alpha1beta1 and integrin-linked kinase interact and modulate ... · alpha1beta1 and integrin-linked...
TRANSCRIPT

lable at ScienceDirect
Atherosclerosis 243 (2015) 477e485
Contents lists avai
Atherosclerosis
journal homepage: www.elsevier .com/locate/atherosclerosis
Alpha1beta1 and integrin-linked kinase interact and modulateangiotensin II effects in vascular smooth muscle cells
Jo~ao Alfredo Moraes a, Ana Clara Frony a, Aline Maria Dias a, Mariana Renovato-Martins a,Genilson Rodrigues a, Cezary Marcinkiewicz b, Jamil Assreuy c, Christina Barja-Fidalgo a, *
a Laboratory of Cellular and Molecular Pharmacology, Department of Cell Biology, IBRAG, Universidade do Estado do Rio de Janeiro (UERJ), Rio de Janeiro, RJ,Brazilb Department of Bioengineering, College of Engineering, Temple University, Philadelphia, PA, USAc Department of Pharmacology, Universidade Federal de Santa Catarina (UFSC), Florian�opolis, SC, Brazil
a r t i c l e i n f o
Article history:Received 8 June 2015Received in revised form2 September 2015Accepted 17 September 2015Available online 25 September 2015
Keywords:Vascular smooth muscle cellAngiotensin IINOXAlpha1beta1 integrinILKObtustatin
* Corresponding author. Laborat�orio de FarmacDepartment of Cell Biology, Universidade do Estado dode Setembro 87fds, Vila Isabel, Rio de Janeiro, RJ, 205
E-mail address: [email protected] (C. Barja-Fid
http://dx.doi.org/10.1016/j.atherosclerosis.2015.09.0260021-9150/© 2015 Elsevier Ireland Ltd. All rights rese
a b s t r a c t
The effects of angiotensin II (Ang II) on vascular smooth muscle cells (VSMC) are modulated by reactiveoxygen species (ROS) and also involve integrin engagement. However, the potential link betweenalpha1beta1 integrin signaling with NOX system and their combined contribution to Ang II effects onVSMC have not been investigated. We aimed to elucidate the moslecular mechanisms underlying theactivation of these two pathways in Ang II effects on VSMC.
Ang II-induced VSMC migration (2-fold increase) and proliferation (2.5-fold increase) is modulated byalpha1beta1 integrin, being inhibited by obtustatin, a specific alpha1beta1 integrin blocker. Ang II alsostimulates ROS production in VSMC (140%) that is NOX1 dependent, being completely inhibited in NOX1silenced cells. The ROS production develops in two peaks, and the second peak is maintained by NOX2activation. Apocynin and obtustatin inhibit the NOX2-associated second peak, but not the first peak ofROS production, which is related to NOX1 activation. Corroborating the involvement of alpha1beta1integrin, the pretreatment of VSMC with obtustatin impaired Ang II-induced FAK phosphorylation, AKTactivation, p21 degradation and the increase of ILK expression. Silencing of ILK blocked cell migration,AKT phosphorylation and the second peak of ROS, but partially inhibits (70%) VSMC proliferation inducedby Ang II.
The data demonstrate a novel role for NOX2 in Ang II effects on VSMC, and suggest alpha1beta1integrin and ILK as target molecules to the development of more effective therapeutic interventions incardiovascular diseases.
© 2015 Elsevier Ireland Ltd. All rights reserved.
1. Introduction
The initial step for the development of cardiovascular diseasesinvolves an inflammatory process that leads to vascular injuryassociated to endothelial damage, extracellular matrix exposure,aggregation and adhesion of platelets and leukocytes, and theactivation of vascular smoothmuscle cells (VSMC). VSMC activationcan be amplified by the release/production of inflammatory me-diators, which in turn induces VSMC migration and proliferation[1,2], leading to neointimal hyperplasia (related to restenosis) [3,4]
ologia Celular e Molecular,Rio de Janeiro (UERJ), Av. 2851-030, Brazil.algo).
rved.
and fibrous cap generation (related to atherosclerosis) [5,6].The increase in angiotensin II (Ang II) levels is an early sign of
cardiovascular disease. Moreover, Ang II levels remain abovenormal even after the disease is established [7]. Several studieshave shown that Ang II activates VSMC, promoting cell migrationand proliferation [8e11]. These effects are mediated by integrinsand rely, to a great extent, on NOX activity [11,12].
NOX4 is reported to be responsible for the homeostatic pro-duction of reactive oxygen species (ROS) [13,14] by VSMC, whereasAng II receptor 1 (AT1R) directly activates NOX1, thus increasingROS production [9]. On the other hand, the involvement NOX2, themain inducible isoform of NOX in inflammatory processes [15], anda putative player this scenario, has not yet been fully described incardiovascular diseases.
Integrins have a crucial role in VSMC activation. The main

J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485478
integrin present in VSMC is alphavbeta3, which binds to vitronectinand osteopontin [16,17], two extracellular matrix proteins involvedin different VSMC functions. The blockage of alphavbeta3 integrinby antibodies or selective peptide ligands (known as disintegrins)[18] prevents VSMC migration and emerge as a promising thera-peutic option to impair the development of neointimal hyperplasia[17e19]. A recent study from our group showed that leukotriene B4,a known pro-inflammatory mediator, activates VSMC through aninside-out alphav-beta3 activation-dependent mechanism [18].Another important integrin present in VSMC is alpha1beta1, whichbinds to collagen type IV, known to be increased in atheromatousplaque [20]. Early studies have suggested that alpha1beta1 integrincould be involved in the effect of Ang II-induced arterial wall hy-pertrophy [21] and VSMC proliferation [22], and also in the stabi-lization of the atheromatous plaque [23].
Integrin activation triggers multi-vectorial signaling pathwaysthat can modulate the cell fate. The activation of Focal AdhesionKinase (FAK) and Integrin Linked Kinase (ILK) are, in many cells, theprimary signals required for integrin link with the cytoskeleton andfor signal transduction [24]. FAK is well described as a scaffold forother signaling accessory proteins and enzymes, including ILK. Thislatter enzymeworks as a double-function protein, acting both as anadaptor molecule of actin cytoskeleton, as well as a kinase. ILK isable to induce AKT phosphorylation and p21 degradation, twopathways involved in VSMC migration and proliferation [25,26].
The putative involvement of alpha1beta1 integrin and ILK inAng II effects on VSMC has been individually reported [21,27].However, there are no studies investigating a potential link be-tween both molecules on Ang-II-mediated VSMC activation,neither their association with NOX activity and with the redoxsensitive mechanisms that may underlie the activation of integrinsignaling triggered by Ang II.
