect, albumin or other colloidshave been used, as well as fresh frozen plas-ma and crystalloids. The purpose of thisprocedure is to eliminate toxic substancesfrom patient plasma, such as autoantibod-ies, alloantibodies, immune complexes, pro-teins or toxins. Plasma exchange is widelyused in the treatment of different patholo-gies. Specifically, this procedure has beenapplied to the following disorders: Guillain-Barré syndrome,79 multiple sclerosis,80
inflammatory demyelinating polyradicu-loneuropathy,81 acute inflammatory de-myelinating disease of the CNS82 and otherperipheral neurological alterations.83
Here, plasma exchange is presented as anovel approach for the treatment of AD witha focus on plasma Aβ clearance, taking intoaccount the fact that 90% of circulating Aβmay be bound to albumin.84 Hence, thepotential mobilization of plasma Aβ boundto therapeutic albumin through plasmaexchange could in turn translate into amobilization of brain Aβ and, as a conse-quence, lead to an improvement of thepatient’s cognitive functions.
With this in mind and taking into accountthat preliminary studies have demonstratedthat Human Albumin Grifols® is able to bindAβ peptide,85 a clinical investigation pro-gram using Human Albumin Grifols througha plasma exchange regimen in patients withmild to moderate AD was carried out.
CLINICAL INVESTIGATION PROGRAM OFAβ MOBILIZATION THROUGH ALBUMINBINDING AND PLASMA EXCHANGE INMILD TO MODERATE AD
Pilot study (proof-of-concept)The first clinical study carried out was a pilotstudy aimed to assess whether HumanAlbumin Grifols was able to mobilize plasmaAβ peptide when used in a therapeutic plas-ma exchange program at a rate of two plas-ma exchanges per week during 3 weeks,
that is, 6 plasma exchanges in total.Furthermore, a possible change in the cog-nitive status was also assessed throughneuropsychological evaluations.
During each plasma exchange procedure, acomplete plasma volume was removed fromthe patient and was simultaneouslyreplaced by a similar volume of 5% HumanAlbumin Grifols, which is a concentration ofalbumin similar to that found naturally inplasma. Preferably, plasma exchanges wereperformed through a double-lumen centralline, although peripheral access was alsopermitted. After each procedure, bloodcount, calcium, activated partial thrombo-plastin time, prothrombin time and fibrino-gen were monitored before patients weredischarged.
Plasma Aβ40 and Aβ42 levels were deter-mined at baseline, before and after eachplasma exchange and once a month during6 months of follow-up. On the other hand,CSF Aβ40 and Aβ42 were determinedthrough a regular spinal tap at baseline, atthe end of the plasma exchange period andat 3 and 6 months after the plasmaexchange period. Determinations of plasmaAβ40 and Aβ42 were carried out with a sand-wich-type ELISA test (β-amyloid [1-40]ELISA kit, Zymed, U.S.A. and Innotestβ-amyloid [1-42] CE, Innogenetics, Belgium)originally commercialized for CSF determi-nations, following a protocol variation rec-ommended by the manufacturer so that itcould be more suitable for plasma determi-nations. It is important to note that at thatmoment a validated ELISA test for plasmaAβ40 and Aβ42 was not commercially avail-able.
In addition to biochemical determinations,cognitive status was evaluated at baselineand at 3 and 6 months after the plasmaexchange period through the Mini-MentalStatus Examination (MMSE)86 and theAlzheimer’s Disease Assessment Scale, cog-nitive subscale (ADAS-Cog) examination.87
Finally, neuroimaging studies were also per-formed. Morphological assessments con-sisted of a magnetic resonance imaging(MRI) performed at baseline and at 3 and 6months after the plasma exchange period toassess changes in the volume of the hip-pocampus, cingulate and other areas ofinterest. Functional neuroimaging assess-ments consisting of a single photon emis-sion computed tomography (SPECT) were
performed at baseline and at 6 months afterthe plasma exchange period to assesschanges in brain perfusion (Neurogam™software, Segami Corp., Columbia, MD,USA).88 A final follow-up visit was sched-uled at 1 year after the enrollment.
This pilot study was performed in a singlecenter (ACE Foundation - Catalan Instituteof Applied Neurosciences, Barcelona,Spain). Before participating, each patientand/or close relative and/or legal repre-sentative signed the correspondinginformed consent. Previously, the studyhad been approved by the local EthicalCommittee and by the Spanish Ministry ofHealth. In addition, the study was conduct-ed according to the Code of EthicalPrinciples for Medical Research InvolvingHuman Subjects of the World MedicalAssociation.
All patients fulfilled DSM-IV (Diagnostic andStatistical Manual of Mental Disorders, 4th
edition) criteria for dementia and were diag-nosed according to the NINCDS-ADRDA(National Institute of Neurological andCommunicative Disorders and theAlzheimer’s Disease and Related DisordersAssociation) criteria for possible and proba-ble AD.89 All patients received a thoroughclinical and neurological examination and acomprehensive neuropsychological evalua-tion including tests for general cognition,memory, language, perceptual and con-structional abilities and executive functions.Complete blood analysis and neuroimagingstudies were performed in all subjects toexclude other potential causes of dementiafollowing the guidelines for the diagnosis ofAD from the Study group on BehavioralNeurology and Dementia of the SpanishNeurological Society.
The patient population consisted of maleand female subjects aged between 55 and85 years, diagnosed with mild to moderateAD (NINCDS-ADRDA criterion) and anMMSE score between 20 and 24. Moreover,patients had to be on stable treatment withdonepezil (6 months) and had to have anMRI or CAT scan performed within 6 monthsprior to participation, with absence of cere-bral-vascular findings.
Pilot study resultsTen patients were included in this pilot studyfollowing a single-arm, open-label design.Seven out of the 10 patients underwent
Drug News Perspect 22(6), July/August 2009 LOOKING AHEAD
329M. Boada et al. pp. 325-339

plasma exchanges with 5% Human AlbuminGrifols. Out of these 7 patients, 3 underwent5 plasma exchanges, 2 underwent 4 plasmaexchanges and 2 underwent 3 plasmaexchanges, during the planned 3-week peri-od. The main reason why not all patientsunderwent the 6 plasma exchanges wasthat the hematology team responsible forthe procedure followed the precautionaryprinciple in this special patient population inrelation with low coagulation parametersafter each plasma exchange and with themild anemia that is common in therapeuticplasma exchange programs. As will be stat-ed later, based on the fact that the proce-dure was shown to be safe during the pilotstudy, an extension of the study was per-formed in which practically all patientsunderwent the 6 plasma exchanges withinthe planned 6 weeks.
Figure 2 shows the average plasma levels ofAβ40 and Aβ42 in the 7 patients that under-went plasma exchanges. Although thereappears to be a slight variation of Aβ40 with-in the plasma exchange period, no clearpattern can be seen. On the other hand, thelack of a variation pattern is even more evi-dent for Aβ42. At that moment, the investi-gators already realized that the lack of areliable ELISA test for plasma Aβ determi-nations did not permit the adequate inter-pretation of plasma results. The methodwas improved during the study extension asshown later.
