advances in immunology volume 93
TRANSCRIPT


Contents
Contributors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ix
Class Switch Recombination: A Comparison BetweenMouse and Human
Qiang Pan-Hammarstrom, Yaofeng Zhao, and Lennart Hammarstrom
Abstract. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 11. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12. Mechanism of CSR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23. Comparison Between Human and Mouse . . . . . . . . . . . . . . . . . . . . 154. Concluding Remarks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
Anti-IgE Antibodies for the Treatment of IgE-MediatedAllergic Diseases
Tse Wen Chang, Pheidias C. Wu, C. Long Hsu, and Alfur F. Hung
Abstract. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 631. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 642. Rationale Leading to the Invention of the Anti-IgE Concept. . . . . 673. Anti-IgE Is Approved for Treating Moderate-to-Severe Asthma . . 734. Studies on Other Allergic Diseases . . . . . . . . . . . . . . . . . . . . . . . . . 785. The Potential of Using Anti-IgE to Assist Allergen-Based
Immunotherapy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 836. Pivotal Roles of IgE and FceRI in Type I Hypersensitivity . . . . . . 857. Neutralization of Free IgE. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 888. Downregulation of FceRI. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 90
v

vi contents
9. Potential Beneficial Effects of IgE:Anti-IgEImmune Complexes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 93
10. Can Anti-IgE Modulate IgE-Committed B Lymphoblasts andMemory B Cell? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
11. Other Immunoregulatory Effects of Anti-IgE . . . . . . . . . . . . . . . . . 9812. Can Anti-IgE Attain a Long-Term Remission State?. . . . . . . . . . . . 10013. Are There Adverse Effects Associated with Anti-IgE Therapy? . . . 10114. Other Approaches for Targeting IgE or IgE-Expressing
B Cells . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10315. Concluding Remarks. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 107
Immune Semaphorins: Increasing Members and TheirDiverse Roles
Hitoshi Kikutani, Kazuhiro Suzuki, and Atsushi Kumanogoh
Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1211. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1212. Sema4D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1223. Sema4A . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1274. Sema6D and Its Receptor Plexin-A1 . . . . . . . . . . . . . . . . . . . . . . . . 1305. Sema7A . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1356. Other Semaphorins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1377. Summary and Perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 138
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
Tec Kinases in T Cell and Mast Cell Signaling
Martin Felices, Markus Falk, Yoko Kosaka, and Leslie J. Berg
Abstract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1451. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1452. Subcellular Localization of Tec Kinases . . . . . . . . . . . . . . . . . . . . . . 1473. Tec Kinases in Signaling Pathways . . . . . . . . . . . . . . . . . . . . . . . . . . 1514. Regulation of Tec Kinase Activation . . . . . . . . . . . . . . . . . . . . . . . . 1605. Distinct Versus Redundant Functions of Tec Kinases . . . . . . . . . . . 1636. Tec Kinases in Mast Cell Signaling . . . . . . . . . . . . . . . . . . . . . . . . . 1667. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172

contents vii
Integrin Regulation of Lymphocyte Trafficking: Lessons fromStructural and Signaling Studies
Tatsuo Kinashi
Abstract. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1851. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1852. Leukocyte Integrins . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1863. Affinity and Valency Regulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1894. Integrin Conformational Changes . . . . . . . . . . . . . . . . . . . . . . . . . . 1895. Integrin-Mediated Adhesion Steps in Lymphocyte Trafficking . . . . 1956. Talin as Intracellular Regulator for Lymphocyte Adhesion
and Migration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2017. Intracellular Signals in Chemokine-Induced Adhesion
and Migration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2038. Inside-Out Signaling Events in TCR-Stimulated Lymphocytes . . . 2119. Concluding Remarks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 215
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216
Regulation of Immune Responses and Hematopoiesis by theRap1 Signal
Nagahiro Minato, Kohei Kometani, and Masakazu Hattori
Abstract. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2291. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2292. General Biology of the Rap1 Signal. . . . . . . . . . . . . . . . . . . . . . . . . 2303. Rap1 Signal in Lymphocyte Development
and Immune Responses . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2374. Rap1 Signal in Hematopoiesis and Leukemia . . . . . . . . . . . . . . . . . 2485. Rap1 Signal in Malignancy: New Aspects in Cancer. . . . . . . . . . . . 2536. Conclusions and Perspectives. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 255
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 256
Lung Dendritic Cell Migration
Hamida Hammad and Bart N. Lambrecht
Abstract. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2651. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2652. Airway DC Subsets: Localization and Phenotype . . . . . . . . . . . . . . 2663. Recruitment of DCs to the Lung. . . . . . . . . . . . . . . . . . . . . . . . . . . 267

vi i i contents
4. Migration of Airway DCs to the LNs. . . . . . . . . . . . . . . . . . . . . . . . 2695. Recruitment of pDCs to the Sites of Inflammation. . . . . . . . . . . . . 2726. Conclusions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 273
Index . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 279Contents of Recent Volumes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 291

Contributors
Numbers in parentheses indicate the pages on which the authors’ contributions begin.
Leslie J. Berg (145), Department of Pathology, University of MassachusettsMedical School, Massachusetts
Tse Wen Chang (63), Genomics Research Center, Academia Sinica,Nankang, Taipei 115, Taiwan
Markus Falk (145), Department of Pathology, University of MassachusettsMedical School, Massachusetts
Martin Felices (145), Department of Pathology, University of MassachusettsMedical School, Massachusetts
Hamida Hammad (265), Department of Pulmonary Medicine, ErasmusMedical Center, Dr Molewaterplein 50, 3015 GE Rotterdam, TheNetherlands
LennartHammarstrom (1), Department of LaboratoryMedicine, Division ofClinical Immunology, Karolinska University Hospital Huddinge, SE-14186Stockholm, Sweden
Masakazu Hattori (229), Department of Immunology and Cell Biology,Graduate School of Biostudies, Kyoto University, Kyoto, Japan
C. Long Hsu (63), Genomics Research Center, Academia Sinica, Nankang,Taipei 115, Taiwan; Department of Life Science, National Tsing HuaUniversity, Hsinchu 300, Taiwan
Alfur F. Hung (63), Genomics Research Center, Academia Sinica, Nankang,Taipei 115, Taiwan; Department of Life Science, National Tsing HuaUniversity, Hsinchu 300, Taiwan
Hitoshi Kikutani (121), Department of Molecular Immunology andCREST Program of JST, Research Institute for Microbial Diseases, OsakaUniversity, Suita, Osaka 5650871, Japan
Tatsuo Kinashi (185), Department of Molecular Genetics, Institute ofBiomedical Science, Kansai Medical University, Kyoto 606, Japan
Kohei Kometani (229), Department of Immunology and Cell Biology,Graduate School of Biostudies, Kyoto University, Kyoto, Japan
ix

x contributors
Yoko Kosaka (145), Department of Pathology, University of MassachusettsMedical School, Massachusetts
Atsushi Kumanogoh (121), Department of Immunopathology, ResearchInstitute for Microbial Diseases, Osaka University, Suita, Osaka 5650871,Japan
Bart N. Lambrecht (265), Department of Pulmonary Medicine, ErasmusMedical Center, Dr Molewaterplein 50, 3015 GE Rotterdam, TheNetherlands
Nagahiro Minato (229), Department of Immunology and Cell Biology,Graduate School of Biostudies, Kyoto University, Kyoto, Japan
Qiang Pan-Hammarstrom (1), Department of Laboratory Medicine, Divisionof Clinical Immunology, Karolinska University Hospital Huddinge, SE-14186Stockholm, Sweden
Kazuhiro Suzuki (121), Department of Molecular Immunology andCREST Program of JST, Research Institute for Microbial Diseases, OsakaUniversity, Suita, Osaka 5650871, Japan
Pheidias C. Wu (63), Genomics Research Center, Academia Sinica, Nankang,Taipei 115, Taiwan; Department of Life Science, National Tsing HuaUniversity, Hsinchu 300, Taiwan
Yaofeng Zhao (1), Department of Laboratory Medicine, Division of ClinicalImmunology, Karolinska University Hospital Huddinge, SE-14186 Stockholm,Sweden

a#
Class Switch Recombination: A Comparison BetweenMouse and Human
Qiang Pan‐Hammarstrom, Yaofeng Zhao, and Lennart Hammarstrom
Department of Laboratory Medicine, Division of Clinical Immunology, KarolinskaUniversity Hospital Huddinge, SE‐14186 Stockholm, Sweden
A
dv2
bstract.............................................................................................................
1ances in immunology, vol. 93 0065-2776/07 $
007 Elsevier Inc. All rights reserved. DOI: 10.1016/S0065-2776(06)93
1
1. I ntroduction ....................................................................................................... 1 2. M echanism of CSR.............................................................................................. 2 3. C omparison Between Human and Mouse ................................................................ 15 4. C oncluding Remarks............................................................................................ 42R
eferences ......................................................................................................... 43Abstract
Humans andmice separatedmore than 60million years ago. Since then, evolutionhas led to a multitude of changes in their genomic sequences. The divergence ofgenes has resulted in differences both in the innate and adaptive immune systems.In this chapter, we focus on species difference with regard to immunoglobulinclass switch recombination (CSR). We have compared the immunoglobulin con-stant region gene loci from human and mouse, with an emphasis on the switchregions, germ line transcription promoters, and 30 enhancers. We have alsocompared pathways/factors that are involved in CSR. Although there are remark-able similarities in the cellular machinery involved in CSR, there are also anumber of unique features in each species.
1. Introduction
Owing to development of the gene ‘‘knockout’’ technology, Mus musculus hasemerged as a leading mammalian system for biomedical research over the pastdecades. Mouse models have served as surrogates for exploring human physi-ology and pathology, leading to major discoveries in many areas of biomedicalresearch, including immunology. The availability of the human, mouse, and ratgenome sequences (Gibbs et al., 2004; Lander et al., 2001; Venter et al., 2001;Waterston et al., 2002) has provided possibilities for cataloging the murineorthologs of human genes and allowed a way to identify and to performfunctional studies on human disease associated genes.
35.00
001-6

2 QIANG PAN ‐HAMMARSTROM ET AL .
One concern, however, is if mouse models faithfully represent human diseaseprocesses or not. Humans and mice separated more than 60 million years ago(Madsen et al., 2001; Murphy et al., 2001). Since then, evolution has led to amultitude of changes in their genomic sequences, includingmutations, insertions,deletions, and duplications. The average divergence rate is about one substitutionfor every two nucleotides (Waterston et al., 2002) and genes encoding variousclasses of proteins have evolved with different paces. One notable set of proteinsthat seem to be under positive or purifying selection, and thus evolves rapidly, arethose implicated in host defense (Waterston et al., 2002).The divergence of genes in the mouse and human genomes has resulted in
differences both in the innate and in the adaptive immune systems, leading todevelopment of different pathways, involving a variety of chemical messengers.In this chapter, we will highlight some of these differences and address speciesdifferences related to immunoglobulin (Ig) class switch recombination (CSR).
2. Mechanism of CSR
2.1. Class Switch Recombination ‘‘ABC’’
The first antibodies produced in a humoral immune response are of theIgM class. Activated B cells subsequently undergo isotype switching to secreteantibodies of different isotypes: IgG, IgA, and IgE. Isotype switching does notaffect the antibody specificity, but alters the effector functions of the antibody.The change in antibody class is effectuated by a deletional recombinationevent called class switch recombination (CSR), where the constant regiongene of the m heavy chain (Cm) is replaced by a downstream CH gene(Cg, Ca, or Ce) and intervening sequences are excised as circular DNA(Iwasato et al., 1990; Matsuoka et al., 1990; von Schwedler et al., 1990).CSR involves DNA regions, called ‘‘switch (S) regions,’’ that are located in the
intron upstream of each C region gene. S regions are composed of tandemlyrepeated sequences that contain common pentamer sequences (GAGCT andGGGGT), but differ in length and degree of sequence similarity with Sm.CSR is a unique form of recombination. It is referred to as a ‘‘region‐specific’’
rather than ‘‘site‐specific’’ process, as no consensus sequence has been identi-fied at the junctions of recombined S regions. It is also distinct from homo-logous recombination (HR), as it does not depend on a long stretch of homologybetween the sequences involved.CSR is influenced in both a positive and a negative manner by a number of
cytokines and B cell activators. The mechanism involved is partly mediatedthrough the ability of cytokines and activators to regulate transcription of unrear-ranged CH genes prior to CSR, yielding what are referred to as germ line (GL)transcripts (Stavnezer‐Nordgren and Sirlin, 1986; Yancopoulos et al., 1986).

CLASS SWITCH RECOMBINATION 3
GL transcripts all have a similar structure, resulting from the initiation of tran-scription from an I (intervening) exon upstream of the S region and are spliced tothe first exon of the correspondingCHgene.GL transcripts are required forCSR,and targeting of CSR to a given C region gene is considered to be tightlycorrelated with transcription from the corresponding upstream GL promoter(Chaudhuri et al., 2004; Stavnezer, 1996).
At the DNA level, CSR is initiated by activation‐induced deaminase (AID;Muramatsu et al., 2000; Revy et al., 2000), probably by deamination of dC residueswithin the S regions. The initial lesions are subsequently processed and DNAdouble strand breaks (DSBs) are introduced that may lead to recombinationof the two S regions involved. These processes require activation of a number ofDNA damage response/repair pathways, including ataxia‐telangiectasia mutated(ATM)/ataxia‐telangiectasia and Rad3‐related (ATR)‐dependent signaling, baseexcision repair (BER), mismatch repair (MMR), and nonhomologous end joining(NHEJ; Chaudhuri and Alt, 2004).
2.2. V(D)J Recombination and CSR
Mammalian organisms require an additional form of DNA recombination,V(D)J recombination, in order to produce functional antibody encodinggenes. V(D)J recombination mediates assembly of the gene segments thatencode the Ig heavy‐ and light‐chain variable domains. It is distinct from CSRin several regards: it occurs early in B cell development in the bone marrow; it isinitiated by the lymphocyte‐specific proteins RAG1 and RAG2 instead of AID;it proceeds through precise DNA cleavage at conserved signal sequences and istherefore a ‘‘site‐specific’’ rather than a ‘‘region‐specific’’ recombination process(Dudley et al., 2005; Jung and Alt, 2004; Schatz, 2004). There are, however,also similarities between the two types of recombination. Both V(D)J recom-bination and CSR involve DNA deletion by a mechanism whereby interven-ing sequences are excised as circular DNA. Moreover, CSR resembles V(D)Jrecombination in that DSBs are generated during the switch reaction (Catalanet al., 2003; Schrader et al., 2005; Wuerffel et al., 1997). Furthermore, compo-nents of the NHEJ machinery are implicated in resolution of the DSBs inboth recombination processes (Chaudhuri and Alt, 2004; Lieber et al., 2004),whereas other DNA repair pathways/factors appear to be more ‘‘CSR specific’’or ‘‘V(D)J specific’’ (see discussion in Section 2.5.3).
2.3. CSR and Somatic Hypermutation
Somatic hypermutation (SHM), a process where point mutations are intro-duced at a high rate into the Ig variable (V) genes, helps shape the Ig repertoireand, similar to CSR, occurs in the germinal center. Both SHM and CSR require

4 QIANG PAN ‐HAMMARSTROM ET AL .
transcription through the targeted regions and are initiated by the B cell‐specificfactor AID (Muramatsu et al., 2000; Revy et al., 2000). Resolution of the initiallesions in the V and S region genes is, however, somewhat different (seediscussion in Section 2.5.3), andDSBs seem not to be prominent intermediates.Instead, single‐strand breaks (SSBs) or single‐strand nicks appear to be essentialin SHM (Faili et al., 2002b; Li et al., 2004b; Neuberger et al., 2005).
2.4. Function of AID
AID was discovered by Honjo and coworkers and shown to be a B cell factorthat is essential for both SHM and CSR (Muramatsu et al., 1999, 2000). AID‐deficient mice are devoid of both SHM and CSR (Muramatsu et al., 2000), asare patients with an autosomal recessive form of the hyper‐IgM syndrome(HIGM2), caused by mutations in the human AID‐encoding gene (Revy et al.,2000). Ectopic expression of AID in nonlymphoid cells is sufficient to induceboth SHM and CSR, suggesting that it is the only B cell‐specific factor neededfor these processes (Martin et al., 2002; Okazaki et al., 2002; Yoshikawa et al.,2002). AID is also essential for gene conversion (Arakawa et al., 2002; Harriset al., 2002), which is the dominant mechanism for V region diversification inselected animal species, including chickens and possibly sheep.AID was initially thought to edit mRNA, as it shares a high degree of
sequence homology with the RNA‐editing enzyme APOBEC‐1 (apolipo-protein B mRNA editing catalytic polypeptide 1). In this model, AID deami-nates cytosines to uracils in the mRNA encoding a ubiquitously expressed, asyet undefined factor(s) that is essential for both SHM and CSR. Althoughthere is some evidence that supports this model (Begum et al., 2004; Doi et al.,2003; Ito et al., 2004), there is an increasing wealth of data supporting a DNAdeamination model, where AID initiates SHM and CSR by converting thecytosines in DNA to uracils (for review see Honjo et al., 2005; Lee et al., 2004).AID preferentially deaminates single‐stranded DNA (ssDNA) in vitro
(Bransteitter et al., 2003; Chaudhuri et al., 2003; Dickerson et al., 2003;Ramiro et al., 2003) and the deamination of C is most prominent withinWRC sequences (Pham et al., 2003; Yu et al., 2004), reflecting the in vivoSHM hotspots (RGYW/WRCY motifs; Milstein et al., 1998). The AID‐mediated cytidine deamination also seems to be targeted by transcription(Chaudhuri et al., 2003; Ramiro et al., 2003), a process that may provideAID substrates by exposing short stretches of ssDNA during elongation orby generating secondary structures like ‘‘R loops,’’ where transcripts hybridizeto the template strand, forming long stretches of single‐stranded regions onthe nontemplate strand. Importantly, these ‘‘R loops’’ have previously beenimplicated in CSR (Yu et al., 2003). No ‘‘R loops’’ can, however, be formed in

CLASS SWITCH RECOMBINATION 5
the V regions during SHM, and another mechanism, involving replication proteinA (RPA), a ssDNA‐binding protein, has been proposed (Chaudhuri et al., 2004).RPA interacts specifically with AID in activated B cells and the RPA–AIDcomplex is thought to bind to and stabilize short ssDNA sequences at the transienttranscription bubbles, with a preference for RGYW motifs (Chaudhuri et al.,2004). RPA also binds to S regions in an AID‐dependent fashion and the RPA–AID complexmay have a potential role in targeting S region sequences, which arealso rich in RGYW motifs (Chaudhuri et al., 2004). These in vitro biochemicalstudies have provided an explanation for the link between the CSR and SHMrequirements for transcription and AID‐dependent DNA deamination.
2.5. dU:dG Mismatches Processing and DNA DSB Resolution in CSR
2.5.1. dU: Mismatches in CSR
The dU:dG mismatches resulting from AID activity can be repaired, replicatedover (introducing transition mutations at G/C sites) or processed to initiateCSR or SHM. Both the base excision (uracil DNA glycosylase, UNG) andMMR (MSH2) pathways can recognize dU:dG pairs, and based on the differ-ent consequences of UNG deficiency (Rada et al., 2002), MSH2 deficiency(Ehrenstein and Neuberger, 1999; Schrader et al., 1999), and UNG–MSH2double deficiency (Rada et al., 2004), the major pathway for CSR has beensuggested to be dependent on UNG activity whereas the MSH2‐dependentpathway serves as a backup. In the UNG‐dependent pathway, the uracil basecan be removed by UNG, generating an abasic site that is then recognized byan apurinic/apyrimidic (AP) endonuclease (APE or APEX), which in turnproduces a nick. Closely positioned nicks on both strands could theoreticallyconvert the SSBs to DSBs that are required for CSR. In the MSH2‐dependentpathway, the dU:dG mismatches would be recognized by the MMR proteinsand single‐strand nicks may be introduced which eventually leads to theformation of DSBs (Stavnezer and Schrader, 2005).
One question that remains is which endonuclease actually cleaves at theabasic site. APE1 (APEX1) is the major APE in mammalian cells (Dempleet al., 1991; Robson and Hickson, 1991; Xanthoudakis and Curran, 1992), butits potential role in CSR has not been documented. A second APE, APEX2,has also been identified (Hadi and Wilson, 2000; Ide et al., 2003). Mice with atargeted inactivation of the APEX2 gene show thymic atrophy, reduced num-ber of B cells, and attenuated immune responses, suggesting that APEX2 mayhave unique functional properties that cannot be compensated by APEX1 (Ideet al., 2004). However, thus far, there is no evidence to support the notionthat APEX2 is involved in CSR. Another pathway, mediated by Mre11/Rad 50,has recently been proposed by Maizels and coworkers. The authors found that

6 QIANG PAN ‐HAMMARSTROM ET AL .
Mre11, rather than APE1, is associated with rearranged Ig genes inhypermutating B cells and that Mre11/Rad50 cleaves at abasic sites withinsingle‐stranded regions of DNA (Larson et al., 2005). Although the Mre11/Rad50/NBS1 complex has previously been implicated in CSR (Kracker et al.,2005; Lahdesmaki et al., 2004; Pan et al., 2002b; Reina‐San‐Martin et al., 2005)and potentially in SHM (Yabuki et al., 2005), it is unclear whether its role inCSR is through cleavage of abasic sites or resolution of the DSBs at a laterstage.
2.5.2. DSB Resolutions in CSR
2.5.2.1. ATM and ATR Signaling in CSR TheDSBs generated in the S regionsduring CSR will activate a number of signal‐transducing and DNA repair path-ways. There are two signal‐transduction pathways, one which depends on ATMand a second that depends on the ATR protein. The ATM‐dependent pathwayplays a major role in the response to DSBs, and the ATM protein has beenimplicated in CSR in both humans (Pan et al., 2002b) and mice (Lumsden et al.,2004; Reina‐San‐Martin et al., 2004). Several components of the ATM‐depen-dent pathway, including H2AX, NBS1, Mre11, and 53BP1, have also beenshown to be involved in CSR (Kracker et al., 2005; Lahdesmaki et al., 2004;Manis et al., 2004; Petersen et al., 2001; Reina‐San‐Martin et al., 2003, 2005;Ward et al., 2004). The ATR‐dependent pathway is activated by ssDNA duringDNA replication or by agents such as UV irradiation that produce bulky lesions.By responding to the ssDNA resulting from processed DSBs, ATR may alsoreinforce the ATM response (Shiloh, 2001; Tibbetts et al., 1999). Furthermore,ATR shares several substrates with ATM (Abraham, 2001), including H2AX and53BP1. A modest role of ATR in CSR has been demonstrated in ATR‐deficientpatients, where a normal number of cells that have switched to IgG and IgAproductionwere observed, but where the pattern of CSR junctions was aberrant(Pan‐Hammarstrom et al., 2006).
2.5.2.2. HR and NHEJ in CSR There are two major types of DSB repairmechanisms: HR and NHEJ. There is thus far no direct evidence showingthat molecules involved in HR, such as Rad51, Rad52, and Rad54, arerequired for CSR, although expression of Rad51 is induced in activatedB cells undergoing CSR (Bross et al., 2003; Li et al., 1996). The resolution ofthe CSR‐specific DSBs mainly requires components of the NHEJ pathway.On the basis of gene targeting studies, three components of the NHEJmachinery have been implicated in CSR in mice: DNA‐PKcs, Ku70,and Ku80 (Casellas et al., 1998; Manis et al., 1998a; Rolink et al., 1996). Theimpact of the other two components, DNA ligase IV and XRCC4, has not been

CLASS SWITCH RECOMBINATION 7
analyzed in knockout models, as disruption of Lig4 or XRCC4 in mice resultsin embryonic lethality (Barnes et al., 1998; Frank et al., 1998; Gao et al.,1998b). Involvement of DNA ligase IV in CSR has, however, been demon-strated in patients who carry hypomorphic mutations in the Lig4 gene, wherean altered pattern of in vivo generated CSR junctions in B cells was observed(Pan‐Hammarstrom et al., 2005). As DNA ligase IV, in contrast to Ku andDNA‐PKcs, has no reported roles outside NHEJ (Chaudhuri and Alt, 2004),the observation in DNA ligase IV deficient (Lig4D) patients links the coreNHEJ machinery to CSR.
Recently, five patients with growth retardation, microcephaly, and immuno-deficiency characterized by a profound T and B lymphocytopenia were des-cribed. This autosomal recessive disorder is caused by mutations in a novelDNA repair factor, Cernunnos (XLF; Ahnesorg et al., 2006; Buck et al., 2006).The clinical phenotype of Cernunnos‐deficient patients shares several char-acteristics with Nijmegen breakage syndrome (NBS) and Lig4D patients. How-ever, Cernunnos deficiency does not lead to impaired cell‐cycle checkpoints,as observed in NBS, but results in a defective V(D)J recombination and animpaired DNA end‐ligation process (Buck et al., 2006), similar to that obser-ved in Lig4D patients. The precise role of Cernunnos in NHEJ remainselusive, although it seems to interact with the XRCC4‐ligase IV complex(Ahnesorg et al., 2006). It is interesting to note that in Cernunnos‐deficientpatients, serum levels of IgG and IgA are low or absent, whereas the level of IgMis normal or even high, suggesting a possible role of Cernunnos in CSR (Bucket al., 2006).
2.5.3. DNA Damage Response/Repair Pathways Utilized inIg Gene Diversification
Table 1 summarizes the current knowledge on DNA damage response/repairfactors utilized in V(D)J recombination, SHM and CSR. In general, factorsthat belong to the same pathway tend to show a similar pattern of involvementin Ig gene diversification. For example, the NHEJ core factors, Ku70 andKu80, DNA‐PKcs, DNA ligase IV, and XRCC4, are all involved in V(D)Jrecombination and CSR, but, most likely, not in SHM. Most of the MMRproteins on the other hand are involved in both CSR and SHM, but may not berequired in V(D)J recombination, whereas the ATM‐dependent factors aremost often involved in CSR but not in SHM (Table 1 and references therein).
There are, however, a few exceptions to the above ‘‘rule.’’ In the BERpathway, UNG thus far seems to be the only glycosylase required for removingthe uracil bases in both the CSR and SHM processes. MSH3 seems to be theonly MMR protein studied to date that is neither involved in CSR nor SHM
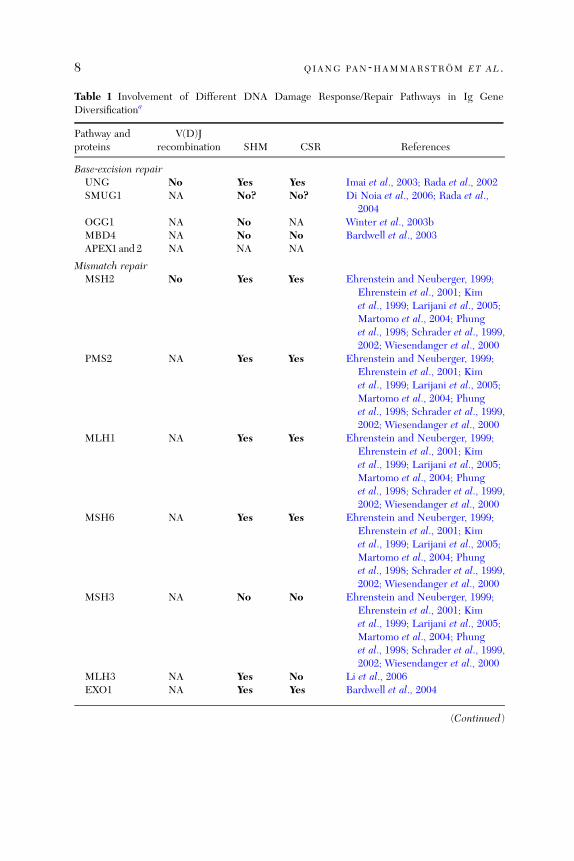
Table 1 Involvement of Different DNA Damage Response/Repair Pathways in Ig GeneDiversificationa
Pathway andproteins
V(D)Jrecombination SHM CSR References
Base‐excision repairUNG No Yes Yes Imai et al., 2003; Rada et al., 2002SMUG1 NA No? No? Di Noia et al., 2006; Rada et al.,
2004OGG1 NA No NA Winter et al., 2003bMBD4 NA No No Bardwell et al., 2003APEX1 and 2 NA NA NA
Mismatch repairMSH2 No Yes Yes Ehrenstein and Neuberger, 1999;
Ehrenstein et al., 2001; Kimet al., 1999; Larijani et al., 2005;Martomo et al., 2004; Phunget al., 1998; Schrader et al., 1999,2002; Wiesendanger et al., 2000
PMS2 NA Yes Yes Ehrenstein and Neuberger, 1999;Ehrenstein et al., 2001; Kimet al., 1999; Larijani et al., 2005;Martomo et al., 2004; Phunget al., 1998; Schrader et al., 1999,2002; Wiesendanger et al., 2000
MLH1 NA Yes Yes Ehrenstein and Neuberger, 1999;Ehrenstein et al., 2001; Kimet al., 1999; Larijani et al., 2005;Martomo et al., 2004; Phunget al., 1998; Schrader et al., 1999,2002; Wiesendanger et al., 2000
MSH6 NA Yes Yes Ehrenstein and Neuberger, 1999;Ehrenstein et al., 2001; Kimet al., 1999; Larijani et al., 2005;Martomo et al., 2004; Phunget al., 1998; Schrader et al., 1999,2002; Wiesendanger et al., 2000
MSH3 NA No No Ehrenstein and Neuberger, 1999;Ehrenstein et al., 2001; Kimet al., 1999; Larijani et al., 2005;Martomo et al., 2004; Phunget al., 1998; Schrader et al., 1999,2002; Wiesendanger et al., 2000
MLH3 NA Yes No Li et al., 2006EXO1 NA Yes Yes Bardwell et al., 2004
(Continued)
8 QIANG PAN ‐HAMMARSTROM ET AL .
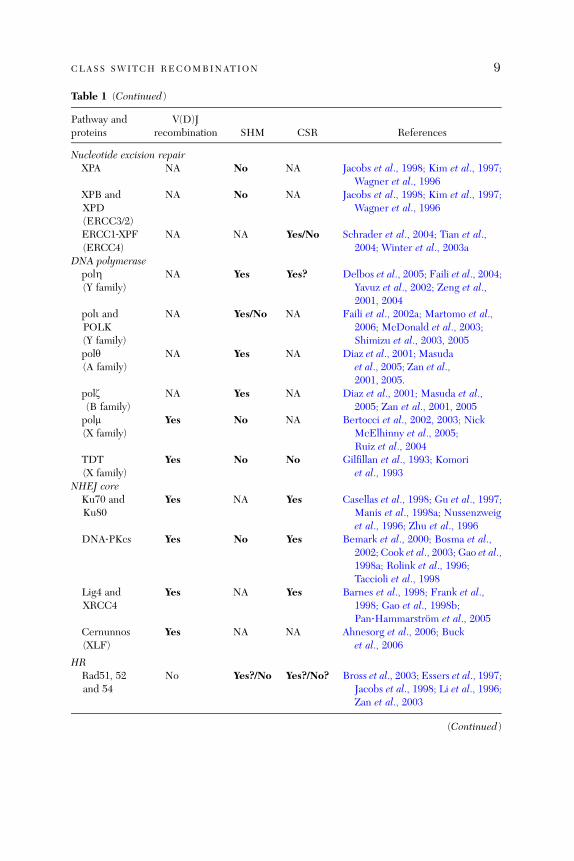
Table 1 (Continued)
Pathway andproteins
V(D)Jrecombination SHM CSR References
Nucleotide excision repairXPA NA No NA Jacobs et al., 1998; Kim et al., 1997;
Wagner et al., 1996XPB andXPD(ERCC3/2)
NA No NA Jacobs et al., 1998; Kim et al., 1997;Wagner et al., 1996
ERCC1‐XPF(ERCC4)
NA NA Yes/No Schrader et al., 2004; Tian et al.,2004; Winter et al., 2003a
DNA polymerasepolZ(Y family)
NA Yes Yes? Delbos et al., 2005; Faili et al., 2004;Yavuz et al., 2002; Zeng et al.,2001, 2004
poli andPOLK(Y family)
NA Yes/No NA Faili et al., 2002a; Martomo et al.,2006; McDonald et al., 2003;Shimizu et al., 2003, 2005
poly(A family)
NA Yes NA Diaz et al., 2001; Masudaet al., 2005; Zan et al.,2001, 2005.
polz(B family)
NA Yes NA Diaz et al., 2001; Masuda et al.,2005; Zan et al., 2001, 2005
polm(X family)
Yes No NA Bertocci et al., 2002, 2003; NickMcElhinny et al., 2005;Ruiz et al., 2004
TDT(X family)
Yes No No Gilfillan et al., 1993; Komoriet al., 1993
NHEJ coreKu70 andKu80
Yes NA Yes Casellas et al., 1998; Gu et al., 1997;Manis et al., 1998a; Nussenzweiget al., 1996; Zhu et al., 1996
DNA‐PKcs Yes No Yes Bemark et al., 2000; Bosma et al.,2002; Cook et al., 2003; Gao et al.,1998a; Rolink et al., 1996;Taccioli et al., 1998
Lig4 andXRCC4
Yes NA Yes Barnes et al., 1998; Frank et al.,1998; Gao et al., 1998b;Pan‐Hammarstrom et al., 2005
Cernunnos(XLF)
Yes NA NA Ahnesorg et al., 2006; Bucket al., 2006
HRRad51, 52and 54
No Yes?/No Yes?/No? Bross et al., 2003; Essers et al., 1997;Jacobs et al., 1998; Li et al., 1996;Zan et al., 2003
(Continued)
CLASS SWITCH RECOMBINATION 9
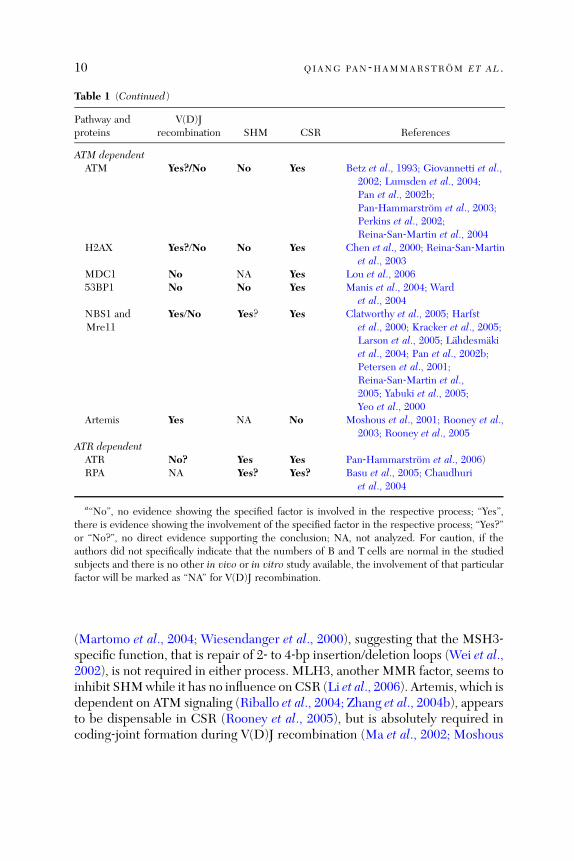
Table 1 (Continued)
Pathway andproteins
V(D)Jrecombination SHM CSR References
ATM dependentATM Yes?/No No Yes Betz et al., 1993; Giovannetti et al.,
2002; Lumsden et al., 2004;Pan et al., 2002b;Pan‐Hammarstrom et al., 2003;Perkins et al., 2002;Reina‐San‐Martin et al., 2004
H2AX Yes?/No No Yes Chen et al., 2000; Reina‐San‐Martinet al., 2003
MDC1 No NA Yes Lou et al., 200653BP1 No No Yes Manis et al., 2004; Ward
et al., 2004NBS1 andMre11
Yes/No Yes? Yes Clatworthy et al., 2005; Harfstet al., 2000; Kracker et al., 2005;Larson et al., 2005; Lahdesmakiet al., 2004; Pan et al., 2002b;Petersen et al., 2001;Reina‐San‐Martin et al.,2005; Yabuki et al., 2005;Yeo et al., 2000
Artemis Yes NA No Moshous et al., 2001; Rooney et al.,2003; Rooney et al., 2005
ATR dependentATR No? Yes Yes Pan‐Hammarstrom et al., 2006)RPA NA Yes? Yes? Basu et al., 2005; Chaudhuri
et al., 2004
a‘‘No’’, no evidence showing the specified factor is involved in the respective process; ‘‘Yes’’,there is evidence showing the involvement of the specified factor in the respective process; ‘‘Yes?’’or ‘‘No?’’, no direct evidence supporting the conclusion; NA, not analyzed. For caution, if theauthors did not specifically indicate that the numbers of B and T cells are normal in the studiedsubjects and there is no other in vivo or in vitro study available, the involvement of that particularfactor will be marked as ‘‘NA’’ for V(D)J recombination.
10 QIANG PAN ‐HAMMARSTROM ET AL .
(Martomo et al., 2004; Wiesendanger et al., 2000), suggesting that the MSH3‐specific function, that is repair of 2‐ to 4‐bp insertion/deletion loops (Wei et al.,2002), is not required in either process. MLH3, another MMR factor, seems toinhibit SHMwhile it has no influence on CSR (Li et al., 2006). Artemis, which isdependent on ATM signaling (Riballo et al., 2004; Zhang et al., 2004b), appearsto be dispensable in CSR (Rooney et al., 2005), but is absolutely required incoding‐joint formation during V(D)J recombination (Ma et al., 2002; Moshous

CLASS SWITCH RECOMBINATION 11
et al., 2001). The Artemis‐dependent ‘‘hairpin opening’’ function may thus bespecific for the V(D)J recombination process.
ATM, ATR, and a few ATM/ATR‐dependent factors, including H2AX,53BP1, and NBS1, are all involved in CSR, but their roles in V(D)J recombi-nation in mammalian cells remain uncertain (Chen et al., 2000; Clatworthyet al., 2005; Hsieh et al., 1993; Yeo et al., 2000). Thus, ATM was thought not tobe required in V(D)J recombination as cells from ataxia‐telangiectasia (A‐T)patients supported normal rearrangement of exogenous substrates (Hsiehet al., 1993), and endogenously rearranged TCRb and IgH genes from A‐Tpatients revealed normal V(D)J coding joints (Giovannetti et al., 2002;Pan‐Hammarstrom et al., 2003). However, similar to patients with Omennssyndrome (RAG2 deficient), A‐T patients display a restricted TCRb repertoire,which may suggest a subtle recombination defect (Giovannetti et al., 2002).Furthermore, both A‐T patients (Taylor et al., 1996) and ATM‐deficient mice(Barlow et al., 1996; Liyanage et al., 2000) are prone to lymphoid malignanciesthat harbor translocations involving V(D)J region genes, although RAG1 andRAG2 seem not to be essential in tumorigenesis in ATM‐deficient mice(Petiniot et al., 2000, 2002). Thus, ATM, and potentially also related factors,may be indirectly involved in the process by sensing the DNA breaks and bysuppression of aberrant V(D)J recombination that may lead to development oflymphoid malignancies (Dudley et al., 2005; Liao and van Dyke, 1999; Perkinset al., 2002).
Two members of the DNA polymerase (pol) X family, TDT and polm, alsoseem to have important, albeit restricted, roles in V(D)J recombination. TDTis crucial for N nucleotide additions at V–D and D–J junctions, whereas polm isrequired in the rearrangement of light chain genes (Bertocci et al., 2003).Members of the DNA pol Y, A and B families, including polZ, poly, and polyz,have all been implicated in SHM (Table 1 and references therein), in particu-lar during the proposed second phase of SHM, where mutations are generatedmainly at A/T pairs (Neuberger et al., 2005; Seki et al., 2005). The role of theseDNA polymerases during CSR is however unclear, although an altered muta-tion pattern in the Sm regions has been observed in xeroderma pigmentosumvariant (XP‐V) patients, who are deficient in DNA polZ (Faili et al., 2004; Zenget al., 2004).
2.6. Regulation of CSR
Cytokines and B cell activators control switching through their ability to re-gulate GL transcription of the CH genes and to induce or suppress the expres-sion of AID. A number of alternative pathways for inducing CSR have alsobeen described.

12 QIANG PAN ‐HAMMARSTROM ET AL .
2.6.1. The ‘‘Accessibility Model’’ and Beyond
Induction or suppression of GL transcription by particular cytokines has beendirectly correlated with subsequent switching to the same isotype after addi-tion of a B cell activator (Stavnezer, 1996, 2000). It has been proposed thatinitiation of GL transcription confers a level of accessibility to the CH locusthat allows binding of additional factors that participate in CSR, that is theaccessibility model of CSR. Indeed, modifications of histone H3 and/or H4,which create localized DNA accessibility to trans‐acting factors, are correlatedwith the level of GL transcription and differential targeting of downstreamS regions during CSR (Li et al., 2004a; Nambu et al., 2003; Wang et al., 2006).Furthermore, the physical association of AID to the S region requires GLtranscription (Nambu et al., 2003). However, histone acetylation alone cannotpromote CSR or GL transcription, and H3 acetylation, in the absence of GLtranscription, does not make S regions accessible to AID binding (Nambuet al., 2003). These studies provide evidence that GL transcription plays animportant role in the regulation of chromatin accessibility during CSR, butalso suggest that GL transcription has functional consequences beyond simplymaking the S region chromatin accessible.As discussed in the previous section, AID preferentially deaminates ssDNA
rather than dsDNA in vitro. Amore direct role of GL transcription has also beenproposed, that is creation of ssDNA within the S regions, through formation oftransient transcription bubbles or R‐loop structures, thus providing targets forAID (for review see Chaudhuri and Alt, 2004; Kaminski and Stavnezer, 2004).
2.6.2. Regulation of AID Expression
Another aspect of ‘‘beyond accessibility’’ is that cytokines and B cell activatorsare able to direct CSR to a particular CH region, not only through regulationof GL transcription but also through their ability to induce the expression ofAID. For example IL‐4, together with TGF‐b and CD40L, is able to induceAID expression in the mouse B cell line CH12F3‐2 and LPS alone, or incombination of IL‐4 or TGF‐b, is able to induce AID in mouse spleen cells(Muramatsu et al., 1999).A B cell‐specific enhancer has been identified in the first intron of the gene
encoding mouse AID, and the transcriptional activity of this enhancer isregulated by E‐proteins (Sayegh et al., 2003). Another putative promoterregion has been identified in a region immediately upstream of the transcrip-tion initiation site and this promoter is not lymphoid specific (Gonda et al.,2003; Yadav et al., 2006). Several transcription factor‐binding sites, includingthose for Pax5 (B cell‐specific activator protein), Sp1, and Sp3, have been

CLASS SWITCH RECOMBINATION 13
identified in this region. However, the data on Pax5 binding are controversial(Gonda et al., 2003; Yadav et al., 2006). STAT6 and NF‐kB p50 are thought tobe required for induction of AID expression by IL‐4 and CD40 engagementand potential binding sites for STAT6 and NF‐kB p50 have been identified in aregion further upstream of the putative promoter of the human AICDA gene(Dedeoglu et al., 2004).
The activity of AID is also regulated at a posttranslational level. The AID–RPA association in activated B cells requires AID phosphorylation (Chaudhuriet al., 2004), and protein kinase A (PKA) was identified as the physiologicalAID kinase (Basu et al., 2005; Pasqualucci et al., 2006). It is possible that AIDmay be sequestered in an inactive state in the cytoplasm, by an as yet unknownmechanism, and with appropriate signaling to the B cells, AID is phosphory-lated by PKA, activated, and subsequently transported to the nucleus (Basuet al., 2005). As multiple signals activate PKA, including cytokines, for instanceTGF‐b‐induced Smad proteins might activate PKA directly (Zhang et al.,2004a), this may add yet another dimension to the regulation of CSR bycytokines and B cell activators. However, the exact signaling that is criticalfor PKA‐mediated regulation of AID, needs to be further investigated.
The mechanism underlying the negative regulation of AID remains to beexplored, although it appears to be B cell specific (Muto et al., 2006). It is alsotempting to hypothesize that inactivation of AID, or retention of AID in thecytoplasm, is due to its interaction with specific inhibitory proteins, a situationthat is reminiscent of the induction of nuclear factor‐kB (NF‐kB) activity (Jimiand Ghosh, 2005; Zhong et al., 1997).
2.6.3. IgH 30 Enhancers
In addition to the promoter elements regulating GL transcription, regionscontaining a series of enhancer elements are located 30 of the human Ca1 andCa2 genes (Chen and Birshtein, 1997; Mills et al., 1997; Pinaud et al., 1997).These regions, similar to the 30 IgH enhancers in the mouse (Dariavach et al.,1991) and rat (Pettersson et al., 1990) Ig heavy chain constant region (IGHC)loci, may constitute a locus control region (LCR; Madisen and Groudine, 1994;Ong et al., 1998; Seidl et al., 1999) that regulates GL transcription and CSR(Cogne et al., 1994; Madisen and Groudine, 1994; Ong et al., 1998; Seidl et al.,1999). Consistent with this hypothesis, we have previously shown that theactivity of the human a1, a2, g3 (Hu et al., 2000; Pan et al., 2000), and g4(Pan‐Hammarstrom et al., unpublished data) GL promoters can be markedlyupregulated in reporter gene assays by DNA segments containing elements ofthe human 30 enhancers.

14 QIANG PAN ‐HAMMARSTROM ET AL .
2.6.4. CD40–CD40L Interaction
CD40 and CD40 ligand (L) interaction is crucial during T‐dependent B cellactivation, and its central role in B cell maturation and CSR is demonstrated inpatients with type I and type III hyper‐IgM syndromes (HIGM) who carrymutated CD40L or CD40 genes (for review see Levy et al., 1997; Lougaris et al.,2005). These patients are characterized by very low levels of serum IgG, IgA,and IgE, with normal or elevated levels of IgM, and associated with a defectivegerminal center formation. In addition to the defects in CSR, SHM is alsosignificantly reduced. However, somatically mutated Ig genes have beenfound in a subset of B cells (IgMþIgDþCD27þ) in these patients, suggestingthat SHM may occur in the absence of classical cognate T–B cell collaboration(Weller et al., 2001).CD40 signaling activates multiple kinases and pathways and eventually leads
to activation of transcription factors, including NF‐kB, NF of activated T cells(NF‐ATs), and activator protein 1 (AP‐1). CD40 signaling is able to direct CSR,by induction of GL transcripts, through the binding of activated NF‐kB tothe corresponding GL promoters (for review see Stavnezer, 2000) or to the30 enhancers (Grant et al., 1996; Sepulveda et al., 2004; Zelazowski et al., 2000).Furthermore, optimal AID induction also requires CD40 signaling (Muramatsuet al., 1999; Zhou et al., 2003).
2.6.5. Alternative Pathways for CSR
In a few CD40L‐deficient patients, where CD40L expression is totally absent,low levels of serum IgA and IgE have still been observed, suggesting thatmechanisms other than CD40–CD40L interaction may also induce CSR (Levyet al., 1997). Indeed, a few alternative CSR pathways have recently beendescribed and are discussed below.
2.6.5.1. BAFF and APRIL The TNF family ligands B cell activation factor ofthe TNF family (BAFF) and a proliferation‐inducing ligand (APRIL) regulatelymphocyte survival and activation. BAFF binds to three receptors that areselectively expressed on B cells; BAFF‐R, transmembrane activator andCAML interactor (TACI) and B cell maturation antigen (BCMA) whereasAPRIL interacts with TACI, BCMA, and proteoglycans (for review seeSchneider, 2005).In the presence of appropriate cytokines, BAFF and APRIL have also been
reported to induce CSR in human B cells (Litinskiy et al., 2002). This findingwas extended by the observation that both ligands can induce CSR in CD40�/�
mouse B cells, suggesting that this form of CSR is not dependent on CD40–CD40L interaction (Castigli et al., 2005b). In this model, TACI and/or BAFF‐R,

CLASS SWITCH RECOMBINATION 15
but not BCMA, seem to be the receptors that mediate CSR by APRIL andBAFF (Castigli et al., 2005b).
2.6.5.2. Toll and Toll‐Like Receptor LPS is known to induce CSR to all iso-types in mouse B cells (Stavnezer, 2000), probably through binding to Toll‐likereceptor 4 (TLR4). A more recently described pathway, involving TLR9 andits ligand, CpG‐containing DNA, has also been shown to induce both mouseand human B cells to undergo CSR to selected Ig isotypes (He et al., 2004; Linet al., 2004; Liu et al., 2003). The TLR9 pathway has received growingattention due to the potential relevance of CpG DNAs in the pathogenesisof autoimmune diseases and as candidates for antiallergens (Klinman, 2004;Peng, 2005).
3. Comparison Between Human and Mouse
3.1. The Constant Region Gene Locus in Human and Mouse
The human IGHC gene locus, localized on chromosome 14, contains ninefunctional genes and two pseudogenes (Cm‐Cd‐Cg3‐Cg1‐Cce‐Ca1‐Ccg‐Cg2‐Cg4‐Ce‐Ca2), organized into two g‐g‐e‐a blocks (Fig. 1). It has evolvedthrough a series of duplications, followed by mutations and specialization ofthe new genes. The locus is still evolving and up to 20% of the Caucasoidpopulation and 44% of the Mongoloid population show duplications of singleor multiple IGHC genes (Rabbani et al., 1996; Fig. 1).
The mouse IGHC locus, localized on chromosome 12, is composed of eightfunctional genes, including four Cg genes but only one Ca gene (Cm‐Cd‐Cg3‐
MouseαεV(D)J γ 1 γ 2aγ 2bγ 3δµ
Eµ 3�αE
Humanα 2εV(D)J γ1 ψε α1 ψγ γ 2 γ 4γ 3δµ
Eµ 3�α1E 3�α2E
α2ε
3�α2EHuman
V(D)J γ 1 ψε α1γ 3δµ
Eµ 3�α1E
α1εψγ γ 2 γ 4
3�α2E
ψγ γ 2 γ 4
α1-ε duplication, 110 kb
Figure 1 The constant region gene locus in human and mouse. Coding regions are shown as filledboxes and pseudogenes are indicated as open boxes. Striped boxes represent the duplicatedconstant regions genes in human Ig locus.

16 QIANG PAN ‐HAMMARSTROM ET AL .
Cg1‐Cg2b‐Cg2a‐Ce‐Ca; Fig. 1). On the basis of sequence homologies, it hasbeen suggested that the ancestral rodent IGHC only contained three Cg genesand that the mouse Cg2b and Cg2a have been generated by a recent duplica-tion (Bruggemann, 1988). It is interesting to note that in one mouse strain(BALB/c), where the IGHC locus has been studied in detail, pseudo‐g‐geneshave also been identified between the g1 and g2b and between the g2b andg2a genes (Cm‐Cd‐Cg3‐Cg1‐Ccg1‐Cg2b‐Ccg2‐Cg2a‐Ce‐Ca; Akahori and Kuro-sawa, 1997). The polarity of the two pseudogenes is, however, opposite to that ofthe functional g genes. Whether similar pseudo‐g‐gene exists in other mousestrains is not known but such information might provide additional clues for theevolution of the mouse IGHC locus.The evolution of the human andmouse IGHC loci after the divergence of the
two species has resulted in differences in the gene organization, the number ofgenes, and the function of selected IGHC genes. Thus, although both specieshave four IgG‐subclass‐encoding genes, a given subclass, for instance humanIgG3, is equivalent neither in terms of structure nor in terms of function to themouse IgG3. Furthermore, CSR to IgG subclasses or IgA is also differentiallyregulated in human and mouse (Sections 3.5 and 3.6).
3.2. Switch Regions in Human and Mouse
3.2.1. Characteristics of S Regions
The S regions for all Ig isotypes, in man and mouse, have previously beenmapped and sequenced. The repetitive sequences of the S regions are mostoften defined by the Sm‐like pentameric repeats, although different standardshave been applied and we have therefore reevaluated the repetitive sequencesusing dot plot analysis (Table 2). One reference sequence from each S regionwas studied. For the mouse S regions, the sequences were all derived from theBALB/c strain, except for Sm, where a complete sequence is only availablefrom the C57BL/6 genome.Structurally, S regions are all composed of tandemly repeated sequences
that contain common pentamer sequences (GAGCT and GGGCT), but differin the length of the repetitive region and the actual sequence of the repeats(summarized in Table 2). Human Sm, Sa, and Se are closely related andcharacterized by a dense clustering of pentameric repeats. No higher orderof these repeats has been identified in the human Sm, Sa, and Se regions,except for five 25‐bp repeats at the 30 border of Sm (Islam et al., 1994; Millset al., 1990; Pan et al., 2001). The four human Sg regions are less related to Sm,and vary considerably in length (Sg1 > Sg3 > Sg2 > Sg4), due to the presenceof different numbers of 79‐bp repeat units (Mills et al., 1995). The human Sg
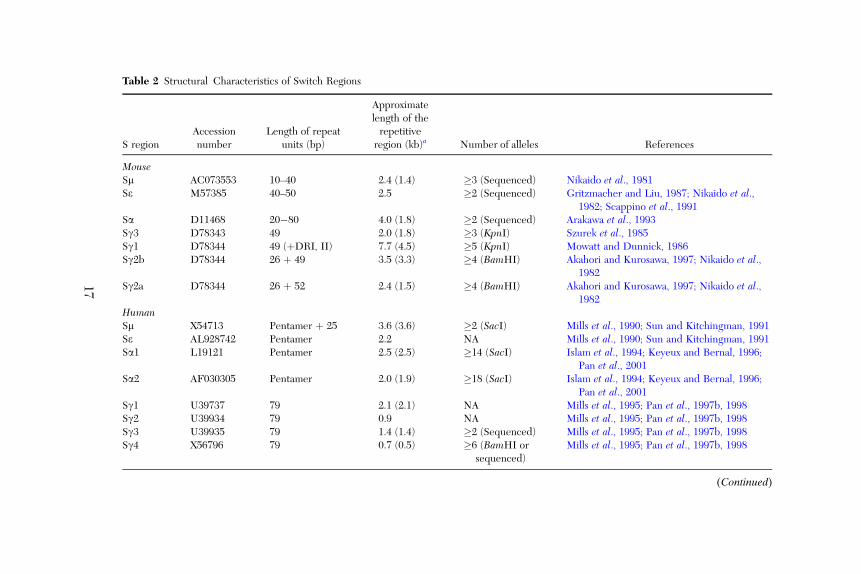
Table 2 Structural Characteristics of Switch Regions
S regionAccessionnumber
Length of repeatunits (bp)
Approximatelength of therepetitive
region (kb)a Number of alleles References
MouseSm AC073553 10–40 2.4 (1.4) �3 (Sequenced) Nikaido et al., 1981Se M57385 40–50 2.5 �2 (Sequenced) Gritzmacher and Liu, 1987; Nikaido et al.,
1982; Scappino et al., 1991Sa D11468 20�80 4.0 (1.8) �2 (Sequenced) Arakawa et al., 1993Sg3 D78343 49 2.0 (1.8) �3 (KpnI) Szurek et al., 1985Sg1 D78344 49 (þDRI, II) 7.7 (4.5) �5 (KpnI) Mowatt and Dunnick, 1986Sg2b D78344 26 þ 49 3.5 (3.3) �4 (BamHI) Akahori and Kurosawa, 1997; Nikaido et al.,
1982Sg2a D78344 26 þ 52 2.4 (1.5) �4 (BamHI) Akahori and Kurosawa, 1997; Nikaido et al.,
1982HumanSm X54713 Pentamer þ 25 3.6 (3.6) �2 (SacI) Mills et al., 1990; Sun and Kitchingman, 1991Se AL928742 Pentamer 2.2 NA Mills et al., 1990; Sun and Kitchingman, 1991Sa1 L19121 Pentamer 2.5 (2.5) �14 (SacI) Islam et al., 1994; Keyeux and Bernal, 1996;
Pan et al., 2001Sa2 AF030305 Pentamer 2.0 (1.9) �18 (SacI) Islam et al., 1994; Keyeux and Bernal, 1996;
Pan et al., 2001Sg1 U39737 79 2.1 (2.1) NA Mills et al., 1995; Pan et al., 1997b, 1998Sg2 U39934 79 0.9 NA Mills et al., 1995; Pan et al., 1997b, 1998Sg3 U39935 79 1.4 (1.4) �2 (Sequenced) Mills et al., 1995; Pan et al., 1997b, 1998Sg4 X56796 79 0.7 (0.5) �6 (BamHI or
sequenced)Mills et al., 1995; Pan et al., 1997b, 1998
(Continued)
17

Length of the repetitive sequences (search length 30 bp, �70% homology)Mouse Sg1 > Sa > Sg2b > Se > Sm ¼ Sg2a > Sg3Human Sm > Sa1 > Se > Sg1 > Sa2 > Sg3 > Sg2 > Sg4
Density of dots corresponding to repeats of similar sequences, Sx/Sx (search length 30 bp, �70% homology)Mouse Sa > Sm > Se > Sg1 > Sg3 > Sg2b > Sg2aHuman Sm > Sa1 > Sa2 > Se > Sg1 > Sg3 > Sg4 > Sg2
Density of dots corresponding to sequence match to Sm, Sx/Sm (search length 30 bp, �70% homology)Mouse Sa > Se > Sg3 > Sg1 ¼ Sg2b > Sg2aHuman Sa1 > Sa2 > Se > Sg4 > Sg2 > Sg1 > Sg3
aThe approximate length of the repetitive sequence of all the S regions listed was estimated by dotplot analysis. The repetitive sequences in a givenS region were defined by running the S region sequences against themselves; the search window is 30 bp and a maximum of 9 (70%) mismatches isallowed. When only three mismatches are allowed (90%), a more dense area of repetitive sequences can be identified and the estimated length wasgiven in parenthesis. Mouse S regions were based on the sequences from BALB/c mice, except for Sm, which was derived from the C57BL/6 mice.
Table 2 (Continued)
18
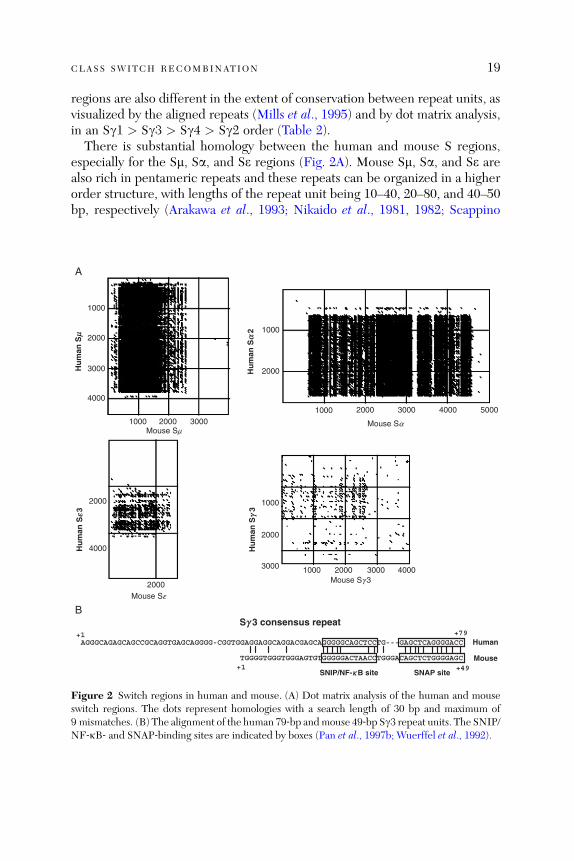
CLASS SWITCH RECOMBINATION 19
regions are also different in the extent of conservation between repeat units, asvisualized by the aligned repeats (Mills et al., 1995) and by dot matrix analysis,in an Sg1 > Sg3 > Sg4 > Sg2 order (Table 2).
There is substantial homology between the human and mouse S regions,especially for the Sm, Sa, and Se regions (Fig. 2A). Mouse Sm, Sa, and Se arealso rich in pentameric repeats and these repeats can be organized in a higherorder structure, with lengths of the repeat unit being 10–40, 20–80, and 40–50bp, respectively (Arakawa et al., 1993; Nikaido et al., 1981, 1982; Scappino
1000
2000
3000
4000
1000
2000
2000
B
A
1000
1000 2000 3000 4000
2000
3000
4000
2000 3000Mouse Sm
Mouse Se
Mouse Sg 3
Mouse Sa
1000
2000
1000 2000 3000 4000 5000
Hu
man
Sm
Hu
man
Sa
2
Hu
man
Se3
Hu
man
Sg
3
Sg 3 consensus repeat
AGGGCAGAGCAGCCGCAGGTGAGCAGGGG-CGGTGGAGGAGGCAGGACGAGCAGGGGGCAGCTCCTG---GAGCTCAGGGGACC
TGGGGTGGGTGGGAGTGTGGGGGACTAACCTGGGACAGCTCTGGGGAGC
+1 +79
+1 +49
Human
Mouse
SNIP/NF-kB site SNAP site
Figure 2 Switch regions in human and mouse. (A) Dot matrix analysis of the human and mouseswitch regions. The dots represent homologies with a search length of 30 bp and maximum of9 mismatches. (B) The alignment of the human 79‐bp andmouse 49‐bp Sg3 repeat units. The SNIP/NF‐kB‐ and SNAP‐binding sites are indicated by boxes (Pan et al., 1997b; Wuerffel et al., 1992).

20 QIANG PAN ‐HAMMARSTROM ET AL .
et al., 1991). The four mouse Sg regions share very little homology with mouseSm (Sg3 > Sg1 ¼ Sg2b > Sg2a), but show homology with human Sg regionsand are organized in 49‐ to 52‐bp repeats (Mowatt and Dunnick, 1986;Nikaido et al., 1982; Szurek et al., 1985). A 26‐bp repeating unit, in additionto a 49‐bp repeating unit, is present in the Sg2a and Sg2b regions (Akahori andKurosawa, 1997). The mouse Sg repeat units and human 79‐bp repeat unitsshow considerable sequence homology, especially with regard to conservationof the A (SNAP binding) and B (SNIP/NF‐kB binding) sites (Akahori andKurosawa, 1997; Pan et al., 1997b; Wuerffel et al., 1992; Fig. 2B). The mouseSg regions are also different with regard to the length of repeat sequences(Sg1 > Sg2b > Sg2a > Sg3) and the degree of conservation between therepeat units (Sg1 > Sg3 > Sg2b > Sg2a). Taken together, the available datasuggests that the Sm and Sgwere probably duplicated from an ancestral sequenceearlier than the human/mouse divergence and structural features unique for Sm(Sa, Se) and Sg have been evolutionarily conserved (Mills et al., 1990).Human Sg1 and mouse Sg1 seem to share several common features, with a
similar location in the Ig locus, most precise repeat units and being the longestSg regions (at least in the BALB/c strain). On the basis of the dot matrixcomparison, the Sg1 repeats appear to be prototypic for units in the other Sgregions in both human and mouse. Other Sg regions, however, do not show acorrelation. For instance, Sg3 is the shortest Sg region in mice but not inhumans. In addition, Sg3 is the only Sg region that shows some degree of homo-logy with Sm in the mouse, whereas in humans, Sg3 shows the least homologywith Sm (Sg4> Sg2> Sg1> Sg3). The most 30 Sg region in human and mouse,Sg4 and Sg2a, share the least homology with other Sg regions. However, thehuman Sg4 is still more related to the ‘‘prototype’’ Sg1, whereas the mouseSg2a is more related to Sg2b, suggesting that both have appeared late inontogeny and subsequently evolved differently.
3.2.2. Polymorphism of S Regions
S regions show extensive polymorphism, both in human and in mouse. In mostcases, however, only restriction fragment length polymorphism (RFLP) data areavailable. The best‐studied human S region is Sg4, where five different BamHIIGHG4 alleles have been characterized by sequencing (Pan et al., 1998). TheseSg4 alleles differ in length due to deletions and insertions of a varying number of79‐bp Sg4 repeat units, ranging from5 to 14 repeats (Pan et al., 1998). In addition,single base substitutions have also been noted in several alleles when comparedwith the prototype Sg4 region (derived from the 9.2‐kb BamHI allele; Pan et al.,1998). In the mouse, a partially sequenced Sg3 from the BAB14 strain has beencompared with the fully sequenced Sg3 from BALB/c, and insertions and

Human Sg4 regionBamHI allele
9.4 kb(12 repeats)
9.2 kb(10 repeats)
IgG4 serum level(g/L)
0.42 ± 0.23
0.26 ± 0.14
Figure 3 Influence of the length of the Sg4 region on serum levels of IgG4. The boxes filled withdifferent patterns represent the individual 79‐bp Sg4 repeat units (Pan et al., 1998).
CLASS SWITCH RECOMBINATION 21
deletions of 49‐bp units were evident (Szurek et al., 1985). A later studyalso suggested that polymorphism in the Sg1 region is due to differences inthe number of 49‐bp elements (Mowatt and Dunnick, 1986). In Table 2, thenumber of possible alleles for each S region, mainly based on RFLP results,is indicated, and the numbers may change when additional sequence databecomes available.
There have been suggestions that the length of the S region correlates toserum levels of a given Ig class. In humans, IgG1 shows the highest serumconcentration, followed by IgG3 (provided that half‐life is taken into account),IgG2, and IgG4, which is in general agreement with the length of the Sg regionspresented in Table 2. We have also shown that the 9.4‐kb BamHI IGHG4 alleleis more productive than the 9.0‐kb allele in normal healthy donors, possibly dueto the extended Sg4 region (Pan et al., 1998; Fig. 3). In BALB/cmice, IgG1 is themost abundant Ig class and Sg1 is indeed the longest among the S regions in thisstrain. By replacing the mouse Sg1 region with synthetic or endogenousS regions of various lengths, Zarrin et al. (2005) have shown that the length ofthe S region directly influences CSR efficiency in vivo, presumably by providingmore substrate for the recombination machinery.
Despite the constant length and organization of human Sg3 regions asso-ciated with both the b and g allotypes, marked differences are noted in the rateof switching (Hassan et al., 1992) and serum IgG3 levels (Morell et al., 1972b).These differences appear to be due to point mutations in a crucial NF‐kB‐binding motif in one of the Sg3 repeats rather than polymorphisms in the GLg3 promoter (Pan et al., 1997a,b).
3.2.3. Secondary Structures in S Regions
As discussed above, CSR is a region rather than a site‐specific event. It istherefore of interest to search for a relation between the recombination break-points and the structural character of the S region. The human and mouseS regions contain a large number of palindromic sequences that may formsecondary ssDNA structures. It has been suggested that CSR preferentially

22 QIANG PAN ‐HAMMARSTROM ET AL .
occurs at transitions from a stem to a loop structure in ssDNA (microsites) inXenopus and in mice (Mussmann et al., 1997). However, a later study basedon large number of human Sm, Sg, and Sa breakpoints did not show a signi-ficant correlation between the breakpoints and such secondary structures(Pan‐Hammarstrom et al., 2004). It is currently unclear whether the differencenoted represents species variations or are due to differences in the methodsused for analysis. Nevertheless, new ways of exploring the role of secondaryand tertiary structures of the S regions are required to fully resolve thequestion.
3.3. 30 Enhancers in Human and Mouse
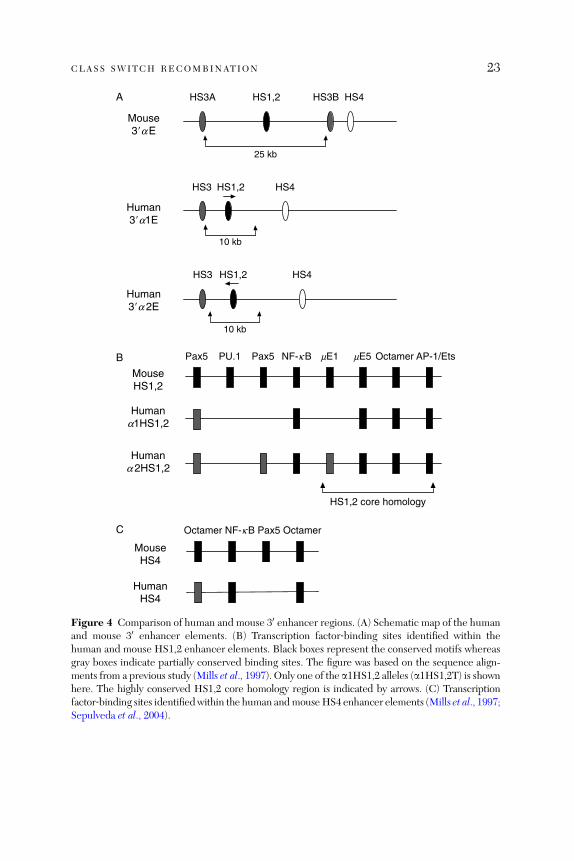
The human and mouse enhancer elements in the 30 a region share significantsequence homology (Mills et al., 1997) and have been proposed to have similarfunctional properties. There are, however, significant differences. First, thereare two 30 enhancer regions in humans and they might be regulated differentlyand could also interact with each other. Second, humans do not have anequivalent for the mouse HS3B enhancer elements and the 30 enhancers areorganized differently (Fig. 4A). The human a1 and a2 HS1,2 enhancers bothreside near the centers of �10‐kb palindromes, with each palindrome closelyflanked by a single copy of HS3 immediately adjacent to the 50 end and an HS4unit located �4‐kb downstream (HS3‐HS1,2‐HS4; Mills et al., 1997). Bycomparison, the mouse HS1,2 is centrally positioned in a much larger(�25 kb) palindrome that contains a copy of HS3 on each end, with HS4located �4‐kb downstream of the palindrome (HS3A‐HS1,2‐HS3B‐HS4;Chauveau and Cogne, 1996; Saleque et al., 1997). Third, certain transcriptionfactor binding sites in the mouse enhancer, including Pax5‐binding sites, donot appear to be conserved in the human HS1,2 or HS4 (Mills et al., 1997;Fig. 4B and C) and transient transfection assays have shown that the activity ofthe human HS4 is indeed regulated differently from that of the equivalentmouse enhancer (Sepulveda et al., 2004).A study has compared the genomic sequences of the entire 30 regulatory
regions, encompassing the known enhancer elements and its downstreamsequences in mice and humans (Sepulveda et al., 2005). Although very limitedsequence identity was observed between these regions, except for the enhan-cers themselves, other features, that is extensive palindromes flanking theHS1,2 enhancer and families of locally repetitive sequences, are conserved(Sepulveda et al., 2005). One interesting difference is that in humans, but notin mice, the locally repetitive sequences contain short tandem repeats thatresemble ‘‘switch’’ sequences. Whether these repetitive sequences have a rolein CSR is unknown but worthy of further investigation.
Human3�a 2E
HS1,2HS3 HS4
10 kb
Human3�a1E
HS1,2HS3 HS4
10 kb
Mouse3�aE
HS3AA HS1,2 HS3B HS4
25 kb
B
MouseHS1,2
Humana1HS1,2
OctamerNF-kBPax5Pax5 PU.1 mE1 mE5 AP-1/Ets
Humana 2HS1,2
HS1,2 core homology
C
MouseHS4
HumanHS4
Octamer NF-kB Pax5 Octamer
Figure 4 Comparison of human and mouse 30 enhancer regions. (A) Schematic map of the humanand mouse 30 enhancer elements. (B) Transcription factor‐binding sites identified within thehuman and mouse HS1,2 enhancer elements. Black boxes represent the conserved motifs whereasgray boxes indicate partially conserved binding sites. The figure was based on the sequence align-ments from a previous study (Mills et al., 1997). Only one of the a1HS1,2 alleles (a1HS1,2T) is shownhere. The highly conserved HS1,2 core homology region is indicated by arrows. (C) Transcriptionfactor‐binding sites identifiedwithin the human andmouseHS4 enhancer elements (Mills et al., 1997;Sepulveda et al., 2004).
CLASS SWITCH RECOMBINATION 23

24 QIANG PAN ‐HAMMARSTROM ET AL .
The 30 enhancer regions in humans are polymorphic. At least four a1HS1,2and two a2HS1,2 alleles have been identified. The allelic polymorphism is dueto varying number of 38‐bp repeats (one to four) immediately 30 of the core ofHS1,2 enhancer, variable spacer elements separating the 38‐bp repeats andvariable external elements bordering the repetition cluster (Denizot et al., 2001;Giambra et al., 2005). The repeat units themselves do not exhibit enhanceractivity and are not conserved between mice and humans (Mills et al., 1997).However, as a number of transcription factor‐binding sites, including NF‐kBand E47, have been identified in these repetitive sequence (Giambra et al.,2005) and increasing the number of these repeats results in an increased levelof HS1,2 enhancer activity in luciferase assays (Denizot et al., 2001), the poly-morphisms in the HS1,2 enhancer might be of functional relevance. Indeed,dysregulation of IgA production in celiac disease and IgA nephropathy has beensuggested to be associated with different a1HS1,2 alleles (Aupetit et al., 2000;Frezza et al., 2004). The human HS3 and HS4 enhancers are not polymorphic(Guglielmi et al., 2004) whereas the mouse 30 enhancers have not been studiedin detail in this respect. However, as the entire 30 regulatory region and down-stream sequences are highly polymorphic, when comparing the 129Sv andC57BL/6 strains (Sepulveda et al., 2005), it will not be surprising if allelicdifferences exist in the enhancer elements.In mice, targeted deletion of the 30 enhancers have shown that HS3A and
HS1,2 are individually dispensable for CSR (Manis et al., 1998b) whereas thejoint deletion of HS3B and HS4 severely affects this process (except CSR toIgG1; Pinaud et al., 2001). In humans, no deletions of these enhancers havebeen reported. However, there is evidence that the mouse HS1,2 enhancerregulates the GL e and g2b promoters (Laurencikiene et al., 2001) whereas thehuman HS1,2 enhancer regulates the GL a and g promoters (Hu et al., 2000;Pan et al., 2000). Both human a1 and a2 HS1,2 fragments show strongenhancer activity on the GL a1 and a2 promoters when transiently transfectedinto human mature B cell lines. HS4 has a modest effect whereas HS3 showsno enhancer activity in these cell lines (Hu et al., 2000). Notably, the combi-nation of HS3‐HS1,2‐HS4 fragments displays a markedly stronger enhanceractivity than the individual fragments, suggesting that they interact synergisti-cally and all three enhancer elements may be needed for the activation ofthe Ca locus before CSR occurs (Hu et al., 2000). Similarly, this combina-tion of enhancer elements also strongly stimulates the GL g3 promoter inan orientation‐independent manner (Pan et al., 2000). Furthermore, theconserved structures flanking the HS1,2 and the highly conserved HS1,2core sequence in mice and humans suggest that the entire 30 enhancer region,rather than the HS3B and HS4 alone, is activated during normal CSR.This hypothesis is supported by an observation that deletion of the entire

CLASS SWITCH RECOMBINATION 25
30 enhancer from a BAC transgene eliminates IgG1 CSR in addition to all theother isotypes (Dunnick et al., 2005).
3.4. AID in Human and Mouse
AID is highly conserved in evolution, from fish to humans (Zhao et al., 2005).At the protein level, human AID and mouse AID shows a sequence identity of91% and thus far no functional differences between these molecules have beennoted.
In human primary B cells, IL‐4 alone is sufficient to drive AID expressionbut CD40 signaling is required for optimal AID production (Zhou et al., 2003).In mouse primary B cells, similar results have been obtained (Dedeoglu et al.,2004), although an earlier study has suggested that IL‐4 alone has no detect-able effect on AID expression and that IL‐4 þ CD40L were not synergistic forAID induction (Muramatsu et al., 1999). LPS is a powerful AID inducer inmice but not in humans (Muramatsu et al., 1999; Zhou et al., 2003), which isconsistent with the previous knowledge that LPS is a strong CSR inducer inmice but not in humans. IL‐4 þ CD40L instead, provides a strong signal forCSR in human B cells.
The intronic enhancer for the AICDA loci in humans and mice share a 70%sequence identity and the two high‐affinity E‐box sites are conserved, as areoctamer, Pax5‐, NF‐kB‐, and Ikaros‐binding sites (Sayegh et al., 2003; Yadavet al., 2006). The upstream promoter region also shares a high degree ofhomology and contains conserved Pax5‐ or Sp1‐binding sites (Gonda et al.,2003; Yadav et al., 2006). The proposed STAT6 and NF‐kB p50 sites upstreamof the promoter region in the human AICDA locus (Dedeoglu et al., 2004) are,however, not conserved in mice. By searching the 8‐kb genomic sequencesupstream of the second exon of the human AICDA loci, we found anotherpotential STAT6‐binding site about 350‐bp upstream of the first E‐box site.However, again, it is not conserved in the mouse locus. Thus, although STAT6and p50 are essential for IL‐4 induction of AID gene expression in mouseB cells (Dedeoglu et al., 2004), the underlying mechanism might still besomewhat different from that in human cells.
Two PKA phosphorylation sites have been identified in the human AIDprotein, threonine 27 (T27) and serine 38 (S38). Both are located within thePKA canonical consensus motifs (RRXS/T; Pasqualucci et al., 2006). In mice,however, T27 is not embedded within a classical PKA motif due to an aminoacid substitution (RHET versus RRET in humans). Although T27 was found tobe a PKA substrate in vitro and a T27A mutant failed to undergo PKAphosphorylation and was not able to rescue CSR in AID�/� mouse cells, S38has been suggested as the residue on mouse AID that is phosphorylated by

26 QIANG PAN ‐HAMMARSTROM ET AL .
PKA (Basu et al., 2005). However, S38 is not conserved in AID from bony fishand yet it can support CSR when transfected into mouse B cells (Barreto et al.,2005). Thus, it is currently unclear how fish AID can ‘‘bypass’’ PKA phosphor-ylation. Alternatively, other PKA site(s), such as T27, which is located in thesame nonconsensus motif as in the mouse (RHET), is utilized in fish AID.
3.5. Regulation of CSR to IgA in Human and Mouse
TGF‐b1 can direct switching from IgM to IgA in both humans and mice byinducing GL transcripts (Islam et al., 1991; Shockett and Stavnezer, 1991)through activation of its corresponding promoter elements in the Ia region(Lin and Stavnezer, 1992; Nilsson and Sideras, 1993). The GL a promoterregions, in particular the TGF‐b1‐responsive element, including binding sitesfor RUNX and SMAD3/4, are highly conserved between humans and mice andthese promoters appear to be regulated similarly (Hanai et al., 1999; Pardaliet al., 2000; Shi and Stavnezer, 1998; Xie et al., 1999).Unlike mice, however, humans express two IgA subclasses, IgA1 and IgA2,
each encoded by a separate gene and directed against different antigens. Thus,IgA1 mainly gives rise to antibodies against protein antigens whereas IgA2 isprimarily directed against polysaccharide antigens. The two human IgA sub-classes are also differentially expressed at different anatomical sites. IgA1comprises about 80–90% of the total IgA in serum and it is predominantlyexpressed in spleen, peripheral lymph nodes, tonsils, nasal mucosa, lacrimalglands, gastric and proximal small intestinal mucosa, whereas IgA2 productionis proportionally larger in the salivary glands and it is the predominant subclassin the large bowel mucosa (Kett et al., 1986). By selective amplification ofrecombined Sa1 or Sa2 regions, we have estimated the proportion of cells thathave switched to IgA1 or IgA2 at different anatomical sites and largely con-firmed the above observations at the DNA level (Pan‐Hammarstrom et al.,unpublished data).The mechanisms underlying the preferential IgA1 or IgA2 responses are still
elusive. The two human GL a promoters are 98% homologous (Nilsson et al.,1991), with identical TGF‐b1‐responsive elements (Nilsson et al., 1995), sug-gesting that the GL a promoters themselves may not contain enough sequenceinformation to ensure subclass‐restricted expression. Additional cis‐elements,such as the 30 enhancers, independent of the TGF‐b1 pathway, may thus berequired. Even though a sequence comparison shows that the a1 and a2 coreenhancer elements are almost identical, there are important structural differ-ences between the two loci where the a2HS1,2 element is inverted relative tothe a1HS1,2 (Fig. 4A). It is also located at a greater distance from the HS3than in the a1 locus (Mills et al., 1997). Indeed, we have shown that the

CLASS SWITCH RECOMBINATION 27
a1HS1,2 element has a stronger effect on the GL g3 promoter than thecorresponding a2 elements (Pan et al., 2000). Thus, the current hypothesissuggests that the 30 a1 and a2 enhancers primarily influence the first (Cg3‐Cg1‐Cce‐Ca1) and second (Cg2‐Cg4‐Ce‐Ca2) block of duplicated IGHCregion genes, respectively. However, the specific factors that would turn onthe a1 or a2 enhancer, and subsequently the expression of the genes in the firstor second block, respectively, remain to be identified.
3.5.1. IgA Production in mMT Mice and Cm‐Deficient Patients
For switching and subsequent production of IgA, additional differences mayexist that may not relate to the difference in the number of IgA genes. Aprevious study from Zinkernagel’s group has shown that IgA is selectivelyexpressed in mMT mice, which lack IgM or IgD expression and have a pro‐Bcell developmental block (Macpherson et al., 2001). The mMT IgA pathwayrequires extrasplenic peripheral lymphoid tissues and has previously beensuggested as an evolutionarily primitive system in which immature B cellscan switch to IgA production at peripheral sites (Macpherson et al., 2001).Patients with mutations or deletions in the Cm gene (IgMD), which preventIgM surface expression on B cells, have also been described previously (LopezGranados et al., 2002; Yel et al., 1996). These patients are highly susceptible toinfections in the respiratory tract and often succumb at an early age, suggestingthat, in contrast to the mouse model described above, serum and secretory IgAmight be low or absent. Indeed, using ELISA, we found that although IgA waspresent in the sera of the patients, it was expressed at about two orders ofmagnitude lower than those found in the mMTmice (Pan et al., 2002a). No IgAwas found in saliva from these patients, nor could any fecal IgA can bedetected (Pan et al., 2002a). While IgAþ cells were detected in histologicalsections taken from the ileum and spleen of mMT mice (Macpherson et al.,2001), no IgAþ cells could be visualized in intestinal biopsy or nasal biopsyfrom IgMD patients (Pan et al., 2002a). The direct IgA switching pathwaydescribed in mice therefore contributes little, if any, to the mucosal defensesystem in humans.
3.6. Regulation of CSR to IgG Subclasses in Human and Mouse
3.6.1. Functional Properties of IgG Subclasses in Human and Mouse
Both human and mouse IgG can be subdivided into four subclasses. However,as diversification of the IgG‐subclass‐encoding genes have occurred after thedivergence of the two species, a given subclass in humans, for instance IgG1 orIgG3, is not the ‘‘functional homologue’’ of mouse IgG1 or IgG3.