In the present report, we have investigated the inside-outsignaling mechanisms involved in the activation of alpha1beta1integrin and its association with the NOX system in VSMC. To thisend, we used a selective alpha1beta1 integrin blocker, namelyobtustatin, a disintegrin isolated from Vipera lebetina obtusa snakevenom [28,29], which helped us to provide hitherto unknown in-formation on the molecular mechanisms involved in integrinsignaling, NOX activation and Ang II effects on VSMC.
2. Material and methods
2.1. Reagents
All reagents are listed in Supplementary methods. Obtustatinwas purified from V. lebetina obtusa venom as early described [29].
2.2. Cell culture
Primary VSMC were isolated from the aortic arch from naïveWistar male rats using an explant method. The protocol wasapproved by the Committee on the Ethics of Animal Experimentalof Universidade do Estado do Rio de Janeiro. (CEUA/003/2013). A7r5VSMC obtained of rat thoracic aorta were originally from theAmerican Type Culture Collection (Rockville, MD USA). Cell cultureis better explained in Supplementary methods.
2.3. mRNA silencing
A7r5 VSMC were transfected with siRNA for 24 h and attenua-tion of ILK, NOX1 or NOX2 expression was verified by Westernblotting. All process of cell silencing is better described inSupplementary methods.
2.4. Cell migration assay
Chemotaxis assay was accomplished in a modified BoydenChamber after 4 h of migration. This assay is better described inSupplementary methods.
2.5. Reactive oxygen species production
ROS production was measured using CM-H2DCFDA probe. ROSassay is better described in Supplementary methods.
2.6. Immunofluorescence microscopy
After 1 h of treatment VSMC were fixed and p47phox, actin andnuclei were stained. p47phox translocation to cell membrane wasquantified in Adobe Photoshop Software. This assay is betterdescribed in Supplementary methods.
2.7. Western blotting
Total protein content in the cell extracts was determined by theBCA method. Samples were submitted to Western blotting assay(described in Supplementary methods).
2.8. Cell proliferation assay
Cell proliferation was measured by cell cycle analysis by flowcytometry and by MTT assay after 24 h of treatment. These assaysare better described in Supplementary methods.
2.9. Statistical analysis
Statistical analysis was done using one-way analysis of variance(ANOVA) followed by Bonferroni post-hoc test and p values < 0.05were considered significant.
3. Results
3.1. Ang II activates vascular smooth muscle cells via alpha1beta1integrin and NOX2
Previous studies have established a role for alpha1beta1 integrinas a modulator of VSMC activation and proliferation [8,21,22]. Toassess the chemotactic effect of Ang II on primary VSMC, we usedmodified Boyden chambers. As shown in Fig. 1A, Ang II had a potentchemotactic activity on VSMC and the treatment of cells withobtustatin, which selectively blocks alpha1beta1 integrin [28,29],inhibited migration towards Ang II.
Ang II increased ROS production in primary VSMC reaching apeak after 1 h and remaining high up to 3 h (Fig. 1B). The pre-treatment with obtustatin did not change ROS production duringthe first 30 min of incubation with Ang II, but levels of ROS weremaintained constant until 1 h and decreased to control. Obtustatinalone was devoid of effect on ROS production. The detailed analysisof ROS production and the effect of obtustatin have shown that ROSproduction induced in smooth muscle cells occurred in two stages,and that obtustatin inhibited only the second part (0.5e1 h) of ROSproduction (see Table 1 in Ref [30]).
To be sure that the source of ROS in smooth muscle cells couldbe ascribed to by NOX, cells treated with Ang II and incubated withDPI, a paninhibitor of NOX, were rendered incapable of ROS pro-duction upon incubationwith Ang II. On the other hand, apocynin, aselective inhibitor of p47phox (a NOX2 subunit), inhibited only thesecond peak of ROS (0.5e1 h) production in Ang II-stimulated cells,an effect similar to that of obtustatin (Fig. 1C; see Fig. 1 in Ref [30]).
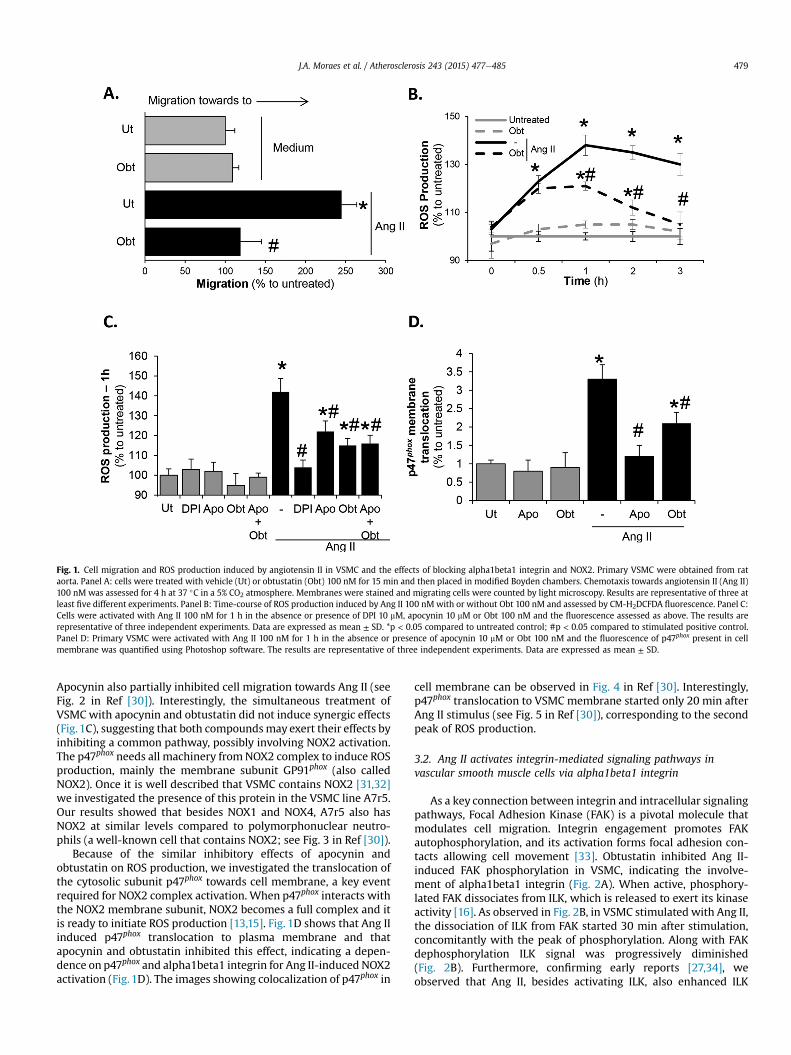
Fig. 1. Cell migration and ROS production induced by angiotensin II in VSMC and the effects of blocking alpha1beta1 integrin and NOX2. Primary VSMC were obtained from rataorta. Panel A: cells were treated with vehicle (Ut) or obtustatin (Obt) 100 nM for 15 min and then placed in modified Boyden chambers. Chemotaxis towards angiotensin II (Ang II)100 nM was assessed for 4 h at 37 �C in a 5% CO2 atmosphere. Membranes were stained and migrating cells were counted by light microscopy. Results are representative of three atleast five different experiments. Panel B: Time-course of ROS production induced by Ang II 100 nMwith or without Obt 100 nM and assessed by CM-H2DCFDA fluorescence. Panel C:Cells were activated with Ang II 100 nM for 1 h in the absence or presence of DPI 10 mM, apocynin 10 mM or Obt 100 nM and the fluorescence assessed as above. The results arerepresentative of three independent experiments. Data are expressed as mean ± SD. *p < 0.05 compared to untreated control; #p < 0.05 compared to stimulated positive control.Panel D: Primary VSMC were activated with Ang II 100 nM for 1 h in the absence or presence of apocynin 10 mM or Obt 100 nM and the fluorescence of p47phox present in cellmembrane was quantified using Photoshop software. The results are representative of three independent experiments. Data are expressed as mean ± SD.