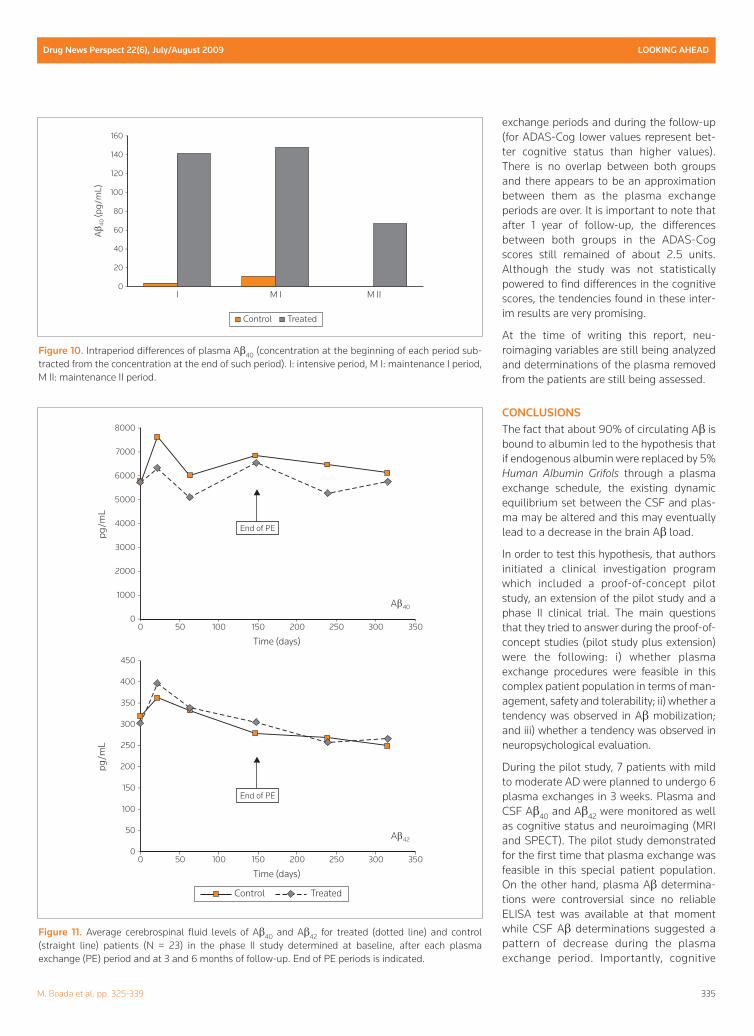
With respect to CSF Aβ40 and Aβ42, Figure 3shows that both peptides follow a similarkinetics: a decrease is observed during theplasma exchange period followed by anincrease after the plasma exchange periodreturning to baseline levels at 6 months offollow-up.
Figure 4 shows the changes from baselineof the scores corresponding to MMSE andADAS-Cog tests measured at 3, 6 and 12months after plasma exchanges. All scores(except obviously that measured at time 0)were assessed after the plasma exchangeperiod (first 3 weeks). From the graphs itclearly appears that the cognitive status ofthe patients as measured by MMSE andADAS-Cog remained stable after 1 year offollow-up.
Regarding MRI findings, the volume of thehippocampus measured at baseline, 3 and 6months suggested a progressive volumeincrease. However, no clear pattern was
LOOKING AHEAD Drug News Perspect 22(6), July/August 2009
330 M. Boada et al. pp. 325-339
Figure 2. Mean plasma levels of Aβ40 and Aβ42 in the 7 patients that underwent plasma exchanges (PE)in the pilot study, determined at baseline, before and after each PE and once a month during 6 monthsof follow-up.
Figure 3. Mean cerebrospinal fluid levels of Aβ40 and Aβ42 in the 7 patients that underwent plasmaexchanges (PE) in the pilot study, determined at baseline, at the end of the PE period and at 3 and 6months after the PE period.
Figure 4. Changes from baseline scores (average from the 7 patients of the pilot study) of the Mini-Mental Status Examination (MMSE) and the Alzheimer’s Disease Assessment Scale, cognitive subscale(ADAS-Cog) measured at 3, 6 and 12 months after plasma exchanges. For clarity, negative values ofADAS-Cog have been represented upwards.

observed for the posterior cingulate and themid frontal gyrus (data not shown). Withrespect to functional neuroimaging(SPECT), 6 out of 7 patients showed a signif-icant perfusion increase in the frontal andtemporal areas (Fig. 5). At 6 months, statis-tical parametric mapping (SPM) analysis88
also showed a significant perfusion increasein both the frontal and temporal areas (datanot shown).
Pilot study conclusions One of the principal conclusions of this pilot(proof-of-concept) study was that treatmentwith 5% Human Albumin Grifols through atherapeutic plasma exchange regimen wasfeasible in mild to moderate AD patients, apatient population in which, to our knowl-edge, this has been the first time that thistherapeutic approach has been carried out.However, an area of uncertainty remainedwith respect to the number of plasmaexchanges to be performed since not allpatients completed the 6-exchange cycle.
Relative to plasma levels of Aβ40 and Aβ42, itwas clear that the lack of a reliable ELISAtest for plasma determinations made theknowledge that could be extracted from thedata obtained very obscure. However, forCSF Aβ40 and Aβ42, a clear pattern of varia-tion was observed for both peptides sug-gesting that CSF Aβ may be mobilized with5% Human Albumin Grifols used in the plas-ma exchanges.
Regarding the neurocognitive scores, thefact that there was a tendency to stabiliza-tion after 1 year of follow-up was interpretedas a promising clinical result, in accordanceto European Medicines Agency (EMEA)guidelines on medicinal products for thetreatment of AD.90 An obvious criticism isthat since the study was open-label, theneurocognitive raters might have set uphigh expectations for the treatment leadingto a bias in the cognitive assessment.Nevertheless, the objective of this pilotstudy was to uncover favorable tendencieswhich could be confirmed in subsequentrandomized, controlled trials.
Very interestingly, once the study was com-pleted, the patients and their families overt-ly expressed their satisfaction with their par-ticipation and requested an additional treat-ment cycle. At that moment, the researchershad improved the curve-fitting method tobe used with the ELISA test and discovered
that the manufacturer had launched animproved test (Innotest β-Amyloid (1-42)RUO, Innogenetics, Belgium) with a higherconcentration of the reference peptide usedin the kit. These circumstances, along withthe observed trend to clinical stabilization at1 year found in the study, were considered tobe sufficient for offering the patients anextension of the original study.
Extension study The extension study was a replica of thepilot study with respect to the number andprocedures of the plasma exchanges andspinal taps, cognitive and neuroimagingassessments. The follow-up period was alsoof 1 year. All the patients that participated in
the pilot study were offered the opportunityto participate in the extension study. Newinformed consents were signed and newapprovals from the local Ethical Committeeand the Spanish Ministry of Health wereobtained.
Six patients previously enrolled in the pilotstudy participated in the extension study. Allpatients except one completed the cycle of6 plasma exchanges in 3 weeks in an outpa-tient regimen. The only patient that did notcomplete the entire cycle underwent 5 con-secutive plasma exchanges. The subject didnot undergo the last plasma exchangebecause the central catheter provided inter-mittent blood flow and had to be removed.