28 QIANG PAN ‐HAMMARSTROM ET AL .
In humans, the four IgG subclasses differ in their relative serum abundance,half‐life, ability to activate complement, affinity for Fcg receptors, and arepreferentially directed against different types of antigens. IgG1 is the predom-inant serum IgG subclass (66%), followed by IgG2 (24%), IgG3 (7%), andIgG4 (3%). IgG1 and IgG3 appear early in ontogeny (Morell et al., 1972a;Oxelius, 1979), are efficient activators of the classical complement pathway(Bruggemann et al., 1987), bind to high‐affinity FcgRI receptors, and aredirected mainly against protein antigens. IgG2 appears much later in ontogenyand is primarily directed against polysaccharide antigens (Hammarstrom et al.,1986). IgG3 is sensitive to proteolytic degradation (Turner et al., 1970) and hasthe shortest half‐life of all subclasses. IgG4 antibodies are functionally mono-valent and do not, under normal circumstances, activate complement. Raisedlevels of IgG4 antibodies are often noted against selected protein antigensafter chronic exposure (Aalberse et al., 1983) and are involved in a variety ofallergic diseases (Djurup, 1985; Perelmutter, 1984).In mice, the four IgG subclasses are also endowed with different biological
and functional properties, although these have not been studied in as muchdetail as in humans. The serum abundance of the four IgG subclasses varies inmice with different genetic backgrounds. In BALB/c, IgG1 is the predominantserum IgG subclass whereas in C57BL/6, IgG2b has the highest serum con-centration (Shimizu et al., 1982). IgG1 is dominant in response to parasiticinfections. It does not activate complement very efficiently but can stimulatephagocytosis through interaction with Fc receptors. IgG2a can efficientlyactivate the complement cascade, binds to high‐affinity Fc receptors, and isimportant for control of viral infections (Coutelier et al., 1987). IgG2b andIgG3 are mainly induced by T‐independent antigens such as polysaccharideantigens and in this respect are the ‘‘functional homologue’’ of human IgG2.
3.6.2. Differential Regulation of CSR to IgG Subclasses by Cytokines
Thus far, no human IgG‐subclass‐specific ‘‘switch factor’’ has been described.In the presence of anti‐CD40 antibodies, IL‐4 can induce CSR to IgG1, IgG3,and IgG4 (Armitage et al., 1993; Fujieda et al., 1995), whereas IL‐10 inducesCSR to IgG1 and IgG3 (Briere et al., 1994; Fujieda et al., 1996). IL‐10 is not aswitch factor for IgG4, but addition of IL‐10 augments IL‐4‐induced g4expression and IgG4 production (Jeannin et al., 1998). Cg3 expression seemsto be upregulated by IL‐4 in the presence of B cell activators such as Staphy-lococcus aureus Cowan I (SAC) or PMA (Kuze et al., 1991), however, neitherSAC nor PMA are switch‐inducing stimuli. IFN‐g has been shown to cooper-ate with IL‐6 to induce IgG2 production in human B cells (Kawano et al.,1994), however, it is unlikely that it acts as a switching factor for IgG2 as its

CLASS SWITCH RECOMBINATION 29
enhancing effect on IgG2 production is not observed when using IgG2‐negative cells (Kawano et al., 1994).
Regulation of CSR to mouse IgG subclasses is different from that in hu-mans. In mouse B cells, IL‐4 directs CSR to IgG1, but not to other IgGsubclasses, whereas IFN‐g selectively induces CSR to IgG2a and, undercertain circumstances, IgG3 (Stavnezer, 2000). Studies have also shown thatIL‐27 can induce CSR to IgG2a in B cells activated with anti‐CD40 antibodyor LPS, in an IFN‐g‐independent manner (Yoshimoto et al., 2004). LPS aloneinduces CSR to IgG2b and IgG3 and addition of TGF‐b or IL‐10 furtherincrease CSR to IgG2b or IgG3, respectively (Stavnezer, 2000).
3.6.3. GL Promoters in Human and Mouse
Sequences located upstream of each Sg region, including the Ig exons and thecorresponding Ig promoter region, are highly conserved among the fourhuman Cg subclasses (Mills et al., 1995). When comparing these sequenceswith mouse GL g2b transcripts, a 175 bp evolutionarily conserved sequence(ECS‐Ig) was previously identified (Mills et al., 1995; Sideras et al., 1989;Fig. 5). The overall homology between human and mouse ECS‐Ig is 65%,which is remarkable for intron sequences, suggesting that it contains importantregulatory elements. Indeed, three NF‐kB‐binding sites have been identifiedin the mouse ECS‐Ig and appear to be required for induction of transcription-al activity of the GL g1 promoter by CD40 engagement (Lin and Stavnezer,1996). A STAT6‐binding site located upstream of the NF‐kB‐binding sites hasalso been identified in the mouse GL g1 promoter and is important forinduction by IL‐4 (Lundgren et al., 1994).
The three NF‐kB‐binding sites, particularly the first one, are highly con-served between human and mouse ECS‐Ig regions (Fig. 5). Encouraged bythis observation, human GL Ig3 (Pan et al., 2000; Schaffer et al., 1999), Ig1(Bhushan and Covey, 2001), and Ig4 (Agresti and Vercelli, 2002) promoterswere subsequently characterized. At least two additional NF‐kB‐binding sitesand one STAT6‐binding site, all located downstream of the first three NF‐kB‐binding sites, are required for activation of the human Ig3 promoters (Panet al., 2000; Schaffer et al., 1999; Fig. 5). These sites are located in the 30 half ofthe ECS‐Ig; however, they are not conserved between species. A 36‐bp regionin the human GL g1 promoter, downstream of the ECS‐Ig, and containingCREB/ATF‐ and NF‐kB‐binding sites, has been shown to contribute to thedifference in expression of g1 and g3 GL transcripts (Bhushan and Covey,2001; Dryer and Covey, 2005). In the human GL g4 promoter, in addition tothe region containing the conserved NF‐kB‐binding sites, an additional IL‐4/CD40 responsive element, that binds STAT6 and interacts selectively with

MouseGL g1 promoter
NF-kBSTAT6/CEBP
1 2 3
1 2 3 4 5Human
GL g 3 promoter
STAT6NF-kB
ECS-Ig
CACCCTCACCCACACATTCACATGAAGTAATCTAAGTCAGGTTTGGACTCCCCCTCACCCTCT
CACCCCCATCCCCACACACCCATGAGGCAGCCTCGGCTGTGTCTGGACTCCCCCTTACCCTGT
GACACAGAAACCCCCAGAATGAAGGGGAACCCTGTCAGGAAATAGCCTTATGCCACCACTGTC
GACACAGAAACCACCAGAAGAAAAGGGAAC..T.TCAGGAAGTAAGC.GGTGCCGCCGGTTTC
AATCCTGTTCTTAGTCAATCATATGATGGAAAGAGGGTGACATTACCTCTCTGGGACAAAGGC
AATCCTGTTCTTAGTC.TTTGCAGCGTGGAGTTCACACCCCTGGGGACCTGAGGGCCGAGCTG
TGTGACTCTGGGAAAGACAAGAGAAGGGCACAG...GAACAGAGACGGCTG
TGATTTCCTAGGAAGACAAATAGCAGCTGACGGCGTGGGCAAGTCTGCCCA
−139ECS-Ig
−229
ECS-Ig
STAT6 NF-kB 1
NF-kB 3 NF-kB 3
STAT6
NF-k B 4 NF-kB 5
+1
+1
Mouse Ig 1
Mouse Ig1
Mouse Ig 1
Mouse Ig 1
Human Ig 3
Human Ig 3
Human Ig 3
Human Ig 3
A
B
Figure 5 Comparison of human and mouse GL g promoters. (A) Schematic map of the humanGL g3 and mouse GL g1 promoters. Black boxes represent the conserved transcription factor‐binding sites whereas gray boxes indicate partially conserved motifs. Striped boxes representtranscription factor‐binding sites that are unique in human GL g3 promoters. (B) Nucleotidesequences of the evolutionarily conserved region. The transcription factor‐binding sites are boxed.The initiation site of the human g3 or mouse g1GL transcript is indicated as ‘‘þ1,’’ above or belowthe sequences.
30 QIANG PAN ‐HAMMARSTROM ET AL .
c‐Rel, has been identified (Agresti and Vercelli, 2002). Regulation of CSR toIgG2 in human B cells is still poorly understood and the GL g2 promoter is theonly GL g promoter that has not been characterized to date. Given the markedhomology of the ECS‐Ig2 with the other human ECS‐Ig regions, a differentialregulation of CSR to IgG2, is most likely determined by regulatory elementslocated outside the ECS‐Ig regions.One concern for the GL promoter studies is that different results are often
obtained when different cell lines are studied (Lin and Stavnezer, 1996; Panet al., 2000; Schaffer et al., 1999; Warren et al., 1999). These differences may

CLASS SWITCH RECOMBINATION 31
be the result of variations in the expression of cellular and nuclear factorsresponsive to CD40 and/or IL‐4 pathway‐associated signals. Furthermore,experiments from transgenic mice suggest that cis‐acting elements upstreamof the three NF‐kB‐ and STAT6‐binding sites might be critical for the GL g1transcription (Berton et al., 2004). Thus, the relevance of studies in B cell linesfor the function of these promoters in normal B cells remains to be elucidated.
3.7. CD40–CD40L Pathway in Human and Mouse
The phenotypes of human patients with HIGM type I (CD40L deficient) andtype III (CD40 deficient) have largely been confirmed in mouse ‘‘knockout’’experiments where either the CD40‐ or the CD40L‐encoding genes have beeninactivated by HR (Castigli et al., 1994; Kawabe et al., 1994; Renshaw et al.,1994; Xu et al., 1994). Unlike ‘‘classical’’ HIGM patients, normal levels ofserum IgM have been observed in both CD40 and CD40L knockout mice.This phenotype was, however, noted in a later study on 56 CD40L‐deficientpatients where the majority (52.7%) had normal IgM serum levels at the timeof diagnosis (Levy et al., 1997). It thus appears that IgM serum levels mayincrease with age, particularly if initiation of IVIG substitution therapy isdelayed (Levy et al., 1997). It would thus be interesting to study the IgMlevel in aged CD40L‐ and CD40‐deficient mice. Another notable differencebetweenCD40L‐ or CD40‐deficientmice andHIGMpatients is that thesemiceshow reduced, albeit detectable, levels of serum IgG with a relatively normallevel of IgG3 (Kawabe et al., 1994; Renshaw et al., 1994), whereas the majorityof patients (92% for CD40L‐deficient and two out of three CD40‐deficientpatients) have undetectable levels of IgG (Ferrari et al., 2001; Levy et al., 1997).This may reflect a difference between Tcell‐independent immune responses ofmice and humans. The CSR pathways other than CD40–CD40L may be moreprominent in mice than in humans.
In mouse B cells, CD40 signaling itself induces GL g1 and e transcriptionand together with IL‐4, it induces CSR to IgG1 and IgE (Stavnezer, 2000). Inthe presence of TGF‐b1, IL‐4, IL‐5, and anti‐d antibody immobilized ondextran, CD40L can also replace LPS to induce CSR to IgA (Snapper et al.,1995). In human B cells, antibodies to CD40 or CD40L have provided the firstrelatively efficient means for induction of CSR. In the presence of IL‐4, anti‐CD40 antibodies induce GL e transcripts and switching to IgE (Gascan et al.,1991). This combination of stimulators also induces secretion of all IgG sub-classes and the respective GL transcripts except IgG2 (Fujieda et al., 1995). Inthe presence of TGF‐b, IL‐10, and SAC, anti‐CD40 antibody‐activated B cellsalso secrete an increased amount of IgA (Defrance et al., 1992). Interestingly,anti‐CD40 antibodies and IL‐10 also induce production of normal, or close to

32 QIANG PAN ‐HAMMARSTROM ET AL .
normal, amounts of IgA and IgG in cells from IgA deficient (IgAD) and asubgroup of common variable immunodeficiency (CVID) patients (Eisensteinet al., 1994; Friman et al., 1996; Nonoyama et al., 1993; Pan‐Hammarstromet al, unpublished data), suggesting that abnormalities in these patients areprobably reversible and regulatory in origin.
3.8. BAFF–APRIL–TACI Pathway in Human and Mouse
BAFF and APRIL activate IgG, IgA, and IgE switching in both human andmouse B cells (Castigli et al., 2005b; Litinskiy et al., 2002). In human B cells,however, secretion of class‐switched antibodies requires additional signals thatinclude cross‐linking of the B cell receptor and cytokines such as IL‐15 or IL‐2(Litinskiy et al., 2002).TACI, which is encoded by TNFRSF13B, binds both BAFF and APRIL. Two
TACI‐deficient mouse models have been described to date. In the first model, asignificant decrease in serum IgM and IgA concentrations were observed,despite an increased number of circulating and splenic B cells. The responsesto T‐independent type II antigens were also abolished (von Bulow et al., 2001).In the second model, TACI deficiency resulted in profound B cell hyperplasia,lymphoma development, and severe autoimmune disease with lupus‐like symp-toms (Seshasayee et al., 2003; Yan et al., 2001). No antibody deficiency wasobserved and the TACI‐deficient B cells instead produced increased amounts ofIgs in vitro (Yan et al., 2001). On the basis of the data from the second mousemodel, TACI was suggested to play an important role in the negative regulationof B cell activation. It is currently unclear why such discrepancies were noted inthe two models, although it might be due to differences in the targetingstrategies (deleting exons 1 and 2 versus sequences that correspond to thetransmembrane and intracellular regions of TACI in the second model) ordifferent genetic backgrounds of the mice.Homozygous or compound heterozygous mutations in TNFRSF13B were
identified in a few patients with CVID (Castigli et al., 2005a; Salzer et al.,2005). These mutations resulted in loss of TACI function, as evidenced byimpaired proliferative responses to IgM‐APRIL costimulation and defectiveCSR induced by APRIL or BAFF. In contrast to the second mouse model,TACI‐deficient patients are characterized by humoral immunodeficiency, withsubstantially reduced serum concentrations of IgM, IgG, and IgA. The totalnumber of peripheral B cells is, however, normal and signs of autoimmunityand lymphoproliferation are not dominant features (Salzer et al., 2005).By analyzing B cells from mice deficient in TACI (the first model), BCMA,
and BAFF‐R, Castigli et al. (2005b) have shown that TACI‐deficient B cellsdo not synthesize IgG1, IgA, and IgE in response to APRIL but are able

CLASS SWITCH RECOMBINATION 33
to produce IgG1 and IgE in response to BAFF. These results suggest thatAPRIL‐induced CSR to all isotypes and BAFF‐induced CSR to IgA are medi-ated by TACI whereas BAFF‐induced CSR to IgG1 and IgE may be mediatedby both TACI and the BAFF‐R. Normal CSR activity was observed in BCMA‐deficient B cells suggesting that signal through this receptor is not requiredfor CSR (Castigli et al., 2005b). However, neither APRIL nor BAFF caninduce CSR and subsequent IgG production in vitro in TACI‐deficienthuman B cells (Salzer et al., 2005). This could potentially explain the observedphenotypic difference, where most of the individuals with TACI deficiencyshow reduced serum levels of both IgA and IgG, whereas TACI‐deficient miceexpress normal levels of IgG.
Taken together, the existing data suggest that TACI can mediate CSRinduced by APRIL/BAFF in both human and mouse B cells, although itmight be of greater importance for IgG production in humans. The humanTACI‐deficient phenotype is also different from the mouse models as neithersevere B cell hyperplasia nor features of systemic lupus erythematosus havebeen observed. The dichotomy between gene deletion (mice) and point muta-tions (humans) cannot fully explain the species difference, as individuals witha complete lack of the TACI protein show a similar phenotype as patientswith hypomorphic homozygous or heterozygous point mutations in the TACI‐encoding gene. Alternatively, species differences might exist in the BAFF–APRIL–TACI system with regard to B cell homeostasis and humoral immuneresponses. This should be taken into account when designing drugs aimedat interfering with the BAFF–APRIL–BAFFR–TACI–BCMA system, aspreviously suggested (Gross et al., 2001).
3.9. Toll and Toll‐Like Receptor in Human and Mouse
In contrast to findings in mice, LPS is not a switch factor for human B cells,probably due to a lack of expression of TLR4 on the B cell surface. However,TLR4 expression can be induced in human B cells by stimulation with IL‐4(Mita et al., 2002), suggesting that human B cells might still be susceptible toLPS–TLR4 signaling under certain conditions. In addition, human monocytesexpress TLR4, which may indirectly influence CSR through secretion ofcytokines. In light of these findings, we may need to reevaluate the role ofthe LPS–TLR4 pathway in CSR in humans. But if there is indeed an effect, itis probably less potent than in mice.
Both human and mouse B cells express TLR9. In the mouse, CpG DNAsdirect CSR and subsequent Ig production to ‘‘Th1‐like’’ Ig isotypes (IgG2a,IgG2b, and IgG3) while suppressing expression of Th2 isotypes (IgG1 andIgE) in a TLR9‐dependent manner (Lin et al., 2004; Liu et al., 2003). A study

34 QIANG PAN ‐HAMMARSTROM ET AL .
further showed that TLR9 signaling is required for CSR to pathogenic IgG2aand IgG2b autoantibodies in a murine SLE model (Ehlers et al., 2006). Inhumans, CpG DNAs can induce CSR to IgG1, IgG2, and IgG3, but sub-sequent IgG production requires additional stimulation by factors such as anti‐BCR, BAFF, or CD40L (He et al., 2004), suggesting that CpG DNAs are lesspotent in human than in murine B cells. The same study also pointed out thatCpG DNAs inhibited IL‐4‐induced Ce GL transcription. However, theauthors did not show whether this would influence subsequent IgE switchingand/or the production of IgE (He et al., 2004).There is thus far no human disease associated with mutations in the gene
encoding TLR9. However, a recent study by Cunningham‐Rundles et al.showed that TLR9 activation is defective in CVID patients. They observedthat CpG DNA did not upregulate expression of CD86 on cells in thesepatients, even when costimulated via the BCR, nor did it induce productionof IL‐6 or IL‐10 as in normal B cells (Cunningham‐Rundles et al., 2006). Inaddition, CpG‐activated dendritic cells from CVID patients produced subnor-mal levels of IFN‐a. The study, however, did not provide evidence that CSR toIgG through the CpG–TLR9 pathway is actually impaired in these patients.
3.10. DNA Repair Factors and CSR
Individuals with defective DNA repair mechanisms display pleiotropic phe-notypes including a predisposition to cancer, neurodegeneration, and develop-mental abnormalities. Immunodeficiency is increasingly recognized as afeature of some of these syndromes and the underlying mechanism appearsto be that the general DNA repair machinery is also required for rearrange-ments of the T and B cell receptor genes, that is V(D)J recombination andCSR. Table 3 highlights the clinical manifestations and the phenotypes of CSRin primary immunodeficiency patients with known defects in DNA repair/recombination. As targeted disruption of the genes encoding ATR, NBS1,Mre11, and DNA ligase IV all result in embryonic lethality, the correspondinghuman disease models have provided unique opportunities to study the role ofthese DNA repair factors in CSR. In the case of H2AX, 53BP1, and MDC1 onthe other hand, only mouse models are available thus far. Below, we will focuson the comparison of a few human diseases and mouse knockout models thatare relevant for CSR.
3.10.1. UNG Deficiency in Human and Mouse
UNG deficiency (HIGM5) has been described in a few patients with a pheno-type resembling AID deficiency (HIGM2), including susceptibility to bacterialinfections, lymphoid hyperplasia, increased serum IgM concentrations, and

Table 3 DNA Repair Defects Affecting CSR, a Comparison Between Human Diseases and Mouse Knockout Models
Gene (syndrome)
Patients carrying mutations Knockout mouse model
ReferencesClinical manifestations CSR Phenotype CSR
UNG (hyper‐IgMsyndrome 5)
Immunodeficiency,
lymphoid hyperplasia
Impaired Normal In vitro CSR
reduced
Imai et al., 2003;Rada et al., 2002
MLH1 Cancer predisposition NA Cancer
predisposition,
infertile
Reduced, altered S
junctional pattern
(" microhomology)
Hackman et al., 1997;Ricciardone et al., 1999;Schrader et al., 1999, 2002;Vilkki et al., 2001;Wei et al., 2002
MSH2 Cancer predisposition
Immunodeficiency?
NA Cancer
predisposition
Reduced,
shift of CSR
breakpoints
Ehrenstein and Neuberger,
1999; Schrader et al., 1999,2002; Wei et al., 2002;Whiteside et al., 2002
ATM (ataxia‐telangiectasia,
A‐T)
Ataxia, telangiectasias,
immunodeficiency,
radiosensitivity, cancer
predisposition
Reduced, altered S
junctional pattern
(" microhomology)
Growth retardation,
immunodeficiency,
thymic lymphoma,
infertile
Reduced, normal or
altered S junctional
pattern
(" microhomology)
Lumsden et al., 2004;Pan et al., 2002b;Reina‐San‐Martin
et al., 2004ATR (Seckel
syndrome,
ATRD)
Microcephaly, growth
retardation,
immunodeficiency?
Altered S junctional
pattern
(" microhomology)
Embryonic lethality NA Pan‐Hammarstrom
et al., 2006
(Continued)
35
Table 3 (Continued)
Gene (syndrome)
Patients carrying mutations Knockout mouse model
ReferencesClinical manifestations CSR Phenotype CSR
NBS1 (p95)(Nijmegen
breakage
syndrome,
NBS)
Microcephaly,
radiosensitivity,
immunodeficiency,
cancer predisposition
Reduced, altered S
junctional pattern
(" microhomology)
NBS1�/�, embryonic
lethality
NA Dumon‐Jones et al., 2003;Kang et al., 2002;Kracker et al., 2005;Lahdesmaki et al., 2004;Pan et al., 2002b;Reina‐San‐Martin et al.,2005; Williams et al.,2002; Zhu et al., 2001
NBS1m/m, growth
retardation,
radiosensitivity,
immunodeficiency,
thymic lymphoma
NA
NBS1DB/DB,
radiosensitivity
NA
NBSD/� Reduced
Mre11 (ataxia‐telangiectasia
like disorder,
ATLD)
Ataxia,
radiosensitivity
Reduced, altered S
junctional pattern
(altered mutation
pattern)
Embryonic lethality NA Lahdesmaki et al., 2004;Xiao and Weaver, 1997
DNA ligase IV(LIG4 syndrome)
Microcephaly growth
retardation,
photosensitivity,
immunodeficiency
Impaired, altered S
junctional pattern
(" microhomology)
Embryonic lethality NA Barnes et al., 1998;Pan‐Hammarstrom
et al., 2005
Artemis (RS‐SCID) Radiosensitivity,
severe combined
immunodeficiency
NA Radiosensitivity,
immunodeficiency
Normal Moshous et al., 2001;Rooney et al., 2003, 2005
36

CLASS SWITCH RECOMBINATION 37
markedly decreased serum levels of IgG and IgA (Imai et al., 2003). Thedeficiency causes a profound impairment in CSR at a DNA precleavage stepand also an altered pattern of SHM (Imai et al., 2003).
The UNG mouse knockout model largely resembles the UNG‐deficientpatient phenotype, although with only a partial defect in CSR (Rada et al.,2002). It is therefore possible that additional DNA glycosylases, such asSMUG1, exert a redundant activity in mice. However, although SMUG1, whenoverexpressed, can partially substitute for UNG in SHM and restore the CSRdefect in Msh2 and UNG double knockout mice, it only plays a minor rolein antibody diversification (Di Noia et al., 2006). Alternatively, the MSH2pathway is more important as a backup pathway in mice during CSR. Inhumans, individuals with heterozygous germ line mutations in one of theMMR genes, including MLH1, MSH2, MSH6, PMS1, and PMS2, are at riskof developing hereditary nonpolyposis colorectal cancer. A few individualswith homozygous or compound heterozygous germ line mutations in theMLH1, MSH2, and MSH6 genes have also been described and these patientssuffer from early‐onset brain tumors, hematological malignancies, or colorectalcarcinomas and adenomas (Bougeard et al., 2003; Hackman et al., 1997;Hegde et al., 2005; Vilkki et al., 2001; Whiteside et al., 2002). Immunodefi-ciency has not been noted in these individuals, with one exception, where achild carrying a homozygous mutation in the MSH2 gene had IgA deficiency(Whiteside et al., 2002). It would thus be of considerable interest to studythe CSR process in cells from MLH1‐, MSH2‐, and MSH6‐deficientindividuals.
3.10.2. APEX in Human and Mouse
To date, two AP endonucleases, APEX1 and APEX2 have been identified inmammalian cells. As discussed above, there is as yet no evidence that either ofthem is involved in CSR, even though such an endonuclease activity is ex-pected to be required in the AID–UNG pathway. Knocking out APEX1 resultsin embryonic lethality (Xanthoudakis et al., 1996) whereas APEX2‐null miceexhibit growth retardation, attenuated immune responses, and radiosensitivity(Ide et al., 2004). No equivalent human disease has been described as yet,although several sequence variants have been identified in the gene encodinghuman APEX1 and four of the variants exhibit reduced DNA repair capacity inan in vitro assay (Hadi et al., 2000).
APEX1 is highly conserved between mouse and human, with a 94% AAsequence identity whereas APEX2 only shows about 80% sequence identity.The highly diversified sequence near the C‐terminus of the latter may provide

38 QIANG PAN ‐HAMMARSTROM ET AL .
multiple protein–protein interacting surfaces, including one for PCNA; someof which may be species specific (Ide et al., 2003).The human APEX2 gene is located on the X chromosome and a human
disease resulting from APEX2 deficiency, as anticipated from the mouseknockout model, might present itself as an X‐linked form of immunodeficiency,with growth retardation and radiosensitivity.
3.10.3. ATM Deficiency in Human and Mouse
ATM deficiency in humans results in a rare, complex, multisystem disorderataxia‐telangiectasia (A‐T), which is characterized by cerebellar degenerationwith ataxia, ocular, and cutaneous telangiectasias, radiosensitivity, chromosom-al instability, and cancer predisposition (Chun and Gatti, 2004). A‐T is alsorecognized as a primary immunodeficiency syndrome involving both humoraland cellular immunity (Peterson et al., 1963; Regueiro et al., 2000). IgAdeficiency has been observed in 60–80% of the patients, and a subgroupsuffers from concomitant IgG subclass deficiency, suggesting a defect in CSRand subsequent production of downstream Ig isotypes. As T cells from A‐Tpatients are abnormal, it is unclear whether the proposed CSR defect is due tointrinsic defects in the B cells, lack of T cell help, or both.By analyzing the in vivo switchedB cells, we have previously demonstrated that
the Sm‐Sa switch recombination junctions in A‐T patients are aberrant and char-acterized by a strong dependence on short sequence homologies (microhomol-ogy; average 7.2 bp versus 1.8 bp in controls, p ¼ 2.6 � 10�7) and a lack ofnormally occurring mutations around the breakpoint (Pan et al., 2002b).These observations have provided the first clue that ATM might be directlyinvolved in the final steps of CSR, including DNA end modification, repair andjoining, and the antibody deficiencies in A‐T patients could thus be due tointrinsic defects in the B cells. We have subsequently shown that the pattern ofmutations in the VH regions is largely normal in A‐T patients, suggesting that theSHM process is unaffected (Pan‐Hammarstrom et al., 2003). Thus, ATM, andATM‐dependent signaling, appears to play a specific role in CSR.ATM‐deficient mice have been generated in several laboratories in the mid‐
1990s (Barlow et al., 1996; Elson et al., 1996; Xu et al., 1996). These miceexhibit growth retardation, infertility, radiosensitivity, defects in T cell matura-tion, and development of thymic lymphomas, features that resemble thehuman disease. However, neither ataxia nor telangiectasias are readily ob-served in these mouse models. Furthermore, levels of serum IgA are normal(Xu et al., 1996), suggesting that B cells may be functionally intact in ATM‐deficient mice.

CLASS SWITCH RECOMBINATION 39
Studies in vitro have, however, shown that B cells from ATM‐deficient micecannot switch to downstream isotopes, including IgA, as efficiently as do wild‐type B cells (Lumsden et al., 2004; Reina‐San‐Martin et al., 2004). The defectin CSR is not due to alterations in GL transcription or DNA damage check-point protein (53BP1) recruitment (Lumsden et al., 2004; Reina‐San‐Martinet al., 2004). Furthermore, the intra‐switch recombination proceeds normally,whereas the long‐range inter‐switch recombination is defective, suggesting arole for ATM in S region synapsis during CSR (Reina‐San‐Martin et al., 2004).
CSR junctions have also been analyzed in ATM‐deficient mouse B cells.However, only one of the two studies showed a small, albeit significant, increasein the length of sequence homology (2.6 bp versus 1.2 bp in wild‐type cells;Lumsden et al., 2004). It should be noted that only Sm–Sg1 junctions have beenanalyzed in these mice, whereas in humans, Sm–Sa and Sm–Sg junctions wereall characterized. The microhomology‐based end joining is much more promi-nent in Sm–Sa as compared to Sm–Sg junctions in A‐T patients (Pan et al., 2002b;Pan‐Hammarstrom et al., 2006). This is also evident in other patients studiedto date, including ATR‐, Ligase IV‐, Mre11‐, and NBS1‐deficient patients(Lahdesmaki et al., 2004; Pan‐Hammarstrom et al., 2004, 2006). This is probablydue to the higher degree of homology between Sm and Sa as compared to Sm andSg regions, where the likelihood of obtaining a 7‐, 10‐, or 15‐bp microhomologybetween the Sm–Sa regions is 8‐, 270‐, and>1000‐fold higher than in the Sm–Sgregions, respectively. Furthermore, in A‐T‐ and ATR‐deficient patients, theSm–Sg junctions tend to use more microhomologies, whereas in Lig4D patients,the Sm–Sg junctions mainly show an increased frequency of 1‐bp insertions(Pan‐Hammarstrom et al., 2005). Moreover, the mutation pattern at, or close to,the Sm–Sg junctions is different from the Sm–Sa junctions in normal controls (Panand Hammarstrom, 1999).
Taken together, the Sm–Sa and Sm–Sg junctions are resolved differently incontrols and patients with various defects in their DNA repair systems. Inmice, the Sa regions also show a much higher degree of homology with Sm,than does Sg, and the microhomology‐based pathway would be a more attrac-tive alternative for Sm/Sa recombination when the normal repair pathway(s) isimpaired. However, as in the case of ATM‐deficient mice, in most mouseknockout models described to date, Sm–Sa junctions have not been analyzedin as much as detail as Sm–Sg1 and Sm–Sg3 junctions (Manis et al., 2004;Reina‐San‐Martin et al., 2003; Schrader et al., 2002; Ward et al., 2004). Thus,when summarizing the human and mouse models, one should be careful not tocompare the human Sm–Sa junctions with the mouse Sm–Sg junctions directlyand conclusions like ‘‘normal CSR junctions’’ based on Sm–Sg data alone mayneed to be reevaluated.

40 QIANG PAN ‐HAMMARSTROM ET AL .
Another point also deserves more attention: in A‐T patients, the CSRjunctions have been amplified from unstimulated PBL, whereas in ATM‐deficient mice, CSR junctions are amplified from either IL‐4 þ CD40L stimu-lated (Lumsden et al., 2004) or IL‐4 þ LPS stimulated (Reina‐San‐Martinet al., 2004) B cells. Our own unpublished data suggest that in normalindividuals, the average length of microhomology at Sm–Sa junctions is signifi-cantly longer in IL‐10 þ CD40L stimulated cells than that in unstimulatedcells (3.9 bp versus 2.0 bp). This suggests that, in addition to the availability ofDNA repair factors and the degree of homology between the S regions,differential stimulation by cytokines or other activators might also alter thebalance between the DNA repair pathways used in CSR.
3.10.4. NBS1 Deficiency in Human and Mouse
NBS1 deficiency in humans results in a rare hereditary disease, NBS, which ischaracterized by immunodeficiency, microcephaly, chromosomal instability,and an extremely high incidence of lymphoid malignancies (Digweed andSperling, 2004). Most NBS patients are of Slavic origin and are homozygousfor the founder NBS1 mutation, 657del5 (Varon et al., 1998). This mutationwas first regarded as a null mutation; however, it was later shown to behypomorphic, and a truncated 70‐kDa NBS1 protein can be produced throughan alternative initiation of translation upstream of the 5‐bp deletion (Maseret al., 2001).The immunodeficiency in NBS is severe and affects both humoral and
cellular immunity (van der Burgt et al., 1996). Absent or low serum levels ofIgG and/or IgA is observed in 80–90% of the patients with IgG4 being mostoften affected (74%), followed by IgG2 (67%), IgG1 (63%), and IgA (50%)(Gregorek et al., 2002; van der Burgt et al., 1996). As in A‐T patients, a CSRdefect has been proposed but it is unclear whether the defect is due to anintrinsic defect in the B cells.By analyzing the in vivo‐switched B cells, we have previously shown a
reduced level of switching to IgA in NBS patients (all homozygous for the657del5; Pan et al., 2002b). There was also a significantly increased length ofsequence homology at the Sm–Sa junctions (average 3.6 bp versus 1.8 bp incontrols, p ¼ 0.028); however, not as dramatic as in those from A‐T patients(Pan et al., 2002b). In addition, a high rate of mutation was observed in theSm–Sa junctions from NBS patients, which is clearly different from those inA‐T patients. Less dependence (but still significant as compared to controls)on microhomology and a normal or high rate of mutation at or close to thebreakpoints were also features of Sm–Sg recombination in NBS patients (Panet al., 2002b). Thus, the NBS1 protein seems to be directly involved in the

CLASS SWITCH RECOMBINATION 41
CSR reaction but it may have other functions in addition to interactingwith ATM.
Targeted disruption of the NBS1 gene (deletion of NBS promoter, exon 1and intron 1, or deletion of exon 6) leads to early embryonic lethality in mice(Dumon‐Jones et al., 2003; Zhu et al., 2001). However, viable knockout micehave been generated by replacing either the NBS1 exons 2 and 3 (NBS1m/m;Kang et al., 2002) or exons 4 and 5 (NBS1DB/DB; Williams et al., 2002) with aneomycin resistance gene. Although both mutants show some of the featuresthat are found in NBS patient cells, including cell‐cycle checkpoint defects,there are also notable phenotypic differences when compared to patients,in particular with regard to humoral immunodeficiency. NBS1m/m mice aregrowth retarded, hypersensitive to ionizing radiation, defective in T‐dependentantibody responses and rapidly develop thymic lymphomas. However, anormal level of serum IgG has been observed (IgA levels were not shown).NBS1DB/DB mice show no immunodeficiency and are not prone to developmentof malignancy. No CSR assay has been performed in either of these modelsto date.
Using a cell‐type‐specific conditional inactivation strategy, two studies haveshown that CSR to various IgG subclasses is reduced in NBS1‐deficient B cells(NBS1D/�, CD19creþNBS1�/LoxP; Kracker et al., 2005; Reina‐San‐Martin et al.,2005). The CSR defect is independent of GL transcription and appears to be aconsequence of inefficient recombination at the DNA level (Reina‐San‐Martinet al., 2005). In both studies, normal CSR junctions, in terms of microhomologyusage and mutation occurrence, were found. However, only Sm–Sg1 junctionswere analyzed and the junctions were derived from cells stimulated by LPS andIL‐4. In addition, in one of the studies, only 75% of the cells were NBS deficientafter tat‐Cre treatment and the ‘‘normal’’ CSR junctions might thus actually bederived from NBS‐proficient cells (Kracker et al., 2005).
A humanized mouse model for NBS has been developed where the human5‐bp deletion hypomorphic allele has been introduced into NBS‐deficientmice (hNBS1657D5mNBS1�/�; Difilippantonio et al., 2005). These mice showcell‐cycle checkpoint defects, T cell developmental defects, and gonadal ab-normalities, which resemble some of the phenotypes in NBS patients. Theserum levels of IgG1 and IgG3 were slightly reduced but B cells from thesemice could produce normal levels of IgG1 and IgG3 after appropriate stimu-lation in vitro. Thus, although both the human studies (Lahdesmaki et al.,2004; Pan et al., 2002b) and the conditional knockout mouse models (Krackeret al., 2005; Reina‐San‐Martin et al., 2005) suggest that NBS1 might be directlyinvolved in CSR, curiously, the humanized mouse model does not show adefect in CSR, at least to IgG. No serum IgA level or IgA switching assay has,however, been reported in these mice. In view of the above reasoning on the

42 QIANG PAN ‐HAMMARSTROM ET AL .
similarities of the Sm and Sa regions, it would thus be of considerable interestto study the Sm–Sa junctions in these mice.
4. Concluding Remarks
In spite of the fact that humans and mice separated in evolution more than60 million years ago, there are still remarkable similarities in the machineryinvolved in CSR. Having said that, there are also distinct characteristics, partic-ularly regarding the regulation of CSR to IgA and various IgG subclasses.Humans and mice also show varying degrees of dependence on alternative/backup systems for CSR to downstream isotypes. The phenotype of knockoutmice most often only reflects part of the clinical picture in patients, which couldbe due to differences in the nature of the gene defect (null or hypomorphic),experimental procedures, and environmental factors. It should also be borne inmind that experiments on humans are carried out on individuals in an outbredpopulation, whereas in mice, in essence, only a single individual is being tested(inbred strain). The different clinical manifestations in patients may reflect acrucial importance of modifying/interacting genes. It is thus interesting to notethat distinct phenotypes have been observed in mice when different parts of thetargeted gene have been inactivated and/or different mouse strains have beenutilized, as exemplified by the TACI‐ and NBS‐deficient mouse models. Thedissimilarities between different knockout models in micemay be a reflection ofthe interaction between the mutated gene and background genes and may thushelp us to understand the full clinical spectrum in patients.We also hope that this chapter will raise awareness on species differences
and that we should be cautious when extrapolating the function of a givenmolecule from one species to another and we need to understand and toacknowledge that each species has evolved unique mechanisms to combatinfections.
Notes added in proof
(A) By analyzing the mutation pattern in the S regions in msh2�/�ung�/�
mice, Xue et al., 2006 have shown that in contrast to the in vitro systems(Bransteitter et al., 2003; Chaudhuri et al., 2003; Dickerson et al., 2003;Ramiro et al., 2003, AID‐catalyzed deamination in vivo occurs in a strand‐symmetric manner, at both donor and acceptor S regions.(B) The phosphatidylinositol 3‐kinase (P13K) signaling has been shown to
suppress CSR by interfering with AID transcription as well as its function(Omori et al., 2006 ).(C) In a study by Honjo and colleagues, UNG is found to play a novel
noncanonical role in a CSR step that follows DNA cleavage (Begum et al., 2006).

CLASS SWITCH RECOMBINATION 43
Acknowledgments
This work was supported by the Swedish Research Council Cancerfonden and the SwedishDoctors Association. We thank Professor J. Stavnezer and Dr. K. Zhang for helpful commentson the manuscript and Professor W. Dunnick for providing mouse Sg region sequences.
References
Aalberse, R. C., van der Gaag, R., and van Leeuwen, J. (1983). Serologic aspects of IgG4 antibodies.I. Prolonged immunization results in an IgG4‐restricted response. J. Immunol. 130, 722–726.
Abraham, R. T. (2001). Cell cycle checkpoint signaling through the ATM and ATR kinases. GenesDev. 15, 2177–2196.
Agresti, A., and Vercelli, D. (2002). c‐Rel is a selective activator of a novel IL‐4/CD40 responsiveelement in the human Ig g4 germline promoter. Mol. Immunol. 38, 849–859.
Ahnesorg, P., Smith, P., and Jackson, S. P. (2006). XLF interacts with the XRCC4‐DNA ligase IVcomplex to promote DNA nonhomologous end‐joining. Cell 124, 301–313.
Akahori, Y., and Kurosawa, Y. (1997). Nucleotide sequences of all the gamma gene loci of murineimmunoglobulin heavy chains. Genomics 41, 100–104.
Arakawa, H., Iwasato, T., Hayashida, H., Shimizu, A., Honjo, T., and Yamagishi, H. (1993). Thecomplete murine immunoglobulin class switch region of the alpha heavy chain gene‐hierarchicrepetitive structure and recombination breakpoints. J. Biol. Chem. 268, 4651–4655.
Arakawa, H., Hauschild, J., and Buerstedde, J. M. (2002). Requirement of the activation‐induceddeaminase (AID) gene for immunoglobulin gene conversion. Science 295, 1301–1306.
Armitage, R. J., Macduff, B. M., Spriggs, M. K., and Fanslow, W. C. (1993). Human B cellproliferation and Ig secretion induced by recombinant CD40 ligand are modulated by solublecytokines. J. Immunol. 150, 3671–3680.
Aupetit, C., Drouet, M., Pinaud, E., Denizot, Y., Aldigier, J. C., Bridoux, F., and Cogne, M. (2000).Alleles of the alpha1 immunoglobulin gene 30 enhancer control evolution of IgA nephropathytoward renal failure. Kidney Int. 58, 966–971.
Bardwell, P. D., Martin, A., Wong, E., Li, Z., Edelmann, W., and Scharff, M. D. (2003). Cuttingedge: The G‐U mismatch glycosylase methyl‐CpG binding domain 4 is dispensable for somatichypermutation and class switch recombination. J. Immunol. 170, 1620–1624.
Bardwell, P. D., Woo, C. J., Wei, K., Li, Z., Martin, A., Sack, S. Z., Parris, T., Edelmann, W., andScharff, M. D. (2004). Altered somatic hypermutation and reduced class‐switch recombinationin exonuclease 1‐mutant mice. Nat. Immunol. 5, 224–229.
Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M., Collins, F., Shiloh, Y., Crawley,J. N., Ried, T., Tagle, D., and Wynshaw‐Boris, A. (1996). Atm‐deficient mice: A paradigm ofataxia telangiectasia. Cell 86, 159–171.
Barnes, D. E., Stamp, G., Rosewell, I., Denzel, A., and Lindahl, T. (1998). Targeted disruptionof the gene encoding DNA ligase IV leads to lethality in embryonic mice. Curr. Biol. 8,1395–1398.
Barreto, V. M., Pan‐Hammarstrom, Q., Zhao, Y., Hammarstrom, L., Misulovin, Z., and Nussenzweig,M. C. (2005). AID from bony fish catalyzes class switch recombination. J. Exp. Med. 202, 733–738.
Basu, U., Chaudhuri, J., Alpert, C., Dutt, S., Ranganath, S., Li, G., Schrum, J. P., Manis, J. P., andAlt, F. W. (2005). The AID antibody diversification enzyme is regulated by protein kinaseA phosphorylation. Nature 438, 508–511.
Begum, N. A., Kinoshita, K., Muramatsu, M., Nagaoka, H., Shinkura, R., and Honjo, T. (2004).De novo protein synthesis is required for activation‐induced cytidine deaminase‐dependent

44 QIANG PAN ‐HAMMARSTROM ET AL .
DNA cleavage in immunoglobulin class switch recombination. Proc. Natl. Acad. Sci. USA 101,13003–13007.
Begum, N. A., Izumi, N., Nishikori, M., Nagaoka, H., Shinkura, R., and Honjo, T. (2006).Requirement of non‐canonical activity of uracil DNA glycosylase for class switch recombination.J. Biol. Chem. (in press).
Bemark, M., Sale, J. E., Kim, H. J., Berek, C., Cosgrove, R. A., and Neuberger, M. S. (2000).Somatic hypermutation in the absence of DNA‐dependent protein kinase catalytic subunit(DNA‐PK(cs)) or recombination‐activating gene (RAG)1 activity. J. Exp. Med. 192, 1509–1514.
Bertocci, B., De Smet, A., Flatter, E., Dahan, A., Bories, J. C., Landreau, C., Weill, J. C., andReynaud, C. A. (2002). Cutting edge: DNA polymerases mu and lambda are dispensable for Iggene hypermutation. J. Immunol. 168, 3702–3706.
Bertocci, B., De Smet, A., Berek, C., Weill, J. C., and Reynaud, C. A. (2003). Immunoglobulink light chain gene rearrangement is impaired in mice deficient for DNA polymerase mu.Immunity 19, 203–211.
Berton, M. T., Linehan, L. A., Wick, K. R., and Dunnick, W. A. (2004). NF‐kB elements associatedwith the Stat6 site in the germline gamma1 immunoglobulin promoter are not necessary for thetranscriptional response to CD40 ligand. Int. Immunol. 16, 1741–1749.
Betz, A. G., Neuberger, M. S., and Milstein, C. (1993). Discriminating intrinsic and antigen‐selected mutational hotspots in immunoglobulin V genes. Immunol. Today 14, 405–411.
Bhushan, A., and Covey, L. R. (2001). CREB/ATF proteins enhance the basal and CD154‐ andIL‐4‐induced transcriptional activity of the human Ig1 proximal promoter. Eur. J. Immunol. 31,653–664.
Bosma, G. C., Kim, J., Urich, T., Fath, D. M., Cotticelli, M. G., Ruetsch, N. R., Radic, M. Z., andBosma, M. J. (2002). DNA‐dependent protein kinase activity is not required for immunoglobulinclass switching. J. Exp. Med. 196, 1483–1495.
Bougeard, G., Charbonnier, F., Moerman, A., Martin, C., Ruchoux, M. M., Drouot, N., andFrebourg, T. (2003). Early onset brain tumor and lymphoma in MSH2‐deficient children. Am.J. Hum. Genet. 72, 213–216.
Bransteitter, R., Pham, P., Scharff, M. D., and Goodman, M. F. (2003). Activation‐induced cytidinedeaminase deaminates deoxycytidine on single‐stranded DNA but requires the action of RNase.Proc. Natl. Acad. Sci. USA 100, 4102–4107.
Briere, F., Servet‐Delprat, C., Bridon, J. M., Saint‐Remy, J. M., and Banchereau, J. (1994). Humaninterleukin 10 induces naive surface immunoglobulin D þ (sIgDþ) B cells to secrete IgG1 andIgG3. J. Exp. Med. 179, 757–762.
Bross, L., Wesoly, J., Buerstedde, J. M., Kanaar, R., and Jacobs, H. (2003). Somatic hypermutationdoes not require Rad54 and Rad54B‐mediated homologous recombination. Eur. J. Immunol. 33,352–357.
Bruggemann, M. (1988). Evolution of the rat immunoglobulin gamma heavy‐chain gene family.Gene 74, 473–482.
Bruggemann,M.,Williams, G. T., Bindon, C. I., Clark,M. R.,Walker,M. R., Jefferis, R.,Waldmann,H., and Neuberger, M. S. (1987). Comparison of the effector functions of human immuno-globulins using a matched set of chimeric antibodies. J. Exp. Med. 166, 1351–1361.
Buck, D., Malivert, L., de Chasseval, R., Barraud, A., Fondaneche, M. C., Sanal, O., Plebani, A.,Stephan, J. L., Hufnagel,M., leDeist, F., Fischer, A., Durandy, A., et al. (2006). Cernunnos, a novelnonhomologous end‐joining factor, is mutated in human immunodeficiency with microcephaly.Cell 124, 287–299.
Casellas, R., Nussenzweig, A.,Wuerffel, R., Pelanda, R., Reichlin, A., Suh,H., Qin, X. F., Besmer, E.,Kenter, A., Rajewsky, K., and Nussenzweig, M. C. (1998). Ku80 is required for immunoglobulinisotype switching. EMBO J. 17, 2404–2411.

CLASS SWITCH RECOMBINATION 45
Castigli, E., Alt, F. W., Davidson, L., Bottaro, A., Mizoguchi, E., Bhan, A. K., and Geha, R. S.(1994). CD40‐deficient mice generated by recombination‐activating gene‐2‐deficient blastocystcomplementation. Proc. Natl. Acad. Sci. USA 91, 12135–12139.
Castigli, E., Wilson, S. A., Garibyan, L., Rachid, R., Bonilla, F., Schneider, L., and Geha, R. S.(2005a). TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat. Genet.37, 829–834.
Castigli, E.,Wilson, S. A., Scott, S., Dedeoglu, F., Xu, S., Lam, K. P., Bram, R. J., Jabara, H., andGeha,R. S. (2005b). TACI and BAFF‐R mediate isotype switching in B cells. J. Exp. Med. 201, 35–39.
Catalan, N., Selz, F., Imai, K., Revy, P., Fischer, A., and Durandy, A. (2003). The block inimmunoglobulin class switch recombination caused by activation‐induced cytidine deaminasedeficiency occurs prior to the generation of DNA double strand breaks in switch mu region.J. Immunol. 171, 2504–2509.
Chaudhuri, J., and Alt, F. W. (2004). Class‐switch recombination: Interplay of transcription, DNAdeamination and DNA repair. Nat. Rev. Immunol. 4, 541–552.
Chaudhuri, J., Tian, M., Khuong, C., Chua, K., Pinaud, E., and Alt, F. W. (2003). Transcription‐targeted DNA deamination by the AID antibody diversification enzyme. Nature 422, 726–730.
Chaudhuri, J., Khuong, C., and Alt, F. W. (2004). Replication protein A interacts with AID topromote deamination of somatic hypermutation targets. Nature 430, 992–998.
Chauveau, C., and Cogne, M. (1996). Palindromic structure of the IgH 30locus control region. Nat.Genet. 14, 15–16.
Chen, C., and Birshtein, B. K. (1997). Virtually identical enhancers containing a segment ofhomology to murine 30IgE‐E(hs1,2) lie downstream of human Ig Ca1 and Ca2 genes.J. Immunol. 159, 1310–1318.
Chen, H. T., Bhandoola, A., Difilippantonio, M. J., Zhu, J., Brown, M. J., Tai, X., Rogakou, E. P.,Brotz, T. M., Bonner, W. M., Ried, T., and Nussenzweig, A. (2000). Response to RAG‐mediatedVDJ cleavage by NBS1 and gamma‐H2AX. Science 290, 1962–1965.
Chun, H. H., and Gatti, R. A. (2004). Ataxia‐telangiectasia, an evolving phenotype. DNA Repair(Amst.) 3, 1187–1196.
Clatworthy, A. E., Valencia‐Burton, M. A., Haber, J. E., and Oettinger, M. A. (2005). The MRE11‐RAD50‐XRS2 complex, in addition to other non‐homologous end‐joining factors, is required forV(D)J joining in yeast. J. Biol. Chem. 280, 20247–20252.
Cogne, M., Lansford, R., Bottaro, A., Zhang, J., Gorman, J., Young, F., Cheng, H. L., and Alt, F. W.(1994). A class switch control region at the 30 end of the immunoglobulin heavy chain locus. Cell77, 737–747.
Cook, A. J., Oganesian, L., Harumal, P., Basten, A., Brink, R., and Jolly, C. J. (2003). Reducedswitching in SCID B cells is associated with altered somatic mutation of recombined S regions.J. Immunol. 171, 6556–6564.
Coutelier, J. P., van der Logt, J. T., Heessen, F. W., Warnier, G., and Van Snick, J. (1987). IgG2arestriction of murine antibodies elicited by viral infections. J. Exp. Med. 165, 64–69.
Cunningham‐Rundles, C., Radigan, L., Knight, A. K., Zhang, L., Bauer, L., andNakazawa, A. (2006).TLR9 activation is defective in common variable immune deficiency. J. Immunol. 176, 1978–1987.
Dariavach, P., Williams, G. T., Campbell, K., Pettersson, S., and Neuberger, M. S. (1991). Themouse IgH 30‐enhancer. Eur. J. Immunol. 21, 1499–1504.
Dedeoglu, F., Horwitz, B., Chaudhuri, J., Alt, F. W., and Geha, R. S. (2004). Induction ofactivation‐induced cytidine deaminase gene expression by IL‐4 and CD40 ligation is dependenton STAT6 and NFkB. Int. Immunol. 16, 395–404.
Defrance, T., Vanbervliet, B., Briere, F., Durand, I., Rousset, F., and Banchereau, J. (1992).Interleukin 10 and transforming growth factor beta cooperate to induce anti‐CD40‐activatednaive human B cells to secrete immunoglobulin A. J. Exp. Med. 175, 671–682.

46 QIANG PAN ‐HAMMARSTROM ET AL .
Delbos, F., De Smet, A., Faili, A., Aoufouchi, S., Weill, J. C., and Reynaud, C. A. (2005).Contribution of DNA polymerase eta to immunoglobulin gene hypermutation in the mouse.J. Exp. Med. 201, 1191–1196.
Demple, B., Herman, T., and Chen, D. S. (1991). Cloning and expression of APE, the cDNAencoding the major human apurinic endonuclease: Definition of a family of DNA repairenzymes. Proc. Natl. Acad. Sci. USA 88, 11450–11454.
Denizot, Y., Pinaud, E., Aupetit, C., Le Morvan, C., Magnoux, E., Aldigier, J. C., and Cogne, M.(2001). Polymorphism of the human a1 immunoglobulin gene 30 enhancer hs1,2 and its relationto gene expression. Immunology 103, 35–40.
Di Noia, J. M., Rada, C., and Neuberger, M. S. (2006). SMUG1 is able to excise uracil fromimmunoglobulin genes: Insight into mutation versus repair. EMBO J. 25, 585–595.
Diaz, M., Verkoczy, L. K., Flajnik, M. F., and Klinman, N. R. (2001). Decreased frequencyof somatic hypermutation and impaired affinity maturation but intact germinal center formationin mice expressing antisense RNA to DNA polymerase zeta. J. Immunol. 167, 327–335.
Dickerson, S. K., Market, E., Besmer, E., and Papavasiliou, F. N. (2003). AID mediates hypermu-tation by deaminating single stranded DNA. J. Exp. Med. 197, 1291–1296.
Difilippantonio, S., Celeste, A., Fernandez‐Capetillo, O., Chen, H. T., Reina San Martin, B., VanLaethem, F., Yang, Y. P., Petukhova, G. V., Eckhaus, M., Feigenbaum, L., Manova, K., Kruhlak, M.,et al. (2005). Role of Nbs1 in the activation of the Atm kinase revealed in humanizedmousemodels.Nat. Cell Biol. 7, 675–685.
Digweed, M., and Sperling, K. (2004). Nijmegen breakage syndrome: Clinical manifestation ofdefective response to DNA double‐strand breaks. DNA Repair (Amst.) 3, 1207–1217.
Djurup, R. (1985). The subclass nature and clinical significance of the IgG antibody response inpatients undergoing allergen‐specific immunotherapy. Allergy 40, 469–486.
Doi, T., Kinoshita, K., Ikegawa, M., Muramatsu, M., and Honjo, T. (2003). De novo proteinsynthesis is required for the activation‐induced cytidine deaminase function in class‐switchrecombination. Proc. Natl. Acad. Sci. USA 100, 2634–2638.
Dryer, R. L., and Covey, L. R. (2005). A novel NF‐kappa B‐regulated site within the human Ig1promoter requires p300 for optimal transcriptional activity. J. Immunol. 175, 4499–4507.
Dudley, D. D., Chaudhuri, J., Bassing, C. H., and Alt, F. W. (2005). Mechanism and control ofV(D)J recombination versus class switch recombination: Similarities and differences. Adv.Immunol. 86, 43–112.
Dumon‐Jones, V., Frappart, P. O., Tong, W. M., Sajithlal, G., Hulla, W., Schmid, G., Herceg, Z.,Digweed, M., and Wang, Z. Q. (2003). Nbn heterozygosity renders mice susceptible to tumorformation and ionizing radiation‐induced tumorigenesis. Cancer Res. 63, 7263–7269.
Dunnick, W. A., Shi, J., Graves, K. A., and Collins, J. T. (2005). The 30 end of the heavy chainconstant region locus enhances germline transcription and switch recombination of the fourgamma genes. J. Exp. Med. 201, 1459–1466.
Ehlers, M., Fukuyama, H., McGaha, T. L., Aderem, A., and Ravetch, J. V. (2006). TLR9/MyD88signaling is required for class switching to pathogenic IgG2a and 2b autoantibodies in SLE.J. Exp. Med. 203, 553–561.
Ehrenstein, M. R., and Neuberger, M. S. (1999). Deficiency in msh2 affects the efficiency and localsequence specificity of immunoglobulin class‐switch recombination: Parallels with somatichypermutation. EMBO J. 18, 3484–3490.
Ehrenstein, M. R., Rada, C., Jones, A. M., Milstein, C., and Neuberger, M. S. (2001). Switchjunction sequences in PMS2‐deficient mice reveal a microhomology‐mediated mechanism of Igclass switch recombination. Proc. Natl. Acad. Sci. USA 98, 14553–14558.
Eisenstein, E. M., Chua, K., and Strober, W. (1994). B cell differentiation defects in commonvariable immunodeficiency are ameliorated after stimulation with anti‐CD40 antibody andIL‐10. J. Immunol. 152, 5957–5968.

CLASS SWITCH RECOMBINATION 47
Elson, A., Wang, Y., Daugherty, C. J., Morton, C. C., Zhou, F., Campos‐Torres, J., and Leder, P.(1996). Pleiotropic defects in ataxia‐telangiectasia protein‐deficient mice. Proc. Natl. Acad. Sci.USA 93, 13084–13089.
Essers, J., Hendriks, R.W., Swagemakers, S.M., Troelstra, C., deWit, J., Bootsma, D., Hoeijmakers,J. H., and Kanaar, R. (1997). Disruption of mouse RAD54 reduces ionizing radiation resistanceand homologous recombination. Cell 89, 195–204.
Faili, A., Aoufouchi, S., Flatter, E., Gueranger, Q., Reynaud, C. A., and Weill, J. C. (2002a).Induction of somatic hypermutation in immunoglobulin genes is dependent on DNA polymer-ase iota. Nature 419, 944–947.
Faili, A., Aoufouchi, S., Gueranger, Q., Zober, C., Leon, A., Bertocci, B., Weill, J. C., and Reynaud,C. A. (2002b). AID‐dependent somatic hypermutation occurs as a DNA single‐strand event inthe BL2 cell line. Nat. Immunol. 3, 815–821.
Faili, A., Aoufouchi, S., Weller, S., Vuillier, F., Stary, A., Sarasin, A., Reynaud, C. A., and Weill, J. C.(2004). DNA polymerase eta is involved in hypermutation occurring during immunoglobulinclass switch recombination. J. Exp. Med. 199, 265–270.
Ferrari, S., Giliani, S., Insalaco, A., Al‐Ghonaium, A., Soresina, A. R., Loubser, M., Avanzini, M. A.,Marconi, M., Badolato, R., Ugazio, A. G., Levy, Y., Catalan, N., et al. (2001). Mutations of CD40gene cause an autosomal recessive form of immunodeficiency with hyper IgM. Proc. Natl. Acad.Sci. USA 98, 12614–12619.
Frank, K. M., Sekiguchi, J. M., Seidl, K. J., Swat, W., Rathbun, G. A., Cheng, H. L., Davidson, L.,Kangaloo, L., and Alt, F. W. (1998). Late embryonic lethality and impaired V(D)J recombinationin mice lacking DNA ligase IV. Nature 396, 173–177.
Frezza, D., Giambra, V., Cianci, R., Fruscalzo, A., Giufre, M., Cammarota, G., Martinez‐Labarga,C., Rickards, O., Scibilia, G., Sferlazzas, C., Bartolozzi, F., Starnino, S., et al. (2004). Increasedfrequency of the immunoglobulin enhancer HS1,2 allele 2 in coeliac disease. Scand. J. Gastro-enterol. 39, 1083–1087.
Friman, V., Hanson, L. A., Bridon, J. M., Tarkowski, A., Banchereau, J., and Briere, F. (1996). IL‐10‐driven immunoglobulin production by B lymphocytes from IgA‐deficient individuals corre-lates to infection proneness. Clin. Exp. Immunol. 104, 432–438.
Fujieda, S., Zhang, K., and Saxon, A. (1995). IL‐4 plus CD40 monoclonal antibody induceshuman B cells gamma subclass‐specific isotype switch: Switching to g1, g3, and g4, but notg2. J. Immunol. 155, 2318–2328.
Fujieda, S., Saxon, A., and Zhang, K. (1996). Direct evidence that gamma 1 and gamma 3 switchingin human B cells is interleukin‐10 dependent. Mol. Immunol. 33, 1335–1343.
Gao, Y., Chaudhuri, J., Zhu, C., Davidson, L., Weaver, D. T., and Alt, F. W. (1998a). A targetedDNA‐PKcs‐nullmutation reveals DNA‐PK‐independent functions for KU in V(D)J recombination.Immunity 9, 367–376.
Gao, Y., Sun, Y., Frank, K. M., Dikkes, P., Fujiwara, Y., Seidl, K. J., Sekiguchi, J. M., Rathbun,G. A., Swat, W., Wang, J., Bronson, R. T., Malynn, B. A., et al. (1998b). A critical role for DNAend‐joining proteins in both lymphogenesis and neurogenesis. Cell 95, 891–902.
Gascan, H., Gauchat, J. F., Aversa, G., Van Vlasselaer, P., and de Vries, J. E. (1991). Anti‐CD40monoclonal antibodies or CD4 þ T cell clones and IL‐4 induce IgG4 and IgE switching inpurified human B cells via different signaling pathways. J. Immunol. 147, 8–13.
Giambra, V., Fruscalzo, A., Giufre, M., Martinez‐Labarga, C., Favaro, M., Rocchi, M., and Frezza, D.(2005). Evolution of human IgH30EX duplicated structures: Both enhancers HS1,2 arepolymorphic with variation of transcription factor’s consensus sites. Gene 346, 105–114.
Gibbs, R. A., Weinstock, G.M.,Metzker, M. L., Muzny, D.M., Sodergren, E. J., Scherer, S., Scott, G.,Steffen, D.,Worley, K. C., Burch, P. E., Okwuonu, G., Hines, S., et al. (2004). Genome sequence ofthe Brown Norway rat yields insights into mammalian evolution. Nature 428, 493–521.

48 QIANG PAN ‐HAMMARSTROM ET AL .
Gilfillan, S., Dierich, A., Lemeur, M., Benoist, C., and Mathis, D. (1993). Mice lacking TdT:Mature animals with an immature lymphocyte repertoire. Science 261, 1175–1178.
Giovannetti, A., Mazzetta, F., Caprini, E., Aiuti, A., Marziali, M., Pierdominici, M., Cossarizza, A.,Chessa, L., Scala, E., Quinti, I., Russo, G., and Fiorilli, M. (2002). Skewed T‐cell receptorrepertoire, decreased thymic output, and predominance of terminally differentiated T cells inataxia telangiectasia. Blood 100, 4082–4089.
Gonda,H., Sugai,M.,Nambu,Y.,Katakai,T.,Agata,Y.,Mori,K. J., Yokota, Y., andShimizu,A. (2003).ThebalancebetweenPax5andId2activities is thekeytoAIDgeneexpression.J.Exp.Med.198,1427–1437.
Grant, P. A., Andersson, T., Neurath, M. F., Arulampalam, V., Bauch, A., Muller, R., Reth, M., andPettersson, S. (1996). A T cell controlled molecular pathway regulating the IgH locus: CD40‐mediated activation of the IgH 30 enhancer. EMBO J. 15, 6691–6700.
Gregorek, H., Chrzanowska, K. H., Michalkiewicz, J., Syczewska, M., and Madalinski, K. (2002).Heterogeneity of humoral immune abnormalities in children with Nijmegen breakage syn-drome: An 8‐year follow‐up study in a single centre. Clin. Exp. Immunol. 130, 319–324.
Gritzmacher, C. A., and Liu, F. T. (1987). Conserved organization of the murine immunoglobulinepsilon gene region: Restriction endonuclease maps and switch‐region nucleotide sequence.J. Immunol. 139, 603–607.
Gross, J. A., Dillon, S. R., Mudri, S., Johnston, J., Littau, A., Roque, R., Rixon, M., Schou, O.,Foley, K. P., Haugen, H., McMillen, S., Waggie, K., et al. (2001). TACI‐Ig neutralizes moleculescritical for B cell development and autoimmune disease: Impaired B cell maturation in micelacking BLyS. Immunity 15, 289–302.
Gu, Y., Seidl, K. J., Rathbun, G. A., Zhu, C., Manis, J. P., van der Stoep, N., Davidson, L., Cheng,H. L., Sekiguchi, J. M., Frank, K., Stanhope‐Baker, P., Schlissel, M. S., et al. (1997). Growthretardation and leaky SCID phenotype of Ku70‐deficient mice. Immunity 7, 653–665.
Guglielmi, L., Truffinet, V., Magnoux, E., Cogne, M., and Denizot, Y. (2004). The polymorphism ofthe locus control region lying downstream the human IgH locus is restricted to hs1,2 but not tohs3 and hs4 enhancers. Immunol. Lett. 94, 77–81.
Hackman, P., Tannergard, P., Osei‐Mensa, S., Chen, J., Kane, M. F., Kolodner, R., Lambert, B.,Hellgren, D., and Lindblom, A. (1997). A human compound heterozygote for two MLH1missense mutations. Nat. Genet. 17, 135–136.
Hadi, M. Z., and Wilson, D. M., III (2000). Second human protein with homology to theEscherichia coli abasic endonuclease exonuclease III. Environ. Mol. Mutagen. 36, 312–324.
Hadi, M. Z., Coleman, M. A., Fidelis, K., Mohrenweiser, H. W., and Wilson, D. M., III (2000).Functional characterization of Ape1 variants identified in the human population. Nucleic AcidsRes. 28, 3871–3879.
Hammarstrom, L., Lefranc, G., Lefranc, M. P., Persson, M. A., and Smith, C. I. (1986). Aberrantpattern of anti‐carbohydrate antibodies in immunoglobulin class or subclass‐deficient donors.Monogr. Allergy 20, 50–56.
Hanai, J., Chen, L. F., Kanno, T., Ohtani‐Fujita, N., Kim,W. Y., Guo,W.H., Imamura, T., Ishidou, Y.,Fukuchi, M., Shi, M. J., Stavnezer, J., Kawabata, M., et al. (1999). Interaction and functionalcooperation of PEBP2/CBF with Smads. Synergistic induction of the immunoglobulin germlineCalpha promoter. J. Biol. Chem. 274, 31577–31582.
Harfst, E., Cooper, S., Neubauer, S., Distel, L., and Grawunder, U. (2000). Normal V(D)J recombi-nation in cells from patients with Nijmegen breakage syndrome.Mol. Immunol. 37, 915–929.
Harris, R. S., Sale, J. E., Petersen‐Mahrt, S. K., and Neuberger, M. S. (2002). AID is essential forimmunoglobulin V gene conversion in a cultured B cell line. Curr. Biol. 12, 435–438.
Hassan, M. S., Islam, K. B., Hammarstrom, L., and Smith, C. I. (1992). Regulation of Cg3expression. Role of switch in the allotype‐associated variation of human serum IgG3 levels.J. Immunol. 148, 2555–2562.

CLASS SWITCH RECOMBINATION 49
He, B., Qiao, X., and Cerutti, A. (2004). CpG DNA induces IgG class switch DNA recombinationby activating human B cells through an innate pathway that requires TLR9 and cooperates withIL‐10. J. Immunol. 173, 4479–4491.
Hegde, M. R., Chong, B., Blazo, M. E., Chin, L. H., Ward, P. A., Chintagumpala, M. M., Kim, J. Y.,Plon, S. E., and Richards, C. S. (2005). A homozygous mutation in MSH6 causes Turcotsyndrome. Clin. Cancer Res. 11, 4689–4693.
Honjo, T., Nagaoka, H., Shinkura, R., and Muramatsu, M. (2005). AID to overcome the limitationsof genomic information. Nat. Immunol. 6, 655–661.
Hsieh, C. L., Arlett, C. F., and Lieber, M. R. (1993). V(D)J recombination in ataxia telangiectasia,Bloom’s syndrome, and a DNA ligase I‐associated immunodeficiency disorder. J. Biol. Chem.268, 20105–20109.
Hu, Y., Pan, Q., Pardali, E.,Mills, F. C., Bernstein, R.M.,Max, E. E., Sideras, P., andHammarstrom,L. (2000). Regulation of germline promoters by the two human Ig heavy chain 30 a enhancers.J. Immunol. 164, 6380–6386.
Ide, Y., Tsuchimoto, D., Tominaga, Y., Iwamoto, Y., and Nakabeppu, Y. (2003). Characterization ofthe genomic structure and expression of the mouse Apex2 gene. Genomics 81, 47–57.
Ide, Y., Tsuchimoto, D., Tominaga, Y., Nakashima, M., Watanabe, T., Sakumi, K., Ohno, M., andNakabeppu, Y. (2004). Growth retardation and dyslymphopoiesis accompanied by G2/M arrestin APEX2‐null mice. Blood 104, 4097–4103.
Imai, K., Slupphaug, G., Lee, W. I., Revy, P., Nonoyama, S., Catalan, N., Yel, L., Forveille, M.,Kavli, B., Krokan, H. E., Ochs, H. D., Fischer, A., et al. (2003). Human uracil‐DNA glycosylasedeficiency associated with profoundly impaired immunoglobulin class‐switch recombination.Nat. Immunol. 4, 1023–1028.
Islam, K. B., Nilsson, L., Sideras, P., Hammarstrom, L., and Smith, C. I. (1991). TGF‐beta 1induces germ‐line transcripts of both IgA subclasses in human B lymphocytes. Int. Immunol. 3,1099–1106.
Islam, K. B., Baskin, B., Nilsson, L., Hammarstrom, L., Sideras, P., and Smith, C. I. (1994).Molecular analysis of IgA deficiency. Evidence for impaired switching to IgA. J. Immunol.152, 1442–1452.
Ito, S., Nagaoka, H., Shinkura, R., Begum, N., Muramatsu, M., Nakata, M., and Honjo, T.(2004). Activation‐induced cytidine deaminase shuttles between nucleus and cytoplasm likeapolipoprotein B mRNA editing catalytic polypeptide 1. Proc. Natl. Acad. Sci. USA 101,1975–1980.
Iwasato, T., Shimizu, A., Honjo, T., and Yamagishi, H. (1990). Circular DNA is excised byimmunoglobulin class switch recombination. Cell 62, 143–149.
Jacobs, H., Fukita, Y., van der Horst, G. T., de Boer, J., Weeda, G., Essers, J., de Wind, N.,Engelward, B. P., Samson, L., Verbeek, S., de Murcia, J. M., de Murcia, G., et al. (1998).Hypermutation of immunoglobulin genes in memory B cells of DNA repair‐deficient mice.J. Exp. Med. 187, 1735–1743.
Jeannin, P., Lecoanet, S., Delneste, Y., Gauchat, J. F., and Bonnefoy, J. Y. (1998). IgE versus IgG4production can be differentially regulated by IL‐10. J. Immunol. 160, 3555–3561.
Jimi, E., and Ghosh, S. (2005). Role of nuclear factor‐kappaB in the immune system and bone.Immunol. Rev. 208, 80–87.
Jung, D., and Alt, F. W. (2004). Unraveling V(D)J recombination; insights into gene regulation. Cell116, 299–311.
Kaminski, D. A., and Stavnezer, J. (2004). Antibody class switching: Uncoupling S region accessi-bility from transcription. Trends Genet. 20, 337–340.
Kang, J., Bronson, R. T., and Xu, Y. (2002). Targeted disruption of NBS1 reveals its roles in mousedevelopment and DNA repair. EMBO J. 21, 1447–1455.

50 QIANG PAN ‐HAMMARSTROM ET AL .
Kawabe, T., Naka, T., Yoshida, K., Tanaka, T., Fujiwara, H., Suematsu, S., Yoshida, N., Kishimoto, T.,and Kikutani, H. (1994). The immune responses in CD40‐deficient mice: Impaired immuno-globulin class switching and germinal center formation. Immunity 1, 167–178.
Kawano, Y., Noma, T., and Yata, J. (1994). Regulation of human IgG subclass production bycytokines. IFN‐gamma and IL‐6 act antagonistically in the induction of human IgG1 butadditively in the induction of IgG2. J. Immunol. 153, 4948–4958.
Kett, K., Brandtzaeg, P., Radl, J., and Haaijman, J. J. (1986). Different subclass distribution of IgA‐producing cells in human lymphoid organs and various secretory tissues. J. Immunol. 136,3631–3635.
Keyeux, G., and Bernal, J. E. (1996). New allele variants of the immunoglobulin switch (Salpha)regions. Hum. Genet. 97, 695–696.
Kim, N., Kage, K., Matsuda, F., Lefranc, M. P., and Storb, U. (1997). B lymphocytes of xerodermapigmentosum or Cockayne syndrome patients with inherited defects in nucleotide excision repairare fully capable of somatic hypermutation of immunoglobulin genes. J. Exp. Med. 186, 413–419.
Kim, N., Bozek, G., Lo, J. C., and Storb, U. (1999). Different mismatch repair deficiencies all havethe same effects on somatic hypermutation: Intact primary mechanism accompanied by second-ary modifications. J. Exp. Med. 190, 21–30.
Klinman, D. M. (2004). Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev.Immunol. 4, 249–258.
Komori, T., Okada, A., Stewart, V., and Alt, F. W. (1993). Lack of N regions in antigen receptorvariable region genes of TdT‐deficient lymphocytes. Science 261, 1171–1175.
Kracker, S., Bergmann, Y., Demuth, I., Frappart, P. O., Hildebrand, G., Christine, R., Wang, Z. Q.,Sperling, K., Digweed, M., and Radbruch, A. (2005). Nibrin functions in Ig class‐switchrecombination. Proc. Natl. Acad. Sci. USA 102, 1584–1589.
Kuze, K., Shimizu, A., and Honjo, T. (1991). Characterization of the enhancer region for germlinetranscription of the gamma 3 constant region gene of human immunoglobulin. Int. Immunol. 3,647–655.
Lander,E. S., Linton,L.M.,Birren,B.,Nusbaum,C., Zody,M.C., Baldwin, J.,Devon,K.,Dewar,K.,Doyle, M., FitzHugh, W., Funke, R., Gage, D., et al. (2001). Initial sequencing and analysis ofthe human genome. Nature 409, 860–921.
Larijani, M., Frieder, D., Basit, W., and Martin, A. (2005). The mutation spectrum of purified AIDis similar to the mutability index in Ramos cells and in ung(‐/‐) msh2(‐/‐) mice. Immunogenetics6, 840–845.
Larson, E. D., Cummings, W. J., Bednarski, D. W., and Maizels, N. (2005). MRE11/RAD50cleaves DNA in the AID/UNG‐dependent pathway of immunoglobulin gene diversification.Mol. Cell 20, 367–375.
Laurencikiene, J., Deveikaite, V., and Severinson, E. (2001). HS1,2 enhancer regulation of germ-line e and g2b promoters in murine B lymphocytes: Evidence for specific promoter‐enhancerinteractions. J. Immunol. 167, 3257–3265.
Lee, G. S., Brandt, V. L., and Roth, D. B. (2004). B cell development leads off with a base hit: dU:dG mismatches in class switching and hypermutation. Mol. Cell 16, 505–508.
Levy, J., Espanol‐Boren, T., Thomas, C., Fischer, A., Tovo, P., Bordigoni, P., Resnick, I., Fasth, A.,Baer, M., Gomez, L., Sanders, E. A., Tabone, M. D., et al. (1997). Clinical spectrum of X‐linkedhyper‐IgM syndrome. J. Pediatr. 131, 47–54.
Li, M. J., Peakman, M. C., Golub, E. I., Reddy, G., Ward, D. C., Radding, C. M., and Maizels, N.(1996). Rad51 expression and localization in B cells carrying out class switch recombination.Proc. Natl. Acad. Sci. USA 93, 10222–10227.

CLASS SWITCH RECOMBINATION 51
Li, Z., Luo, Z., and Scharff, M. D. (2004a). Differential regulation of histone acetylation andgeneration of mutations in switch regions is associated with Ig class switching. Proc. Natl. Acad.Sci. USA 101, 15428–15433.
Li, Z., Woo, C. J., Iglesias‐Ussel, M. D., Ronai, D., and Scharff, M. D. (2004b). The generation ofantibody diversity through somatic hypermutation and class switch recombination. Genes Dev.18, 1–11.
Li, Z., Peled, J. U., Zhao, C., Svetlanov, A., Ronai, D., Cohen, P. E., and Scharff, M. D. (2006).A role for Mlh3 in somatic hypermutation. DNA Repair (Amst.) 5, 675–682.
Liao, M. J., and van Dyke, T. (1999). Critical role for Atm in suppressing V(D)J recombination‐driven thymic lymphoma. Genes Dev. 13, 1246–1250.
Lieber, M. R., Ma, Y., Pannicke, U., and Schwarz, K. (2004). The mechanism of vertebrate nonhomol-ogous DNA end joining and its role in V(D)J recombination. DNA Repair (Amst.) 3, 817–826.
Lin, L., Gerth, A. J., and Peng, S. L. (2004). CpG DNA redirects class‐switching towards ‘‘Th1‐like’’ Ig isotype production via TLR9 and MyD88. Eur. J. Immunol. 34, 1483–1487.
Lin, S. C., and Stavnezer, J. (1996). Activation of NF‐kB/Rel by CD40 engagement induces themouse germ line immunoglobulin Cgamma1 promoter. Mol. Cell. Biol. 16, 4591–4603.
Lin, Y. C., and Stavnezer, J. (1992). Regulation of transcription of the germ‐line Ig a constantregion gene by an ATF element and by novel transforming growth factor‐beta 1‐responsiveelements. J. Immunol. 149, 2914–2925.
Litinskiy, M. B., Nardelli, B., Hilbert, D. M., He, B., Schaffer, A., Casali, P., and Cerutti, A. (2002).DCs induce CD40‐independent immunoglobulin class switching through BLyS and APRIL.Nat. Immunol. 3, 822–829.
Liu, N., Ohnishi, N., Ni, L., Akira, S., and Bacon, K. B. (2003). CpG directly induces T‐betexpression and inhibits IgG1 and IgE switching in B cells. Nat. Immunol. 4, 687–693.
Liyanage,M.,Weaver, Z., Barlow, C., Coleman, A., Pankratz, D. G., Anderson, S.,Wynshaw‐Boris, A.,and Ried, T. (2000). Abnormal rearrangement within the alpha/delta T‐cell receptor locus inlymphomas from Atm‐deficient mice. Blood 96, 1940–1946.
Lopez Granados, E., Porpiglia, A. S., Hogan, M. B., Matamoros, N., Krasovec, S., Pignata, C.,Smith, C. I., Hammarstrom, L., Bjorkander, J., Belohradsky, B. H., Casariego, G. F., GarciaRodriguez, M. C., et al. (2002). Clinical and molecular analysis of patients with defects in muheavy chain gene. J. Clin. Invest. 110, 1029–1035.
Lou, Z., Minter‐Dykhouse, K., Franco, S., Gostissa, M., Rivera, M. A., Celeste, A., Manis, J. P., vanDeursen, J., Nussenzweig, A., Paull, T. T., Alt, F. W., and Chen, J. (2006). MDC1 maintainsgenomic stability by participating in the amplification of ATM‐dependent DNA damage signals.Mol. Cell 21, 187–200.
Lougaris, V., Badolato,R., Ferrari, S., andPlebani, A. (2005).Hyper immunoglobulinM syndromedueto CD40 deficiency: Clinical, molecular, and immunological features. Immunol. Rev. 203, 48–66.
Lumsden, J. M., McCarty, T., Petiniot, L. K., Shen, R., Barlow, C., Wynn, T. A., Morse, H. C., III,Gearhart, P. J., Wynshaw‐Boris, A., Max, E. E., and Hodes, R. J. (2004). Immunoglobulin classswitch recombination is impaired in Atm‐deficient mice. J. Exp. Med. 200, 1111–1121.
Lundgren, M., Larsson, C., Femino, A., Xu, M., Stavnezer, J., and Severinson, E. (1994).Activation of the Ig germ‐line gamma 1 promoter. Involvement of C/enhancer‐bindingprotein transcription factors and their possible interaction with an NF‐IL‐4 site. J. Immunol.153, 2983–2995.
Lahdesmaki, A., Taylor, A. M., Chrzanowska, K. H., and Pan‐Hammarstrom, Q. (2004). Delinea-tion of the role of the Mre11 complex in class switch recombination. J. Biol. Chem. 279,16479–16487.