J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485 479
Apocynin also partially inhibited cell migration towards Ang II (seeFig. 2 in Ref [30]). Interestingly, the simultaneous treatment ofVSMC with apocynin and obtustatin did not induce synergic effects(Fig.1C), suggesting that both compoundsmay exert their effects byinhibiting a common pathway, possibly involving NOX2 activation.The p47phox needs all machinery fromNOX2 complex to induce ROSproduction, mainly the membrane subunit GP91phox (also calledNOX2). Once it is well described that VSMC contains NOX2 [31,32]we investigated the presence of this protein in the VSMC line A7r5.Our results showed that besides NOX1 and NOX4, A7r5 also hasNOX2 at similar levels compared to polymorphonuclear neutro-phils (a well-known cell that contains NOX2; see Fig. 3 in Ref [30]).
Because of the similar inhibitory effects of apocynin andobtustatin on ROS production, we investigated the translocation ofthe cytosolic subunit p47phox towards cell membrane, a key eventrequired for NOX2 complex activation. When p47phox interacts withthe NOX2 membrane subunit, NOX2 becomes a full complex and itis ready to initiate ROS production [13,15]. Fig. 1D shows that Ang IIinduced p47phox translocation to plasma membrane and thatapocynin and obtustatin inhibited this effect, indicating a depen-dence on p47phox and alpha1beta1 integrin for Ang II-induced NOX2activation (Fig. 1D). The images showing colocalization of p47phox in
cell membrane can be observed in Fig. 4 in Ref [30]. Interestingly,p47phox translocation to VSMC membrane started only 20 min afterAng II stimulus (see Fig. 5 in Ref [30]), corresponding to the secondpeak of ROS production.
3.2. Ang II activates integrin-mediated signaling pathways invascular smooth muscle cells via alpha1beta1 integrin
As a key connection between integrin and intracellular signalingpathways, Focal Adhesion Kinase (FAK) is a pivotal molecule thatmodulates cell migration. Integrin engagement promotes FAKautophosphorylation, and its activation forms focal adhesion con-tacts allowing cell movement [33]. Obtustatin inhibited Ang II-induced FAK phosphorylation in VSMC, indicating the involve-ment of alpha1beta1 integrin (Fig. 2A). When active, phosphory-lated FAK dissociates from ILK, which is released to exert its kinaseactivity [16]. As observed in Fig. 2B, in VSMC stimulated with Ang II,the dissociation of ILK from FAK started 30 min after stimulation,concomitantly with the peak of phosphorylation. Along with FAKdephosphorylation ILK signal was progressively diminished(Fig. 2B). Furthermore, confirming early reports [27,34], weobserved that Ang II, besides activating ILK, also enhanced ILK
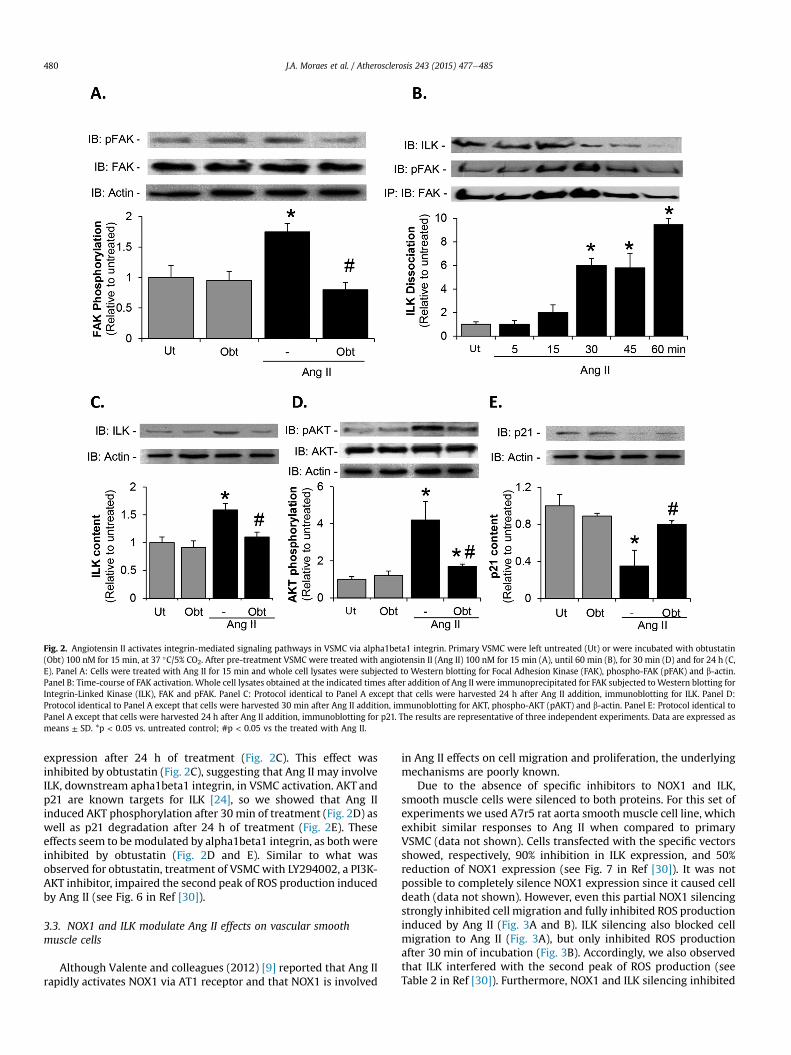
Fig. 2. Angiotensin II activates integrin-mediated signaling pathways in VSMC via alpha1beta1 integrin. Primary VSMC were left untreated (Ut) or were incubated with obtustatin(Obt) 100 nM for 15 min, at 37 �C/5% CO2. After pre-treatment VSMC were treated with angiotensin II (Ang II) 100 nM for 15 min (A), until 60 min (B), for 30 min (D) and for 24 h (C,E). Panel A: Cells were treated with Ang II for 15 min and whole cell lysates were subjected to Western blotting for Focal Adhesion Kinase (FAK), phospho-FAK (pFAK) and b-actin.Panel B: Time-course of FAK activation. Whole cell lysates obtained at the indicated times after addition of Ang II were immunoprecipitated for FAK subjected toWestern blotting forIntegrin-Linked Kinase (ILK), FAK and pFAK. Panel C: Protocol identical to Panel A except that cells were harvested 24 h after Ang II addition, immunoblotting for ILK. Panel D:Protocol identical to Panel A except that cells were harvested 30 min after Ang II addition, immunoblotting for AKT, phospho-AKT (pAKT) and b-actin. Panel E: Protocol identical toPanel A except that cells were harvested 24 h after Ang II addition, immunoblotting for p21. The results are representative of three independent experiments. Data are expressed asmeans ± SD. *p < 0.05 vs. untreated control; #p < 0.05 vs the treated with Ang II.