Drug News Perspect 22(6), July/August 2009 LOOKING AHEAD
331M. Boada et al. pp. 325-339
Figure 5. Neurogam™ results from 2 representative patients with a different number of plasmaexchanges (PE) performed: patient with 5 PE (left panels) had an improved perfusion and patient with3 PE had an impaired perfusion (right panels). Upper and middle images: before and after treatmentcompared with a database of healthy subjects (hot colors [red and white] mean hyperperfusion and coldcolors [blue and green] mean hypoperfusion). Bottom images: differences observed before and aftertreatment for each patient (hot colors [yellow, red and white] mean improved perfusion and cold colors[green, blue and purple] mean impaired perfusion).

Extension study results With the improved curve-fitting for theELISA test (basically the improvement wasthat fitting was performed according to a 4-parameter-log model instead of a straight-line model) and the use of the new test witha higher concentration of the reference pep-tide, the levels of plasma Aβ40 (hAmyloidb40 ELISA [HS], The Genetics Company,Switzerland) and Aβ42 (Innotest β-amyloid[1-42] RUO, Innogenetics, Belgium) yieldedthe results shown in Figure 6. It is worthmentioning that the difference in plasmaconcentration between Aβ40 and Aβ42 is ofabout one order of magnitude as reflectedin the graph. If one focuses on the plasmaexchange period (central segment of thegraph), a clear saw-tooth pattern isobserved, although it is more apparent forAβ40 than for Aβ42 due to the differences inconcentration previously mentioned. Thispattern was so regular and consistent forboth peptides and so reproducible in rela-tion with each plasma exchange that thereis little doubt that it is related to the mech-anism of action of albumin through plasmaexchange on Aβ.
On the other hand, Aβ40 and Aβ42 in the CSFdid not show the variation found in the pilotstudy but rather a tendency to remain stableand even a trend towards an increase in thecase of Aβ42 (data not shown).
Taking into account the total of 2 years offollow-up from both the pilot and extensionstudies, the neurocognitive scores yieldedthe results shown in Figure 7.
In both graphs, the upper line representsthe patient’s actual scores (changes frombaseline) while the lower straight line repre-sents the expected progression for this typeof patient at 2 years of follow-up. Therefore,the surface lying in between can be consid-ered a kind of “improvement area” and givesan idea of the tendency of the patientstreated with 5% Human Albumin Grifols toremain more stable than what was expect-ed. Finally, results of neuroimaging studiesshowed a similar trend to that observed inthe pilot study.
After obtaining these results, it was clearthat a phase II, randomized and controlledclinical trial was warranted to assesswhether the behavior of one group ofpatients treated with albumin and plasmaexchange was different from that of a group
of nontreated patients in terms of biochem-ical, clinical and neuroimaging outcomes.
Phase II clinical trialA phase II, randomized, controlled, parallel,single-blind clinical trial was carried out tocompare the mobilization of CSF and plas-ma Aβ, cognitive status and neuroimagingbetween a group of patients treated with5% Human Albumin Grifols in a plasma
exchange regimen and a group of nontreat-ed patients. In addition, Aβ40 and Aβ42determinations were planned to be carriedout from the plasma removed from thetreated patients.
The patient population consisted of maleand female subjects, aged between 55 and85 years, diagnosed with mild to moderateAD (NINCDS-ADRDA criterion) and anMMSE score between 18 and 26. Moreover,
LOOKING AHEAD Drug News Perspect 22(6), July/August 2009
332 M. Boada et al. pp. 325-339
Figure 6. Mean plasma levels of Aβ40 and Aβ42 in the 6 patients that underwent plasma exchanges (PE)in the extension study, determined at baseline, before and after each PE exchange and once a monthduring 6 months of follow-up.
Figure 7. Changes from baseline scores (average from the patients included in both the pilot and theextension study) of the Mini-Mental Status Examination (MMSE) and the Alzheimer’s DiseaseAssessment Scale, cognitive subscale (ADAS-Cog) measured during 2 years of follow-up at weeks 15,27, 52, 60, 75 and 87. For clarity, the negative values of ADAS-Cog have been represented upwards. Thedotted line represents the expected progression for this type of Alzheimer’s disease patients at 2 yearsof follow-up. The surface lying between the two lines can be considered as an indicator of improvement.

patients were required to be on stable treat-ment with acetylcholinesterase inhibitors (3months) and had to have an MRI or CATscan performed within the 12 months priorto participation, with absence of cerebral-vascular findings.
In this phase II study, 36 evaluable patientswere planned to be enrolled in 4 centers, 2in Spain and 2 centers in the United States.However, the total sample size wasincreased to 42 patients in expectation of adropout rate of 15% approximately.
Given the fact that there was approximatelya 1-year delay in site initiations in the U.S. ascompared with Spain, it was decided to per-form an interim analysis with the first 29patients (80% of the 36 evaluable patientsplanned) recruited in Spain (at that time,there was only one patient included in theU.S.). Out of the 29 patients included inSpain, 27 had completed the whole study. Atthe time of writing this interim analysis, thestudy continues including patients in onecenter in Spain and in the two U.S. centers.
As with the pilot and the extension studies,each patient and/or close relative and/orlegal representative signed the correspon-ding informed consent before participation.Beforehand, the study had been approvedby the corresponding local EthicalCommittee and by the Spanish Ministry ofHealth. In the U.S. the study was approvedby the local Independent Review Boardsand was submitted to the Food & DrugAdministration.
Patients were randomly assigned to either aplasma exchange (removal of one completeplasma volume with simultaneous substitu-tion with 5% Human Albumin Grifols) groupor a control group. The treatment groupunderwent the same plasma exchange pro-cedure previously described for the pilot andextension studies. The control group did notundergo real plasma exchanges but a shamprocedure consisting of first inserting a cutcatheter under the skin at the sameanatomical location as the treated patients.Through the sham central line, controlpatients were apparently connected to theplasma exchange machine which apparent-ly worked in the same way as the real proce-dure by circulating a colored liquid in aclose-circuit manner. Therefore, the plasmaexchange procedure resembled that of thetreated patients although no plasma wasremoved from the subject and no albumin
was infused. The sham procedure was test-ed several times until the investigatorsreached the consensus that only an expertin the field could realize that the procedurewas not real.
In this phase II study there were 3 plasmaexchange periods: 1) an intensive period inwhich patients underwent 6 plasmaexchanges in 3 weeks (2 per week, same asin pilot study); 2) a maintenance period I inwhich there were 6 plasma exchanges in 6weeks (1 per week); and 3) a maintenanceperiod II in which there were 6 plasmaexchanges in 12 weeks (1 every 2 weeks).There were 1–2 weeks of rest between peri-ods. Control group patients underwent thesame number of sham procedures.
For both treatment and control groups,plasma Aβ40 and Aβ42 were determined atbaseline, before and after each plasmaexchange and at 3 and 6 months of follow-up. On the other hand, CSF Aβ40 and Aβ42were determined through a regular spinaltap at baseline, between each plasmaexchange period and at 3 and 6 monthsafter the plasma exchange periods.