52 QIANG PAN ‐HAMMARSTROM ET AL .
Ma, Y., Pannicke, U., Schwarz, K., and Lieber, M. R. (2002). Hairpin opening and overhangprocessing by an Artemis/DNA‐dependent protein kinase complex in nonhomologous endjoining and V(D)J recombination. Cell 108, 781–794.
Macpherson, A. J., Lamarre, A., McCoy, K., Harriman, G. R., Odermatt, B., Dougan, G.,Hengartner, H., and Zinkernagel, R. M. (2001). IgA production without mu or delta chainexpression in developing B cells. Nat. Immunol. 2, 625–631.
Madisen, L., and Groudine, M. (1994). Identification of a locus control region in the immunoglob-ulin heavy‐chain locus that deregulates c‐myc expression in plasmacytoma and Burkitt’slymphoma cells. Genes Dev. 8, 2212–2226.
Madsen, O., Scally, M., Douady, C. J., Kao, D. J., DeBry, R. W., Adkins, R., Amrine, H. M.,Stanhope, M. J., de Jong, W. W., and Springer, M. S. (2001). Parallel adaptive radiations in twomajor clades of placental mammals. Nature 409, 610–614.
Manis, J. P., Gu, Y., Lansford, R., Sonoda, E., Ferrini, R., Davidson, L., Rajewsky, K., and Alt, F. W.(1998a). Ku70 is required for late B cell development and immunoglobulin heavy chain classswitching. J. Exp. Med. 187, 2081–2089.
Manis, J. P., van der Stoep, N., Tian, M., Ferrini, R., Davidson, L., Bottaro, A., and Alt, F. W.(1998b). Class switching in B cells lacking 30 immunoglobulin heavy chain enhancers. J. Exp.Med. 188, 1421–1431.
Manis, J. P., Morales, J. C., Xia, Z., Kutok, J. L., Alt, F. W., and Carpenter, P. B. (2004). 53BP1 linksDNA damage‐response pathways to immunoglobulin heavy chain class‐switch recombination.Nat. Immunol. 5, 481–487.
Martin, A., Bardwell, P. D.,Woo, C. J., Fan,M., Shulman,M. J., and Scharff,M.D. (2002). Activation‐induced cytidine deaminase turns on somatic hypermutation in hybridomas.Nature 415, 802–806.
Martomo, S. A., Yang, W. W., and Gearhart, P. J. (2004). A role for msh6 but not msh3 in somatichypermutation and class switch recombination. J. Exp. Med. 200, 61–68.
Martomo, S. A., Yang, W. W., Vaisman, A., Maas, A., Yokoi, M., Hoeijmakers, J. H., Hanaoka, F.,Woodgate, R., and Gearhart, P. J. (2006). Normal hypermutation in antibody genes fromcongenic mice defective for DNA polymerase iota. DNA Repair (Amst.) 5, 392–398.
Maser, R. S., Zinkel, R., and Petrini, J. H. (2001). An alternative mode of translation permitsproduction of a variant NBS1 protein from the common Nijmegen breakage syndrome allele.Nat. Genet. 27, 417–421.
Masuda, K., Ouchida, R., Takeuchi, A., Saito, T., Koseki, H., Kawamura, K., Tagawa,M., Tokuhisa, T.,Azuma, T., and O‐Wang, J. (2005). DNA polymerase theta contributes to the generationof C/G mutations during somatic hypermutation of Ig genes. Proc. Natl. Acad. Sci. USA 102,13986–13991.
Matsuoka, M., Yoshida, K., Maeda, T., Usuda, S., and Sakano, H. (1990). Switch circular DNAformed in cytokine‐treated mouse splenocytes: Evidence for intramolecular DNA deletion inimmunoglobulin class switching. Cell 62, 135–142.
McDonald, J. P., Frank, E. G., Plosky, B. S., Rogozin, I. B.,Masutani, C., Hanaoka, F., Woodgate, R.,and Gearhart, P. J. (2003). 129‐derived strains of mice are deficient in DNA polymerase iota andhave normal immunoglobulin hypermutation. J. Exp. Med. 198, 635–643.
Mills, F. C., Brooker, J. S., and Camerini‐Otero, R. D. (1990). Sequences of human immunoglobulinswitch regions: Implications for recombination and transcription.NucleicAcids Res.18, 7305–7316.
Mills, F. C., Mitchell, M. P., Harindranath, N., and Max, E. E. (1995). Human Ig S gamma regionsand their participation in sequential switching to IgE. J. Immunol. 155, 3021–3036.
Mills, F. C., Harindranath, N., Mitchell, M., and Max, E. E. (1997). Enhancer complexes locateddownstream of both human immunoglobulin Ca genes. J. Exp. Med. 186, 845–858.
Milstein, C., Neuberger, M. S., and Staden, R. (1998). Both DNA strands of antibody genes arehypermutation targets. Proc. Natl. Acad. Sci. USA 95, 8791–8794.

CLASS SWITCH RECOMBINATION 53
Mita, Y., Dobashi, K., Endou, K., Kawata, T., Shimizu, Y., Nakazawa, T., and Mori, M. (2002).Toll‐like receptor 4 surface expression on human monocytes and B cells is modulated by IL‐2and IL‐4. Immunol. Lett. 81, 71–75.
Morell, A., Skvaril, F., Hitzig, W. H., and Barandun, S. (1972a). IgG subclasses: Development ofthe serum concentrations in ‘‘normal’’ infants and children. J. Pediatr. 80, 960–964.
Morell, A., Skvaril, F., Steinberg, A. G., Van Loghem, E., and Terry, W. D. (1972b). Correlationsbetween the concentrations of the four sub‐classes of IgG and Gm Allotypes in normal humansera. J. Immunol. 108, 195–206.
Moshous, D., Callebaut, I., de Chasseval, R., Corneo, B., Cavazzana‐Calvo, M., Le Deist, F.,Tezcan, I., Sanal, O., Bertrand, Y., Philippe, N., Fischer, A., and de Villartay, J. P. (2001).Artemis, a novel DNA double‐strand break repair/V(D)J recombination protein, is mutated inhuman severe combined immune deficiency. Cell 105, 177–186.
Mowatt, M. R., and Dunnick, W. A. (1986). DNA sequence of the murine gamma 1 switch segmentreveals novel structural elements. J. Immunol. 136, 2674–2683.
Muramatsu, M., Sankaranand, V. S., Anant, S., Sugai, M., Kinoshita, K., Davidson, N. O., andHonjo, T. (1999). Specific expression of activation‐induced cytidine deaminase (AID), a novelmember of the RNA‐editing deaminase family in germinal center B cells. J. Biol. Chem. 274,18470–18476.
Muramatsu, M., Kinoshita, K., Fagarasan, S., Yamada, S., Shinkai, Y., and Honjo, T. (2000). Classswitch recombination and hypermutation require activation‐induced cytidine deaminase (AID),a potential RNA editing enzyme. Cell 102, 553–563.
Murphy, W. J., Eizirik, E., Johnson, W. E., Zhang, Y. P., Ryder, O. A., and O’Brien, S. J. (2001).Molecular phylogenetics and the origins of placental mammals. Nature 409, 614–618.
Mussmann, R., Courtet, M., Schwager, J., and Du Pasquier, L. (1997). Microsites for immuno-globulin switch recombination breakpoints from Xenopus to mammals. Eur. J. Immunol. 27,2610–2619.
Muto, T., Okazaki, I. M., Yamada, S., Tanaka, Y., Kinoshita, K., Muramatsu, M., Nagaoka, H., andHonjo, T. (2006). Negative regulation of activation‐induced cytidine deaminase in B cells. Proc.Natl. Acad. Sci. USA 103, 2752–2757.
Nambu, Y., Sugai, M., Gonda, H., Lee, C. G., Katakai, T., Agata, Y., Yokota, Y., and Shimizu, A.(2003). Transcription‐coupled events associating with immunoglobulin switch region chromatin.Science 302, 2137–2140.
Neuberger, M. S., Di Noia, J. M., Beale, R. C., Williams, G. T., Yang, Z., and Rada, C. (2005).Somatic hypermutation at A.T pairs: Polymerase error versus dUTP incorporation. Nat. Rev.Immunol. 5, 171–178.
Nick McElhinny, S. A., Havener, J. M., Garcia‐Diaz, M., Juarez, R., Bebenek, K., Kee, B. L.,Blanco, L., Kunkel, T. A., and Ramsden, D. A. (2005). A gradient of template dependencedefines distinct biological roles for family X polymerases in nonhomologous end joining. Mol.Cell 19, 357–366.
Nikaido, T., Nakai, S., and Honjo, T. (1981). Switch region of immunoglobulin Cmu gene iscomposed of simple tandem repetitive sequences. Nature 292, 845–848.
Nikaido, T., Yamawaki‐Kataoka, Y., and Honjo, T. (1982). Nucleotide sequences of switch regionsof immunoglobulin Ce and C g genes and their comparison. J. Biol. Chem. 257, 7322–7329.
Nilsson, L., and Sideras, P. (1993). The human Ia1 and Ia2 germline promoter elements: Proximalpositive and distal negative elements may regulate the tissue specific expression of Ca1 and Ca2germline transcripts. Int. Immunol. 5, 271–282.
Nilsson, L., Islam, K. B., Olafsson, O., Zalcberg, I., Samakovlis, C., Hammarstrom, L., Smith, C. I.,and Sideras, P. (1991). Structure of TGF‐beta 1‐induced human immunoglobulin Ca1 and Ca2germ‐line transcripts. Int. Immunol. 3, 1107–1115.

54 QIANG PAN ‐HAMMARSTROM ET AL .
Nilsson, L., Grant, P., Larsson, I., Pettersson, S., and Sideras, P. (1995). The human I alpha 1region contains a TGF‐beta 1 responsive enhancer and a putative recombination hotspot. Int.Immunol. 7, 1191–1204.
Nonoyama, S., Farrington, M., Ishida, H., Howard, M., and Ochs, H. D. (1993). Activated B cellsfrom patients with common variable immunodeficiency proliferate and synthesize immuno-globulin. J. Clin. Invest. 92, 1282–1287.
Nussenzweig, A., Chen, C., da Costa Soares, V., Sanchez, M., Sokol, K., Nussenzweig, M. C., andLi, G. C. (1996). Requirement for Ku80 in growth and immunoglobulin V(D)J recombination.Nature 382, 551–555.
Okazaki, I. M., Kinoshita, K., Muramatsu, M., Yoshikawa, K., and Honjo, T. (2002). The AIDenzyme induces class switch recombination in fibroblasts. Nature 416, 340–345.
Omori, S. A., Cato, M. H., Anzelon‐Mills, A., Puri, K. D., Shapiro‐Shelef, M., Calame, K., andRickert, R. C. (2006). Regulation of class‐switch recombination and plasma cell differentiationby phosphatidylinositol 3‐kinase signaling. Immunity 25, 545–557.
Ong, J., Stevens, S., Roeder, R. G., and Eckhardt, L. A. (1998). 30 IgH enhancer elements shiftsynergistic interactions during B cell development. J. Immunol. 160, 4896–4903.
Oxelius, V. A. (1979). IgG subclass levels in infancy and childhood. Acta Paediatr. Scand. 68,23–27.
Pan, Q., and Hammarstrom, L. (1999). Targeting of human switch recombination breakpoints:Implications for the mechanism of m‐g isotype switching. Eur. J. Immunol. 29, 2779–2787.
Pan, Q., Lindersson, Y., Sideras, P., and Hammarstrom, L. (1997a). Structural analysis of humangamma 3 intervening regions and switch regions: Implication for the low frequency of switchingin IgG3‐deficient patients. Eur. J. Immunol. 27, 2920–2926.
Pan, Q., Rabbani, H., Mills, F. C., Severinson, E., and Hammarstrom, L. (1997b). Allotype‐associated variation in the human g3 switch region as a basis for differences in IgG3 production.J. Immunol. 158, 5849–5859.
Pan, Q., Rabbani, H., and Hammarstrom, L. (1998). Characterization of human g4 switch regionpolymorphisms suggests a meiotic recombinational hot spot within the Ig locus: Influence of Sregion length on IgG4 production. J. Immunol. 161, 3520–3526.
Pan, Q., Petit‐Frere, C., Stavnezer, J., and Hammarstrom, L. (2000). Regulation of the promoterfor human immunoglobulin g3 germ‐line transcription and its interaction with the 30a enhancer.Eur. J. Immunol. 30, 1019–1029.
Pan, Q., Petit‐Frere, C., Dai, S., Huang, P., Morton, H. C., Brandtzaeg, P., and Hammarstrom, L.(2001). Regulation of switching and production of IgA in human B cells in donors withduplicated alpha1 genes. Eur. J. Immunol. 31, 3622–3630.
Pan, Q., Matamoros, N., Bjorkander, J., Conley, M. E., and Hammarstrom, L. (2002a). Lack of IgAin Cm‐deficient patients. Nat. Immunol. 3, 595.
Pan, Q., Petit‐Frere, C., Lahdesmaki, A., Gregorek, H., Chrzanowska, K. H., and Hammarstrom, L.(2002b). Alternative end joining during switch recombination in patients with ataxia‐telangiectasia. Eur. J. Immunol. 32, 1300–1308.
Pan‐Hammarstrom, Q., Dai, S., Zhao, Y., van Dijk‐Hard, I. F., Gatti, R. A., Børresen‐Dale, A. L.,and Hammarstrom, L. (2003). ATM is not required in somatic hypermutation of VH, but isinvolved in the introduction of mutations in the switch m region. J. Immunol. 170, 3707–3716.
Pan‐Hammarstrom, Q., Zhao, Y., and Hammarstrom, L. (2004). Lack of association betweenhuman switch recombination breakpoints and the secondary structure of targeted DNA regions.J. Immunol. 172, 2727.
Pan‐Hammarstrom, Q., Jones, A. M., Lahdesmaki, A., Zhou, W., Gatti, R. A., Hammarstrom, L.,Gennery, A. R., and Ehrenstein, M. R. (2005). Impact of DNA ligase IV on nonhomologous

CLASS SWITCH RECOMBINATION 55
end joining pathways during class switch recombination in human cells. J. Exp. Med. 201,189–194.
Pan‐Hammarstrom, Q., Lahdesmaki, A., Zhao, Y., Du, L., Zhao, Z., Wen, S., Ruiz‐Perez, V. L.,Dunn‐Walters, D. K., Goodship, J. A., and Hammarstrom, L. (2006). Disparate roles of ATR andATM in immunoglobulin class switch recombination and somatic hypermutation. J. Exp. Med.203, 99–110.
Pardali, E., Xie, X. Q., Tsapogas, P., Itoh, S., Arvanitidis, K., Heldin, C. H., ten Dijke, P.,Grundstrom, T., and Sideras, P. (2000). Smad and AML proteins synergistically confertransforming growth factor beta1 responsiveness to human germ‐line IgA genes. J. Biol. Chem.275, 3552–3560.
Pasqualucci, L., Kitaura, Y., Gu, H., and Dalla‐Favera, R. (2006). PKA‐mediated phosphorylationregulates the function of activation‐induced deaminase (AID) in B cells. Proc. Natl. Acad. Sci.USA 103, 395–400.
Peng, S. L. (2005). Signaling in B cells via Toll‐like receptors. Curr. Opin. Immunol. 17, 230–236.Perelmutter, L. (1984). IgG4: Non‐IgE mediated atopic disease. Ann. Allergy 52, 64–69.Perkins, E. J., Nair, A., Cowley, D. O., van Dyke, T., Chang, Y., and Ramsden, D. A. (2002). Sensing
of intermediates in V(D)J recombination by ATM. Genes Dev. 16, 159–164.Petersen, S., Casellas, R., Reina‐San‐Martin, B., Chen, H. T., Difilippantonio, M. J., Wilson, P. C.,
Hanitsch, L., Celeste, A., Muramatsu, M., Pilch, D. R., Redon, C., Ried, T., et al. (2001). AID isrequired to initiate Nbs1/g‐H2AX focus formation and mutations at sites of class switching.Nature 414, 660–665.
Peterson, R. D. A., Blaw, M., and Good, R. A. (1963). Ataxia‐telangiectasia: A possible clinicalcounterpart of the animals rendered immunologically incompetent by thymectomy. J. Pediatr.63, 701–703.
Petiniot, L. K., Weaver, Z., Barlow, C., Shen, R., Eckhaus, M., Steinberg, S. M., Ried, T.,Wynshaw‐Boris, A., and Hodes, R. J. (2000). Recombinase‐activating gene (RAG) 2‐mediatedV(D)J recombination is not essential for tumorigenesis in Atm‐deficient mice. Proc. Natl. Acad.Sci. USA 97, 6664–6669.
Petiniot, L. K., Weaver, Z., Vacchio, M., Shen, R., Wangsa, D., Barlow, C., Eckhaus, M., Steinberg,S. M., Wynshaw‐Boris, A., Ried, T., and Hodes, R. J. (2002). RAG‐mediated V(D)J recombina-tion is not essential for tumorigenesis in Atm‐deficient mice. Mol. Cell. Biol. 22, 3174–3177.
Pettersson, S., Cook, G. P., Bruggemann, M., Williams, G. T., and Neuberger, M. S. (1990).A second B cell‐specific enhancer 30 of the immunoglobulin heavy‐chain locus. Nature 344,165–168.
Pham, P., Bransteitter, R., Petruska, J., andGoodman,M. F. (2003). Processive AID‐catalysed cytosinedeamination on single‐stranded DNA simulates somatic hypermutation. Nature 424, 103–107.
Phung, Q. H., Winter, D. B., Cranston, A., Tarone, R. E., Bohr, V. A., Fishel, R., and Gearhart, P. J.(1998). Increased hypermutation at G and C nucleotides in immunoglobulin variable genes frommice deficient in the MSH2 mismatch repair protein. J. Exp. Med. 187, 1745–1751.
Pinaud, E., Aupetit, C., Chauveau, C., and Cogne, M. (1997). Identification of a homolog of the Ca30/hs3 enhancer and of an allelic variant of the 30IgE/hs1,2 enhancer downstream of the humanimmunoglobulin alpha 1 gene. Eur. J. Immunol. 27, 2981–2985.
Pinaud, E., Khamlichi, A. A., Le Morvan, C., Drouet, M., Nalesso, V., Le Bert, M., and Cogne, M.(2001). Localization of the 30 IgH locus elements that effect long‐distance regulation of classswitch recombination. Immunity 15, 187–199.
Rabbani, H., Pan, Q., Kondo, N., Smith, C. I., and Hammarstrom, L. (1996). Duplicationsand deletions of the human IGHC locus: Evolutionary implications. Immunogenetics 45,136–141.

56 QIANG PAN ‐HAMMARSTROM ET AL .
Rada, C., Williams, G. T., Nilsen, H., Barnes, D. E., Lindahl, T., and Neuberger, M. S. (2002).Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG‐deficient mice. Curr. Biol. 12, 1748–1755.
Rada, C., Di Noia, J. M., and Neuberger, M. S. (2004). Mismatch recognition and uracil excisionprovide complementary paths to both Ig switching and the A/T‐focused phase of somaticmutation. Mol. Cell 16, 163–171.
Ramiro, A. R., Stavropoulos, P., Jankovic, M., and Nussenzweig, M. C. (2003). Transcriptionenhances AID‐mediated cytidine deamination by exposing single‐stranded DNA on the non-template strand. Nat. Immunol. 4, 452–456.
Regueiro, J. R., Porras, O., Lavin, M., and Gatti, R. A. (2000). Ataxia‐telangiectasia: A primaryimmunodeficiency revisited. Immunol. Allergy Clin. North Am. 20, 177–205.
Reina‐San‐Martin, B., Difilippantonio, S., Hanitsch, L., Masilamani, R. F., Nussenzweig, A., andNussenzweig, M. C. (2003). H2AX is required for recombination between immunoglobulinswitch regions but not for intra‐switch region recombination or somatic hypermutation. J. Exp.Med. 197, 1767–1778.
Reina‐San‐Martin, B., Chen, H. T., Nussenzweig, A., and Nussenzweig, M. C. (2004). ATM isrequired for efficient recombination between immunoglobulin switch regions. J. Exp. Med. 200,1103–1110.
Reina‐San‐Martin, B., Nussenzweig, M. C., Nussenzweig, A., and Difilippantonio, S. (2005).Genomic instability, endoreduplication, and diminished Ig class‐switch recombination in B cellslacking Nbs1. Proc. Natl. Acad. Sci. USA 102, 1590–1595.
Renshaw, B. R., Fanslow, W. C., Armitage, R. J., Campbell, K. A., Liggitt, D., Wright, B., Davison,B. L., and Maliszewski, C. R. (1994). Humoral immune responses in CD40 ligand‐deficientmice. J. Exp. Med. 180, 1889–1900.
Revy, P., Muto, T., Levy, Y., Geissmann, F., Plebani, A., Sanal, O., Catalan, N., Forveille, M.,Dufourcq‐Labelouse, R., Gennery, A., Tezcan, I., Ersoy, F., et al. (2000). Activation‐inducedcytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper‐IgMsyndrome (HIGM2). Cell 102, 565–575.
Riballo, E., Kuhne, M., Rief, N., Doherty, A., Smith, G. C., Recio, M. J., Reis, C., Dahm, K.,Fricke, A., Krempler, A., Parker, A. R., Jackson, S. P., et al. (2004). A pathway of double‐strandbreak rejoining dependent upon ATM, Artemis, and proteins locating to g‐H2AX foci. Mol. Cell16, 715–724.
Ricciardone, M. D., Ozcelik, T., Cevher, B., Ozdag, H., Tuncer, M., Gurgey, A., Uzunalimoglu, O.,Cetinkaya, H., Tanyeli, A., Erken, E., and Ozturk, M. (1999). Human MLH1 deficiencypredisposes to hematological malignancy and neurofibromatosis type 1. Cancer Res. 59,290–293.
Robson, C. N., and Hickson, I. D. (1991). Isolation of cDNA clones encoding a human apurinic/apyrimidinic endonuclease that corrects DNA repair and mutagenesis defects in E. coli xth(exonuclease III) mutants. Nucleic Acids Res. 19, 5519–5523.
Rolink, A., Melchers, F., and Andersson, J. (1996). The SCID but not the RAG‐2 gene product isrequired for Sm‐Se heavy chain class switching. Immunity 5, 319–330.
Rooney, S., Alt, F. W., Lombard, D., Whitlow, S., Eckersdorff, M., Fleming, J., Fugmann, S.,Ferguson, D. O., Schatz, D. G., and Sekiguchi, J. (2003). Defective DNA repair and increasedgenomic instability in Artemis‐deficient murine cells. J. Exp. Med. 197, 553–565.
Rooney, S., Alt, F. W., Sekiguchi, J., and Manis, J. P. (2005). Artemis‐independent functions ofDNA‐dependent protein kinase in Ig heavy chain class switch recombination and development.Proc. Natl. Acad. Sci. USA 102, 2471–2475.

CLASS SWITCH RECOMBINATION 57
Ruiz, J. F., Lucas, D., Garcia‐Palomero, E., Saez, A. I., Gonzalez,M. A., Piris, M. A., Bernad, A., andBlanco, L. (2004). Overexpression of human DNA polymerase mu (Pol m) in a Burkitt’slymphoma cell line affects the somatic hypermutation rate. Nucleic Acids Res. 32, 5861–5873.
Saleque, S., Singh, M., Little, R. D., Giannini, S. L., Michaelson, J. S., and Birshtein, B. K. (1997).Dyad symmetry within the mouse 30 IgH regulatory region includes two virtually identicalenhancers (Ca30E and hs3). J. Immunol. 158, 4780–4787.
Salzer, U., Chapel, H. M., Webster, A. D., Pan‐Hammarstrom, Q., Schmitt‐Graeff, A., Schlesier,M., Peter, H. H., Rockstroh, J. K., Schneider, P., Schaffer, A. A., Hammarstrom, L., andGrimbacher, B. (2005). Mutations in TNFRSF13B encoding TACI are associated with commonvariable immunodeficiency in humans. Nat. Genet. 37, 820–828.
Sayegh, C. E., Quong, M. W., Agata, Y., and Murre, C. (2003). E‐proteins directly regulateexpression of activation‐induced deaminase in mature B cells. Nat. Immunol. 4, 586–593.
Scappino, L. A., Chu, C., and Gritzmacher, C. A. (1991). Extended nucleotide sequence of theswitch region of the murine gene encoding immunoglobulin E. Gene 99, 295–296.
Schaffer, A., Cerutti, A., Shah, S., Zan, H., and Casali, P. (1999). The evolutionarily conservedsequence upstream of the human Ig heavy chain Sg3 region is an inducible promoter: Synergis-tic activation by CD40 ligand and IL‐4 via cooperative NF‐kB and STAT‐6 binding sites.J. Immunol. 162, 5327–5336.
Schatz, D. G. (2004). V(D)J recombination. Immunol. Rev. 200, 5–11.Schneider, P. (2005). The role of APRIL and BAFF in lymphocyte activation. Curr. Opin.
Immunol. 17, 282–289.Schrader, C. E., Edelmann, W., Kucherlapati, R., and Stavnezer, J. (1999). Reduced isotype switch-
ing in splenic B cells frommice deficient in mismatch repair enzymes. J. Exp. Med. 190, 323–330.Schrader, C. E., Vardo, J., and Stavnezer, J. (2002). Role for mismatch repair proteins Msh2, Mlh1,
and Pms2 in immunoglobulin class switching shown by sequence analysis of recombinationjunctions. J. Exp. Med. 195, 367–373.
Schrader, C. E., Vardo, J., Linehan, E., Twarog, M. Z., Niedernhofer, L. J., Hoeijmakers, J. H., andStavnezer, J. (2004). Deletion of the nucleotide excision repair gene Ercc1 reduces immuno-globulin class switching and alters mutations near switch recombination junctions. J. Exp. Med.200, 321–330.
Schrader, C. E., Linehan, E. K., Mochegova, S. N., Woodland, R. T., and Stavnezer, J. (2005).Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J. Exp. Med. 202,561–568.
Seidl, K. J., Manis, J. P., Bottaro, A., Zhang, J., Davidson, L., Kisselgof, A., Oettgen, H., and Alt,F. W. (1999). Position‐dependent inhibition of class‐switch recombination by PGK‐neor cas-settes inserted into the immunoglobulin heavy chain constant region locus. Proc. Natl. Acad. Sci.USA 96, 3000–3005.
Seki, M., Gearhart, P. J., and Wood, R. D. (2005). DNA polymerases and somatic hypermutation ofimmunoglobulin genes. EMBO Rep. 6, 1143–1148.
Sepulveda, M. A., Emelyanov, A. V., and Birshtein, B. K. (2004). NF‐kB and Oct‐2 synergize toactivate the human 30 Igh hs4 enhancer in B cells. J. Immunol. 172, 1054–1064.
Sepulveda, M. A., Garrett, F. E., Price‐Whelan, A., and Birshtein, B. K. (2005). Comparativeanalysis of human and mouse 30 Igh regulatory regions identifies distinctive structural features.Mol. Immunol. 42, 605–615.
Seshasayee, D., Valdez, P., Yan, M., Dixit, V. M., Tumas, D., and Grewal, I. S. (2003). Loss of TACIcauses fatal lymphoproliferation and autoimmunity, establishing TACI as an inhibitory BLySreceptor. Immunity 18, 279–288.

58 QIANG PAN ‐HAMMARSTROM ET AL .
Shi, M. J., and Stavnezer, J. (1998). CBF a3 (AML2) is induced by TGF‐b1 to bind and activate themouse germline Ig a promoter. J. Immunol. 161, 6751–6760.
Shiloh, Y. (2001). ATM and ATR: Networking cellular responses to DNA damage. Curr. Opin.Genet. Dev. 11, 71–77.
Shimizu, A., Takahashi, N., Yaoita, Y., and Honjo, T. (1982). Organization of the constant‐regiongene family of the mouse immunoglobulin heavy chain. Cell 28, 499–506.
Shimizu, T., Shinkai, Y., Ogi, T., Ohmori, H., and Azuma, T. (2003). The absence of DNApolymerase kappa does not affect somatic hypermutation of the mouse immunoglobulin heavychain gene. Immunol. Lett. 86, 265–270.
Shimizu, T., Azuma, T., Ishiguro, M., Kanjo, N., Yamada, S., and Ohmori, H. (2005). Normalimmunoglobulin gene somatic hypermutation in Pol kappa‐Pol iota double‐deficient mice.Immunol. Lett. 98, 259–264.
Shockett, P., and Stavnezer, J. (1991). Effect of cytokines on switching to IgA and alpha germlinetranscripts in the B lymphoma I.29 mu. Transforming growth factor‐beta activates transcriptionof the unrearranged C alpha gene. J. Immunol. 147, 4374–4383.
Sideras, P., Mizuta, T. R., Kanamori, H., Suzuki, N., Okamoto, M., Kuze, K., Ohno, H., Doi, S.,Fukuhara, S., and Hassan, M. S. (1989). Production of sterile transcripts of Cg genes in an IgM‐producing human neoplastic B cell line that switches to IgG‐producing cells. Int. Immunol. 1,631–642.
Snapper, C. M., Kehry, M. R., Castle, B. E., and Mond, J. J. (1995). Multivalent, but not divalent,antigen receptor cross‐linkers synergize with CD40 ligand for induction of Ig synthesis and classswitching in normal murine B cells. A redefinition of the TI‐2 vs T cell‐dependent antigendichotomy. J. Immunol. 154, 1177–1187.
Stavnezer, J. (1996). Antibody class switching. Adv. Immunol. 61, 79–146.Stavnezer, J. (2000). Molecular processes that regulate class switching. Curr. Top. Microbiol.Immunol. 245, 127–168.
Stavnezer, J., and Schrader, C. E. (2005). Mismatch repair converts AID‐instigated nicks todouble‐strand breaks for antibody class‐switch recombination. Trends Genet. 22, 23–28.
Stavnezer‐Nordgren, J., and Sirlin, S. (1986). Specificity of immunoglobulin heavy chainswitch correlates with activity of germline heavy chain genes prior to switching.EMBO J. 5, 95–102.
Sun, Z. J., and Kitchingman, G. R. (1991). Sequencing of selected regions of the human immuno-globulin heavy‐chain gene locus that completes the sequence from JH through the deltaconstant region. DNA Seq. 1, 347–355.
Szurek, P., Petrini, J., and Dunnick, W. (1985). Complete nucleotide sequence of the murine g3switch region and analysis of switch recombination sites in two gamma 3‐expressing hybridomas.J. Immunol. 135, 620–626.
Taccioli, G. E., Amatucci, A. G., Beamish, H. J., Gell, D., Xiang, X. H., Torres Arzayus, M. I.,Priestley, A., Jackson, S. P., Marshak Rothstein, A., Jeggo, P. A., and Herrera, V. L. (1998).Targeted disruption of the catalytic subunit of the DNA‐PK gene in mice confers severecombined immunodeficiency and radiosensitivity. Immunity 9, 355–366.
Taylor, A. M., Metcalfe, J. A., Thick, J., and Mak, Y. F. (1996). Leukemia and lymphoma in ataxiatelangiectasia. Blood 87, 423–438.
Tian, M., Shinkura, R., Shinkura, N., and Alt, F. W. (2004). Growth retardation, early death, andDNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol. Cell.Biol. 24, 1200–1205.
Tibbetts, R. S., Brumbaugh, K. M., Williams, J. M., Sarkaria, J. N., Cliby, W. A., Shieh, S. Y.,Taya, Y., Prives, C., and Abraham, R. T. (1999). A role for ATR in the DNA damage‐inducedphosphorylation of p53. Genes. Dev. 13, 152–157.
Turner, M. W., Bennich, H. H., and Natvig, J. B. (1970). Simple method of subtyping humanG‐myeloma proteins based on sensitivity to pepsin digestion. Nature 225, 853–855.

CLASS SWITCH RECOMBINATION 59
van der Burgt, I., Chrzanowska, K. H., Smeets, D., and Weemaes, C. (1996). Nijmegen breakagesyndrome. J. Med. Genet. 33, 153–156.
Varon, R., Vissinga, C., Platzer, M., Cerosaletti, K. M., Chrzanowska, K. H., Saar, K., Beckmann,G., Seemanova, E., Cooper, P. R., Nowak, N. J., Stumm, M., Weemaes, C. M., et al. (1998).Nibrin, a novel DNA double‐strand break repair protein, is mutated in Nijmegen breakagesyndrome. Cell 93, 467–476.
Venter, J. C., Adams, M. D., Myers, E. W., Li, P. W., Mural, R. J., Sutton, G. G., Smith, H. O.,Yandell, M., Evans, C. A., Holt, R. A., Gocayne, J. D., Amanatides, P., et al. (2001). Thesequence of the human genome. Science 291, 1304–1351.
Vilkki, S., Tsao, J. L., Loukola, A., Poyhonen, M., Vierimaa, O., Herva, R., Aaltonen, L. A., andShibata, D. (2001). Extensive somatic microsatellite mutations in normal human tissue. CancerRes. 61, 4541–4544.
von Bulow, G. U., van Deursen, J. M., and Bram, R. J. (2001). Regulation of the T‐independenthumoral response by TACI. Immunity 14, 573–582.
von Schwedler, U., Jack, H. M., and Wabl, M. (1990). Circular DNA is a product of theimmunoglobulin class switch rearrangement. Nature 345, 452–456.
Wagner, S. D., Elvin, J. G., Norris, P., McGregor, J. M., and Neuberger, M. S. (1996). Somatichypermutation of Ig genes in patients with xeroderma pigmentosum (XP‐D). Int. Immunol.8, 701–705.
Wang, L., Whang, N., Wuerffel, R., and Kenter, A. L. (2006). AID‐dependent histone acetylation isdetected in immunoglobulin S regions. J. Exp. Med. 203, 215–226.
Ward, I. M., Reina‐San‐Martin, B., Olaru, A., Minn, K., Tamada, K., Lau, J. S., Cascalho, M.,Chen, L., Nussenzweig, A., Livak, F., Nussenzweig, M. C., and Chen, J. (2004). 53BP1 isrequired for class switch recombination. J. Cell. Biol. 165, 459–464.
Warren, W. D., Roberts, K. L., Linehan, L. A., and Berton, M. T. (1999). Regulation of thegermline immunoglobulin Cg1 promoter by CD40 ligand and IL‐4: Dual role for tandemNF‐kB binding sites. Mol. Immunol. 36, 31–44.
Waterston, R. H., Lindblad‐Toh, K., Birney, E., Rogers, J., Abril, J. F., Agarwal, P., Agarwala, R.,Ainscough, R., Alexandersson, M., An, P., Antonarakis, S. E., Attwood, J., et al. (2002). Initialsequencing and comparative analysis of the mouse genome. Nature 420, 520–562.
Wei, K., Kucherlapati, R., and Edelmann, W. (2002). Mouse models for human DNA mismatch‐repair gene defects. Trends Mol. Med. 8, 346–353.
Weller, S., Faili, A., Garcia, C., Braun, M. C., Le Deist, F. F., de Saint Basile, G. G., Hermine, O.,Fischer, A., Reynaud, C. A., and Weill, J. C. (2001). CD40‐CD40L independent Ig genehypermutation suggests a second B cell diversification pathway in humans. Proc. Natl. Acad.Sci. USA 98, 1166–1170.
Whiteside, D., McLeod, R., Graham, G., Steckley, J. L., Booth, K., Somerville, M. J., and Andrew,S. E. (2002). A homozygous germ‐line mutation in the human MSH2 gene predisposes tohematological malignancy and multiple cafe‐au‐lait spots. Cancer Res. 62, 359–362.
Wiesendanger, M., Kneitz, B., Edelmann, W., and Scharff, M. D. (2000). Somatic hypermutation inMutS homologue (MSH)3‐,MSH6‐, andMSH3/MSH6‐deficientmice reveals a role for theMSH2‐MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 191, 579–584.
Williams, B. R., Mirzoeva, O. K., Morgan, W. F., Lin, J., Dunnick, W., and Petrini, J. H. (2002).A murine model of Nijmegen breakage syndrome. Curr. Biol. 12, 648–653.
Winter, A. G., Samuel, K., Hsia, K. T., and Melton, D. W. (2003a). The repair and recombinationenzyme ERCC1 is not required for immunoglobulin class switching. DNA Repair (Amst.) 2,561–569.
Winter, D. B., Phung, Q. H., Zeng, X., Seeberg, E., Barnes, D. E., Lindahl, T., and Gearhart, P. J.(2003b). Normal somatic hypermutation of Ig genes in the absence of 8‐hydroxyguanine‐DNAglycosylase. J. Immunol. 170, 5558–5562.

60 QIANG PAN ‐HAMMARSTROM ET AL .
Wuerffel, R., Jamieson, C. E., Morgan, L., Merkulov, G. V., Sen, R., and Kenter, A. L. (1992).Switch recombination breakpoints are strictly correlated with DNA recognition motifs forimmunoglobulin Sg3 DNA‐binding proteins. J. Exp. Med. 176, 339–349.
Wuerffel, R. A., Du, J., Thompson, R. J., and Kenter, A. L. (1997). Ig Sg3 DNA‐specifc doublestrand breaks are induced in mitogen‐activated B cells and are implicated in switch recombina-tion. J. Immunol. 159, 4139–4144.
Xanthoudakis, S., and Curran, T. (1992). Identification and characterization of Ref‐1, a nuclearprotein that facilitates AP‐1 DNA‐binding activity. EMBO J. 11, 653–665.
Xanthoudakis, S., Smeyne, R. J., Wallace, J. D., and Curran, T. (1996). The redox/DNA repairprotein, Ref‐1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA93, 8919–8923.
Xiao, Y., and Weaver, D. T. (1997). Conditional gene targeted deletion by Cre recombinasedemonstrates the requirement for the double‐strand break repair Mre11 protein in murineembryonic stem cells. Nucleic Acids Res. 25, 2985–2991.
Xie, X. Q., Pardali, E., Holm, M., Sideras, P., and Grundstrom, T. (1999). AML and Ets proteinsregulate the I alpha1 germ‐line promoter. Eur. J. Immunol. 29, 488–498.
Xu, J., Foy, T. M., Laman, J. D., Elliott, E. A., Dunn, J. J., Waldschmidt, T. J., Elsemore, J., Noelle,R. J., and Flavell, R. A. (1994). Mice deficient for the CD40 ligand. Immunity 1, 423–431.
Xu, Y., Ashley, T., Brainerd, E. E., Bronson, R. T., Meyn, M. S., and Baltimore, D. (1996). Targeteddisruption of ATM leads to growth retardation, chromosomal fragmentation during meiosis,immune defects, and thymic lymphoma. Genes Dev. 10, 2411–2422.
Xue, K., Rada, C., and Neuberger, M. S. (2006). The in vivo pattern of AID targeting toimmunoglobulin switch regions deduced from mutation spectra in msh2/‐ ung‐/‐ mice. J. Exp.Med. 203, 2085–2094.
Yabuki, M., Fujii, M. M., and Maizels, N. (2005). The MRE11‐RAD50‐NBS1 complex acceleratessomatic hypermutation and gene conversion of immunoglobulin variable regions. Nat. Immunol.6, 730–736.
Yadav, A., Olaru, A., Saltis, M., Setren, A., Cerny, J., and Livak, F. (2006). Identification of aubiquitously active promoter of the murine activation‐induced cytidine deaminase (AICDA)gene. Mol. Immunol. 43, 529–541.
Yan, M., Wang, H., Chan, B., Roose‐Girma, M., Erickson, S., Baker, T., Tumas, D., Grewal, I. S.,and Dixit, V. M. (2001). Activation and accumulation of B cells in TACI‐deficient mice. Nat.Immunol. 2, 638–643.
Yancopoulos, G. D., DePinho, R. A., Zimmerman, K. A., Lutzker, S. G., Rosenberg, N., and Alt,F. W. (1986). Secondary genomic rearrangement events in pre‐B cells: VHDJH replacement by aLINE‐1 sequence and directed class switching. EMBO J. 5, 3259–3266.
Yavuz, S., Yavuz, A. S., Kraemer, K. H., and Lipsky, P. E. (2002). The role of polymerase eta insomatic hypermutation determined by analysis of mutations in a patient with xeroderma pig-mentosum variant. J. Immunol. 169, 3825–3830.
Yel, L., Minegishi, Y., Coustan‐Smith, E., Buckley, R. H., Trubel, H., Pachman, L. M., Kitchingman,G. R., Campana, D., Rohrer, J., and Conley, M. E. (1996). Mutations in the mu heavy‐chain genein patients with agammaglobulinemia. N. Engl. J. Med. 335, 1486–1493.
Yeo, T. C., Xia, D., Hassouneh, S., Yang, X. O., Sabath, D. E., Sperling, K., Gatti, R. A., Concannon,P., and Willerford, D. M. (2000). V(D)J rearrangement in Nijmegen breakage syndrome. Mol.Immunol. 37, 1131–1139.
Yoshikawa, K., Okazaki, I. M., Eto, T., Kinoshita, K., Muramatsu, M., Nagaoka, H., and Honjo, T.(2002). AID enzyme‐induced hypermutation in an actively transcribed gene in fibroblasts.Science 296, 2033–2036.

CLASS SWITCH RECOMBINATION 61
Yoshimoto, T., Okada, K., Morishima, N., Kamiya, S., Owaki, T., Asakawa,M., Iwakura, Y., Fukai, F.,andMizuguchi, J. (2004). Induction of IgG2a class switching in B cells by IL‐27. J. Immunol. 173,2479–2485.
Yu, K., Chedin, F., Hsieh, C. L., Wilson, T. E., and Lieber, M. R. (2003). R‐loops at immunoglobulinclass switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 4, 442–451.
Yu, K., Huang, F. T., and Lieber, M. R. (2004). DNA substrate length and surrounding sequenceaffect the activation‐induced deaminase activity at cytidine. J. Biol. Chem. 279, 6496–6500.
Zan, H., Komori, A., Li, Z., Cerutti, A., Schaffer, A., Flajnik, M. F., Diaz, M., and Casali, P. (2001).The translesion DNA polymerase zeta plays a major role in Ig and bcl‐6 somatic hypermutation.Immunity 14, 643–653.
Zan, H., Wu, X., Komori, A., Holloman, W. K., and Casali, P. (2003). AID‐dependent generation ofresected double‐strand DNA breaks and recruitment of Rad52/Rad51 in somatic hypermuta-tion. Immunity 18, 727–738.
Zan, H., Shima, N., Xu, Z., Al‐Qahtani, A., Evinger Iii, A. J., Zhong, Y., Schimenti, J. C., and Casali,P. (2005). The translesion DNA polymerase theta plays a dominant role in immunoglobulin genesomatic hypermutation. EMBO J. 24, 3757–3769.
Zarrin, A. A., Tian, M., Wang, J., Borjeson, T., and Alt, F. W. (2005). Influence of switch regionlength on immunoglobulin class switch recombination. Proc. Natl. Acad. Sci. USA 102,2466–2470.
Zelazowski, P., Shen, Y., and Snapper, C. M. (2000). NF‐kB/p50 and NF‐kB/c‐Rel differentiallyregulate the activity of the 30aE‐hsl,2 enhancer in normal murine B cells in an activation‐dependent manner. Int. Immunol. 12, 1167–1172.
Zeng, X., Winter, D. B., Kasmer, C., Kraemer, K. H., Lehmann, A. R., and Gearhart, P. J. (2001).DNA polymerase Z is an A‐T mutator in somatic hypermutation of immunoglobulin variablegenes. Nat. Immunol. 2, 537–541.
Zeng, X., Negrete, G. A., Kasmer, C., Yang, W. W., and Gearhart, P. J. (2004). Absence of DNApolymerase Z reveals targeting of C mutations on the nontranscribed strand in immunoglobulinswitch regions. J. Exp. Med. 199, 917–924.
Zhang, L., Duan, C. J., Binkley, C., Li, G., Uhler, M. D., Logsdon, C. D., and Simeone, D. M.(2004a). A transforming growth factor beta‐induced Smad3/Smad4 complex directly activatesprotein kinase A. Mol. Cell. Biol. 24, 2169–2180.
Zhang, X., Succi, J., Feng, Z., Prithivirajsingh, S., Story, M. D., and Legerski, R. J. (2004b). Artemisis a phosphorylation target of ATM and ATR and is involved in the G2/M DNA damagecheckpoint response. Mol. Cell. Biol. 24, 9207–9220.
Zhao, Y., Pan‐Hammarstrom, Q., Zhao, Z., and Hammarstrom, L. (2005). Identification of theactivation‐induced cytidine deaminase gene from zebrafish: An evolutionary analysis. Dev.Comp. Immunol. 29, 61–71.
Zhong, H., SuYang, H., Erdjument‐Bromage, H., Tempst, P., and Ghosh, S. (1997). The transcrip-tional activity of NF‐kappaB is regulated by the IkappaB‐associated PKAc subunit through acyclic AMP‐independent mechanism. Cell 89, 413–424.
Zhou, C., Saxon, A., and Zhang, K. (2003). Human activation‐induced cytidine deaminase isinduced by IL‐4 and negatively regulated by CD45: Implication of CD45 as a Janus kinasephosphatase in antibody diversification. J. Immunol. 170, 1887–1893.
Zhu, C., Bogue, M. A., Lim, D. S., Hasty, P., and Roth, D. B. (1996). Ku86‐deficient mice exhibitsevere combined immunodeficiency and defective processing of V(D)J recombination inter-mediates. Cell 86, 379–389.
Zhu, J., Petersen, S., Tessarollo, L., and Nussenzweig, A. (2001). Targeted disruption of theNijmegen breakage syndrome gene NBS1 leads to early embryonic lethality in mice. Curr. Biol.11, 105–109.

a#
Anti‐IgE Antibodies for the Treatment of IgE‐MediatedAllergic Diseases
Tse Wen Chang,* Pheidias C. Wu,*,† C. Long Hsu,*,† and Alfur F. Hung*,†
*Genomics Research Center, Academia Sinica, Nankang, Taipei 115, Taiwan†Department of Life Science, National Tsing Hua University, Hsinchu 300, Taiwan
A
dva20
bstract ...........................................................................................................
63nces in immunology, vol. 93 0065-2776/07 $
07 Elsevier Inc. All rights reserved. DOI: 10.1016/S0065-2776(06)93
63
1. I ntroduction ..................................................................................................... 64 2. R ationale Leading to the Invention of the Anti‐IgE Concept ..................................... 67 3. A nti‐IgE Is Approved for Treating Moderate‐to‐Severe Asthma.................................. 73 4. S tudies on Other Allergic Diseases........................................................................ 78 5. T he Potential of Using Anti‐IgE to Assist Allergen‐Based Immunotherapy ................... 83 6. P ivotal Roles of IgE and FceRI in Type I Hypersensitivity ........................................ 85 7. N eutralization of Free IgE .................................................................................. 88 8. D ownregulation of FceRI.................................................................................... 90 9. P otential Beneficial Effects of IgE:Anti‐IgE Immune Complexes................................ 93 10. C an Anti‐IgE Modulate IgE‐Committed B Lymphoblasts and Memory B Cell?............. 96 11. O ther Immunoregulatory Effects of Anti‐IgE.......................................................... 98 12. C an Anti‐IgE Attain a Long‐Term Remission State? ................................................. 1 00 13. A re There Adverse Effects Associated with Anti‐IgE Therapy?................................... 1 01 14. O ther Approaches for Targeting IgE or IgE‐Expressing B Cells ................................. 1 03 15. C oncluding Remarks .......................................................................................... 1 06R
eferences ....................................................................................................... 1 07Abstract
The pharmacological purposes of the anti‐IgE therapy are to neutralize IgE and toinhibit its production to attenuate type I hypersensitivity reactions. The therapy isbased on humanized IgG1antibodies that bind to free IgE and to membrane‐bound IgE on B cells, but not to IgE bound by the high‐affinity IgE.Fc receptorson basophils andmast cells or by the low‐affinity IgE.Fc receptors on B cells. Afternearly 20 years since inception, therapeutic anti‐IgE antibodies (anti‐IgE) havebeen studied in about 30 Phase II and III clinical trials in many allergy indica-tions, and a lead antibody, omalizumab, has been approved for treating patients(12 years and older) with moderate‐to‐severe allergic asthma. Anti‐IgE has con-firmed the roles of IgE in the pathogenesis of asthma and helped define the concept‘‘allergic asthma’’ in clinical practice. It has been shown to be safe and efficaciousin treating pediatric allergic asthma and treating allergic rhinitis and is beinginvestigated for treating peanut allergy, atopic dermatitis, latex allergy, andothers. It has potential for use to combine with specific and rush immunotherapyfor increased safety and efficacy. Anti‐IgE thus appears to provide a prophylacticand therapeutic option for moderate to severe cases of many allergic diseases and
35.00
002-8

64 TSE WEN CHANG ET AL .
conditions inwhich IgE plays a significant role. This chapter reviews the evolutionof the anti‐IgE concept and the clinical studies of anti‐IgE on various diseaseindications, and presents a comprehensive analysis on the multiple intricateimmunoregulatory pharmacological effects of anti‐IgE. Finally, it reviews otherapproaches that target IgE or IgE‐expressing B cells.
1. Introduction
1.1. The Current Status of the Anti‐IgE Development
A therapeutic anti‐IgE antibody is a monoclonal antibody (MAb) designed totarget IgE and IgE‐expressing B cells without the complication of its cross‐linking IgE bound by the high‐affinity IgE.Fc receptors (also called type I IgE.Fc receptors, FceRI) on basophils and mast cells. Such an antibody is distinc-tively different from a common anti‐IgE antibody, which can bind to and cross‐link IgE‐FceRI and hence sensitize the effector cells bearing them to dischargevarious pharmacological mediators. In this chapter, a therapeutic anti‐IgEantibody designed above is referred to as ‘‘anti‐IgE,’’ as has been used routinelyin the allergy and asthma fields.Historically, research on IgE has arrived at an interesting juncture. IgE was
discovered in 1967 by Joh ansson (Johansson, 1967) and Ishizakas (Ishiza ka andIshizaka, 1967), and 20 ye ars late r in early 1987, the an ti ‐Ig E con cept wasinvented by one of the authors (Chang) of this chapter. In 2007, it will be20 years since the anti‐IgE concept was proposed and research on developingthe first antib ody pro totype initiated (Cha ng et al ., 1990). Today, vario usimmunoassys relating to IgE are now essential tools in the care of allergicdiseases, and the initial application of the anti‐IgE therapy for treatingmoderate‐to‐severe allergic asthma has been approved by health agencies inmany countries—it has been used to treat more than 60,000 patients withdifficult‐to‐treat allergic asthma.The diagram in Fig. 1 summarizes the main events in developing the anti‐IgE
program. Initially, CGP51901 (a chimeric anti‐IgE derived from mouse MAbTESC‐21; Davis et al., 1993), which was studied in a Phase I and II clinical trial(Corne et al., 1997; Racine‐Poon et al., 1997), and CGP56901 (or TNX‐901, ahu ma ni ze d a nt i‐IgE based on TESC‐21; Kolbinger et a l., 1993) were developedin one corporate program. Later, omalizumab (a humanized anti‐IgE, alsoreferred to as E25) emerged in another corporate program (Presta et al., 1993).In 1996, the two programs were combined and omalizumab was chosen for fur-ther development on the basis of its superior manufacturing process. TNX‐901,which was shown to be safe and efficacious in a Phase II trial on allergic rhinitis(using CGP51901) and in a Phase II trial on peanut allergy (Leung et al ., 2003),should serve as a backup drug candidate.

Invention
1987
phase III trial
Phase II trial
Phase I trial
1990
Chimeric MAbCGP51901
1994
19961999 2002 2003 2005 2006
19931991
Humanized MAbTNX-901
Humanized MAbomalizumab
Tanox/Novartis
First
Genentech
Omalizumab
TNX-901
USAapproval
for asthma
Australiaapproval
for asthma
EUapproval
for asthma
>30 phase II, III trials on asthma,allergic rhinitis, latex allergy, and so on
60,000 asthmatic patients treated
Phase II trial on peanut allergy
Figure 1 The major events in the development of the anti‐IgE therapeutic approach.
THERAPEUTIC ANTI- IgE ANTIBODIES 65
Among the key events in the clinical development of the anti‐IgE conceptare that omalizumab was approved by the United States in 2003 and by theEuropean Union in 2005 for use in treating patients with moderate‐to‐severeallergic asthma. Omalizumab has now been studied in nearly 30 Phase II andIII human trials in various allergic diseases and conditions (Sections 3–5).
1.2. The Main Chemical Features of the Anti‐IgE Therapeutic
For most clinicians treating allergic diseases, omalizumab would appear as a verydifferent drug among the battery of drugs for treating allergic diseases. An anti‐IgE is a protein, a macromolecular (�150,000 Da) biological substance; it is ahumanized IgG (g1,k) antibody or a recombinant antibody, which has beensubstantially improved by genetic engineering methodologies for in vivo use inhuman patients. In overall structure, chemical and physical properties (such askinetic properties), and ability to mediate a wide spectrum of Fc‐related immunemechanisms, a recombinant, humanized anti‐IgE IgG1 is similar to an authentichuman IgG1.Only the three short complementarity‐determining regions (CDRs)in the VH domain and the three CDRs in the Vk domain from the parental mouseantibody are retained; nearly the entire four framework segments in each of theVH and the Vk are derived from sequence‐matched human VH and Vk; the entireCH1, CH2, and CH3 domains of the heavy chain are from human g1 and the Ck

66 TSE WEN CHANG ET AL .
domain of the light chain is from human k (Kolbinger et al., 1993; Presta et al.,1993). A striking feature of omalizumab and TNX‐901 is that, like a human IgG1,they circulate in the treated patients with a half‐life of about 21 days.Omalizumab is presently provided by the manufacturers in a dry powder
formulation, which requires the reconstitution with water to resume a solutionform for subcuta neous injection (Stru nk an d Bloomb erg, 2006). Oth er formu -lations such as a solution in prefilled syringes for easier administration areunder development. Unlike other small molecular compounds synthesized inorganic chemical plants, omalizumab is produced by a host Chinese hamsterovary (CHO) cell line in 12,000‐ or 15,000‐liter bioreactor tanks. CHO cell linehas become a standard for producing protein pharmaceuticals for humanapplicat ions (Wurm, 2004). In our case, the CHO cell line was engin eeredby transfecting with the recombinant ‘‘humanized’’ genes coding the improvedg1 and k chains. The CHO cells express the exogenously introduced antibodygenes and produce the humanized antibody in very high yields.
1.3. IgE ‐ Mediated Allerg y Is a Vast Me dical Field
IgE is well known for its roles in mediating type I hypersensitivity reaction(Gould et al., 2003; Janeway et al., 2005). Through the work of many resear-chers in the last few deca des an d the clinic al studies of an ti‐ IgE more rec ently,IgE is now known to play important roles in many allergic diseases. Allergicdiseases are generally defined as significant pathological changes that arecaused by excessive reactions of the immune system to innocuous substanceswhich the patients are exposed to. While allergic reactions to some substancesinvolves nearl y entir ely type II, III, or IV hypersensiti vity reaction s (Janewayet al ., 2005), most allerg ic reactions to inhaled or ingested pro tein substa ncesinvolves at le ast partly typ e I hypersen sitivity reaction and IgE (Oettgen andGeha, 1999). The main allerg ic disea ses or cond itions that involve IgE by someextent include allergic asthma (Menz et al., 1998; Oettgen and Geha, 2001),allergic rhinitis (Bodtger et al., 2006; Tschopp et al., 1998) and conjunctivitis(Mimura et al., 2004), allerg ic or anaphylac tic reaction s to certain foods (suchas peanuts, tree nuts, shell fish, and so on; Sabr a et al ., 2003), allerg ic reactionsto certain drugs (such as protamin and heparin; Sicherer and Leung, 2005;Weiss et al., 1989), allergic reactions to insect bites (especially wasp and fireant stings; King and Spangfort, 2000; Schafer and Przybilla, 1996), atopicdermatitis (Leung, 1993), allergic reactions to natural rubber latex (Ebo andStevens, 2002), allergic reactions to certain raw materials (such as papain, sub-tilisin, yeasts, and so on) (Baur, 1979; Lemiere et al., 1996) or products in factor-ies (Bernstein, 1997), and allergic reactions to other less common harmlesssubstances.

THERAPEUTIC ANTI- IgE ANTIBODIES 67
Many of the allergic diseases mentioned above affect ever‐increasing popu-lation in most regio ns of the world (Isol auri et al., 2004). The preva lence isrelated to economica l develo pment (Section 7.1) (Gold and Wright, 2005) andin developed countries the aggregate rates of cases of allergic diseases that areserious enough to seek doctors’ help are more than 10% generally and maybe20�30% in some regions. These diseases affect the health and the quality oflife (sleep, school, work, family, and so on) of millions of patients and consumelarge amounts of healthcare resources.
1.4. The Scope of This Chapter
An earlier review (Chang, 2000) by the lead author of this chapter presented anoverview of the rationale and pharmacological basis of the anti‐IgE therapy. Sincethen, a lot of progress relating to anti‐IgE has been made. A large number ofreview articles have been published on anti‐IgE, especially reviews summarizingthe clinical trials on allergic asthma and discussing the utility of this treatmentmodality in managing asthma (Busse, 2001; Holgate et al., 2005a; Lanier et al.,2003;Milgrom, 2004; Strunk and Bloomberg, 2006). In this chapter, wewill focuson aspects of the anti‐IgE concept and development, which have been largely leftout by most previous anti‐IgE reviews. We will discuss the rationale behindthe anti‐IgE invention, the clinical development of anti‐IgE on various diseaseindications, and present a comprehensive analysis on the multiple intricateimmunoregulatory pharmacological effects of anti‐IgE. We will also discussthe development of other approaches that target IgE and IgE‐expressing B cells.
2. Rationale Leading to the Invention of the Anti‐IgE Concept
2.1. IgE Isotype‐Specific Control and IgE Targeting
The idea of isotype‐specific suppression of antibodies had already been pur-sued academically (Bich‐Thuy et al., 1984; Hoover and Lynch, 1983) byresearchers before the anti‐IgE approach was conceptualized. In the field ofIgE suppression, a group led by Haba and Nisonoff investigated the potentialof inducing intolerance to IgE in mice by administering IgE within a few daysafter birth when the mice did not produce any IgE. This could suppress theproduction of IgE even when the mice grew to adulthood. After the anti‐IgEconcept became known, Haba’s group identified syngeneic anti‐IgE antibodiesin the mice that had been injected with IgE at a neonatal age. They also foundthat IgE‐secreting B cells in these mice were substantially reduced (Haba andNisonoff, 1990, 1994).
Among the antibody isotypes, substantial suppression would not seem logi-cal except perhaps for IgE. While there is a general belief that IgE plays a role

68 TSE WEN CHANG ET AL .
in the defense against parasitic worms, especially helminthes (Capron andCapron, 1994), the results from many studies could best be grossly characterizedas suggestive, but not conclusive (Pritchard, 1993; to be discussed in more detailin Section 13.1). Up until 1980s and even until now, most of the research done onIgE since its discovery in 1967 had been related to the adverse effects of IgE onallergic diseases. The activity ofmast cells and basophils in inflammatoryprocessesassociated with allergenic responses (Lewis and Austen, 1981; Wasserman, 1989)and the roles of IgE in sensitizing those inflammatory cells had become estab-lished (Metzger et al., 1982; Razin et al., 1983). Thus, therapeutic approachesthat could decrease the production of IgE would seem to be very logical.The concentrations of IgE in most patients with allergy are minute, ranging
from less than 3 0–1000 interna tional units (IU ) per millilite r (1 IU ¼ 2.4 ng),which corre sponds to a total amou nt of 12 mg or less in the entir e body(calculated based on a total volume of 5 liters of body fluid in a person), formore than 90% of the patie nts (Manz et al . , 2005). The B cells, whi ch exp ressmIgE as part of the B cell receptor and can potentially differentiate to IgE‐producing plasma cells, also account for minute proportions among lympho-cytes (Ka saian et al ., 199 5). Thus, it appear s that IgE and mIgE ‐ expres singB cells are very attractive therapeutic targets.A few years before setting foot on the anti‐IgE program, the lead author
(Chang) of this chapter, had been carrying out research on the OKT3 antibody,a mouse MAb against a human T cell surface component, in another corpora-tion. Chan g et al . (1981) pro posed tha t the an tigen recog nized by OKT 3 is partof then yet unidentified T cell receptor linking to a signal‐transducing process.In the next few years, the a/b and d/g chains of the T cell receptors werediscovered and OKT3 antigen, which was later renamed as CD3 in an Inter-national Leukocyte Association, was found to be indeed part of the T cellreceptor. While Chang’s interest on OKT3 was mainly academic, to his amaze-ment, the antibody was developed by the corporation as an immunosuppres-sive agent and approved by the US Food and Drug Administration (FDA) in1986 for use in preven ting org an rejection in kidney transplantati on (Weimaret al., 1988). OKT 3 is the firs t approved therapeutic antibod y. It acts on theT cell receptor and causes T cell depletion. In the spring allergy season in 1987,Chang, being affected by severe allergic rhinitis himself, drew from theexperience on OKT3 and designed the anti‐IgE approach.
2.2. The Unique Set of Anti‐IgE‐Binding Specificities
On the basis of the rationale in the Section 2.1, free IgE in the blood and ininterstitial fluid and mIgE‐expressing B cells, which include mIgE‐committedB lymphoblasts and memory B cells, are very attractive therapeutic targets by

THERAPEUTIC ANTI- IgE ANTIBODIES 69
specific immunological agents. On the surface of mIgE‐expressing B cells, aspecific cell surface marker that is unique for those B cells and that isreachable by an immunological agent had not been identified, except perhapsmIgE itself. mIgE is part of the B cell receptor, which is linked via associatedsubunits to signal‐transducing pathways (DeFranco, 1993; Niiro and Clark,2002), on B cells expressing mIgE. This makes mIgE a viable immunologicalsite to target for modulating mIgE‐expressing B cells. Thus, an antibody couldpotentially be designed to target free IgE in the body fluid and mIgE on B cellssimultaneously, because the relatively small amounts of soluble, free IgEshould not consume or neutralize a potential targeting antibody prohibitively.
The trick in designing an antibody to target IgE and mIgE was that theantibody must not bind to IgE that is already bound by FceRI. It had beenknown that antibodies made against IgE could sensitize basophils and mastcells and, in fact, several laboratories had used these antibodies to study themechanisms of activation of those effector cells (Conroy et al., 1977; Ishizakaet al., 1984). By applying hybridoma methodology and a series of screeningprocedures, hybridoma clones secreting MAbs with the set of desired specifi-cities wer e id entified. Figure 2 sum marizes the bi nding speci ficities of anti ‐ IgEfound at the time of its discovery and in later studies.
The selected MAbs have high binding affinity to human IgE (Kd about 1 �10–10 M), for they are designed to compete with the high‐affinity IgE receptor,FceRI, for binding to IgE (Corne et al., 1997; Davis et al., 1993). The MAbshave no crossbinding activities toward other serum substances and other celltypes. They were examined for crossbinding activities to various histologicaltissue sections, which represented all tissues in the human body, and found tobe free of such activities.
2.3. Structural Basis of the Unique Binding Specificities
The mouse antihuman IgE MAbs with the required set of binding specificitieswere obtained before the 3D structures of IgE, its high‐affinity receptor,FceRI, and low‐affinity receptor, FceRII, had been solved. The bent confor-mation of IgE and of the 1:1 binding stoichiometry between the a chain ofFceRI and IgE were elucidated by various approaches (Keown et al., 1997;Zheng et al., 1991). The X‐ray structures of the FceRI a chain, free (PDB entry1f2q, 2.4 A ; Garma n et al., 1998) and comp lexed with human Ig E CH3– CH4domain s (PDB entry 1f6a, 3.5 A ; Garma n et al ., 2000), have pro vided astructural basis for understanding the binding properties of the anti‐IgEMAbs (see below). The structure of FceRII, also known as CD23, andthe characteristics of its binding to IgE have also been elucidated in detail(Hibbert et al., 2005; Wurzburg et al., 2006).

lgE
In blood,interstitial fluid
IgE on FceRI
On mast cells,basophils
lgE on CD23
On B cells
mIgE
On B lymphoblasts,memory B cells
lgE on sol. CD23
In blood, interstitialfluid
lgE: anti-lgEcomplexes
In blood,interstitial fluid
t1/2 ~ 1−2 days
t1/2 ~ 21days
t1/2 ~ 2−3 weeks
X
X
X
Anti-lgEA humanized
lgG1,k
Figure 2 The unique set of binding specificities an anti‐IgE antibody. The half‐lives of IgE,anti‐IgE, and the immune‐complexes of IgE and anti‐IgE are also indicated.
70 TSE WEN CHANG ET AL .
It is now clear that both omalizumab and TNX‐901 IgG1 antibodies, theFceRI a chain, and CD23 bind to the CH3 domain of IgE. They affect thebinding of each other, suggesting that their binding sites on IgE either overlapor are probably in proximity to one another. The binding of IgE by anti‐IgEwill prevent IgE from binding to FceRI and to CD23 on B cells. Conversely,the binding of IgE by FceRI will prevent anti‐IgE from binding to IgE. Thebinding of IgE by CD23 will also block anti‐IgE binding to IgE. At first glance,such an explanation seems straight forward, based on a simple understandingthat the binding of IgE by one molecule will sterically hinder the access byanother. In other words, the binding of IgE by one entity masks the bindingsite for another. This would be perfectly rationale, had there been only oneCH3 domain in one IgE molecule. However, there are two CH3 domains (twoe chains) in one IgE molecule.The Fc (CH2–CH3–CH4 region) of IgE exists in a bent conformation, with
the CH2 lobes bending toward one side by a large angle against the two CH3lobes and hanging not distant from the CH4 lobes (Wan et al., 2 002). The twotrunks of CH2–CH3–CH4 domains do not have an axis of twofold symmetrybetween them. The two CH3 domains have a pseudo dyad axis between them;

THERAPEUTIC ANTI- IgE ANTIBODIES 71
the two CH4 domains also have one; the two dyad axes are offset by 3�. Thetwo CH2 domains are linked to their respective CH3 domains by linkersegments that are part of CH3 and lay crossed. Looking down from atop, thetwo CH2 domains and two CH3 domains are like a bent letter ‘‘X.’’
The fact that one IgE molecule can be bound by two anti‐IgE moleculesmay explain why anti‐IgE blocks IgE binding to CD23 or FceRI. Themembrane‐bound form of CD23 exists on the cell surface as a trimer, eachof which extends a C‐type lectin‐like ‘‘head’’ domain through a long ‘‘stalk’’ anda ‘‘neck ’’ structure (Hibbe rt et al., 2005). The head dom ain binds to the outerside of a CH3 domain of IgE in a Ca2þ‐dependent, nonlectin‐like manner(independent of carbohydrates). A single head domain of CD23 binds to IgEwith very low affinity (Kd ¼ 10–5 M) and hence two of the heads must bind totwo CH3 domains to hold the IgE in place (Kd ¼ 10 –8 M). Thus, the bindingby CD23 precludes anti‐IgE to bind to IgE simultaneously.
Figure 3 sho ws that the two CH3 domains an d two CH4 domain s form arhombic shape with a large opening in the center. The a chain of FceRI bindsto both CH3 domains (indicated by shaded parts), which is uncommon inreceptor–ligand interaction. The wide opening of the two CH3 lobes allows thea chain to bind to the inner sides of the CH3 domains. Even more strikingly,the two sites on the CH3 domains bound by the a chain of FceRI, with onelarger than the other, share 4‐amino acid residues between their respectiveCH3 peptide chains. The binding by a chain of FceRI causes significantconformational change pushing apart the two CH3 domains and enlargingthe open ing between t hem (com pare Fig. 3A and C, also D and E).
It is not yet solved where on the dimeric CH3 domains the two anti‐IgEmolecules bind to. Presumably, they are near the binding site for the a chain ofFceRI such that binding by the a chain of FceRI would hinder both anti‐IgEmolecules to bind to. However, the fact that CH2 domains bend to and hangover closely one side of the CH3 domains would suggest that the binding sitesfor anti‐IgE should not be near the inner sides of the CH3 lobes (because twoanti‐IgE molecules can bind to one IgE molecule). A crystal structure of CH3and the Fv of anti‐IgE would be of great interest.
2.4. Prevailing Concepts at the Time of the Invention
At the present time, the prospects of anti‐IgE as a treatment option for manyallergic diseases look promising. However, leaders in the field of allergy andimmunology were largely skeptical about the approach in the early years of theprogram. In the realm of pharmaceutical development in the late 1980s, thethrust was to identify, by a combination of compound screening and organicsynthesis, small molecules (as opposed to protein drugs) that could block

A
Ce3 A
Ce4 A Ce4 B
Ce3 B
B C
D E
Figure 3 The binding site of the a chain of FceRI on CH3 domains. (A) The two CH3 lobes andtwo CH4 lobes form a rhombic shape with an opening in the center; the binding site for a chain ison the inner sides of both CH3 lobes, near the junctions with CH2 lobes. (B) The ‘‘frontal’’ view ofCH3 domains before a chain binding. (C) The frontal view of CH3 domains after a chain binding.(D) The ‘‘top’’ view of CH3 domains before a chain binding. (E) The top view of CH3 domainsafter a chain binding. The structures show that the binding by a chain causes the opening of thetwo CH3 lobes wider. The binding sites for FceRII (CD23) and potential binding sites for anti‐IgEare discussed in the text.
72 TSE WEN CHANG ET AL .

THERAPEUTIC ANTI- IgE ANTIBODIES 73
targeted biological processes. In the field of allergy, most of the pharmaceuticalresearch was to develop drugs that could be used to block leukotrienes,tryptase , or other inflam matory mediators (Barn es, 2004 ). Res earche rs hadalso started to screen compound libraries for drug leads that can bind to thea chain of FceRI and block its binding by IgE. The notion of using anantibody to treat allergic disease was beyond the imagination of most academicresea rchers and clinic ians then (Chang, 2006).
An immediate feeling among scientists first told of the anti‐IgE approach wasthat other anti‐IgE antibodies had already been known to be potent inducers ofbasophil andmast cell activation, andwould cause anaphylactic reactions, if theywere injected into a person. Hence, the idea of injecting an anti‐IgE antibody,albeit different from ordinary anti‐IgE antibodies, was met with skepticism.Among other major arguments put forth by immunologists was that even if IgEcould be neutralized to better than 99%, the remainder would still be sufficientto charge the receptors on basophils and mast cells, keeping them sensitive toallerge ns (Pruza nsky and Patter son, 1988). Anothe r thought was that as IgE isbound by the high‐affinity receptors on mast cells for many months or perhapsmore than a year, the neutralization of free IgE by administrating anti‐IgEwould not affect the sensitivity of those cells. Yet another argument was thatIgE is essential for immune defense functions and cannot be compromised(Capron and Capron, 1994; Capron and Dessaint, 1992). Despite these overalldubious sentiments, the new therapeutic concept eventually found a few sup-porters from a large pharmaceutical company and has been actively developed,although bumps were hit on the way, in the last 20 years. Officials and scientistsat the FDA in the United States and at the Medicines Control Agency (nowreorganized) in United Kingdom encouraged the development of this drastical-ly different experimental drug for allergic diseases with cautious interest andprovided guidance to us in preparing for the first human experimentation. Thechimeric anti‐IgE antibody, CGP51901, was first administered to patients inSouthampton, United Kingdom in 1991. Initially, while we had difficulty dis-secting the biological results, mainly the rapidly rising total IgE (accumulatingimmune complexes), safety was not a major concern.
3. Anti‐IgE Is Approved for Treating Moderate‐to‐Severe Asthma
In 1994�1995, the first Phase II trial of anti‐IgE was performed withCGP51901 at three medical centers in Central Texas on patients with severeallergic rhinitis caused by mountain cedar pollens. The patients were rando-mized in four groups and given weekly injections of placebo or CGP51901 at

74 TSE WEN CHANG ET AL .
15‐, 30‐, or 60‐mg dose. The results revealed a robustly clear dose response,showing that the antibody was safe and efficacious in alleviating nasal andocular allergic symptoms (results not published; the lead author of this chapterparticipated in the study).In 1996, the two corporate anti‐IgE development programs were combined
(Fig. 1). The new partn ership plan ned clinic al develo pm ent paths for omali-zumab and decided that several Phase II trials be carried out on both allergicasthma and allergic rhinitis. As the development program proceeded in thefollowing years, increased emphasis was placed on allergic asthma. It wasreasoned that the incidences of asthma, especially pediatric asthma, wererising in most regions of the world at alarming rates (Cookson, 1999; Kussinand Fulkerson, 1995). Also, the most severe cases (10%) of asthma are life‐threatening and consume large amounts of healthcare resources. In the newawareness of pharmacoeconomic environment, the costs of a drug per patientper year must be weighed against the therapeutic benefits achieved and thehealthcare dollar saved (Hoskins et al., 2000).Many excellent review articles have been written by clinical investigators
summarizing and analyzing in great detail the results obtained in the series oftrials of omalizumab on allergic rhinitis. To avoid repeating such a task, thissection will summarize the clinical studies succinctly for immunologists, whonormally do not survey clinically oriented journals, and discuss aspects of theeffects of anti‐IgE on asthma that are not covered by other reviews.
3.1. ‘‘Allergic Asthma’’ Has Been Adopted as a New Clinical Indication
‘‘Allergic asthma,’’ a term that had already appeared in the literature in the1950s (Prickma n, 1951) but used spa ringly in the ensuing few deca des, hasemerged as an important concept as the anti‐IgE concept gradually takes hold.Practically, most clinicians treating asthma patients do not need to knowpossible immunological or allergic involvement in the asthmatic disease theytreat. They use corticosteroids to suppress inflammatory responses and b2adrenergic receptor agonists to relax constricted airway smooth muscles.In pharmaceutical development, a candidate drug is studied for use to treat
a defined clinical indication. Normally, a drug being developed for asthma,such as a new chemical entity or a new formulation of b2 agonist, antileuko-triene, or corticosteroid, is tested for treating asthma without considering theasthmatic cases’ possible allergic nature. The frequent news and officialreports of the clinical study results of the anti‐IgE drug candidates in the pastdecade have gradually brought an awakening among clinicians treating asthmapatients that in a large pro portion of asthma cases, allergy is involved (Holga teet al., 2005a) . In the clinical trials of anti ‐ IgE, a ski n prick test was r equired to

THERAPEUTIC ANTI- IgE ANTIBODIES 75
screen patients for positive reactivity toward at least one allergen suspected tobe the cause of potential allergy in the examined patient. Now that omalizu-mab has been approved for marketing for ‘‘allergic asthma,’’ clinicians usepositive skin prick test reactivity as one criterion to determine the suitability ofan asthma patient to receive omalizumab medication.
3.2. Clinical Parameters Determined in the Clinical Studies
The clinical studies of omalizumab for treating patients with allergic asthma,with either moderate‐to‐severe or severe disease, in either adolescent and adultor pediatric populations, with or without the combination of other drugs, alladop ted a gene ral pro tocol design (Fig. 4; Buh l et al ., 2002; Milgrom et al .,2001; Soler et al., 2001), which has been regarded as highly effective. As a majorclinical objective of the new anti‐IgE drug was to reduce corticosteroid use,a key component in the clinical study design was that it enabled the exam-ination whether the patients’ dependence on corticosteroids can be partiallyreduced or withdrawn entirely.
In the multicenter, double‐blinded clinical studies, patients with allergicasthma were recruited with a set of screening criteria and randomized intoplacebo and treatment groups. Generally, patients were allowed a 4‐ to 6‐week‘‘run‐in’’ period to establish their use of adequate amounts of corticosteroids,then a 28‐week core‐study or ‘‘add‐on’’ period, in which the patients weregiven placebo or omalizumab of 150‐ or 300‐mg dose every 2 or 4 weeksaccording to their plasma IgE levels and body weights, and lastly, a 4‐, 6‐, or12 ‐ month ‘‘extensio n period’’ for continu al observation (Fig. 4). In the firs t16 weeks of the 28‐week add‐on period, patients were maintained at theirregular doses of corticosteroid; in the last 12 weeks of the 28‐week add‐onperiod, a ‘‘steroid reduction’’ schedule was implemented for each patient.Beginning at the starting point of the 12‐week period, a quarter of thestabilized amount of corticosteroid was taken off. If, in the 2‐week interval,the patient maintained symptom‐free, another quarter amount of the steroidwould be reduced, whereas if a patient became symptomatic as defined by thechart in Fig. 4, the ster oid would be resum ed to the dose of 2 weeks before andkept at that dose throughout the remaining period of the trial (Buhl et al.,2002; Milgrom et al., 2001; Soler et al., 2001).
To date, a total of 17 clinical studies of omalizumab on allergic asthma,covering adult, adolescent, and pediatric patients, with varying degrees ofdisease severity, and with different accompanying medications, have beenperformed and published. Those results clearly indicate that anti‐IgE waseffective in reducing the numbers of asthma attacks (exacerbations) at thesame time when corticosteroids were substantially reduced or completely

Study time frame
Run-in Add-on Extension Phase
4−6 weeks 28 weeks
16 weeks 12 weeks
16 weeks to 12 months
Anti-lgE add-on period
Steroid reduction scheme
Steroid maintenance Steroid reduction
(Dosage)
100%
75%
50%
25%
0%
S
NS
NS
NS
S
S
S
0 2 4 6 8 10 12Weeks
S, symptomatic; NS, non-symptomatic
Symptomatic criteria (as appeared in Holgate et al., 2004):
1. >50% increase in 24-h rescue medication use on at least 2 of any 3 consecutive days compared to mean use over the last 7 days of the preceding phase.2. Mean daily asthma symptom score 4 over the previous 7 days. > −3. Fall in morning peak expiratory flow (PEF) of >20% on at least 2 of any 3 consecutive days relative to the mean morning PEF over the last 7 days of the proceeding phase. 4. Worsening of disease between visits requiring an unscheduled practitioner or hospital visit.5. At least 2 of 3 consecutive nights with awakenings due to asthma symptoms requiring rescue medication.6. An asthma exacerbation.
Figure 4 The clinical trial protocol used in most of the trials of omalizumab in allergic asthma.
76 TSE WEN CHANG ET AL .
withdrawn in most of the patients (Bousquet et al., 2005; Soler et al., 2001).The numbers of unscheduled doctor’s office visits, emergency departmentvisits, days of hospitalization were also significantly reduced (Bousquet et al.,2005; Humbert et al., 2005). Lung functions, in terms of forced expiratory

THERAPEUTIC ANTI- IgE ANTIBODIES 77
volume , wer e increased (Ayre s et al., 2004). The other measure ments, whichwere of concern to the patients and the clinicians and whose changes werestatistically significant, included many quality‐of‐life measurements, includingreduction of missing days to school or to work and loss of sleep (Ayres et al.,2004; Buhl et al., 2002; Lemanske et al., 2002).
3.3. The Clinical Studies Confirm the Roles of IgE in thePathogenesis of Asthma
At the time when the anti‐IgE therapeutic concept was conceptualized andduring its initial clinical development, it was still debated whether IgE isinvolved in asthma. The situation was different in allergic rhinitis, wherethere was a general belief that IgE is involved in the disease. In the UnitedStates and Western European countries, clinicians treating moderate to severecases of allergic rhinitis were mostly allergists, who were trained in thespecialty of allergy. These clinicians were familiar with allergen‐based immu-notherapy and applied it to treat patients. They used patch tests and skin pricktests to gauge immune responses to suspected allergens and measured plasmatotal IgE and allergen‐specific IgE levels to assist their therapeutic practice.Immunology and IgE have been a big part of the knowledge and tool base ofthe medicine applied for allergic rhinitis (Sections 4.1 and 5).
Asthma patients were generally not treated by allergists, but by pulmonaryor thoracic physicians, or internists, who did not receive substantial amounts ofimmunology in their training. These clinicians treated mainly the inflammationand bronchial constriction associated with asthma. The knowledge on possibleinvolvement of the immune system or IgE in asthma would not matter in theircare of asthmatic patients. Very few cases of asthma are treated with immuno-therapy. Perhaps the most relevant research linking IgE to asthma were anumber of surveys investigating the correlation between (1) asthma and serumIgE levels (Bur rows et al., 1989) and (2) the sever ity of asth ma an d serum IgElevels (Borish et al ., 2005). Ho wever, the overall clinic al sett ing and the lack ofexperimental evidence linking directly IgE activities to asthma left open anunsettled dispute whether IgE is involved in the pathogenesis of asthma.
The clinical studies of anti‐IgE demonstrate that anti‐IgE, by virtue of itsability to incapacitate IgE, can not only inhibit the early‐phase reactions,occurring within the first 4 h of an asthma attack, but also the late‐phasereactions, occurring beyond the first 4 h (Boulet et al., 1997; Fahy et al., 1997).These inhibitory effects of anti‐IgE result in improving various symptoms ofallergic asthma, as shown convincingly in more than 15 Phase II and III clinicalstudies. Thus, anti‐IgE has helped to confirm unambiguously the roles of IgEin the pa thogenes is of allergic asthma (Holgate et al ., 200 5a).