J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485480
expression after 24 h of treatment (Fig. 2C). This effect wasinhibited by obtustatin (Fig. 2C), suggesting that Ang II may involveILK, downstream apha1beta1 integrin, in VSMC activation. AKT andp21 are known targets for ILK [24], so we showed that Ang IIinduced AKT phosphorylation after 30 min of treatment (Fig. 2D) aswell as p21 degradation after 24 h of treatment (Fig. 2E). Theseeffects seem to bemodulated by alpha1beta1 integrin, as both wereinhibited by obtustatin (Fig. 2D and E). Similar to what wasobserved for obtustatin, treatment of VSMCwith LY294002, a PI3K-AKT inhibitor, impaired the second peak of ROS production inducedby Ang II (see Fig. 6 in Ref [30]).
3.3. NOX1 and ILK modulate Ang II effects on vascular smoothmuscle cells
Although Valente and colleagues (2012) [9] reported that Ang IIrapidly activates NOX1 via AT1 receptor and that NOX1 is involved
in Ang II effects on cell migration and proliferation, the underlyingmechanisms are poorly known.
Due to the absence of specific inhibitors to NOX1 and ILK,smooth muscle cells were silenced to both proteins. For this set ofexperiments we used A7r5 rat aorta smooth muscle cell line, whichexhibit similar responses to Ang II when compared to primaryVSMC (data not shown). Cells transfected with the specific vectorsshowed, respectively, 90% inhibition in ILK expression, and 50%reduction of NOX1 expression (see Fig. 7 in Ref [30]). It was notpossible to completely silence NOX1 expression since it caused celldeath (data not shown). However, even this partial NOX1 silencingstrongly inhibited cell migration and fully inhibited ROS productioninduced by Ang II (Fig. 3A and B). ILK silencing also blocked cellmigration to Ang II (Fig. 3A), but only inhibited ROS productionafter 30 min of incubation (Fig. 3B). Accordingly, we also observedthat ILK interfered with the second peak of ROS production (seeTable 2 in Ref [30]). Furthermore, NOX1 and ILK silencing inhibited
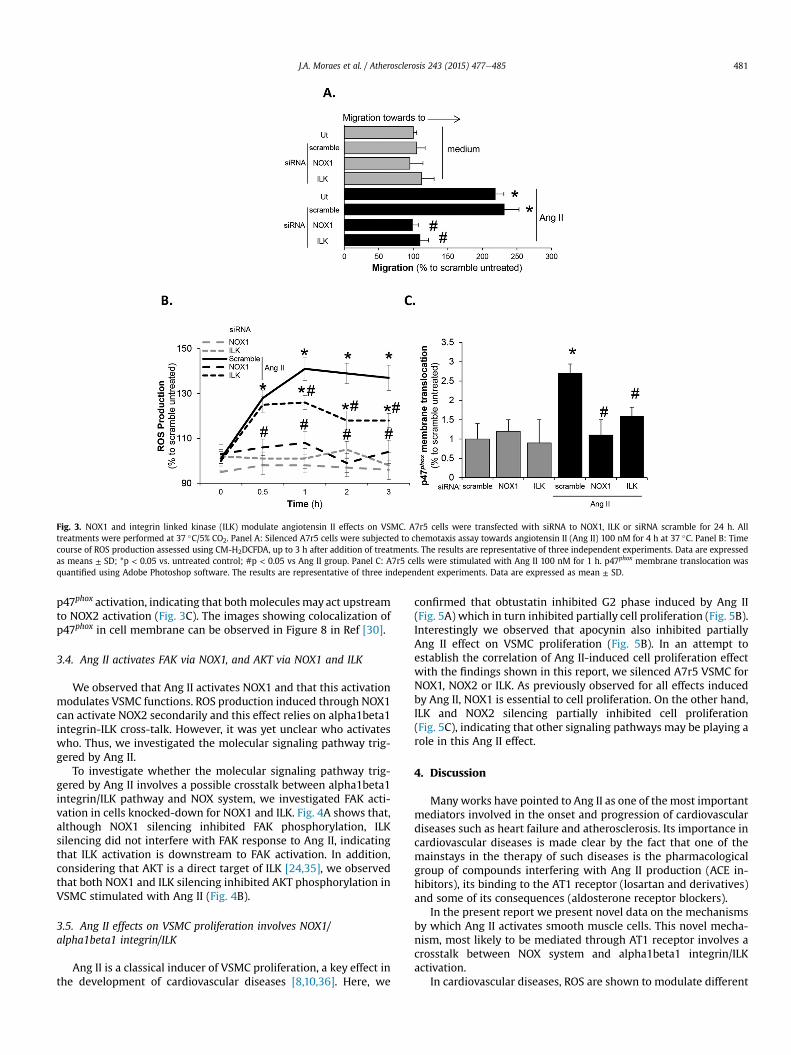
Fig. 3. NOX1 and integrin linked kinase (ILK) modulate angiotensin II effects on VSMC. A7r5 cells were transfected with siRNA to NOX1, ILK or siRNA scramble for 24 h. Alltreatments were performed at 37 �C/5% CO2. Panel A: Silenced A7r5 cells were subjected to chemotaxis assay towards angiotensin II (Ang II) 100 nM for 4 h at 37 �C. Panel B: Timecourse of ROS production assessed using CM-H2DCFDA, up to 3 h after addition of treatments. The results are representative of three independent experiments. Data are expressedas means ± SD; *p < 0.05 vs. untreated control; #p < 0.05 vs Ang II group. Panel C: A7r5 cells were stimulated with Ang II 100 nM for 1 h. p47phox membrane translocation wasquantified using Adobe Photoshop software. The results are representative of three independent experiments. Data are expressed as mean ± SD.