Cognitive status was evaluated throughMMSE and the ADAS-Cog tests at baseline,between each plasma exchange period andat 3 and 6 months after the plasmaexchange periods except for MMSE whichhad just 5 determinations since this test wasnot performed between the intensive periodand the maintenance I period. Cognitiveassessments were performed by independ-ent neuropsychologists not directly involvedin the trial and blinded to study treatment.
Neuroimaging assessments consisted of anMRI performed at baseline and at the end ofthe 3 plasma exchange periods and at 6months after the plasma exchange periods.Functional neuroimaging (SPECT) was per-formed at baseline and at the end of main-tenance I and maintenance II treatmentperiod and at 3 and 6 months after the plas-ma exchange periods. A final follow-up visitwas scheduled at 6 months after the plasmaexchange periods (approximately 1 yearafter recruitment).
Phase II study interim resultsOf the 29 patients included in this interimanalysis, 14 were randomly assigned to thetreatment group and 15 to the controlgroup. In the treatment group, all except 2
Drug News Perspect 22(6), July/August 2009 LOOKING AHEAD
333M. Boada et al. pp. 325-339
patients underwent all the planned plasmaexchanges corresponding to the 3 periods.One patient underwent only the 3 first plas-ma exchanges because the family decidedto withdraw the patient from the study.Another patient also underwent only the 3first plasma exchanges because of the pres-entation of transient aphasia with noischemic findings on cerebral MRI. However,the investigators recommended thispatient’s withdrawal.
Figure 8 shows the average plasma levels ofAβ40 and Aβ42 for treated and controlgroups corresponding to the 23 patientswith available data at the time of writing thisreport. Similarly to what occurred in theextension study, there is a clear saw-toothpattern for Aβ40 in the treated group whichis not present in the control group, stronglysuggesting that the changes found in theextension study that are now reproduced inthe phase II study are associated with theplasma exchange procedure and relatedwith its mechanism of action. Of note, oncethe plasma exchange period is over, there isa return to levels similar to those of the con-trol group. For Aβ42 there is also a saw-toothpattern in the treated group, although thecontrol group shows a similar behavior andan evident overlap exists between bothgroups. Again, since the plasma Aβ42 con-centration is much lower than that of Aβ40 itlies near the detection limit of the tech-nique. From these graphs it appears reason-able to think that Aβ40 is the peptide thatmost reliably represents the kinetics of plas-ma Aβ during plasma exchange.
Very interesting information arose by work-ing out the average rate of change of Aβ40for each plasma exchange period, that is,dividing the change of Aβ40 concentration ineach period by the time period in days. Theresult, shown in Figure 9, is a measure of themobilization of Aβ40 in pg/mL/day, indicat-ing the average amount of Aβ40 that ismobilized per unit volume and unit time.
The difference between the treatment andcontrol groups in the graph is remarkable:While the control group does not presentany change in Aβ40 mobilization, the treat-ment group presents a clear pattern in rela-tion with the plasma exchange periods.During the intensive period, there is a high-er Aβ40 mobilization (about 8 pg/mL/day)than that during the maintenance period I(about 4 pg/mL/day), and the lowest mobi-

lization was during the maintenance periodII (about 1 pg/mL/day). Beyond the figures,what is most important of this variable is thefact that in treated patients there appears tobe a relation between the magnitude of
Aβ40 mobilization and the “plasmaexchange dose” (2 plasma exchanges perweek in the intensive period, 1 plasmaexchange per week in the maintenance peri-od I and 1 plasma exchange every 2 weeks in
the maintenance period II). On the otherhand, there is no variation observed in thecontrol group regarding Aβ40 mobilization.
However, in order to assess whether there isa plasma exchange period which is the bestin terms of absolute change of Aβ40 concen-tration, the authors subtracted the concen-tration at the beginning of each period fromthe concentration at the end of such period.These results are shown in Figure 10.
The interpretation of the results is very help-ful in order to decide if a more favorableplasma exchange schedule could be select-ed: The change in plasma Aβ40 concentra-tion produced by 2 plasma exchanges perweek for 3 weeks (intensive) is similar to thatproduced by 1 plasma exchange per weekfor 6 weeks (maintenance I) and twice thechange produced by 1 plasma exchangeevery 2 weeks for 12 weeks (maintenance II).Therefore, if a therapeutic schedule were tobe selected based on these data, 1 plasmaexchange per week for 6 weeks should bethe same as 2 plasma exchanges per weekfor 3 weeks.
Figure 11 shows CSF Aβ40 and Aβ42 for treat-ed and control groups corresponding to the23 patients with available data at the timeof writing this report. While for Aβ40 there isno overlap between both groups and levelsof the treated group remain lower thanthose of the control group throughout theplasma exchange period, this pattern is notso clear for Aβ42. However, it must be kept inmind that CSF Aβ42 concentration is oneorder of magnitude lower than that of Aβ40,making it difficult to reliably assess differ-ences since Aβ42 levels are closer to thedetection limit of the technique.
Finally, results of cognitive scores are shownin Figure 12. Differences from baselineregarding the MMSE scores are representedon the first graph. The treated group scoreson average are better than those of the con-trol group during all the plasma exchangeperiods and during the follow-up (for MMSEhigher values represent better cognitive sta-tus than lower values). There is no overlapbetween both groups and there appears tobe a tendency towards impairment after thefinalization of plasma exchanges. On theother hand, differences from baseline for theADAS-Cog scores are represented in thesecond graph. Again, the treated groupscores on average are better than those ofthe control group during all plasma
LOOKING AHEAD Drug News Perspect 22(6), July/August 2009
334 M. Boada et al. pp. 325-339
Figure 8. Average plasma levels of Aβ40 and Aβ42 of treated and control patients (N = 23) in the phaseII study, determined at baseline, before and after each plasma exchange (PE) during the 3 treatmentperiods and at 3 and 6 months of follow-up. The three segments limited by vertical lines indicate the 3PE periods: intensive period (2 PE per week), maintenance I period (1 PE per week) and maintenance IIperiod (1 PE every 2 weeks). End of PE periods is indicated.
Figure 9. Plasma Aβ40 mobilization (changes of average Aβ40 concentration in pg/mL per day, duringeach plasma exchange period) for treated and control patients during the 3 plasma exchange periods(I: intensive period, M I: maintenance I period, M II: maintenance II period).
exchange periods and during the follow-up(for ADAS-Cog lower values represent bet-ter cognitive status than higher values).There is no overlap between both groupsand there appears to be an approximationbetween them as the plasma exchangeperiods are over. It is important to note thatafter 1 year of follow-up, the differencesbetween both groups in the ADAS-Cogscores still remained of about 2.5 units.Although the study was not statisticallypowered to find differences in the cognitivescores, the tendencies found in these inter-im results are very promising.
At the time of writing this report, neu-roimaging variables are still being analyzedand determinations of the plasma removedfrom the patients are still being assessed.
CONCLUSIONSThe fact that about 90% of circulating Aβ isbound to albumin led to the hypothesis thatif endogenous albumin were replaced by 5%Human Albumin Grifols through a plasmaexchange schedule, the existing dynamicequilibrium set between the CSF and plas-ma may be altered and this may eventuallylead to a decrease in the brain Aβ load.