78 TSE WEN CHANG ET AL .
3.4. Analyses of Good Responders Among Asthma Patients
For an expensive drug like omalizumab, a diagnostic or screening procedurethat helps identify patients who are likely to respond well to and benefitsubstan tially from the drug is highly desir able (Heane y an d Robinson , 2005).Asthmatic patients with protracting conditions present variable sets of patho-genic components and probably respond to anti‐IgE therapy with differentdegrees of improvement. In addition to a positive skin test to allergens, criteriasuch as a suitable range of IgE would be highly desirable for identifyingpotential good responders.The various clinical trials of omalizumab on asthma and the corporations
marketing omalizumab have indicated that omalizumab achieved response ratesin the range of 60–90%. It appeared thatwhen the studied or targeted patients aremore severe, the response rates are higher. Statistical analyses performed on theaggregated results of clinical trials on asthma indicate that anti‐IgE benefit bestthe most severe, difficult‐to‐treat patients, coincidentally matching a pharmacoe-conomic criterion (Oba and Salzman, 2004). Most of those difficult‐to‐treatasthma patients cannot be controlled even with very high doses of inhaled andinjectable corticosteroid (BDP > 800 mg/day) and have poor lung functions(FEV1 < 65%) (Bousquet et al., 2004). Anti‐IgE appears to be able to removethe root cause of inflammation in those difficult‐to‐treat patients (Ayres et al.,2004; Humbert et al., 2005). Clinicians have also suggested that asthma patientswho have concomitant allergic rhinitis also respond well to anti‐IgE treatment(Vi gno la et a l., 2004).The basal levels of IgE do not seem to correlate with responsiveness or
degrees of efficacy. In the large number of clinical trials on omalizumab,patients with plasma IgE outside the 30�700 IU range were excluded. Thedosing schedule allowed omalizumab to be in excess of replenished IgE duringthe clinical trial periods and hence IgE levels within this range should make nodifference to the effectiveness of anti‐IgE. That plasma IgE levels affect thedensity of FceRI on effector cells and the accumulation of anti‐IgE:IgEimmune complexes will be discussed in Sections 7–9.
4. Studies on Other Allergic Diseases
Among the large populations of patients affected by allergic rhinitis, foodallergy (especially peanut allergy), or atopic dermatitis are severe cases,which cannot be adequately treated with currently available drugs and are inneed of better medicine. There are smaller patient populations who areaffected by sensitivity to occupation‐related materials, such as natural rubberlatex, papain, yeast, and so on, to certain drugs, or to insect stings. While it is

THERAPEUTIC ANTI- IgE ANTIBODIES 79
clear that IgE is involved in the pathogenesis of allergic rhinitis, the informa-tion in the literature is not indicative of whether IgE is a dominant factor inother allergic diseases or conditions. Thus, anti‐IgE offers not only a valuabletool to study the extent of IgE’s involvement in these allergic diseases but also apossible treatment modality for the severe cases of these clinical indications.
4.1. Clinical Applications in Treating Allergic Rhinitis
Ten to forty percent of the general population is affected by allergic rhinitis indifferent regions of the world (Asher et al., 2006; Schoenwetter, 2000). Propor-tions of those populations have substantial symptoms and use over‐the‐countermedicine or seek physicians’ help. In the United States, patients visit doctors’offices more times for allergic rhinitis than for any other disease. There aremore than 10,000 licensed allergists in the United States (information fromAmerican Academy of Allergy, Asthma, and Immunology), who treat mainlypatients with allergic rhinitis with immunotherapy.
Unlike most allergic asthmatic cases, which are caused by indoor allergenssuch as dust mites and cat dander, most allergic rhinitis cases are caused byoutdoor airborne protein antigens such as tree pollens in the spring and grasspollens in the late summer and fall (Boulet et al., 1997; Leynaert et al., 1999).This interesting dichotomy is a curious subject for investigation. By mechan-isms that are yet not well understood, the exposure of the body to minuteamounts of protein antigen at the mucosal surface in the respiratory and thegastrointestinal tracts can induce antigen‐specific IgE production in somepeople. Some patients have sensitivity toward one or very few allergens,while others are sensitive to a large numbers of allergens. Some patientshave very defined allergic rhinitis, while others have concomitantly asthmaticand dermatitis symptoms. Some patients have distinctive seasonal patterns,suffering seasonal allergic rhinitis only in the spring when tree pollens aredense, or only in the fall when grass pollens are rampant, or in both seasons,whereas others have perennial allergic rhinitis and are affected by a number ofallergens that together are present throughout the whole year (Beck andLeung, 2000; Braunstahl, 2005).
Altogether more than a dozen Phase II and III clinical trials have beenperformed on allergic rhinitis in the United States, Germany, Scandinaviancountries, Australia, New Zealand, Japan, and other countries. These includestudies that investigated the efficacy and safety of (1) CGP51901 on patientswith severe allergic reactions to mountain cedar pollens and (2) omalizumabon patients with sensitivity on birch pollens, ragweed pollens, pollens of othergrasses, and others (Adelroth et al., 2000; Casale, 2001; Casale et al., 1997,2001; Nayak et al., 2003). In both clinical trials on seasonal allergic rhinitis and

80 TSE WEN CHANG ET AL .
on perennial allergic rhinitis, anti‐IgE was shown to be effective in improvingallergic rhinitis symptoms, reducing the inflammation and mucous secretions inthe nasal and conjunctiva linings and reducing sneezing attacks. The treatmentalso improved quality of life measurements.These studies have amply demonstrated that IgE and type I hypersensitivity
are clearly a dominant mechanism for most cases of allergic rhinitis. Despitethe robust data on the safety and efficacy of anti‐IgE in treating allergicrhinitis, governmental regulatory approval of marketing omalizumab for aller-gic rhinitis indication is not imminent. It appears that FDA requires a suffi-ciently long postmarketing period to ensure that the long‐term use of anti‐IgEby asthma patients does not cause undesirable side effects.
4.2. Anti‐IgE Studies on Treating Peanut Sensitivity
Researchers working on anti‐IgE recognized early the importance of investigat-ing the utility of anti‐IgE in treating sensitivity or anaphylactic reactions topeanuts. In economically developed countries, some people, mostly children,develop extre me sensitivity to peanu ts (Sk olnick et al., 2 001). They mou ntsevere allergic reactions, often in the form of anaphylaxis, from ingesting orinhaling peanut product at an amount as small as half a peanut. The incidencesare often accidental and life‐threatening, hence peanut allergy often causesconstant fear in the patients and their families. The treatment of peanut allergyis strict avoidance of peanut‐containing food product. Patients and their rela-tives are advised to carry a prefilled epinephrine (b2 agonist) syringe forrelieving anaphylactic reactions induced by inadvertent exposures. It is alsoknown that allergen‐based immunotherapy and corticosteroids do not treatpeanut allergy effectively. Thus, a therapy that can substantially reduce patient’ssensitivity to peanut would be of great relief for these patients and their families.TNX ‐ 901 was studi ed in a Phase II clinic al trial on peanu t allerg y (Leun g
et al., 2003). In the study, 8 4 volunteer ing pa tients wer e rand omized in to fourgroups and given placebo or TNX‐901 at 150, 300, or 450 mg subcutaneouslyevery 4 weeks for four times. Two to three weeks after the last injection, thepatients were challenged with increasing amounts of encapsulated peanut flour,which the patients swallowed. It was shown that TNX‐901 could increase thetolerable amounts with increasing doses of the anti‐IgE. At the 450‐mgdose, thetolerability was increased from an average of half a peanut to 28 peanuts inmost patients. These results do not suggest that the patients can venture outeating peanut packages or peanut‐containing food, but the anti‐IgE drugwill protect patients from anaphylactic reactions, when they accidentally eatpeanut‐containing meals or food products or breathe in peanut‐containingpowders.

THERAPEUTIC ANTI- IgE ANTIBODIES 81
A Phase II trial of a similar design was also carried out for omalizumab.However, it was terminated in the middle of the trial, because a few patientsdeveloped severe hypersensitivity reactions, while they were tested for theirbaseline levels of sensitivity to peanuts by taking in peanut flour, prior toreceiving omalizumab. A new study with a different design will be startedlater this year (information from news releases from the corporations develop-ing omalizumab). Furthermore, the favorable results of TNX‐901 on peanutallergy should encourage clinical studies on allergy toward other foods.
4.3. Clinical Application in Latex Sensitivity
Some people are sensitized to natural rubber latex and products made from it,especially latex gloves for healthcare workers (physicians, dentists, nurses, andso on) and balloons for childre n (Sussman et al., 2002). Pat ients are expose d tonatural rubber latex by direct skin contact to latex products and by inhalinglatex‐containing powders used for packing and aiding use of latex products.The most frequent symptoms associated with latex are respiratory and skincomplic ations (Fi sh, 2002). Most patients develo p IgE response s to proteinscontained in natural rubber latex. This raises the following intriguing question:if a patient is exposed to latex products mainly by direct skin contact, how dothe proteins contained in the latex powder or in the latex sheets induce IgEimmune response?
In addition to avoidance of latex products, immunotherapy using naturalrubber latex extracts have also been tested on children with latex sensitivityand found to have partial response. A small Phase II trial was performedinvestigating the efficacy of omalizumab on 18 healthcare workers with allergyto latex pro ducts (Leynadi er et al., 2004). The pa tients developed nasal andocular symptoms and mild‐to‐moderate asthma on using latex gloves and theirsera were shown to contain IgE specific for latex proteins. In the double‐blinded study, patients received omalizumab (150–750 mg/month) or placebofor 16 weeks. They were evaluated in terms of ‘‘conjunctival challenge test totalscores’’ after 8 and 16 weeks of treatment. The results indicated very convinc-ingly that anti‐IgE helps alleviate allergic reactions to natural rubber latex.
4.4. Allergic Diseases or Conditions for Which Anti‐IgE Has BeenTested in Case Studies
As the safety and efficacy of anti‐IgE have been well established in allergicasthma and allergic rhinitis, clinicians have become adventurous in testing theeffects of anti‐IgE on diseases that manifest allergic symptoms and are knownto be associated with elevated levels of IgE. These tests have not been

82 TSE WEN CHANG ET AL .
performed systematically and generally involve only one or a few patients.Most of these tests have not been published, but a few have been published ascase reports. Among many other allergic diseases that are prevalent and havean apparent association with IgE is atopic dermatitis (also called urticaria;Leung and Soter, 2001; Shehade et al., 1988), which affects mainly children atan alarming rate in economically advanced countries (Eigenmann et al., 1998;Novak et al., 2003a). The hallmark of atopic dermatitis is the generally veryhigh blood IgE levels, which are in excess of 1000 IU/ml in most patients andreach more than 10,000 IU/ml in a sizable fraction of patients (Laske et al.,2003; Somos et al., 2001).The clinical studies of omalizumab only included patients with plasma IgE
in the range of 30–700 IU/ml. It was estimated that administrating anti‐IgE atthe designed protocol would maintain anti‐IgE in excess of newly synthesizedIgE in the treatment period. Thus, the high IgE in atopic dermatitis patientswould dissuade use of anti‐IgE, which administered at comparable doses couldnot neutralize the newly synthesized IgE and hence would not be effective.The three reports of off‐label use of omalizumab on 3, 3, and 7 patients,respectively, yielded mixed, but promising results. In the first report, thethree adult patients aged 34�48, who had baseline serum IgE at 23,000,5440, and 24,400 IU/ml, respectively, failed to benefit from omalizumabadministere d at 450 ‐ mg dos e ev ery other week for 4 mon ths (Krathe n andHsu, 2005). In the second report , three youn g rec alcitrant atopic dermatitispatients 11�13 in age, who had baseline serum IgE in the range of 1990–6120IU/ml, received omalizumab at 150–450 mg every other week for 22 weeksand experienced substantial improvement. In the third report, seven patients,7�58 in age, who had persistent asthma and severe atopic dermatitis andbaseline serum IgE in the range of 265–2020 IU/ml, received omalizumab at375 mg every other week for 7 months (two patients got omalizumab for only3 months because their insurance companies refused to cover the costs of thetreatment). All patients improved in their eczema severity scores measuredat 3 months (for all seven patients) and 7 months (for five patients) after treat-ment (Lane et al., 2006; Vigo et al., 2006). It is not clear why the first patientgroup responded differently from the other two; IgE levels might be a factor(Section 9.2).Two other very interesting clinical indications have been tested with omali-
zumab, for which the results have been published. There was a case reporton a patien t with seri ous cold ‐ induced urticari a/anaphy laxis (Boyce, 2006).A regimen of 375 mg of omalizumab biweekly resolved the patient’s urticaria,asthma, and rhinitis symptoms, with noticeable improvement occurring afterthe second injection of omalizumab. There was also a case report on a patientwith serious interstitial cystitis, which does not seem to be an allergic disease

THERAPEUTIC ANTI- IgE ANTIBODIES 83
(Lee et al ., 2006). Becaus e the patien t also had allergic rhi nitis and asth ma,specific immunotherapy (SIT) was recommended for her. However, she woulddevelop anaphylactic reactions even to 1000‐fold diluted antigens, and henceomalizumab was suggested to tame the anaphylactic reactions. The patient wasgiven omalizumab at 300 mg every 4 weeks. To the surprise of the patient andthe treating clinician, the patient’s interstitial cystitis was improved on the firstinjection. While both of those cases cannot be used to support that anti‐IgE beapproved for these disease indications, they encourage further investigation onthe roles of IgE in these diseases and the potential of anti‐IgE in treating them.The case reports also provide extraordinary insights into the pharmacologicalmechanisms of anti‐IgE.
5. The Potential of Using Anti‐IgE to Assist Allergen‐Based Immunotherapy
In economically advanced countries in North America and Western Europe,allergy is a well‐defined medical specialty serving substantial medical needs.In this specialty, vast amounts of research and development have been carriedout and clinical tools accumulated. Undoubtedly, the most significant body ofknowledge base in the allergy specialty is the know‐how on immunotherapy.Since immunotherapy was introduced by Noon nearly a century ago (in 1911),a sea of experience has been organized for clinical practice. Clinicians arefamiliar with the hundreds of allergens present in the local area where theypractice. They have established mechanisms for acquiring, stocking, and usinglarge numbers of antigen preparations for performing patch or prick skin testsand for providing allergen‐based hyposensitization immunotherapy. They alsouse various immunoassays to measure total IgE and allergen‐specific IgE inpatients to assist therapeutic procedures.
Immunotherapy is often chosen by patients to treat severe allergic rhinitisand veno m anaph ylaxis (Ross et al., 2000). A major reason is tha t it cansometimes achieve cure. The immunization with allergens adopted in immu-notherapy drives an array of effects, including the production of protectiveIgG4 antibodies, the shift from TH2 to TH1, and others, which dampen theactions mediated by allergen‐specific IgE (Carlsen, 2004; Ross et al., 2000; Tillet al., 2004). The shortcomings of immunotherapy are that it does not alleviatethe allergic symptoms in about half of the patients who receive the treatmentand is generally not effective for treating asthma (Bousquet et al., 1991; Limbet al., 2006; Nelson, 2003). Another main drawback is that the administrationof allergens can sometimes cause anaphylactic reactions (Bernstein et al., 2004;Borchers et al., 2004), especially if the doses of allergens are increased and theinjection schedule is compressed. Thus, it is a rational proposition that immu-notherapy and anti‐IgE therapy are combined to enhance the advantages of

84 TSE WEN CHANG ET AL .
immunoth erapy and to minimize its sho rtcomi ngs (Cha ng, 2000). Thi s mayexpand the practice of immunotherapy to cover broader patient populations(Parks and Casa le, 2 006).
5.1. The Combination of Anti‐IgE and SIT
In four well‐designed clinical studies carried out in Germany (Bez et al., 2004;Kopp et al., 2003; Kuehr et al., 2002; Rolinck‐Werninghaus et al., 2004), theeffects of omalizumab were investigated for augmenting the efficacy of SIT onpolysensitized allergic rhinitis patients, who were allergic to birch pollens inthe initial period and subsequently to grass pollens in the following periodduring the pollen season from approximately February to July, depending onthe locations of the patients. The birch and grass pollen periods were distinctlyseparated by 1�2 weeks and nonoverlapping. The grass pollen antigen used inthe trial was prepared by mixing six grass pollens and secale pollens. In the firstsegment (period) of the trial, grass antigen was used as an irrelevant antigencontrol, and in the second segment (period), birch pollen antigen was used as acontrol. The results indicated that omalizumab alone could achieve an efficacycomparable to that of SIT, and the combination of SIT and omalizumab couldachieve an efficacy better than either SIT or omalizumab alone.
5.2. Priming Patients with Anti‐IgE for Rush Immunotherapy
In a typical rush immunotherapy (RIT) protocol, a patient receives severalantigen injections in steeply escalating doses within a few hours on the firsttreatment day. In some but not all protocols, the patient continues to receiveseveral antigen injections in still sharply increasing doses within a few hoursin each of the follow ing few days in an inpa tient pro cedure. In the follow ingseveral months, the patient receives weekly increasing doses of antigen(Sharke y and Portnoy, 1996). RIT, which can be perfor med in an ‘‘off ‐ season ’’or an ‘‘off‐site,’’ is suggested for patients with severe allergy that needs to betreated promptly. Because the rates of increments of antigen in RIT are muchmore aggressive than those in a typical SIT, the incidences of severe reactions,including anaph ylaxis, are inevitably much higher (Greine der, 1 996). Thus,while RIT is highly desirable for resolving the allergic problems of patients inshort time frames, it is highly risky.In a recently published double‐blinded, placebo‐controlled clinical study
investigating the utility of anti‐IgE in protecting patients receiving RIT fromanaphylactic reactions, the patients, who had ragweed pollen‐induced seasonalallergic rhinitis, were pretreated with three doses of 4‐weekly or five doses ofbiweekly omalizumab injections, started 9 weeks before the RIT protocol.

THERAPEUTIC ANTI- IgE ANTIBODIES 85
One week after the last omalizumab injection, patients were given six injec-tions of Amb a 1 antigen, a major ragweed pollen antigen (100‐fold increase indosages) in 3 h or eight injections (330‐fold increase in dosages) in 5 h. In thefollowing 12 weeks, the patients received increasing doses of the ragweedpollen antigen. The results indicated that pretreatment with omalizumabcould reduce the incidences of anaphylactic reactions by 80%, while signifi-cantly reducing severity scores of allergic s ymptoms (Casale et al ., 2006).
6. Pivotal Roles of IgE and Fc«RI in Type I Hypersensitivity
6.1. Stages Along IgE‐Mediated Allergic Pathway
Omalizumab has already been approved in many countries for treating allergicasthma and has potential for broad use in treating severe allergic rhinitis andseveral other IgE‐mediated allergic diseases. In order to understand how anti‐IgE renders its pharmacological mechanisms to achieve the effects of alleviat-ing allergic symptoms, it is important to dissect the IgE‐mediated allergicpathway and analyze the intricate interactions among its elements and relatedfactors . Figure 5 sho ws that the IgE ‐m ediated pathw ay can be divide d in tosensitization, triggering, and manifestation stages. In these three stages, thekey elements are also indicated.
The sensitization stage in a patient may take many months to several years(Zeiger and Heller, 1993), depe nding on a complex set of intrinsic and extrinsic(environmental) factors particular to the patient. The intrinsic factors includegenetic and nongenetic ones (Cookson, 1999; Novak and Bieber, 2003). In theprogressive process of sensitization in a patient toward certain foreign, harm-less, mostly protein antigens encountered by the patient, the B cells are influ-enced to switch to Ig E‐ expre ssing B cells (Klei nJan et al., 2000). The end resul tof this sensitization stage is the continual production of IgE specific to theseantigens, consequently creating a sensitive state, in which the patient may betriggered to mount IgE‐mediated reactions on exposure to the antigens.
In the triggering stage of a patient sensitized to an allergen, the allergen‐specific IgE is present at a significant proportion in total IgE (more detail inSection 6). The allerg en‐ speci fic IgE occu pies a significan t number of Fc e RIon mast cells, basophils, and activated eosinophils. These effector cells are saidto be in an ‘‘armed’’ state and ready to ‘‘fire’’ anytime a threshold number ofallergen‐specific IgE‐charged FceRI on them is cross‐linked and aggregatedby allergen molecu les (Ishiza ka et al. , 1984). The trig gering stag e in an allergicresponse can be an astonishingly fast process. For example, in an allergic res-ponse to tree pollens or house dust mites, a patient takes in the pollen particlesor fecal particles of mites through inhalation. The allergen particles arebrought into mucous fluid in the nasal lining or lower airway and broken

Resting lgM/lgD B cell
Allergens Sensitization
lgE B cell lgE plasma cell
Histamine, tryptase
leukotrienes
cytokines, chemokinesMastcell/basophil
Triggering
Inflammation bronchoconstrictionAllergic rhinitis asthma anaphylaxis
Manifestion
a-E
a-E
a-E
a-E
a-E
lgE
Figure 5 The IgE‐mediated allergic pathway. Sites denoted by a circled aE indicated the stepswhere anti‐IgE has been shown to act or can potentially act at.
86 TSE WEN CHANG ET AL .
apart and the proteins contained therein dissolved. Certain (not all) proteinsfind ways to get across the mucosal epithelia into the basal side, where theallergenic proteins bind to the allergen‐specific IgE on mast cells, cross‐linkingthe IgE and thereby aggregating the underlying FceRI, initiating a signal‐transducing cascade, leading to Ca2þ mobilization and exocytotic processes, andultimately the discharge of pharmacological mediators from mediator‐packed

THERAPEUTIC ANTI- IgE ANTIBODIES 87
granules (Janeway et al., 2005; Kinet, 1999). The entire process can sometimestake less than a minute. The triggering process involving many mast cells in thelocal area will continue during the period a patient is exposed to over‐thresholdconcentration of allergens.
The mediators released from sensitized mast cells and basophils are of threecategories (Bingham and Austen, 2000; Serafin and Austen, 1987). The pre-packed mediators include histamines, tryptase, and chymase. In the secondcategory are lipid mediators such as leukotrienes, which are synthesized withinminutes after the sensitization of the cells. The third category includes cytokinesand chemokines (Bingham and Austen, 2000; Williams and Galli, 2000), whosegenes are activated and expressed as the cells are activated. These proteinfactors are synthesized and released after cells are activated for about 4 h.
The manifestation stage, which may last from minutes to hours to days, canbe divided into periods of early‐ and late‐phase reactions, which are mediatedlargely by the prepacked mediators and small molecular mediators and by thecytokines and chemokines, respectively. The mediators released from mastcells, basophils, and activated eosinophils bind to respective receptors onvarious cell types and cause inflammatory processes directly or indirectly,seen in all types of allergic responses. The factors also bind to smooth musclesof the airway and cause bronchoconstriction. In an anaphylaxis, a systematic,sometimes violent, reaction occurs and can pose life‐threatening situations.
Numerous elements, including histamines, tryptase, leukotrienes, cytokines,and oth er mediators, as w ell as the ‘‘insta bility’’ of mast cells (Section 8.2), inthe IgE‐mediated allergic pathway are targets of therapeutic intervention forthe purpose of attenuating allergic responses. On the basis of the pivotal role ofIgE in arming mast cells and basophils, IgE is a rational target for therapeuticintervention, because blockage at this step shuts off the later steps, includingthe release of all mediators by mast cells and basophils.
6.2. Direct Pharmacological Effects of Anti‐IgE
IgE mediates its broad range of effects via binding to FceRI and FceRIIexpressed on various cell types. The interaction of IgE with FceRI in the IgE‐mediated allergic pathway is central to almost all effects leading to the manifes-tation of allergic symptoms. The most direct effect resulting from the binding ofanti‐IgE to free IgE is that IgE is blocked from binding to FceRI on basophils,mast cells, and activated eosinophils, which should turn off the IgE‐mediatedpathw ay shown in Fig. 5. The oth er di rect effec ts are that IgE is block edfrom binding to FceRII (membrane‐bound CD23) on B cells, macrophages,granulocytes, platelets, and many other cell types and to soluble CD23 inthe blood and interstitial space. How the inhibitory effect on IgE binding to

88 TSE WEN CHANG ET AL .
FceRI will disarm the FceRI‐equipped effectors must be assessed in terms ofextent and timing as to be di scussed in Secti on 8. How bl ocking IgE binding toFceRII will affect the activity of the IgE‐mediated allergic pathway and overallimmune activ ity will be discusse d in Section 1 1.2.
7. Neutralization of Free IgE
Because IgE is a key molecule in the IgE‐mediated allergic pathway, neutra-lizing IgE and decreasing its synthesis would seem to be a logical approach tomitigate the IgE‐mediated allergic pathway. In applying anti‐IgE to neutralizethe activity of IgE in the blood and interstitial space in a patient, a questionstands out: to how low should the free IgE be brought down? In a few earlyclinical trials, answers in terms of the percentages of the baseline levels ofplasma IgE were provided. However, those data are probably not correct,because the immunoassays for determining free IgE involved high dilutionof plasma samples, which would liberate IgE from the immune complexes,resulting in an overestimation of the free IgE levels. In addition, in consideringthe great variations of total IgE and allergen‐specific IgE concentrationsamong different patients, there is probably no simple answer. This sectionintends to analyze these issues in detail.
7.1. Total IgE and the Proportion of Allergen‐Specific IgE
Anti‐IgE targets the entire isotype of IgE in a patient. Now that anti‐IgE isknown to be capable of achieving therapeutic efficacy in treating IgE‐mediatedallergic diseases, one would logically ask to how low IgE should be reduced forcausing a significant extent of loss of sensitivity of mast cells and basophils. Thesensitivity of mast cells and basophils is a function of a host of factors. Thischapter proposes that among these factors, the most critical ones are (1) thedensity of FceRI occupied by allergen‐specific IgE on the surface of mast cellsand basophils and (2) the concentration of allergens in the fluid surroundingthe cells (at the particular tissue site and at the moment of concern). Further-more, the density of FceRI occupied by allergen‐specific IgE on the surface ofmast cells and basophils are a function of two variables: (1) the proportion ofallergen‐specific IgE in total IgE and (2) the concentration of total IgE.Different IgE molecules bind through their common Fc to FceRI with an
equal affinity, regardless of their antigenic specificities. Over time, the propor-tion of FceRI that is occupied by allergen‐specific IgE is the same as that of thisallergen‐specific IgE in total IgE. In the other part of the equation, the serumIgE concentration correlates with the density of FceRI (occupied by IgE) onbasophils. In a classical study published by Litchenstein and colleagues in the

THERAPEUTIC ANTI- IgE ANTIBODIES 89
late 1970s (Mal veaux et al ., 1978), it was found that among 26 donors analyzed ,the serum IgE concentrations ranged from 2.6 to 5500 ng/ml (about 2000‐foldsin range), and the numbers of surface IgE molecules per basophil ranged in awell‐correlating fashion from 6000 to 600,000 (about 100‐folds in range). Thus,the density of FceRI occupied by allergen‐specific IgE on the surface ofbasophils and probably mast cells are determined by the above two variables.
Parenthetically, the above concept provides a molecular basis for explainingthe ‘‘hygiene hypothesis,’’ which states that people who lack frequent microbialinfection during infancy and early childhood, as the result of much improvedhygienic conditions, have increased likelihood of developing allergic diseases.A popular explanation is that microbial infections during the first years of lifecondition the immune system toward the TH1 mode and hence weaken theimmune activities stimulated by IL‐4, Il‐5, and other drivers of the TH2branch, and conversely, in the absence of frequent microbial infections theimmune system is skewed toward the TH2 mode. It is now established thatIgE is involved in a dominant fashion in the pathogenesis of allergic asthma,allergic rhinitis, and probably several major allergic diseases. Therefore, thehygiene hypothesis must be explained in terms of how the IgE‐mediatedallergic pathway is enhanced in some individuals, who had few infections inearly childhood. We believe that a plausible molecular elucidation of thehygiene hypothesis can be made by analyzing how the decrease of infectionsby microorganisms, viruses, and helminthic worms, and the increase of expo-sure to harmless environmental antigens affect in patients the total IgEconcentration, the proportion of allergen‐specific IgE in total IgE, and theconcentration of allergens in the body fluids the inflammatory cells encounter.
7.2. IgE Concentration versus IgE Occupancy of FceRI
The density of allergen‐specific IgE‐occupied FceRI on mast cells will bereduced , if t he total density of Fc e RI is reduced . Figure 6 exami nes thepercentages of FceRI on a mast cell that are occupied by IgE over a broadrange (5 logs) of IgE concentrations, from 0.1 to 10,000 IU/ml. This analysisindicates that in almost the entire range of IgE concentration present in thehuman population, the FceRI are essentially all occupied. Only at the extremelylow range of IgE levels, the percentage of FceRI occupancy is reducedsignificantly: at 3 IU/ml of IgE, the proportion of occupied FceRI is about25%; at 1 IU/ml of IgE, the occupancy of FceRI is about 10%. This suggeststhat for patients with baseline IgE levels higher than the average 300 IU/ml ofasthmatic patients, IgE levels must be reduced to more than 99% for therecepto r occup ancy to fall below 25%. In Secti on 8, the stabilit y of IgE ‐occupied and ‐unoccupied FceRI and the turnover of FceRI will be discussed.

100%
75%
50%
25%
0%0.1 1 10 100
100
50
150
0
200
1000 10,000
lgE concentration (lU/ml)
Rec
epto
r oc
cupa
ncy
Num
ber
of o
ccup
ied
rece
ptor
per
cell
(�10
−3)
Figure 6 The relationship between IgE concentration and occupancy rate of FceRI on basophils.The calculation is based on Kd of 1 � 10–10 M for anti‐IgE’s binding to IgE and 220,000 FceRI(receptors) per basophil.
90 TSE WEN CHANG ET AL .
8. Downregulation of Fc«RI
8.1. The Dynamical Relationship Between Free IgE and FceRI
The above analysis that IgE must be reduced to >99% in order for FceRI onmast cells and basophils to become significantly unoccupied is based on steadystate kinetic properties, reflecting the concentration of IgE and the density ofFceRI and the interaction between them. However, on the surface of livingmast cells and basophils, the presence of IgE‐unoccupied and ‐occupiedFceRI is highly dynamic and regulated, and not merely dictated by thechemical kinetics between IgE and FceRI.As discusse d in Secti on 7.1, the density of Fce RI on basophi ls is relate d to
the conce ntration of IgE in the blood (Mal veaux et al ., 1978). The me ticulousresearch carried out by the groups of MacGlashan, Casale, and others on theeffects of anti‐IgE on downregulating FceRI on basophils (Lin et al., 2004;MacGlashan et al., 1997 ), mast ce lls (Beck et al ., 2004), and dendritic cells(Prussin et al., 2003) ha s pro vided in sight into the dy namical relationsh ipbetween IgE and FceRI. New molecules of FceRI are being synthesized androuted to the surface by basophils (used in most studies), and FceRI moleculeson the cell surface are rerouted back (internalized) and degraded. On the cellsurface, FceRI are occupied by IgE according to the kinetics of binding (Kd,kon, and koff) between IgE and FceRI. Statistically, when the IgE concentrationis not limiting, a bare FceRI is occupied by IgE swiftly and does not stay

THERAPEUTIC ANTI- IgE ANTIBODIES 91
unoccupied for a long time. When the IgE concentration becomes limiting, abare FceRI, which is freshly synthesized and routed to the cell surface or haslost its bound IgE through thermodynamic process, may not be reoccupied byIgE for a long time (MacGlashan et al., 1998, 1999).
As a part of the mechanism evolved to regulate the concentration of FceRIon basophils, IgE‐occupied FceRI are stable and maintained on the surface.FceRI that are not occupied by IgE are structurally unstable and recognizedand inter nalized for degradati on (Kubo et al ., 2001). Thus, as fr ee IgE isreduced to under a certain concentration, the replenishment of newly synthe-sized FceRI does not compensate for the degradation of FceRI, resulting in agradual loss of FceRI on the cell surface, until a balance between replenishmentand degradation is reached.
The kinetics of maintaining FceRI on basophils is further compounded bythe fact that basophils have a life span of 1�2 weeks. As old basophils die, allFceRI are degraded; as new basophils are generated via the differentiation ofprecursor cells, FceRI are synthesized. However, if IgE concentration is verylow, the synthesized FceRI on the newly generated basophils are not occupiedand are degraded rapidly, leaving a low number of FceRI on the cell surface(Mac Glashan, 2004). Unlik e basophi ls, mast cells have a longer life span ofsever al weeks to several months (Fodi nger et al., 1 994), hence the renewalprocess of these cells plays a lesser factor in the kinetics of FceRI maintenanceon the cells (Bork owski et al ., 2001).
8.2. Anti‐IgE as a Mast ‘‘Cell‐Stabilizing’’ Agent
A subcutaneous injected anti‐IgE antibody (humanized IgG1) should diffuseinto the vasculature and get dissipated via the blood circulation and distributedin various tissues in the body within a few days. On the basis of the high affinityof anti‐IgE for binding to IgE, if anti‐IgE is provided in large excess over thefree IgE in a person, the free IgE will be reduced to more than 99% in hoursor within 1� 2 days (Co rne et al., 1997). Such a depletion of f ree Ig E shouldinitiate the gradual loss of FceRI on basophils and mast cells via the molecularand cellular dyna mics discusse d in Section 8.1. Fc e RI on basophils are report -edly reduced by 70% in 2 weeks and by more than 97% in 3 months (Lin et al.,2004; MacGlashan et al., 1997). The downregulation of FceRI on mast cellsfollows slower kinetics. Nonetheless, mast cells in patients treated with anti‐IgE gradually lose their sensitivity to allergen stimulation, as much higheramoun ts of allerge n are requ ired to in duce a positive skin prick test (Becket al ., 2004).
In the IgE‐mediated allergic pathway, mast cells are the major source ofpharmacological mediators in various allergic responses, perhaps most vividly

92 TSE WEN CHANG ET AL .
displayed in the nasal linings, the lower airway, conjunctiva, and skin, as observedin allergic rhinitis, asthma, and atopic dermatitis. Thus, a general hyposensitiza-tion ofmast cells by ‘‘mast cell‐stabilizing’’ agents (a somewhat imprecise term, forit assumes that the mast cells in the patients are unstable) has been an attractivetherapeutic approach for decades. Since as early as in the 1970s, a class ofmolecules, known as cromones, including disodium cromoglycogate and sodiumnedocromil, has been used clinically and being investigated to treat asthma,allergic rhinitis, and allergic conjunctivitis. Cromones have been shown to beable to retard the processes of exocytosis, degranulation, and lipid mediatorsynthesis induced by allergen‐initiated, FceRI‐mediated cascades (Edwards,2005; Storms and Kaliner, 2005; van Cauwenberge et al., 2000).In a rough sense, the mast cells in an allergic patient are either too sensitive
or undesirably too ‘‘potent.’’ Anti‐IgE, by depleting IgE, indirectly causes theloss of FceRI on these mast cells and renders them insensitive to stimulationby allergens. Cromones, which are generally mild in action, work by modulat-ing the lipid membrane and slowing Ca2þ mobilization, which is requiredfor the exocytotic process. Thus, cromones make mast cells impotent, whileanti ‐Ig E rende rs them insensi tive (Chang and Shiung , 200 6).
8.3. How Low Should FceRI‐IgE Fall for a Mast Cell to Become Insensitive?
In relating the density of allergen‐specific IgE‐occupied FceRI to the sensitiv-ity of mast cells, one inevitably encounters the question whether there is athreshold of density of such receptors, which could be expressed in terms of acertain number per cell. In reality, there is no specific number, because thecells are exposed to a horde of factors, all of which contribute in large or smallparts to determining the threshold level of allergen‐specific IgE‐occupiedFce RI of a mast cell. On close r exami nation, among the vario us factors , theconcentratio n of allergen s in the fluid surrou nding the mast cell (Section 7. 1) isprobably a dominant one. This factor is obviously highly variable amongpatients, as they are sensitized to different sets of allergens by differentdegrees. The amounts of allergens in the air or food also vary at differenttime points. Furthermore, the mast cells located in different mucosal areas inthe body also receive different amounts of allergens.The quantification of threshold densities of allergen‐specific IgE‐occupied
receptors has been approached in cell culture using a hapten‐specific mono-specific IgE. In the estimation, when the concentration of allergens was notlimited, a minimal number of 100�200 FceRI–IgE complexes would beenough to sensitize a basophil (Maeyama et al., 1986; Posner et al., 2002). Ifallergen‐specific IgE accounts for 10% of the total IgE, aminimal of 1000�2000FceRI molecules would be sufficient for the basophil to be sensitive to allergen

THERAPEUTIC ANTI- IgE ANTIBODIES 93
stimulat ion (Mac Glasha n et al., 1997). Thi s w ould suggest tha t unde r theparticular set of conditions, in which allergen‐specific IgE accounts for 10%of total IgE and allergen concentration is not limiting, the total IgE concen-tration in the particular patient must be reduced to below 0.8 IU/ml (based onFig. 5). In an asth matic patien t with an average plasma Ig E concen tration of300 IU/ml, this represents a 99.7% reduction of total IgE. It should be notedthat this analysis accounts for steady state kinetics only, without consideringthe dynamic FceRI regulation on the cell.
The above data might lead to the notion that the FceRI density must fall tovery low levels for the basophils and mast cells to lose sensitivity in the patientstreated with anti‐IgE. This notion may not be correct, because when theproportion of allergen‐specific IgE in total IgE is small and the concentrationof allergens in the blood or particular mucosal areas is limiting, the density ofallergen‐specific IgE‐charged FceRI on the basophils or mast cells may benear a threshold level, even though the total FceRI on these effector cells havenot fallen to drastically low levels.
9. Potential Beneficial Effects of IgE:Anti‐IgE Immune Complexes
The precipitous drop of FceRI on basophils and mast cells has provided aplausible, convincing explanation for the pharmacological effect of anti‐IgE inimproving IgE‐mediated allergic symptoms. Indeed, as basophils and mastcells are rendered insensitive to allergen stimulation, the discharge of media-tors by these cells on exposure to allergens will be retarded and hencemanifestation of allergic symptoms greatly diminished. Can this FceRI down-regulation effect provides the whole explanation for the pharmacologicalbenefit of anti‐IgE? In Sections 9–11, we will discuss other potential pharma-cological effects of anti‐IgE, which may also contribute to its therapeuticeffects.
9.1. How Soon Can Clinical Improvement Be Observed?
Understanding the kinetics of the course of symptom improvement on anti‐IgE treatment is obviously very important for clinicians to apply such atreatment for allergic patients. It is also important to delineate the variouspharmacological mechanisms that contribute to the therapeutic effects of anti‐IgE. Although a large number of trials have been performed, there has notbeen a detailed analysis on the kinetics of improvement, especially in the initialweeks of anti‐IgE treatment. In the unpublished first Phase II trial of anti‐IgE,namely, the trial of CGP51901 on mountain cedar pollens caused allergicrhinitis in Central Texas in 1994�1995, weekly symptom severity scores were

94 TSE WEN CHANG ET AL .
obtained and plotted. This was possible because the trial design adopted aweekly injection schedule and allowed the study investigators to examine thetrial patients on weekly time points. There was a significant drop in the overallseverity score 1 week after the first injection of anti‐IgE. These results wereconsistent with the observations made in another clinical study on allergicrhinitis, in which patients were found to make reduced response to allergenchallenge within 7� 14 days after the in itial treatme nt with omali zumab (Li net al ., 200 4).A detailed analysis on how soon clinical symptoms improved in asthmatic
patients treated with anti‐IgE was also performed, although the trial designprovided mostly 4‐weekly data. It was found that among the patients whoeventually responded to omalizumab, 61% in 4 weeks and 87% in 12 weeksshowed significan t improveme nt (Bousq uet et al., 2004). In the case report ofthree patients with rec alcitrant atop ic derma titis (Section 4.4), the pat ientsresponded favora bly within 2 weeks of omali zumab treatme nt (La ne et al.,2006). In the case report of one patient with inter stitial cystit is (Section 4.4),the pa tient imp roved imme diately after the firs t anti ‐ IgE injection (Lee et al.,2006). In the case study of one pa tient with cold ‐in duced urticari a (Section4.4), the patien t improve d afte r the secon d bi weekly anti ‐ IgE injection (Boyce,2006).It is possible that the molecular and cellular pharmacological mechanisms of
anti‐IgE progress in a similar time frame in patients with allergic rhinitis,allergic asthma, or other IgE‐mediated allergic diseases. However, the broaderinvolvement of inflammation and tissue damages in modest‐to‐severe asthmamay take longer time to heal and to become clinically evident.It was fou nd in the study by Lin et al . (2004) tha t 1 week afte r the subcuta -
neous injection of omalizumab, the basophils in the treated allergic rhinitispatients, who were sensitive to ragweed pollens, lost about 70% of the FceRIon cell surface. It is conceivable that the mast cells in the nasal lining of thesepatients should not have lost as much FceRI as basophils. However, a nasalantigen challenge test indicated that the allergic response in the nasal lininghad already been attenuated at day 7, suggesting that the mast cells in thearea had already been attenuated.Could the loss of less than 70% of FceRI on mast cells render those cells
insensitive? One would reason that the observed 70% loss of FceRI should notbe sufficient for the mast cells in the nasal lining of those patients to losesensitivity to respond to inhaled allergens. The counts of pollen particles, suchas those of ragweed and birch, per unit volume per day of air may rise 5�10times in a 1‐week period during the pollen season of a plant type (informationfrom daily pollen counts reported in metropolitan areas). If an allergic rhinitispatient already responds to the specific pollens and is symptomatic in the

THERAPEUTIC ANTI- IgE ANTIBODIES 95
beginn ing of the week, and fails to respond to the polle ns at 5 � 10 time s theconcen tration at the end of the week due to a treatme nt, the 70% dro p in Fc e RIon those mast cells is not the likely me chanism accoun ting f or the aforeme n-tione d attenua ted allerg ic response . Chang (2000) prese nted a rational hypo th-esis tha t other phar macolog ical me chanisms con tribute to the thera peuticeffec ts of anti ‐ IgE, espe cially in the ea rly stag es of the treatmen t.
9.2. The Rapid ly Accumu lated Immu ne Complex es May Ser ve asAntige n Trapp ers
The half ‐ life of IgE in huma ns is 1� 2 days, that o f anti ‐ IgE (a huma n IgG1)abou t 21 days, and that of anti ‐ IgE:IgE comp lexes ab out 20 da ys (Fi g. 2; Foxet al., 1996; Lanier, 2003 ). The ad ministra tion of anti ‐ IgE ei ther subc utane-ously or intraven ously bri ngs free IgE to near zero con centrations within a fewdays. How ever, the IgE ‐sec reting plasma cells, whi ch are not targ ets of anti ‐IgE an d have a life span of sever al month s or longer, cont inue to sec rete IgE(Section 10.1). This causes a rap id accumula tion of anti ‐Ig E:IgE immu necomplexes to 5–10 times the baseline levels of IgE within a week (Corneet al., 1997; Milgrom et al., 2001). One IgE molecule has two antigenic sitesfor anti‐IgE and can be bound by two anti‐IgE molecules at the same time; oneanti‐IgE molecule has two antigen‐binding arms and can bind to two IgEmolecules at the same time. It is hence peculiar (and scientifically fascinating)that anti‐IgE and IgE form small complexes both in the test tubes and in theblood of patients treated with anti‐IgE, with the largest being a hexamercomple x, f ormed by three anti ‐ IgE an d three IgE mole cules (Liu et al .,1995). The an ti ‐Ig E:IgE comp lexes are soluble and do not precip itate in thekidney and do not cau se immune comp lex pro blems (Fox et al ., 1996).
Both anti‐IgE (an IgG) and IgE are freely diffusible across the vascularcapillaries and should equilibrate between the vascular and extravascularspaces. The immune complexes are stable, owing to the high binding affinityof anti‐IgE for IgE, cannot cross blood capillaries, and should remain in theblood circulation or local tissue sites, where they are formed. Thus, the anti‐IgE:IgE immune complexes will be accumulated to high concentrations in theblood or in local tissue sites such as in the mucosal linings of the nasal passageand the lower airway.
The IgE comprising the immune complexes have their antigen‐binding sitesavailable for binding to antigens. Because the IgE can no longer bind to FceRI,it should function as a blocking, protective IgG. In this way, anti‐IgE convertspotentially adverse, effector cell‐sensitizing IgE molecules into beneficial,antigen‐blocking IgE species. Thus, the rapidly accumulating immune com-plexes probably can serve as antigen‐sweeping agents. As the allergenmolecules

96 TSE WEN CHANG ET AL .
break through the mucosal epithelia and get into the interstitial fluid onthe basal side, they are bound by the immune complexes, before reaching toIgE bound by FceRI on mast cells residing in the area. While direct experimen-tal evidence has yet to be obtained, it is a rational hypothesis that anti‐IgE:IgEimmune complexes should contribute to the therapeutic effects of anti‐IgEwithin the first and second weeks after the first anti‐IgE injection. It remainsa curious question as to whether on anti‐IgE treatment patients with high IgElevels fare better in the first 1 or 2 weeks than patients with much lowerIgE levels.In the off‐label study of omalizumab on three patients with atopic dermatitis
(Krathe n and Hsu , 20 05) discu ssed in Secti on 4.4, one of the patien ts hadconcomitant severe asthma and a baseline IgE level of 24,400 IU/ml.A regimen of 450 mg of omalizumab every 2 weeks for 4 months did notrelieve her atopic dermatitis, but her asthma was significantly improved afterstarting omalizumab. It was estimated that the patient had a total of �290‐mgIgE in her body and synthesized �145‐mg IgE every 1�2 days. Clearly, dosingof anti‐IgE at 450 mg every other week cannot neutralize all of the IgE in herbody. Also, even a 99.9% drop in total basal IgE would still leave 24 IU/ml,which is sufficient to charge and maintain normal levels of FceRI on basophilsand mast cells. These numbers suggest that the patient’s asthma was improvedby mechanisms other than the depletion of IgE and the downregulation ofFceRI. Perhaps, the effect of immune complexes is one of those mechanisms.The immune complexes may possibly function in another aspect. The
trapping of incoming allergens by the immune complexes should prevent theallergens from interacting with the mIgE expressed on mIgE‐committedB lymphoblasts and memory B cells. This neutralization of the antigen shouldinhibit allergen‐driven, IgE‐destined immune response (Hamelmann et al.,1997; Takhar et al., 2005) and hence have long‐term attenuating effects onallergic response.
10. Can Anti‐IgE Modulate IgE‐Committed B Lymphoblasts andMemory B Cell?
One of the most intriguing issues that remain to be addressed definitively iswhether anti‐IgE can modulate mIgE‐expressing B cells. This question is ofgreat interest, because if anti‐IgE can inhibit mIgE‐committed B memory cellsor B lymphoblasts, the generation of allergen‐specific IgE that is induced bythe new exposure to allergens should be intercepted, resulting in a profoundattenuation of the allergic pathway. Several lines of in vitro and animal modelstudies amply support that anti‐IgE can inhibit IgE production by B cells andcause the lysis of IgE‐expressing B cells (Davis et al., 1993; Haak‐Frendscho

THERAPEUTIC ANTI- IgE ANTIBODIES 97
et al., 1994). In a cell culture system, where human peripheral mononuclearcells were driven by IL‐4 and other factors to increase IgE‐committed B cells,so that the e chain mRNA expression and IgE production became measurable,anti‐IgE (CGP51901) inhibited the synthesis of e chain mRNA and IgE. In anin vivo mouse model, in which BALB/c mice were transplanted with cells ofSE44 cell lin e (Sun et al., 1991), which was an NS0 myeloma that had beentransfected with human e chain DNA and expressed human mIgE on surface,TESC ‐ 21 (mouse M Ab an ti‐ IgE, see Secti on 1.1) stopped SE44 tumor growth(Davis et al., 1993). Studie s by oth er gro ups also showed in mice that endog e-nously produced syngeneic anti‐IgE antibodies could suppress IgE synthesisand decreased IgE‐secreting plaques, probably by eliminating IgE‐expressingB ce lls before they matu red to plasma cells (Haba and Nisonoff, 1994).In contrast to the abundant results obtained in animal model and in vitrostudies, similar potential effects of anti‐IgE have not been revealed resolutelyin human patients.
That mIgE is part of the B cell receptor on IgE‐committed B cells explainsthe in vitro and animal model results. Studies with IgM‐expressing B cells alsodemonstrate that anti‐IgM antibodies, in the absence of costimulatory factors,cause apoptosis (Chan et al., 1990; Mayumi et al., 1996) and mediatedantibod y ‐ dependen t cell cytoto xicity (Walke r et al., 1985) and comp lement ‐mediated cell lysis of the IgM‐expressing B cells (Caraux and Weigle, 1983a,b).The possible decrease of IgE synthesis has been difficult to observe in vivo inhumans because the long‐living IgE‐secreting plasma cells, which express verylow levels of mIgE and are not targets of anti‐IgE, in the bone marrow andother lymph oid tissue s continu e to secrete IgE (Shap iro ‐She lef and Calame ,2005). Thus, ev en if the generatio n of allerge n‐ specifi c mIgE ‐ expressing B cellsand IgE‐secreting plasma cells is interrupted, the effect is masked by theproduction of total IgE.
The inhibition or downregulation of mIgE‐expressing B cells by anti‐IgE issuggestive in some observations. In the case report of an interstitial cysti-tis patien t treated with ant i‐ IgE (Lee et al., 2006; Secti on 4.4), the initialbaseline total serum IgE concentration was 653 IU/ml. Seven months afterthe anti‐IgE treatment, the total IgE (the sum of IgE in immune complexesand free IgE) decreased to 168 IU/ml. This drop of IgE suggests that existingIgE‐secreting plasma cells were gradually dying off and new plasma cells werenot replenished. In the clinical study of anti‐IgE on 225 pediatric patients withasthma, it was found that total IgE increased to multiples of baseline levelsand then gradua lly decl ined afte r 1 6 weeks of anti ‐ IgE treatmen t (B erger et al .,2003). In anoth er trial, a significant pro portion of patient s had IgE levels afte r6 months of treatment lower than their baseline IgE levels before omaliz-umab (Lan ier, 2006). These data indicate that at least in some pa tients, the

98 TSE WEN CHANG ET AL .
generatio n of new IgE ‐ secreting plasma cells appears to be inhi bited (Infu hret al ., 200 5).
11. Other Immunoregulatory Effects of Anti‐IgE
11.1. Anti‐IgE Should Neutralize the Cytokinergic Properties of IgE
IgE is one of the five classes of antibodies. Its overall structure is very similar tothat of antibodies from other classes and its antigen‐binding domains are fromthe pool of variable regions shared by other classes of antibodies. Its produc-tion is induced by exposure to antigens, albeit preferentially by parasitic worms(Capron et al., 1987) an d a wide range of envir onmenta l an tigens. T he immu-nological mechanisms of IgE leading to the protective and pathological effectsare initiated and aggravated by its interaction with the respective antigens.Thus, in almost all regards, IgE is a bona fide antibody class that has evolved tobear a set of immune defense functions.Aside from the ability to interact with antigens for initiating immunological
processes, IgE has a characteristic that is strikingly different from those of theother antibody classes—it can bind to its receptor (FceRI) with very high affinityby itself without prior engagement with its antigen (Metzger et al., 1986). Thisbinding to the receptor initiates an array of effects on mast cells without antigenparticipation, thus enabling IgE to function like a cytokine‐like substance andexhibit the following cytokinergic properties. Studies by several research groupshave shown that IgE alone can initiate FceRI‐mediated signal‐transducing cas-cade (Kawakami and Kitaura, 2005), leading to Ca2þ influx (Lam et al., 2003;Pandey et al., 2004; Tanaka et al., 2005), histamine release, leukotriene synthesisand release, and synthesis of IL‐6 and other cytokines (Kalesnikoff et al., 2001;Kohno et al., 2005), although such activities are much less intense than thoseinduced by allergens and IgE combined. As for the effects on the survival (Asaiet al., 2001; Kalesnikoff et al., 2001; Kawakami andGalli, 2002), histamine content(Tanaka et al ., 2002), and migratory activity of mast cells (Kitaura et al., 2005),IgE’s effect is no less than that of IgE‐allergen complexes. Thus, IgE by itselfpotentiates mast cells in inflammatory processes (Kawakami and Kitaura, 2005).Monoclonal IgE antibodies have been shown to be either highly or poorly
cytokinergic. The difference appears to lie in the variable domains (Foote, 2003;James et al., 2003) and the molecular mechanisms concerning such a bafflingdifference remain to be elucidated. The native polyclonal IgE as a whole in ahuman body resembles highly cytokinergic IgE (Kitaura et al ., 2003). By virtue ofits ability to bind to IgE and block IgE binding to FceRI, anti‐IgE can impede thevarious cytokinergic activities of IgE, damping the inflammatory makeup in theimmune system.

THERAPEUTIC ANTI- IgE ANTIBODIES 99
11.2. The Overall Attenuation of Immune Reactivity
The IgE‐mediated allergic pathway, which generates mediators manifestingallergic symptoms, flames up the immune activities broadly in the local areasor systematically. The wide spectrum of cytokines, such as TNF‐a, inter-feron‐a, IL‐4, and IL‐5, and various chemokines, secreted by mast cells andbasophils are inflammatory in nature. These factors not only cause symptomsdirectly but also activate and recruit various cell types augmenting the inflam-matory state. Anti‐IgE, by tying up IgE, eventually attenuates the IgE‐mediated pathway and hence the inflammatory conditions. Such an inhibitoryeffect can even be observed by the overall decrease of T and B cells in theimmune system (Holgate et al., 2005b; Noga et al., 2003; Ong et al., 2005).
CD23 on cell surface and soluble CD23 play multiple roles in regulatingIgE synthesis and in inducing immune response to allergens. IgE:allergencomplexes bind to membrane CD23 (via Fc of IgE) and facilitate antigenpresentation (Getahun et al., 2005; Kilmon et al., 2004). Simultaneous bindingof soluble CD23 to mIgE and CD21 on B cells will lead to the enhancement ofIgE production (Aubry et al., 1992; Reljic et al., 1997). It has been found thatin the gastrointestinal tract, CD23 on mucosal epithelial cells can transportIgE from the basal side to the lumen through tran scytosis (Tu et al., 2005).This IgE in the lumen binds to food‐derived antigens and facilitates its entryacross the mucosal epitheli al layer (Li et al., 2006). Anti ‐Ig E block s Ig Ebinding to CD23 and hence inhibits many of these processes.
The depletion of free IgE by anti‐IgE has a profound, fast effect on down-regula ting Fc e RI on dend ritic cells (Prus sin et al., 2003). Seven days afte radministrating anti‐IgE, the FceRI on both subsets of precursor dendritic cellswere already decreased significantly (estimated 30–50%). This should inhibitthe an tigen ‐ presenting pro cess going t hrough IgE an d dendritic cells ( Kraftet al., 2001; Novak et al., 2003b).
11.3. Local Environment in Disease‐Affected Tissues is of Utmost Interest
Much of our understanding on the dynamic interactions between IgE andanti‐IgE has been derived from studies analyzing such interactions in theblood. However, mast cells, which are key players in allergic rhinitis, asthma,dermatitis, reside in extravascular space in disease‐affected tissues (Brody andMetcalfe, 1998; Liu, 1997). It is believed that mast cells establish residency inspecifi c locat ions and develo p diff erent sets of ch aracteristics (Church andLevi ‐ Schaffer, 1997). It is conce ivable that mast cells residing in the nasallining, lower airway, other areas of the mucosal tracts, and in the skin, differ intryptase and chymase content, sensitivity, receptor regulation, and life span(Chang and Shiung, 2006; Galli and Hammel, 1994; Irani and Schwartz, 1994;

100 TSE WEN CHANG ET AL .
Lowman et al., 1988; Miller and Schwartz, 1989; Peng et al., 2003). Interest-ingly, clinicians participating in the anti‐IgE clinical trials observed that thesymptoms of allergic rhinitis or allergic asthma were improved much soonerthan skin pri ck test reactiv ity (Be ck et al., 2004). Are the mast ce lls in theairway and na sal mu cosal linin gs diffe rent from tho se in the skin (Pawankaret al ., 199 7)?The local microenvironment in the allergic disease‐affected tissues is of
great interest, awaiting vigorous investigation. Airborne allergen particles aretrapped at the mucosal surfaces in the nasal lining and the bronchial tracts andthe allergen molecules contained therein find ways to get through the mucosalepithelia to the basal side. Here the allergen molecules interact with manycell types, including mast cells, dendritic cells, and probably IgE‐ and IgG‐committed lymphoblasts and memory B cells. It is now known that allergen‐specific IgE‐secreting plasma cells in the local mucosa exist (KleinJan et al.,2000; Smurthwaite et al., 2001) and that allergen‐specific B cells are driven byallergens to switch to IgE‐expressing B cells and to undergo affinity maturation(class switch recombination) in situ in the mucosa (Coker et al., 2003; Tanakaet al., 2005; Wilson et al., 2002). The IgE secreted by those plasma cells in thelocal area can sensitize mast cells in the same site.On subcutaneous injection of anti‐IgE, the anti‐IgE molecules diffuse into
the vascular space and are dissipated by the blood circulation. They diffuseinto the local microenvironment of the affected tissues and bind to free IgE,forming immune complexes, which can no longer diffuse into the vascularspace. Because the locally synthesized IgE may be rich in allergen‐specificIgE, the accumulated immune complexes should contain IgE that can helpcapture the incoming allerge ns (Section 9.2).
12. Can Anti‐IgE Attain a Long‐Term Remission State?
The value and impact of the anti‐IgE therapy will be greatly expanded, if it canachieve a long‐term remission state at least in some of the treated patients. Theclinical trials performed so far have not incorporated a study segment toaddress this aspect for a few considerations. A major concern among officialsat governmental regulatory agencies and researchers developing the anti‐IgEprogram during the early phase of the clinical development of anti‐IgE wasthat the effect of anti‐IgE in depleting IgE is not reversible. Since the initialconcept was to use anti‐IgE to target or deplete IgE‐expressing B cells andsince IgE was believed to be required for certain immune defense functionssuch a concern was logical. Now that the depletion of IgE for 1 or 2 years isknown not to pose great risks (Ayres et al., 2004; Berger et al., 2003; Finn et al.,2003; Lanier et al., 2003), we should address whether anti‐IgE can (1) have

THERAPEUTIC ANTI- IgE ANTIBODIES 101
long‐term effects on the functions of IgE and (2) attain long‐term remissionstate in the treated patients.
Information concerning whether anti‐IgE can attain a long‐term remissionstate originates from anecdotal, nonsystematic observations made by clinicianstreating asthma patients. Clearly, some patients appeared to have achievedremission state. However, there are no statistical data on the proportion ofcases and the duration of such remission states. There is no information as towhat makes an anti‐IgE treatment to achieve a remission state. Nonetheless,anti‐IgE, when used in chronic protracting allergic asthma, helps tame aninflammatory state and tune‐up or rebuild a healthy airway.
If anti‐IgE can drive toward a remission state, it probably causes a shift inthe balance between IgE‐mediated and non‐IgE‐mediated responses. Manypharmacological mechanisms of anti‐IgE should contribute to such a shift.Among them, potentially the most potent one is the modulation and down-regula tion of Ig E‐ commi tted B cells by anti ‐ IgE, as di scussed in Secti on 10. Ina patient, the generation of IgE‐committed allergen‐specific memory B cells isan important end result of the sensitization process that may take as long as afew years (Weissman an d Le wis, 2002). Thus, if an ti‐ Ig E can downregulat eor eliminate IgE‐committed B memory cells, it should have very profound,long‐term attenuating effects on the state of sensitivity against allergens.
When a patient is under anti‐IgE treatment, the IgE‐mediated allergicpathway is eventually blocked off and the numerous IgE‐related activitiesdiscussed in Sections 9–11 inhibited. Since the patient is exposed to allergensas usual, the allergens should induce other immune responses, such as protec-tive IgG, against the allergens. These responses should gradually drive a shiftin the balance between IgE versus non‐IgE‐related responses toward the non‐IgE compartments. When a substantial shift is created, the patient shouldachieve a remission state. Along this line of rational ization (Chang, 2000), sincethe shift to favorable non‐IgE‐related responses may be induced by allergen‐based immu nothera py (SIT or RIT, di scussed in Secti on 5), a combinati on ofanti‐IgE with SIT or RIT may help patients achieve long‐term remission stateor even cure more effectively and safely than with SIT or RIT alone.
13. Are Ther e Ad verse Effect s Associ ated with Ant i‐IgE The rapy?
13.1. Is Immune Defense Function Compromised?
It is a rational assumption that the IgE antibody class had evolved in a branchof the vertebra te specie s for immun e defense (Warr et al., 199 5). While thereis not a large body of evidence in the literature supporting IgE’s roles inimmune defense, a significant number of papers suffice to indicate that IgEcontributes in part to the defense of various infectious agents, especially

102 TSE WEN CHANG ET AL .
parasitic worms, in many animal species. Because immunity against infectiousagents is critical for the survival of many animal species, the immune systemhas evolved to be highly redundant, so that multiple immune mechanismscorrobora te to defend against an invading infectiou s pathogen (Litman et al.,2005). Thus, IgE is probabl y still essentia l for animal s and even humans livingin primitive habitats. However, for humans living in many regions of the worldtoday, or for pets, laboratory animals, even live stocks housed in relativelyclean, confi ned facilities, IgE appear s to have become nonessen tial (La nierand Chang, 2004). Thi s unde rstanding is supported by the follow ing resea rchfindings.The roles of IgE in immune defense, especially against parasitic worms,
have been studied in various types of laboratory mice, including wild‐type onesproducing normal ranges of IgE and biologically or genetically manipulatedones, which are devoid of either IgE or IgE‐mediated effector functions. Insome studies, wild‐type mice were treated with anti‐IL‐4 antibody to abolishtheir ability to produce any IgE. In other studies (Madden et al., 1991; Sheret al., 1990), two strains of mice, which failed to produce any IgE, wereemployed: in one strain the gene for the IgE e chain was knocked out (Gurishet al., 2004; King et al., 1997), while in the other strain, the IL‐4 gene wasimpaired (El Ridi et al., 1998; Watanabe et al., 1988, 1993). Other studies usedanother strain of mice, in which the gene of the FceRIa subunit was knockedout (Jankovi c et al ., 1997), and IgE could not sensi tize the mast cells an dbasophils in them. In another set of experiments, mice were treated withpolyclona l antibod ies tha t neutral ize IgE (Am iri et al., 1994).Studies have been performed by a number of groups to investigate whether
these mice incapable of producing IgE or devoid of FceRI were weakened intheir ability to defend the challenges of various parasites, including Schistoso-ma masoni, Nippostrongylus brasiliensis, and Trichinella spiralis. The resultsas a whole were inconsistent and failed to prove that a lack of IgE pathwaysimpairs the ability to eliminate parasites. One of the anti‐IgE clinical trialson asthma, which was carried out in Brazil, had monitored parasite infectionas part of the study protocol. No increased rate of parasite infection wasobserved.The question, whether IgE is involved in the immune surveillance of malig-
nantly transformed cells and whether anti‐IgE compromises such a function,had been raised. On the basis of the numbers of malignancies identified duringthe anti‐IgE treatment period, it was cautioned that an apparent increase ofmalignancy in cidence s was found in the anti ‐ IgE ‐ treated popula tion (Strun kand Bloomb erg, 2006). Si nce the overall rate was within the range fou nd in thegeneral population, and the malignancies found in these mostly older allergypatients were of heterogeneous tissue origins, governmental regulatory agencies

THERAPEUTIC ANTI- IgE ANTIBODIES 103
did not sound an alarm for such a concern, but required that a multiyearpostmarket follow‐up be performed.
13.2. Observed Adverse Reactions
Aside from the concern that anti‐IgEmay compromise certain immune functions,anti‐IgE is well tolerated and causes relatively few serious adverse effects (Strunkand Bloomberg, 2006). The various symptoms of discomfort, such as fever,headache, injection site rashes, are common for a subcutaneous injectable andfor an antibody drug, since the rates of these complications were similar inpatients receiving anti‐IgE treatment or placebo. There have been a few casesof anaphylactic reactions among patients with very sensitive disposition andcomplex, multiple diseases. Anti‐IgE may still benefit those highly sensitivepatients, but caution must be taken while administering the drug (Dreyfus andRandolph, 2006). There has been no report of immune complex diseases or serumsickness disease. Omalizumab was found not to induce antibody response againstitself.
14. Other Approaches for Targeting IgE or IgE‐Expressing B Cells
14.1. Approaches for Attenuating IgE‐Mediated Allergic Pathway
Various therapeutic approaches have been developed to modulate the immunesystem to inhibit IgE synthesis, to inhibit TH2 response, or to drive a shiftfrom TH2 to TH1 response (St okes an d Casa le, 2004). These includ e anti ‐ IL ‐ 4,anti‐IL‐5, and IL‐4 and IL‐5 receptor antagonists (Barnes, 2002; Kips et al.,2001; Yamagata and Ichinose, 2006). These immune modulators appear to bevery attractive agents for attenuating the IgE‐mediated pathway and of theTH2 arm. However, the results from clinical studies are not satisfactory interms of their abilities to alleviate clinical symptoms. Anti‐CD23 has also beenstudied in human clinical studies and been shown to downregulate blood IgElevels by 50% (Nakamura et al., 2000; Rosenwasser et al., 2003). The initialassessment was that anti‐CD23 probably cannot improve clinical symptoms(Poole et al., 2005).
The ineffectiveness of these various approaches may be due to their inabilityto downregulate IgE to a great extent. On the basis of the results of anti‐IgE onneutralizing IgE and on downregulating FceRI on basophils and mast cells, formost patients with IgE concentrations above 10 IU/ml, if the IgE concentra-tion is not reduced by more than 95% or 99%, downregulation of FceRI wouldnot result. Since anti‐IL‐4‐treated and IL‐4 gene knocked out mice do notproduce any IgE (Watanab e et al., 1988), why can anti ‐ IL‐ 4 an d solub le IL ‐ 4

104 TSE WEN CHANG ET AL .
receptor not lower IgE to near zero levels (Borish et al., 2001; Hart et al.,2002)? One plausible explanation is the fact that IL‐4 acts at very low con-centrations in short di stances in the lymph oid microen vironmen t (de Vrieset al., 1993) and therefore , it requires ve ry large amou nts of an ti‐ IL ‐ 4 to flushthe local microenvironments.
14.2. An Approach to Target a Unique Epitope on mIgE
The many molecular attributes of anti‐IgE, such as having a long half‐life,forming small immune complexes, not inducing antibody response, and multi-ple proven and potential pharmacological mechanisms, are very difficult toattain with another therapeutic. However, anti‐IgE is expensive and willprobably not be afford able for most patie nts with very high IgE (Ames et al. ,2004) such as those with plasma IgE higher than 1000 IU/ml. Therefore, anapproach that can target IgE‐committed B cells directly and inhibit thesynthesis of IgE substan tially will be ve ry desir able (Cha ng et al. , 1990).IgE‐committed B lymphoblasts and memory B cells express mIgE. IgE‐
expressing B lymphoblasts are in the differentiation and maturation process tobecome IgE‐producing plasma B cells. In addition, mIgE is not expressed inother cell types. Thus, mIgE appears to be an ideal target for immunologicalagents that aim at modulating mIgE‐expressing B cells. Our group had made aseemingly unlikely discovery (Peng et al. , 1992) tha t the e chain of huma nmIgE contains a 52‐amino acid peptide segment (referred to as the CemXdomain) between the CH4 domain and the C‐terminal membrane‐anchoringtransmemb rane peptide (Fig. 7). Using mouse me mbrane e chain as a refer-ence, CemX is resulted from an alternative splicing using an acceptor site 156‐base pairs upstream of the previous known site. In human mIgE, e chainswithout CemX, while faintly expressed at the mRNA level, are not detectableat the protein le vel (P eng et al ., 1992).The sequence of CemX shares no significant homology with sequences in
the entire DNA and protein databases. Thus, the uniqueness of CemX hasprovided an attractive site for targeting mIgE and mIgE‐expressing B cells.Because mIgE is part of the B cell receptor, antibodies targeting mIgE shouldactivate signal‐tranducing process, leading to the anergization or apoptoticpathway of those cells, in the absence of costimulators (Donjerkovic andScott, 2000; Gauld et al., 2005; Tighe et al., 1997). Mouse anti‐CemX MAbs,such as a20, have been prepared (Chen et al. , 2002) and are in the proce ss ofbeing humanized in our laboratory. It is possible that anti‐CemX antibodiescan be used in combination with anti‐IgE for treating patients with veryhigh IgE. The potential advantage of an anti‐CemX antibody is that becausethe antibodies are not neutralized by IgE, a much smaller amount of the
mlgE
Anti-lgE omalizumab TNX-901
Anti-CεmXCεmX
GLAGGSAQSQ RAPDRVLCHS GQQQGLPRAA GGSVPHPRCH CGAGRADWPG PP
Plasma membrane
B cellCytoplasm
s s
s s
Figure 7 The location and sequence of CemX domain in mIgE. The antigenic sites targeted byanti‐IgE MAbs, such as omalizumab and TNX‐901, and by anti‐CemX MAbs, such as a20, areindicated.
THERAPEUTIC ANTI- IgE ANTIBODIES 105
antibody will be required for each administration. If IgE‐committed B cells areindeed purged by the anti‐CemX antibody, the new synthesis of allergen‐specific IgE will be interrupted, resulting in a long‐term attenuating effecton the sensitization state of patients.
Perhaps after a decade of use of anti‐IgE, the concern over long‐termdepletion of IgE will subside, and a therapeutic approach for developing avaccine‐like product based on the CemX peptide should become attractive.The CemX peptide can be linked to a T cell reactive foreign peptide and usedas an immunogen to elicit in patients antibodies that, like the passively admi-nistered anti‐CemX antibodies, bind to mIgE and modulate mIgE‐expressingB cells. Such a treatment will not require large amounts of the immunogenproduct and may have long‐term effects on suppressing IgE production.

106 TSE WEN CHANG ET AL .
15. Concluding Remarks
The clinical utility of omalizumab for pediatric asthma, allergic rhinitis, peanutallergy, atopic dermatitis, and others, and for combining with SIT and RITwillprobably take another 5–10 years to develop for most regions of the world. Thetherapeutic efficacy exhibited by omalizumab and TNX‐901 in about 30 PhaseII and III clinical trials amply demonstrates that IgE plays significant roles inthe pathogenesis of not only allergic rhinitis but also allergic asthma, peanutallergy, and probably atopic dermatitis. The clinical trial results also establishthat IgE depletion is a feasible strategy for treating various IgE‐mediatedallergic diseases. Furthermore, the results from anti‐IgE and from other exper-imental drugs, such as anti‐IL‐5 and anti‐CD23, also reveal that the IgEdepletion should be nearly complete and should last for a long term, at leastseveral months.The successful development of anti‐IgE has provided a treatment option for
many patients with difficult‐to‐treat allergic asthma and in the future, anti‐IgEmay also provide a treatment option for patients with other IgE‐mediatedallergic diseases that are severe and difficult to treat. Anti‐IgE will not be anaffordable treatment option for most patients in economically less developedcountries. The yearly requirement of omalizumab for an asthma patient isabout 2–8 g, which represents substantial production costs. Even with suchsizable quantities, they are only suitable for those patients whose serum IgElevels are less than 700 IU/ml. About 10–15% of patients have IgE levels above700 IU/ml. Thus, for economically less fortunate patients and those with highIgE, including most atopic dermatitis patients, a rather different thinking inproviding anti‐IgE or and on the understanding of the diseases must bestimulated. It should be noted that while excluding patients with IgE above700 IU/ml had a rationale basis, excluding patients with IgE below 30 IU/mldid not have sou nd ba sis (Lanier, 2006). As lon g as their allergic disease s areIgE‐mediated, those patients should respond to anti‐IgE well.While many researchers have unraveled multiple intriguing pharmacological
effects of anti‐IgE, we still fall short in elucidating definitively the pharmaco-logical mechanisms responsible for the therapeutic efficacy of anti‐IgE. Whilethe downregulation of FceRI antibodies on basophils and mast cells has beenestablished, numerous other pharmacological mechanisms, as outlined herein(Sections 9–12) have not yet been convincingly validated. These other mechan-isms very likely play important roles, especially in the first few weeks, before thedownregulation of FceRI has reached physiologically significant levels.Among many of the questions that remain to be resolved regarding
the pharmacological effects and therapeutic efficacy of anti‐IgE is whether anti‐IgE can modulate or inhibit IgE‐committed B lymphoblasts and memory B cells.

THERAPEUTIC ANTI- IgE ANTIBODIES 107
Such an effect is critical for anti‐IgE to (1) intercept the new synthesis of allergen‐specific IgE, (2) cause a decisive shift from TH2 to TH1 conditions, and hence(3) achieve a long‐term effect on attenuating IgE‐mediated diseases. In thisaspect, different patients may respond differently. Also, different anti‐IgE anti-bodies, such as omalizumab and TNX‐901, may act differently. In this regard, thedevelopment of an anti‐IgE antibody with an affinity much higher (by say 100‐fold) than omalizumab and TNX‐901 will be of great interest. Thus, whileapplications based on the anti‐IgE concept has been in active development fornearly 20 years, much research is needed to address the many questions posedherein.
Acknowledgments
Supported by grant no. 94–2320‐B007–004 from the National Science Council, Taiwan.
References
Adelroth, E., Rak, S., Haahtela, T., Aasand, G., Rosenhall, L., Zetterstrom, O., Byrne, A., Cham-pain, K., Thirlwell, J., Cioppa, G. D., and Sandstrom, T. (2000). Recombinant humanized mAb‐E25, an anti‐IgE mAb, in birch pollen‐induced seasonal allergic rhinitis. J. Allergy Clin.Immunol. 106, 253–259.
Ames, S. A., Gleeson, C. D., and Kirkpatrick, P. (2004). Omalizumab. Nat. Rev. Drug. Discov. 3,199–200.
Amiri, P., Haak‐Frendscho, M., Robbins, K., McKerrow, J. H., Stewart, T., and Jardieu, P. (1994).Anti‐immunoglobulin E treatment decreases worm burden and egg production in Schistosomamansoni‐infected normal and interferon gamma knockout mice. J. Exp. Med. 180, 43–51.
Asai, K., Kitaura, J., Kawakami, Y., Yamagata, N., Tsai, M., Carbone, D. P., Liu, F. T., Galli, S. J.,and Kawakami, T. (2001). Regulation of mast cell survival by IgE. Immunity 14, 791–800.
Asher, M. I., Montefort, S., Bjorksten, B., Lai, C. K., Strachan, D. P., Weiland, S. K., and Williams,H. (2006). Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinocon-junctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross‐sectional surveys. Lancet 368, 733–743.
Aubry, J. P., Pochon, S., Graber, P., Jansen, K. U., and Bonnefoy, J. Y. (1992). CD21 is a ligand forCD23 and regulates IgE production. Nature 358, 505–507.
Ayres, J. G., Higgins, B., Chilvers, E. R., Ayre, G., Blogg, M., and Fox, H. (2004). Efficacy andtolerability of anti‐immunoglobulin E therapy with omalizumab in patients with poorly con-trolled (moderate‐to‐severe) allergic asthma. Allergy 59, 701–708.
Barnes, P. J. (2002). Cytokine modulators as novel therapies for asthma. Annu. Rev. Pharmacol.Toxicol. 42, 81–98.
Barnes, P. J. (2004). New drugs for asthma. Nat. Rev. Drug Discov. 3, 831–844.Baur, X. (1979). Studies on the specificity of human IgE‐antibodies to the plant proteases papain
and bromelain. Clin. Allergy 9, 451–457.Beck, L. A., and Leung, D. Y. (2000). Allergen sensitization through the skin induces systemic
allergic responses. J. Allergy Clin. Immunol. 106, S258–S263.

108 TSE WEN CHANG ET AL .
Beck, L. A., Marcotte, G. V., MacGlashan, D., Togias, A., and Saini, S. (2004). Omalizumab‐induced reductions in mast cell Fce psilon RI expression and function. J. Allergy Clin. Immunol.114, 527–530.
Berger, W., Gupta, N., McAlary, M., and Fowler‐Taylor, A. (2003). Evaluation of long‐term safetyof the anti‐IgE antibody, omalizumab, in children with allergic asthma. Ann. Allergy AsthmaImmunol. 91, 182–188.
Bernstein, D. I. (1997). Allergic reactions to workplace allergens. JAMA 278, 1907–1913.Bernstein, D. I., Wanner, M., Borish, L., and Liss, G. M. (2004). Twelve‐year survey of fatalreactions to allergen injections and skin testing: 1990–2001. J. Allergy Clin. Immunol. 113,1129–1136.
Bez, C., Schubert, R., Kopp, M., Ersfeld, Y., Rosewich, M., Kuehr, J., Kamin, W., Berg, A. V.,Wahu, U., and Zielen, S. (2004). Effect of anti‐immunoglobulin E on nasal inflammation inpatients with seasonal allergic rhinoconjunctivitis. Clin. Exp. Allergy 34, 1079–1085.
Bich‐Thuy, L. T., Banchereau, J., and Revillard, J. P. (1984). Suppression of polyclonal humanB cell activation by IgG binding factors: Interference with the maturation of Ig‐containing cellsinto Ig‐secreting cells. Cell. Immunol. 87, 231–239.
Bingham, C. O.,III, and Austen, K. F. (2000). Mast‐cell responses in the development of asthma.J. Allergy Clin. Immunol. 105, S527–S534.
Bodtger, U., Poulsen, L. K., and Linneberg, A. (2006). Rhinitis symptoms and IgE sensitization asrisk factors for development of later allergic rhinitis in adults. Allergy 61, 712–716.
Borchers, A. T., Keen, C. L., and Gershwin, M. E. (2004). Fatalities following allergen immuno-therapy. Clin. Rev. Allergy Immunol. 27, 147–158.
Borish, L., Chipps, B., Deniz, Y., Gujrathi, S., Zheng, B., and Dolan, C. M. (2005). Total serum IgElevels in a large cohort of patients with severe or difficult‐to‐treat asthma. Ann. Allergy AsthmaImmunol. 95, 247–253.
Borish, L. C., Nelson, H. S., Corren, J., Bensch, G., Busse, W. W., Whitmore, J. B., and Agosti,J. M. (2001). Efficacy of soluble IL‐4 receptor for the treatment of adults with asthma. J. AllergyClin. Immunol. 107, 963–970.
Borkowski, T. A., Jouvin, M. H., Lin, S. Y., and Kinet, J. P. (2001). Minimal requirements for IgE‐mediated regulation of surface Fc epsilon RI. J. Immunol. 167, 1290–1296.
Boulet, L. P., Turcotte, H., Laprise, C., Lavertu, C., Bedard, P. M., Lavoie, A., and Hebert, J.(1997). Comparative degree and type of sensitization to common indoor and outdoor allergensin subjects with allergic rhinitis and/or asthma. Clin. Exp. Allergy 27, 52–59.
Bousquet, J., Becker, W. M., Hejjaoui, A., Chanal, I., Lebel, B., Dhivert, H., and Michel, F. B.(1991). Differences in clinical and immunologic reactivity of patients allergic to grass pollens andto multiple‐pollen species. II. Efficacy of a double‐blind, placebo‐controlled, specific immuno-therapy with standardized extracts. J. Allergy Clin. Immunol. 88, 43–53.
Bousquet, J., Wenzel, S., Holgate, S., Lumry, W., Freeman, P., and Fox, H. (2004). Predictingresponse to omalizumab, an anti‐IgE antibody, in patients with allergic asthma. Chest 125,1378–1386.
Bousquet, J., Cabrera, P., Berkman, N., Buhl, R., Holgate, S., Wenzel, S., Fox, H., Hedgecock, S.,Blogg, M., and Cioppa, G. D. (2005). The effect of treatment with omalizumab, an anti‐IgEantibody, on asthma exacerbations and emergency medical visits in patients with severe persis-tent asthma. Allergy 60, 302–308.
Boyce, J. A. (2006). Successful treatment of cold‐induced urticaria/anaphylaxis with anti‐IgE.J. Allergy Clin. Immunol. 117, 1415–1418.
Braunstahl, G. J. (2005). The unified immune system: Respiratory tract‐nasobronchial interactionmechanisms in allergic airway disease. J. Allergy Clin. Immunol. 115, 142–148.

THERAPEUTIC ANTI- IgE ANTIBODIES 109
Brody, D., and Metcalfe, D. D. (1998). Mast cells: A unique and functional diversity. Clin. Exp.Allergy 28, 1167–1170.
Buhl, R., Hanf, G., Soler, M., Bensch, G., Wolfe, J., Everhard, F., Champain, K., Fox, H., andThirlwell, J. (2002). The anti‐IgE antibody omalizumab improves asthma‐related quality of lifein patients with allergic asthma. Eur. Respir. J. 20, 1088–1094.
Burrows, B., Martinez, F. D., Halonen, M., Barbee, R. A., and Cline, M. G. (1989). Association ofasthma with serum IgE levels and skin‐test reactivity to allergens. N. Engl. J. Med. 320, 271–277.
Busse, W. W. (2001). Anti‐immunoglobulin E (omalizumab) therapy in allergic asthma. Am.J. Respir. Crit. Care Med. 164, S12–S17.
Capron, A., and Dessaint, J. P. (1992). Immunologic aspects of schistosomiasis. Annu. Rev. Med.43, 209–218.
Capron, M., and Capron, A. (1994). Immunoglobulin E and effector cells in schistosomiasis.Science 264, 1876–1877.
Capron, A., Dessaint, J. P., Capron, M., Ouma, J. H., and Butterworth, A. E. (1987). Immunity toschistosomes: Progress toward vaccine. Science 238, 1065–1072.
Caraux, J., and Weigle, W. O. (1983a). Anti‐idiotype antibody‐dependent cell‐mediated cytotoxicity(ADCC) against idiotype‐bearing cells. Cell. Immunol. 78, 23–32.
Caraux, J., andWeigle,W.O. (1983b). Surface immunoglobulins as targets for anti‐immunoglobulin‐dependent cell‐mediated lysis of B cells. Cell. Immunol. 76, 372–378.
Carlsen, K. H. (2004). Therapeutic strategies for allergic airways diseases. Paediatr. Respir. Rev. 5,45–51.
Casale, T. B. (2001). Anti‐immunoglobulin E (omalizumab) therapy in seasonal allergic rhinitis.Am. J. Respir. Crit. Care Med. 164, S18–S21.
Casale, T. B., Bernstein, I. L., Busse, W. W., LaForce, C. F., Tinkelman, D. G., Stoltz, R. R.,Dockhorn, R. J., Reimann, J., Su, J. Q., Fick, R. B., Jr., and Adelman, D. C. (1997). Use of ananti‐IgE humanized monoclonal antibody in ragweed‐induced allergic rhinitis. J. Allergy Clin.Immunol. 100, 110–121.
Casale, T. B., Condemi, J., LaForce, C., Nayak, A., Rowe, M., Watrous, M., McAlary, M., Fowler‐Taylor, A., Racine, A., Gupta, N., Fick, R., and Della Cioppa, G. (2001). Effect of omalizumab onsymptoms of seasonal allergic rhinitis: A randomized controlled trial. JAMA 286, 2956–2967.
Casale, T. B., Busse, W. W., Kline, J. N., Ballas, Z. K., Moss, M. H., Townley, R. G., Mokhtarani,M., Seyfert‐Margolis, V., Asare, A., Bateman, K., and Deniz, Y. (2006). Omalizumab pretreat-ment decreases acute reactions after rush immunotherapy for ragweed‐induced seasonal allergicrhinitis. J. Allergy Clin. Immunol. 117, 134–140.
Chan, C. S., Soehnlen, F., and Schechter, G. P. (1990). Differential response of malignant humanB‐cells to anti‐IgM immunoglobulin (anti‐mu) and B‐cell growth factor: Unique direct cytotoxi-city of anti‐mu on prolymphocytic leukemia cells. Blood 76, 1601–1606.
Chang, T. W. (2000). The pharmacological basis of anti‐IgE therapy. Nat. Biotechnol. 18, 157–162.Chang, T. W. (2006). Developing antibodies for targeting immunoglobulin and membrane‐bound
immunoglobulin E. Allergy Asthma Proc. 27, S7–S14.Chang, T. W., and Shiung, Y. Y. (2006). Anti‐IgE as a mast cell‐stabilizing therapeutic agent.
J. Allergy Clin. Immunol. 117, 1203–1212.Chang, T. W., Kung, P. C., Gingras, S. P., and Goldstein, G. (1981). Does OKT3 monoclonal
antibody react with an antigen‐recognition structure on human T cells? Proc. Natl. Acad. Sci.USA 78, 1805–1808.
Chang, T. W., Davis, F. M., Sun, N. C., Sun, C. R., MacGlashan, D. W., Jr., and Hamilton, R. G.(1990). Monoclonal antibodies specific for human IgE‐producing B cells: A potential therapeuticfor IgE‐mediated allergic diseases. Biotechnology (NY) 8, 122–126.