J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485 481
p47phox activation, indicating that bothmoleculesmay act upstreamto NOX2 activation (Fig. 3C). The images showing colocalization ofp47phox in cell membrane can be observed in Figure 8 in Ref [30].
3.4. Ang II activates FAK via NOX1, and AKT via NOX1 and ILK
We observed that Ang II activates NOX1 and that this activationmodulates VSMC functions. ROS production induced through NOX1can activate NOX2 secondarily and this effect relies on alpha1beta1integrin-ILK cross-talk. However, it was yet unclear who activateswho. Thus, we investigated the molecular signaling pathway trig-gered by Ang II.
To investigate whether the molecular signaling pathway trig-gered by Ang II involves a possible crosstalk between alpha1beta1integrin/ILK pathway and NOX system, we investigated FAK acti-vation in cells knocked-down for NOX1 and ILK. Fig. 4A shows that,although NOX1 silencing inhibited FAK phosphorylation, ILKsilencing did not interfere with FAK response to Ang II, indicatingthat ILK activation is downstream to FAK activation. In addition,considering that AKT is a direct target of ILK [24,35], we observedthat both NOX1 and ILK silencing inhibited AKT phosphorylation inVSMC stimulated with Ang II (Fig. 4B).
3.5. Ang II effects on VSMC proliferation involves NOX1/alpha1beta1 integrin/ILK
Ang II is a classical inducer of VSMC proliferation, a key effect inthe development of cardiovascular diseases [8,10,36]. Here, we
confirmed that obtustatin inhibited G2 phase induced by Ang II(Fig. 5A) which in turn inhibited partially cell proliferation (Fig. 5B).Interestingly we observed that apocynin also inhibited partiallyAng II effect on VSMC proliferation (Fig. 5B). In an attempt toestablish the correlation of Ang II-induced cell proliferation effectwith the findings shown in this report, we silenced A7r5 VSMC forNOX1, NOX2 or ILK. As previously observed for all effects inducedby Ang II, NOX1 is essential to cell proliferation. On the other hand,ILK and NOX2 silencing partially inhibited cell proliferation(Fig. 5C), indicating that other signaling pathways may be playing arole in this Ang II effect.
4. Discussion
Manyworks have pointed to Ang II as one of the most importantmediators involved in the onset and progression of cardiovasculardiseases such as heart failure and atherosclerosis. Its importance incardiovascular diseases is made clear by the fact that one of themainstays in the therapy of such diseases is the pharmacologicalgroup of compounds interfering with Ang II production (ACE in-hibitors), its binding to the AT1 receptor (losartan and derivatives)and some of its consequences (aldosterone receptor blockers).
In the present report we present novel data on the mechanismsby which Ang II activates smooth muscle cells. This novel mecha-nism, most likely to be mediated through AT1 receptor involves acrosstalk between NOX system and alpha1beta1 integrin/ILKactivation.
In cardiovascular diseases, ROS are shown to modulate different
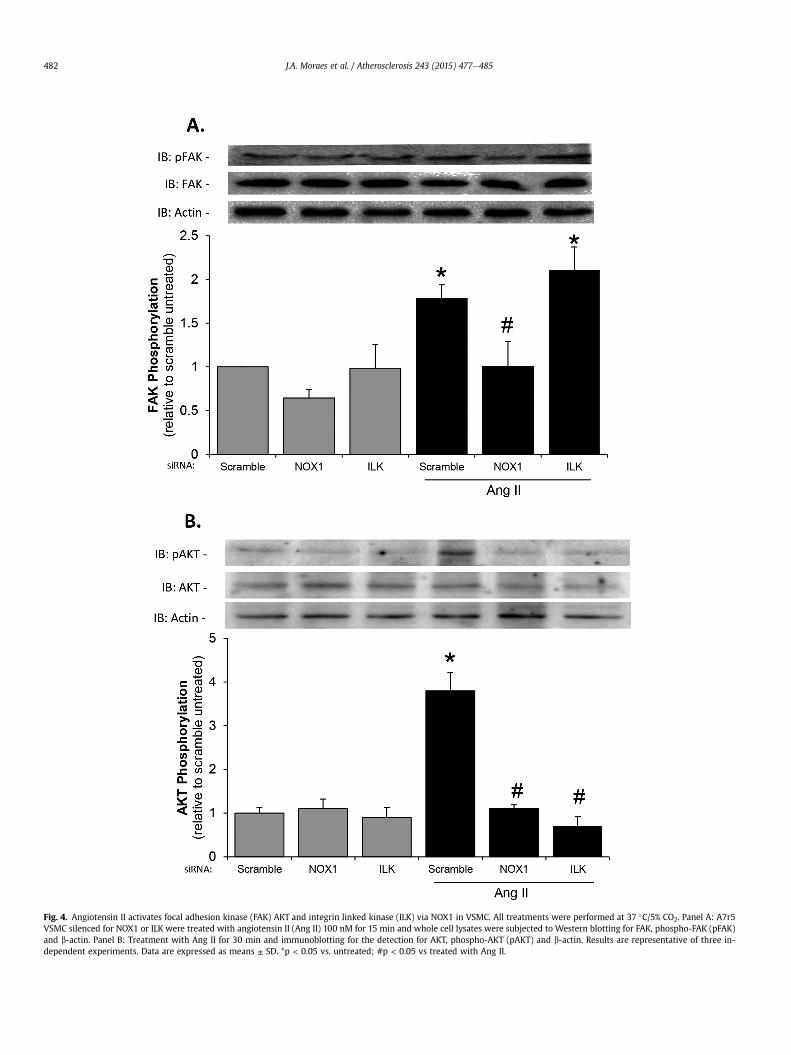
Fig. 4. Angiotensin II activates focal adhesion kinase (FAK) AKT and integrin linked kinase (ILK) via NOX1 in VSMC. All treatments were performed at 37 �C/5% CO2. Panel A: A7r5VSMC silenced for NOX1 or ILK were treated with angiotensin II (Ang II) 100 nM for 15 min and whole cell lysates were subjected to Western blotting for FAK, phospho-FAK (pFAK)and b-actin. Panel B: Treatment with Ang II for 30 min and immunoblotting for the detection for AKT, phospho-AKT (pAKT) and b-actin. Results are representative of three in-dependent experiments. Data are expressed as means ± SD. *p < 0.05 vs. untreated; #p < 0.05 vs treated with Ang II.