In order to test this hypothesis, that authorsinitiated a clinical investigation programwhich included a proof-of-concept pilotstudy, an extension of the pilot study and aphase II clinical trial. The main questionsthat they tried to answer during the proof-of-concept studies (pilot study plus extension)were the following: i) whether plasmaexchange procedures were feasible in thiscomplex patient population in terms of man-agement, safety and tolerability; ii) whether atendency was observed in Aβ mobilization;and iii) whether a tendency was observed inneuropsychological evaluation.
During the pilot study, 7 patients with mildto moderate AD were planned to undergo 6plasma exchanges in 3 weeks. Plasma andCSF Aβ40 and Aβ42 were monitored as wellas cognitive status and neuroimaging (MRIand SPECT). The pilot study demonstratedfor the first time that plasma exchange wasfeasible in this special patient population.On the other hand, plasma Aβ determina-tions were controversial since no reliableELISA test was available at that momentwhile CSF Aβ determinations suggested apattern of decrease during the plasmaexchange period. Importantly, cognitive
Drug News Perspect 22(6), July/August 2009 LOOKING AHEAD
335M. Boada et al. pp. 325-339
Figure 10. Intraperiod differences of plasma Aβ40 (concentration at the beginning of each period sub-tracted from the concentration at the end of such period). I: intensive period, M I: maintenance I period,M II: maintenance II period.
Figure 11. Average cerebrospinal fluid levels of Aβ40 and Aβ42 for treated (dotted line) and control(straight line) patients (N = 23) in the phase II study determined at baseline, after each plasmaexchange (PE) period and at 3 and 6 months of follow-up. End of PE periods is indicated.

scores (MMSE and ADAS-Cog) remainedstable at 1 year of follow-up.
Due to the tendencies observed in the pilotstudy and the requests formalized by thepatients and their families, an extensionstudy was performed following the sameprocedures as those of the pilot study andthe patients were followed up for 1 moreyear. In the extension study a new andimproved ELISA test was used and plasmaAβ determinations demonstrated that varia-tions in levels were associated with the plas-ma exchanges, a pattern more apparent forAβ40 than for Aβ42. However, CSF Aβ levelsdid not confirm the pattern found in thepilot study. When cognitive status wasassessed at 2 years of follow-up, scores weremuch more stable than expected for thispatient population.
Taken together, the results from the pilotstudy and its extension suggest that thereplacement of endogenous albumin with5% Human Albumin Grifols through plasmaexchange was able to produce alterations inthe plasma Aβ kinetics and that this findingmay be related to a tendency towards thestabilization of cognitive scores.
The above conclusion led to a phase II, ran-domized and controlled clinical trial inwhich a group of patients treated with 5%Human Albumin Grifols through a plasmaexchange program was compared with acontrol group of patients not treated withplasma exchanges. Thirty-six evaluablepatients were planned for this trial but aninterim analysis with the first 29 patients(80%) recruited has been performed. Threedifferent plasma exchange schedules were
assessed: 2 plasma exchanges per week for3 weeks, 1 plasma exchange per week for 6weeks and 1 plasma exchange every 2 weeksfor 12 weeks. The total follow-up, includingthe treatment period, was of approximately1 year. CSF Aβ40 levels suggested differ-ences between groups but this was not thecase for Aβ42. Plasma Aβ40 levels were againfound to be associated with plasmaexchanges. Interestingly, Aβ40 mobilizationmeasured in pg/mL/day showed that theschedule of 1 plasma exchange per week for6 weeks was similar to that of 2 plasmaexchanges per week for 3 weeks, suggestingthat both schedules may be equivalent.Changes from baseline in both cognitivescoring systems (MMSE and ADAS-Cog)measured at 1 year showed differencesbetween treated and control groups, withpatients belonging to the treatment groupscoring better.
Although the phase II clinical trial is stillongoing, the interim results presented in thisreport together with the previous resultsobtained during the pilot study and its exten-sion suggest that the novel approach oftreating mild to moderate AD with thereplacement of endogenous albumin with5% Human Albumin Grifols through a plas-ma exchange program is not only feasible inthis group of patients but it shows a tenden-cy to arrest cognitive impairment at the timeperiod studied. As a consequence, thisapproach may have a promising role in thefuture.
However, some uncertain areas still remainand will need further clarification: i) a morereliable method of measuring plasma andCSF Aβ42 is needed; ii) the role of MRI andfunctional neuroimaging needs clarificationin terms of their utility to compare treatedwith nontreated patients; iii) a consistentpattern of variation in CSF Aβ has not beenobserved, a result which needs further justi-fication; and iv) the confirmation of thepromising cognitive outcomes observedduring our long-term follow-up studies ispending the finalization of the current studyand will probably need larger randomizedclinical trials.
ACKNOWLEDGEMENTSThe authors are grateful to Dr. Oscar L.Lopez and Dr. James T. Becker from theUniversity of Pittsburgh for their collabora-tion in the early stages of the study design.
LOOKING AHEAD Drug News Perspect 22(6), July/August 2009
336 M. Boada et al. pp. 325-339
Figure 12. Differences from baseline scores (mean ± standard error) of the Mini-Mental StatusExamination (MMSE) and the Alzheimer's Disease Assessment Scale, cognitive subscale (ADAS-Cog) intreated (dotted line) and control (straight line) patients, measured between each plasma exchange (PE)period and at 3 and 6 months of follow-up (MMSE was not determined between the intensive periodand the maintenance I period). End of PE periods is indicated.

Dr. Becker has also collaborated in the MRIanalyses.
DISCLOSURESThis work was supported by Instituto GrifolsS.A. Laura Núñez and Antonio Páez areemployees of the sponsor of the study. Theother authors declare no potential conflictsof interest relevant to this paper.
REFERENCES1. Ferri, C.P., Prince, M., Brayne, C. et al.
Alzheimer’s Disease International. Globalprevalence of dementia: a Delphi consensusstudy. Lancet 2005, 366(9503): 2112-7.
2. Wimo, A., Winblad, B. and Jönsson, L. An esti-mate of the total worldwide societal costs ofdementia in 2005. Alzheimers Dement 2007,3(2): 81-91.
3. Crutcher, K.A. Apolipoprotein E is a prime sus-pect, not just an accomplice, in Alzheimer’sdisease. J Mol Neurosci 2004, 23(3): 181-8.
4. Hirschhorn, J.N., Lohmueller, K., Byrne, E.and Hirschhorn, K. A comprehensive review ofgenetic association studies. Genet Med 2002,4(2): 45-61.
5. Feldman, H.H., Jacova, C., Robillard, A. et al.Diagnosis and treatment of dementia: 2.Diagnosis. CMAJ 2008, 178(7): 825-36.
6. Visser, P.J., Verhey, F.R., Boada, M. et al.Development of screening guidelines and clin-ical criteria for predementia Alzheimer’s dis-ease. The DESCRIPA Study. Neuro-epidemiology 2008, 30(4): 254-65.
7. Dubois, B., Feldman, H.H., Jacova, C. et al.Research criteria for the diagnosis ofAlzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol 2007, 6(8):734-46.