110 TSE WEN CHANG ET AL .
Chen,H. Y., Liu, F. T., Hou, C.M.,Huang, J. S., Sharma, B. B., andChang, T.W. (2002).Monoclonalantibodies against the C(epsilon)mX domain of human membrane‐bound IgE and their potentialuse for targeting IgE‐expressing B cells. Int. Arch. Allergy Immunol. 128, 315–324.
Church, M. K., and Levi‐Schaffer, F. (1997). The human mast cell. J. Allergy Clin. Immunol. 99,155–160.
Coker, H. A., Durham, S. R., and Gould, H. J. (2003). Local somatic hypermutation and classswitch recombination in the nasal mucosa of allergic rhinitis patients. J. Immunol. 171,5602–5610.
Conroy, M. C., Adkinson, N. F., Jr., and Lichtenstein, L. M. (1977). Measurement of IgE on humanbasophils: Relation to serum IgE and anti‐IgE‐induced histamine release. J. Immunol. 118,1317–1321.
Cookson, W. (1999). The alliance of genes and environment in asthma and allergy. Nature 402,B5–B11.
Corne, J., Djukanovic, R., Thomas, L., Warner, J., Botta, L., Grandordy, B., Gygax, D., Heusser, C.,Patalano, F., Richardson, W., Kilchherr, E., Staehelin, T., et al. (1997). The effect of intravenousadministration of a chimeric anti‐IgE antibody on serum IgE levels in atopic subjects: Efficacy,safety, and pharmacokinetics. J. Clin. Invest. 99, 879–887.
Davis, F. M., Gossett, L. A., Pinkston, K. L., Liou, R. S., Sun, L. K., Kim, Y. W., Chang, N. T.,Chang, T. W., Wagner, K., Bews, J., Brinkmann, V., Towbin, H., et al. (1993). Can anti‐IgE beused to treat allergy? Springer Semin. Immunopathol. 15, 51–73.
DeFranco, A. L. (1993). Structure and function of the B cell antigen receptor. Annu. Rev. Cell Biol.9, 377–410.
de Vries, J. E., Punnonen, J., Cocks, B. G., de Waal Malefyt, R., and Aversa, G. (1993). Regulationof the human IgE response by IL4 and IL13. Res. Immunol. 144, 597–601.
Donjerkovic, D., and Scott, D. W. (2000). Activation‐induced cell death in B lymphocytes. Cell Res.10, 179–192.
Dreyfus, D. H., and Randolph, C. C. (2006). Characterization of an anaphylactoid reaction toomalizumab. Ann. Allergy Asthma Immunol. 96, 624–627.
Ebo, D. G., and Stevens, W. J. (2002). IgE‐mediated natural rubber latex allergy: Practicalconsiderations for health care workers. Ann. Allergy Asthma Immunol. 88, 568–575.
Edwards, A. M. (2005). The discovery of cromolyn sodium and its effect on research and practicein allergy and immunology. J. Allergy Clin. Immunol. 115, 885–888.
Eigenmann, P. A., Sicherer, S. H., Borkowski, T. A., Cohen, B. A., and Sampson, H. A. (1998).Prevalence of IgE‐mediated food allergy among children with atopic dermatitis. Pediatrics 101,8–13.
El Ridi, R., Ozaki, T., and Kamiya, H. (1998). Schistosoma mansoni infection in IgE‐producing andIgE‐deficient mice. J. Parasitol. 84, 171–174.
Fahy, J. V., Fleming, H. E., Wong, H. H., Liu, J. T., Su, J. Q., Reimann, J., Fick, R. B., Jr., andBoushey, H. A. (1997). The effect of an anti‐IgE monoclonal antibody on the early‐ and late‐phase responses to allergen inhalation in asthmatic subjects. Am. J. Respir. Crit. Care Med. 155,1828–1834.
Finn, A., Gross, G., van Bavel, J., Lee, T., Windom, H., Everhard, F., Fowler‐Taylor, A., Liu, J., andGupta, N. (2003). Omalizumab improves asthma‐related quality of life in patients with severeallergic asthma. J. Allergy Clin. Immunol. 111, 278–284.
Fish, J. E. (2002). Occupational asthma and rhinoconjunctivitis induced by natural rubber latexexposure. J. Allergy Clin. Immunol. 110, S75–S81.
Fodinger, M., Fritsch, G., Winkler, K., Emminger, W., Mitterbauer, G., Gadner, H., Valent, P., andMannhalter, C. (1994). Origin of human mast cells: Development from transplanted hemato-poietic stem cells after allogeneic bone marrow transplantation. Blood 84, 2954–2959.

THERAPEUTIC ANTI- IgE ANTIBODIES 111
Foote, J. (2003). Immunology. Isomeric antibodies. Science 299, 1327–1328.Fox, J. A., Hotaling, T. E., Struble, C., Ruppel, J., Bates, D. J., and Schoenhoff, M. B. (1996).
Tissue distribution and complex formation with IgE of an anti‐IgE antibody after intravenousadministration in cynomolgus monkeys. J. Pharmacol. Exp. Ther. 279, 1000–1008.
Galli, S. J., and Hammel, I. (1994). Mast cell and basophil development. Curr. Opin. Hematol. 1,33–39.
Garman, S. C., Kinet, J. P., and Jardetzky, T. S. (1998). Crystal structure of the human high‐affinityIgE receptor. Cell 95, 951–961.
Garman, S. C., Wurzburg, B. A., Tarchevskaya, S. S., Kinet, J. P., and Jardetzky, T. S. (2000).Structure of the Fc fragment of human IgE bound to its high‐affinity receptor Fc epsilonRIalpha. Nature 406, 259–266.
Gauld, S. B., Benschop, R. J., Merrell, K. T., and Cambier, J. C. (2005). Maintenance of B cellanergy requires constant antigen receptor occupancy and signaling. Nat. Immunol. 6,1160–1167.
Getahun, A., Hjelm, F., and Heyman, B. (2005). IgE enhances antibody and T cell responsesin vivo via CD23þ B cells. J. Immunol. 175, 1473–1482.
Gold, D. R., and Wright, R. (2005). Population disparities in asthma. Annu. Rev. Public Health 26,89–113.
Gould, H. J., Sutton, B. J., Beavil, A. J., Beavil, R. L., McCloskey, N., Coker, H. A., Fear, D., andSmurthwaite, L. (2003). The biology of IGE and the basis of allergic disease. Annu. Rev.Immunol. 21, 579–628.
Greineder, D. K. (1996). Risk management in allergen immunotherapy. J. Allergy Clin. Immunol.98, S330–S334.
Gurish, M. F., Bryce, P. J., Tao, H., Kisselgof, A. B., Thornton, E. M., Miller, H. R., Friend, D. S.,and Oettgen, H. C. (2004). IgE enhances parasite clearance and regulates mast cell responses inmice infected with Trichinella spiralis. J. Immunol. 172, 1139–1145.
Haak‐Frendscho, M., Robbins, K., Lyon, R., Shields, R., Hooley, J., Schoenhoff, M., and Jardieu, P.(1994). Administration of an anti‐IgE antibody inhibits CD23 expression and IgE productionin vivo. Immunology 82, 306–313.
Haba, S., and Nisonoff, A. (1990). Inhibition of IgE synthesis by anti‐IgE: Role in long‐terminhibition of IgE synthesis by neonatally administered soluble IgE. Proc. Natl. Acad. Sci. USA87, 3363–3367.
Haba, S., and Nisonoff, A. (1994). Effects of syngeneic anti‐IgE antibodies on the development ofIgE memory and on the secondary IgE response. J. Immunol. 152, 51–57.
Hamelmann, E., Oshiba, A., Schwarze, J., Bradley, K., Loader, J., Larsen, G. L., and Gelfand,E. W. (1997). Allergen‐specific IgE and IL‐5 are essential for the development of airwayhyperresponsiveness. Am. J. Respir. Cell Mol. Biol. 16, 674–682.
Hart, T. K., Blackburn, M. N., Brigham‐Burke, M., Dede, K., Al‐Mahdi, N., Zia‐Amirhosseini, P.,and Cook, R. M. (2002). Preclinical efficacy and safety of pascolizumab (SB 240683):A humanized anti‐interleukin‐4 antibody with therapeutic potential in asthma. Clin. Exp.Immunol. 130, 93–100.
Heaney, L. G., and Robinson, D. S. (2005). Severe asthma treatment: Need for characterisingpatients. Lancet 365, 974–976.
Hibbert, R. G., Teriete, P., Grundy, G. J., Beavil, R. L., Reljic, R., Holers, V. M., Hannan, J. P.,Sutton, B. J., Gould, H. J., and McDonnell, J. M. (2005). The structure of human CD23 and itsinteractions with IgE and CD21. J. Exp. Med. 202, 751–760.
Holgate, S., Casale, T., Wenzel, S., Bousquet, J., Deniz, Y., and Reisner, C. (2005a). The anti‐inflammatory effects of omalizumab confirm the central role of IgE in allergic inflammation.J. Allergy Clin. Immunol. 115, 459–465.

112 TSE WEN CHANG ET AL .
Holgate, S. T., Djukanovic, R., Casale, T., and Bousquet, J. (2005b). Anti‐immunoglobulin Etreatment with omalizumab in allergic diseases: An update on anti‐inflammatory activity andclinical efficacy. Clin. Exp. Allergy 35, 408–416.
Hoover, R. G., and Lynch, R. G. (1983). Isotype‐specific suppression of IgA: Suppression of IgAresponses in BALB/c mice by T alpha cells. J. Immunol. 130, 521–523.
Hoskins, G., McCowan, C., Neville, R. G., Thomas, G. E., Smith, B., and Silverman, S. (2000).Risk factors and costs associated with an asthma attack. Thorax 55, 19–24.
Humbert, M., Beasley, R., Ayres, J., Slavin, R., Hebert, J., Bousquet, J., Beeh, K. M., Ramos, S.,Canonica, G. W., Hedgecock, S., Fox, H., Blogg, M., et al. (2005). Benefits of omalizumab asadd‐on therapy in patients with severe persistent asthma who are inadequately controlleddespite best available therapy (GINA 2002 step 4 treatment): INNOVATE. Allergy 60, 309–316.
Infuhr, D., Crameri, R., Lamers, R., and Achatz, G. (2005). Molecular and cellular targets of anti‐IgE antibodies. Allergy 60, 977–985.
Irani, A. M., and Schwartz, L. B. (1994). Human mast cell heterogeneity. Allergy Proc. 15,303–308.
Ishizaka, K., and Ishizaka, T. (1967). Identification of gamma‐E‐antibodies as a carrier of reaginicactivity. J. Immunol. 99, 1187–1198.
Ishizaka, T., Conrad, D. H., Schulman, E. S., Sterk, A. R., Ko, C. G., and Ishizaka, K. (1984). IgE‐mediated triggering signals for mediator release from human mast cells and basophils. Fed. Proc.43, 2840–2845.
Isolauri, E., Huurre, A., Salminen, S., and Impivaara, O. (2004). The allergy epidemic extendsbeyond the past few decades. Clin. Exp. Allergy 34, 1007–1010.
James, L. C., Roversi, P., and Tawfik, D. S. (2003). Antibody multispecificity mediated by confor-mational diversity. Science 299, 1362–1367.
Janeway, C. A., Jr., Travers, P., Walport, M., and Shlomchik, M. J. (2005). ‘‘Immunobiology: TheImmune System in Health and Disease.’’ Garland Science Publishing, New York.
Jankovic, D., Kullberg, M. C., Dombrowicz, D., Barbieri, S., Caspar, P., Wynn, T. A., Paul, W. E.,Cheever, A. W., Kinet, J. P., and Sher, A. (1997). Fc epsilonRI‐deficient mice infected withSchistosoma mansoni mount normal Th2‐type responses while displaying enhanced liver pathol-ogy. J. Immunol. 159, 1868–1875.
Johansson, S. G. (1967). Raised levels of a new immunoglobulin class (IgND) in asthma. Lancet 2,951–953.
Kalesnikoff, J., Huber, M., Lam, V., Damen, J. E., Zhang, J., Siraganian, R. P., and Krystal, G.(2001). Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokineproduction and cell survival. Immunity 14, 801–811.
Kasaian, M. T., Meyer, C. H., Nault, A. K., and Bond, J. F. (1995). An increased frequency of IgE‐producing B cell precursors contributes to the elevated levels of plasma IgE in atopic subjects.Clin. Exp. Allergy 25, 749–755.
Kawakami, T., and Galli, S. J. (2002). Regulation of mast‐cell and basophil function and survival byIgE. Nat. Rev. Immunol. 2, 773–786.
Kawakami, T., and Kitaura, J. (2005). Mast cell survival and activation by IgE in the absence ofantigen: A consideration of the biologic mechanisms and relevance. J. Immunol. 175,4167–4173.
Keown, M. B., Ghirlando, R., Mackay, G. A., Sutton, B. J., and Gould, H. J. (1997). Basis of the 1:1stoichiometry of the high affinity receptor Fc epsilon RI‐IgE complex. Eur. Biophys. J. 25,471–476.
Kilmon, M. A., Shelburne, A. E., Chan‐Li, Y., Holmes, K. L., and Conrad, D. H. (2004). CD23trimers are preassociated on the cell surface even in the absence of its ligand, IgE. J. Immunol.172, 1065–1073.

THERAPEUTIC ANTI- IgE ANTIBODIES 113
Kinet, J. P. (1999). The high‐affinity IgE receptor (Fc epsilon RI): From physiology to pathology.Annu. Rev. Immunol. 17, 931–972.
King, C. L., Xianli, J., Malhotra, I., Liu, S., Mahmoud, A. A., and Oettgen, H. C. (1997). Mice witha targeted deletion of the IgE gene have increased worm burdens and reduced granulomatousinflammation following primary infection with Schistosoma mansoni. J. Immunol. 158, 294–300.
King, T. P., and Spangfort, M. D. (2000). Structure and biology of stinging insect venom allergens.Int. Arch. Allergy Immunol. 123, 99–106.
Kips, J. C., Tournoy, K. G., and Pauwels, R. A. (2001). New anti‐asthma therapies: Suppression ofthe effect of interleukin (IL)‐4 and IL‐5. Eur. Respir. J. 17, 499–506.
Kitaura, J., Song, J., Tsai, M., Asai, K., Maeda‐Yamamoto, M., Mocsai, A., Kawakami, Y., Liu, F. T.,Lowell, C. A., Barisas, B. G., Galli, S. J., and Kawakami, T. (2003). Evidence that IgE moleculesmediate a spectrum of effects on mast cell survival and activation via aggregation of theFcepsilonRI. Proc. Natl. Acad. Sci. USA 100, 12911–12916.
Kitaura, J., Kinoshita, T., Matsumoto, M., Chung, S., Kawakami, Y., Leitges, M., Wu, D., Lowell,C. A., and Kawakami, T. (2005). IgE� and IgEþ Ag‐mediated mast cell migration in anautocrine/paracrine fashion. Blood 105, 3222–3229.
KleinJan, A., Vinke, J. G., Severijnen, L. W., and Fokkens, W. J. (2000). Local production anddetection of (specific) IgE in nasal B‐cells and plasma cells of allergic rhinitis patients. Eur.Respir. J. 15, 491–497.
Kohno, M., Yamasaki, S., Tybulewicz, V. L., and Saito, T. (2005). Rapid and large amount ofautocrine IL‐3 production is responsible for mast cell survival by IgE in the absence of antigen.Blood 105, 2059–2065.
Kolbinger, F., Saldanha, J., Hardman, N., and Bendig, M. M. (1993). Humanization of a mouseanti‐human IgE antibody: A potential therapeutic for IgE‐mediated allergies. Protein Eng. 6,971–980.
Kopp, M. V., Mayatepek, E., Engels, E., Brauburger, J., Riedinger, F., Ihorst, G., Wahn, U., andKuehr, J. (2003). Urinary leukotriene E4 levels in children with allergic rhinitis treated withspecific immunotherapy and anti‐IgE (Omalizumab). Pediatr. Allergy Immunol. 14, 401–404.
Kraft, S., Katoh, N., Novak, N., Koch, S., and Bieber, T. (2001). Unexpected functions ofFcepsilonRI on antigen‐presenting cells. Int. Arch. Allergy Immunol. 124, 35–37.
Krathen, R. A., and Hsu, S. (2005). Failure of omalizumab for treatment of severe adult atopicdermatitis. J. Am. Acad. Dermatol. 53, 338–340.
Kubo, S., Matsuoka, K., Taya, C., Kitamura, F., Takai, T., Yonekawa, H., and Karasuyama, H.(2001). Drastic up‐regulation of Fcepsilonri on mast cells is induced by IgE binding throughstabilization and accumulation of Fcepsilonri on the cell surface. J. Immunol. 167, 3427–3434.
Kuehr, J., Brauburger, J., Zielen, S., Schauer, U., Kamin, W., Von Berg, A., Leupold, W., Berg-mann, K. C., Rolinck‐Werninghaus, C., Grave, M., Hultsch, T., and Wahn, U. (2002). Efficacy ofcombination treatment with anti‐IgE plus specific immunotherapy in polysensitized childrenand adolescents with seasonal allergic rhinitis. J. Allergy Clin. Immunol. 109, 274–280.
Kussin, P. S., and Fulkerson, W. J. (1995). The rising tide of asthma. Trends in the epidemiology ofmorbidity and mortality from asthma. Respir. Care Clin. North Am. 1, 163–175.
Lam, V., Kalesnikoff, J., Lee, C. W., Hernandez‐Hansen, V., Wilson, B. S., Oliver, J. M., andKrystal, G. (2003). IgE alone stimulates mast cell adhesion to fibronectin via pathways similarto those used by IgEþ antigen but distinct from those used by Steel factor. Blood 102,1405–1413.
Lane, J. E., Cheyney, J. M., Lane, T. N., Kent, D. E., and Cohen, D. J. (2006). Treatment ofrecalcitrant atopic dermatitis with omalizumab. J. Am. Acad. Dermatol. 54, 68–72.
Lanier, B. (2006). Unanswered clinical questions and speculation about the role of anti‐immunoglobulin E in atopic and nonatopic disease. Allergy Asthma Proc. 27, S37–S42.

114 TSE WEN CHANG ET AL .
Lanier, B. Q. (2003). Newer aspects in the treatment of pediatric and adult asthma: Monoclonalanti‐IgE. Ann. Allergy Asthma Immunol. 90, 13–15.
Lanier, B. Q., and Chang, T. W. (2004). Will anti‐IgE therapy compromise normal immunefunctions? Allergy Clin. Immunol. Int.: J. World Allergy Org. 16, 237–240.
Lanier, B. Q., Corren, J., Lumry, W., Liu, J., Fowler‐Taylor, A., and Gupta, N. (2003). Omalizumabis effective in the long‐term control of severe allergic asthma. Ann. Allergy Asthma Immunol. 91,154–159.
Laske, N., Bunikowski, R., and Niggemann, B. (2003). Extraordinarily high serum IgE levels andconsequences for atopic phenotypes. Ann. Allergy Asthma Immunol. 91, 202–204.
Lee, J., Doggweiler‐Wiygul, R., Kim, S., Hill, B. D., and Yoo, T. J. (2006). Is interstitial cystitis anallergic disorder? A case of interstitial cystitis treated successfully with anti‐IgE. Int. J. Urol. 13,631–634.
Lemanske, R. F., Jr., Nayak, A., McAlary, M., Everhard, F., Fowler‐Taylor, A., and Gupta, N.(2002). Omalizumab improves asthma‐related quality of life in children with allergic asthma.Pediatrics 110, 55–59.
Lemiere, C., Cartier, A., Dolovich, J., and Malo, J. L. (1996). Isolated late asthmatic reaction afterexposure to a high‐molecular‐weight occupational agent, subtilisin. Chest 110, 823–824.
Leung, D. Y. (1993). Role of IgE in atopic dermatitis. Curr. Opin. Immunol. 5, 956–962.Leung, D. Y., and Soter, N. A. (2001). Cellular and immunologic mechanisms in atopic dermatitis.J. Am. Acad. Dermatol. 44, S1–S12.
Leung, D. Y., Sampson, H. A., Yunginger, J. W., Burks, A. W., Jr., Schneider, L. C., Wortel, C. H.,Davis, F. M., Hyun, J. D., and Shanahan, W. R., Jr. (2003). Effect of anti‐IgE therapy in patientswith peanut allergy. N. Engl. J. Med. 348, 986–993.
Lewis, R. A., and Austen, K. F. (1981). Mediation of local homeostasis and inflammation byleukotrienes and other mast cell‐dependent compounds. Nature 293, 103–108.
Leynadier, F., Doudou, O., Gaouar, H., Le Gros, V., Bourdeix, I., Guyomarch‐Cocco, L., andTrunet, P. (2004). Effect of omalizumab in health care workers with occupational latex allergy.J. Allergy Clin. Immunol. 113, 360–361.
Leynaert, B., Bousquet, J., Neukirch, C., Liard, R., and Neukirch, F. (1999). Perennial rhinitis: Anindependent risk factor for asthma in nonatopic subjects: Results from the European Commu-nity Respiratory Health Survey. J. Allergy Clin. Immunol. 104, 301–304.
Li, H., Nowak‐Wegrzyn, A., Charlop‐Powers, Z., Shreffler, W., Chehade, M., Thomas, S., Roda, G.,Dahan, S., Sperber, K., and Berin, M. C. (2006). Transcytosis of IgE‐antigen complexes byCD23a in human intestinal epithelial cells and its role in food allergy. Gastroenterology 131,47–58.
Limb, S. L., Brown, K. C., Wood, R. A., Eggleston, P. A., Hamilton, R. G., and Adkinson, N. F., Jr.(2006). Long‐term immunologic effects of broad‐spectrum aeroallergen immunotherapy. Int.Arch. Allergy Immunol. 140, 245–251.
Lin, H., Boesel, K. M., Griffith, D. T., Prussin, C., Foster, B., Romero, F. A., Townley, R., andCasale, T. B. (2004). Omalizumab rapidly decreases nasal allergic response and FcepsilonRI onbasophils. J. Allergy Clin. Immunol. 113, 297–302.
Litman, G. W., Cannon, J. P., and Dishaw, L. J. (2005). Reconstructing immune phylogeny: Newperspectives. Nat. Rev. Immunol. 5, 866–879.
Liu, F. T. (1997). Truly MASTerful cells: Mast cells command B cell IgE synthesis. J. Clin. Invest.99, 1465–1466.
Liu, J., Lester, P., Builder, S., and Shire, S. J. (1995). Characterization of complex formation byhumanized anti‐IgE monoclonal antibody and monoclonal human IgE. Biochemistry 34,10474–10482.

THERAPEUTIC ANTI- IgE ANTIBODIES 115
Lowman, M. A., Rees, P. H., Benyon, R. C., and Church, M. K. (1988). Human mast cellheterogeneity: Histamine release from mast cells dispersed from skin, lung, adenoids, tonsils,and colon in response to IgE‐dependent and nonimmunologic stimuli. J. Allergy Clin. Immunol.81, 590–597.
MacGlashan, D. (2004). Loss of receptors and IgE in vivo during treatment with anti‐IgE antibody.J. Allergy Clin. Immunol. 114, 1472–1474.
MacGlashan,D.W., Jr., Bochner, B. S., Adelman,D.C., Jardieu, P.M., Togias, A.,McKenzie‐White,J., Sterbinsky, S. A., Hamilton, R. G., and Lichtenstein, L. M. (1997). Down‐regulation of Fc(epsilon)RI expression on human basophils during in vivo treatment of atopic patients withanti‐IgE antibody. J. Immunol. 158, 1438–1445.
MacGlashan, D., Jr., McKenzie‐White, J., Chichester, K., Bochner, B. S., Davis, F. M., Schroeder,J. T., and Lichtenstein, L. M. (1998). In vitro regulation of FcepsilonRIalpha expression onhuman basophils by IgE antibody. Blood 91, 1633–1643.
MacGlashan, D., Jr., Lichtenstein, L. M., McKenzie‐White, J., Chichester, K., Henry, A. J., Sutton,B. J., and Gould, H. J. (1999). Upregulation of FcepsilonRI on human basophils by IgE antibodyis mediated by interaction of IgE with FcepsilonRI. J. Allergy Clin. Immunol. 104, 492–498.
Madden, K. B., Urban, J. F., Jr., Ziltener, H. J., Schrader, J. W., Finkelman, F. D., and Katona, I. M.(1991). Antibodies to IL‐3 and IL‐4 suppress helminth‐induced intestinal mastocytosis.J. Immunol. 147, 1387–1391.
Maeyama, K., Hohman, R. J., Metzger, H., and Beaven, M. A. (1986). Quantitative relationshipsbetween aggregation of IgE receptors, generation of intracellular signals, and histamine secre-tion in rat basophilic leukemia (2H3) cells. Enhanced responses with heavy water. J. Biol. Chem.261, 2583–2592.
Malveaux, F. J., Conroy, M. C., Adkinson, N. F., Jr., and Lichtenstein, L. M. (1978). IgE receptorson human basophils. Relationship to serum IgE concentration. J. Clin. Invest. 62, 176–181.
Manz, R. A., Hauser, A. E., Hiepe, F., and Radbruch, A. (2005). Maintenance of serum antibodylevels. Annu. Rev. Immunol. 23, 367–386.
Mayumi, M., Sumimoto, S., Kanazashi, S., Hata, D., Yamaoka, K., Higaki, Y., Ishigami, T., Kim,K. M., Heike, T., and Katamura, K. (1996). Negative signaling in B cells by surface immunoglo-bulins. J. Allergy Clin. Immunol. 98, S238–S247.
Menz, G., Ying, S., Durham, S. R., Corrigan, C. J., Robinson, D. S., Hamid, Q., Pfister, R.,Humbert, M., and Kay, A. B. (1998). Molecular concepts of IgE‐initiated inflammation in atopicand nonatopic asthma. Allergy 53, 15–21.
Metzger, H., Goetze, A., Kanellopoulos, J., Holowka, D., and Fewtrell, C. (1982). Structure of thehigh‐affinity mast cell receptor for IgE. Fed. Proc. 41, 8–11.
Metzger, H., Alcaraz, G., Hohman, R., Kinet, J. P., Pribluda, V., and Quarto, R. (1986). Thereceptor with high affinity for immunoglobulin E. Annu. Rev. Immunol. 4, 419–470.
Milgrom, H. (2004). Anti‐IgE therapy in children with asthma. Minerva Pediatr. 56, 469–479.Milgrom, H., Berger, W., Nayak, A., Gupta, N., Pollard, S., McAlary, M., Taylor, A. F., and Rohane,
P. (2001). Treatment of childhood asthma with anti‐immunoglobulin E antibody (omalizumab).Pediatrics 108, 36–45.
Miller, J. S., and Schwartz, L. B. (1989). Human mast cell proteases and mast cell heterogeneity.Curr. Opin. Immunol. 1, 637–642.
Mimura, T., Amano, S., Funatsu, H., Yamagami, S., Araie, M., Kaji, Y., Arimoto, A., Ishida, Y., Usui,T., and Okamoto, S. (2004). Correlations between allergen‐specific IgE serum levels in patientswith allergic conjunctivitis in spring. Ocul. Immunol. Inflamm. 12, 45–51.
Nakamura, T., Kloetzer, W. S., Brams, P., Hariharan, K., Chamat, S., Cao, X., LaBarre, M. J.,Chinn, P. C., Morena, R. A., Shestowsky, W. S., Li, Y. P., Chen, A., et al. (2000). In vitro IgE

116 TSE WEN CHANG ET AL .
inhibition in B cells by anti‐CD23 monoclonal antibodies is functionally dependent on theimmunoglobulin Fc domain. Int. J. Immunopharmacol. 22, 131–141.
Nayak, A., Casale, T., Miller, S. D., Condemi, J., McAlary, M., Fowler‐Taylor, A., Della Cioppa, G.,and Gupta, N. (2003). Tolerability of retreatment with omalizumab, a recombinant humanizedmonoclonal anti‐IgE antibody, during a second ragweed pollen season in patients with seasonalallergic rhinitis. Allergy Asthma Proc. 24, 323–329.
Nelson, H. S. (2003). Advances in upper airway diseases and allergen immunotherapy. J. AllergyClin. Immunol. 111, S793–S798.
Niiro, H., and Clark, E. A. (2002). Regulation of B‐cell fate by antigen‐receptor signals. Nat. Rev.Immunol. 2, 945–956.
Noga, O., Hanf, G., and Kunkel, G. (2003). Immunological and clinical changes in allergicasthmatics following treatment with omalizumab. Int. Arch. Allergy Immunol. 131, 46–52.
Novak, N., and Bieber, T. (2003). Allergic and nonallergic forms of atopic diseases. J. Allergy Clin.Immunol. 112, 252–262.
Novak, N., Bieber, T., and Leung, D. Y. (2003a). Immune mechanisms leading to atopic dermatitis.J. Allergy Clin. Immunol. 112, S128–S139.
Novak, N., Kraft, S., and Bieber, T. (2003b). Unraveling the mission of FcepsilonRI on antigen‐presenting cells. J. Allergy Clin. Immunol. 111, 38–44.
Oba, Y., and Salzman, G. A. (2004). Cost‐effectiveness analysis of omalizumab in adults andadolescents with moderate‐to‐severe allergic asthma. J. Allergy Clin. Immunol. 114, 265–269.
Oettgen, H. C., and Geha, R. S. (1999). IgE in asthma and atopy: Cellular and molecularconnections. J. Clin. Invest. 104, 829–835.
Oettgen, H. C., and Geha, R. S. (2001). IgE regulation and roles in asthma pathogenesis. J. AllergyClin. Immunol. 107, 429–440.
Ong, Y. E., Menzies‐Gow, A., Barkans, J., Benyahia, F., Ou, T. T., Ying, S., and Kay, A. B. (2005).Anti‐IgE (omalizumab) inhibits late‐phase reactions and inflammatory cells after repeat skinallergen challenge. J. Allergy Clin. Immunol. 116, 558–564.
Pandey, V., Mihara, S., Fensome‐Green, A., Bolsover, S., and Cockcroft, S. (2004). Monomeric IgEstimulates NFAT translocation into the nucleus, a rise in cytosol Ca2þ, degranulation, andmembrane ruffling in the cultured rat basophilic leukemia‐2H3 mast cell line. J. Immunol.172, 4048–4058.
Parks, K. W., and Casale, T. B. (2006). Anti‐immunoglobulin E monoclonal antibody administeredwith immunotherapy. Allergy Asthma Proc. 27, S33–S36.
Pawankar, R., Okuda, M., Yssel, H., Okumura, K., and Ra, C. (1997). Nasal mast cells in perennialallergic rhinitics exhibit increased expression of the Fc epsilonRI, CD40L, IL‐4, and IL‐13, andcan induce IgE synthesis in B cells. J. Clin. Invest. 99, 1492–1499.
Peng, C., Davis, F. M., Sun, L. K., Liou, R. S., Kim, Y. W., and Chang, T. W. (1992). A new isoformof human membrane‐bound IgE. J. Immunol. 148, 129–136.
Peng, Q., McEuen, A. R., Benyon, R. C., and Walls, A. F. (2003). The heterogeneity of mast celltryptase from human lung and skin. Eur. J. Biochem. 270, 270–283.
Poole, J. A., Meng, J., Reff, M., Spellman, M. C., and Rosenwasser, L. J. (2005). Anti‐CD23monoclonal antibody, lumiliximab, inhibited allergen‐induced responses in antigen‐presentingcells and T cells from atopic subjects. J. Allergy Clin. Immunol. 116, 780–788.
Posner, R. G., Savage, P. B., Peters, A. S., Macias, A., DelGado, J., Zwartz, G., Sklar, L. A., andHlavacek, W. S. (2002). A quantitative approach for studying IgE‐FcepsilonRI aggregation.Mol.Immunol. 38, 1221–1228.
Presta, L. G., Lahr, S. J., Shields, R. L., Porter, J. P., Gorman, C. M., Fendly, B. M., and Jardieu,P. M. (1993). Humanization of an antibody directed against IgE. J. Immunol. 151, 2623–2632.
Prickman, L. E. (1951). Allergic asthma. Minn. Med. 34, 755–756.

THERAPEUTIC ANTI- IgE ANTIBODIES 117
Pritchard, D. I. (1993). Immunity to helminths: Is too much IgE parasite—rather than host‐protective? Parasite Immunol. 15, 5–9.
Prussin, C., Griffith, D. T., Boesel, K. M., Lin, H., Foster, B., and Casale, T. B. (2003). Omalizumabtreatment downregulates dendritic cell FcepsilonRI expression. J. Allergy Clin. Immunol. 112,1147–1154.
Pruzansky, J. J., and Patterson, R. (1988). Limiting concentrations of human basophil‐bound IgEantibody required for histamine release. Immunology 64, 307–310.
Racine‐Poon, A., Botta, L., Chang, T. W., Davis, F. M., Gygax, D., Liou, R. S., Rohane, P.,Staehelin, T., van Steijn, A. M., and Frank, W. (1997). Efficacy, pharmacodynamics, andpharmacokinetics of CGP 51901, an anti‐immunoglobulin E chimeric monoclonal antibody, inpatients with seasonal allergic rhinitis. Clin. Pharmacol. Ther. 62, 675–690.
Razin, E., Mencia‐Huerta, J. M., Stevens, R. L., Lewis, R. A., Liu, F. T., Corey, E., and Austen,K. F. (1983). IgE‐mediated release of leukotriene C4, chondroitin sulfate E proteoglycan, beta‐hexosaminidase, and histamine from cultured bone marrow‐derived mouse mast cells. J. Exp.Med. 157, 189–201.
Reljic, R., Cosentino, G., and Gould, H. J. (1997). Function of CD23 in the response of humanB cells to antigen. Eur. J. Immunol. 27, 572–575.
Rolinck‐Werninghaus, C., Hamelmann, E., Keil, T., Kulig, M., Koetz, K., Gerstner, B., Kuehr, J.,Zielen, S., Schauer, U., Kamin, W., Von Berg, A., Hammermann, J., et al. (2004). The co‐seasonalapplication of anti‐IgE after preseasonal specific immunotherapy decreases ocular and nasalsymptom scores and rescue medication use in grass pollen allergic children. Allergy 59,973–979.
Rosenwasser, L. J., Busse, W. W., Lizambri, R. G., Olejnik, T. A., and Totoritis, M. C. (2003).Allergic asthma and an anti‐CD23 mAb (IDEC‐152): Results of a phase I, single‐dose, dose‐escalating clinical trial. J. Allergy Clin. Immunol. 112, 563–570.
Ross, R. N., Nelson, H. S., and Finegold, I. (2000). Effectiveness of specific immunotherapy in thetreatment of hymenoptera venom hypersensitivity: A meta‐analysis. Clin. Ther. 22, 351–358.
Sabra, A., Bellanti, J. A., Rais, J. M., Castro, H. J., de Inocencio, J. M., and Sabra, S. (2003). IgEand non‐IgE food allergy. Ann. Allergy Asthma Immunol. 90, 71–76.
Schafer, T., and Przybilla, B. (1996). IgE antibodies to Hymenoptera venoms in the serum arecommon in the general population and are related to indications of atopy. Allergy 51, 372–377.
Schoenwetter, W. F. (2000). Allergic rhinitis: Epidemiology and natural history. Allergy AsthmaProc. 21, 1–6.
Serafin, W. E., and Austen, K. F. (1987). Mediators of immediate hypersensitivity reactions.N. Engl. J. Med. 317, 30–34.
Shapiro‐Shelef, M., and Calame, K. (2005). Regulation of plasma‐cell development. Nat. Rev.Immunol. 5, 230–242.
Sharkey, P., and Portnoy, J. (1996). Rush immunotherapy: Experience with a one‐day schedule.Ann. Allergy Asthma Immunol. 76, 175–180.
Shehade, S. A., Layton, G. T., and Stanworth, D. R. (1988). IgG4 and IgE antibodies in atopicdermatitis and urticaria. Clin. Exp. Dermatol. 13, 393–396.
Sher, A., Coffman, R. L., Hieny, S., and Cheever, A. W. (1990). Ablation of eosinophil and IgEresponses with anti‐IL‐5 or anti‐IL‐4 antibodies fails to affect immunity against Schistosomamansoni in the mouse. J. Immunol. 145, 3911–3916.
Sicherer, S. H., and Leung, D. Y. (2005). Advances in allergic skin disease, anaphylaxis, andhypersensitivity reactions to foods, drugs, and insects. J. Allergy Clin. Immunol. 116,153–163.
Skolnick, H. S., Conover‐Walker, M. K., Koerner, C. B., Sampson, H. A., Burks, W., and Wood,R. A. (2001). The natural history of peanut allergy. J. Allergy Clin. Immunol. 107, 367–374.

118 TSE WEN CHANG ET AL .
Smurthwaite, L., Walker, S. N., Wilson, D. R., Birch, D. S., Merrett, T. G., Durham, S. R., andGould, H. J. (2001). Persistent IgE synthesis in the nasal mucosa of hay fever patients.Eur. J. Immunol. 31, 3422–3431.
Soler, M., Matz, J., Townley, R., Buhl, R., O’Brien, J., Fox, H., Thirlwell, J., Gupta, N., and DellaCioppa, G. (2001). The anti‐IgE antibody omalizumab reduces exacerbations and steroidrequirement in allergic asthmatics. Eur. Respir. J. 18, 254–261.
Somos, S., Schneider, I., and Farkas, B. (2001). Immunoglobulins in tears and sera in patients withatopic dermatitis. Allergy Asthma Proc. 22, 81–86.
Stokes, J., and Casale, T. B. (2004). Rationale for new treatments aimed at IgE immunomodula-tion. Ann. Allergy Asthma Immunol. 93, 212–217.
Storms, W., and Kaliner, M. A. (2005). Cromolyn sodium: Fitting an old friend into current asthmatreatment. J. Asthma 42, 79–89.
Strunk, R. C., and Bloomberg, G. R. (2006). Omalizumab for asthma. N. Engl. J. Med. 354,2689–2695.
Sun, L. K., Liou, R. S., Sun, N. C., Gossett, L. A., Sun, C., Davis, F. M., MacGlashan, D. W., Jr.,and Chang, T. W. (1991). Transfectomas expressing both secreted and membrane‐bound formsof chimeric IgE with anti‐viral specificity. J. Immunol. 146, 199–205.
Sussman, G. L., Beezhold, D. H., and Kurup, V. P. (2002). Allergens and natural rubber proteins.J. Allergy Clin. Immunol. 110, S33–S39.
Takhar, P., Smurthwaite, L., Coker, H. A., Fear, D. J., Banfield, G. K., Carr, V. A., Durham, S. R.,and Gould, H. J. (2005). Allergen drives class switching to IgE in the nasal mucosa in allergicrhinitis. J. Immunol. 174, 5024–5032.
Tanaka, S., Takasu, Y., Mikura, S., Satoh, N., and Ichikawa, A. (2002). Antigen‐independentinduction of histamine synthesis by immunoglobulin E in mouse bone marrow‐derived mastcells. J. Exp. Med. 196, 229–235.
Tanaka, S., Mikura, S., Hashimoto, E., Sugimoto, Y., and Ichikawa, A. (2005). Ca2þ influx‐mediatedhistamine synthesis and IL‐6 release in mast cells activated by monomeric IgE. Eur. J. Immunol.35, 460–468.
Tighe, H., Warnatz, K., Brinson, D., Corr, M., Weigle, W. O., Baird, S. M., and Carson, D. A.(1997). Peripheral deletion of rheumatoid factor B cells after abortive activation by IgG. Proc.Natl. Acad. Sci. USA 94, 646–651.
Till, S. J., Francis, J. N., Nouri‐Aria, K., and Durham, S. R. (2004). Mechanisms of immunotherapy.J. Allergy Clin. Immunol. 113, 1025–1034.
Tschopp, J. M., Sistek, D., Schindler, C., Leuenberger, P., Perruchoud, A. P., Wuthrich, B.,Brutsche, M., Zellweger, J. P., Karrer, W., and Brandli, O. (1998). Current allergic asthma andrhinitis: Diagnostic efficiency of three commonly used atopic markers (IgE, skin prick tests, andPhadiatop). Results from 8329 randomized adults from the SAPALDIA Study. Swiss Study onAir Pollution and Lung Diseases in Adults. Allergy 53, 608–613.
Tu, Y., Salim, S., Bourgeois, J., Di Leo, V., Irvine, E. J., Marshall, J. K., and Perdue, M. H. (2005).CD23‐mediated IgE transport across human intestinal epithelium: Inhibition by blocking sitesof translation or binding. Gastroenterology 129, 928–940.
van Cauwenberge, P., Bachert, C., Passalacqua, G., Bousquet, J., Canonica, G. W., Durham, S. R.,Fokkens, W. J., Howarth, P. H., Lund, V., Malling, H. J., Mygind, N., Passali, D., et al. (2000).Consensus statement on the treatment of allergic rhinitis. European Academy of Allergology andClinical Immunology. Allergy 55, 116–134.
Vignola, A. M., Humbert, M., Bousquet, J., Boulet, L. P., Hedgecock, S., Blogg, M., Fox, H., andSurrey, K. (2004). Efficacy and tolerability of anti‐immunoglobulin E therapy with omalizumabin patients with concomitant allergic asthma and persistent allergic rhinitis: SOLAR. Allergy 59,709–717.

THERAPEUTIC ANTI- IgE ANTIBODIES 119
Vigo, P. G., Girgis, K. R., Pfuetze, B. L., Critchlow, M. E., Fisher, J., and Hussain, I. (2006).Efficacy of anti‐IgE therapy in patients with atopic dermatitis. J. Am. Acad. Dermatol. 55,168–170.
Walker, K. Z., Boux, H. A., Hayden, G. E., Goodnow, C. C., and Raison, R. L. (1985).A monoclonal antibody with selectivity for human kappa myeloma and lymphoma cells whichhas potential as a therapeutic agent. Adv. Exp. Med. Biol. 186, 833–841.
Wan, T., Beavil, R. L., Fabiane, S. M., Beavil, A. J., Sohi, M. K., Keown, M., Young, R. J., Henry,A. J., Owens, R. J., Gould, H. J., and Sutton, B. J. (2002). The crystal structure of IgE Fc revealsan asymmetrically bent conformation. Nat. Immunol. 3, 681–686.
Warr, G. W., Magor, K. E., and Higgins, D. A. (1995). IgY: Clues to the origins of modernantibodies. Immunol. Today 16, 392–398.
Wasserman, S. I. (1989). Mast cell‐mediated inflammation in asthma. Ann. Allergy 63, 546–550.Watanabe, N., Katakura, K., Kobayashi, A., Okumura, K., and Ovary, Z. (1988). Protective
immunity and eosinophilia in IgE‐deficient SJA/9 mice infected with Nippostrongylus brasilien-sis and Trichinella spiralis. Proc. Natl. Acad. Sci. USA 85, 4460–4462.
Watanabe, N., Ishiwata, K., Kaneko, S., Oku, Y., Kamiya, M., and Katakura, K. (1993). Immunedefense and eosinophilia in congenitally IgE‐deficient SJA/9 mice infected with Angiostrongyluscostaricensis. Parasitol. Res. 79, 431–434.
Weimar, W., Baumgartner, D., Hendriks, G. F., Hesse, C. J., Balk, A. H., Simoons, M. L., and Bos,E. (1988). The prophylactic use of Orthoclone OKT3 in kidney and heart transplantation.Transplant. Proc. 20, 96–100.
Weiss, M. E., Nyhan, D., Peng, Z. K., Horrow, J. C., Lowenstein, E., Hirshman, C., and Adkinson,N. F., Jr. (1989). Association of protamine IgE and IgG antibodies with life‐threatening reactionsto intravenous protamine. N. Engl. J. Med. 320, 886–892.
Weissman, D. N., and Lewis, D. M. (2002). Allergic and latex‐specific sensitization: Route,frequency, and amount of exposure that are required to initiate IgE production. J. Allergy Clin.Immunol. 110, S57–S63.
Williams, C. M., and Galli, S. J. (2000). The diverse potential effector and immunoregulatory rolesof mast cells in allergic disease. J. Allergy Clin. Immunol. 105, 847–859.
Wilson, D. R., Merrett, T. G., Varga, E. M., Smurthwaite, L., Gould, H. J., Kemp, M., Hooper, J.,Till, S. J., and Durham, S. R. (2002). Increases in allergen‐specific IgE in BAL after segmentalallergen challenge in atopic asthmatics. Am. J. Respir. Crit. Care Med. 165, 22–26.
Wurm, F. M. (2004). Production of recombinant protein therapeutics in cultivated mammaliancells. Nat. Biotechnol. 22, 1393–1398.
Wurzburg, B. A., Tarchevskaya, S. S., and Jardetzky, T. S. (2006). Structural changes in the lectindomain of CD23, the low‐affinity IgE receptor, upon calcium binding. Structure 14, 1049–1058.
Yamagata, T., and Ichinose, M. (2006). Agents against cytokine synthesis or receptors. Eur.J. Pharmacol. 533, 289–301.
Zeiger, R. S., and Heller, S. (1993). Development of nasal basophilic cells and nasal eosinophilsfrom age 4 months through 4 years in children of atopic parents. J. Allergy. Clin Immunol. 91,723–734.
Zheng, Y., Shopes, B., Holowka, D., and Baird, B. (1991). Conformations of IgE bound to itsreceptor Fc epsilon RI and in solution. Biochemistry 30, 9125–9132.

Immune Semaphorins: Increasing Membersand Their Diverse Roles
Hitos hi Kikutani ,* Kazuhiro Suzuk i,* and Atsushi Kuma nogoh †
*Department of Molecular Immunology and CREST Program of JST, Research Institute forMicrobial Diseases, Osaka University, Suita, Osaka 5650871, Japan
†Department of Immunopathology, Research Institute for Microbial Diseases,Osaka University, Suita, Osaka 5650871, Japan
A
ad#
bstract............................................................................................................. 1
121vances in immunology, vol. 93 0065-2776/07
2007 Elsevier Inc. All rights reserved. DOI: 10.1016/S0065-2776(06)9
21
1. I ntroduction ....................................................................................................... 1 21 2. S ema4D ............................................................................................................ 1 22 3. S ema4A............................................................................................................. 1 27 4. S ema6D and Its Receptor Plexin‐A1 ....................................................................... 1 30 5. S ema7A............................................................................................................. 1 35 6. O ther Semaphorins.............................................................................................. 1 37 7. S ummary and Perspectives.................................................................................... 1 38R
eferences ......................................................................................................... 1 39Abstract
The semaphorin family consists of soluble and membrane‐bound proteinsoriginally identified as axonal guidance cues functioning during neuronaldevelopment. However, it is becoming increasingly clear that semaphorinsplay diverse roles in organogenesis, vascular growth, and tumor progression.In addition, emerging evidence indicates that several semaphorins, called‘‘immune semaphorins,’’ play crucial roles also during immune responses.Extensive studies on the immune semaphorins have revealed not only parallelsbut also differences in the semaphorin functions between the immune andnervous systems, providing unexpected but meaningful insights into thebiological activities of these molecules. This chapter focuses on our currentunderstanding of the roles of semaphorins and their receptors in the immunesystem.
1. Introduction
The semaphorins comprise a large family of phylogenetically conserved pro-teins, and more than 20 members have been identified in a variety of speciesfrom viruses to humans. Many members act as axon guidance cues duringneuronal development (Pasterkamp and Kolodkin, 2003; Tessier‐Lavigne andGoodman, 1996; Yu and Kolodkin, 1999). Both soluble and membrane‐boundsemaphorins have been identified, and they have been categorized into eight
$35.00
3003-X

122 HITOSHI KIKUTANI ET AL .
subclasses based on sequence similarity and distinctive structural features.Semaphorin subclasses I and II are found in invertebrate species, and sub-classes III–VII are expressed in vertebrates (Pasterkamp and Kolodkin, 2003;Tessier‐Lavigne and Goodman, 1996; Yu and Kolodkin, 1999). Additionally,some nonneurotropic DNA viruses encode functional semaphorin molecules,which are classified into class VIII (Spriggs, 1999).Two families of semaphorin receptors have been identified including plexins
and neuropilins (Comeau et al., 1998; He and Tessier‐Lavigne, 1997; Kolodkinet al., 1997; Takahashi et al., 1999; Tamagnone and Comoglio, 2000; Tamagnoneet al., 1999; Winberg et al., 1998). Most membrane‐bound vertebrate sema-phorins directly bind plexins, while class III secreted semaphorins requireneuropilins as obligate coreceptors. However, studies have demonstrated thatsemaphorin receptor usage is more complex than previously thought. Sema3Esignals independently of neuropilin through Plexin‐D1 (Gu et al., 2005), andglycosylphosphatidylinositol (GPI)‐linked Sema7A binds a b1 integrin receptorindependently of plexins (Pasterkamp et al., 2003). Additionally, two moleculesthat are unrelated to plexins and neuropilins, CD72 and Tim‐2, functionallyinteract with class IV transmembrane semaphorins in the immune system(Kumanogoh and Kikutani, 2003; Kumanogoh et al., 2000, 2002a).Although functions for semaphorins were originally identified in the nervous
system, semaphorins are now thought to fulfil diverse physiological roles un-related to axon guidance, including organogenesis, vascularization, angiogenesis,neuronal apoptosis, and neoplastic transformation (Kitsukawa et al., 1995; Krugeret al., 2005; Sekido et al., 1996). Additionally, studies have revealed that severalsemaphorins are crucially involved in various phases of immune responses. Inparticular, two class IV semaphorins, Sema4D and Sema4A, play important rolesin the immune system (Delaire et al., 2001; Kumanogoh et al., 2000, 2002a), andtheir physiological importance was clearly demonstrated through in vivo analysesusing gene‐disrupted mice. Additionally, it is becoming clear that other sema-phorins such as Sema6D (Takegahara et al., 2006) and Sema7A (Czopik et al.,2006; Holmes et al., 2002) also play immunoregulatory roles. In this chapter, wewill discuss the recent advances related to the biological functions of semaphorinsin the immune system.
2. Sema4D
Sema4D is the first semaphorin molecule identified with a functional role inthe immune system (Bougeret et al., 1992; Delaire et al., 1998). It wasoriginally defined as CD100, a differentiation antigen expressed on humanT cells recognized by a panel of monoclonal antibodies (Bougeret et al., 1992).Subsequent molecular cloning revealed that this molecule belonged to the

IMMUNE SEMAPHORINS 123
class IV transmembrane semaphorin subfamily (Furuyama et al., 1996; Hallet al., 1996). In the nervous system, Sema4D binds Plexin‐B1, a member of theplexin family, and is chemorepulsive to various neurons (Swiercz et al., 2002;Tamagnone et al., 1999). However, Sema4D binds CD72, a negative regulatorof B cells, and enhances the activation of B cells and dendritic cells (DCs) inthe immune system (Kumanogoh et al., 2000, 2002b). Analysis of Sema4D�/�
mice revealed that Sema4D plays pivotal roles not only in fine‐tuning B cellantigen receptor (BCR) signaling but also in the generation of antigen‐specificT cells (Kumanogoh et al., 2002b; Shi et al., 2000). However, no apparentdefect was found in the nervous system of these mutant mice, despite the factthat Sema4D–Plexin‐B1 signaling is a well‐studied axon guidance signal(Kruger et al., 2005; Negishi et al., 2005).
2.1. Sema4D–CD72 Interactions in B Cell Signaling
In the immune system, Sema4D is constitutively expressed on Tcells (Bougeretet al., 1992; Delaire et al., 1998). B cells weakly express Sema4D, but itsexpression is significantly elevated by the treatment with inflammatory stimulisuch as anti‐CD40 (Kumanogoh et al., 2000). Treatment of mouse B cells withthe extracellular domain of mouse Sema4D fused to the Fc portion of IgGSema4D�Fc or exogenous expression of Sema4D significantly enhances theCD40‐induced proliferation and differentiation in mouse B cells, but Sema4Ddoes not affect mouse T cell function (Kumanogoh et al., 2000). Sema4D�/�
mice develop decreased T‐dependent antibody responses (Shi et al., 2000).Human Sema4D�Fc protein also enhances human B cell activation (Ishidaet al., 2003). Cells exogenously expressing human Sema4D promote theaggregation and survival of human B cells in vitro (Hall et al., 1996).
In the immune system, CD72 appears to be the major receptor for Sema4D(Kumanogoh et al., 2000), and Sema4D specifically binds CD72with a relativelylow affinity (Kd ¼ �3 � 10–7 M). Agonistic anti‐CD72 mAbs mimic the effectof Sema4D binding on B cells. CD72, a C‐type lectin family, contains twoimmunoreceptor tyrosine‐based inhibitory motifs (ITIMs) in its cytoplasmicdomain that recruit the tyrosine phosphatase SHP‐1 and functions as a negativeregulator of B cells (Adachi et al., 1998, 2001). Indeed, B cells from CD72�/�
mice are hyperresponsive to BCR stimulation (Pan et al., 1999). Several linesof evidence indicate that Sema4D turns off an inhibitory signal originatingat CD72, thereby enhancing B cell activation. Both Sema4D�Fc and anti‐CD72 mAb block tyrosine phosphorylation and the association of SHP‐1 withCD72 in anti‐m‐stimulated B cells (Adachi et al., 1998; Kumanogoh et al., 2000;Wu et al., 1998). Additionally, CD72 is tyrosine‐phosphorylated and associateswith SHP‐1 when transiently expressed in COS7 cells. In these cells, CD72

124 HITOSHI KIKUTANI ET AL .
tyrosine‐phosphorylation and SHP‐1 association are blocked by coincubationwith Sema4D�Fc (Kumanogoh et al., 2000). Furthermore, CD72 is tyrosine‐phosphorylated and constitutively associated with SHP‐1 in B cells fromSema4D�/� mice (Shi et al., 2000). Finally, the phenotype of Sema4D�/�
B cells (hyporesponsive) is almost the opposite of that seen in CD72�/� B cells(hyperresponsive; Shi et al., 2000).The mechanism by which the Sema4D–CD72 interaction regulates BCR
signaling is now becoming clear, but it remains unknown how Sema4D isinvolved in the regulation of CD40 and TLR4 signals. CD72 is constitutivelyassociated with the BCR complex. Anti‐m stimulation activates downstreamtyrosine kinases such as Blk, Fyn, and Lyn (Kurosaki, 2002). Among thesekinases, Lyn phosphorylates the ITIMs of CD72 (Fusaki et al., 2000; Wuet al., 1998), thereby recruiting SHP‐1. SHP‐1 is thought to counteract theaction of the tyrosine kinases activated following BCR stimulation. Interestingly,Sema4D induces the dissociation of CD72 from the BCR complex (Kumanogohet al., 2005b), and the sequestration of CD72 from the BCR signalosome, whichis rich in tyrosine kinases, likely facilitates the SHP‐1‐mediated dephosphorya-tion of the CD72 ITIMs and their subsequent dissociation. Indeed, tyrosinephosphorylation of downstream signaling molecules such as Ig‐b, Syk, Erk, andBLNK and Ca2þ mobilization were severely impaired in Sema4D�/� B cellsafter anti‐m stimulation. These deficiencies were likely due to the constitutiveinhibitory signals arising from CD72 in the absence of Sema4D (Kumanogohet al., 2005b). Additionally, anti‐m‐stimulated Sema4D�/� B cells proliferated toa lesser extent than wild‐type B cells, a result consistent with decreased BCRsignaling. Furthermore, hyper‐cross‐linking of the BCR by anti‐m antibodiescaused less apoptotic cell death in Sema4D�/� B cells (Kumanogoh et al.,2005b), an additional sign of impaired signaling. These findings indicate thatSema4D is critically involved in tuning the strength of BCR signals via itsinteraction with CD72 (Fig. 1).
2.2. Sema4D–CD72 Interactions in Maintaining B Cell Homeostasis
Several studies examining mice with mutated signaling molecules downstreamof the BCR have clearly demonstrated that changes in the threshold of BCRtriggering affect the in vivo survival and turnover of B cells (Niiro and Clark,2002). BrdU uptake by B cells in Sema4D�/� mice was considerably slowerthan that seen in wild‐type mice fed with BrdU (Kumanogoh et al., 2005b).Furthermore, BrdU‐labeled B cells disappeared from Sema4D�/� mice moreslowly than from wild‐type mice. Thus, the overall turnover of B cells inSema4D�/� mice is reduced compared with wild‐type mice.

Sema4D
BCR
CD72
SHP-1
P
SHP-1
Enhancement of BCR signals
Figure 1 Sema4D turns off the negative signaling of CD72. In the absence of Sema4D, CD72is constitutively associated with the BCR complex, and recruits SHP‐1 to the tyrosine‐phospho-rylated ITIM. SHP‐1 dephosphorylates and inactivates several signaling molecules, including Fynand Lyn. Binding of Sema4D induces dissociation of CD72 from BCR complex, leading to thedephosphorylation of the CD72 ITIMs, dissociation of SHP‐1 from CD72 and augmentation ofBCR signaling. Under homeostatic conditions, Sema4D, which is weakly expressed on resting Bcells, maintains certain B cell subsets by fine‐tuning BCR signals. In this setting, Sema4D signalingmay occur through B cell–B cell interactions and/or an autocrine fashion. On the other hand, insecondary lymphoid organs during humoral immune responses, where numerous interactionsbetween B cells and antigen‐specific helper T cells occur, Sema4D abundantly expressed onT cells strongly enhances BCR signals through CD72, accomplishing full activation of B cells.
IMMUNE SEMAPHORINS 125
In young Sema4D�/� mice, the population of CD5þ B1 cells is significantlyreduced, although other B cell subsets such as conventional follicular B cellsand marginal zone B cells appear to be normal (Shi et al., 2000). However, asSema4D�/� mice aged, the proportion of CD21highCD23low marginal zoneB cells gradually increased (Kumanogoh et al., 2005b). The expansion ofmarginal zone B cells is sometimes observed in mice with defective BCRsignaling (Kurosaki, 2002; Niiro and Clark, 2002), whereas the numbers ofB1 cells are increased in mice lacking inhibitory receptors such as CD22 andCD72 (O’Keefe et al., 1996; Pan et al., 1999; Sato et al., 1996), suggesting thatthe requirements for BCR signaling differ among B cell subsets. Therefore, ahigher BCR‐signaling threshold may promote the development and/or survival

126 HITOSHI KIKUTANI ET AL .
of marginal zone B cells but may be detrimental for the development ofB1 cells in Sema4D�/� mice.Interestingly, the expansion of marginal zone B cells in Sema4D�/� mice was
accompanied by the production of a variety of autoantibodies, including anti‐ss-DNA, anti‐dsDNA, RFs, anti‐Sjogren’s syndrome A, and anti‐ribonucleoprotein,although such autoantibodieswere not detectable by 25weeks of age (Kumanogohet al., 2005b). Furthermore, marked perivascular leukocytic infiltration in severaltissues, including the salivary gland, liver, and kidney, was also observed in agedSema4D�/� mice. The majority of infiltrating cells in the salivary glands wereB cells with a CD21highCD23low phenotype (Kumanogoh et al., 2005b). In addi-tion, whenCD21highCD23lowmarginal zone orCD21lowCD23high follicular B cellspurified from Sema4D�/� mice were cultured in vitro, the former cells predomi-nantly produced autoantibodies. Mice lacking both Sema4D and CD72 or onlyCD72 showed no evidence of autoimmune disease or expansion of marginal zoneB cells, although a limited number of CD72�/� mice exhibited substantialamounts of autoantibodies accompanied with a significant increase of B1 B cellsover 1 year of age (Kumanogoh et al., 2005b). These observations suggest thatthe constitutive association of CD72 with the BCR may promote the expans-ion of marginal zone B cells and the development of autoimmunity in agedSema4D�/� mice.
2.3. Sema4D in T Cell‐Mediated Immunity
As described above, Sema4D�/� B cells are hyporesponsive to anti‐m and anti‐CD40 stimulation. Anti‐CD40‐induced expression of costimulatory moleculesand MHC class II as well as the production of cytokines are also impaired inSema4D�/� DCs (Kumanogoh et al., 2002b). Endogenous Sema4D, which isweakly expressed on B cells and DCs, appears to contribute to setting theactivation threshold of DCs as well as B cells (Kumanogoh et al., 2002b,2005b). In the immune system, however, the major Sema4D‐producing cellsare T cells. Exogenous Sema4D not only restores the responsiveness of B cellsand DCs of Sema4D�/� mice but also further enhances the activation of wild‐type DCs (Kumanogoh et al., 2002b; Shi et al., 2000; Watanabe et al., 2001).Therefore, antigen‐mediated interactions with T cells may lower the activationthreshold of B cells and DCs by increasing the local concentrations ofSema4D. In particular, the enhanced DC function induced by Sema4D maybe critical for the development of T cell‐mediated immunity. The importanceof T cell‐derived Sema4D has been addressed using an in vitro system(Kumanogoh et al., 2002b). Naive CD100þ/þ TCR‐transgenic CD4þ T cellsdifferentiate normally into cytokine‐secreting effector cells when cultured withantigens and CD100�/� antigen‐presenting cells (APCs), whereas CD100�/�

IMMUNE SEMAPHORINS 127
TCR‐transgenic T cells fail to differentiate even in the presence of CD100þ/þ
APCs. Consistent with this data, the in vivo generation of antigen‐specificT cells is also profoundly impaired in Sema4D�/� mice. Thus, the enhance-ment of DC function by T cell‐derived Sema4D may be necessary for efficientestablishment of T cell‐mediated immunity.
3. Sema4A
Sema4A is the second semaphorin identified with a function in the immunesystem. Like Sema4D, it is also a member of the class IV transmembranesemaphorin subfamily (Kumanogoh et al., 2002a). However, unlike Sema4D,Sema4A contributes to the regulation of immune responses by directly actingon T cells (Kumanogoh et al., 2002a). Tim‐2, which belongs to the T cell,immunoglobulin, and mucin domains protein (Tim) family was identified as areceptor for Sema4A (Kumanogoh et al., 2002a). Analyses of Sema4A�/� micerevealed that Sema4A plays critical roles not only in T cell priming but also inthe regulation of Th1/Th2 differentiation (Kumanogoh et al., 2005a).
3.1. Distinct Roles of DC‐Derived and T Cell‐Derived Sema4A inImmune Responses
Sema4A was originally cloned from mouse DCs (Kumanogoh et al., 2002a),and all mouse DC subsets express high levels of Sema4A. Although B cellsexpress low levels of Sema4A under resting conditions, its expression isenhanced by various stimuli (Kumanogoh et al., 2002a). The expression ofSema4A on T cells is uniquely controlled (Kumanogoh et al., 2005a). AlthoughSema4A is barely detectable on resting T cells, stimulation of T cells with anti‐CD3 and anti‐CD28 induces a transient Sema4A expression within 24 h, butits expression level rapidly decreases. However, when T cells are stimulatedwith Th1‐inducing conditions including IL‐12 and anti‐IL‐4, high levels ofSema4A expression are induced and maintained throughout the culture peri-od. In contrast, stimulation of T cells with Th2‐inducing conditions includ-ing IL‐4 and anti‐IFN‐g induces only the transient expression of Sema4A.Furthermore, Sema4A is preferentially expressed on terminally differentiatedTh1 cells or cells of Th1 clones but not on their Th2 counterparts.
The expression pattern of Sema4A suggests that DC‐ and Th1 cell‐expressedSema4A may play distinct roles in the development of immune responses.Sema4A can provide a costimulatory signal to T cells; incubation of T cells withanti‐CD3 and Sema4A�Fc fusion protein significantly enhances proliferationand IL‐2 production (Kumanogoh et al., 2002a). These data strongly suggestthat Sema4A contributes to T cell activation through T cell–DC interactions.

128 HITOSHI KIKUTANI ET AL .
DCs derived from Sema4A�/�mice poorly stimulate allogeneic Tcells in a mixedlymphocyte culture compared with DCs from wild‐type littermates, despite thefact that Sema4A�/� DCs express costimulatory molecules, such as CD80 andCD86 andMHC class II molecules, and produce cytokines normally in responseto anti‐CD40 or LPS stimulation (Kumanogoh et al., 2005a).T cell‐expressed Sema4A is required for in vitro Th1 differentiation. When
CD62LhighCD4þ naive T cells purified from Sema4A�/� mice or wild‐typelittermates are stimulated with anti‐CD3 and anti‐CD28 in the presenceof IL‐12 and anti‐IL‐4, Sema4A�/� T cells fail to differentiate into IFN‐g‐producing Th1 cells. In contrast, Sema4A�/� Tcells differentiate normally intoIL‐4‐producing Th2 cells when cultured with IL‐4 and anti‐IL‐12/anti‐IFN‐g.The selective defect in Th1 differentiation of Sema4A�/� T cells is furtherunderscored by the decreased expression of IL‐12 receptor b2 chain andT‐bet, an essential transcription factor for proper Th1 cell development andhomeostasis (Szabo et al., 2000). Interestingly, normal Th1 differentiation ofSema4A�/� T cells is fully restored by either the addition of Sema4A�Fc orcoculture with wild‐type T cells (Kumanogoh et al., 2005a). Thus, it appearsthat Sema4A contributes to in vitro Th1 differentiation at least in part throughcognate T cell–T cell interactions.Sema4A clearly functions in in vitro systems of T cell stimulation and
differentiation, and a role for it in vivo has also been observed. Sema4A�/�
mice poorly generate antigen‐specific T cells following immunization withvarious antigens (Kumanogoh et al., 2005a). Notably, the generation ofIFN‐g producing antigen‐specific T cells is severely impaired in Sema4A�/�
mice immunized with Th1‐inducing antigens such as heat‐killed Propionibac-terium acnes. In contrast, when infected with Nippostrongylus brasiliensis,a Th2‐inducing intestinal nematode, Sema4A�/� mice mount an enhancedTh2 immune responses compared with infected wild‐type mice. Thus, thein vivo immune responses of Sema4A�/� mice reflect the defects in T cellpriming and Th1 differentiation observed in vitro.Transfer experiments using antigen‐pulsed DCs have been utilized to dis-
sect the role of DC‐derived and T cell‐derived Sema4A in each phase of in vivoimmune responses (Kumanogoh et al., 2005a). The transfer of antigen‐pulsedDCs from Sema4A�/� or Sema4Aþ/þ mice into Sema4A�/� or Sema4Aþ/þ
mice creates four possible combinations with respect to Sema4A expression—Sema4Aþ/þ DCs/Sema4Aþ/þ T cells, Sema4A�/� DCs/Sema4Aþ/þ T cells,Sema4Aþ/þ DCs/Sema4A�/� T cells, and Sema4A�/� DCs/Sema4A�/�
T cells. The transfer of Sema4Aþ/þ DCs into Sema4Aþ/þ mice results in thegreatest proliferation and IL‐2 production by antigen‐specific T cells and Th1‐biased cytokine production, while these T cell responses are severely impairedin Sema4A�/� mice receiving Sema4A�/� DCs. However, when Sema4A�/�
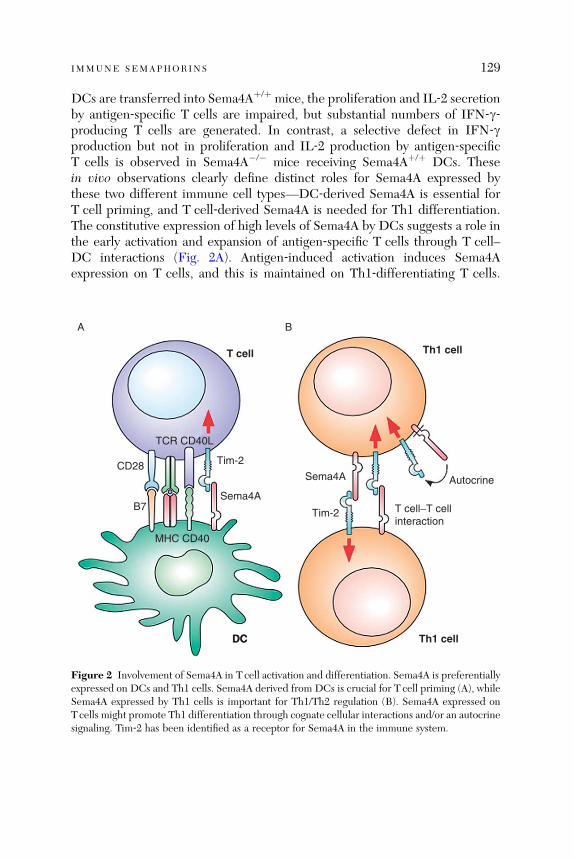
IMMUNE SEMAPHORINS 129
DCs are transferred into Sema4Aþ/þ mice, the proliferation and IL‐2 secretionby antigen‐specific T cells are impaired, but substantial numbers of IFN‐g‐producing T cells are generated. In contrast, a selective defect in IFN‐gproduction but not in proliferation and IL‐2 production by antigen‐specificT cells is observed in Sema4A�/� mice receiving Sema4Aþ/þ DCs. Thesein vivo observations clearly define distinct roles for Sema4A expressed bythese two different immune cell types—DC‐derived Sema4A is essential forT cell priming, and T cell‐derived Sema4A is needed for Th1 differentiation.The constitutive expression of high levels of Sema4A by DCs suggests a role inthe early activation and expansion of antigen‐specific T cells through T cell–DC interactions (Fig. 2A). Antigen‐induced activation induces Sema4Aexpression on T cells, and this is maintained on Th1‐differentiating T cells.
T cell
DC
TCR CD40L
CD28
B7
A B
Tim-2
Autocrine
Tim-2 T cell−T cellinteraction
DC
MHC CD40
Sema4A
Sema4A
Th1 cell
Th1 cell
Figure 2 Involvement of Sema4A in T cell activation and differentiation. Sema4A is preferentiallyexpressed on DCs and Th1 cells. Sema4A derived from DCs is crucial for T cell priming (A), whileSema4A expressed by Th1 cells is important for Th1/Th2 regulation (B). Sema4A expressed onTcells might promote Th1 differentiation through cognate cellular interactions and/or an autocrinesignaling. Tim‐2 has been identified as a receptor for Sema4A in the immune system.

130 HITOSHI KIKUTANI ET AL .
Thus, T cell‐expressed Sema4A may enhance Th1 differentiation throughT cell–T cell interactions and/or an autocrine pathway (Fig. 2B).
3.2. Receptors for Sema4A in the Immune System
Tim‐2 was identified as a Sema4A‐binding protein by expression cloning usinga cDNA library derived from mouse T cells (Kumanogoh et al., 2002a).The surface plasmon resonance assay revealed that the dissociation constant forSema4A binding to Tim‐2 is 7� 10–8 M. Since Sema4A binding induces tyrosinephosphorylation of the cytoplasmic tail of Tim‐2, Tim‐2 appears to transduceSema4A signals. Other Tim family members, Tim‐1 and Tim‐3, were previouslyshown to be involved in the regulation of helper T cell activity (McIntire et al.,2001; Monney et al., 2002), and genetic polymorphisms in both the mouse andhuman Tim loci (particularly Tim‐1 genes) correlate with susceptibility to mouseairway hypersensitivity and human asthma (McIntire et al., 2001, 2003). Addi-tionally, Tim‐3 is preferentially expressed on Th1 cells, and a role for Tim‐3 inregulating Th1 function has been suggested (Monney et al., 2002).Mice injected with the extracellular domain of Tim‐2 fused to the Fc portion of
IgG (Tim‐2‐Fc) exhibit in reduced Th1 and enhanced Th2 responses, and admin-istration of Tim‐2‐Fc suppresses the development of experimental autoimmuneencephalomyelitis (EAE) in SJLmice immunized with proteolipid protein (PLP)139–151 peptide (Chakravarti et al., 2005). Lung inflammation is exacerbated inTim‐2‐deficient mice in a model of airway atopy, and this is accompanied bydysregulated Th2 responses (Rennert et al., 2006). The phenotypes are similar tothose observed in Sema4A�/�mice.However, Tcells fromboth Tim‐2‐Fc‐treatedand Tim‐2‐deficient mice exhibit increased in vitro basal proliferation in theabsence of exogenous antigen, and this is not seen in Sema4A�/� mice. Thus,these findings support a role for Tim‐2 as a functional receptor for Sema4A, butthey also suggest that Sema4A or Tim‐2 may have another functional bindingpartner in the immune system. Specifically, Tim‐2 may bind another ligand thathas a constitutive inhibitory effect on T cells. Like other class IV semaphorins,Sema4A may bind members of the Plexin‐B subfamily, some of which areexpressed by activated T cells (A.K. and H.K., unpublished data). Consideringthat Plexin‐A1 plays a critical role during the development of immune responsesas described later, Sema4A may also have an effect on T cell function throughinteractions with plexin family members.
4. Sema6D and Its Receptor Plexin‐A1
The secreted semaphorin, Sema3A, binds a receptor complex composed ofthe signal transducing Plexin‐A1 and ligand‐binding Neuropilin‐1 to induce che-morepulsive signals (Pasterkamp and Kolodkin, 2003; Takahashi et al., 1999).