J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485482
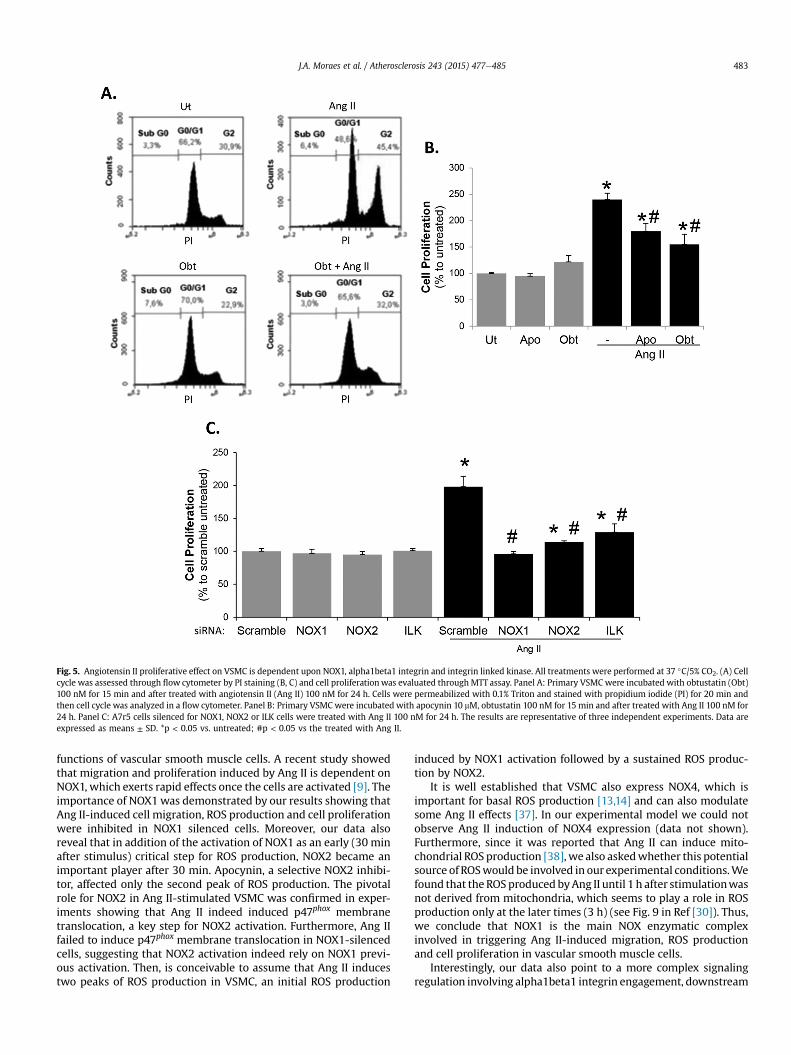
Fig. 5. Angiotensin II proliferative effect on VSMC is dependent upon NOX1, alpha1beta1 integrin and integrin linked kinase. All treatments were performed at 37 �C/5% CO2. (A) Cellcycle was assessed through flow cytometer by PI staining (B, C) and cell proliferationwas evaluated through MTT assay. Panel A: Primary VSMC were incubated with obtustatin (Obt)100 nM for 15 min and after treated with angiotensin II (Ang II) 100 nM for 24 h. Cells were permeabilized with 0.1% Triton and stained with propidium iodide (PI) for 20 min andthen cell cycle was analyzed in a flow cytometer. Panel B: Primary VSMC were incubated with apocynin 10 mM, obtustatin 100 nM for 15 min and after treated with Ang II 100 nM for24 h. Panel C: A7r5 cells silenced for NOX1, NOX2 or ILK cells were treated with Ang II 100 nM for 24 h. The results are representative of three independent experiments. Data areexpressed as means ± SD. *p < 0.05 vs. untreated; #p < 0.05 vs the treated with Ang II.
J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485 483
functions of vascular smooth muscle cells. A recent study showedthat migration and proliferation induced by Ang II is dependent onNOX1, which exerts rapid effects once the cells are activated [9]. Theimportance of NOX1was demonstrated by our results showing thatAng II-induced cell migration, ROS production and cell proliferationwere inhibited in NOX1 silenced cells. Moreover, our data alsoreveal that in addition of the activation of NOX1 as an early (30 minafter stimulus) critical step for ROS production, NOX2 became animportant player after 30 min. Apocynin, a selective NOX2 inhibi-tor, affected only the second peak of ROS production. The pivotalrole for NOX2 in Ang II-stimulated VSMC was confirmed in exper-iments showing that Ang II indeed induced p47phox membranetranslocation, a key step for NOX2 activation. Furthermore, Ang IIfailed to induce p47phox membrane translocation in NOX1-silencedcells, suggesting that NOX2 activation indeed rely on NOX1 previ-ous activation. Then, is conceivable to assume that Ang II inducestwo peaks of ROS production in VSMC, an initial ROS production
induced by NOX1 activation followed by a sustained ROS produc-tion by NOX2.
It is well established that VSMC also express NOX4, which isimportant for basal ROS production [13,14] and can also modulatesome Ang II effects [37]. In our experimental model we could notobserve Ang II induction of NOX4 expression (data not shown).Furthermore, since it was reported that Ang II can induce mito-chondrial ROS production [38], we also askedwhether this potentialsource of ROSwould be involved in our experimental conditions.Wefound that the ROSproduced byAng II until 1 h after stimulationwasnot derived from mitochondria, which seems to play a role in ROSproduction only at the later times (3 h) (see Fig. 9 in Ref [30]). Thus,we conclude that NOX1 is the main NOX enzymatic complexinvolved in triggering Ang II-induced migration, ROS productionand cell proliferation in vascular smooth muscle cells.