8. Poulin, P. and Zakzanis, K.K. In vivo neu-roanatomy of Alzheimer’s disease: evidencefrom structural and functional brain imaging.Brain Cogn 2002, 49(2): 220-5.
9. Delacourte, A., Sergeant, N., Champain, D. etal. Nonoverlapping but synergetic tau andAPP pathologies in sporadic Alzheimer’s dis-ease. Neurology 2002, 59(3): 398-407.
10. Cummings, J.L. Alzheimer’s disease. N Eng JMed 2004, 351(1): 56-67.
11. Markesbery, W.R. and Carney, J.M. Oxidativealterations in Alzheimer’s disease. BrainPathol 1999, 9(1): 133-46.
12. Sastre, M., Klockgether, T. and Heneka, M.T.Contribution of inflammatory processes toAlzheimer’s disease: molecular mechanisms.Int J Dev Neurosci 2006, 24(2-3): 167-76.
13. Yankner, B.A. Mechanisms of neuronal degen-eration in Alzheimer’s disease. Neuron 1996,16(5): 921-32.
14. Stadelmann, C., Deckwerth, T.L., Srinivasan,A., Bancher, C., Brôck, W. and Lassmann, H.Activation of caspase-3 in single neurons andautophagic granules of granulovacuolardegeneration in Alzheimer’s disease. Evidencefor apoptotic cell death. Am J Pathol 1999,155(5): 1459-66.
15. Selkoe, D.J. The cell biology of β-amyloid pre-cursor protein and presenilin in Alzheimer’sdisease. Trends Cell Biol 1998, 8(11): 447-53.
16. Yoshikai, S., Sasaki, H., Doh-ura, K., Furuya,H. and Sakaki, Y. Genomic organization of thehuman amyloid β-protein precursor gene.Gene 1990, 87(2): 257-63.
17. Furukawa, K., Sopher, B.L., Rydel, R.E. et al.Increased activity-regulating and neuroprotec-tive efficacy of α-secretase-derived secretedamyloid precursor protein conferred by a C-ter-minal heparin-binding domain. J Neurochem1996, 67(5): 1882-96.
18. Fawzi, N.L., Okabe, Y., Yap, E.H. and Head-Gordon, T. Determining the critical nucleusand mechanism of fibril elongation of theAlzheimer’s Aβ(1-40) peptide. J Mol Biol 2007,365(2): 535-50.
19. Sawaya, M.R., Sambashivan, S., Nelson, R. etal. Atomic structures of amyloid cross-β spinesreveal varied steric zippers. Nature 2007,447(7143): 453-7.
20. Deane, R. and Zlokovic, B.V. Role of the blood-brain barrier in the pathogenesis of Alzheimer’sdisease. Curr Alzheimer Res 2007, 4(2): 191-7.
21. Johnson, G.V. and Stoothoff, W.H. Tau phos-phorylation in neuronal cell function and dys-function. J Cell Sci 2004, 117(Pt 24): 5721-9.
22. Kins, S. and Beyreuther, K. Teasing out thetangles. Nat Med 2006, 12(7): 764-5.
23. Mazanetz, M.P. and Fischer, P.M. Untanglingtau hyperphosphorylation in drug design forneurodegenerative diseases. Nat Rev DrugDiscov 2007, 6(6): 464-79.
24. Goodman, Y. and Mattson, M.P. Secretedforms of β-amyloid precursor protein protecthippocampal neurons against amyloid β-pep-tide-induced oxidative injury. Exp Neurol1994, 128(1): 1-12.
25. Dickson, D.W., Crystal, H.A., Bevona, C.,Honer, W., Vincent, I. and Davies, P.Correlations of synaptic and pathologicalmarkers with cognition of the elderly.Neurobiol Aging 1995, 16(3): 285-98.
26. Delaère, P., He, Y., Fayet, G., Duyckaerts, C.and Hauw, J.J. β A4 deposits are constant inthe brain of the oldest old: an immunocyto-chemical study of 20 French centenarians.Neurobiol Aging 1993, 14(2): 191-4.
27. Troy, C.M., Rabacchi, S.A., Xu, Z., Maroney,A.C., Connors, T.J., Shelanski, M.L. andGreene, L.A. β-amyloid-induced neuronal
apoptosis requires c-Jun N-terminal kinaseactivation. J Neurochem 2001, 77(1): 157-64.
28. Kajkowski, E.M., Lo, C.F., Ning, X. et al.β-amyloid peptide-induced apoptosis regulat-ed by a novel protein containing a G proteinactivation module. J Biol Chem 2001, 276(22):18748-56.
29. Chen, X. and Yan, S.D. Mitochondrial Aβ: apotential cause of metabolic dysfunction inAlzheimer’s disease. IUBMB Life 2006, 58(12):686-94.
30. Caricasole, A., Copani, A., Caruso, A. et al.The Wnt pathway, cell-cycle activation and β-amyloid: novel therapeutic strategies inAlzheimer’s disease? Trends Pharmacol Sci2003, 24(5): 233-8.
31. Reddy, P.H., McWeeney, S., Park, B.S. et al.Gene expression profiles of transcripts in amy-loid precursor protein transgenic mice: up-reg-ulation of mitochondrial metabolism andapoptotic genes is an early cellular change inAlzheimer’s disease. Hum Mol Genet 2004 15,13(12): 1225-40.
32. Kagan, B.L., Hirakura, Y., Azimov, R.,Azimova, R. and Lin, M.C. The channelhypothesis of Alzheimer’s disease: current sta-tus. Peptides 2002, 23(7): 1311-5.
33. D’Ambrosio, C., Arena, S., Fulcoli, G.,Scheinfeld, M.H., Zhou, D., D’Adamio, L. andScaloni, A. Hyperphosphorylation of JNK-interacting protein 1, a protein associated withAlzheimer disease. Mol Cell Proteomics 2006,5(1): 97-113.
34. Bamberger, M.E. and Landreth, G.E.Microglial interaction with β-amyloid: implica-tions for the pathogenesis of Alzheimer’s dis-ease. Microsc Res Tech 2001, 54(2): 59-70.
35. Liu, R., Liu, I.Y., Bi, X., Thompson, R.F.,Doctrow, S.R., Malfroy, B. and Baudry, M.Reversal of age-related learning deficits andbrain oxidative stress in mice with superoxidedismutase/catalase mimetics. Proc Natl AcadSci U S A 2003, 100(14): 8526-31.
36. Gervais, F.G., Xu, D., Robertson, G.S. et al.Involvement of caspases in proteolytic clae-vage of Alzheimer’s amyloid-β precursor pro-tein and amyloidogenic A-β peptide formation.Cell 1999, 97(3): 395-406.
37. Francis, P.T. The interplay of neurotransmittersin Alzheimer’s disease. CNS Spectr 2005,10(11 Suppl 18): 6-9.
38. Black, S.E., Doody, R., Li, H. et al. Donepezilpreserves cognition and global function inpatients with severe Alzheimer disease.Neurology 2007, 69(5): 459-69.