IMMUNE SEMAPHORINS 131
Additionally, the class VI transmembrane semaphorin, Sema6D, exerts multiplebiological activities not only during embryonic development but also in theregulation of immune responses through interactions with Plexin‐A1 (Takegaharaet al., 2006; Toyofuku et al., 2004a,b).
4.1. Sema6D–Plexin‐A1 Interactions in Cardiac Development
Sema6DmRNA is abundantly expressed in cells of the neural crest and heart ofboth mouse and chick embryos (Toyofuku et al., 2004a). The in vitro bindingassays clearly demonstrated that Sema6D strongly binds Plexin‐A1‐expressingcells, weakly binds Plexin‐A4‐expressing cells, but does not bind cells expressingother plexin molecules (Toyofuku et al., 2004a).
Overexpression of Sema6D in chick embryos enhances the right bending ofthe cardiac tube and expansion of the ventricular region. In contrast, RNAi‐mediated knockdown of either Sema6D or Plexin‐A1 impairs cardiac tubebending. Sema6D inhibits the migration of ventricular endocardial cells, butconversely, the migration of cells of the conotruncus region is enhanced(Toyofuku et al., 2004a). Additionally, knockdown of Plexin‐A1 abolishes boththe inhibitory and enhancing activities of Sema6D on cells from these twodistinct regions. Interestingly, Plexin‐A1 differentially associates with two re-ceptor type tyrosine kinases; off‐track (PTK7) at the ventricle and vascularendothelial growth factor receptor type 2 (VEGFR2) at the conotruncusregion. Knockdown of off‐track renders the cells of the ventricle but not ofthe conotruncus region unresponsive to Sema6D, while the expression of adominant negative form of VEGFR2 abolishes the responsiveness of the cellsof the conotruncus region but not of the ventricle to Sema6D (Toyofuku et al.,2004a). Thus, Sema6D exerts opposite biological activities at distinct regionsthrough two different receptor complexes, and this appears to contribute toorganized cardiac morphogenesis.
4.2. Sema6D–Plexin‐A1 Interactions in DC Function
Ting and colleagues have shown that Plexin‐A1 is one of the gene productsinduced by CIITA transcription factor that is expressed in DCs and involved inthe interaction of DCs with T cells (Wong et al., 2003). Knockdown of Plexin‐A1 induced by the expression of short‐hairpin RNA suppresses the ability ofcells of DC lines to prime T cells in vitro and in vivo. Additionally, relativelyhigh levels of Sema6D mRNA are expressed in different lymphocyte popula-tions including T cells, B cells, and NK cells. Sema6D�Fc fusion proteinstimulates bone marrow‐derived DCs to produce various cytokines such as

132 HITOSHI KIKUTANI ET AL .
IL‐12 and to increase the expression of MHC class II molecules (Takegaharaet al., 2006).Roles for the interaction of Sema6D with Plexin‐A1 in DC function have
been revealed through the generation and analysis of Plexin‐A1�/� mice(Takegahara et al., 2006). The nervous and cardiovascular development pro-ceed normally in Plexin‐A1�/� mice, indicating that other plexin family mem-bers might compensate for the absence of Plexin‐A1 in these tissues. However,T cell‐mediated immunity is severely impaired in Plexin‐A1�/� mice. Thesemice are resistant to myelin oligodendrocyte protein (MOG)‐induced EAEbecause of the defective generation of MOG‐specific T cells. The defectiveT cell immunity is at least in part attributable to the impaired DC function inthese mice. Plexin‐A1�/� DCs poorly stimulate OT‐II TCR transgenic T cellsin the presence of ovalbumin‐derived peptides or allogeneic T cells comparedto wild‐type DCs. The ability of Plexin‐A1�/� DCs to bind and respond toSema6D is also severely impaired. These observations suggest that Sema6D onT cells may stimulate DCs through Plexin‐A1 during T cell–DC interactions,and this interaction may be required for the efficient generation of antigen‐specific T cells.
4.3. Sema6D–Plexin‐A1 Interactions in Osteoclastogenesis
Plexin‐A1�/� mice develop striking osteopetrosis. Osteopetrosis can be causedby the overactivation of osteoblasts and/or defective function of osteoclasts(Theill et al., 2002). Although osteoblast function in Plexin‐A1�/� mice isnormal, osteoclast differentiation is severely impaired in these mice. Thebones of Plexin‐A1�/� mice have decreased numbers of osteoclasts and re-duced osteoclast surface ratios, and these mice also exhibit decreased boneresorption markers such as deoxypyridinoline and collagen type I fragments.In vitro osteoclastogenesis from Plexin‐A1�/� bone marrow is reduced com-pared to wild‐type bone marrow. Furthermore, Sema6D�Fc enhances thein vitro induction of osteoclasts from bone marrow cells in the presence ofM‐CSF and RANKL (Takegahara et al., 2006).
4.4. Plexin‐A1 Forms a Receptor Complex with TREM‐2 and DAP12 inDCs and Osteoclasts
During chick cardiac morphogenesis, Plexin‐A1 differentially associates witheither off‐track or VEGFR2 and exerts two distinct biological activities(Toyofuku et al., 2004a). However, neither off‐track nor VEGFR2 is expressedin DCs and osteoclasts. In these cells, Plexin‐A1 associates with the triggeringreceptor expressed on myeloid cell‐2 (TREM‐2)–DAP12 complex instead of

IMMUNE SEMAPHORINS 133
off‐track or VEGFR2 (Takegahara et al., 2006). DAP12 contains an immunor-eceptor tyrosine‐based activation motif (ITAM) in its cytoplasmic region andrecruits Src‐like tyrosine kinases such as ZAP‐70 and Syk. DAP12 forms acomplex with activating NK receptors including Ly49D, CD94/NKG2C, andKIR2DS and acts as a signaling adaptor molecule for these receptors (Lanierand Bakker, 2000). DAP12 also forms a complex with TREM‐1 or TREM‐2although the functions and ligands of TREM molecules remain unknown(Colonna, 2003). Interestingly, the phenotypes of DAP12�/� mice are some-what similar to those of Plexin‐A1�/� mice. DAP12�/� mice not only have adefect in T cell priming but also develop osteopetrosis that is caused bydefective osteoclastogenesis (Bakker et al., 2000; Kaifu et al., 2003). Addition-ally, the genetic mutations of DAP12 as well as the TREM‐2 result in defectiveosteoclast differentiation (Paloneva et al., 2000, 2002, 2003).
Coimmunoprecipitation analysis revealed that Plexin‐A1 associates directlywith TREM‐2 through the T cell, immunoglobulin‐like (TIG) domain of itsextracellular region (Takegahara et al., 2006). The association of DAP12 with itscounterparts such as TREMs and activating NK receptors is dependent on theinteraction between a negatively charged residue in the transmembrane domainof DAP12 and a positively charged residue in the transmembrane domain of itscounterparts. However, Plexin‐A1 does not contain such charged amino acidresidues in its transmembrane domain. When cells transfected with Plexin‐A1,TREM‐2, and DAP12 were stimulated with Sema6D, tyrosine phosphorylationof DAP12 was observed. Sema6D‐induced IL‐12 production is much lower inDCs treated with TREM‐2‐specific siRNA, and DAP12�/� DCs have a similarphenotype (Takegahara et al., 2006). Thus, DAP12 appears to associate withPlexin‐A1 indirectly through TREM‐2 and to deliver costimulatory signals toDCs. Given the impaired osteoclast function caused by the defect of the DAP12or TREM‐2 gene, these adaptor molecules might mediate Plexin‐A1 signalingalso in osteoclasts (Fig. 3).
The knockdown of TREM‐2 or the targeted mutation of the DAP12 genesubstantially reduces but does not completely abolish Sema6D‐induced IL‐12production in DCs (Takegahara et al., 2006). Thus, some other molecules inaddition to DAP12 may mediate Sema6D signaling. Many semaphorins mod-ulate the activities of Rho‐like small GTPases to induce growth cone collapseor axon turning (Kruger et al., 2005; Pasterkamp and Kolodkin, 2003).For example, Sema3A activates Rac GTPase in dorsal root ganglion (DRG)neurons through the neuropilin‐1–Plexin‐A1 complex. Interestingly, Sema6Dalso induces Rac GTPase activation in DCs (Takegahara et al., 2006), and thisis not affected by DAP12 deficiency. Therefore, there may be at least twosignaling pathways: one involves DAP12 and Src‐like kinases and the other RacGTPase (Fig. 3). Although it is unclear whether there is a cross talk between

Sema6D
Plexin-A1
TREM-2
DAP12
ITAM
Src tyrosine kinasesPP
RacRac
Actin dynamics?DC activation
osteoclastogenesis
GDPGTP
Figure 3 Sema6D acts on DCs and osteoclasts through the Plexin‐A1–TREM‐2–DAP12 complex.In DCs and osteoclasts, Plexin‐A1 associates with TREM‐2, linking signals to the ITAM‐containingadaptor protein DAP12. The binding of Sema6D to Plexin‐A1 induces phosphorylation of DAP12,leading to the recruitment of Src family tyrosine kinases. The Sema6D signal mediated by thisreceptor complex results in the activation of DCs and osteoclastogenesis. Sema6D also induces RacGTPase activation in DCs, suggesting that the interaction of Sema6D with Plexin‐A1 may beinvolved in cytoskeletal rearrangements and DC motility.
134 HITOSHI KIKUTANI ET AL .
these two signaling pathways, the Rac GTPase pathway suggests that theSema6D–Plexin‐A1 interaction may also regulate DC motility.Mature DCs engulf T cells after establishing initial contact between their
dendrites and T cells. Interestingly, DCs that are deficient in both Rac1 andRac2 fail to migrate toward and engulf T cells, and they also poorly prime naiveT cells (Benvenuti et al., 2004). These data suggest that Rac GTPases areessential for the motility of DCs during their initial encounters with T cells.The extension of dendrites and cell movement involve the polymerization ofactin cytoskeleton and the formation of focal adhesions at the leading edge.Rac GTPases enhance actin polymerization together with Cdc42 (Fukata et al.,2003; Raftopoulou and Hall, 2004), thus suggesting an attractive scenariowherein Sema6D‐induced Rac activation may be involved in dendrite extensionand displacement of DCs during their interactions with T cells. However, a

IMMUNE SEMAPHORINS 135
role for Rac GTPases in semaphorin signaling is still controversial. Sema3A andSema6D induce Rac activation in DRG neurons and ventricular endocardiaccells (Pasterkamp andKolodkin, 2003; Toyofuku et al., 2004a), leading to growthcone collapse, repulsion of DRG neurons and inhibition of cardiac cell migra-tion, respectively. Additionally, Sema3A‐induced Rac activation appears neces-sary to trigger signals downstream of Plexin‐A1 and suppress cell adhesion anddepolymerization of actin cytoskeleton in DRG neurons (Toyofuku et al., 2005).Further studies are needed to more clearly determine the role of Sema6D‐induced Rac activation in DC function.
5. Sema7A
The GPI‐anchored semaphorin Sema7A, also known as CD108, was identifiedin a search for vertebrate homologues of AHVsema encoded by alcelaphineherpesvirus (Lange et al., 1998; Xu et al., 1998). Sema7A transcripts aredetectable in the nervous system during embryonic development, as well asin various adult tissues, including brain, spinal cord, lung, testis, spleen, andthymus (Mine et al., 2000; Xu et al., 1998; Yamada et al., 1999). In hemato-poietic cells, Sema7A is expressed in erythrocytes and is also known as theJohn‐Milton‐Hagen factor defining a human blood group (Bobolis et al., 1992).Its expression is also observed on activated peripheral blood lymphocytes(Yamada et al., 1999) and thymocytes doubly positive for CD4 and CD8(Mine et al., 2000). An earlier in vitro‐binding study identified Plexin‐C1 asa receptor for Sema7A (Tamagnone et al., 1999). Notably, however, Sema7Acontains an arginine‐glycine‐aspartate (RGD) sequence, a well‐conservedintegrin‐binding motif (Hynes, 2002). Indeed, a neurological study demon-strated that Sema7A promotes axon outgrowth through b1 integrin receptors,but not Plexin‐C1, and contributes to the lateral olfactory tract formation(Pasterkamp et al., 2003).
5.1. Sema7A as a Monocyte Stimulator
An immune function for Sema7A was first described in monocytes (Holmeset al., 2002). Recombinant soluble Sema7A protein stimulates human periph-eral blood monocytes to induce superoxide release and proinflammatory cyto-kine production, including IL‐1b, IL‐6, TNF‐a, and IL‐8. The recombinantprotein also acts as a chemoattractant for monocytes with much more potencythan canonical chemokines. Although the mechanism underlying these activ-ities of Sema7A was unclear, we recently found that Sema7A binds andactivates human monocytes and mouse macrophages through an integrinreceptor (K. S. and H. K., unpublished data). This suggests that Sema7A

GPI-linkage
Sema7A
A B
TCR
CD3
Sema7A
T cell
Cytokine productionchemotaxis
Downregulation of TCR signals
Integrin
Monocyte
RGD motifa
b
Figure 4 Sema7A activates monocytes but negatively regulates T cells. (A) Sema7A stimulatesmonocytes to produce proinflammatory cytokines and undergo chemotaxis. In line with thegrowth‐promoting effect on neuronal axons, monocyte activation by Sema7A is mediated probablyby an integrin receptor. (B) In T cells, Sema7A is supposed to associate the TCR complex on thecell surface and downregulate signals emanating from the TCR. The opposite regulatory roles ofSema7A in immune cells indicate the need for further studies of this protein.
136 HITOSHI KIKUTANI ET AL .
may utilize the identical receptor and signal transduction machinery in boththe nervous and immune systems (Fig. 4A).
5.2. Sema7A as a Negative Regulator for T Cells
Medzhitov and colleagues reported that Sema7A negatively regulates T cell‐mediated immune responses in a T cell autonomous manner (Czopik et al.,2006). They showed that Sema7A�/� T cells were hyperproliferative both inresponse to antigenic stimuli and under homeostatic conditions. Consistentwith this, Sema7A�/� mice experienced highly aggressive course of MOG‐induced EAE compared with wild‐type animals. However, the mechanismby which Sema7A deficiency leads to T cell hyperactivity remains unclear.Although TCR internalization following anti‐CD3 antibody cross‐linking wasdefective in Sema7A�/� T cells, only a slight enhancement in TCR‐mediatedsignaling was seen in these cells. Since GPI‐anchored proteins reside inspecialized signaling modules, lipid rafts, where immune receptors accumu-late, it has been assumed that Sema7A interacts in cis with TCR complexes in

IMMUNE SEMAPHORINS 137
the lipid rafts and downmodulates TCR‐mediated T cell responses by unde-fined mechanisms (Fig. 4B).
The reported phenotype of Sema7A�/� mice appears inconsistent with theability of Sema7A to potently stimulate monocytes/macrophages, major effec-tor cells in T cell mediated immunity. Indeed, in our experiments, Sema7A�/�
mice are impaired in their ability to develop not only EAE but also contacthypersensitivity (K. S. and H. K., unpublished data). Thus, further studies areneeded to clarify the role of Sema7A in immune responses.
6. Other Semaphorins
6.1. Viral Semaphorins
Viruses encode proteins within their own genome that facilitate infectiousprocesses or support viral transmission. Interestingly, not only the aforemen-tioned alcelaphine herpesvirus but also numerous poxviruses encode sema-phorins (Ensser and Fleckenstein, 1995; Kolodkin et al., 1993). Vaccinia virussemaphorin A39R binds Plexin‐C1, which is also a receptor for AHVsema, andinduces robust responses in human monocytes, including cell aggregation,proinflammatory cytokine production, and upregulation of the monocyte cellsurface marker CD54 (ICAM‐1; Comeau et al., 1998). Additionally, studieshave demonstrated that A39R suppresses integrin‐mediated adhesion andmigration (Walzer et al., 2005a) and attenuates the phagocytotic capacity ofDCs (Walzer et al., 2005b). These observations suggest that viral semaphorinsmight play dual roles in the host. They can enhance inflammation and deterio-rate the disease by activating the host immune system, but in contrast, they canbe a means for viruses to evade immune surveillance by suppressing immunecell functions.
6.2. Class III Semaphorins
Sema3A is the human homologue of collapsin‐1, the first identified vertebratesemaphorin (Kolodkin et al., 1993). Extensive studies in the nervous systemhave established a role for Sema3A as an axonal guidance factor duringdevelopment. Interestingly, several lines of evidence suggest that Sema3Aalso affects immune cell functions. Consistent with its chemorepulsive activityon neurons, Delaire (2001) reported that Sema3A inhibited the spontaneousmigration of monocytes in a transwell assay. Studies have also demonstratedthat Sema3A is secreted from activated T cells, DCs (Lepelletier et al., 2006)and various types of tumor cells (Catalano et al., 2006). Additionally, treatmentof T cells with recombinant or tumor cell‐derived Sema3A inhibited

138 HITOSHI KIKUTANI ET AL .
TCR‐mediated proliferation and cytokine production by downregulating theMAPK signaling cascades (Catalano et al., 2006; Lepelletier et al., 2006).These observations suggest a possibility that Sema3A may serve as a negativeregulator for T cells in physiological and pathological immune responses.Although the inhibition of T cell function is mediated by neuropilin‐1, itscoreceptor on neuronal cells, Plexin‐A1, is not expressed by T cells (Catalanoet al., 2006). Thus, Sema3A might act on T cells through other members of thePlexin‐A subfamily.
7. Summary and Perspectives
Accumulating evidence has established the semaphorin family as a novel classof immunoregulatory molecules. Although detailed investigations have beenperformed on only a limited number of the family members, they are clearlyinvolved in various phases of the immune response through several distinctmechanisms. Each of two class IV semaphorins, Sema4D and Sema4A, makesa respective contribution to homeostatic maintenance of B cell subsets anddifferentiation of effector T cells. Sema6D as well as both of the class IVsemaphorins participate in cognate interactions between T cells and DCs,enabling optimal T cell priming against antigens. In addition, considering thepotent stimulatory capacity on monocytes, Sema7A might play a role in theeffector phases of immune responses. Moreover, several family members arecrucially involved in the development or progress of animal models of autoim-mune and inflammatory diseases. For example, antibody blockade or geneticdeletion of Sema4D (Kumanogoh et al., 2002b), Sema4A (Kumanogoh et al.,2002a), or Sema6D (Takegahara et al., 2006) ameliorates the clinical course ofEAE. Conversely, Sema4D‐deficient mice develop autoimmune diseases alongwith elevated levels of various types of autoantibodies (Kumanogoh et al.,2005b). Thus, the manipulation of the semaphorin functions could providenovel avenues for therapeutic strategies for the treatment of these immunedisorders.Early studies on class IV semaphorins, Sema4D and Sema4A, have revealed
that they exert costimulation‐like activities during immune responses throughreceptor systems different from those in the nervous system (Kumanogoh et al.,2000, 2002a). However, as mentioned above, a study has demonstrated thatSema6D functions in the immune system through Plexin‐A1 (Takegahara et al.,2006), which is a prototype of semaphorin receptors identified in the nervoussystem, suggesting that certain semaphorins may exert immunological activitiesthrough the mechanisms similar to those seen in the nervous system. Indeed,semaphorins have been shown to regulate migration of immune cells (Delaireet al., 2001; Holmes et al., 2002). Therefore, the mechanisms by which

IMMUNE SEMAPHORINS 139
semaphorins are involved in immune regulation will be more complex thanpreviously expected. Further studies will be required to outline the roles ofsemaphorin molecules in the immune system. Finally, increased understandingof the ‘‘immune semaphorins’’ could provide valuable insights into the potentialfunction of semaphorins in the other systems as well as facilitate the creation ofa comprehensive model detailing the wide array of physiological processesregulated by this interesting family of proteins.
Acknowledgments
We thank K. Kubota for excellent secretarial assistance. We are also grateful to N. Takegahara andM. Mizui for the creation of artworks. This study was supported by the following funding agencies:The Ministry of Education, Culture, Sports, Science, and Technology, Japan and The CoreResearch for Evolutional Science and Technology (CREST) program of the Japanese Scienceand Technology Agency (JST) to A.K. and H.K, and Research Fellowships of the Japan Society forthe Promotion of Science for Young Scientists to K.S.
References
Adachi, T., Flaswinkel, H., Yakura, H., Reth, M., and Tsubata, T. (1998). The B cell surface proteinCD72 recruits the tyrosine phosphatase SHP‐1 upon tyrosine phosphorylation. J. Immunol. 160,4662–4665.
Adachi, T., Wienands, J., Wakabayashi, C., Yakura, H., Reth, M., and Tsubata, T. (2001). SHP‐1requires inhibitory co‐receptors to down‐modulate B cell antigen receptor‐mediated phosphor-ylation of cellular substrates. J. Biol. Chem. 276, 26648–26655.
Bakker, A. B., Hoek, R. M., Cerwenka, A., Blom, B., Lucian, L., McNeil, T., Murray, R., Phillips,L. H., Sedgwick, J. D., and Lanier, L. L. (2000). DAP12–deficient mice fail to developautoimmunity due to impaired antigen priming. Immunity 13, 345–353.
Benvenuti, F., Hugues, S., Walmsley, M., Ruf, S., Fetler, L., Popoff, M., Tybulewicz, V. L., andAmigorena, S. (2004). Requirement of Rac1 and Rac2 expression by mature dendritic cells forT cell priming. Science 305, 1150–1153.
Bobolis, K. A., Moulds, J. J., and Telen, M. J. (1992). Isolation of the JMH antigen on a novelphosphatidylinositol‐linked human membrane protein. Blood 79, 1574–1581.
Bougeret, C., Mansur, I. G., Dastot, H., Schmid, M., Mahouy, G., Bensussan, A., and Boumsell, L.(1992). Increased surface expression of a newly identified 150‐kDa dimer early after humanT lymphocyte activation. J. Immunol. 148, 318–323.
Catalano, A., Caprari, P., Moretti, S., Faronato, M., Tamagnone, L., and Procopio, A. (2006).Semaphorin‐3A is expressed by tumor cells and alters T‐cell signal transduction and function.Blood 107, 3321–3329.
Chakravarti, S., Sabatos, C. A., Xiao, S., Illes, Z., Cha, E. K., Sobel, R. A., Zheng, X. X., Strom,T. B., and Kuchroo, V. K. (2005). Tim‐2 regulates T helper type 2 responses and autoimmunity.J. Exp. Med. 202, 437–444.
Colonna, M. (2003). TREMs in the immune system and beyond. Nat. Rev. Immunol. 3, 445–453.Comeau, M. R., Johnson, R., DuBose, R. F., Petersen, M., Gearing, P., VandenBos, T., Park, L.,
Farrah, T., Buller, R.M., Cohen, J. I., Strockbine, L.D., Rauch, C., et al. (1998). A poxvirus‐encoded

140 HITOSHI KIKUTANI ET AL .
semaphorin induces cytokine production frommonocytes and binds to a novel cellular semaphorinreceptor, VESPR. Immunity 8, 473–482.
Czopik, A. K., Bynoe, M. S., Palm, N., Raine, C. S., and Medzhitov, R. (2006). Semaphorin 7A is anegative regulator of T cell responses. Immunity 24, 591–600.
Delaire, S., Elhabazi, A., Bensussan, A., and Boumsell, L. (1998). CD100 is a leukocyte sema-phorin. Cell. Mol. Life Sci. 54, 1265–1276.
Delaire, S., Billard, C., Tordjman, R., Chedotal, A., Elhabazi, A., Bensussan, A., and Boumsell, L.(2001). Biological activity of soluble CD100. II. Soluble CD100, similarly to H‐SemaIII, inhibitsimmune cell migration. J. Immunol. 166, 4348–4354.
Ensser, A., and Fleckenstein, B. (1995). Alcelaphine herpesvirus type 1 has a semaphorin‐likegene. J. Gen. Virol. 76, 1063–1067.
Fukata, M., Nakagawa, M., and Kaibuchi, K. (2003). Roles of Rho‐family GTPases in cell polarisa-tion and directional migration. Curr. Opin. Cell Biol. 15, 590–597.
Furuyama, T., Inagaki, S., Kosugi, A., Noda, S., Saitoh, S., Ogata, M., Iwahashi, Y., Miyazaki, N.,Hamaoka, T., and Tohyama, M. (1996). Identification of a novel transmembrane semaphorinexpressed on lymphocytes. J. Biol. Chem. 271, 33376–33381.
Fusaki, N., Tomita, S., Wu, Y., Okamoto, N., Goitsuka, R., Kitamura, D., and Hozumi, N. (2000).BLNK is associated with the CD72/SHP‐1/Grb2 complex in the WEHI231 cell line aftermembrane IgM cross‐linking. Eur. J. Immunol. 30, 1326–1330.
Gu, C., Yoshida, Y., Livet, J., Reimert, D. V., Mann, F., Merte, J., Henderson, C. E., Jessell, T. M.,Kolodkin, A. L., and Ginty, D. D. (2005). Semaphorin 3E and plexin‐D1 control vascular patternindependently of neuropilins. Science 307, 265–268.
Hall, K. T., Boumsell, L., Schultze, J. L., Boussiotis, V. A., Dorfman, D. M., Cardoso, A. A.,Bensussan, A., Nadler, L. M., and Freeman, G. J. (1996). Human CD100, a novel leukocytesemaphorin that promotes B‐cell aggregation and differentiation. Proc. Natl. Acad. Sci. USA 93,11780–11785.
He, Z., and Tessier‐Lavigne, M. (1997). Neuropilin is a receptor for the axonal chemorepellentSemaphorin III. Cell 90, 739–751.
Holmes, S., Downs, A., Fosberry, A., Hayes, P., Michalovich, D., Murdoch, P., Moores, K., Fox, J.,Deen, K., Pettman, G., Wattam, T., and Lewis, C. (2002). Sema7A is a potent monocytestimulator. Scand. J. Immunol. 56, 270–275.
Hynes, R. O. (2002). Integrins: Bidirectional, allosteric signaling machines. Cell 110, 673–687.Ishida, I., Kumanogoh, A., Suzuki, K., Akahani, S., Noda, K., and Kikutani, H. (2003). Involvementof CD100, a lymphocyte semaphorin, in the activation of the human immune system via CD72:Implications for the regulation of immune and inflammatory responses. Int. Immunol. 15,1027–1034.
Kaifu, T., Nakahara, J., Inui, M., Mishima, K., Momiyama, T., Kaji, M., Sugahara, A., Koito, H.,Ujike‐Asai, A., Nakamura, A., Kanazawa, K., Tan‐Takeuchi, K., et al. (2003). Osteopetrosis andthalamic hypomyelinosis with synaptic degeneration in DAP12‐deficient mice. J. Clin. Invest.111, 323–332.
Kitsukawa, T., Shimono, A., Kawakami, A., Kondoh, H., and Fujisawa, H. (1995). Overexpressionof a membrane protein, neuropilin, in chimeric mice causes anomalies in the cardiovascularsystem, nervous system and limbs. Development 121, 4309–4318.
Kolodkin, A. L., Matthes, D. J., and Goodman, C. S. (1993). The semaphorin genes encode a familyof transmembrane and secreted growth cone guidance molecules. Cell 75, 1389–1399.
Kolodkin, A. L., Levengood, D. V., Rowe, E. G., Tai, Y. T., Giger, R. J., and Ginty, D. D. (1997).Neuropilin is a semaphorin III receptor. Cell 90, 753–762.
Kruger, R. P., Aurandt, J., and Guan, K. L. (2005). Semaphorins command cells to move. Nat. Rev.Mol. Cell. Biol. 6, 789–800.

IMMUNE SEMAPHORINS 141
Kumanogoh, A., and Kikutani, H. (2003). Immune semaphorins: A new area of semaphorinresearch. J. Cell Sci. 116, 3463–3470.
Kumanogoh, A., Watanabe, C., Lee, I., Wang, X., Shi, W., Araki, H., Hirata, H., Iwahori, K.,Uchida, J., Yasui, T., Matsumoto, M., Yoshida, K., et al. (2000). Identification of CD72 as alymphocyte receptor for the class IV semaphorin CD100: A novel mechanism for regulating Bcell signaling. Immunity 13, 621–631.
Kumanogoh, A., Marukawa, S., Suzuki, K., Takegahara, N., Watanabe, C., Ch’ng, E., Ishida, I.,Fujimura, H., Sakoda, S., Yoshida, K., and Kikutani, H. (2002a). Class IV semaphorin Sema4Aenhances T‐cell activation and interacts with Tim‐2. Nature 419, 629–633.
Kumanogoh, A., Suzuki, K., Ch’ng, E., Watanabe, C., Marukawa, S., Takegahara, N., Ishida, I.,Sato, T., Habu, S., Yoshida, K., Shi, W., and Kikutani, H. (2002b). Requirement for thelymphocyte semaphorin, CD100, in the induction of antigen‐specific T cells and the maturationof dendritic cells. J. Immunol. 169, 1175–1181.
Kumanogoh, A., Shikina, T., Suzuki, K., Uematsu, S., Yukawa, K., Kashiwamura, S., Tsutsui, H.,Yamamoto, M., Takamatsu, H., Ko‐Mitamura, E. P., Takegahara, N., Marukawa, S., et al.(2005a). Nonredundant roles of Sema4A in the immune system: Defective T cell priming andTh1/Th2 regulation in Sema4A‐deficient mice. Immunity 22, 305–316.
Kumanogoh, A., Shikina, T., Watanabe, C., Takegahara, N., Suzuki, K., Yamamoto, M., Takamatsu,H., Prasad, D. V., Mizui, M., Toyofuku, T., Tamura, M., Watanabe, D., et al. (2005b). Require-ment for CD100–CD72 interactions in fine‐tuning of B‐cell antigen receptor signaling andhomeostatic maintenance of the B‐cell compartment. Int. Immunol. 17, 1277–1282.
Kurosaki, T. (2002). Regulation of B cell fates by BCR signaling components. Curr. Opin.Immunol. 14, 341–347.
Lange, C., Liehr, T., Goen,M., Gebhart, E., Fleckenstein, B., and Ensser, A. (1998). New eukaryoticsemaphorins with close homology to semaphorins of DNA viruses. Genomics 51, 340–350.
Lanier, L. L., and Bakker, A. B. (2000). The ITAM‐bearing transmembrane adaptor DAP12 inlymphoid and myeloid cell function. Immunol. Today 21, 611–614.
Lepelletier, Y., Moura, I. C., Hadj‐Slimane, R., Renand, A., Fiorentino, S., Baude, C., Shirvan, A.,Barzilai, A., and Hermine, O. (2006). Immunosuppressive role of semaphorin‐3A on T cellproliferation is mediated by inhibition of actin cytoskeleton reorganization. Eur. J. Immunol. 36,1782–1793.
McIntire, J. J., Umetsu, S. E., Akbari, O., Potter, M., Kuchroo, V. K., Barsh, G. S., Freeman, G. J.,Umetsu, D. T., and DeKruyff, R. H. (2001). Identification of Tapr (an airway hyperreactivityregulatory locus) and the linked Tim gene family. Nat. Immunol. 2, 1109–1116.
McIntire, J. J., Umetsu, S. E., Macaubas, C., Hoyte, E. G., Cinnioglu, C., Cavalli‐Sforza, L. L.,Barsh, G. S., Hallmayer, J. F., Underhill, P. A., Risch, N. J., Freeman, G. J., et al. (2003).Immunology: Hepatitis A virus link to atopic disease. Nature 425, 576.
Mine, T., Harada, K., Matsumoto, T., Yamana, H., Shirouzu, K., Itoh, K., and Yamada, A. (2000).CDw108 expression during T‐cell development. Tissue Antigens 55, 429–436.
Monney, L., Sabatos, C. A., Gaglia, J. L., Ryu, A., Waldner, H., Chernova, T., Manning, S.,Greenfield, E. A., Coyle, A. J., Sobel, R. A., Freeman, G. J., and Kuchroo, V. K. (2002). Th1–specific cell surface protein Tim‐3 regulates macrophage activation and severity of an auto-immune disease. Nature 415, 536–541.
Negishi, M., Oinuma, I., and Katoh, H. (2005). R‐ras as a key player for signaling pathway ofplexins. Mol. Neurobiol. 32, 217–222.
Niiro, H., and Clark, E. A. (2002). Regulation of B‐cell fate by antigen‐receptor signals. Nat. Rev.Immunol. 2, 945–956.
O’Keefe, T. L., Williams, G. T., Davies, S. L., and Neuberger, M. S. (1996). Hyperresponsive Bcells in CD22–deficient mice. Science 274, 798–801.

142 HITOSHI KIKUTANI ET AL .
Paloneva, J., Kestila, M., Wu, J., Salminen, A., Bohling, T., Ruotsalainen, V., Hakola, P., Bakker,A. B., Phillips, J. H., Pekkarinen, P., Lanier, L. L., Timonen, T., et al. (2000). Loss‐of‐functionmutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat. Genet. 25,357–361.
Paloneva, J., Manninen, T., Christman, G., Hovanes, K., Mandelin, J., Adolfsson, R., Bianchin, M.,Bird, T., Miranda, R., Salmaggi, A., Tranebjaerg, L., Konttinen, Y., et al. (2002). Mutations in twogenes encoding different subunits of a receptor signaling complex result in an identical diseasephenotype. Am. J. Hum. Genet. 71, 656–662.
Paloneva, J., Mandelin, J., Kiialainen, A., Bohling, T., Prudlo, J., Hakola, P., Haltia, M., Konttinen,Y. T., and Peltonen, L. (2003). DAP12/TREM2 deficiency results in impaired osteoclast differ-entiation and osteoporotic features. J. Exp. Med. 198, 669–675.
Pan, C., Baumgarth, N., and Parnes, J. R. (1999). CD72–deficient mice reveal nonredundant rolesof CD72 in B cell development and activation. Immunity 11, 495–506.
Pasterkamp, R. J., and Kolodkin, A. L. (2003). Semaphorin junction: Making tracks toward neuralconnectivity. Curr. Opin. Neurobiol. 13, 79–89.
Pasterkamp, R. J., Peschon, J. J., Spriggs, M. K., and Kolodkin, A. L. (2003). Semaphorin 7Apromotes axon outgrowth through integrins and MAPKs. Nature 424, 398–405.
Raftopoulou, M., and Hall, A. (2004). Cell migration: Rho GTPases lead the way. Dev. Biol. 265,23–32.
Rennert, P. D., Ichimura, T., Sizing, I. D., Bailly, V., Li, Z., Rennard, R., McCoon, P., Pablo, L.,Miklasz, S., Tarilonte, L., and Bonventre, J. V. (2006). T cell, Ig domain, mucin domain‐2 gene‐deficient mice reveal a novel mechanism for the regulation of TH2 immune responses andairway inflammation. J. Immunol. 177, 4311–4321.
Sato, S., Miller, A. S., Inaoki, M., Bock, C. B., Jansen, P. J., Tang, M. L., and Tedder, T. F. (1996).CD22 is both a positive and negative regulator of B lymphocyte antigen receptor signaltransduction: Altered signaling in CD22–deficient mice. Immunity 5, 551–562.
Sekido, Y., Bader, S., Latif, F., Chen, J. Y., Duh, F. M., Wei, M. H., Albanesi, J. P., Lee, C. C.,Lerman, M. I., and Minna, J. D. (1996). Human semaphorins A(V) and IV reside in the 3p21.3small cell lung cancer deletion region and demonstrate distinct expression patterns. Proc. Natl.Acad. Sci. USA 93, 4120–4125.
Shi, W., Kumanogoh, A., Watanabe, C., Uchida, J., Wang, X., Yasui, T., Yukawa, K., Ikawa, M.,Okabe, M., Parnes, J. R., Yoshida, K., and Kikutani, H. (2000). The class IV semaphorin CD100plays nonredundant roles in the immune system: Defective B and T cell activation in CD100–deficient mice. Immunity 13, 633–642.
Spriggs, M. K. (1999). Shared resources between the neural and immune systems: Semaphorinsjoin the ranks. Curr. Opin. Immunol. 11, 387–391.
Swiercz, J. M., Kuner, R., Behrens, J., and Offermanns, S. (2002). Plexin‐B1 directly interacts withPDZ‐RhoGEF/LARG to regulate RhoA and growth cone morphology. Neuron 35, 51–63.
Szabo, S. J., Kim, S. T., Costa, G. L., Zhang, X., Fathman, C. G., and Glimcher, L. H. (2000). Anovel transcription factor, T‐bet, directs Th1 lineage commitment. Cell 100, 655–669.
Takahashi, T., Fournier, A., Nakamura, F., Wang, L. H., Murakami, Y., Kalb, R. G., Fujisawa, H.,and Strittmatter, S. M. (1999). Plexin‐neuropilin‐1 complexes form functional semaphorin‐3Areceptors. Cell 99, 59–69.
Takegahara, N., Takamatsu, H., Toyofuku, T., Tsujimura, T., Okuno, T., Yukawa, K., Mizui, M.,Yamamoto, M., Prasad, D. V., Suzuki, K., Ishii, M., Terai, K., et al. (2006). Plexin‐A1 and itsinteraction with DAP12 in immune responses and bone homeostasis. Nat. Cell Biol. 8, 615–622.
Tamagnone, L., and Comoglio, P. M. (2000). Signalling by semaphorin receptors: Cell guidanceand beyond. Trends Cell Biol. 10, 377–383.

IMMUNE SEMAPHORINS 143
Tamagnone, L., Artigiani, S., Chen, H., He, Z., Ming, G. I., Song, H., Chedotal, A., Winberg,M. L., Goodman, C. S., Poo, M., Tessier‐Lavigne, M., and Comoglio, P. M. (1999). Plexins area large family of receptors for transmembrane, secreted, and GPI‐anchored semaphorins invertebrates. Cell 99, 71–80.
Tessier‐Lavigne, M., and Goodman, S. C. (1996). The molecular biology of axon guidance. Science274, 1123–1133.
Theill, L. E., Boyle, W. J., and Penninger, J. M. (2002). RANK‐L and RANK: T cells, bone loss, andmammalian evolution. Annu. Rev. Immunol. 20, 795–823.
Toyofuku, T., Zhang, H., Kumanogoh, A., Takegahara, N., Suto, F., Kamei, J., Aoki, K., Yabuki, M.,Hori, M., Fujisawa, H., and Kikutani, H. (2004a). Dual roles of Sema6D in cardiac morphogen-esis through region‐specific association of its receptor, Plexin‐A1, with off‐track and vascularendothelial growth factor receptor type 2. Genes Dev. 18, 435–447.
Toyofuku, T., Zhang, H., Kumanogoh, A., Takegahara, N., Yabuki, M., Harada, K., Hori, M., andKikutani, H. (2004b). Guidance of myocardial patterning in cardiac development by Sema6Dreverse signalling. Nat. Cell Biol. 6, 1204–1211.
Toyofuku, T., Yoshida, J., Sugimoto, T., Zhang, H., Kumanogoh, A., Hori, M., and Kikutani, H.(2005). FARP2 triggers signals for Sema3A‐mediated axonal repulsion. Nat. Neurosci. 8,1712–1719.
Walzer, T., Galibert, L., Comeau, M. R., and De Smedt, T. (2005a). Plexin C1 engagement onmouse dendritic cells by viral semaphorin A39R induces actin cytoskeleton rearrangement andinhibits integrin‐mediated adhesion and chemokine‐induced migration. J. Immunol. 174, 51–59.
Walzer, T., Galibert, L., and De Smedt, T. (2005b). Poxvirus semaphorin A39R inhibits phagocyto-sis by dendritic cells and neutrophils. Eur. J. Immunol. 35, 391–398.
Watanabe, C., Kumanogoh, A., Shi, W., Suzuki, K., Yamada, S., Okabe, M., Yoshida, K., andKikutani, H. (2001). Enhanced immune responses in transgenic mice expressing a truncatedform of the lymphocyte semaphorin CD100. J. Immunol. 167, 4321–4328.
Winberg, M. L., Noordermeer, J. N., Tamagnone, L., Comoglio, P. M., Spriggs, M. K., Tessier‐Lavigne, M., and Goodman, C. S. (1998). Plexin A is a neuronal semaphorin receptor thatcontrols axon guidance. Cell 95, 903–916.
Wong, A. W., Brickey, W. J., Taxman, D. J., van Deventer, H. W., Reed, W., Gao, J. X., Zheng, P.,Liu, Y., Li, P., Blum, J. S., McKinnon, K. P., and Ting, J. P. (2003). CIITA‐regulated plexin‐A1affects T‐cell‐dendritic cell interactions. Nat. Immunol. 4, 891–898.
Wu, Y., Nadler, M. J., Brennan, L. A., Gish, G. D., Timms, J. F., Fusaki, N., Jongstra‐Bilen, J., Tada,N., Pawson, T., Wither, J., Neel, B. G., and Hozumi, N. (1998). The B‐cell transmembraneprotein CD72 binds to and is an in vivo substrate of the protein tyrosine phosphatase SHP‐1.Curr. Biol. 8, 1009–1017.
Xu, X., Ng, S., Wu, Z. L., Nguyen, D., Homburger, S., Seidel‐Dugan, C., Ebens, A., and Luo, Y.(1998). Human semaphorin K1 is glycosylphosphatidylinositol‐linked and defines a new sub-family of viral‐related semaphorins. J. Biol. Chem. 273, 22428–22434.
Yamada, A., Kubo, K., Takeshita, T., Harashima, N., Kawano, K., Mine, T., Sagawa, K., Sugamura,K., and Itoh, K. (1999). Molecular cloning of a glycosylphosphatidylinositol‐anchored moleculeCDw108. J. Immunol. 162, 4094–4100.
Yu, H. H., and Kolodkin, A. L. (1999). Semaphorin signaling: A little less per‐plexin. Neuron 22,11–14.

a#
Tec Kinases in T Cell and Mast Cell Signaling
Martin Felices, Markus Falk, Yoko Kosaka, and Leslie J. Berg
Department of Pathology, University of Massachusetts Medical School, Massachusetts
A
dv2
bstract............................................................................................................. 1
145ances in immunology, vol. 93 0065-2776/07 $
007 Elsevier Inc. All rights reserved. DOI: 10.1016/S0065-2776(06)93
45
1. I ntroduction ....................................................................................................... 1 45 2. S ubcellular Localization of Tec Kinases ................................................................... 1 47 3. T ec Kinases in Signaling Pathways .......................................................................... 1 51 4. R egulation of Tec Kinase Activation........................................................................ 1 60 5. D istinct Versus Redundant Functions of Tec Kinases ................................................. 1 63 6. T ec Kinases in Mast Cell Signaling ......................................................................... 1 66 7. C onclusions........................................................................................................ 1 72R
eferences ......................................................................................................... 1 72Abstract
The Tec family of tyrosine kinases consists of five members (Itk, Rlk, Tec, Btk,and Bmx) that are expressed predominantly in hematopoietic cells. The excep-tions, Tec and Bmx, are also found in endothelial cells. Tec kinases constitutethe second largest family of cytoplasmic protein tyrosine kinases. While B cellsexpress Btk and Tec, and T cells express Itk, Rlk, and Tec, all four of thesekinases (Btk, Itk, Rlk, and Tec) can be detected in mast cells. This chapter willfocus on the biochemical and cell biological data that have been accumulatedregarding Itk, Rlk, Btk, and Tec. In particular, distinctions between the differ-ent Tec kinase family members will be highlighted, with a goal of providinginsight into the unique functions of each kinase. The known functions of Teckinases in T cell and mast cell signaling will then be described, with a particularfocus on T cell receptor and mast cell FceRI signaling pathways.
1. Introduction
1.1. Overview of Tec Kinases
Five Tec kinase family members have been described in mammalian cells.These kinases are highly expressed in hematopoietic cells, including B cells(Mano et al., 1993; Rawlings et al., 1993; Tsukada et al., 1993; Vetrie et al.,1993), T cells (Haire et al., 1994; Heyeck and Berg, 1993; Hu et al., 1995;Mano et al., 1993; Siliciano et al., 1992; Yamada et al., 1993), and mastcells (Kawakami et al., 1994, 1995; Sommers et al., 1995). The Tec kinasefamily members share a number of structural features. Each has a C‐terminalkinase domain, followed by an Src homology (SH)2 domain, and an SH3 do-main, much like Src family protein tyrosine kinases (PTKs) (Smith et al., 2001).
35.00
004-1

146 MARTIN FELICES ET AL .
However, unlike the Src kinases, Tec family kinases, with the exception of Bmx,possess a proline‐rich region (PRR) at the N‐terminal side of the SH3 domain;interestingly, Btk andTec have two of these regions,whereas Itk andRlk have onlyone. At the N‐terminal side of the PRR, in Itk, Btk, and Tec, there is a Zn2þ‐binding region known as the Btk homology (BH) motif. The combination of theBH and the PRR has been labeled the Tec homology (TH) domain. Finally, at theN‐terminal end, all Tec family kinases, with the exception of Rlk, possess apleckstrin homology (PH) domain; in Rlk, the N‐terminal region contains acysteine‐string motif (Berg et al., 2005). These structural differences, specificallythe difference in PRRs and N‐terminal domains, may contribute to the distinctfunctions of each Tec kinase.
1.2. Regulation of Tec Kinase Expression Levels
As a group, the Tec family kinases are predominantly expressed in hematopoie-tic cells; however, each individual Tec kinase has a distinct cell type‐specificpattern of expression. In addition, each cell type has a hierarchy of expressionlevels and functions for the Tec kinases expressed in that cell. Of the three Teckinases expressed in T cells, Itk, Rlk, and Tec, Itk appears to have the predomi-nant role in T cell receptor (TCR) signaling. Itk is expressed in thymocytesand mature T cells, and is found at maximal levels in the mature adult thymus(Gibson et al., 1993; Heyeck and Berg, 1993; Siliciano et al., 1992; Tanakaet al., 1993; Yamada et al., 1993). Similar to Itk, Rlk is expressed in thymocytesand mature resting T cells; however, Rlk mRNA levels are 3‐ to 10‐fold lowerthan the levels of Itk mRNA in resting T cells (Colgan et al., 2004; Hu et al.,1995; Miller et al., 2004; Sommers et al., 1995). Furthermore, unlike Itk, Rlkis dramatically downregulated at both the mRNA and protein levels on TCRstimulation (Hu et al., 1995; Sommers et al., 1995). The third Tec kinaseexpressed in T cells, Tec, is expressed at much lower levels in resting T cells,with mRNA levels �100‐fold lower than that of Itk (Berg et al., 2005). Inter-estingly, Tec is upregulated in T cells 2–3 days following their stimulation,suggesting a more important role for Tec in T cell effector function andrestimulation, rather than in Tcell development or initial activation (Tomlinsonet al., 2004b).Tec kinase levels are also individually regulated during T helper cell differ-
entiation. When naive CD4þ T cells differentiate into T helper (Th) cells, Itklevels increase approximately two‐ to threefold in Th2 cells versus Th1 cells,consistent with the known role of Itk in Th2 responses (Colgan et al., 2004;Miller et al., 2004). Similar to Itk, Tec is expressed at twofold higher levels inTh2 cells than in Th1 cells; however, in this latter case, the functional signifi-cance of this differential expression is not known, as Tec‐deficient mice have

Tec KINASES IN T CELL AND MAST CELL SIGNALING 147
no reported T cell signaling defects (Tomlinson et al., 2004b). In contrast to Itkand Tec, Rlk is downregulated following naive CD4þ T cell activation, and isreexpressed in Th1 cells, but not Th2 cells; these data have suggested a specificrole for Rlk in Th1 responses (Hu et al., 1995; Kashiwakura et al., 1999; Milleret al., 2004). For an in‐depth review of Tec kinases and their roles in T helpercell differentiation, please refer to articles by Schwartzberg et al. (2005) andKosaka et al. (2006).
2. Subcellular Localization of Tec Kinases
2.1. Regulation of Membrane Recruitment
Each Tec family kinase shows a distinct pattern of subcellular localization(Fig. 1). At steady state levels Itk and Tec are found in the cytoplasm; followingactivation of phosphalidylinositol (PI)‐3‐kinase (PI3K) and the generation ofPI(3,4,5)P3 (PIP3) at the plasma membrane, Itk and Tec are recruited to themembrane via their PH domains (Ching et al., 1999; Shan et al., 2000; Yanget al., 2001). In contrast, Rlk, which lacks a PH domain, is constitutively ass-ociated with the plasma membrane via its palmitoylated cysteine‐string motif.Thus, while Itk and Tec both require PI3K activity for plasma membraneassociation, Rlk lipid raft association is PI3K‐independent (Chamorro et al.,2001).
Following TCR stimulation and the activation of PI3K, Itk recruitment tothe membrane requires its PH domain and is independent of other domaininteractions (Bunnell et al., 2000; Ching et al., 1999). Deletion of the Itk PHdomain abolishes TCR activation‐induced colocalization of Itk with the TCRcomplex at the plasma membrane and also prevents the subsequent tyrosinephosphorylation and activation of Itk. Substitution of the PH domain of Itkwith a membrane localization (e.g., myristylation) sequence from Lck restoresItk membrane localization, but does not allow TCR signal‐induced tyrosinephosphorylation of Itk, indicating a more complex role for the PH domain thansimple plasma membrane association (Ching et al., 1999). Possible roles for thePH domain include recruitment to the immediate vicinity of the activatedreceptor, or a more structural role in Itk activation.
The regulation of Tec recruitment to the plasma membrane following TCRstimulation has some distinct features compared to Itk. Like Itk, Tec can berecruited to the membrane through the interaction of its PH domain withPIP3. However, the interaction of the Tec PH domain with PIP3 must bedifferent from that of the other Tec kinases, as illustrated by the behavior ofPH domain substitution mutants. For instance, substitution of the glutamicacid residue at position 41 with lysine in the Btk PH domain has been shown

Figure 1 Membrane localization of Tec family kinases. (A) The larger isoform of Rlk (RLK‐L) isconstitutively localized to the plasma membrane through palmitoylation of the cysteine string motifat the amino terminus (1). On T cell receptor (TCR) engagement (2), Rlk proteins in the vicinity ofthe TCR are activated by Fyn through phosphorylation of the Rlk kinase domain. (B) Prior to B cellreceptor (BCR) engagement, Btk is cytosolic. Following BCR stimulation (1), Lyn activates PI3K(2), leading to the production of PIP3 (3). Btk is then recruited to the plasma membrane throughinteraction of its PH domain with PIP3 (4). Lyn phosphorylates and activates Btk (5). (C) Prior toTCR engagement, Itk is cytosolic. Following TCR stimulation (1), Lck activates PI3K (2), leadingto the production of PIP3 (3). Itk is recruited to the plasma membrane through interaction of itsPH domain with PIP3 (4). Lck then phosphorylates and activates Itk (5). (D) Following TCRengagement (1), Lck activates PI3K (2), leading to the production of PIP3 (3). Tec is then recruitedinto vesicles at the plasma membrane that contain signaling components such as Lck and PLC‐g(4). Tec is then recruited to PIP3 through its PH domain (5) where it can be activated (presumablyby Lck in T cells and Lyn in B cells) (6).
148 MARTIN FELICES ET AL .
to increase Btk binding to PIP3; in contrast, the comparable mutation inthe PH domain of Tec (E42K) reduces Tec binding to PIP3 (Yang et al.,2001). In addition, while the Tec PH domain is required for tyrosine phos-phorylation of Tec, membrane recruitment could also be mediated by the TecSH2 domain (Yang et al., 2001). Finally, one study found that the PH domainof Tec is dispensable for Tec accumulation at the plasma membrane andinstead identified the SH3 domain as essential for accumulation of Tec at the

Tec KINASES IN T CELL AND MAST CELL SIGNALING 149
immunological synapse (Garcon et al., 2004a). Interestingly, these authors alsofound that a functional Tec PH domain is required for proper activation of Tecfollowing TCR stimulation, but suggest that membrane accumulation of Tec isnot PI3K‐dependent.
An interesting report indicates that in B cells, Btk can promote its ownsustained activation by a positive feedback loop (Saito et al., 2003). Btk bindsto PI5‐kinase, transporting it to the membrane following activation; at themembrane, PI5K converts PI(4)P into PI(4,5)P2, thereby providing a renewablesource of substrate for PI3K, and prolonging the activation signal.
Since membrane localization is a prerequisite for Tec kinase function inantigen receptor signaling pathways, these signals can be terminated by inhibi-tion of membrane recruitment. For Itk, recruitment to the plasma membraneis negatively regulated by the lipid phosphatase, phosphatase and tensin homo-logue deleted on chromosome 10 (PTEN), which removes phosphates from theD3 position of phosphoinositides, and thereby reduces the levels of PIP3 at themembrane (Shan et al., 2000). In PTEN‐deficient cells, such as the Jurkat Tcelltumor line, Itk is constitutively localized to the plasma membrane and hyper-responsive to TCR stimulation (Shan et al., 2000). This mechanism for negativeregulation sets Itk apart from Rlk, which has no dependence on phosphoinosi-tides for membrane localization. Membrane localization of Tec, in contrast, isregulated by the src homology 2‐containing inositol‐5‐phosphatase (SHIP)family of inositol phosphatases (Tomlinson et al., 2004a). The proposed mech-anism for this regulation is similar to that of PTEN, involving dephosphoryla-tion of PIP3 leading to decreased PH domain‐mediated recruitment of Tec tothe plasma membrane. However, unlike the indirect regulation of Itk byPTEN, Tec also seems to directly interact with SHIP1 and SHIP2, an interac-tion that is dependent on the Tec SH3 domain (Tomlinson et al., 2004a). On thebasis of this observation, it is possible that in conditions of PI3K‐independentrecruitment of Tec to the membrane, interaction of SHIP with the Tec SH3domain might be sufficient to preclude Tec membrane localization. One finalfeature that sets Tec apart from other family members is its unique subcellularlocalization on TCR‐induced activation. Whereas Itk appears diffusely loca-lized at the immunological synapse, Tec has a more punctuate localizationpattern at the T cell–APC interface, indicative of its presence in vesicularstructures (Tomlinson et al., 2004b). Formation and maintenance of theseTec‐containing vesicles require TCR signaling through Src‐family kinases andPI3K (Kane and Watkins, 2005). These vesicles also contain the early endoso-mal antigen 1 (EEA1) marker as well as signaling components such as Lck andthe Tec substrate PLC‐g1. In theory, this packaging of signaling componentsfacilitates signaling through Tec. No such assembly of signaling componentshas been described for any of the other Tec family kinases.

150 MARTIN FELICES ET AL .
2.2. Nuclear Localization and Functions of Tec Kinases
Several reports have also indicated that Tec kinasesmay have an important role inthe nucleus (Fig. 2). In this regard, the data for Rlk is the most compelling. Twoisoforms of Rlk, which arise from alternative sites of translation initiation on thesame mRNA have been described (Debnath et al., 1999). The larger 58‐kDaisoform is cytoplasmic and localizes to lipid rafts through the cysteine‐stringmotifon palmitoylation. The shorter 52‐kDa isoform lacks the cysteine‐stringmotif andlocalizes to the nucleus when expressed in the absence of the larger form.Consistent with these data, a mutation that abolishes palmitoylation in thecysteine‐string motif of the larger isoform allows this protein to migrate to thenucleus (Debnath et al., 1999). However, in spite of the fact that both isoformscontain a nuclear localization sequence (residues 57–71), both proteins are foundonly in the cytoplasm when coexpressed, suggesting a direct physical interactionbetween the two isoforms. While the data demonstrating the ability of Rlk totraffic to the nucleus are compelling, little is known about the function of nuclearRlk. One report indicated that Rlk could bind to the IFN‐g promoter region.Combined with its up‐regulation in Th1 cells, these findings suggest an importantfunction for Rlk in Th1 cell development and signaling (Kashiwakura et al., 1999).
Figure 2 Proposed mechanisms of Tec family kinase nuclear localization. (A) On TCR engage-ment (1), the short form of Rlk (RLK‐S), usually found in a complex with the long form (RLK‐L),migrates to the nucleus (2). Once in the nucleus RLK‐S binds to the promoter region of the IFN‐ggene (3). (B) On TCR engagement (1), the SH3 region of Itk interacts with the PRR region of thenuclear transporter Karyopherin alpha (Rch1a) (2). The Itk–Rch1a complex translocates intothe nucleus (3). In the nucleus, Itk may bind T‐bet, a master regulator of IFN‐g transcription (4).(C)OnBCRengagement, Btk phosphorylates the transcription factor TFII‐I (1). TFII‐I translocatesinto the nucleus (2) and activates transcription (3). Btk also translocates to the nucleus (4). Exportof Btk out of the nucleus is regulated by Btk binding to the export protein exportin‐1 (Expt‐1) (5).

Tec KINASES IN T CELL AND MAST CELL SIGNALING 151
Some data also indicate that Itk may traffic to the nucleus. In CD3‐stimulatedJurkat T cells, a small proportion of the total Itk protein was found in thenucleus. In this case, the nuclear localization was mediated by karyopherinalpha (Rch1a), a nuclear transporter binding to the Itk SH3 domain via itsPRR (Perez‐Villar et al., 2001). This finding is somewhat surprising, as Rch1ais generally required for the nuclear import of proteins containing a basic‐type (classical) nuclear localization signal, which is lacking in Itk. Nonetheless,expression of a PRR mutant of Rch1a in Jurkat cells prevented nuclear tran-slocation of Itk as well as mitogen‐induced IL‐2 production by these T cells.These data suggest that the nuclear fraction of Itk may play a role during T cellactivation (Perez‐Villar et al., 2001). Consistent with this notion, one reportindicates that Itk might directly interact with and phosphorylate T‐bet, aconstitutively nuclear transcription factor that regulates IFN‐g transcription(Hwang et al., 2005).
The ability to shuttle between the cytoplasm and the nucleus has also beendemonstrated for Btk (Mohamed et al., 2000). Deletion of the Btk SH3 domainled to Btk localization predominantly in the nucleus. Furthermore, inhibiting anuclear export protein, exportin‐1, also resulted in nuclear accumulation ofBtk. However, potential nuclear targets of Btk and the exact mechanism of Btkshuttling remain to be determined. Additional data suggest a close link betweenBtk and the regulation of gene transcription. In this context, it is worth notingthat Btk has been implicated in NF‐kB activation in B cells, indirectly linkingBtk to nuclear signaling (Petro et al., 2000). In addition, TFII‐I, a versatiletranscription initiation factor, has been shown to be tyrosine phosphorylated byBtk shortly after B cell receptor (BCR) stimulation. Consistent with this, over-expression of wild‐type Btk induced TFII‐I‐dependent transcriptional activa-tion in COS7 cells (Novina et al., 1999). TFII‐I was also found to associate withthe Btk PH and kinase domains. These data suggest that Btk phosphorylation ofBAP/TFII‐I provides a link between BCR engagement and the modulation ofgene expression (Egloff and Desiderio, 2001). In addition, the chromatin‐remodeling protein, Bam11, has been shown to bind to the Btk PH domainand to inhibit Btk kinase activity (Kikuchi et al., 2000). These data suggesta model in which Btk activates BAM11 and the SWI/SNF transcriptionalcomplex via TFII‐I activation in B cells (Hirano et al., 2004).
3. Tec Kinases in Signaling Pathways
3.1. Antigen Receptor Signaling Pathways
Although some of the details vary, the antigen receptor signaling pathways inT cells, B cells, and mast cells share a similar overall scheme. Briefly, followingreceptor engagement, a Src family tyrosine kinase is activated and phosphorylates

152 MARTIN FELICES ET AL .
receptor subunits, leading to the recruitment and activation of an Syk familykinase (Dal Porto et al., 2004; Gilfillan and Tkaczyk, 2006; Kane et al., 2000;Samelson, 2002). Activated Tec family kinases are then recruited to the re-ceptor signaling complexes through interactions with adapter proteins of theSLP‐76 (SH2‐domain‐containing leukocyte protein of 76 kDa) and LAT (linkerfor activation of T cells) families (Bunnell et al., 2000; Ching et al., 2000; Shanand Wange, 1999; Su et al., 1999). Once at the membrane, Tec family kinasesare activated through phosphorylation by Src family kinases, and in turn,phosphorylate and activate PLC‐g (Fluckiger et al., 1998; Liu et al., 1998;Schaeffer et al., 1999; Takata and Kurosaki, 1996). PLC‐g catalyzes catabolismof PI(4,5)P2 into inositol‐1,4,5‐triphosphate (IP3) and diacylglycerol (DAG)(Rhee, 2001). IP3 is required for intracellular Ca2þ release, which triggerssustained calcium influx that activates downstream effectors like the NFATtranscription factors (Crabtree and Olson, 2002; Lewis and Cahalan, 1995).DAG activates members of the PKC (protein kinase C) family, as well asRASGRP (RAS guanyl‐releasing protein), leading to the activation of JNK(JUN amino‐terminal kinase) and ERK1/ERK2 (extracellular‐signal‐regulatedkinase) and thereby regulating the transcription factor, AP‐1 (Newton, 2004;Samelson, 2002). This model is supported by data from Tec kinase‐deficientlymphocytes and mast cells, which show defects in antigen receptor mediatedPLC‐g phosphorylation, IP3 production, Ca
2þ influx, ERK and JNK activation,and the downstream activation of transcription factors, NFAT and AP1 (DalPorto et al., 2004; Fowell et al., 1999; Gilfillan and Tkaczyk, 2006; Liu et al.,1998; Miller and Berg, 2002; Schaeffer et al., 1999, 2001). A more detaileddiscussion of Tec kinase signaling in mast cells can be found below.The most prominent known substrate of Tec kinases is PLC‐g, which is
phosphorylated following antigen receptor stimulation. Tec kinases are broughtinto proximity with PLC‐g via interactions with adapter molecules. Specifically,some Tec kinases have been shown to associate with adapters, SLP‐76 andSLP‐65, and the membrane‐bound linker molecule, LAT. In T cells, theadapters LAT and SLP‐76 couple Itk to PLC‐g1 via interactions mediated bythe Itk SH2 and SH3 domains (Bunnell et al., 2000; Chan et al., 1999; Chinget al., 2000; Su et al., 1999). In B cells, where Itk is not expressed, Btk iscoupled to PLC‐g2 by the BLNK/SLP‐65 adapter complex (Su et al., 1999).The function of Tec kinases in antigen receptor signaling pathways is also
dependent on protein–protein interactions mediated by the nonenzymaticdomains of the kinases. Studies addressing the binding partners of Tec kinaseswere initiated more than 10 years ago, with the finding that the Src familytyrosine kinases, Lyn, Fyn, and Hck, could bind to the Btk TH domain(Cheng et al., 1994). At that time, these data provided the first evidence linkingBtk, and potentially other Tec kinases, to established signaling pathways inB lymphocytes and other leukocytes. Subsequently, similar associations of Src
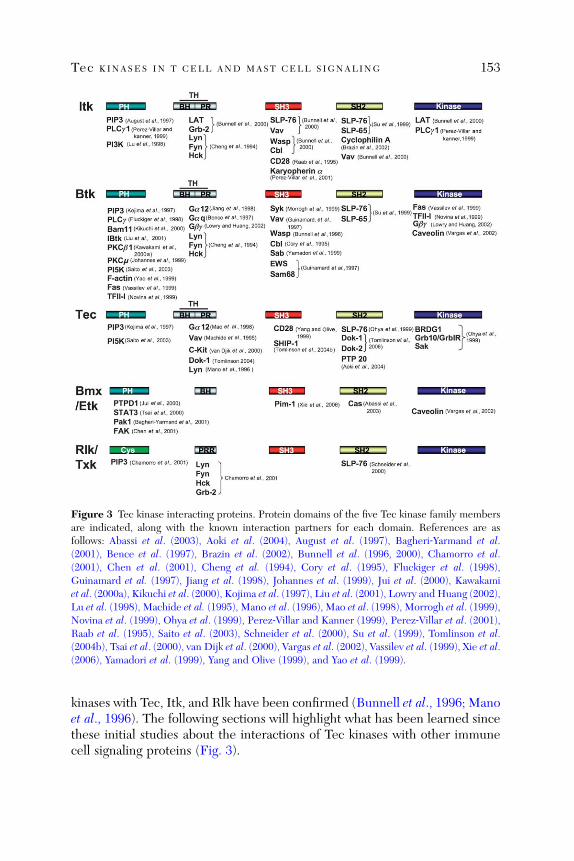
Figure 3 Tec kinase interacting proteins. Protein domains of the five Tec kinase family membersare indicated, along with the known interaction partners for each domain. References are asfollows: Abassi et al. (2003), Aoki et al. (2004), August et al. (1997), Bagheri‐Yarmand et al.(2001), Bence et al. (1997), Brazin et al. (2002), Bunnell et al. (1996, 2000), Chamorro et al.(2001), Chen et al. (2001), Cheng et al. (1994), Cory et al. (1995), Fluckiger et al. (1998),Guinamard et al. (1997), Jiang et al. (1998), Johannes et al. (1999), Jui et al. (2000), Kawakamiet al. (2000a), Kikuchi et al. (2000), Kojima et al. (1997), Liu et al. (2001), Lowry and Huang (2002),Lu et al. (1998), Machide et al. (1995), Mano et al. (1996), Mao et al. (1998), Morrogh et al. (1999),Novina et al. (1999), Ohya et al. (1999), Perez‐Villar and Kanner (1999), Perez‐Villar et al. (2001),Raab et al. (1995), Saito et al. (2003), Schneider et al. (2000), Su et al. (1999), Tomlinson et al.(2004b), Tsai et al. (2000), van Dijk et al. (2000), Vargas et al. (2002), Vassilev et al. (1999), Xie et al.(2006), Yamadori et al. (1999), Yang and Olive (1999), and Yao et al. (1999).
Tec KINASES IN T CELL AND MAST CELL SIGNALING 153
kinases with Tec, Itk, and Rlk have been confirmed (Bunnell et al., 1996; Manoet al., 1996). The following sections will highlight what has been learned sincethese initial studies about the interactions of Tec kinases with other immunecell signaling proteins (Fig. 3).

154 MARTIN FELICES ET AL .
3.2. Interactions with Negative Regulators of Signaling
Since the activation of tyrosine kinases leads to dramatic changes in cell phy-siology, patterns of gene expression, and proliferative state, it is critical thatthese signals be terminated at later times following antigen receptor stimula-tion. One route to termination of signaling is via the interaction of kinases withnegative regulatory molecules. As mentioned above, one such mechanisminvolves activation of the SH2‐containing inositol phosphatases, (SHIP)‐1and ‐2. The main function of SHIP proteins, like that of PTEN, is to counter-act PI3K activity, thereby diminishing recruitment of signaling molecules thatare dependent on PH domain‐mediated interactions with PIP3.Consistent with this indirect mode of regulation, SHIP‐1 and ‐2 have been
found to directly interact with, and functionally inhibit, Tec in vitro (Tomlinsonet al., 2004a). Two regions in SHIP‐1 have been shown to mediate interactionswith the Tec SH3 domain, suggesting the possibility that two Tec moleculescan bind simultaneously to SHIP‐1. Tec‐induced NFAT activation is potentlyinhibited by SHIP‐1 and ‐2, and requires intact SHIP phosphatase activity.Overall, this pathway is thought to operate by diminishing Tec membranelocalization (Tomlinson et al., 2004a).Another mechanism by which SHIP downregulates signaling is via interac-
tions with Dok proteins, negative regulatory adapter molecules that reduce Erkactivation. Specifically, the NPyX motif within the C‐terminus of SHIP‐1 inter-acts with the phosphotyrosine‐binding domains of Dok‐1, ‐2, and ‐3 (Robsonet al., 2004; Tamir et al., 2000). In addition, the SH2 domain of Tec interacts withphosphotyrosine motifs in Dok‐1 and Dok‐2 (Gerard et al., 2004; Yoshidaet al., 2000), while the PRR of Tec also binds to Dok‐1 (Tomlinson et al.,2004a). On the basis of these data, a Tec/SHIP/Dok complex has been proposedand is thought to inhibit Tec kinase activity. Consistent with this notion, a stablecomplex between Tec and Dok‐1, together with the Lyn tyrosine kinase, hasbeen observed. Further, Tec and Lyn can each phosphorylate Dok‐1 (Lianget al., 2002; Tomlinson et al., 2004a; van Dijk et al., 2000). PhosphorylatedDok‐1 has also been shown to bind to the SH2 domains of several signalingmolecules activated by stem cell factor (SCF), suggesting that Dok‐1, andtherefore indirectly Tec, may function to modulate signaling through c‐kit(van Dijk et al., 2000). Interactions similar to those of Tec with the SHIP/Dokpathway have not been observed to date for the other Tec family kinases.Additional proteins have been identified as binding partners for Tec family
kinases that result in inhibition of their activity. For example, the tyrosinephosphatase, PTP20, is tyrosine‐phosphorylated by Tec and coimmunopreci-pitates with Tec from a B cell line (Aoki et al., 2004). A role for PTP20 in the

Tec KINASES IN T CELL AND MAST CELL SIGNALING 155
negative regulation of Tec is further supported by the observation that PTP20can inhibit Tec signaling following BCR stimulation in the Ramos B cell line.Several negative regulators of Btk have also been described. Hirano et al.(2004) reported a Btk‐associated molecule, BAM11 that binds to the PHdomain of Btk and prevents Btk recruitment to the membrane. In addition,a small 203aa protein, inhibitor of Btk (IBtk), was found in a yeast two‐hybridscreen as a binding partner for the Btk PH domain. IBtk inhibits Btk kinaseactivity and interferes with BCR‐mediated calcium mobilization and NF‐kBactivation (Liu et al., 2001). In another study, Yamadori et al. (1999) detectedan SH3‐domain binding protein that preferentially associates with Btk, calledSab. Overexpression of Sab in B cells reduces BCR‐induced Btk tyrosine phos-phorylation, calcium mobilization, and PIP3 generation, clearly suggesting arole for Sab as a negative regulator of Btk‐mediated BCR signaling.
PKC has also been proposed as a negative regulator of Tec kinase activity.Two reports document an association between Btk and PKC, mediated by theBtk PH and TH domains. Johannes et al. (1999) first described an interactionbetween PKCm and the PH–TH region of Btk. Although PKCm is ubiquitouslyexpressed, high levels are found in the thymus and in hematopoietic cells. In asecond report, Kawakami et al. (2000a) demonstrate binding between Btk andPKCb1 in mast cells following FceRI stimulation. This latter interaction wasshown to downregulate Btk kinase activity, resulting in a negative feedback loop.Interestingly, in contrast to Btk, Itk binds to a broad spectrum of PKC iso-forms in FceRI‐stimulated mast cells, including PKC‐a, ‐bI, ‐bII, ‐e, ‐z, and ‐y(Kawakami et al., 1995).
A unique mode of negative regulation has been proposed for Itk based onobservations of a cis–trans proline isomerization that occurs in the Itk SH2domain, but not in the SH2 domains of the other Tec kinases (Brazin et al.,2002; Mallis et al., 2002). This proline isomerization produces two differentconformations of the Itk SH2 domain. The trans SH2 conformer is involvedin phospholigand binding and is therefore implicated in activation of Itk(Breheny et al., 2003). The cis SH2 conformer is involved in phosphotyrosine‐independent binding to the SH3 domain of another molecule of Itk, thusforming a homodimer that is likely to interfere with Itk activation (Brazinet al., 2000; Breheny et al., 2003). The peptidyl‐prolyl isomerase, cyclophilinA, which catalyses cis–trans proline isomerization, has been to shown to bind toItk and inhibit Itk kinase activity (Brazin et al., 2002). Treatment with cyclo-sporin A, which inhibits cyclophilin A, increases Itk activation and phosphory-lation of Itk substrates. Consistent with these findings, T cells from cyclophilinA‐deficient mice are hypersensitive to TCR stimulation due to increased Itkactivity (Colgan et al., 2004).

156 MARTIN FELICES ET AL .
3.3. Interactions with the Cytoskeletal Components
One major outcome of antigen receptor signaling is the reorganization of theactin cytoskeleton. This structural reorganization is essential for the recruit-ment of signaling molecules to the immunological synapse, where activationpathways are initiated. Intriguingly, the first observation linking Tec kinaseswith the actin cytoskeleton came from studies in insects. Tec29, the Tec familykinase found inDrosophila melanogaster, is an essential protein required duringembryonic development. In Drosophila, Tec29 is required for the actin‐dependent growth of ring canals, the intracellular bridges between nurse cellsand the oocyte (Guarnieri et al., 1998; Roulier et al., 1998). More recently,support for Tec kinases as modulators of the cytoskeleton in mammalian cellshas emerged, with implications for signaling, migration, and cell adhesion.Much of the data in this area focuses on Bmx, which is highly expressed in
cells with a strong migratory potential, including endothelial cells and meta-static carcinoma cell lines. Studies of Bmx have shown that this Tec kinasemember is activated by extracellular matrix proteins (Chen et al., 2001). Bmxactivation is dependent on focal adhesion kinase (FAK), which functions as akey mediator in integrin signaling and is thought to mediate cell migration byrecruitment and phosphorylation of the docking protein, p130Cas (Cas). FAKcontrols cellular responses to the extracellular matrix and binds via its FERMdomain to the PH domain of Bmx (Chen et al., 2001). This interaction activatesBmx, thereby linking Bmx to integrin signaling and cell adhesion. Anotherreport gives mechanistic insight into this process by providing evidence thatBmx interacts with Cas at membrane ruffles—sites of active actin remodelingin motile cells (Abassi et al., 2003). Specifically, Cas binds to the SH2 domainof Bmx. Furthermore, Bmx phosphorylates Cas and induces binding to anoth-er docking protein, Crk. The Cas–Crk complex connects several extracellularstimuli to the regulation of the actin cytoskeleton and cell motility.Among Tec family kinases, a role in actin modulation is not restricted to
Bmx. Several lines of evidence indicate a role for Itk in regulating actin poly-merization, largely based on data showing impaired TCR‐mediated actinreorganization in T cells from Itk‐deficient mice (Labno et al., 2003; Woodset al., 2001). This defect has been linked to reduced activation of Cdc42 andWASP in the absence of Itk and Rlk. Consistent with these findings, WASPbinds to the SH3 domains of Itk and Btk, directly linking these Tec kinases tocytoskeletal rearrangements (Bunnell et al., 1996). Additional evidence for Teckinase interactions with the cytoskeleton comes from studies on the rat baso-philic cell line, RBL‐2H3. In these cells, a fraction of Btk colocalizes with actinfibers following stimulation through FceRI. This interaction is mediated by thePH domain of Btk (Yao et al., 1999). Interestingly Btk is also found at high

Tec KINASES IN T CELL AND MAST CELL SIGNALING 157
levels in platelets, where 30% of total Btk is associated with actin filamentsafter stimulation of the thrombin receptor (Mukhopadhyay et al., 2001).
Further links between Tec family kinases and the actin cytoskeleton aresuggested by interactions with Vav, a guanine nucleotide exchange factor forthe Rho family GTPases, Rac and Cdc42. These GTPases are important inactin remodeling during polarization and migration processes in leukocytes(Turner and Billadeau, 2002). Loss of Itk expression in Jurkat tumor cells aswell as primary T cells reduced TCR‐mediated actin polarization, correlatingwith impaired recruitment of Vav to the activated receptor. Both the ItkSH2 and SH3 domains bind to Vav (Bunnell et al., 2000; Dombroski et al.,2005; Kline et al., 2001) and similar interactions have been reported for Vavand the Btk SH3 domain (Guinamard et al., 1997). Although the precisefunctional outcome of these interactions is not understood, these data providea biochemical link between Tec kinases and cytoskeletal remodeling.
3.4. Btk and Toll‐Like Receptor Signaling
Several lines of evidence suggest a role for Btk in Toll‐like receptor (TLR)signaling pathways. TLRs 4, 6, 8, and 9 have been shown to bind to Btk(Jefferies et al., 2003). In addition, Btk is activated following lipopolysacchar-ide (LPS) stimulation of peripheral blood mononuclear cells, and is requiredfor LPS‐induced TNFa production (Horwood et al., 2006). Consistent withthese findings, peripheral blood mononuclear cells from patients with X‐linkedagammaglobulinemia (a deficiency in Btk) produce less TNFa and IL‐1bfollowing TLR4 or TLR2 stimulation than cells from healthy controls. Greyet al. (2006) also showed that Btk phosphorylates the MyD88 adapter‐likeprotein, Mal, in stimulated monocytes, further supporting the involvement ofBtk in TLR signaling pathways. Finally, coimmunoprecipitation experimentsdemonstrated an interaction of Btk with MyD88 itself, and with interleukin‐1receptor‐associated kinase (IRAK)‐1, key molecules in TLR4 signal transduc-tion (Jefferies et al., 2003). Together these findings provide strong support forBtk as a mediator of LPS‐induced NF‐kB activation.
3.5. Associations with Additional Signaling Proteins
3.5.1. CD28 and CD2
Itk can also be activated and recruited to the plasma membrane followingstimulation of CD2 or CD28 (August et al., 1994; King et al., 1996, 1998;Marengere et al., 1997; Tanaka et al., 1997). In the case of CD28 cross‐linking,

158 MARTIN FELICES ET AL .
membrane recruitment requires the Itk SH3 domain (Ching et al., 1999;Marengere et al., 1997). A similar result has also been reported for Tec,where targeting of Tec to activated CD28 is dependent on an intact Tec SH3domain (Garcon et al., 2004b).
3.5.2. Heterotrimeric G‐Proteins
In parallel to the well‐documented membrane recruitment of Btk via PHdomain‐mediated PIP3 binding, an additional mechanism of Btk membranelocalization has been proposed based on a PH/TH domain‐mediated associa-tion of Btk with heterotrimeric G‐protein b and g subunits (Gbg). Binding ofGbg to Btk leads to enhanced Btk kinase activity (Lowry and Huang, 2002).Furthermore, the purified a subunit of the G(q) class of heterotrimericG‐proteins (Gaq) has also been shown to bind to the Btk PH domain, and tocause activation of Btk in vivo (Bence et al., 1997). This latter observationstrengthens the role of Btk as a direct effector of G‐proteins. Finally, anotherG‐protein subunit, Ga12, also binds Btk, triggering Btk kinase activity (Jianget al., 1998). Little is known about potential interactions between other Teckinase members and heterotrimeric G‐proteins, although Itk has also beenshown to bind to Gbg (Langhans‐Rajasekaran et al., 1995).
3.5.3. Pim‐1
Biochemical evidence has indicated an interaction between Bmx and the44‐kDa serine/threonine kinase Pim‐1, a protein that has been implicated intumorigenesis (Amson et al., 1989; Breuer et al., 1989; Dhanasekaran et al.,2001; van Lohuizen et al., 1989). Specifically, the N‐terminal proline‐rich motifof the 44‐kDa isoform of Pim‐1 was shown to compete with the proline‐richmotifs of p53 for binding to the Bmx SH3 domain. These data have suggested amodel where the disruption of p53 binding to Bmx by Pim‐1 leads to enhancedBmx activity in prostate cancer cells, thereby conferring resistance of thesetumor cells to chemotherapeutic drugs (Xie et al., 2006). Similarly, Bmx has alsobeen implicated in the progression of breast cancer. In these studies, p21‐activated kinase 1 (PAK1), a CDC42/Rac‐dependent serine/threonine kinase,binds to Bmx via the Bmx PH domain leading to PAK1 phosphorylation andactivation. Consistent with these data, a kinase‐inactive mutant of Bmx expressedin human cancer cells reduced proliferation and tumorigenicity, suggesting animportant role for Bmx in tumor growth (Bagheri‐Yarmand et al., 2001).
3.5.4. Fas
Btk has been shown to bind to the intracellular domain of Fas, linking Btk tothe apoptotic cell death pathway in B cells. This interaction occurs via the Btk

Tec KINASES IN T CELL AND MAST CELL SIGNALING 159
PH and kinase domains. Furthermore, Btk binding to Fas inhibits the Fas–FADD interaction, thereby preventing initiation of the apoptotic cascade thatnormally occurs following FADD recruitment of caspase‐8. As predicted bythese data, B cell susceptibility to Fas‐mediated apoptosis is increased in theabsence of Btk (Vassilev et al., 1999).
3.5.5. BRDG1, Grb10/GrbIR, Sak
To determine downstream effectors of Tec, Ohya et al. (1999) performed ayeast two‐hybrid screen for interacting partners of the Tec kinase domain. Thisapproach identified the Sak kinase, Grb10/Grb1R, and a previously unknowndocking protein, BRDG1 (BCR downstream signaling 1) (Ohya et al., 1999).Of these, BRDG1 has been shown to be phosphorylated directly by Tecin vitro and to increase Tec activity in a positive feedback loop (Ohya et al.,1999). Sak, a poorly characterized serine/threonine kinase thought to par-ticipate in cell cycle control, is tyrosine phosphorylated by Tec in kidney293 cells (Yamashita et al., 2001). Grb10 is an adapter molecule involved ininsulin receptor signaling and implicated in c‐fos activation (Mano et al., 1998).Overexpression of Grb10 suppresses Tec‐mediated activation of the c‐fos pro-moter, indicating a novel role for Grb10 as an effector molecule downstream ofTec (Mano et al., 1998).
3.5.6. PTPD1
As described above for Tec, yeast two‐hybrid screens have been performed toidentify potential interaction partners of Bmx. This approach identifiedprotein‐tyrosine phosphatase D1 (PTPD1) as a binding partner of the BmxPH domain (Jui et al., 2000). PTPD1 was found to enhance Bmx kinaseactivity, as well as STAT3 activity. In addition, PTPD1 can activate Tec, sug-gesting a more general role of PTPD1 in the regulation of Tec kinases (Juiet al., 2000). Interestingly, a direct Bmx–STAT3 interaction has also beenreported. Further, overexpression of a dominant‐negative form of Bmxreduced v‐Src‐mediated activation of STAT3 in a rat liver cell line (Tsai et al.,2000). Together, these finding suggest an important role for Bmx in STAT3activation and cell transformation.
3.5.7. c‐cbl
Several Tec kinases bind to the proto‐oncogene, c‐cbl, a ubiquitin‐ligase. c‐cblcontains a unique tyrosine kinase binding (TKB) domain that recognizesphosphotyrosine residues on activated tyrosine kinases (Meng et al., 1999).Generally, c‐cbl exerts its regulatory capacity through interactions with recep-tor tyrosine kinases. However, both Itk and Btk have been shown to interact

160 MARTIN FELICES ET AL .
with c‐cbl via their SH3 domains binding to proline‐rich motfis in c‐cbl(Bunnell et al., 1996; Cory et al., 1995).
3.5.8. Caveolin‐1
One report describes an interaction between Btk and Bmx with themembrane‐organizing coat protein, caveolin‐1 (Vargas et al., 2002). Caveolincomplexes with membrane components such as cholesterol and sphingolipidsto form caveolae, subsets of lipid rafts (Harris et al., 2002). In the case of Btk,the kinase domain was verified as the critical mediator of this interaction.Together with the finding that caveolin‐1 downregulates tyrosine phosphoryla-tion of Btk as well as Btk in vitro autokinase activity, these data suggest thatcaveolins may function as negative regulators of antigen receptor signaling.
4. Regulation of Tec Kinase Activation
4.1. Regulation by Tyrosine Phosphorylation
In addition to membrane localization and interaction with adapter molecules,Tec family kinase activity is regulated by tyrosine phosphorylation. As with Srcfamily kinases, phosphorylation occurs in a conserved activation loop in thekinase domain that generally results in a conformational change allowingsubstrate access to the catalytic site of the kinase domain (Berg et al., 2005;Roskoski, 2005). Unlike Src family kinases, Tec family kinases do not autopho-sphorylate initially and instead require transphosphorylation in the kinasedomain by Src family tyrosine kinases (Chamorro et al., 2001; Debnath et al.,1999; Heyeck et al., 1997; Mahajan et al., 1995; Mano et al., 1996; Rawlingset al., 1996). Following phosphorylation by Src family kinases, Tec familykinases, at least Btk and Itk, then undergo autophosphorylation (Park et al.,1996; Rawlings et al., 1996; Wilcox and Berg, 2003).The initial transphosphorylation, which augments enzymatic activity of the
Tec kinases, has been studied primarily for Btk and Itk, and to a lesser extentfor Rlk and Tec. Mutational analyses of Itk and Rlk show that replacement ofthese tyrosine residues with phenylalanine (Itk‐Y511 and Rlk‐Y420) reducesin vitro kinase activity (Chamorro et al., 2001; Heyeck et al., 1997). Lck seemsto be crucial for the transphosphorylation for Itk, as Jurkat T cells lacking Lckexhibit no TCR‐induced tyrosine phosphorylation of Itk (Gibson et al., 1996;Heyeck et al., 1997). In contrast, immunoprecipitation analysis together within vitro kinase assays indicate that Rlk is transphosphorylated by Fyn, and notLck (Chamorro et al., 2001; Debnath et al., 1999). Tec is activated by Lyn(Mano et al., 1996), whereas Btk seems to require both Lyn and Syk (Kurosakiand Kurosaki, 1997).

Tec KINASES IN T CELL AND MAST CELL SIGNALING 161
Following transphosphorylation, Itk, Btk, and Bmx autophosphorylate onconserved residues in the SH3 domain (Park et al., 1996; Rawlings et al., 1996;Wilcox and Berg, 2003) (Itk Y180, Btk Y223, and Bmx Y215, the Tec SH3domain is tyrosine phosphorylated at a nonhomologous site (Y206) (Nore et al.,2003). Mutational analyses of these autophosphorylation sites have beensomewhat confusing. Substitution of Btk Y223 with phenylalanine has littleeffect on Btk activity in DT‐40 B cells, but seems to promote Btk activity infibroblasts (Kurosaki and Kurosaki, 1997; Park et al., 1996). In contrast,mutation of Itk Y180 impairs Itk function in the TCR response (Wilcox andBerg, 2003). It is important to keep in mind that these residues are located inthe SH3 domain, a region required for inter‐ and intramolecular interactions;thus, the consequences of these amino acid substitutions may, in part, be dueto effects on protein–protein interactions rather than on catalytic activity.
4.2. Inter‐ and Intramolecular Domain Interactions
The regulation of many PTKs is mediated by conformational changes thatdictate the catalytic activity of the enzyme. For instance, Src family kinases aremaintained in an inactive state prior to signaling via intramolecular interac-tions that stabilize binding of the SH2 domain to the C‐terminal negativeregulatory phosphotyrosine (Roskoski, 2004). Tec family kinases lack thisC‐terminal autoinhibitory sequence, and thus, are likely regulated by a distinctmechanism from that of the Src family kinases.
The three‐dimensional X‐ray crystal structures of the Itk kinase domain inboth the active (phosphorylated) and inactive (unphosphorylated) forms havebeen solved, and interestingly, are virtually identical; these findings suggestthat additional domains of Itk beyond the kinase domain are required for theregulation of Itk activity (Brown et al., 2004). Several lines of evidence furthersuggest that conformational changes involving multiple protein domains arecritical for the regulation of Tec family kinases (Fig. 4). For instance, the SH3domains of Itk, Tec, and Btk, but not Rlk, can each interact with theirneighboring PRR, precluding interactions of these domains with alternativeligands (Andreotti et al., 1997; Laederach et al., 2002, 2003; Pursglove et al.,2002). Tec family kinases favor intermolecular formation of homodimers overintramolecular interactions. Rlk, Tec, and Btk from homodimers via theirPRR and SH3 domains, while Itk, distinctly, forms homodimers via its SH2and SH3 domains (Brazin et al., 2000). While not yet definitively demon-strated, it seems likely that these interdomain interactions yield inactiveforms of the Tec kinases.
Differences in the regulatory mechanisms mediated by intra‐ and intermo-lecular domain interactions have been observed between the Tec kinase family

Figure 4 Models of Tec kinase regulation via intra‐ and intermolecular interactions. (A) Unlikethe other Tec family kinases, Rlk does not appear to undergo intramolecular interactions. Instead,at steady state levels, Rlk may exist as an intermolecular homodimer engaged through SH3–PRRinteractions. On TCR activation, Rlk is proposed to undergo a conformational change prompted bytransphosphorylation of its kinase domain and autophosphorylation of the SH3 domain. This wouldinhibit homodimerization and lead to interactions of the SH2 and SH3 domains with othersignaling components (adapters). (B) Itk is proposed to undergo intramolecular interactions viathe SH3 and PRR regions. However, at steady state levels, the preferred conformation is anintermolecular homodimer mediated by SH2 and SH3 binding. This mode of interaction is distinctfrom the other Tec family kinases, which form homodimers through SH3–PRR interactions. OnTCR activation, Itk may undergo a conformational change prompted by transphosphorylation of
162 MARTIN FELICES ET AL .

Tec KINASES IN T CELL AND MAST CELL SIGNALING 163
members. For instance, disruption of SH3–PRR interaction, which dramati-cally increases kinase activity of Btk and Tec, has minimal effects on Itk kinaseactivity (Ching et al., 2000; Hao and August, 2002; Li et al., 1995; Wilcox andBerg, 2003; Yamashita et al., 1996). These data fit models of Btk regulation,since the Btk PRR binds to Src family kinases, possibly providing an activationsignal; in contrast, the effect of altered accessibility of the Itk PRR has notbeen elucidated (Bunnell et al., 2000; Cheng et al., 1994; Yang et al., 1995).Nonetheless, these data suggest that Itk uses a distinct mechanism of regulatingligand binding.
5. Distinct Versus Redundant Functions of Tec Kinases
5.1. Tec Kinases in T Cell Development
Several lines of evidence indicate a role for Tec family kinases in T celldevelopment and selection. Here again, Itk appears to have the principalrole, with Rlk and Tec having little to no known effects (Berg et al., 2005;Liao and Littman, 1995; Liu et al., 1998; Schaeffer et al., 1999). Experimentsusing Itk‐deficient mice crossed to TCR transgenic lines demonstrated a rolefor Itk in positive selection (Liao and Littman, 1995; Lucas et al., 2002;Schaeffer et al., 2000). Specifically, positive selection into the CD4 lineage isless efficient when Itk is absent (Lucas et al., 2002). Other studies haveindicated that a deficiency in Itk, or both Itk and Rlk, leads to impairednegative selection; in some cases, a deficiency in Itk may cause a delay, ratherthan a block in negative selection (Lucas et al., 2003; Schaeffer et al., 2000).Consistent with these defects, Itk‐deficient mice also have a mild defect innatural killer‐T (NKT) cell development that increases in severity as the miceage (Gadue and Stein, 2002).
Of interest, recent publications have demonstrated a role for Itk and Rlk inCD8 T cell development and function (Atherly et al., 2006a,b; Broussard et al.,2006; Dubois et al., 2006). Mice deficient in Itk have increased numbers and
the kinase domain and autophosphorylation of the SH3 domain. This conformational switch isaccompanied by cis–trans proline isomerization in the SH2 domain of Itk. These changes inhibitdimerization and lead to SH2 and SH3 domains accessibility for binding to other signalingcomponents (adapters). (C, D) Tec and Btk can undergo intramolecular interactions via SH3–PRR binding. However, at steady state levels, intermolecular homodimers, mediated by transSH3–PRR binding, are the preferred conformations. On antigen receptor activation, Tec andBtk may undergo conformational changes prompted by transphosphorylation of the kinasedomains and autophosphorylation of the SH3 domains. These changes would inhibit dimerizationand lead to SH2 and SH3 domains interactions with other signaling components (adapters).