Interestingly, our data also point to a more complex signalingregulation involving alpha1beta1 integrin engagement, downstream

J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485484
to NOX1 and upstream to NOX2. Obtustatin, which selectively bindsto that integrin, inhibited only the second peak of ROS production,and partially inhibited VSMCmigration and proliferation induced byAng II. It is important to remark thatwepreviously observed that AngII treatment was able to activate alpha1beta1 integrin, leading toVSMC adhesion to collagen IV, and obtustatin inhibited this effect(data not shown).
Previous evidence has shown a pivotal role for alpha1beta1integrin in the development and progression of atherosclerosis.Due to its property of selectively binding to collagen, alpha1beta1integrin contributes to smoothmuscle cell proliferation and arterialstiffness [21e23]. In this scenario, ILK emerges as an attractivemolecule to be investigated since its activation by integrins canintegrate many signaling pathways associated to different cell re-sponses of smooth muscle cells [16,24,27,39].
We propose that Ang II induces NOX1 activation, which directlyleads to the engagement of alpha1beta1 integrin that in turn leadsto FAK activation. The increase in FAK phosphorylation is transient(with a peak in 30 min) and it is related to its dissociation from ILKthat becomes active, and thus induces AKT phosphorylation atSer473. The integrin activation in many cell types is well known toinduce, via PI3K-PDK, the phosphorylation of AKT at Thr308, whichis involved in the modulation of cell migration [10,25]. However,AKT further phosphorylation is a key step to ensure full AKTactivity[39].We observed that AKT phosphorylation at Ser473 was inhibitedin ILK-silenced cells. Furthermore, the treatment of VSMC with thePI3K inhibitor LY294002 inhibited only the second peak of ROSinduced by Ang II (see Fig. 6 in Ref [30]). Together, these resultssuggest that a full AKT activation by ILK is necessary tomaintain theendogenous production of ROS, which in turn is known to modu-late VSMC proliferation [11]. Confirming the importance of ILKactivation in these processes, Ang II-induced cell migration wasinhibited by obtustatin and in ILK-silenced cells. Moreover, simi-larly to obtustatin or apocynin, the silencing of ILK inhibited onlythe second peak of ROS production associated to NOX2 activation,and reduced cell proliferation. Then, our data reveals a very intri-cate signaling pathway underlying the activation of alpha1beta1integrin and ILK to trigger NOX2 activation to maintain the pro-duction of ROS, and stimulate VSMC proliferation after Ang IIchallenge.
The ability of Ang II to induce the transactivation of other typesof receptor was early described. Ang II was shown to promote theinside-out activation of epidermal growth factor receptor, whichinduces the amplification of ROS production in VSMC. However, incontrast with our study, no further details were provided in regardto NOXs involvement.
In summary, we showed that Ang II-induced ROS production isNOXs dependent and the initial ROS production induced by NOX1leads to NOX2 activation, which sustains the effect. We also showthat alpha1beta1 integrin/ILK crosstalk is a link between NOX1 andNOX2 activation, which in turn is responsible for Ang II-inducedVSMC migration and proliferation (see Fig. 10 in Ref [30]). Theseresults point to alpha1beta1 integrin as a promising target fortherapeutic treatment in cardiovascular diseases and ILK as apossible target to be considered in pharmacological interventions.
Conflict of interest
The authors report no conflict of interest.
Acknowledgments
This work was supported by grants from the Coordenaç~ao deAperfeiçoamento de Pessoal de Nível Superior (CAPES), Fundaç~aode Amparo �a Pesquisa do Estado do Rio de Janeiro (FAPERJ) and
Conselho Nacional de Desenvolvimento Científico e Tecnol�ogico(CNPq). We thank Gabriele Muniz for technical assistance.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.atherosclerosis.2015.09.026.
References
[1] M. Back, D.X. Bu, R. Branstrom, et al., Leukotriene B4 signaling through NF-kappaB-dependent BLT1 receptors on vascular smooth muscle cells inatherosclerosis and intimal hyperplasia, Proc. Natl. Acad. Sci. U. S. A. 102(2005) 17501e17506.
[2] A.I. Willis, D. Pierre-Paul, B.E. Sumpio, et al., Vascular smooth muscle cellmigration: current research and clinical implications, Vasc. Endovascular Surg.38 (2004) 11e23.
[3] A. Curcio, D. Torella, C. Indolfi, Mechanisms of smooth muscle cell prolifera-tion and endothelial regeneration after vascular injury and stenting: approachto therapy, Circ. J. Off. J. Jpn. Circ. Soc. 75 (2011) 1287e1296.
[4] S.O. Marx, H. Totary-Jain, A.R. Marks, Vascular smooth muscle cell prolifera-tion in restenosis, Circ. Cardiovasc. Interv. 4 (2011) 104e111.
[5] J.L. Johnson, Emerging regulators of vascular smooth muscle cell function inthe development and progression of atherosclerosis, Cardiovasc. Res. 103(2014) 452e460.
[6] P. Libby, Inflammation in atherosclerosis, Arterioscler. Thromb. Vasc. Biol. 32(2012) 2045e2051.
[7] A.C. Montezano, A. Nguyen Dinh Cat, F.J. Rios, et al., Angiotensin II andvascular injury, Curr. Hypertens. Rep. 16 (2014) 431.
[8] M.A. Bunni, Kramarenko II, L. Walker, et al., Role of integrins in angiotensin II-induced proliferation of vascular smooth muscle cells, Am. J. Physiol. CellPhysiol. 300 (2011) C647eC656.
[9] A.J. Valente, T. Yoshida, S.N. Murthy, et al., Angiotensin II enhances AT1-Nox1binding and stimulates arterial smooth muscle cell migration and prolifera-tion through AT1, Nox1, and interleukin-18, Am. J. Physiol. Heart Circ. Physiol.303 (2012) H282eH296.
[10] Y.J. Shen, X.X. Zhu, X. Yang, et al., Cardamonin inhibits angiotensin II-inducedvascular smooth muscle cell proliferation and migration by downregulatingp38 MAPK, Akt, and ERK phosphorylation, J. Nat. Med. 68 (2014) 623e629.
[11] A. Cave, Selective targeting of NADPH oxidase for cardiovascular protection,Curr. Opin. Pharmacol. 9 (2009) 208e213.
[12] K.K. Griendling, D. Sorescu, M. Ushio-Fukai, NAD(P)H oxidase: role in car-diovascular biology and disease, Circ. Res. 86 (2000) 494e501.
[13] K. Schroder, Isoform specific functions of Nox protein-derived reactive oxygenspecies in the vasculature, Curr. Opin. Pharmacol. 10 (2010) 122e126.