39. Winblad, B., Cummings, J., Andreasen, N. etal. A six-month doubleblind, randomized,placebo-controlled study of a transdermalpatch in Alzheimer’s disease-rivastigminepatch versus capsule. Int J Geriatr Psychiatry2007, 22(5): 456-67.
Drug News Perspect 22(6), July/August 2009 LOOKING AHEAD
337M. Boada et al. pp. 325-339

40. Burns, A., Bernabei, R., Bullock, R. et al.Safety and efficacy of galantamine (Reminyl) insevere Alzheimer’s disease (the SERAD study):a randomised, placebo-controlled, double-blind trial. Lancet Neurol 2009, 8(1): 39-47.
41. Boada Rovira, M., Brodaty H., Cras P. et al.Efficacy and safety of donepezil in patientswith Alzheimer’s disease. Results of a global,multinational, clinical experience study. DrugsAging 2004, 21(1): 43-53.
42. Reisberg, B., Doody, R., Stoffler, A., Schmitt,F., Ferris, S. and Mobius, H.J. Memantine inmoderate-to-severe Alzheimer’s disease. NEngl J Med 2003, 348(14): 1333-41.
43. Selkoe, D.J. and Schenk, D. Alzheimer’s dis-ease: Molecular understanding predicts amy-loid-based therapeutics. Annu Rev PharmacolToxicol 2003, 43: 545-84.
44. Sorbera, L.A., Bozzo, J. and Serradell, N.Alzheimer’s disease one century later: Thesearch for effective therapeutic targets contin-ues. Drugs Fut 2007, 32(7): 625-34.
45. Smith, W.W., Gorospe, M. and Kusiak, J.W.Signaling mechanisms underlying Aβ toxicity:potential therapeutic targets for Alzheimer’sdisease. CNS Neurol Disord Drug Targets2006, 5(3): 355-61.
46. Aisen, P.S. The development of anti-amyloidtherapy for Alzheimer’s disease: from secretasemodulators to polymerisation inhibitors. CNSDrugs 2005, 19(12): 989-96.
47. Shaw, K.T., Utsuki, T., Rogers, J. et al.Phenserine regulates translation of β-amyloidprecursor protein mRNA by a putative inter-leukin-1 responsive element, a target for drugdevelopment. Proc Natl Acad Sci U S A 2001,98(13): 7605-10.
48. Venti, A., Giordano, T., Eder, P., Bush, A.I.,Lahiri, D.K., Greig, N.H. and Rogers, J.T. Theintegrated role of desferrioxamine andphenserine targeted to an iron-responsive ele-ment in the APP-mRNA 5’-untranslatedregion. Ann N Y Acad Sci 2004, 1035: 34-48.
49. Allinson, T.M., Parkin, E.T., Turner, A.J. andHooper, N.M. ADAMs family members as amy-loid precursor protein α-secretases. J NeurosciRes 2003, 74(3): 342-52.
50. Tanabe, C., Hotoda, N., Sasagawa, N.,Sehara-Fujisawa, A., Maruyama, K. andIshiura, S. ADAM19 is tightly associated withconstitutive Alzheimer’s disease APP α-secre-tase in A172 cells. Biochem Biophys ResCommun 2007, 352(1): 111-7.
51. Fahrenholz, F. α-Secretase as a therapeutictarget. Curr Alzh Res 2007, 4: 412-7.
52. Postina, R., Schroeder, A., Dewachter, I. et al.A disintegrin-metalloproteinase prevents amy-loid plaque formation and hippocampaldefects in an Alzheimer disease mouse model.J Clin Invest 2004, 113(10): 1456-64.
53. Kojro, E., Gimpl, G., Lammich, S., Marz, W.and Fahrenholz, F. Low cholesterol stimulatesthe nonamyloidogenic pathway by its effect onthe α-secretase ADAM 10. Proc Natl Acad SciU S A 2001, 98(10): 5815-20.
54. Kirsch, C., Eckert, G.P., Koudinov, A.R. andMüller, W.E. Brain cholesterol, statins andAlzheimer’s sisease. Pharmacopsychiatry2003, 36(Suppl 2): S113-9.
55. Pogacic, V. and Herrling, P. List of drugs indevelopment for neurodegenerative diseases.Update June 2007. Neurodegener Dis 2007,4(6): 443-86.
56. De Strooper, B., Craessaerts, K., Van Leuven,F. and Van Den Berghe, H. Exchanging theextracellular domain of amyloid precursor pro-tein for horseradish peroxidase does not inter-fere with α-secretase cleavage of the β-amy-loid region, but randomizes secretion inMadin-Darby canine kidney cells. J Biol Chem1995, 270(51): 30310-4.
57. Venugopal, C., Demos, C.M., Rao, K.S.,Pappolla, M.A. and Sambamurti, K. β-secre-tase: structure, function, and evolution. CNSNeurol Disord Drug Targets 2008, 7(3): 278-94.
58. Willem, M., Lammich, S. and Haass, C.Function, regulation and therapeutic proper-ties of β-secretase (BACE1). Semin Cell DevBiol 2009, 20(2): 175-82.
59. John, V. Human β-secretase (BACE) and BACEinhibitors: progress report. Curr Top MedChem 2006, 6(6): 569-78.
60. Safety study of CTS21166 to treat Alzheimerdisease. ClinicalTrials.gov IdentifierNCT00621010. Accessed July 27, 2009.
61. Godin, C., Auclair, A., Ferland, M., Hebert,S.S., Carreau, M. and Levesque, G. Presenilin-1 is indirectly implicated in Notch1 cleavage.Neuroreport 2003, 14(12): 1613-6.
62. Searfoss, G.H., Jordan, W.H., Calligaro, D.O. etal. Adipsin, a biomarker of gastrointestinal toxi-city mediated by a functional γ-secretaseinhibitor. J Biol Chem 2003, 278(46): 46107-16.
63. Wolfe, M.S. Selective amyloid-β loweringagents. BMC Neurosci 2008, 9(Suppl 2): S4-7.
64. Lorenzo, A. and Yankner, B.A. β-amyloid neu-rotoxicity requires fibril formation and is inhib-ited by congo red. Proc Natl Acad Sci U S A1994, 91(25): 12243-7.
65. Yamin, G., Ono, K., Inayathullah, M. andTeplow, D.B. Amyloid β-protein assembly as atherapeutic target of Alzheimer’s disease. CurrPharm Des 2008, 14(30): 3231-46.
66. Woodhouse, A., Dickson, T.C. and Vickers, J.C.Vaccination strategies for Alzheimer’s disease:A new hope? Drugs Aging 2007, 24(2): 107-19.
67. Orgogozo, J.M., Gilman, S., Dartigues, J.F., etal. Subacute meningoencephalitis in a subset
of patients with AD after Aβ immunization.Neurology 2003, 61(1): 46-54.
68. Ferrer, I., Boada, M., Sánchez, M.L., Rey, M.J.and Costa-Jussá, F. Neuropathology andpathogenetics of encephalitis following amy-loid-β immunization in Alzheimer’s Disease.Brain Pathol 2004, 14(1): 11-20.
69. Solomon, B., Koppel, R., Frankel, D. andHanan-Aharon, E. Disaggregation ofAlzheimer β-amyloid by site-directed mAb.Proc Natl Acad Sci U S A 1997, 94(8): 4109-12.