164 MARTIN FELICES ET AL .
percentages of CD8 single positive (SP) T cells in the thymus (Lucas et al.,2002). Three studies have analyzed these cells more closely and have concludedthat they share features with lineages of innate and memory CD8þ T cells(Atherly et al., 2006b; Broussard et al., 2006; Dubois et al., 2006). Specifically,CD8þ Tcells in mice lacking Itk, or both Itk and Rlk, are CD44hi, CD122þ, andNK1.1þ; further, these cells produce Interferon‐g directly ex vivo, and can beselected on bone marrow‐derived major histocompatibility complex (MHC)class I molecules—all characteristics of innate T cells such NKT cells andMHC class‐Ib‐restricted T cells (Kronenberg, 2005; Rodgers and Cook,2005). Differences between the Itk�/� and the Itk/Rlk�/� CD8þ T cells areminimal, indicating a role for Itk, but not Rlk, in regulating the development ofthese cells. Given that Tcell development appears normal in Tec‐deficient mice,it is not likely that Tec contributes significantly to the development of CD8þ Tcells (Ellmeier et al., 2000). Taken together, these findings suggest a role for Teckinases in the development of both adaptive and innate lymphocytes.
5.2. Tec Kinases in Other Cell Types
There is much less data that establishes the functional importance of Tecfamily kinases that are found in hematopoietic cells other than lymphocytes(for review, see Schmidt et al., 2004). Btk is the best‐studied, and is present inmany hematopoietic lineages. For instance, monocytes/macrophages devoid ofBtk have been shown to be impaired in TNF production, phagocytosis, andnitric oxide production (Horwood et al., 2003; Mukhopadhyay et al., 2002).Functions of platelets derived from signals through the GPVI also seem to beimpaired with Btk and Btk/Tec deficiency (Atkinson et al., 2003; Quek et al.,1998). Contrary to these reports that indicate a positive influence of Btk inthese cells, one study demonstrates that Btk may play a negative role in thedevelopment and function of dendritic cells (Kawakami et al., 2006).
5.3. Redundancy Among Tec Kinase Family Members
As described above, T cells, B cells, and mast cells each express multiple Teckinase family members (Fig. 5). In T cells, Itk appears to be the predominantTec kinase expressed, based on biochemical analyses of TCR signaling pathwaysin T cells lacking each of the Tec family kinases. The basic conclusions fromthese studies were that Tec and Rlk play little essential role in TCR signaling;further, these studies indicated that a deficiency in both Itk and Rlk created themost severe defect (Schaeffer et al., 1999). The hierarchy of impaired signalingcorrelates with the expression levels of each Tec kinase in naıve T cells. Inaddition, the enhanced defect in the Itk/Rlk‐deficient T cells indicates some

Figure 5 Differential expression of Tec family kinases in T cells, B cells, and mast cells. Itk, Rlk,and Tec are expressed in T cells, while Btk and Tec are expressed in B cells. Current data indicatethat Tec kinases in T cells and B cells mediate activation of PLC‐g downstream of Src kinasesfollowing antigen receptor stimulation. In mast cells, which express all of these Tec kinases, onlythe positive role of Btk has been established.
Tec KINASES IN T CELL AND MAST CELL SIGNALING 165
redundancy between these two family members in TCR signaling (Schaefferet al., 1999). In contrast, the role of Tec in TCR signaling has beenmore difficultto discern. While Tec‐deficient primary T cells are unimpaired (Ellmeier et al.,2000), overexpression of Tec in T cell tumor lines enhances TCR signalingleading to increased NFAT activation (Tomlinson et al., 2004b). Tec may alsohave a more prominent role in signaling in previously activated T cells (Altmanet al., 2004).
In B cells, an analogous situation has been observed with Btk and Tec. Whileprimary B cells lacking Btk are severely impaired in BCR signaling (Takata andKurosaki, 1996), B cells lacking Tec are not (Ellmeier et al., 2000). However,in the absence of both Btk and Tec, BCR signaling is further impaired to theextent that doubly deficient mice totally lack mature B cells, suggestingredundant functions for these two Tec kinases in B cells (Ellmeier et al.,2000). Interestingly, reconstitution studies performed in Btk‐deficient DT‐40B cells indicate that Rlk, Tec, and Itk can each restore PLC‐g phosphorylationand Ca2þ mobilization in these cells in response to BCR stimulation; however,Itk and Tec, but not Rlk are also able to restore BCR‐induced apoptosis(Tomlinson et al., 1999).
While evidence for redundancies in function among Tec kinases is apparent,additional data indicate that each Tec kinase also shows unique characteristics.For instance, in in vitro studies, overexpression of Tec and Rlk augment PLC‐gphosphorylation and downstream events; in contrast, overexpression of Itk

166 MARTIN FELICES ET AL .
seems to have no effect ( Somme rs et al., 1999; Tomlinson et al., 2004b ). It maybe that diffe rences in intermole cular interactio ns and modes of regula tion ofthe Tec family memb ers account for these di sparate outcome s. Furthe rmore,overexpress ion of Btk ha s no ability to enh ance T cell signaling , in spite o f thesimilar role of Btk in activ ating PLC ‐ g in B cells and mast cells (Tomlinsonet al., 2004b ); thi s failure may resul t from an in ability of Btk to in teractproperly with T cell ‐ specifi c adap ters and oth er signaling pro teins.Thus far, mast cells are the only cell type known to exp ress the four
predomi nant Tec kinase s, Btk, Itk , Rlk, and Tec ( Somme rs et al., 1995; Yamadaet al., 1993 ; an d our unpublish ed obs ervations) . Thus, althou gh little is cu r-rently known abou t Itk, Rlk, or Tec in these cells, studies of mast cells presentan opport unity to exami ne the inter play betwe en these Tec kinase s resid ing ina singl e cell typ e.
6. Tec Kinases in Mast Cell Signaling
6.1. Over view of Mast Cells
Mast cells are wid ely accepte d as effec tor cells in allergic responses and in nateimmunity (for recent compre hensiv e reviews, see Galli et al., 2005a; Ok ayamaand Kawakami, 2006 ). Mou nting evidenc e has esta blished a role for mast cellsin ad aptive immun e responses (Galli et al., 2005b). Overal l, these cells arecapable of wide spread influence due to the impre ssive variety of mediators,both pro‐ and an ti‐ in flammato ry, that they release upon stimu lation.Du ring in nate immu ne responses, mast ce lls are highly specialized to
respond rapidly upon stimu lation, an d can serve as ‘‘sen tinels’’ that alert thelocal surrou ndings to rapidly counter pot entially detrime ntal challeng es. Thisis due to the presence of numerous granul es containing prefor med factors thatare relea sed by exocy tosis w ithin sec onds to min utes follow ing mast celltriggering. Man y di fferent factors are sequeste red in these granul es, includ inghistamine, seroton in, a variety of pro teases, and a numbe r of cytokines. Inaddition to degranulation, activated mast cells also respond within minutes ofstimulation by producing proinflammatory arachidonic acid metabolites suchas leukotrienes and prostaglandins. These early‐phase mediators not onlyenhance responses such as vascular permeability and smooth muscle contrac-tion but also have direct effects on immune cells that express the appropriaterecepto rs (Ga lli et al., 2005b).The recent escalated interest in mast cells is based largely on a growing
appreciation for the later responses of these cells, specifically, the de novosynthesis of cytokines and chemokines that drive adaptive immune responsesand of which mast cells are known to produce a large variety. The use of mast
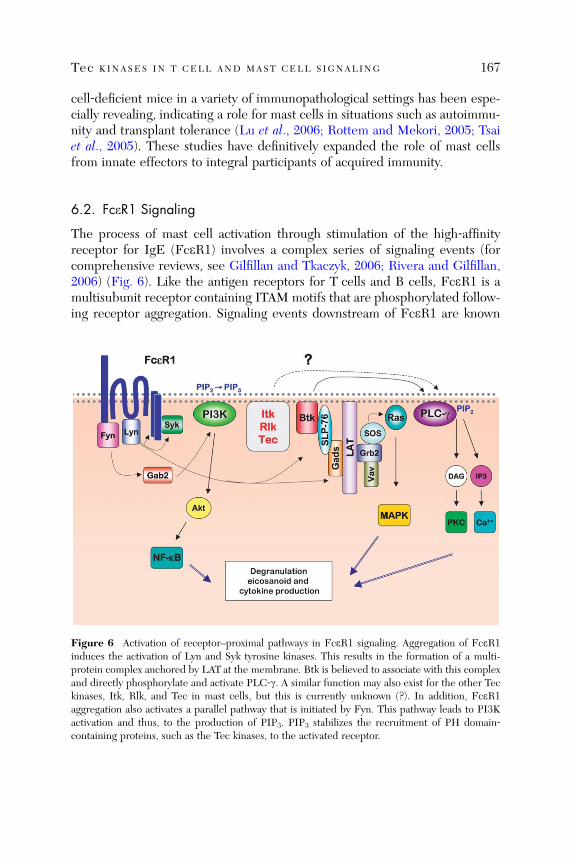
Tec KINASES IN T CELL AND MAST CELL SIGNALING 167
cell‐deficient mice in a variety of immunopathological settings has been espe-cially revealing, indicating a role for mast cells in situations such as autoimmu-nity and transplant tolerance (Lu et al., 2006; Rottem and Mekori, 2005; Tsaiet al., 2005). These studies have definitively expanded the role of mast cellsfrom innate effectors to integral participants of acquired immunity.
6.2. FceR1 Signaling
The process of mast cell activation through stimulation of the high‐affinityreceptor for IgE (FceR1) involves a complex series of signaling events (forcomprehensive reviews, see Gilfillan and Tkaczyk, 2006; Rivera and Gilfillan,2006) (Fig. 6). Like the antigen receptors for T cells and B cells, FceR1 is amultisubunit receptor containing ITAM motifs that are phosphorylated follow-ing receptor aggregation. Signaling events downstream of FceR1 are known
Figure 6 Activation of receptor–proximal pathways in FceR1 signaling. Aggregation of FceR1induces the activation of Lyn and Syk tyrosine kinases. This results in the formation of a multi-protein complex anchored by LAT at the membrane. Btk is believed to associate with this complexand directly phosphorylate and activate PLC‐g. A similar function may also exist for the other Teckinases, Itk, Rlk, and Tec in mast cells, but this is currently unknown (?). In addition, FceR1aggregation also activates a parallel pathway that is initiated by Fyn. This pathway leads to PI3Kactivation and thus, to the production of PIP3. PIP3 stabilizes the recruitment of PH domain‐containing proteins, such as the Tec kinases, to the activated receptor.

168 MARTIN FELICES ET AL .
to utilize intermediates that are common to both B cells and T cells, andtherefore, it is not surprising that many of the biochemical pathways describedin lymphocyte signaling are shared with mast cells.The earliest FceR1 activation‐induced signaling events require the recruit-
ment and activation of tyrosine kinases. In mast cells, the Src‐family kinase Lynbinds to FceR1 and phosphorylates the b and g chain ITAMs of FceR1, whichinduces the recruitment and activation of Syk. This potentiates the formationof a complex of proteins that are brought into close proximity by their interac-tion, directly or indirectly, with the adaptor molecule LAT (Rivera et al., 2001).Components of this complex include Grb2, Gads, PLC‐g1, PLC‐g2, SLP‐76,and Vav. Btk (and other Tec kinases) is also assumed to participate in this LAT‐anchored complex in mast cells, given their key role in regulating PLC‐g inlymphocytes. Similar to lymphocytes, PLC‐g activation is a crucial outcome ofassembling this complex in mast cells, as indicated by the reduced levels ofFceR1‐induced PLC‐g activation and Ca2þ release observed in mast cells thatlack proteins associated with this complex (Manetz et al., 2001; Pivniouk et al.,1999; Saitoh et al., 2000). Signaling defects in these mutant mast cells alsotranslate to impairments in degranulation and cytokine responses.In addition to signaling processes that are known to stem directly from PLC‐g
activation, PI3K‐dependent pathways also play an essential role in FceR1‐induced signaling (Fukao et al., 2003). As discussed above, PI3K‐catalyzedPIP3 production is important for recruitment of PH domain‐containing pro-teins, including Tec family kinases. However, the PI3K pathway has beenpostulated to be distinct, although functionally complementary to the LAT–PLC‐g pathway (Parravicini et al., 2002). This pathway is linked to the Srckinase Fyn, which, like Lyn, also associates with FceR1. Fyn activation leadsto Gab2 phosphorylation and association with the p85 subunit of PI3K,enabling PI3K activity. Evidence suggests that this axis is particularly impor-tant in sustaining Ca2þ signals necessary for degranulation responses of mastcells, but it is clear that PI3K‐mediated pathways are necessary for overalloptimal mast cell function, since cytokine responses are also attenuated inmice that lack molecules involved in this pathway (Gu et al., 2001; Parraviciniet al., 2002).FceR1‐proximal events culminate in the activation of multiple downstream
pathways, including theMAP kinases p38, ERK, and JNK, as well as the NF‐kBand NFAT family transcription factors. As expected, these activation events arealso coupled to the induction of negative regulatory events. For instance,deficiencies in SHIP or PTEN in mast cells results in functional hyper-responsiveness (Furumoto et al., 2006; Huber et al., 1998; Kalesnikoff et al.,2002). Additionally, it is well established that Lyn phosphorylation of ITIM

Tec KINASES IN T CELL AND MAST CELL SIGNALING 169
motifs in receptors such as FcgRIIb is critical (Ali et al., 2004; Gomez et al.,2005; Gu et al., 2001), as highlighted by the hyperresponsiveness of Lyn�/�
mast cells (Hernandez‐Hansen et al., 2003; Malbec et al., 1998; Odom et al.,2004). How extracellular stimuli, as well as the coinduction of inhibitoryreceptors, elicit distinct functional responses from mast cells is still poorlyunderstood.
6.3. Btk in Mast Cell Function and Signaling
The key role of Btk in mast cell activation has been established by extensivestudies conducted by Kawakami and colleagues. Initially, these investigatorsshowed that Btk is phosphorylated following FceR1 stimulation, suggesting afunctional role for Btk in this pathway (Kawakami et al., 1994). Indeed, Btk‐deficient and xid (expressing functionally inactive Btk protein) mast cells dem-onstrate multiple defects in effector function. Specifically, in vitro degranula-tion responses have been found to be modestly impaired (Hata et al., 1998;Heinonen et al., 2002; Kawakami et al., 2000b; Setoguchi et al., 1998). Thisfinding was supported by studies demonstrating defective anaphylactic reac-tions elicited in vivo in Btk�/� mice (Hata et al., 1998). Leukotriene produc-tion by Btk�/� mast cells is also impaired (Kawakami et al., 2000b). Further,Btk is critical in regulating de novo cytokine production; antigen‐inducedsecretion of GM‐CSF, TNF‐a, IL‐2, and IL‐6 are all reduced with Btk�/�
mast cells (Hata et al., 1998). In the case of IL‐2 and TNF‐a, deficient cytokinesecretion was found to be due to impaired transcription of these genes.
The biochemical consequences of the Btk deficiency in mast cells closelyparallel that seen in B cells following BCR stimulation (Fig. 5). For example,Btk�/� mast cells are impaired in PLC‐g2 activation, IP3 generation, and Ca2þ
mobilization (Kawakami et al., 2000b). Additionally, Akt phosphorylation isreduced in Btk�/� mast cells, indicating that PI3K activity is impaired (Kitauraet al., 2000). These defects are accompanied by a failure to activate the JNKand p38 MAPK pathways to normal levels (Kawakami et al., 1997).
However, in contrast to the defects in B cell development observed in theabsence of Btk, mast cell development both in vivo and in vitro appearsnormal. Interestingly, in vitro culture of Btk�/� bone marrow cells with IL‐3produces increased numbers of mast cells; this increase is associated with adefect in JNK‐dependent apoptosis on IL‐3 withdrawal (Kawakami et al.,1997). These data suggest that Btk is required for signaling events distinctfrom those initiated by FceR1. In this regard, Btk has been found to associatewith c‐kit and to be involved in mast cell c‐kit signaling (Iwaki et al., 2005;Setoguchi et al., 1998).

170 MARTIN FELICES ET AL .
6.4. Possible Role of Itk in Mast Cells
Initial studies carried out by the Kawakami laboratory indicate that Itkparticipates, albeit in an unknown capacity, in mast cell signaling pathways(Kawakami et al., 1995; Yamada et al., 1993). As in T cells, endogenous Itk isphosphorylated and activated in FceR1‐stimulated bone marrow‐derived mastcells. Furthermore, like Btk, Itk was found to interact with multiple PKCmembers in mast cells (Kawakami et al., 1995).To date, one report has addressed the functional responses of mast cells in
Itk‐deficient mice. Although this study indicated that mast cells in Itk�/� micewere present in normal numbers with unaltered morphology, these cellsexhibited more profound defects than mast cells lacking Btk (Forssell et al.,2005). Itk�/� mast cells were found to be significantly impaired in an allergicairway inflammation model in which the extent of degranulation can bemeasured by plasma extravasation and direct visualization of mast cell granules.An issue to consider with these (and other) in vivo studies is the potential
contribution of the overall environment in intact Itk�/� mice. Since mast cellsdevelop from precursors that leave the bone marrow and differentiate in theperiphery, the possibility that systemic differences between wild type and Itk‐deficient mice could influence in vivo mast cell biology cannot be ruled out.For instance, it has been noted that Itk�/� mice have elevated levels of serumIgE (Schaeffer et al., 2001). IgE alone, in the absence of antigen, has beenshown to generate signals that can modify the characteristics of mast cells byenhancing their survival and increasing surface FceR1 expression (Kawakamiand Galli, 2002). The possibility also exists that the high levels of spontaneousIgE bound to mast cells might prevent antigen‐specific IgE from aggregatingand inducing degranulation in the allergy model used. Despite these concerns,the in vivo data described above are noteworthy; it will be interesting todetermine whether future in vitro studies will provide data to support theseconclusions.
6.5. Potential Positive Roles of Multiple Tec Kinases in Mast Cells
Thus far, potential functional redundancy of Tec kinases in mast cells has notbeen addressed. Given the positive role of Itk in promoting optimal activationof PLC‐g1 in T cells, as well as the known function of Btk in mast cells, it ispossible that Itk also promotes PLC‐g activation in mast cells. This type ofredundancy might be revealed by studies of Itk�/� mast cells, or possibly, onlyby analyses of doubly deficient Itk/Btk�/� mast cells. As mentioned above,T cells from Rlk�/� mice are nearly normal, whereas T cells lacking both Itkand Rlk exhibit defects more severe than T cells lacking Itk alone. These datahave suggested that Rlk is partially able to compensate for the absence of Itk

Tec KINASES IN T CELL AND MAST CELL SIGNALING 171
(Schaeffer et al., 1999). A similar situation has been observed for Btk and Tecin B cells (Ellmeier et al., 2000). Given the defects seen in Btk‐deficient mastcells, it is clear that the presence of Itk, Rlk, and Tec at endogenous levels isinsufficient to fully compensate for the loss of Btk. However, despite thecritical role of Btk in regulating PLC‐g2, a deficiency in Btk does not rendermast cells as severely compromised as does a deficiency in LAT or Syk(Costello et al., 1996; Saitoh et al., 2000), suggesting the possibility that theother Tec kinases may partially compensate for the absence of Btk. Consistentwith this notion, when Itk was overexpressed in Btk�/� mast cells, a partialrestoration of responses was observed (Hata et al., 1998). Similarly, it has beenshown that overexpression of Itk, Tec, or Rlk can restore function to Btk�/�
B cells (Fluckiger et al., 1998; Tomlinson et al., 1999). As the enzymatic activityof Itk has been reported to be less potent than that of Btk (Hawkins and Marcy,2001; Lowry et al., 2001), it is possible that even higher levels of Itk may benecessary to functionally replace Btk in mast cells.
In addition to differences in the expression levels and catalytic activities ofBtk, Itk, Rlk, and Tec, it is likely that these kinases also have different functionsbased on their nonenzymatic domains. Although Rlk�/� T cells and mice havefew obvious defects, studies suggest that Itk and Rlk have distinct functions inT cells. For example, Itk and Rlk display differential expression in T helper‐type 1 (Th1) and Th2 cells (Miller et al., 2004). Itk, Rlk, and Tec also havedifferent localization patterns, as described above. Therefore, the possibilityexists that Btk and Itk are also important for different types of effectorfunctions or at different stages of development in mast cells.
6.6. Potential Negative Roles for Tec Kinases in Mast Cell Signaling
Although the current evidence suggests that Itk plays a positive role in mast cells,it is worth considering a potential negative regulatory role for Itk in these cells. Asmentioned briefly above, an interesting study suggests that Btk performs anegative regulatory function in dendritic cells, despite the established positivefunction it has in B cells and mast cells (Kawakami et al., 2006). Such dataopen up the possibility that Tec family kinases may have unexpected roles indistinct cell types. A precedent for this has been seen in recent studiesdemonstrating hyperresponsiveness of mast cells lacking particular intracellu-lar signaling molecules, suggesting a role for these proteins in antagonizingpositive responses. For instance, intriguing data has emerged from the study ofLATand its structural homologues, LAT2 (LAB, NTAL), and LAX in mast cells(Saitoh et al., 2000; Volna et al., 2004; Zhu et al., 2004, 2006). LAT is anecessary component of positive signaling in T cells, functioning as a scaffold-ing protein that links upstream phosphorylation events to PLC‐g‐dependent

172 MARTIN FELICES ET AL .
signaling. As might be expected from its positive role in T cells, LAT�/� mastcells are significantly impaired in effector function. LAT2 shares homologywith LAT but importantly, does not bind to PLC‐g. Upon disruption of LAT2,B cells behave relatively normally, but surprisingly, LAT2�/� mast cells showedenhanced responses compared to wild‐type, suggesting a negative role forLAT2 in mast cell signaling (Janssen et al., 2003). Interestingly, when bothLAT2 and LAT are absent, mast cell responses are more defective than in theabsence of LAT alone. Therefore, LAT2 may function as an inefficient replace-ment for LAT when LAT is absent, but may act predominantly to negativelyregulate LAT when both family members are present. Furthermore, LAX�/�
mast cells also share a phenotype similar to that of LAT2�/� mast cells. Themechanism underlying these latter observations is still not established; LAT2may compete with LAT for localization and signaling partners, or may becritical in a distinct pathway that interferes with positive LAT signals. Regard-less of the mechanism, a similar scenario can be envisioned for the Tec kinasesin mast cells, where Btk is the dominant kinase based on high levels ofexpression and/or kinase activity, and Itk (or the others) has a predominantlynegative regulatory role.
7. Conclusions
This chapter has summarized the known functions of Tec family tyrosinekinases in T cell and mast cell signaling, focusing on the biochemical interac-tions and modes of regulation of Itk, Btk, Rlk, and Tec. Overall, the currentdata indicate some degree of redundancy among these kinases, based onobservations of modest deficiencies in cells lacking a single Tec kinase, andmore severe deficiencies in the absence of two family members. Nonetheless,distinct activities, binding partners, subcellular localization, and intra‐ andintermolecular domain interactions have been seen for each individual Teckinase. These findings highlight the fact that, while each Tec kinase familymember shares some functional activities with the others members, a uniquerole for each kinase in each distinct cell type is likely to be found.
Acknowledgments
This work was supported by grants from the NIH (AI37584 and AI66118) and the Center forDisease Control (CI000101).
References
Abassi, Y. A., Rehn, M., Ekman, N., Alitalo, K., and Vuori, K. (2003). p130Cas couples the tyrosinekinase Bmx/Etk with regulation of the actin cytoskeleton and cell migration. J. Biol. Chem. 278,35636–35643.

Tec KINASES IN T CELL AND MAST CELL SIGNALING 173
Ali, K., Bilancio, A., Thomas, M., Pearce, W., Gilfillan, A. M., Tkaczyk, C., Kuehn, N., Gray, A.Giddings, J., Peskett, E., Fox, R., Bruce, I., et al. (2004). Essential role for the p110deltaphosphoinositide 3‐kinase in the allergic response. Nature 431, 1007–1011.
Altman, A., Kaminski, S., Busuttil, V., Droin, N., Hu, J., Tadevosyan, Y., Hipskind, R. A., andVillalba, M. (2004). Positive feedback regulation of PLCgamma1/Ca(2þ) signaling byPKCtheta in restimulated T cells via a Tec kinase‐dependent pathway. Eur. J. Immunol. 34,2001–2011.
Amson, R., Sigaux, F., Przedborski, S., Flandrin, G., Givol, D., and Telerman, A. (1989). Thehuman protooncogene product p33pim is expressed during fetal hematopoiesis and in diverseleukemias. Proc. Natl. Acad. Sci. USA 86, 8857–8861.
Andreotti, A. H., Bunnell, S. C., Feng, S., Berg, L. J., and Schreiber, S. L. (1997). Regulatoryintramolecular association in a tyrosine kinase of the Tec family. Nature 385, 93–97.
Aoki, N., Ueno, S., Mano, H., Yamasaki, S., Shiota, M., Miyazaki, H., Yamaguchi‐Aoki, Y.,Matsuda, T., and Ullrich, A. (2004). Mutual regulation of protein‐tyrosine phosphatase 20and protein‐tyrosine kinase Tec activities by tyrosine phosphorylation and dephosphorylation.J. Biol. Chem. 279, 10765–10775.
Atherly, L. O., Brehm, M. A., Welsh, R. M., and Berg, L. J. (2006a). Tec kinases Itk and Rlk arerequired for CD8þ T cell responses to virus infection independent of their role in CD4þ T cellhelp. J. Immunol. 176, 1571–1581.
Atherly, L. O., Lucas, J. A., Felices, M., Yin, C. C., Reiner, S. L., and Berg, L. J. (2006b). The Tecfamily tyrosine kinases Itk and Rlk regulate the development of conventional CD8þ T cells.Immunity 25, 79–91.
Atkinson, B. T., Ellmeier, W., and Watson, S. P. (2003). Tec regulates platelet activation by GPVI inthe absence of Btk. Blood 102, 3592–3599.
August, A., Gibson, S., Kawakami, Y., Kawakami, T., Mills, G. B., and Dupont, B. (1994). CD28 isassociated with and induces the immediate tyrosine phosphorylation and activation of the Tecfamily kinase ITK/EMT in the human Jurkat leukemic T‐cell line. Proc. Natl. Acad. Sci. USA 91,9347–9351.
August, A., Sadra, A., Dupont, B., and Hanafusa, H. (1997). Src‐induced activation of inducibleT cell kinase (ITK) requires phosphatidylinositol 3‐kinase activity and the Pleckstrin homologydomain of inducible T cell kinase. Proc. Natl. Acad. Sci. USA 94, 11227–11232.
Bagheri‐Yarmand, R., Mandal, M., Taludker, A. H., Wang, R. A., Vadlamudi, R. K., Kung, H. J.,and Kumar, R. (2001). Etk/Bmx tyrosine kinase activates Pak1 and regulates tumorigenicity ofbreast cancer cells. J. Biol. Chem. 276, 29403–29409.
Bence, K., Ma, W., Kozasa, T., and Huang, X. Y. (1997). Direct stimulation of Bruton’s tyrosinekinase by G(q)‐protein alpha‐subunit. Nature 389, 296–299.
Berg, L. J., Finkelstein, L. D., Lucas, J. A., and Schwartzberg, P. L. (2005). Tec family kinases inT lymphocyte development and function. Annu. Rev. Immunol. 23, 549–600.
Brazin, K. N., Fulton, D. B., and Andreotti, A. H. (2000). A specific intermolecular associationbetween the regulatory domains of a Tec family kinase. J. Mol. Biol. 302, 607–623.
Brazin, K. N., Mallis, R. J., Fulton, D. B., and Andreotti, A. H. (2002). Regulation of the tyrosinekinase Itk by the peptidyl‐prolyl isomerase cyclophilin A. Proc. Natl. Acad. Sci. USA 99,1899–1904.
Breheny, P. J., Laederach, A., Fulton, D. B., and Andreotti, A. H. (2003). Ligand specificitymodulated by prolyl imide bond Cis/Trans isomerization in the Itk SH2 domain: A quantitativeNMR study. J. Am. Chem. Soc. 125, 15706–15707.
Breuer, M. L., Cuypers, H. T., and Berns, A. (1989). Evidence for the involvement of pim‐2, a newcommon proviral insertion site, in progression of lymphomas. EMBO J. 8, 743–748.

174 MARTIN FELICES ET AL .
Broussard, C., Fleischecker, C.,Horai, R., Chetana,M., Venegas, A.M., Sharp, L. L., Hedrick, S.M.,Fowlkes, B. J., and Schwartzberg, P. L. (2006). Altered development of CD8þ T cell lineages inmice deficient for the Tec kinases Itk and Rlk. Immunity 25, 93–104.
Brown, K., Long, J.M., Vial, S. C., Dedi, N., Dunster, N. J., Renwick, S. B., Tanner, A. J., Frantz, J. D.,Fleming, M. A., and Cheetham, G. M. (2004). Crystal structures of interleukin‐2 tyrosine kinaseand their implications for the design of selective inhibitors. J. Biol. Chem. 279, 18727–18732.
Bunnell, S. C., Henry, P. A., Kolluri, R., Kirchhausen, T., Rickles, R. J., and Berg, L. J. (1996).Identification of Itk/Tsk Src homology 3 domain ligands. J. Biol. Chem. 271, 25646–25656.
Bunnell, S. C., Diehn, M., Yaffe, M. B., Findell, P. R., Cantley, L. C., and Berg, L. J. (2000).Biochemical interactions integrating Itk with the T cell receptor‐initiated signaling cascade.J. Biol. Chem. 275, 2219–2230.
Chamorro, M., Czar, M. J., Debnath, J., Cheng, G., Lenardo, M. J., Varmus, H. E., andSchwartzberg, P. L. (2001). Requirements for activation and RAFT localization of theT‐lymphocyte kinase Rlk/Txk. BMC Immunol. 2, 3.
Chan, T.O., Rittenhouse, S. E., and Tsichlis, P. N. (1999). AKT/PKBand otherD3 phosphoinositide‐regulated kinases: Kinase activation by phosphoinositide‐dependent phosphorylation. Annu.Rev. Biochem. 68, 965–1014.
Chen, R., Kim, O., Li, M., Xiong, X., Guan, J. L., Kung, H. J., Chen, H., Shimizu, Y., and Qiu, Y.(2001). Regulation of the PH‐domain‐containing tyrosine kinase Etk by focal adhesion kinasethrough the FERM domain. Nat. Cell Biol. 3, 439–444.
Cheng, G., Ye, Z. S., and Baltimore, D. (1994). Binding of Bruton’s tyrosine kinase to Fyn, Lyn, orHck through a Src homology 3 domain‐mediated interaction. Proc. Natl. Acad. Sci. USA 91,8152–8155.
Ching, K. A., Kawakami, Y., Kawakami, T., and Tsoukas, C. D. (1999). Emt/Itk associates withactivated TCR complexes: Role of the pleckstrin homology domain. J. Immunol. 163,6006–6013.
Ching, K. A., Grasis, J. A., Tailor, P., Kawakami, Y., Kawakami, T., and Tsoukas, C. D. (2000). TCR/CD3‐Induced activation and binding of Emt/Itk to linker of activated T cell complexes:Requirement for the Src homology 2 domain. J. Immunol. 165, 256–262.
Colgan, J., Asmal, M., Neagu, M., Yu, B., Schneidkraut, J., Lee, Y., Sokolskaja, E., Andreotti, A.,and Luban, J. (2004). Cyclophilin A regulates TCR signal strength in CD4þ Tcells via a proline‐directed conformational switch in Itk. Immunity 21, 189–201.
Cory, G. O., Lovering, R. C., Hinshelwood, S., MacCarthy‐Morrogh, L., Levinsky, R. J., andKinnon, C. (1995). The protein product of the c‐cbl protooncogene is phosphorylated afterB cell receptor stimulation and binds the SH3 domain of Bruton’s tyrosine kinase. J. Exp. Med.182, 611–615.
Costello, P. S., Turner, M., Walters, A. E., Cunningham, C. N., Bauer, P. H., Downward, J., andTybulewicz, V. L. (1996). Critical role for the tyrosine kinase Syk in signalling through the highaffinity IgE receptor of mast cells. Oncogene 13, 2595–2605.
Crabtree, G. R., and Olson, E. N. (2002). NFAT signaling: Choreographing the social lives of cells.Cell 109(Suppl.), S67–S79.
Dal Porto, J. M., Gauld, S. B., Merrell, K. T., Mills, D., Pugh‐Bernard, A. E., and Cambier, J.(2004). B cell antigen receptor signaling 101. Mol. Immunol. 41, 599–613.
Debnath, J., Chamorro, M., Czar, M. J., Schaeffer, E. M., Lenardo, M. J., Varmus, H. E., andSchwartzberg, P. L. (1999). rlk/TXK encodes two forms of a novel cysteine string tyrosine kinaseactivated by Src family kinases. Mol. Cell. Biol. 19, 1498–1507.
Dhanasekaran, S. M., Barrette, T. R., Ghosh, D., Shah, R., Varambally, S., Kurachi, K., Pienta,K. J., Rubin, M. A., and Chinnaiyan, A. M. (2001). Delineation of prognostic biomarkers inprostate cancer. Nature 412, 822–826.

Tec KINASES IN T CELL AND MAST CELL SIGNALING 175
Dombroski, D., Houghtling, R. A., Labno, C. M., Precht, P., Takesono, A., Caplen, N. J., Billadeau,D. D., Wange, R. L., Burkhardt, J. K., and Schwartzberg, P. L. (2005). Kinase‐independentfunctions for Itk in TCR‐induced regulation of Vav and the actin cytoskeleton. J. Immunol. 174,1385–1392.
Dubois, S., Waldmann, T. A., and Muller, J. R. (2006). ITK and IL‐15 support two distinct subsetsof CD8þ T cells. Proc. Natl. Acad. Sci. USA 103, 12075–12080.
Egloff, A. M., and Desiderio, S. (2001). Identification of phosphorylation sites for Bruton’s tyrosinekinase within the transcriptional regulator BAP/TFII‐I. J. Biol. Chem. 276, 27806–27815.
Ellmeier, W., Jung, S., Sunshine, M. J., Hatam, F., Xu, Y., Baltimore, D., Mano, H., and Littman,D. R. (2000). Severe B cell deficiency in mice lacking the tec kinase family members Tec andBtk. J. Exp. Med. 192, 1611–1624.
Fluckiger, A. C., Li, Z., Kato, R. M., Wahl, M. I., Ochs, H. D., Longnecker, R., Kinet, J. P., Witte,O. N., Scharenberg, A. M., and Rawlings, D. J. (1998). Btk/Tec kinases regulate sustainedincreases in intracellular Ca2þ following B‐cell receptor activation. EMBO J. 17, 1973–1985.
Forssell, J., Sideras, P., Eriksson, C., Malm‐Erjefalt, M., Rydell‐Tormanen, K., Ericsson, P. O., andErjefalt, J. S. (2005). Interleukin‐2‐inducible T cell kinase regulates mast cell degranulation andacute allergic responses. Am. J. Respir. Cell Mol. Biol. 32, 511–520.
Fowell, D. J., Shinkai, K., Liao, X. C., Beebe, A. M., Coffman, R. L., Littman, D. R., and Locksley,R. M. (1999). Impaired NFATc translocation and failure of Th2 development in Itk‐deficientCD4þ T cells. Immunity 11, 399–409.
Fukao, T., Terauchi, Y., Kadowaki, T., and Koyasu, S. (2003). Role of phosphoinositide 3‐kinasesignaling in mast cells: New insights from knockout mouse studies. J. Mol. Med. 81, 524–535.
Furumoto, Y., Brooks, S., Olivera, A., Takagi, Y., Miyagishi, M., Taira, K., Casellas, R., Beaven,M. A., Gilfillan, A. M., and Rivera, J. (2006). Cutting Edge: Lentiviral short hairpin RNAsilencing of PTEN in human mast cells reveals constitutive signals that promote cytokinesecretion and cell survival. J. Immunol. 176, 5167–5171.
Gadue, P., and Stein, P. L. (2002). NK T cell precursors exhibit differential cytokine regulation andrequire Itk for efficient maturation. J. Immunol. 169, 2397–2406.
Galli, S. J., Kalesnikoff, J., Grimbaldeston, M. A., Piliponsky, A. M., Williams, C. M., and Tsai, M.(2005a). Mast cells as ‘‘tunable’’ effector and immunoregulatory cells: Recent advances. Annu.Rev. Immunol. 23, 749–786.
Galli, S. J., Nakae, S., and Tsai, M. (2005b). Mast cells in the development of adaptive immuneresponses. Nat. Immunol. 6, 135–142.
Garcon, F., Bismuth, G., Isnardon, D., Olive, D., and Nunes, J. A. (2004a). Tec kinase migrates tothe T cell‐APC interface independently of its pleckstrin homology domain. J. Immunol. 173,770–775.
Garcon, F., Ghiotto, M., Gerard, A., Yang, W. C., Olive, D., and Nunes, J. A. (2004b). The SH3domain of Tec kinase is essential for its targeting to activated CD28 costimulatory molecule. Eur.J. Immunol. 34, 1972–1980.
Gerard, A., Favre, C., Garcon, F., Nemorin, J. G., Duplay, P., Pastor, S., Collette, Y., Olive, D., andNunes, J. A. (2004). Functional interaction of RasGAP‐binding proteins Dok‐1 and Dok‐2 withthe Tec protein tyrosine kinase. Oncogene 23, 1594–1598.
Gibson, S., Leung, B., Squire, J. A., Hill, M., Arima, N., Goss, P., Hogg, D., and Mills, G. B. (1993).Identification, cloning, and characterization of a novel human T‐cell‐specific tyrosine kinaselocated at the hematopoietin complex on chromosome 5q. Blood 82, 1561–1572.
Gibson, S., August, A., Kawakami, Y., Kawakami, T., Dupont, B., and Mills, G. B. (1996). TheEMT/ITK/TSK (EMT) tyrosine kinase is activated during TCR signaling: LCK is required foroptimal activation of EMT. J. Immunol. 156, 2716–2722.

176 MARTIN FELICES ET AL .
Gilfillan, A. M., and Tkaczyk, C. (2006). Integrated signalling pathways for mast‐cell activation.Nat. Rev. Immunol. 6, 218–230.
Gomez, G., Gonzalez‐Espinosa, C., Odom, S., Baez, G., Cid, M. E., Ryan, J. J., and Rivera, J.(2005). Impaired FcepsilonRI‐dependent gene expression and defective eicosanoid and cytokineproduction as a consequence of Fyn deficiency in mast cells. J. Immunol. 175, 7602–7610.
Gray, P., Dunne, A., Brikos, C., Jefferies, C. A., Doyle, S. L., and O’Neill, L. A. (2006). MyD88adapter‐like (Mal) is phosphorylated by Bruton’s tyrosine kinase during TLR2 and TLR4 signaltransduction. J. Biol. Chem. 281, 10489–10495.
Gu, H., Saito, K., Klaman, L. D., Shen, J., Fleming, T., Wang, Y., Pratt, J. C., Lin, G., Lim, B.,Kinet, J. P., and Neel, B. G. (2001). Essential role for Gab2 in the allergic response. Nature 412,186–190.
Guarnieri, D. J., Dodson, G. S., and Simon, M. A. (1998). SRC64 regulates the localization of aTec‐family kinase required for Drosophila ring canal growth. Mol. Cell 1, 831–840.
Guinamard, R., Fougereau, M., and Seckinger, P. (1997). The SH3 domain of Bruton’s tyrosinekinase interacts with Vav, Sam68 and EWS. Scand. J. Immunol. 45, 587–595.
Haire, R. N., Ohta, Y., Lewis, J. E., Fu, S. M., Kroisel, P., and Litman, G. W. (1994). TXK, a novelhuman tyrosine kinase expressed in T cells shares sequence identity with Tec family kinases andmaps to 4p12. Hum. Mol. Genet. 3, 897–901.
Hao, S., and August, A. (2002). The proline rich region of the Tec homology domain of ITKregulates its activity. FEBS Lett. 525, 53–58.
Harris, J., Werling, D., Hope, J. C., Taylor, G., and Howard, C. J. (2002). Caveolae and caveolin inimmune cells: Distribution and functions. Trends Immunol. 23, 158–164.
Hata, D., Kawakami, Y., Inagaki, N., Lantz, C. S., Kitamura, T., Khan, W. N., Maeda‐Yamamoto,M., Miura, T., Han, W., Hartman, S. E., Yao, L., Nagai, H., et al. (1998). Involvement of Bruton’styrosine kinase in FcepsilonRI‐dependent mast cell degranulation and cytokine production.J. Exp. Med. 187, 1235–1247.
Hawkins, J., and Marcy, A. (2001). Characterization of Itk tyrosine kinase: Contribution of non-catalytic domains to enzymatic activity. Protein Expr. Purif. 22, 211–219.
Heinonen, J. E., Smith, C. I., and Nore, B. F. (2002). Silencing of Bruton’s tyrosine kinase (Btk)using short interfering RNA duplexes (siRNA). FEBS Lett. 527, 274–278.
Hernandez‐Hansen, V., Mackay, G. A., Lowell, C. A., Wilson, B. S., and Oliver, J. M. (2004). TheSrc kinase Lyn is a negative regulator of mast cell proliferation. J. Leukoc. Biol. 75, 143–151.
Heyeck, S. D., and Berg, L. J. (1993). Developmental regulation of a murine T‐cell‐specifictyrosine kinase gene, Tsk. Proc. Natl. Acad. Sci. USA 90, 669–673.
Heyeck, S. D., Wilcox, H. M., Bunnell, S. C., and Berg, L. J. (1997). Lck phosphorylates theactivation loop tyrosine of the Itk kinase domain and activates Itk kinase activity. J. Biol. Chem.272, 25401–25408.
Hirano, M., Kikuchi, Y., Nisitani, S., Yamaguchi, A., Satoh, A., Ito, T., Iba, H., and Takatsu, K.(2004). Bruton’s tyrosine kinase (Btk) enhances transcriptional co‐activation activity of BAM11,a Btk‐associated molecule of a subunit of SWI/SNF complexes. Int. Immunol. 16, 747–757.
Horwood, N. J., Mahon, T., McDaid, J. P., Campbell, J., Mano, H., Brennan, F. M., Webster, D.,and Foxwell, B. M. (2003). Bruton’s tyrosine kinase is required for lipopolysaccharide‐inducedtumor necrosis factor alpha production. J. Exp. Med. 197, 1603–1611.
Horwood, N. J., Page, T. H., McDaid, J. P., Palmer, C. D., Campbell, J., Mahon, T., Brennan, F. M.,Webster, D., and Foxwell, B. M. (2006). Bruton’s tyrosine kinase is required for TLR2 andTLR4‐induced TNF, but not IL‐6, production. J. Immunol. 176, 3635–3641.
Hu, Q., Davidson, D., Schwartzberg, P. L., Macchiarini, F., Lenardo, M. J., Bluestone, J. A., andMatis, L. A. (1995). Identification of Rlk, a novel protein tyrosine kinase with predominantexpression in the T cell lineage. J. Biol. Chem. 270, 1928–1934.

Tec KINASES IN T CELL AND MAST CELL SIGNALING 177
Huber, M., Helgason, C. D., Damen, J. E., Liu, L., Humphries, R. K., and Krystal, G. (1998).The src homology 2‐containing inositol phosphatase (SHIP) is the gatekeeper of mast celldegranulation. Proc. Natl. Acad. Sci. USA 95, 11330–11335.
Hwang, E. S., Szabo, S. J., Schwartzberg, P. L., and Glimcher, L. H. (2005). T helper cell fatespecified by kinase‐mediated interaction of T‐bet with GATA‐3. Science 307, 430–433.
Iwaki, S., Tkaczyk, C., Satterthwaite, A. B., Halcomb, K., Beaven, M. A., Metcalfe, D. D., andGilfillan, A. M. (2005). Btk plays a crucial role in the amplification of Fc epsilonRI‐mediatedmast cell activation by kit. J. Biol. Chem. 280, 40261–40270.
Janssen, E., Zhu, M., Zhang, W., and Koonpaew, S. (2003). LAB: A new membrane‐associatedadaptor molecule in B cell activation. Nat. Immunol. 4, 117–123.
Jefferies, C. A., Doyle, S., Brunner, C., Dunne, A., Brint, E., Wietek, C., Walch, E., Wirth, T., andO’Neill, L. A. (2003). Bruton’s tyrosine kinase is a Toll/interleukin‐1 receptor domain‐bindingprotein that participates in nuclear factor kappaB activation by Toll‐like receptor 4. J. Biol.Chem. 278, 26258–26264.
Jiang, Y., Ma, W., Wan, Y., Kozasa, T., Hattori, S., and Huang, X. Y. (1998). The G proteinG alpha12 stimulates Bruton’s tyrosine kinase and a rasGAP through a conserved PH/BMdomain. Nature 395, 808–813.
Johannes, F. J., Hausser, A., Storz, P., Truckenmuller, L., Link, G., Kawakami, T., and Pfizenmaier,K. (1999). Bruton’s tyrosine kinase (Btk) associates with protein kinase C mu. FEBS Lett. 461,68–72.
Jui, H. Y., Tseng, R. J., Wen, X., Fang, H. I., Huang, L. M., Chen, K. Y., Kung, H. J., Ann, D. K.,and Shih, H. M. (2000). Protein‐tyrosine phosphatase D1, a potential regulator and effector forTec family kinases. J. Biol. Chem. 275, 41124–41132.
Kalesnikoff, J., Baur, N., Leitges, M., Hughes, M. R., Damen, J. E., Huber, M., and Krystal, G.(2002). SHIP negatively regulates IgEþ antigen‐induced IL‐6 production in mast cells byinhibiting NF‐kappa B activity. J. Immunol. 168, 4737–4746.
Kane, L. P., and Watkins, S. C. (2005). Dynamic regulation of Tec kinase localization in membrane‐proximal vesicles of a T cell clone revealed by total internal reflection fluorescence and confocalmicroscopy. J. Biol. Chem. 280, 21949–21954.
Kane, L. P., Lin, J., and Weiss, A. (2000). Signal transduction by the TCR for antigen. Curr. Opin.Immunol. 12, 242–249.
Kashiwakura, J., Suzuki, N., Nagafuchi, H., Takeno, M., Takeba, Y., Shimoyama, Y., and Sakane, T.(1999). Txk, a nonreceptor tyrosine kinase of the Tec family, is expressed in T helper type 1 cellsand regulates interferon gamma production in human T lymphocytes. J. Exp. Med. 190,1147–1154.
Kawakami, T., and Galli, S. J. (2002). Regulation of mast‐cell and basophil function and survival byIgE. Nat. Rev. Immunol. 2, 773–786.
Kawakami, Y., Yao, L., Miura, T., Tsukada, S., Witte, O. N., and Kawakami, T. (1994). Tyrosinephosphorylation and activation of Bruton tyrosine kinase upon Fc epsilon RI cross‐linking. Mol.Cell. Biol. 14, 5108–5113.
Kawakami, Y., Yao, L., Tashiro, M., Gibson, S., Mills, G. B., and Kawakami, T. (1995). Activationand interaction with protein kinase C of a cytoplasmic tyrosine kinase, Itk/Tsk/Emt, on Fcepsilon RI cross‐linking on mast cells. J. Immunol. 155, 3556–3562.
Kawakami, Y., Miura, T., Bissonnette, R., Hata, D., Khan, W. N., Kitamura, T., Maeda‐Yamamoto,M., Hartman, S. E., Yao, L., Alt, F. W., and Kawakami, T. (1997). Bruton’s tyrosine kinaseregulates apoptosis and JNK/SAPK kinase activity. Proc. Natl. Acad. Sci. USA 94, 3938–3942.
Kawakami, Y., Kitaura, J., Hartman, S. E., Lowell, C. A., Siraganian, R. P., and Kawakami, T.(2000a). Regulation of protein kinase CbetaI by two protein‐tyrosine kinases, Btk and Syk. Proc.Natl. Acad. Sci. USA 97, 7423–7428.

178 MARTIN FELICES ET AL .
Kawakami, Y., Kitaura, J., Satterthwaite, A. B., Kato, R. M., Asai, K., Hartman, S. E.,Maeda‐Yamamoto, M., Lowell, C. A., Rawlings, D. J., Witte, O. N., and Kawakami, T.(2000b). Redundant and opposing functions of two tyrosine kinases, Btk and Lyn, in mast cellactivation. J. Immunol. 165, 1210–1219.
Kawakami, Y., Inagaki, N., Salek‐Ardakani, S., Kitaura, J., Tanaka, H., Nagao, K., Kawakami, Y.,Xiao, W., Nagai, H., Croft, M., and Kawakami, T. (2006). Regulation of dendritic cell maturationand function by Bruton’s tyrosine kinase via IL‐10 and Stat3. Proc. Natl. Acad. Sci. USA 103,153–158.
Kikuchi, Y., Hirano, M., Seto, M., and Takatsu, K. (2000). Identification and characterization of amolecule, BAM11, that associates with the pleckstrin homology domain of mouse Btk. Int.Immunol. 12, 1397–1408.
King, P. D., Sadra, A., Han, A., Liu, X. R., Sunder‐Plassmann, R., Reinherz, E. L., and Dupont, B.(1996). CD2 signaling in T cells involves tyrosine phosphorylation and activation of the Tecfamily kinase, EMT/ITK/TSK. Int. Immunol. 8, 1707–1714.
King, P. D., Sadra, A., Teng, J. M., Bell, G. M., and Dupont, B. (1998). CD2‐mediated activation ofthe Tec‐family tyrosine kinase ITK is controlled by proline‐rich stretch‐4 of the CD2 cytoplasmictail. Int. Immunol. 10, 1009–1016.
Kitaura, J., Asai, K., Maeda‐Yamamoto, M., Kawakami, Y., Kikkawa, U., and Kawakami, T. (2000).Akt‐dependent cytokine production in mast cells. J. Exp. Med. 192, 729–740.
Kline, J. B., Moore, D. J., and Clevenger, C. V. (2001). Activation and association of the Tectyrosine kinase with the human prolactin receptor: Mapping of a Tec/Vav1‐receptor binding site.Mol. Endocrinol. 15, 832–841.
Kojima, T., Fukuda, M., Watanabe, Y., Hamazato, F., and Mikoshiba, K. (1997). Characterization ofthe pleckstrin homology domain of Btk as an inositol polyphosphate and phosphoinositidebinding domain. Biochem. Biophys. Res. Commun. 236, 333–339.
Kosaka, Y., Felices, M., and Berg, L. J. (2006). Itk and Th2 responses: Action but no reaction.Trends Immunol. 27, 453–460.
Kronenberg, M. (2005). Toward an understanding of NKT cell biology: Progress and paradoxes.Annu. Rev. Immunol. 23, 877–900.
Kurosaki, T., and Kurosaki, M. (1997). Transphosphorylation of Bruton’s tyrosine kinase ontyrosine 551 is critical for B cell antigen receptor function. J. Biol. Chem. 272, 15595–15598.
Labno, C. M., Lewis, C. M., You, D., Leung, D. W., Takesono, A., Kamberos, N., Seth, A.,Finkelstein, L. D., Rosen, M. K., Schwartzberg, P. L., and Burkhardt, J. K. (2003). Itk functionsto control actin polymerization at the immune synapse through localized activation of Cdc42 andWASP. Curr. Biol. 13, 1619–1624.
Laederach, A., Cradic, K. W., Brazin, K. N., Zamoon, J., Fulton, D. B., Huang, X. Y., and Andreotti,A. H. (2002). Competing modes of self‐association in the regulatory domains of Bruton’s tyrosinekinase: Intramolecular contact versus asymmetric homodimerization. Protein Sci. 11, 36–45.
Laederach, A., Cradic, K. W., Fulton, D. B., and Andreotti, A. H. (2003). Determinants of intraversus intermolecular self‐association within the regulatory domains of Rlk and Itk. J. Mol. Biol.329, 1011–1020.
Langhans‐Rajasekaran, S. A., Wan, Y., and Huang, X. Y. (1995). Activation of Tsk and Btk tyrosinekinases by G protein beta gamma subunits. Proc. Natl. Acad. Sci. USA 92, 8601–8605.
Lewis, R. S., and Cahalan, M. D. (1995). Potassium and calcium channels in lymphocytes. Annu.Rev. Immunol. 13, 623–653.
Li, T., Tsukada, S., Satterthwaite, A., Havlik, M. H., Park, H., Takatsu, K., and Witte, O. N. (1995).Activation of Bruton’s tyrosine kinase (BTK) by a point mutation in its pleckstrin homology (PH)domain. Immunity 2, 451–460.

Tec KINASES IN T CELL AND MAST CELL SIGNALING 179
Liang, X., Wisniewski, D., Strife, A., Shivakrupa, R., Clarkson, B., and Resh,M.D. (2002). Phospha-tidylinositol 3‐kinase and Src family kinases are required for phosphorylation and membranerecruitment of Dok‐1 in c‐Kit signaling. J. Biol. Chem. 277, 13732–13738.
Liao, X. C., and Littman, D. R. (1995). Altered T cell receptor signaling and disrupted T celldevelopment in mice lacking Itk. Immunity 3, 757–769.
Liu, K. Q., Bunnell, S. C., Gurniak, C. B., and Berg, L. J. (1998). T cell receptor‐initiated calciumrelease is uncoupled from capacitative calcium entry in Itk‐deficient T cells. J. Exp. Med. 187,1721–1727.
Liu, W., Quinto, I., Chen, X., Palmieri, C., Rabin, R. L., Schwartz, O. M., Nelson, D. L., and Scala,G. (2001). Direct inhibition of Bruton’s tyrosine kinase by IBtk, a Btk‐binding protein. Nat.Immunol. 2, 939–946.
Lowry, W. E., and Huang, X. Y. (2002). G Protein beta gamma subunits act on the catalytic domainto stimulate Bruton’s agammaglobulinemia tyrosine kinase. J. Biol. Chem. 277, 1488–1492.
Lowry, W. E., Huang, J., Lei, M., Rawlings, D., and Huang, X. Y. (2001). Role of the PHTHmodule in protein substrate recognition by Bruton’s agammaglobulinemia tyrosine kinase.J. Biol. Chem. 276, 45276–45281.
Lu, L. F., Lind, E. F., Gondek, D. C., Bennett, K. A., Gleeson, M. W., Pino‐Lagos, K., Scott, Z. A.,Coyle, A. J., Reed, J. L., Van Snick, J., Strom, T. B., Zheng, X. X., et al. (2006). Mast cells areessential intermediaries in regulatory T‐cell tolerance. Nature 442, 997–1002.
Lu, Y., Cuevas, B., Gibson, S., Khan, H., LaPushin, R., Imboden, J., and Mills, G. B. (1998).Phosphatidylinositol 3‐kinase is required for CD28 but not CD3 regulation of the TEC familytyrosine kinase EMT/ITK/TSK: Functional and physical interaction of EMT with phosphatidy-linositol 3‐kinase. J. Immunol. 161, 5404–5412.
Lucas, J. A., Atherly, L. O., and Berg, L. J. (2002). The absence of Itk inhibits positive selectionwithout changing lineage commitment. J. Immunol. 168, 6142–6151.
Lucas, J. A., Miller, A. T., Atherly, L. O., and Berg, L. J. (2003). The role of Tec family kinases inT cell development and function. Immunol. Rev. 191, 119–138.
Machide, M., Mano, H., and Todokoro, K. (1995). Interleukin 3 and erythropoietin induceassociation of Vav with Tec kinase through Tec homology domain. Oncogene 11, 619–625.
Mahajan, S., Fargnoli, J., Burkhardt, A. L., Kut, S. A., Saouaf, S. J., and Bolen, J. B. (1995). Srcfamily protein tyrosine kinases induce autoactivation of Bruton’s tyrosine kinase. Mol. Cell. Biol.15, 5304–5311.
Malbec, O., Fong, D. C., Turner, M., Tybulewicz, V. L., Cambier, J. C., Fridman,W. H., andDaeron,M. (1998). Fc epsilon receptor I‐associated lyn‐dependent phosphorylation of Fc gamma receptorIIB during negative regulation of mast cell activation. J. Immunol. 160, 1647–1658.
Mallis, R. J., Brazin, K. N., Fulton, D. B., and Andreotti, A. H. (2002). Structural characterizationof a proline‐driven conformational switch within the Itk SH2 domain. Nat. Struct. Biol. 9,900–905.
Manetz, T. S., Gonzalez‐Espinosa, C., Arudchandran, R., Xirasagar, S., Tybulewicz, V., and Rivera,J. (2001). Vav1 regulates phospholipase cgamma activation and calcium responses in mast cells.Mol. Cell. Biol. 21, 3763–3774.
Mano, H., Mano, K., Tang, B., Koehler, M., Yi, T., Gilbert, D. J., Jenkins, N. A., Copeland, N. G.,and Ihle, J. N. (1993). Expression of a novel form of Tec kinase in hematopoietic cells andmapping of the gene to chromosome 5 near Kit. Oncogene 8, 417–424.
Mano, H., Yamashita, Y., Miyazato, A., Miura, Y., and Ozawa, K. (1996). Tec protein‐tyrosine kinaseis an effector molecule of Lyn protein‐tyrosine kinase. FASEB J. 10, 637–642.
Mano, H., Ohya, K., Miyazato, A., Yamashita, Y., Ogawa, W., Inazawa, J., Ikeda, U., Shimada, K.,Hatake, K., Kasuga, M., Ozawa, K., and Kajigaya, S. (1998). Grb10/GrbIR as an in vivo substrateof Tec tyrosine kinase. Genes Cells 3, 431–441.

180 MARTIN FELICES ET AL .
Mao, J., Xie, W., Yuan, H., Simon, M. I., Mano, H., and Wu, D. (1998). Tec/Bmx non‐receptortyrosine kinases are involved in regulation of Rho and serum response factor by Galpha12/13.EMBO J. 17, 5638–5646.
Marengere, L. E., Okkenhaug, K., Clavreul, A., Couez, D., Gibson, S., Mills, G. B., Mak, T. W., andRottapel, R. (1997). The SH3 domain of Itk/Emt binds to proline‐rich sequences in thecytoplasmic domain of the T cell costimulatory receptor CD28. J. Immunol. 159, 3220–3229.
Meng, W., Sawasdikosol, S., Burakoff, S. J., and Eck, M. J. (1999). Structure of the amino‐terminaldomain of Cbl complexed to its binding site on ZAP‐70 kinase. Nature 398, 84–90.
Miller, A. T., and Berg, L. J. (2002). Defective Fas ligand expression and activation‐induced celldeath in the absence of IL‐2‐inducible T cell kinase. J. Immunol. 168, 2163–2172.
Miller, A. T., Wilcox, H. M., Lai, Z., and Berg, L. J. (2004). Signaling through Itk promotes T helper2 differentiation via negative regulation of T‐bet. Immunity 21, 67–80.
Mohamed, A. J., Vargas, L., Nore, B. F., Backesjo, C. M., Christensson, B., and Smith, C. I. (2000).Nucleocytoplasmic shuttling of Bruton’s tyrosine kinase. J. Biol. Chem. 275, 40614–40619.
Morrogh, L. M., Hinshelwood, S., Costello, P., Cory, G. O., and Kinnon, C. (1999). The SH3domain of Bruton’s tyrosine kinase displays altered ligand binding properties when auto‐phosphorylated in vitro. Eur. J. Immunol. 29, 2269–2279.
Mukhopadhyay, S., Ramars, A. S., and Dash, D. (2001). Bruton’s tyrosine kinase associates with theactin‐based cytoskeleton in activated platelets. J. Cell. Biochem. 81, 659–665.
Mukhopadhyay, S., Mohanty, M.,Mangla, A., George, A., Bal, V., Rath, S., and Ravindran, B. (2002).Macrophage effector functions controlled by Bruton’s tyrosine kinase are more crucial than thecytokine balance of T cell responses for microfilarial clearance. J. Immunol. 168, 2914–2921.
Newton, A. C. (2004). Diacylglycerol’s affair with protein kinase C turns 25. Trends Pharmacol. Sci.25, 175–177.
Nore, B. F., Mattsson, P. T., Antonsson, P., Backesjo, C. M., Westlund, A., Lennartsson, J.,Hansson, H., Low, P., Ronnstrand, L., and Smith, C. I. (2003). Identification of phosphorylationsites within the SH3 domains of Tec family tyrosine kinases. Biochem. Biophys. Acta 1645,123–132.
Novina, C. D., Kumar, S., Bajpai, U., Cheriyath, V., Zhang, K., Pillai, S., Wortis, H. H., and Roy,A. L. (1999). Regulation of nuclear localization and transcriptional activity of TFII‐I by Bruton’styrosine kinase. Mol. Cell. Biol. 19, 5014–5024.
Odom, S., Gomez, G., Kovarova,M., Furumoto, Y., Ryan, J. J.,Wright, H. V., Gonzalez‐Espinosa, C.,Hibbs, M. L., Harder, K. W., and Rivera, J. (2004). Negative regulation of immunoglobulinE‐dependent allergic responses by Lyn kinase. J. Exp. Med. 199, 1491–1502.
Ohya, K., Kajigaya, S., Kitanaka, A., Yoshida, K.,Miyazato, A., Yamashita, Y., Yamanaka, T., Ikeda, U.,Shimada, K., Ozawa, K., and Mano, H. (1999). Molecular cloning of a docking protein, BRDG1,that acts downstream of the Tec tyrosine kinase. Proc. Natl. Acad. Sci. USA 96, 11976–11981.
Okayama, Y., and Kawakami, T. (2006). Development, migration, and survival of mast cells.Immunol. Res. 34, 97–115.
Park, H., Wahl, M. I., Afar, D. E., Turck, C. W., Rawlings, D. J., Tam, C., Scharenberg, A. M.,Kinet, J. P., and Witte, O. N. (1996). Regulation of Btk function by a major autophosphorylationsite within the SH3 domain. Immunity 4, 515–525.
Parravicini, V., Gadina, M., Kovarova, M., Odom, S., Gonzalez‐Espinosa, C., Furumoto, Y., Saitoh, S.,Samelson, L. E., O’Shea, J. J., and Rivera, J. (2002). Fyn kinase initiates complementary signalsrequired for IgE‐dependent mast cell degranulation. Nat. Immunol. 3, 741–748.
Perez‐Villar, J. J., and Kanner, S. B. (1999). Regulated association between the tyrosine kinaseEmt/Itk/Tsk and phospholipase‐C gamma 1 in human T lymphocytes. J. Immunol. 163,6435–6441.

Tec KINASES IN T CELL AND MAST CELL SIGNALING 181
Perez‐Villar, J. J., O’Day, K., Hewgill, D. H., Nadler, S. G., and Kanner, S. B. (2001). Nuclearlocalization of the tyrosine kinase Itk and interaction of its SH3 domain with karyopherin alpha(Rch1alpha). Int. Immunol. 13, 1265–1274.
Petro, J. B., Rahman, S. M., Ballard, D. W., and Khan, W. N. (2000). Bruton’s tyrosine kinase isrequired for activation of IkappaB kinase and nuclear factor kappaB in response to B cellreceptor engagement. J. Exp. Med. 191, 1745–1754.
Pivniouk, V. I., Martin, T. R., Lu‐Kuo, J. M., Katz, H. R., Oettgen, H. C., and Geha, R. S. (1999).SLP‐76 deficiency impairs signaling via the high‐affinity IgE receptor in mast cells. J. Clin.Invest. 103, 1737–1743.
Pursglove, S. E., Mulhern, T. D., Mackay, J. P., Hinds, M. G., and Booker, G. W. (2002). Thesolution structure and intramolecular associations of the Tec kinase SRC homology 3 domain.J. Biol. Chem. 277, 755–762.
Quek, L. S., Bolen, J., and Watson, S. P. (1998). A role for Bruton’s tyrosine kinase (Btk) in plateletactivation by collagen. Curr. Biol. 8, 1137–1140.
Raab, M., Cai, Y. C., Bunnell, S. C., Heyeck, S. D., Berg, L. J., and Rudd, C. E. (1995). p56Lck andp59Fyn regulate CD28 binding to phosphatidylinositol 3‐kinase, growth factor receptor‐boundprotein GRB‐2, and T cell‐specific protein‐tyrosine kinase ITK: Implications for T‐cellcostimulation. Proc. Natl. Acad. Sci. USA 92, 8891–8895.
Rawlings, D. J., Saffran, D. C., Tsukada, S., Largaespada, D. A., Grimaldi, J. C., Cohen, L., Mohr,R. N., Bazan, J. F., Howard, M., Copeland, N. G., Jenkins, N. A., and Witte, O. N. (1993).Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science261, 358–361.
Rawlings, D. J., Scharenberg, A. M., Park, H., Wahl, M. I., Lin, S., Kato, R. M., Fluckiger, A. C.,Witte, O. N., and Kinet, J. P. (1996). Activation of BTK by a phosphorylation mechanisminitiated by SRC family kinases. Science 271, 822–825.
Rhee, S. G. (2001). Regulation of phosphoinositide‐specific phospholipase C. Annu. Rev. Biochem.70, 281–312.
Rivera, J., and Gilfillan, A. M. (2006). Molecular regulation of mast cell activation. J. Allergy Clin.Immunol. 117, 1214–1225; quiz 1226.
Rivera, J., Arudchandran, R., Gonzalez‐Espinosa, C., Manetz, T. S., and Xirasagar, S. (2001).A perspective: Regulation of IgE receptor‐mediated mast cell responses by a LAT‐organizedplasma membrane‐localized signaling complex. Int. Arch. Allergy Immunol. 124, 137–141.
Robson, J. D., Davidson, D., and Veillette, A. (2004). Inhibition of the Jun N‐terminal proteinkinase pathway by SHIP‐1, a lipid phosphatase that interacts with the adaptor molecule Dok‐3.Mol. Cell. Biol. 24, 2332–2343.
Rodgers, J. R., and Cook, R. G. (2005). MHC class Ib molecules bridge innate and acquiredimmunity. Nat. Rev. Immunol. 5, 459–471.
Roskoski, R., Jr. (2004). Src protein‐tyrosine kinase structure and regulation. Biochem. Biophys.Res. Commun. 324, 1155–1164.
Roskoski, R., Jr. (2005). Src kinase regulation by phosphorylation and dephosphorylation. Biochem.Biophys. Res. Commun. 331, 1–14.
Rottem, M., and Mekori, Y. A. (2005). Mast cells and autoimmunity. Autoimmun. Rev. 4, 21–27.Roulier, E. M., Panzer, S., and Beckendorf, S. K. (1998). The Tec29 tyrosine kinase is required
during Drosophila embryogenesis and interacts with Src64 in ring canal development.Mol. Cell1, 819–829.
Saito, K., Tolias, K. F., Saci, A., Koon, H. B., Humphries, L. A., Scharenberg, A., Rawlings, D. J.,Kinet, J. P., and Carpenter, C. L. (2003). BTK regulates PtdIns‐4,5‐P2 synthesis: Importance forcalcium signaling and PI3K activity. Immunity 19, 669–678.

182 MARTIN FELICES ET AL .
Saitoh, S., Arudchandran, R., Manetz, T. S., Zhang, W., Sommers, C. L., Love, P. E., Rivera, J., andSamelson, L. E. (2000). LAT is essential for Fc(epsilon)RI‐mediated mast cell activation.Immunity 12, 525–535.
Samelson, L. E. (2002). Signal transduction mediated by the T cell antigen receptor: The role ofadapter proteins. Annu. Rev. Immunol. 20, 371–394.
Schaeffer, E. M., Debnath, J., Yap, G., McVicar, D., Liao, X. C., Littman, D. R., Sher, A., Varmus,H. E., Lenardo, M. J., and Schwartzberg, P. L. (1999). Requirement for Tec kinases Rlk and Itkin T cell receptor signaling and immunity. Science 284, 638–641.
Schaeffer, E. M., Broussard, C., Debnath, J., Anderson, S., McVicar, D. W., and Schwartzberg,P. L. (2000). Tec family kinases modulate thresholds for thymocyte development and selection.J. Exp. Med. 192, 987–1000.
Schaeffer, E. M., Yap, G. S., Lewis, C. M., Czar, M. J., McVicar, D. W., Cheever, A. W., Sher, A.,and Schwartzberg, P. L. (2001). Mutation of Tec family kinases alters T helper cell differentia-tion. Nat. Immunol. 2, 1183–1188.
Schmidt, U., Boucheron, N., Unger, B., and Ellmeier, W. (2004). The role of Tec family kinases inmyeloid cells. Int. Arch. Allergy Immunol. 134, 65–78.
Schneider, H., Guerette, B., Guntermann, C., and Rudd, C. E. (2000). Resting lymphocyte kinase(Rlk/Txk) targets lymphoid adaptor SLP‐76 in the cooperative activation of interleukin‐2transcription in T‐cells. J. Biol. Chem. 275, 3835–3840.
Schwartzberg, P. L., Finkelstein, L. D., and Readinger, J. A. (2005). TEC‐family kinases:Regulators of T‐helper‐cell differentiation. Nat. Rev. Immunol. 5, 284–295.
Setoguchi, R., Kinashi, T., Sagara, H., Hirosawa, K., and Takatsu, K. (1998). Defective degranula-tion and calcium mobilization of bone‐marrow derived mast cells from Xid and Btk‐deficientmice. Immunol. Lett. 64, 109–118.
Shan, X., and Wange, R. L. (1999). Itk/Emt/Tsk activation in response to CD3 cross‐linking inJurkat T cells requires ZAP‐70 and Lat and is independent of membrane recruitment. J. Biol.Chem. 274, 29323–29330.
Shan, X., Czar, M. J., Bunnell, S. C., Liu, P., Liu, Y., Schwartzberg, P. L., and Wange, R. L. (2000).Deficiency of PTEN in Jurkat T cells causes constitutive localization of Itk to the plasmamembrane and hyperresponsiveness to CD3 stimulation. Mol. Cell. Biol. 20, 6945–6957.
Siliciano, J. D., Morrow, T. A., and Desiderio, S. V. (1992). itk, a T‐cell‐specific tyrosine kinase geneinducible by interleukin 2. Proc. Natl. Acad. Sci. USA 89, 11194–11198.
Smith, C. I., Islam, T. C., Mattsson, P. T., Mohamed, A. J., Nore, B. F., and Vihinen, M. (2001). TheTec family of cytoplasmic tyrosine kinases: Mammalian Btk, Bmx, Itk, Tec, Txk and homologs inother species. Bioessays 23, 436–446.
Sommers, C. L., Huang, K., Shores, E. W., Grinberg, A., Charlick, D. A., Kozak, C. A., and Love,P. E. (1995). Murine txk: A protein tyrosine kinase gene regulated by T cell activation. Oncogene11, 245–251.
Sommers, C. L., Rabin, R. L., Grinberg, A., Tsay, H. C., Farber, J., and Love, P. E. (1999). A rolefor the Tec family tyrosine kinase Txk in T cell activation and thymocyte selection. J. Exp. Med.190, 1427–1438.
Su, Y. W., Zhang, Y., Schweikert, J., Koretzky, G. A., Reth, M., and Wienands, J. (1999). Interactionof SLP adaptors with the SH2 domain of Tec family kinases. Eur. J. Immunol. 29, 3702–3711.
Takata, M., and Kurosaki, T. (1996). A role for Bruton’s tyrosine kinase in B cell antigen receptor‐mediated activation of phospholipase C‐gamma 2. J. Exp. Med. 184, 31–40.
Tamir, I., Stolpa, J. C., Helgason, C. D., Nakamura, K., Bruhns, P., Daeron, M., and Cambier, J. C.(2000). The RasGAP‐binding protein p62dok is a mediator of inhibitory FcgammaRIIB signalsin B cells. Immunity 12, 347–358.

Tec KINASES IN T CELL AND MAST CELL SIGNALING 183
Tanaka, N., Asao, H., Ohtani, K., Nakamura, M., and Sugamura, K. (1993). A novel human tyrosinekinase gene inducible in T cells by interleukin 2. FEBS Lett. 324, 1–5.
Tanaka, N., Abe, H., Yagita, H., Okumura, K., Nakamura, M., and Sugamura, K. (1997). Itk, aT cell‐specific tyrosine kinase, is required for CD2‐mediated interleukin‐2 promoter activationin the human T cell line Jurkat. Eur. J. Immunol. 27, 834–841.
Tomlinson, M. G., Kurosaki, T., Berson, A. E., Fujii, G. H., Johnston, J. A., and Bolen, J. B. (1999).Reconstitution of Btk signaling by the atypical tec family tyrosine kinases Bmx and Txk. J. Biol.Chem. 274, 13577–13585.
Tomlinson, M. G., Heath, V. L., Turck, C. W., Watson, S. P., and Weiss, A. (2004a). SHIP familyinositol phosphatases interact with and negatively regulate the Tec tyrosine kinase. J. Biol. Chem.279, 55089–55096.
Tomlinson, M. G., Kane, L. P., Su, J., Kadlecek, T. A., Mollenauer, M. N., and Weiss, A. (2004b).Expression and function of Tec, Itk, and Btk in lymphocytes: Evidence for a unique role for Tec.Mol. Cell. Biol. 24, 2455–2466.
Tsai, M., Grimbaldeston, M. A., Yu, M., Tam, S. Y., and Galli, S. J. (2005). Using mast cell knock‐inmice to analyze the roles of mast cells in allergic responses in vivo. Chem. Immunol. Allergy 87,179–197.
Tsai, Y. T., Su, Y. H., Fang, S. S., Huang, T. N., Qiu, Y., Jou, Y. S., Shih, H. M., Kung, H. J., andChen, R. H. (2000). Etk, a Btk family tyrosine kinase, mediates cellular transformation by linkingSrc to STAT3 activation. Mol. Cell. Biol. 20, 2043–2054.
Tsukada, S., Saffran, D. C., Rawlings, D. J., Parolini, O., Allen, R. C., Klisak, I., Sparkes, R. S.,Kubagawa, H., Mohandas, T., Quan, S., Belmont, J. W., Cooper, M. D., et al. (1993). Deficientexpression of a B cell cytoplasmic tyrosine kinase in human X‐linked agammaglobulinemia. Cell72, 279–290.
Turner, M., and Billadeau, D. D. (2002). VAV proteins as signal integrators for multi‐subunitimmune‐recognition receptors. Nat. Rev. Immunol. 2, 476–486.
van Dijk, T. B., van Den Akker, E., Amelsvoort, M. P., Mano, H., Lowenberg, B., and von Lindern, M.(2000). Stem cell factor induces phosphatidylinositol 30‐kinase‐dependent Lyn/Tec/Dok‐1complex formation in hematopoietic cells. Blood 96, 3406–3413.
van Lohuizen, M., Verbeek, S., Krimpenfort, P., Domen, J., Saris, C., Radaszkiewicz, T., and Berns, A.(1989). Predisposition to lymphomagenesis in pim‐1 transgenic mice: Cooperation with c‐mycand N‐myc in murine leukemia virus‐induced tumors. Cell 56, 673–682.
Vargas, L., Nore, B. F., Berglof, A., Heinonen, J. E.,Mattsson, P. T., Smith, C. I., andMohamed, A. J.(2002). Functional interaction of caveolin‐1 with Bruton’s tyrosine kinase and Bmx. J. Biol. Chem.277, 9351–9357.
Vassilev, A., Ozer, Z., Navara, C., Mahajan, S., and Uckun, F. M. (1999). Bruton’s tyrosine kinase asan inhibitor of the Fas/CD95 death‐inducing signaling complex. J. Biol. Chem. 274, 1646–1656.
Vetrie, D., Vorechovsky, I., Sideras, P., Holland, J., Davies, A., Flinter, F., Hammarstrom, L.,Kinnon, C., Levinsky, R., Bobrow, M., Edvard Smith, C. I., and Bentley, D. R. (1993). The geneinvolved in X‐linked agammaglobulinaemia is a member of the src family of protein‐tyrosinekinases. Nature 361, 226–233.
Volna, P., Lebduska, P., Draberova, L., Simova, S., Heneberg, P., Boubelik, M., Bugajev, V.,Malissen, B., Wilson, B. S., Horejsi, V., Malissen, M., and Draber, P. (2004). Negative regulationof mast cell signaling and function by the adaptor LAB/NTAL. J. Exp. Med. 200, 1001–1013.
Wilcox, H. M., and Berg, L. J. (2003). Itk phosphorylation sites are required for functional activityin primary T cells. J. Biol. Chem. 278, 37112–37121.
Woods, M. L., Kivens, W. J., Adelsman, M. A., Qiu, Y., August, A., and Shimizu, Y. (2001). A novelfunction for the Tec family tyrosine kinase Itk in activation of beta 1 integrins by the T‐cellreceptor. EMBO J. 20, 1232–1244.