[14] A. Nguyen Dinh Cat, A.C. Montezano, D. Burger, et al., Angiotensin II, NADPHoxidase, and redox signaling in the vasculature, Antioxid. Redox Signal. 19(2013) 1110e1120.
[15] O. Sareila, T. Kelkka, A. Pizzolla, et al., NOX2 complex-derived ROS as immuneregulators, Antioxid. Redox Signal. 15 (2011) 2197e2208.
[16] J.J. Li, M. Han, J.K. Wen, et al., Osteopontin stimulates vascular smooth musclecell migration by inducing FAK phosphorylation and ILK dephosphorylation,Biochem. Biophys. Res. Commun. 356 (2007) 13e19.
[17] J. Varadarajulu, M. Laser, M. Hupp, et al., Targeting of alpha(v) integrins in-terferes with FAK activation and smooth muscle cell migration and invasion,Biochem. Biophys. Res. Commun. 331 (2005) 404e412.
[18] J. Moraes, J. Assreuy, C. Canetti, et al., Leukotriene B4 mediates vascularsmooth muscle cell migration through alphavbeta3 integrin transactivation,Atherosclerosis 212 (2010) 406e413.
[19] E.T. Choi, M.F. Khan, J.E. Leidenfrost, et al., Beta3-integrin mediates smoothmuscle cell accumulation in neointima after carotid ligation in mice, Circu-lation 109 (2004) 1564e1569.
[20] S. Katsuda, Y. Okada, T. Minamoto, et al., Collagens in human atherosclerosis.Immunohistochemical analysis using collagen type-specific antibodies, Arte-rioscler. Thromb. J. Vasc. Biol. Am. Heart Assoc. 12 (1992) 494e502.
[21] H. Louis, A. Kakou, V. Regnault, et al., Role of alpha1beta1-integrin in arterialstiffness and angiotensin-induced arterial wall hypertrophy in mice, Am. J.Physiol. Heart Circ. Physiol. 293 (2007) H2597eH2604.
[22] T.T. Nguyen, J.P. Ward, S.J. Hirst, beta1-Integrins mediate enhancement ofairway smooth muscle proliferation by collagen and fibronectin, Am. J. Respir.Crit. Care Med. 171 (2005) 217e223.
[23] K. Schapira, E. Lutgens, A. de Fougerolles, et al., Genetic deletion or antibodyblockade of alpha1beta1 integrin induces a stable plaque phenotype inApoE�/� mice, Arterioscler. Thromb. Vasc. Biol. 25 (2005) 1917e1924.
[24] J. Qin, C. Wu, ILK: a pseudokinase in the center stage of cell-matrix adhesionand signaling, Curr. Opin. Cell Biol. 24 (2012) 607e613.
[25] Y. Imai, D.R. Clemmons, Roles of phosphatidylinositol 3-kinase and mitogen-activated protein kinase pathways in stimulation of vascular smooth musclecell migration and deoxyriboncleic acid synthesis by insulin-like growthfactor-I, Endocrinology 140 (1999) 4228e4235.
[26] Z.Y. Yang, R.D. Simari, N.D. Perkins, et al., Role of the p21 cyclin-dependentkinase inhibitor in limiting intimal cell proliferation in response to arterial

J.A. Moraes et al. / Atherosclerosis 243 (2015) 477e485 485
injury, Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 7905e7910.[27] S.I. Bettink, C. Werner, C.H. Chen, et al., Integrin-linked kinase is a central
mediator in angiotensin II type 1- and chemokine receptor CXCR4 signaling inmyocardial hypertrophy, Biochem. Biophys. Res. Commun. 397 (2010)208e213.
[28] C. Marcinkiewicz, P.H. Weinreb, J.J. Calvete, et al., Obtustatin: a potent se-lective inhibitor of alpha1beta1 integrin in vitro and angiogenesis in vivo,Cancer Res. 63 (2003) 2020e2023.
[29] M.P. Moreno-Murciano, D. Monleon, J.J. Calvete, et al., Amino acid sequenceand homology modeling of obtustatin, a novel non-RGD-containing shortdisintegrin isolated from the venom of Vipera lebetina obtusa, Protein Sci.Publ. Protein Soc. 12 (2003) 366e371.
[30] J.A. Moraes, A.C. Frony, A.M. Dias, et al., Data in support of alpha1beta1 andintegrin-linked kinase interact and modulate angiotensin II effects in vascularsmooth muscle cells, Data Brief (2015) (in press).
[31] C. Marshall, A.J. Mamary, A.J. Verhoeven, et al., Pulmonary artery NADPH-oxidase is activated in hypoxic pulmonary vasoconstriction, Am. J. Respir.Cell Mol. Biol. 15 (5) (1996) 633e644.
[32] H.D. Qin, D. Huang, X.D. Weng, et al., Upregulation of peroxisome proliferator-activated receptor-gamma and NADPH oxidases are involved in restenosis
after balloon injury, J. Cell. Physiol. 221 (2) (2009) 387e393.[33] S.T. Arold, How focal adhesion kinase achieves regulation by linking ligand
binding, localization and action, Curr. Opin. Struct. Biol. 21 (2011) 808e813.[34] S.Y. Han, Y.S. Kang, Y.H. Jee, et al., High glucose and angiotensin II increase
beta1 integrin and integrin-linked kinase synthesis in cultured mouse podo-cytes, Cell Tissue Res. 323 (2006) 321e332.
[35] S. Persad, S. Attwell, V. Gray, et al., Regulation of protein kinase B/Akt-serine473 phosphorylation by integrin-linked kinase: critical roles for kinase ac-tivity and amino acids arginine 211 and serine 343, J. Biol. Chem. 276 (2001)27462e27469.
[36] P. Lacolley, V. Regnault, A. Nicoletti, et al., The vascular smooth muscle cell inarterial pathology: a cell that can take on multiple roles, Cardiovasc. Res. 95(2012) 194e204.
[37] K.J. Massey, N.J. Hong, J.L. Garvin, Angiotensin II stimulates superoxide pro-duction in the thick ascending limb by activating NOX4, Am. J. Physiol. CellPhysiol. 303 (2012) C781eC789.
[38] S.I. Dikalov, Z. Ungvari, Role of mitochondrial oxidative stress in hypertension,Am. J. Physiol. Heart Circ. Physiol. 305 (2013) H1417eH1427.
[39] T.N. Yoganathan, P. Costello, X. Chen, et al., Integrin-linked kinase (ILK): a“hot” therapeutic target, Biochem. Pharmacol. 60 (2000) 1115e1119.