70. Banks, W.A., Terrell, B., Farr, S.A., Robinson,S.M., Nonaka, N. and Morley, J.E. Passage ofamyloid β protein antibody across the blood-brain barrier in a mouse model of Alzheimer’sdisease. Peptides 2002, 23(12): 2223-6.
71. Deane, R., Sagare, A., Hamm, K. et al. IgG-assisted age-dependent clearance ofAlzheimer’s amyloid β peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci2005, 25(50): 11495-503.
72. Lannfelt, L., Blennow, K., Zetterberg, H. et al.Safety, efficacy, and biomarker findings ofPBT2 in targeting Aβ as a modifying therapyfor Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial.Lancet Neurol 2008, 7(9): 779-86.
73. Cuajungco, M.P., Frederickson, C.J. and BushAI. Amyloid-β metal interaction and metalchelation. Subcell Biochem 2005, 38: 235-54.
74. Shibata, M., Yamada, S., Kumar, S.R. et al.Clearance of Alzheimer’s amyloid-ss(1-40)peptide from brain by LDL receptor-relatedprotein-1 at the blood-brain barrier. J ClinInvest 2000, 106(12): 1489-99.
75. Deane, R., Du Yan, S., Submamaryan, R.K. etal. RAGE mediates amyloid-β peptide trans-port across the blood-brain barrier and accu-mulation in brain. Nat Med 2003, 9(7): 907-13.
76. Grimmer, T., Riemenschneider, M., Förstl, H.et al. Beta amyloid in Alzheimer’s disease:increased deposition in brain is reflected inreduced concentration in cerebrospinal fluid.Biol Psychiatry 2009, 65(11): 927-34.
77. Deane, R., Wu, Z., Zlokovic, B.V. RAGE (yin)versus LRP (yang) balance regulates alzheimeramyloid β-peptide clearance through trans-port across the blood-brain barrier. Stroke2004, 35(11 Suppl 1): 2628-31.
78. DeMattos, R.B., Bales, K.R., Cummins, D.J.,Dodart, J.C., Paul, S.M., Holtzman, D.M.Peripheral anti-A β antibody alters CNS andplasma A β clearance and decreases brain A βburden in a mouse model of Alzheimer’s dis-ease. Proc Natl Acad Sci U S A 2001, 98(15):8850-5.
79. Bambauer, R. and Arnold, A. Plasmapheresiswith a substitution solution of human serumprotein (5%) versus plasmapheresis with asubstitution solution of human albumin (5%)
LOOKING AHEAD Drug News Perspect 22(6), July/August 2009
338 M. Boada et al. pp. 325-339

in patients suffering from autoimmune dis-eases. Artif Organs 1999, 23(12): 1079-87.
80. Meca-Lallana, J.E., Rodríguez-Hilario, H.,Martínez-Vidal, S. et al. Plasmapheresis: itsuse in multiple sclerosis and other demyelinat-ing processes of the central nervous system. Anobservation study. Rev Neurol 2003, 37(10):917-26.
81. Dyck, P.J., Litchy, W.J., Kratz, K.M. et al. Aplasma exchange versus immune globulininfusion trial in chronic inflammatory demyeli-nating polyradiculoneuropathy. Ann Neurol1994, 36(6): 838-45.
82. Weinshenker, B.G., O’Brien, P.C., Petterson,T.M. et al. A randomized trial of plasmaexchange in acute central nervous systeminflammatory demyelinating disease. AnnNeurol 1999, 46(6): 878-86.
83. Mazzi, G., Raineri, A., Zucco, M., Passadore,P., Pomes, A., Orazi, B.M. Plasma exchange inchronic peripheral neurological disorders. Int JArtif Organs 1999, 22(1): 40-6.
84. Biere, A.L., Ostaszewski, B., Stimson, E.R.,Hyman, B.T., Maggio, J.E., Selkoe, D.J.Amyloid β-peptide is transported on lipopro-teins and albumin in human plasma. J BiolChem 1996, 271(51): 32916-22.
85. Costa, M., Ortiz, A.M. and Jorquera, J.I.Binding of a β-amyloid 1-42 peptide to HumanAlbumin Grifols®. Alzheimers Dement 2009,5(4): P417.
86. Giménez-Roldán, S., Novillo, M.J., Navarro,E., Dobato, J.L. and Giménez-Zuccarelli, M.Mini-mental state examination: proposal ofprotocol to be used. Rev Neurol 1997, 25(140):576-83.
87. Wouters, H., van Gool, W.A., Schmand, B. andLindeboom, R. Revising the ADAS-cog for amore accurate assessment of cognitive impair-ment. Alzheimer Dis Assoc Disord 2008,22(3): 236-44.
88. Junghöfer, M., Peyk, P., Flaisch, T., Schupp,H.T. Neuroimaging methods in affective neuro-science: selected methodological issues. ProgBrain Res 2006, 156: 123-43.
89. McKhann, G., Drachman, D., Folstein, M.,Katzman, R., Price, D. and Stadlan, E.M.Clinical diagnosis of Alzheimer’s disease:report of the NINCDS-ADRDA Work Groupunder the auspices of Department of Healthand Human Services Task Force on Alzheimer’sdisease. Neurology 1984, 34(7): 939-44.
90. European Medicines Agency. Committee forMedicinal Products for Human Use (CHMP).
Guidelines on Medicinal Products for theTreatment of Alzheimer’s Disease and OtherDementias. London, 24 July 2008, Doc. Ref.CPMP/EWP/553/95 Rev. 1.
Mercè Boada* works at the ACE Foundation of theCatalan Institute of Applied Neurosciences and theNeurology Service of University General Hospital Valld’Hebron, Barcelona, Spain. Isabel Hernández, LluísTárraga and Mar Buendia also work for the ACEFoundation of the Catalan Institute of AppliedNeurosciences in Barcelona. Pilar Ortiz, Joan Muñoz andRamón P. Pla work at the Blood and Tissue Bank of theUniversity General Hospital Vall d’Hebron, Barcelona,Spain. Fernando Anaya works for the Nephrology Serviceof University Hospital Gregorio Marañón in Madrid,Spain. Laura Núñez and Antonio Páez work in theClinical Trials Department of Instituto Grifols S.A.,Barcelona, Spain. Javier Olazarán works at theNeurology Service of the University Hospital GregorioMarañón, Madrid, Spain. Isabel Roca works in theNuclear Medicine Service at Corachan Clinic, Barcelona,Spain. Gemma Cuberas works in the Nuclear MedicineService at University General Hospital Vall d’Hebron,Barcelona, Spain. Isidre Ferrer works at theNeuropathology Institute of the University GeneralHospital Bellvitge, Barcelona, Spain. *Correspondence:Mercè Boada, Fundació ACE, Institut Català deNeurociències Aplicades, Marquès de Sentmenat, 57,08029 Barcelona, Spain. Tel.: +34 934 447 318; Fax: +34934 101 701; E-mail: [email protected]
Drug News Perspect 22(6), July/August 2009 LOOKING AHEAD
339M. Boada et al. pp. 325-339