184 MARTIN FELICES ET AL .
Xie, Y., Xu, K., Dai, B., Guo, Z., Jiang, T., Chen, H., and Qiu, Y. (2006). The 44 kDa Pim‐1 kinasedirectly interacts with tyrosine kinase Etk/BMX and protects human prostate cancer cells fromapoptosis induced by chemotherapeutic drugs. Oncogene 25, 70–78.
Yamada, N., Kawakami, Y., Kimura, H., Fukamachi, H., Baier, G., Altman, A., Kato, T., Inagaki, Y.,and Kawakami, T. (1993). Structure and expression of novel protein‐tyrosine kinases, Emb andEmt, in hematopoietic cells. Biochem. Biophys. Res. Commun. 192, 231–240.
Yamadori, T., Baba, Y., Matsushita, M., Hashimoto, S., Kurosaki, M., Kurosaki, T., Kishimoto, T.,and Tsukada, S. (1999). Bruton’s tyrosine kinase activity is negatively regulated by Sab, theBtk‐SH3 domain‐binding protein. Proc. Natl. Acad. Sci. USA 96, 6341–6346.
Yamashita, Y., Miyazato, A., Ohya, K., Ikeda, U., Shimada, K., Miura, Y., Ozawa, K., and Mano, H.(1996). Deletion of Src homology 3 domain results in constitutive activation of Tec protein‐tyrosine‐kinase. Jpn. J. Cancer Res. 87, 1106–1110.
Yamashita, Y., Kajigaya, S., Yoshida, K., Ueno, S., Ota, J., Ohmine, K., Ueda, M., Miyazato, A.,Ohya, K., Kitamura, T., Ozawa, K., and Mano, H. (2001). Sak serine‐threonine kinase acts as aneffector of Tec tyrosine kinase. J. Biol. Chem. 276, 39012–39020.
Yang, W., Malek, S. N., and Desiderio, S. (1995). An SH3‐binding site conserved in Bruton’styrosine kinase and related tyrosine kinases mediates specific protein interactions in vitro andin vivo. J. Biol. Chem. 270, 20832–20840.
Yang, W. C., and Olive, D. (1999). Tec kinase is involved in transcriptional regulation of IL‐2 andIL‐4 in the CD28 pathway. Eur. J. Immunol. 29, 1842–1849.
Yang, W. C., Ching, K. A., Tsoukas, C. D., and Berg, L. J. (2001). Tec kinase signaling in T cells isregulated by phosphatidylinositol 3‐kinase and the Tec pleckstrin homology domain. J. Immunol.166, 387–395.
Yao, L., Janmey, P., Frigeri, L. G., Han, W., Fujita, J., Kawakami, Y., Apgar, J. R., and Kawakami, T.(1999). Pleckstrin homology domains interact with filamentous actin. J. Biol. Chem. 274,19752–19761.
Yoshida, K., Yamashita, Y., Miyazato, A., Ohya, K., Kitanaka, A., Ikeda, U., Shimada, K., Yamanaka, T.,Ozawa, K., and Mano, H. (2000). Mediation by the protein‐tyrosine kinase Tec of signalingbetween the B cell antigen receptor and Dok‐1. J. Biol. Chem. 275, 24945–24952.
Zhu, M., Liu, Y., Koonpaew, S., Granillo, O., and Zhang, W. (2004). Positive and negativeregulation of FcepsilonRI‐mediated signaling by the adaptor protein LAB/NTAL. J. Exp. Med.200, 991–1000.
Zhu, M., Rhee, I., Liu, Y., and Zhang, W. (2006). Negative regulation of Fc epsilonRI‐mediatedsignaling and mast cell function by the adaptor protein LAX. J. Biol. Chem. 281, 18408–18413.

a#
Integrin Regulation of Lymphocyte Trafficking: Lessons fromStructural and Signaling Studies
Tatsuo Kinashi
Department of Molecular Genetics, Institute of Biomedical Science,Kansai Medical University, Kyoto 606, Japan
A
dv2
bstract............................................................................................................. 1
185ances in immunology, vol. 93 0065-2776/07 $
007 Elsevier Inc. All rights reserved. DOI: 10.1016/S0065-2776(06)93
85
1. I ntroduction ....................................................................................................... 1 85 2. L eukocyte Integrins ............................................................................................. 1 86 3. A ffinity and Valency Regulation.............................................................................. 1 89 4. I ntegrin Conformational Changes........................................................................... 1 89 5. I ntegrin‐Mediated Adhesion Steps in Lymphocyte Trafficking...................................... 1 95 6. T alin as Intracellular Regulator for Lymphocyte Adhesion and Migration....................... 2 01 7. I ntracellular Signals in Chemokine‐Induced Adhesion and Migration............................ 2 03 8. I nside‐Out Signaling Events in TCR‐Stimulated Lymphocytes ..................................... 2 11 9. C oncluding Remarks............................................................................................ 2 15R
eferences ......................................................................................................... 2 16Abstract
High trafficking capability of lymphocytes is crucial in immune surveillance andantigen responses. Central to this regulatory process is a dynamic control oflymphocyte adhesion behavior regulated by chemokines and adhesion receptorssuch as integrins. Modulation of lymphocyte adhesive responses occurs in a widerange of timewindow from less than a second tohours, enabling rolling lymphocyteto attach to andmigrate through endothelium and interactwith antigen‐presentingcells. While there has been a rapid progress in the understanding of integrinstructure, elucidation of signaling events to relay extracellular signaling to integrinsin physiological contexts has recently emerged from studies using gene‐targetingand gene‐silencing technique. Regulatory molecules critical for integrin activitycontrol distribution of integrins, polarized cellmorphology andmotility, suggestinga signaling network that coordinates integrin function with lymphocyte migration.Here, I review recent studies of integrin structural changes and intracellular signalmolecules that trigger integrin activation (inside‐out signals), and discuss mole-cular mechanisms that control lymphocyte integrins and how inside‐out signalscoordinately modulate adhesive reactions and cell shape and migration.
1. Introduction
Immune cells are the most motile cells in the body. Multiphoton microscopyhas been used to reveal a vivid picture of the robust motility that occurs duringlymphocyte interactions with dendritic cells (DCs) in peripheral lymphoid
35.00
005-3

186 TATSUO KINASHI
tissues (Mempel et al., 2004; Miller et al., 2002). The dynamic regulation ofimmune cell adhesive interactions is fundamentally integrated into immuno-logical responses, and the integrin adhesion receptors play critical roles in thisprocess (Butcher and Picker, 1996; Butcher et al., 1999; Springer, 1990, 1995).Integrins constitute a large family of surface glycoproteins, and they arecomposed of a and b subunits (Hemler, 1990). In particular, leukocyte integ-rins, such as lymphocyte function‐associated antigen (LFA‐1; aL /b2) and thea4 integrins are important in lymphocyte trafficking to peripheral lymphoidtissues through binding to the endothelial ligands ICAM (intercellular adhe-sion molecule)‐1 and ICAM‐2 for LFA‐1 and VCAM (vascular cell adhesionmolecule)‐1 and MAdCAM (mucosal addressin cell adhesion molecule)‐1 fora4 integrins. LFA‐1 and a4 integrins mediate firm attachment of lymphocytesto high endothelial venules (HEV) and facilitate subsequent migration intotissues (Butcher et al., 1999). LFA‐1 is also the critical adhesion moleculein the immunological synapse, a specialized adhesion complex between T cellsand antigen‐presenting cells (APC), and these interactions promote theactivation of naive T cells (Sims and Dustin, 2002).The ability of cells to modulate the strength of integrin adhesion in response to
extracellular stimuli such as antigen or chemokines is essential to proper immunefunction. This activation process, referred to as ‘‘inside‐out signaling’’ (Dustin andSpringer, 1989), ultimately modulates integrin adhesiveness through affinitymodulation (Carman and Springer, 2003), in which ligand‐binding affinity isaltered, and avidity modulation, in which integrin cell surface diffusion andclustering are modified (van Kooyk and Figdor, 2000), which is referred to asvalency modulation here. Recently, our understanding of these phenomena hasbeen facilitated by three‐dimensional structures of integrins. Distinct conforma-tional changes in the integrin extracellular domains are clearly associated withaffinity changes on ligand binding or cell activation (Shimaoka et al., 2002; Takagiand Springer, 2002). Although integrin activation following physiological adhe-sion has been documented, the molecules that relay the inside‐out signal and themechanism(s) by which affinity and valency modulation are regulated have beenelusive. In this article, I review the recent studies of lymphocyte integrin regula-tions from viewpoints of structure and valency, intracellular signaling, and theirphysiological relevancies in lymphocyte trafficking.
2. Leukocyte Integrins
Integrin adhesion molecules are a large family of 24 members of hetero-dimeric cell‐surface receptors composed of 18 a and 8 b subunits (Fig. 1).Cell–matrix and cell–cell interactions mediated by integrins are central to

b2
aL*
aM*
aX*
aD*
b1aV
a1*a2*a3
a4
a5
a6
a8 a9
b4
b7aE
b3
b5
b6b8
a IIba7
a11*
a10*
Figure 1 Integrin family. The associations between the 18 a chains and 8 chains form at least24 integrins. Leukocyte integrins are in gray. The integrins that contain the aI domain are indicatedwith asterisks (�).
INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 187
many fundamental biological processes such as embryogenesis, angiogenesis,organ formation, and immune functions (Hynes, 2002). The leukocyte integ-rin, LFA‐1 (aLb2) is a member of the b2 integrins exclusively expressed onleukocytes, and shares a common b2 subunit with Mac‐1 (aMb2), p150/95(aXb2), and a D (aDb2). LFA‐1 plays important roles in binding to endotheli-um during leukocyte extravasation, lymphocyte homing, and in immunologicalsynapse formation between T cells and APC (Sims and Dustin, 2002). Lym-phocytes also express a4 integrins (a4b1, a4b7) that contribute to lymphocytetrafficking to inflamed or peripheral lymphoid tissues. Ligands for LFA‐1include ICAM‐1, ‐2, ‐3, and junctional adhesion molecule‐1 (JAM‐1) that areexpressed on endothelium or APC. aMb2 and aXb2, also known as comple-ment receptor (CR)‐3 and ‐4, bind to iC3b‐coated particles in addition toICAM‐1, and mediate phagocytosis of microbes. A hereditary defect in theb2 subunit that impairs expressions of leukocyte integrins causes a life‐threatening immunodeficiency, leukocyte‐adhesion deficiency (LAD; Etzioni,1996). b2 and a4 integrins have been attractive drug targets for inhibition ofinflammatory or autoimmune diseases (Staunton et al., 2006).
In the absence of activation, aLb2 has low affinity for ligand. In inside‐outsignaling, signals received by other receptors activate intracellular signalingpathways that impinge on integrin cytoplasmic domains and make the extra-cellular domain competent for ligand binding on a timescale of less than 1s.This property enables leukocytes to rapidly respond to signals in the environ-ment, such as cognate antigen or chemoattractant, to activate adhesion, anddirect cell migration. Rapid progress in the integrin extracellular structurehas recently made, which provides important clues how the integrin confor-mational changes are propagated through the cytoplasmic domain to the legdomains to the ligand‐binding headpiece (Carman and Springer, 2003; Dustinet al., 2004).
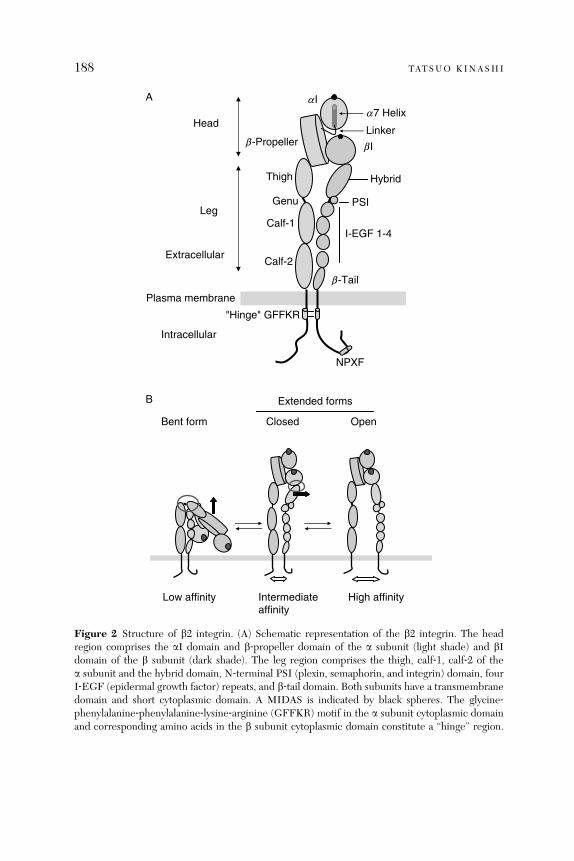
"Hinge" GFFKR
NPXF
a I
b -Propeller
Thigh
Calf-1
Calf-2
Hybrid
I-EGF 1-4
b -Tail
b I
Genu
Extracellular
Intracellular
Plasma membrane
a7 Helix
PSI
LinkerHead
A
B
Leg
Low affinity Intermediateaffinity
High affinity
Bent form Closed Open
Extended forms
Figure 2 Structure of b2 integrin. (A) Schematic representation of the b2 integrin. The headregion comprises the aI domain and b‐propeller domain of the a subunit (light shade) and bIdomain of the b subunit (dark shade). The leg region comprises the thigh, calf‐1, calf‐2 of thea subunit and the hybrid domain, N‐terminal PSI (plexin, semaphorin, and integrin) domain, fourI‐EGF (epidermal growth factor) repeats, and b‐tail domain. Both subunits have a transmembranedomain and short cytoplasmic domain. A MIDAS is indicated by black spheres. The glycine‐phenylalanine‐phenylalanine‐lysine‐arginine (GFFKR) motif in the a subunit cytoplasmic domainand corresponding amino acids in the b subunit cytoplasmic domain constitute a ‘‘hinge’’ region.
188 TATSUO KINASHI

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 189
3. Affinity and Valency Regulation
Before going into details of integrin structural changes and regulatory signalingpathways, how integrin controls total cell adhesiveness (avidity) is briefly con-sidered. Theoretically, avidity is related to affinity and surface density (valency)of integrins. Inside‐out signals are thought to modulate either or both of theseparameters and act in a timewindow ranging from less than a second tominutes.Initial attachments are often transient and weak, and stabilization subsequentlyoccurs by ligand‐induced conformational changes, linkage to the cytoskeleton,or cell spreading. It is difficult to distinguish avidity changes by inside‐outsignals from those induced by ligand occupancy even in a short time window.To distinguish inside‐out signals from postadhesion events, integrin affinity orvalency changes should be examined before cell adhesion. Since conventionalaffinity measurements are not sensitive enough to detect affinity changes inmicromolar ranges, affinity modulation has been underestimated. Thus, valencyregulation has been often inferred when activators induce adhesion withoutdetectable affinity changes. Availability of monoclonal antibodies to detectdistinct conformations of integrins eases this difficulty, and more sensitivemethods to detect affinity (Chan et al., 2003; Lollo et al., 1993), conformation(Chigaev et al., 2003; Larson et al., 2005), and mobility (Cairo et al., 2006) arereported and shown to be instrumental to dissect effects of inside‐out signals onintegrins (Sections 4 and 5). These materials and methods are expected to beexploited widely to study roles of inside‐out signals.
4. Integrin Conformational Changes
4.1. Global Changes of Extracellular Domains in IntegrinsThat Lack the aI Domains
The overall integrin structure resembles a ‘‘head’’ connected to ‘‘two legs’’(Fig. 2A). The a subunit comprises an N‐terminal b‐propeller at the top,followed by three b‐sandwich modules (thigh, calf‐1, and calf‐2). The b subunitcomprises an N‐terminal PSI (plexin, semaphorin, and integrin) domain, fol-lowed by a b‐sandwich hybrid domain, a bI domain (von Willebrand factor
The b cytoplasmic domain contains a talin‐binding asparagine‐proline‐(any amino acid)‐phenylalanine (NPXF)motif. (B) Equilibrium of the bent (low affinity) and extended conformationswith the ‘‘closed’’ (intermediate affinity) and the ‘‘open’’ (high affinity) states triggered by separationof the cytoplasmic tails. The extended‐open, high‐affinity conformation is induced/stabilized byseparation of the a and b cytoplasmic and leg regions. The flexible joints at the genu and betweenI‐EGF‐1 and I‐EGF‐2, and the bI/hybrid domain interface are indicated by circles. The upright andoutward motions of the extracellular domains and the hybrid domain in transition from the bent tothe extended and from the closed to the open states are indicated by thick arrows.

190 TATSUO KINASHI
A domain), four epidermal growth factor (EGF) repeats, and a b‐tail domain.Half of the 18 integrin a subunits (a1, a2, a10, a11, aL, aM, aX, aD, and aE)also include an I domain in their a subunits (aI‐domain) inserted through shortlinkers into the upper face of the b‐propeller. Where present, this domain is themajor site of ligand binding. The major sites of ligand recognition of integrinsthat lack the aI domain are the top face of the bI domain and the loops on theupper surface of the b‐propeller. Both the aI and bI domains contain a metalion‐dependent adhesion site (MIDAS), where a divalent metal is coordinated bya ligand’s acidic residue (Hynes, 2002).Recent structural studies of integrins that lack the I domain have led to a
general model of integrin conformational changes; in the low‐affinity confor-mation, the leg region is acutely bent at the ‘‘genu’’ (knee) between the thighand calf‐1 domains and between the I‐EGF‐1 and I‐EGF‐2, with the ligand‐binding headpiece in proximity to the membrane proximal leg region, topo-logically pointing toward the plasma membrane (Xiong et al., 2001, 2002;Fig. 2B). The electron microscopic analysis of negatively stained solublerecombinant integrins together with mutational studies and physicochemicalmeasurement elegantly demonstrate that the switch blade‐like extension of theleg regions shifts the molecule to the intermediate or high‐affinity conforma-tions in a manner dependent on the orientation of the bI domain and hybriddomain. In a ‘‘closed’’ conformation, the bI makes an acute angle with thehybrid domain, and in an ‘‘open’’ high‐affinity conformation, the outwardmotion of the hybrid domain occurs, making an obtuse angle with the bIdomain (Takagi et al., 2002). Therefore, the extension of the legpiece andthe orientation between the hybrid and bI domains of the headpiece are thekey translator for converting global conformational changes into regulation ofaffinity (Takagi and Springer, 2002). Although a bent conformation may not beequated with low‐affinity binding in all situations (Adair et al., 2005; Xionget al., 2002), the extension is thought to be particularly relevant in cell–celladhesion mediated by leukocyte integrins. Indeed, it has been suggested frommany studies using monoclonal antibodies that integrins undergo dynamicconformational changes in the legpiece (Lu et al., 2001a) as well as theheadpiece (Humphries, 2004; Lu et al., 2001b, 2004), depending on divalentmetals, ligand binding, or inside‐out signals.
4.2. Extensions of Extracellular Domains of b2 Integrins
It has been recently demonstrated by using soluble recombinant aXb2 andaLb2 that b2 integrins also show three distinct conformations: a bent confor-mation, extended conformations with closed or open states of the headpiece(Nishida et al., 2006; Fig. 2B), as seen in integrins that lack I domains (Takagi

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 191
et al., 2002). When the entire extracellular domains of a and b subunits arelinked via a disulfide bond and coiled‐coil sequences fused at the C‐terminalends (clasped form), aXb2 predominantly showed V‐shaped bent forms inphysiological Mg2þ and Ca2þ concentrations. Compared with clasped aXb2,clasped aLb2 appears to be more relaxed in conformation, showing both thebent (55%) and extended‐closed (45%). This is in line with the characteristicsof aXb2, which requires stronger cellular activation for adhesion than othermembers of b2 integrins. Removal of C‐terminal clasp (unclasped) of aXb2increased extends forms with the closed (50%) and open (25%) headpiece withthe rest remained bent. Unclasping of aLb2 also increased the extended‐openconformation. These results are in an excellent agreement with those ofintegrins without the aI‐domain, and support a coherent model of integrinconformational changes through the bent to the extended‐closed to theextended‐open states. Since these distinct states can coexist under the definedconditions, the conformational changes are not all‐or‐none responses, butshould be regarded as equilibria among multiple states (Carman and Springer,2003). Thus, in basal states integrin molecules are continually flexing (breath-ing) to some degree, and stabilization of the legpiece prefers the bent form,and its separation shifts an equilibrium toward the extended forms. Theequilibrium points likely differ in integrin family members. A b2 monoclonalantibody (CBR‐LFA1/2; Petruzzelli et al., 1995), which induces high‐affinitystates and stimulates adhesion by binding an epitope in the I‐EGF3 domain,separates a and b leg regions and induced or stabilized extended conforma-tions. Thus, disruption of the interaction of the a and b cytoplasmic tails byinside‐out signals probably leads to a loss of the interactions between the legregions, resulting in repositioning of the ligand‐binding headpiece pointingaway from the plasma membrane (Fig. 2B). This model is consistent withstudies on epitopes of stimulatory mAb that have now been shown to lie in theknee or leg regions (Lu et al., 2001a; Xie et al., 2004). Exposure of theseepitopes is low in the bent state of the integrin (where they are masked) buthigh in the extended state (Lu et al., 2001a; Xie et al., 2004). The bI domainappears to play a regulatory role in this conformational change relay. Thetreatment of the clasped and unclasped aXb2 with a small molecule antagonist,XVA143, greatly increased extended conformations predominantly with theopen state (Nishida et al., 2006). This is consistent with the proposed mecha-nism of XVA143, acting on the MIDAS of the bI domain, leading to thebI activation with the hybrid domain swing‐out, while inhibiting activation ofthe aI domain (Shimaoka et al., 2003a). The bI and hybrid domains may serveas a switch in transmitting the conformational signals from the ligand‐bindingaI domain to the C‐terminal regions on ligand binding and from the cytoplasmictails to the aI domain by inside‐out signals.

192 TATSUO KINASHI
4.3. Multiple Affinity States of the aI Domain
The I domain was crystallized in three major forms: closed (low affinity,�2mM),intermediate (3–9 mM), and open (high affinity, �0.2 mM; Shimaoka et al., 2002,2003b). The major conformational changes during the transition from the closedto open states include a rearrangement of the cation‐coordinating residues in theMIDAS site, accompanied by a small inwardmovement of the a1 helix and a largedownward shift of the mobile C‐terminal (a7) helix. Crystal structures of aL Idomain reveal that the a7 helix can adopt three different positions. An inter-mediate state between the fully closed and fully open forms of the domaininvolves a downward shift halfway to that observed for the fully open state.Thus, rearrangement of the MIDAS into the ligand‐binding configuration istightly coupled to a downward movement of the C‐terminal helix.
4.4. Regulation of the aI Domain Conformations by the bI Domain
The open and closed conformations of the aI domain are regulated by interac-tion of the C‐terminal linker with the bI domain. A conserved glutamic acid inthe C‐terminal linker appears to act as an internal ligand to the bI domain andplays important role in conformational changes of the aI domain (Huth et al.,2000; Yang et al., 2004b). Mutations of the glutamic acid in the linker or aminoacids constituting the MIDAS of the bI domain result in the low‐affinity state ofthe aI domain. Furthermore, double mutation of these residues to cysteine,allowing formation of the disulfide bond between the linker and bI, results in aconstitutive high‐affinity state of aI (Yang et al., 2004b). These results supportthe hypothesis that the bI domain regulates the activity of aI by pulling down onthe linker region leading to a downward movement of the C‐terminal a helix byexertion of a bell‐rope‐like pull on a segment within the C‐terminal linker region(Carman and Springer, 2003). Because this site is equivalent to the ligand‐binding site in integrins that lack I domains, the interaction of the linker withthe MIDAS of the bI domain may occur that are analogous to those thatregulated interactions with ligands in integrins that lack I domains. Thus, thethree headpiece units, the aI, b‐propeller, and bI domains, make a ternaryinteraction interface where structural rearrangements of the latter two domainsaffect the conformation of the aI domain.
4.5. Regulation of the bI Domain by Extensions and Divalent Metals
Affinity regulation of the bI domain is thought to occur by the same mecha-nism as that regulating the aI domain. Both a1 and a7 helix movements arecritical for bI domain regulation generating low‐ and high‐affinity states (Luo

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 193
et al., 2004; Mould et al., 2002, 2003b; Yang et al., 2004a). The outward motionof the hybrid domain is linked to a7 helix movements presumably because thehybrid domain exerts a downward pull on this structural element. The orien-tation between the hybrid and bI domains is therefore thought to be a keytranslator for converting global conformational changes into regulation ofaffinity. However, the high‐affinity state of the b3 integrin locked by a disulfidebond between the b6 and a7 loop remains in the bent conformer. This suggeststhat a7 helix downward movement of the bI domain leading to the high‐affinitystates does not necessarily lead to extended conformers (Luo et al., 2004).
It has been well known that divalent metal ions affect integrin activities,depending on concentrations andmetal species. For examples,Mn2þ has a potentstimulatory effect on integrin activity, and Mg2þ and Ca2þ are stimulatory andinhibitory on lymphocyte integrins, depending on concentrations, respectively(Dransfield et al., 1992; Shimizu and Mobley, 1993). The major sites of themodulatory effects of the divalent metals are in the bI domain. The bI domaincontains a MIDAS (b MIDAS) centered between two other metal‐binding sites,the adjacent MIDAS, ADMIDAS, and the ligand‐induced metal‐binding site,LIMBS (Xiong et al., 2001). Ligand‐binding activity of the bI domain is regulatedby variable divalent cation occupancy.Occupation of theADMIDAS in highCa2þ
decreases ligand binding, whereas replacement by competing Mn2þ activatesligand binding. Low Ca2þ, with Ca2þ occupancy at the LIMBS, may synergizewith Mg2þ to support ligand binding (Chen et al., 2003; Mould et al., 2003a).A mutation of the LIMBS site in a4b7 results in a low‐affinity state, capable ofsupporting lymphocyte rolling, whereas mutation of the ADMIDAS results in ahigh‐affinity state, supporting firm adhesion of lymphocytes (Chen et al., 2003),suggesting that the ADMIDAS and LIMBS also have global effects on integrinbent/extension conformations.
4.6. Cytoplasmic Domain
Both a and b subunits have short cytoplasmic domains (Sastry and Horwitz,1993). The cytoplasmic domains have categorically three functions: a /b het-erodimer formation, signaling interface from the inside and outside, andintegrin endocytosis/recycling. It is becoming clear that these functions maycross‐talk in lymphocyte trafficking.
From the studies using soluble extracellular regions of integrins, a physicalassociation of C‐terminal regions induces or stabilizes bent conformations andits separation trigger the extension and affinity upregulation. The membraneproximal glycine‐phenylalanine‐phenylalanine‐lysine‐arginine (GFFKR) motifof the a subunit, referred to as the ‘‘hinge’’ domain, is conserved throughout allintegrin families. This motif acts as a negative regulatory sequence suppressing

194 TATSUO KINASHI
integrin adhesion (Hughes et al., 1996); deletion of the motif converts inactiveLFA‐1 into constitutively active LFA‐1 (Lu and Springer, 1997). The arginine inthis GFFKR motif and an aspartic acid at the corresponding position in the bchain form a salt bridge, placing the a and b cytoplasmic regions in closejuxtaposition. Thismay stabilize bent conformations of integrins,making adhesiveactivities low. Consistently, lymphocytes generated from ‘‘knock‐in’’ mice expres-sing theaL subunit that lacks theGFFKRmotif showhigher basal adhesion levelsthan wild‐type lymphocytes (Semmrich et al., 2005). Deletion of the GFFKRmotif also lowers surface amounts of LFA‐1, but not other integrins members,supporting an important role of the GFFKRmotif in heterodimer formation withthe b2 subunit (Lu and Springer, 1997). Thus, the GFFKR motif facilitates aheterodimer formation, which is a requisite process to transport an a /b hetero-dimer to cell surface as an inactive, perhaps bent conformer. On the other hand,regulatory functions through the GFFKR motif in inside‐out signaling, oroutside‐in signaling are less clear, compared to its structural requirement forinactive integrin on cell surface. The LFA‐1 that lacks the GFFKR motif stillresponds to inside‐out signals including the TCR complex for attachment toICAM‐1, and activation of JNK or Erk is not altered on binding to ICAM‐1(Semmrich et al., 2005). Interestingly, lymphocyte expressing LFA‐1 that lacksthe GFFKR motif is impaired in detachment on ICAM‐1. This defect mayunderlie hypoplastic peripheral lymph nodes, and impaired humoral responsesto Tcell‐dependent antigen and leukocyte recruitment into inflamed peritoneum(Semmrich et al., 2005).The integrin cytoplasmic domains play crucial roles in transmitting the
inside‐out signals to the extracellular domains as well as outside‐in signalsfrom ligand‐bound I domains, through binding to cytoskeletal linker proteinsand intracellular proteins to the distinct sites of the cytoplasmic domains(Calderwood, 2004; Liu et al., 2000). The NPxY/F is well conserved in allb integrins and is shown to interact with an actin cytoskeleton linker protein,talin (Calderwood, 2004). In addition, phosphorylation of amino acids in thecytoplasmic domains is increased by inside‐out signals or after ligand binding(Fagerholm et al., 2004). Both aL and b2 cytoplasmic phosphorylations aretriggered by inside‐out signals or modulates integrin functions (Fagerholmet al., 2005; Hibbs et al., 1991), perhaps through recruitment of binding proteinsthat recognize phosphorylated amino acids.The integrin surface distribution is thought to be regulated by lateral
diffusion through linkage of cytoplasmic domains to the cytoskeleton. Distinctconformations of LFA‐1 are shown to have different surface mobilitymeasured by single‐particle tracking (Cairo et al., 2006; Kucik et al., 1996).Intracellular transport also plays an important role of integrin distribution.Integrins are endocytosed and recycled back between the plasma membrane

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 195
and intracellular pools. The critical role of the cytoplasmic domain is demon-strated for polarized endocytic recycle of avb3 to the migrating front inneutrophils and fibroblasts (Jones et al., 2006; Lawson and Maxfield, 1995).The mutation of the membrane proximal endocytosis motif Y735xxF (F,a bulky hydrophobic amino acid) of the human b2 subunit inhibits internaliza-tion of LFA‐1, and impairs detachment (Tohyama et al., 2003) and transportingof LFA‐1 to the ruffling membrane (Fabbri et al., 1999), resulting in defectivemigration. Endocytic recycling pathways of LFA‐1 are different from conven-tional clathrin‐mediated pathways and depend on lipid raft (Fabbri et al.,2005). The polarized redistribution of LFA‐1 to the leading edge after chemo-kine stimulation is also inhibited by mutations of the aL cytoplasmic regionafter the GFFKR motif (Katagiri et al., 2003). These mutations make LFA‐1 inlow‐affinity bent conformations with a low exposure of a NKI‐L16 epitope(Tohyama et al., 2003), a legpiece extension reporter antibody that recognizesthe interface the aL genu and thigh domains (Xie et al., 2004). Thus, thecytoplasmic regions also have key roles in endocytosis and recycling of leuko-cyte integrins and link affinity/conformational changes with spatial regulation.
In summary, the extension and the hybrid domain–bI interface can act asflexible joints and may adopt distinct positions, each of which is likely to have aglobal effect on the overall ligand‐binding affinity of the integrin. Integrinextension may affect cell adhesion through two distinct modes: accessibility toligands by extending the head region into a position appropriate for recogni-tion of extracellular ligands, and affinity to ligands by freeing the hybriddomain from the structural restrain. Association and separation of a /b cyto-plasmic domains play regulatory roles in transmitting signals from inside‐outand outside‐in signals and also control integrin distribution coupled withcytoskeleton and endocytic recycling.
5. Integrin‐Mediated Adhesion Steps in Lymphocyte Trafficking
It has been becoming apparent that chemokines and antigens play decisiveroles in adhesive interactions with endothelial cells and APC. In this section,I focus the critical steps of lymphocyte trafficking from attachment and migra-tion through endothelial venules to interactions with APC (Fig. 3), and discusshow lymphocyte adhesiveness is modulated by chemokines and antigens interms of integrin affinity/conformation and spatial regulation.
5.1. Conversion of Rolling to Firm Adhesion by Chemokines
During entry into peripheral lymph nodes, naive T cells interact with HEV in aprocess involving sequential adhesion steps: (1) tether (capture) or roll onHEV through selectin‐mediated interactions, (2) arrest (stop) mediated by
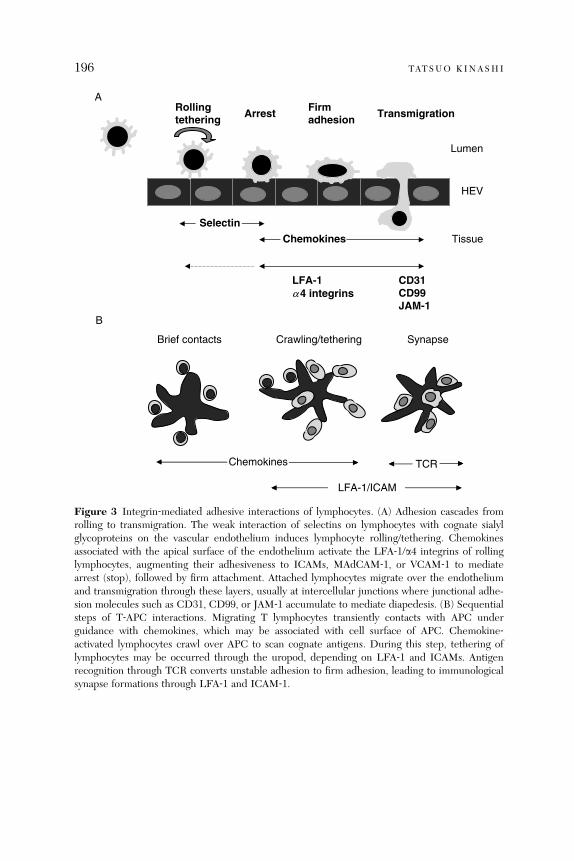
Rollingtethering
Firmadhesion TransmigrationArrest
A
B
Selectin
LFA-1a4 integrins
CD31CD99JAM-1
Chemokines
HEV
Lumen
Tissue
Brief contacts Crawling/tethering Synapse
LFA-1/ICAM
TCRChemokines
Figure 3 Integrin‐mediated adhesive interactions of lymphocytes. (A) Adhesion cascades fromrolling to transmigration. The weak interaction of selectins on lymphocytes with cognate sialylglycoproteins on the vascular endothelium induces lymphocyte rolling/tethering. Chemokinesassociated with the apical surface of the endothelium activate the LFA‐1/a4 integrins of rollinglymphocytes, augmenting their adhesiveness to ICAMs, MAdCAM‐1, or VCAM‐1 to mediatearrest (stop), followed by firm attachment. Attached lymphocytes migrate over the endotheliumand transmigration through these layers, usually at intercellular junctions where junctional adhe-sion molecules such as CD31, CD99, or JAM‐1 accumulate to mediate diapedesis. (B) Sequentialsteps of T‐APC interactions. Migrating T lymphocytes transiently contacts with APC underguidance with chemokines, which may be associated with cell surface of APC. Chemokine‐activated lymphocytes crawl over APC to scan cognate antigens. During this step, tethering oflymphocytes may be occurred through the uropod, depending on LFA‐1 and ICAMs. Antigenrecognition through TCR converts unstable adhesion to firm adhesion, leading to immunologicalsynapse formations through LFA‐1 and ICAM‐1.
196 TATSUO KINASHI

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 197
LFA‐1 and a4 integrins on lymphocytes activated by chemokines throughbinding to ICAM‐1 and MAdCAM‐1 on the endothelium, and (3) diapedesis(transendothelial migration; Fig. 3A). Although rolling is facilitated by lympho-cyte expression of L‐selectin, with a minor contribution from LFA‐1 anda4 integrins, LFA‐1 and a4b7 have major roles in the firm attachment oflymphocytes to HEVs of peripheral lymph nodes and Peyer’s patches (Butcheret al., 1999). Chemokines, including C‐C‐chemokine ligand 21 (CCL21), CXC‐chemokine ligand 12 (CXCL12), and CXCL13 (Ebisuno et al., 2003; Okadaet al., 2002; Stein et al., 2000; Warnock et al., 2000), localized on the apicalendothelial surface rapidly increase integrin avidity, resulting in lymphocytearrest. Since upregulation of lymphocyte adhesion by chemokines are transientand affinity changes likely occur in micromolar ranges, it is technically difficultto detect affinity modulation using conventional assays. Thus, it has been oftencontroversial whether affinity modulation occurs in physiological contexts.Nonetheless, the modality of integrin avidity regulation in this step has beenreported to involve both affinity and valency regulation. Stimulation of primaryT cells with chemokines induces the patch‐like clustering of LFA‐1 and themicroclustering of a4b1, which correlate respectively with increased cellularadhesion to low‐density ICAM‐1 (Constantin et al., 2000) and transient teth-ering and firm adhesion under shear flow (Grabovsky et al., 2000). LFA‐1 anda4 integrin affinity is also augmented by chemokine signaling (Chan et al.,2003; Grabovsky et al., 2000; Shimaoka et al., 2006) and is important forlymphocyte homing to peripheral lymph nodes (Constantin et al., 2000).
In terms of currently understood integrin conformations, the conformationmediating rolling appears to most closely correspond to lymphocyte integrinsin an extended conformation with a low‐affinity I domain, whereas the confor-mation that mediates arrest adhesion appears to most closely correspond to anextended conformation with an intermediate or high‐affinity I domain. Theexperiments using K562 cells reconstituted LFA‐1 carrying a locked aLI domain with either low, intermediate, or high‐affinity state demonstratethat rolling adhesion occurs when extension of low‐affinity LFA‐1 is inducedby the treatment with XVA143 and that the shift from low to intermediateaffinity transforms rolling adhesion to firm adhesion in shear flow (Salas et al.,2002, 2004, 2006). In neutrophil LFA‐1, a shift from low to intermediateaffinity stabilized by IC487475, an aL I domain allosteric antagonist, supportsrolling, whereas high affinity is associated with shear‐resistant leukocyte arrest(Green et al., 2006). Thus, extension with affinity changes is thought to be akey step in transition of lymphocyte rolling to arrest triggered by chemokines.
Does chemokines actually induce extension of integrins and affinity changesduring a shift from rolling? It has been shown that immobilized chemokinesinduce extended conformations under physiological shear flow, whereas

198 TATSUO KINASHI
soluble chemokines do not (Shamri et al., 2005). Immobilized chemokinesinduce lymphocyte b2 legpiece extension recognized by a reporter antibodyKIM127 (Robinson et al., 1992; Xie et al., 2004) with intermediate affinitychanges under shear flow (Shamri et al., 2005). The extension depends onGi‐coupled signaling, suggesting endothelial chemokines induce extension ofLFA‐1 with affinity changes in less than a second (Shamri et al., 2005). Thisstudy proposes a model, in which extension of bent LFA‐1 is the critical firststep of lymphocyte arrest, making LFA‐1 accessible to surface ICAM‐1, lead-ing to ICAM‐1‐induced high‐affinity LFA‐1 conformations and stabilization ofcell adhesion under flow (Shamri et al., 2005). The whole process occurswithin a second at restricted sites on the lymphocyte surface and requirescooperation of inside‐out and outside‐in signaling.To examine unbending more directly, conformational changes in cell surface
a4b1 are probed using fluorescent resonance energy transfer (FRET) betweenan FITC‐labelled ligand peptide donor and rhodamine B acceptors in theplasma membrane. Stimulation with Mn2þ induced a high affinity to ligandand placing the headpiece of the resting integrin near the membrane surfaceallows for an extension of theMn2þ activated headpiece�50 A from the surface.This distance is approximately one‐half that expected if the integrin moleculeundergoes the conformational change from the fully folded to the fully extendedconformation. The activation of the integrin by inside‐out signaling through aG‐protein–coupled receptor, resulting in the intermediate affinity, leads to thehead region moving away from the surface by �25 A after stimulation by achemoattractant. The distance change is correlated with ligand‐binding affinity.The half‐time corresponding the diminution of FRET due to activation ofthe integrin is less than 30 s. The results indicate that there is a coordinationbetween extension of the ligand‐binding headpiece away from the cell surfaceand affinity to ligand, and the fully extended conformation were not observedwith this method. Although it could be possible that the extended‐open con-formations are not induced by inside‐out signals alone, the fully extendedconformation may exist at the moment of the engagement of the integrin bythe natural endothelial ligand under shear flow. The transient bond formation inrolling would allow the forces to induce a molecular extension, and a ligand‐bound I domain induces or stabilizes fully extended conformations with theopen headpiece, resulting in arrest and firm adhesion (Shamri et al., 2005).Interestingly, a study using FRET technology demonstrates that separation of
the aL and b2 cytoplasmic regions occurs following chemokine stimulation andligand binding (Kim et al., 2003). This study indicates that separation of cyto-plasmic regions occurs by inside‐out and outside‐in signaling and support thenotion that chemokine‐stimulated inside‐out signals inhibit close associations of

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 199
a and b cytoplasmic regions, which likely releases a restrain on bent conforma-tions and induce unbending of the extracellular domains to mediate lymphocytearrest.
5.2. Transmigration
Given the presence of chemokines on the apical side of the endothelium, it isunlikely that a chemokine gradient across the endothelium stimulate transmi-gration of T cells in vivo. Thus, apical chemokines arrest rolling lymphocytesand subsequently stimulate cell motility over the endothelium. When migrat-ing lymphocytes reach intercellular junctions, they begin diapedesis betweenapposed endothelial cells (Johnson‐Leger et al., 2000). b1 and b2 integrins areinvolved in transmigration step, in addition to adhesion molecules such asCD31, CD99, or JAM‐1 (Muller, 2003). In addition, shear flow is required forefficient lymphocyte transmigration (Cinamon et al., 2001). Lymphocyte trans-migration needs apical chemokines and Gi‐dependent signaling under shearflow conditions (Cinamon et al., 2001). Requirement of shear stress in lym-phocyte transmigration suggest a mechanosensitive regulatory process (Vogeland Sheetz, 2006) acting on migrating lymphocytes and endothelial barriersthrough activation of intracellular signaling such as focal adhesion kinases (Liet al., 1997) and small GTPases (de Bruyn et al., 2003; Tamada et al., 2004).This may cause enhancement lymphocyte adhesion and motility in verticaldirections, or modulation of junctional permeability.
In addition to paracellular pathways of transmigration, lymphocyte maymigrate in a transcellular fashion (Carman and Springer, 2004), as reportedin leukocytes in vivo (Feng et al., 1998). During transcellular migration,cuplike‐endothelial projections enriched for ICAM‐1 and VCAM‐1 surroundleukocytes. b2 and a4 integrins are distributed in linear clustering patternsoriented parallel to the direction of diapedesis (Carman and Springer, 2004).Vimentin is also involved in transcellular machineries (Nieminen et al., 2006).Apparently, transcellular migration does not require chemokine gradients, andarrested lymphocytes may go through endothelial barrier directly, perhapsskipping firm attachment and migration steps. Further studies are necessaryusing recent advances in imaging technology to address the site of lymphocytetransmigration in vivo.
5.3. Interstitial Migration in Lymphoid Tissues
In situ imaging techniques using multiphoton microscopy have revealedrobust interstitial migration of naive lymphocytes into peripheral lymphnodes (Miller et al., 2002), probably under the control of chemokines and

200 TATSUO KINASHI
integrins. Lymphocyte migration behavior appears to be a random walk, butwith spatial restrictions; the movement of T cells is confined to subcorticalT cell areas and they are excluded from B cell follicles. The high motilityof naive T cells enables them to encounter the rare population of antigen‐presenting DCs that have migrated into draining lymph nodes from peripheraltissues. Specific targeting of lymphocyte migration to T cell areas dependson chemokines such as CCL21 and CXCL12, and migration to B cell folliclesdepends on CXCL13 (Moser et al., 2004), and lymphocyte homing to splenicwhite pulp has been shown to depend on LFA‐1 and a4 integrins (Lo et al.,2003). Soluble chemokine gradients might direct lymphocyte migrationto specific compartments through chemotaxis in lymphoid tissues. But thereare little convincing data to indicate gradient distributions of chemokinesin vivo. Chemokines are highly charged molecules and readily associ-ated with extracellular matrix (ECM) proteins and cell surface via heparansulfates or glycosaminoglycans, as seen in HEV (Miyasaka and Tanaka, 2004).Chemokines bound to ECM or cell surface of reticular stromal cells couldguide lymphocyte interstitial migration in chemokinetic (migration dependenton nongradient chemoattaractants) and haptokinetic (migration dependent onsubstrates) fashions, independently of chemokine gradients.
5.4. Interactions with APC
The transient activation of integrins by chemokines enables lymphocytes toscan for cognate antigen during brief contacts with APCs (Fig. 3B). LFA‐1activation by inside‐out signaling is demonstrated for binding of T cells toICAM‐1 on stimulation with TCR ligation (Dustin and Springer, 1989). OnceT cells recognize cognate antigen through peptide–MHC ligation of the TCR,a dynamic redistribution of TCR and LFA‐1 to the contact site occurs withinminutes (Grakoui et al., 1999); LFA‐1 then translocates from the center of thecontact site to the periphery, accompanied by the reciprocal movement of theTCR complex. These events lead to the formation of a stable adhesion termedimmunological synapses (IS), or supramolecular activating clusters (SMACs), acharacteristic structure in which an external LFA‐1 ring surrounds centralTCR clusters (cSMAC; Monks et al., 1998). In the peripheral SMAC(pSMAC), ICAM‐1‐engaged LFA‐1 is colocalized with talin, and likely takesextended conformations. Thus, segregation of LFA‐1 to the peripheral isstructurally relevant distribution so that large extracellular domains of LFA‐1and ICAM‐1 do not sterically hinder relatively small TCR and antigen–peptide–MHC complex, allowing stable antigen recognition (Sims and Dustin,2002). However, the mechanisms underlying the SMAC, especially how LFA‐1molecules are redistributed as a ring remains elusive.

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 201
It is becoming clear that T cells, particularly naive cells are activated duringshort contacts with antigen peptide‐MHC bearing APC. For example, T cellsin a collagen matrix stop very infrequently but still get activated and proliferate(Gunzer et al., 2000). Multiphoton scanning laser microscopy have shown thatafter encountering APC‐presented cognate antigen in vivo, T cells undergodistinct changes in their adhesion patterns (Bousso and Robey, 2003; Mempelet al., 2004; Miller et al., 2004): initial short‐lived contacts with antigen‐presenting DCs followed by the formation of stable T cell–APC conjugates,which eventually lead to autonomous T cells migration and cell division.Integrins and intracellular signaling from receptors for chemokines and anti-gens likely regulate these changes in T cell behavior after activation (Friedmanet al., 2005). It has been recently demonstrated that CCL21 is bound to cellsurface of CD11cþ dendritic APC (Friedman et al., 2006). Interestingly,chemokines bound to APC stimulate the initial short‐lived interactions oflymphocytes via LFA‐1 and ICAMs, and enhances the subsequent formationsof an antigen‐dependent stable adhesion. During initial contacts, a T cellmoves over a chemokine‐bound APC, which often results in tethering aturopod, while the leading edge is active and the cell often appears to crawlaway from the APC. The uropod tethering occurs depending on LFA‐1 andICAMs. When the leading edge subsequently engages with antigen‐bearingcell surface of the same (in cis) or a neighboring cell (in trans), the uropodtether rapidly released, concomitant with initiation of Ca2þ influx and ISformation, indicating antigen recognition and activation (Friedman et al.,2006). The initial transient interaction and tethering may help keep lympho-cytes in proximity to APC until LFA‐1 is sufficiently activated. Antigen engage-ment triggers TCR‐mediated inside‐out signals and further stabilizesattachments and initiates IS formation (Fig. 3B). Thus, this two‐step ‘‘tether‐to‐synapse’’ dynamic is mediated by sequential activation of LFA‐1 by surface‐bound chemokines and cognate antigens and may correspond to a transitionfrom a ‘‘swarming’’ pattern of lymphocyte interactions with antigen‐bearingAPC to stopping and IS formation in vivo (Miller et al., 2004).
6. Talin as Intracellular Regulator for Lymphocyte Adhesion and Migration
Talin is a 250‐kDa cytoskeletal protein that links integrins and the actin cyto-skeleton (Horwitz et al., 1986). It is a component of focal adhesion complexes infibroblasts (Burridge and Connell, 1983). Talin has an additional functionin regulating cadherin gene expression, which is independent of integrins(Becam et al., 2005). Talin is localized in the leading edge of chemokine‐stimulated lymphocytes (Foger et al., 2006; Gomez‐Mouton et al., 2001) andin immunological synapse (Monks et al., 1998). In addition to linking integrins

202 TATSUO KINASHI
with actin cytoskeleton, it has been proposed that talin serves as inside‐outsignals (Calderwood, 2004). Talin has an N‐terminal integrin‐binding FERM(4.1 ezrin, radixin, moesin) domain and a C‐terminal actin‐binding tail domainand serves as a linker between integrins and actin cytoskeleton. The FERMdomain contains a region that directly associates with the NPXY/F motif, whichis conserved in the b chains of most integrins (Calderwood, 2004). Overexpres-sion of this domain activates b,b,and b3 integrins (Calderwood et al., 1999; Kimet al., 2003). Knockdown of talin by small interference RNA inhibits b3 integrinactivation (Tadokoro et al., 2003). Proteolytic cleavage of talin by calpainproduces talin head fragments (Yan et al., 2001). Separation of the cytoplasmicdomains of aLb2 is detected by FRET by overexpression of the talin head(Kim et al., 2003). Thus, generation of this talin fragment potentially serves asan inside‐out signal to modulate LFA‐1 affinity by separation of LFA‐1 cyto-plasmic domains. However, it was reported that calpain inhibitors reduce TCR‐stimulated LFA‐1‐mediated adhesion of lymphocytes to ICAM‐1 (Stewartet al., 1998). But this study did not focus on either talin cleavage or affinityregulation of LFA‐1. Rather it was proposed that calpain induced release ofLFA‐1 from a cytoskeletal restraint that prevents lateral diffusion and cluster-ing. Thus, the result was interpreted in terms of valency regulation. In supportof a negative function for talin, LFA‐1 is constitutively associated with talinin resting neutrophils, but after activation LFA‐1 dissociates from talin andassociates with a‐actinin (Sampath et al., 1998). Furthermore, treatment with alow dose of cytochalasin D, which inhibits actin polymerization, upregulatesintegrin surface diffusion and adhesion (Kucik et al., 1996). Latrunclin A,which also prevents actin polymerization by binding to actin monomers, in-creases rolling and firm adhesion by LFA‐1 (Salas et al., 2002). Although talinand actin cytoskeletons are important in postadhesion events by strengtheningof adhesion complex or cell migration (Smith et al., 2005), cleavage of talin isnot involved in transition from rolling adhesion to firm adhesion (Constantinet al., 2000; Shamri et al., 2005). Knockdown of talin lowers chemokine‐triggered lymphocyte interactions to low‐density, but not high‐density ICAM‐1in shear flow, indicating an important role of talin in adhesion strengthening(Shamri et al., 2005).Interestingly, in migrating lymphocytes ICAM‐1‐engaged, high‐affinity con-
formations of LFA‐1 recognized by mAb24 is localized in the midbody areatermed fo cal zone and coloc alized with talin (S mith et al. , 2005). Inter nalreflection microscopy shows that a cell attaches strongly in the focal zoneand to a lesser extent at the leading edge, but not in the uropod. Thus, thissuggests that LFA‐1 affinity/conformation changes are spatially regulated: highin the focal zone, low in the uropod, perhaps intermediate in the leading edge.ICAM‐1‐engaged, high‐affinity LFA‐1 is low mobility on the cell surface,

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 203
suggestin g a linkage of this subp opulation of LFA ‐ 1 to cytoske leton via talin.This study shows that the LFA‐ 1–tali n comple x formati on is required forefficie nt migratio n on ICAM ‐ 1 (Smith i, 2005).
7. Intr acellular Signals in Chemo kine ‐ Induced A dhesion and Migrat ion
Chemokines activate multiple signaling pathways, including the phosphatidyli-nositol 3‐kinase (PI3K), phospholipase C (PLC), Ras/Rho family of smallGTPases, and mitogen activated protein (MAP) kinase cascades, each of whichhas been implicated in the inside‐out signaling cascades that control integrinaffinity and valency regulation and the associated changes in cytoskeleton, cellpolarity, and morphology, which regulate lymphocyte migration (Fig. 4).
7.1. PI3K Pathways
PI3K plays a cru cial role in chem otaxis or di rected migratio n along a chem okinegradie nt (Ward, 2004 ). Experi ments using PI3 K inhibi tors have shown thatinhibi tion of PI3K activ ity bl ocks chemok ine‐ trig gered LFA ‐ 1 clusterin g andadhesi on to low density ICAM ‐ 1. Howe ver, treatmen t with PI3 K inhibitors didnot block ad hesion to high density ICA M‐ 1 or in viv o lymph ocyte hom ing
GPCR
RhoA
ROCK
PI3Kg
Rap1
RAPLMst1
DOCK2
Rac
Leading edge Uropod
Rap1-GEF(CalDAG-GEF, C3G)
Integrin activation
PKCz
Gi G12/13
PIP3
Cytohesin-1RhoH
(B cell)
PKCz
Rho-GEF(Lsc)
Vav
Rac-GEF
SWAP-70
Figure 4 GPCR‐triggered signals lead to integrin activation and development of the leading edgeand uropod structures.

204 TATSUO KINASHI
(Constantin et al., 2000). Therefore, whereas it is clear that activation of PI3Kcan function in inside‐out signaling following ligation of costimulatory mole-cules (Shimizu et al., 1995; Zell et al., 1996) or activation of c‐kit (Kinashi et al.,1995), and that a constitutively active form of the PI3Ka catalytic subunitincreases ligand‐binding affinity and the observed conformational changes inLFA‐1 (Katagiri et al., 2000), the contribution of PI3K signaling pathways tointegrin activation by chemokines remain ill‐defined. Indeed, although PI3K isrequired for efficient chemotaxis of myeloid cells (Hirsch et al., 2000; Li et al.,2000) and lymphocytes (Reif et al., 2004), studies using gene targeting ofthe catalytic subunits of PI3Kg, PI3Kd, or other isoforms did not show anydefects indicative of reduced integrin function (Nombela‐Arrieta et al., 2004;Okkenhaug and Vanhaesebroeck, 2003). These results suggest that PI3K iscritical in directional sensing but does not play a major role in integrinactivation in lymphocytes.Cytohesin‐1, isolated in a yeast two‐hybrid screen using the b2 integrin
cytoplasmic region as bait (Kolanus et al., 1996), has a plekstrin homology(PH) domain that binds to phosphatidylinositol (3,4,5) triphosphate (PIP3).This protein also functions as a guanine exchange factor (GEF) for members ofthe ADP ribosylation factor (ARF) family of GTPases (Meacci et al., 1997).Overexpression and mutational analyses showed that cytohesin‐1 upregulatesLFA‐1 adhesion through valency modulation in a manner dependent on thePH domain and its association with LFA‐1. These experiments indicate a rolefor cytohesin‐1 in leukocyte arrest and transmigration, which also requires theactin regulator ARF6 (Weber et al., 2001). Cytohesin‐1 is reportedly involvedin outside‐in signaling pathways leading to MAP kinase activation (Perez et al.,2003); however, its physiological function as an inside‐out signaling moleculehas yet to be demonstrated in vivo.
7.2. Rho Pathways
RhoA is involved in lymphoid polarization and chemotaxis (Sanchez‐Madridand del Pozo, 1999). Rho signaling is thought to be critical in both chemokine‐triggered LFA‐1 activation and LFA‐1 mediated lymphocyte homing in vivo(Constantin et al., 2000; Laudanna et al., 1996). In a transgenic mouse model, aconstitutively active mutant of RhoA increase basal adhesion of thymocytesand peripheral lymphocytes to VCAM‐1, ICAM‐1, and fibronectin (Vielkindet al., 2005). The distinct RhoA effector regions differentially modulate LFA‐1affinity and valency regulation by chemokines (Giagulli et al., 2004). Forexample, the high‐affinity form of LFA‐1 induced by chemokines is inhibitedby the peptide containing RhoA amino acids 24–40, which also impairs lym-phocyte adhesion to high‐density ICAM‐1 and in vivo homing to Peyer’s

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 205
patches HEVs. Chemokine‐stimulated PKC‐z kinase activity and its transloca-tion to the plasma membrane depends on PI3K partially and the RhoA 24–40effector region, respectively. ROCK, a serine/threonine kinase effector mole-cule of RhoA, is ruled out in these processes using specific inhibitors. Theeffector molecule interacting with the RhoA 24–40 region responsible for thegeneration of high affinity LFA‐1 or PKC‐z translocation has not yet beenidentified.
Rho signaling also has a negative role in integrin‐mediated adhesion. RhoA isrequired to retract the tail of themigrating lymphocytes andmonocytes throughROCK (Smith et al., 2003; Worthylake et al., 2001). Inhibition of RhoA orROCK increases LFA‐1‐mediated adhesion through valency modulation inhuman T cells (Rodriguez‐Fernandez et al., 2001). Lsc, a murine homologueof human p115 Rho GEF, is specifically expressed in hematopoietic‐lineagecells and is shown to be critical in lymphocyte motility and antigen responses(Girkontaite et al., 2001; Rubtsov et al., 2005). Lsc‐deficient B cells, especially amarginal zone B (MZB) cells display enhanced chemotactic responses to serumand sphingosine 1‐phosphate (S1P), but not chemokines. Furthermore, S1P‐induced attachment to ICAM‐1 and VCAM‐1 is increased in Lsc‐deficientMZBcells, which display defective detachment at the trailing edge (Rubtsov et al.,2005). The chemotactic response of MZB cells to S1P is largely mediated byS1P3 (Cinamon et al., 2004), a seven transmembrane receptor coupled with Gias well as G13 and Gq (Windh et al., 1999); the latter two activate RhoA (Sahet al., 2000). Lsc contains a regulator of G‐protein signaling (RGS) domain thatdownmodulates heterotrimeric G‐proteins, especially Ga13 (Hart et al., 1998;Kozasa et al., 1998). Lsc deficiency may result in sustained activation of Ga13and impaired Rho activation at the trailing edge. Thus, ‘‘wiring’’ of Rho signalingto upstream and downstream elements may vary in distinct subcellular regions,generating positive and negative influences on integrin‐mediated adhesion andmigration.
7.3. Rap1 Pathways
The small GTPase Rap proteins have emerged as an important regulator ofintegrin adhesiveness (Bos et al., 2001). The Rap1 family consists of two highlyhomologous rap1a and rap1b, and two related rap2 genes. Constitutively activemutants of Rap1A and Rap1B potently increase b1, b2, and b3 integrin (Bertoniet al., 2002; Caron et al., 2000; Katagiri et al., 2000; Reedquist et al., 2000;Sebzda et al., 2002). Rap2 is also stimulatory in B cell migration (McLeodet al., 2002). Rap1 is activated by the chemokines CCL21, CXCL12, CXCL13(Durand et al., 2006; McLeod et al., 2002; Shimonaka et al., 2003). Inhibition ofRap1 abrogates chemokine‐stimulated adhesionmediated by LFA‐1 and VLA‐4

206 TATSUO KINASHI
(Shimonaka et al., 2003), indicating an important role for Rap1 in inside‐outsignaling triggered by chemokines. Rap1 activation is also required for achemoattractant S1P (Rosen and Goetzl, 2005) to induce B cell migration andadhesion to ICAM‐1 and VCAM‐1 (Durand et al., 2006).Rap1 upregulates ligand‐binding affinity to soluble dimeric ICAM‐1‐Fc and
induces extended conformations of LFA‐1 detected with NKI L‐16 mAb(Katagiri et al., 2000; Reedquist et al., 2000; Tohyama et al., 2003) and alsostimulates LFA‐1 clustering (Katagiri et al., 2003; Sebzda et al., 2002). Acti-vated Rap1 also robustly stimulates lymphocyte migration on ICAM‐1 andtransendothelial migration under shear flow (Shimonaka et al., 2003). Theadhesion‐stimulatory effect of Rap1 requires the aL cytoplasmic domain,especially, the lysine residues at positions 1097 and 1099 after the GFFKRmotif; replacement of these lysines with alanines suppressed the increases inLFA‐1 affinity and the accompanying conformational changes impairs LFA‐1activation on stimulation with chemokines or TCR cross‐linking (Tohyamaet al., 2003), emphasizing the physiological importance of this region intransmitting inside‐out signals to the extracellular region.In agreement with the proposed importance of Rap1 in chemokine‐mediated
integrin activation, defective regulation of Rap1 occurs in Epstein‐Barr virus(EBV)‐transformed lymphocytes derived from some patients with LAD (Kinashiet al., 2004). Although Rap1 activation by chemokines is a pertussis toxin‐sensitive Gi/o‐dependent process (Shimonaka et al., 2003), regulatory processesto trigger Rap1 activation including GDP/GTP exchange factors are not clear.A Rap1 exchange factor CalDAG‐GEFI and Rap1b are critically importantfor platelet aggregation and thrombus formation via aIIbb3 (Chrzanowska‐Wodnicka et al., 2005; Crittenden et al., 2004), but their importance on leukocytetrafficking are not reported.Chat‐H, a hematopoietic‐specific isoform of a Cas family protein (Sakakibara
et al., 2003), is shown to be involved in Rap1 activation by chemokines inlymphocytes (Regelmann et al., 2006). Knockdown of Chat‐H by lentivirus‐mediated RNA interference impairs chemokine‐stimulated Rap1 activation andadhesion mediated by LFA‐1. Chat‐H deficient lymphocytes are also defectivein lymphocyte trafficking to peripheral lymph nodes. Chat‐H localization withthe plasmamembrane and association with an adaptor protein CasL are requiredfor Tcell migration. Chat‐H knockdown neither affects Rac activation by chemo-kines nor impairs Rap1 activation by TCR ligation (Regelmann et al., 2006).Chat‐H may act as a critical signaling molecule upstream of Rap1 to regulatechemokine‐induced adhesion and migration.A Rap1‐binding protein, RAPL (regulator of adhesion and cell polarization
enriched in lymphoid tissues) is isolated in a yeast two‐hybrid screen usingRap1V12 as bait (Katagiri et al., 2003). RAPL possesses a central Ras/Rap

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 207
associ ation (RA) domain, which has a pro tein ‐ in teracting coiled ‐ coil region atthe C ‐ terminu s. RAPL is an alternativ ely splice d form of Rassf5 (also known asNore 1), which belongs to the Rassf tumor suppressor family ( Tomm asi et al .,2002 ). RAPL, whi ch is highly expresse d in lymphocy tes, bi nds to active R ap1 ‐GTP, but not inactive R ap1 ‐ GDP, and associ ates with Rap1 on lymphocy testimulat ion with CXCL12 or follow ing TCR ligatio n, and ove rexpression ofRAPL was sho wn to increase LFA‐ 1 avidity by bot h affinity and valen cymodul ation (Kata giri et al., 2003 ). Activated Rap 1 or R APL ove rexpressionalso induces cell polariza tion simil ar to that seen in chem okine ‐ stimulat edlympho cytes, showing membran e ruffli ng at one end (the leadin g edge) andformati on of a uro pod at the rear. Furthe rmore, RAPL forms a comp lex withLFA ‐ 1, the formati on of which is depen dent on Rap 1 activ ation as well as thepresence of lysines 1097 an d 1099 in the a L ch ain. The association o f RAPLand LFA ‐ 1 is spatial ly regu lated; on stimulat ion with chemok ines or theintrod uction of activated Rap1, RAPL associ ates with LFA ‐ 1 and relocates tothe le ading edge, forming large pa tch ‐ like clusters ( Katagiri et al., 2003 ). Thus,affinity and valency modulation s by Rap1 and R APL are concurren t andcoordin ated with cell polariza tion. This result is furt her supporte d by studiesof lymphocy tes derive d fr om RAPL ‐ deficien t mice (Katagiri, 2 004a); RAPL ‐deficien t T and B ce lls wer e defecti ve in chem okine ‐stimu lated ad hesion, aproce ss dep endent on LFA‐ 1 and VLA ‐ 4. T hese ce lls exhib ited poorly polar-ized mo rpholog y and min imal LFA ‐ 1 clustering . Studie s of RAPL ‐ deficien tmice have also shown a crucia l role for RAP L in other integrin ‐ dependen tproce sses controll ed by ch emokine stimulat ion, such as the t rafficking oflympho cytes and DCs to periphe ral lymph nodes an d the spleen (Ka tagiri,2004). Rec ently, mamma lian Ste20 ‐ like kinase MST1 /STK4 is identifie d as acritical effec tor of RAPL. RAPL regula tes the local ization and kinase activity ofMst1 (Kata giri et al., 20 06 ). Knockdown of Mst1 demons trates its requ irementfor the in duction of both a polarize d morph ology and integri n LFA ‐ 1 cluster-ing and ad hesion triggered by chem okines and TCR ligatio n. RAPL and M st1localize to vesicula r comp artments and dynamica lly transloc ate with LFA‐ 1 tothe leading edge on Rap1 activ ation, sugge sting the regula tory role of RAPL–Mst1 comple x in intracell ular transport of LFA ‐ 1 (Kata giri et al ., 2006 ).
Rap1 ‐ interacting ad aptor molecule (RIAM also known as RARP1; Inag akiet al., 2003) is isolated by yeast two‐hybrid screening with an active Rap1mutantas bait (Lafuente et al., 2004). RIAM is a proline‐rich 100‐kDa protein bearingRA and PH domains. RIAM interacts with the active Rap1 mutant but notH‐Ras mutant in two‐hybrid assays. RIAM interacts with actin‐regulating en-abled (Ena)/vasodilator‐stimulated phosphoprotein (VASP) proteins and profi-lin, and belongs to the MRL (Mig10/RIAM/Lamillipodin) family of proteins(Legg and Machesky, 2004). Overexpression of RIAM‐induced conformational

208 TATSUO KINASHI
changes of b1 and b2 integrins and augmented cell spreading and adhesion ofJurkat T cells to fibronectin and ICAM‐1. Knockdown of RIAM expression byRNA interference reduces levels of polymerized actin and impairs Rap1‐in-duced adhesion. Interestingly, RIAM knockdown displaces active Rap1 fromthe plasma membrane. Actin polymerization by RIAM required Ena–VASPinteractions, but the pro‐adhesive effect does not require these interactions,suggesting that actin polymerization is not involved in integrin activation. RIAMis also shown to enhance talin‐dependent aIIbb3 activation (Han et al., 2006). Itis not known whether RIAM regulates integrin‐mediated lymphocyte adhesionby chemokines or TCR.A murine orthologue of human RIAM is independently identified by cross‐
reactivity of an antibody that binds a proline‐rich sequence of zyxin, andtermed proline‐rich EVH1 ligand 1 (PREL1; Jenzora et al., 2005). PREL1modestly associates with an active H‐Ras mutant, but not other Ras/Rho familymembers, including Rap1 by pulldown assays using the RA domain of PREL1,or coimmunoprecipitation with the full‐length PREL1. PREL1 is shown torelocate to the tips of circular lamellipodia and focal adhesion, and colocalizedwith VASP transiently in a time course similar to that of H‐Ras activation(Jenzora et al., 2005). Although the discrepancy regarding the specificities ofsmall GTPases to which RIAM and PREL1 interact is not clear, the conservedfunction of RIAM and PREIL appears to translocate and activate actin‐remodeling machineries. Further studies are required to clarify the relationshipof RIAM/PREL1 and Rap1‐regulated adhesion.
7.4. Rac Pathways
The deletion of both Rac1 and Rac2 genes leads to a massive egress of hemato-poietic stem/progenitor cells (HSC/Ps) into the blood. HSC/Ps deficient for Rac1and Rac2 displays decreased adhesion to fibronectin, defective chemotaxis toCXCL12, and a failure of bonemarrowengraftment (Gu et al., 2003), suggesting acritical role of Rac1 and Rac2 in integrin‐mediated stem cell adhesion. Rac2‐deficient leukocytes are defective in shear‐dependent L‐selectin‐mediated cap-ture on Glycam‐1 as well as F‐actin generation, and chemoattractant‐stimulatedMAP kinase activation (Roberts et al., 1999). Neutrophils deficient for both Rac1and Rac2 show normal integrin‐mediated adhesion but markedly reduced migra-tion and defective cell spreading (Gu et al., 2003). The effects ofRac deficiency onlymphocytes trafficking has not been described yet.DOCK2, a hematopoietic‐specific member of the CDM (Ced‐5, DOCK180,
Myoblast city) family of proteins, regulates Rac activation in lymphocytes (Fukuiet al., 2001).Deficiency inDOCK2 severely impairsRac1 andRac2 activation and

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 209
defective development of the actin cytoskeleton in lymphocytes and neutrophilsstimulated by chemokines (Fukui et al., 2001; Kunisaki et al., 2006; Sanui et al.,2003). As a consequence, both in vitro lymphocyte chemotactic responses andin vivo trafficking to peripheral lymphoid organs are severely diminished in thesemice (Nombela‐Arrieta et al., 2004). DOCK2 deficiency differentially affectsintegrin activity in T and B cells; adhesive responses to chemokines and phorbolesters through LFA‐1 and a4 integrins are diminished in B cells, but not in Tcells(Nombela‐Arrieta et al., 2004). This effect was also observed in vivo usingintravital microcopy to show that firm attachment to peripheral lymph nodevenules was only impaired for B cells. Changes in affinity for ligand, however, isnot observed in DOCK2‐deficient B cells. Furthermore, as LFA‐1 clusteringoccurs normally in CXCL13‐stimulated B cells, it remains unclear if DOCK2deficiency affects integrin function directly. In in vitro experiments, DOCK2‐deficient T cells are normal in arrest and firm attachment but defective in laterallymphocyte motility before and after transendothelial migration under flow(Shulman et al., 2006). Since actin polymerization triggered by chemokines isdefective in both T and B cells from DOCK2‐deficient mice (Fukui et al., 2001;Shulman et al., 2006), but the integrin defect was seen only in DOCK2‐deficientB, it is possible that actin polymerization is not an effector executing lymphocyteintegrin activation. Instead, DOCK2 may serve a regulatory role controlling Racactivation or a cytoskeleton‐independent function, which could affect otherinside‐out signaling molecules in a B cell‐specific manner. Further investigationto clarify the defect in B cell integrins activation will provide insight into lineage‐specific integrin modulation.
Vav1, a hematopoietic exchange factor for Rac is activated by CXCL12(Vicente‐Manzanares et al., 2005) in human peripheral blood lymphocytes,and overexpression of a dominant‐negative form of Vav1 abolish lymphocytepolarization, actin polymerization, and migration. In one study (Garcia‐Bernalet al., 2005), Vav1 and Rac are shown to be a critical role in CXCL12‐triggereda4b1 integrin activation to mediate VCAM‐1‐dependent attachment of Molt4and primary T cells under static and shear flow conditions. Vav1 is also shownto be involved in LFA‐1 activation by TCR (Krawczyk et al., 2002) (see below).In contrast, neutrophils deficient for both Vav1 and Vav3, major isoformsexpressed in neutrophils, are shown to be normal in chemotaxis and attach-ment on ICAM‐1 under shear flow or on inflamed venule in vivo but aredefective in stable attachment and spreading (Gakidis et al., 2004). Activationof Rac1 and Rac2 by a chemoattractant f‐MLP are normal in Vav1/Vav3‐deficient neutrophils, but signaling through aMb2 to activate protein kinasesincluding PAK are severely diminished, indicating a prominent role of Vavproteins in outside‐in signaling (Gakidis et al., 2004).

210 TATSUO KINASHI
SWAP‐70 is a B‐cell specific Rac exchange factor that binds to PIP3 andF‐actin through a PH domain and C‐terminal region, respectively (Ihara et al.,2006; Shinohara et al., 2002). SWAP‐70‐deficient mice does not show anyabnormalities in homeostatic lymphocyte trafficking. However, lymphocytemigration to inflamed lymph nodes is reduced (Pearce et al., 2006). SWAP‐70‐deficient B blasts show impairment in cell polarization displaying defectiveuropod formation and enhanced cell spreading on anti‐CD44 antibody cross‐linking. Although chemokine‐stimulated adhesive responses to ICAM‐1 andMAdCAM‐1 are normal, lymphocyte polarization of SWAP‐70‐deficientB cells by chemokines is also impaired with enhanced cell spreading. SWAP‐70‐deficient B cells normally adhere to HEV but do not enter into lymph nodetissues efficiently (Pearce et al., 2006). Since SWAP‐70 is shown to modulate atransitional subset of actin filaments in fibroblastic motile cells (Hilpela et al.,2003), SWAP‐70 may have a similar role in B cells and control lymphocytepolarization and migration that is necessary during diapedesis.Coronin1, an inhibitory protein opposing actin‐polymerizing Arp2/3, has
important roles in T cell morphology and migration (Foger et al., 2006).Coronin1‐deficient mice display reduced T cell numbers in peripheral bloodand lymph nodes and spleens. Coronin1‐deficient T cells do not developchemokine‐stimulated polarized cell shapes with talin segregation to the lead-ing edge. Moreover, steady‐state F‐actin is reduced and Rac1 activation bychemokine stimulation is diminished. Consequently, coronin1‐deficient T cellsare defective in chemotaxis and reduced trafficking to peripheral lymph nodes(Foger et al., 2006), but it is not reported whether coronin1 modulate integrinfunctions.
7.5. RhoH
The small GTPase RhoH is identified as a negative regulator of LFA‐1 avidity(Cherry et al., 2004). Gene inactivation of RHOH by insertional mutagenesisor knockdown of mRNA expression with RHOH‐specific siRNA induces con-stitutive activation of LFA‐1 and the structural changes associated with high‐affinity or extended LFA‐1 conformations (Cherry et al., 2004). Inactivation ofRHOH also activates a4b1. These findings strongly suggest that the lowadhesive state of lymphocyte integrins is actively controlled and maintainedby RhoH. RhoH is a leukocyte‐specific inhibitory Rho family member knownto suppress the effects of Rac, Cdc42, and RhoA on nuclear factor‐kB, or p38MAP kinase activation. This protein does not appear to have an effect onassembly of the actin cytoskeleton induced by activated RhoA or platelet‐derived growth factor (PDGF; Li et al., 2002), but reduced CXCL12‐stimu-lated F‐actin and chemotaxis, and also impairs proliferation of HSC and Rac
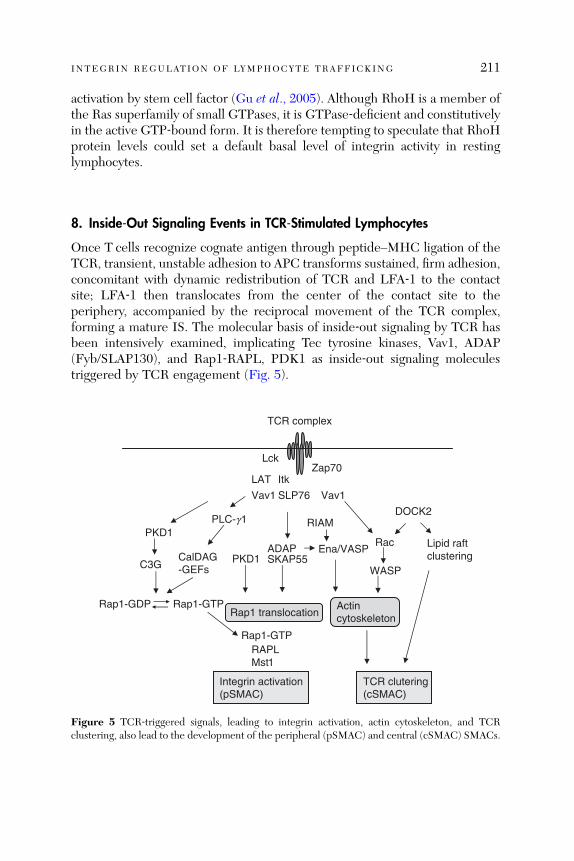
INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 211
activation by stem cell factor (Gu et al., 2005). Although RhoH is a member ofthe Ras superfamily of small GTPases, it is GTPase‐deficient and constitutivelyin the active GTP‐bound form. It is therefore tempting to speculate that RhoHprotein levels could set a default basal level of integrin activity in restinglymphocytes.
8. Inside‐Out Signaling Events in TCR‐Stimulated Lymphocytes
Once T cells recognize cognate antigen through peptide–MHC ligation of theTCR, transient, unstable adhesion to APC transforms sustained, firm adhesion,concomitant with dynamic redistribution of TCR and LFA‐1 to the contactsite; LFA‐1 then translocates from the center of the contact site to theperiphery, accompanied by the reciprocal movement of the TCR complex,forming a mature IS. The molecular basis of inside‐out signaling by TCR hasbeen intensively examined, implicating Tec tyrosine kinases, Vav1, ADAP(Fyb/SLAP130), and Rap1‐RAPL, PDK1 as inside‐out signaling moleculestriggered by TCR engagement (Fig. 5).
TCR complex
PLC-g1
CalDAG-GEFsC3G
DOCK2
Rac
SLP76
ADAPSKAP55
Rap1-GTP
RAPLMst1
Vav1
Itk
WASP
TCR clutering(cSMAC)
Integrin activation(pSMAC)
Vav1
LckZap70
Ena/VASP
LAT
Lipid raftclustering
Actincytoskeleton
Rap1-GDP
RIAM
Rap1-GTP
Rap1 translocation
PKD1
PKD1
Figure 5 TCR‐triggered signals, leading to integrin activation, actin cytoskeleton, and TCRclustering, also lead to the development of the peripheral (pSMAC) and central (cSMAC) SMACs.

212 TATSUO KINASHI
8.1. Tec Family Kinases
The Tec family kinases Itk, Rlk, and Tec are important mediators of TCRsignaling pathways that regulate T cell activation and differentiation (Lucaset al., 2003). For example, TCR‐triggered Itk activation stimulates b1 integrin‐dependent T cell adhesion, which requires PI3K‐dependent Itk membranelocalization and kinase activity (Woods et al., 2001). Itk activation also inducesactin polymerization triggered by TCR ligation (Woods et al., 2001) or chemo-kine stimulation (Takesono et al., 2004). Consistent with this, T cells from Itk�/�
mice are defective in IS formation, adhesion through b1 and b2 integrins,calcium flux, and actin polymerization (Labno et al., 2003). Itk has also beenshown to be required for chemokine‐stimulated lymphocyte migration(Fischer et al., 2004), and adhesion of chemokine‐stimulated Itk�/� thymo-cytes to fibronectin was impaired, indicating that Itk is also involved in integrinregulation by chemokines (Fischer et al., 2004). Mechanistically, Itk is shownto be required for activation of WASP and Cdc42 at the IS, likely explainingthe defect in actin polymerization and IS formation in Itk‐deficient T cells(Labno et al., 2003). However, since WASP deficiency does not affect integrinactivation (Krawczyk et al., 2002), defective actin polymerization is not likely acause of impaired integrin activity in Itk�/� Tcells. In contrast, Itk is importantin formation of the LAT, Vav1, and SLP‐76 signaling complex and activation ofPLCg‐1 (Lucas et al., 2003), indicating that inside‐out signaling triggered byItk is mediated by these downstream elements.
8.2. Rac Signaling Pathways
The importance of Vav1 in LFA‐1 activation is demonstrated by gene targeting(Krawczyk et al., 2002). Vav1‐deficient thymocytes and peripheral T cells showimpaired TCR‐dependent LFA‐1 activation and IS formation, concurrent withboth defective actin cytoskeleton assembly and TCR clustering. In addition,Vav1‐deficient thymocytes and T cells exhibit deficiencies in TCR‐triggeredcalcium flux and PLCg‐1 and PI3K activation, leading to inhibition of T celland thymocyte growth and differentiation (Tybulewicz et al., 2003). Thesepleiotropic defects in Vav1‐deficient mice make it difficult to identify thesignaling molecule downstream of Vav1 crucial for LFA‐1 activation. As Vav1is a GEF for Rho family GTPases, especially Rac, it was thought that defects inthe actin cytoskeleton in Vav1‐deficient T cells impaired LFA‐1 function.However, deficiency in the Rac effector WASP affects only TCR clustering,and not LFA‐1 activation, indicating distinct pathways control LFA‐1 and TCRsurface distribution (Krawczyk et al., 2002). This conclusion is further sup-ported by studies with DOCK2‐deficient lymphocytes, which demonstrate

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 213
impaired cluste ring of TCR molecu les. In con trast, DOC K2 deficien cy hadlittle effect on the formati on of the LFA ‐ 1 rin g, suggesting tha t the actincytoske leton is di spensable for LFA ‐ 1 rin g formati on (Sa nui et al., 2003 ).
8.3. Rap1 Signaling Pathways
Althoug h previ ous s tudies have indicated that enhan ced R ap1 activ ity isassoci ated with T ce ll anergy ( Boussiotis et al., 1997 ), Rap1 ha s bee n shownto positively regula te LFA ‐ 1 avidity and T cell–A PC con jugate formati on( Katagiri et al., 2002 ). Con sistently, di sruption of the rap1a gene reducedRap1 activ ation by TCR ligation concomi tant with modes tly impaired LFA ‐ 1clusterin g an d ad hesion ( Duchniew icz et al., 2006 ). Con siderable Rap1 activa-tion rem ained in Rap1A ‐ deficien t lymphocy tes is lik ely contribu ted by Rap1B .On TCR engag ement, Rap1 is activ ated, alte ring its local ization at the T ‐ cell–APC in terface (Kata giri et al., 2002 ) and plasma me mbrane ( Bivona et al .,2004 ). R ap1 associ ates with RAPL and quic kly initiates transloc ation from theperinucl ear region to the peripheral boun daries of the immun ologica l synapse( Katagiri et al., 2003 ). Dominan t ‐negati ve RAPL inhi bits TCR ‐ induced upre-gulation of LFA ‐ 1 avidity and T cell–A PC conjugate formati on. MST 1/STK 4isolat ed as a RAP L‐ binding partner is also coloc alized at the T ce ll–APCinter face and is require d by TCR ‐ stimulat ed adhesion to ICAM ‐ 1 (Katagi riet al., 2006 ).
In Jur kat T cells, PL Cg‐ 1 is required for Rap1 activation, suggesting that thecalciu m an d diacylgl ycerol (DA G)‐ respons ive Cal DAG ‐ GEF famil y of pro -teins, which include the Rap1 GEFs, are perhaps involved in signaling down-stream of PLC g‐ 1 (Katagiri et al., 2004b). The requireme nt for PLC g‐ 1 forRap1 activation in T cells is in line with studies demonstrating a requirementfor PLCg‐2 in Rap1 activation following B cell receptor ligation (McLeod andGold, 2001). Another Rap1 GEF, C3G, may also contribute to TCR‐stimulatedRap1 activation, particularly in thymocytes (Amsen et al., 2000), anergic T cells(Boussiotis et al., 1997), and Cbl‐b‐deficient T cells (Zhang et al., 2003).PLCg‐1 activation is regulated by a complex containing linker for activatedT cells (LAT), the adaptor proteins Gads and SLP‐76, and Itk (Samelson,2002). Consistent with this, signaling molecules required for TCR‐stimulatedPLC‐g1 activation, such as ZAP‐70, SLP‐76, and Itk, are critical for theactivation of b1 integrins (Kellermann et al., 2002). Vav1 is also involvedin PLC‐g1 activation following TCR stimulation through its association withSLP‐76 and also activation by Itk (Reynolds et al., 2002), placing Rap1 andRAPL downstream of Vav1, SLP‐76, and Itk activation. This organizationraises the possibility that defective LFA‐1 activation in Vav1‐deficient cellsmay result from insufficient Rap1 activation.

214 TATSUO KINASHI
8.4. PKD1
A serine/threonine kinase PKD1 (PKCm) is shown to form a complex withRap1 and plays an important role in b1 integrin activation (Medeiros et al.,2005). PDK1 is a DAG‐responsive PKC, which is activated by PKC� and avariety of external stimuli, including TCR (Rozengurt et al., 2005). PDK1associates with active Rap1, but not inactive Rap1, through a PH domainof PKD1 (Medeiros et al., 2005). PKD1 further forms a complex with theb1 integrin subunit, depending on the C‐terminal five amino acids of the b1subunit that are required in activation‐dependent adhesion (Romzek et al.,1998). This tertiary complex is formed in Jurkat and primary T cells andtranslocated to the plasma membrane on stimulation with phorbol esterPMA and TCR ligation (Medeiros et al., 2005). Furthermore, PKD1 associateswith C3G, suggesting that PKD1 acts upstream of Rap1. Surprisingly, Rap1activation also depends on the b1 integrin expression, as Rap1 activation isdiminished in b1‐deficient Jurkat cells, which is restored by the b1 expression.Knockdown of PKD1 expression reduces b1 integrin clustering and adhesionto fibronectin. A mutant PKD1 lacking the PH domain (PKD1DPH) alsodecreases adhesion to fibronectin as well as Rap1 activation and membranetranslocation, presumably through an abortive complex formation with theb1 integrin and C3G. The kinase activity of PKD1DPH is not required forthe inhibitory effects. Collectively, these results support the notion that PKD1acts as an adaptor to localize Rap1 activation to b1 integrin (Medeiros et al.,2005). It is also reported that PKD1 associates with avb3 integrin by binding tothe b3 integrin C‐terminus, and thereby promotes recycling of avb3 to newlyforming focal adhesion, suggesting a regulatory role of PDK1 in vesicle trans-port of integrins (Woods et al., 2004). This is in agreement with a proposedfunction of the Rap1/RAPL/Mst1 signaling in polarized LFA‐1 transport to theleading edge (Katagiri et al., 2006) and could be also involved in PDK1regulation of b1 integrins.
8.5. Adhesion and Degranulation Adaptor Protein
Unexpected effects of adhesion and degranulation adaptor protein (ADAP;Fyb/SLAP‐130) gene targeting on LFA‐1 activation indicate that it is animportant inside‐out signaling molecule. ADAP‐deficient lymphocytes exhibitimpaired TCR‐triggered b1 and b2 integrin‐dependent adhesion, defectiveinterleukin‐2 production, and decreased proliferation, despite normal calciumflux and MAP kinase activation (Griffiths et al., 2001; Peterson, 2003). ADAPdoes not appear to be involved in chemokine‐stimulated integrin activation.ADAP is a hematopoietic adaptor protein, which associates with SLP‐76 on

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 215
TCR engagement, although it can also associate with the Fyn, the Ena/VASPfamily of actin regulators and SKAP‐55 (Peterson, 2003). ADAP deficiencyimpairs TCR‐stimulated LFA‐1 clustering, but does not affect TCR clusteringitself or assembly of the actin cytoskeleton (Griffiths et al., 2001; Petersonet al., 2001). ADAP colocalizes with LFA‐1 in IS (Wang et al., 2004). Itsassociation with SLP‐76 appears to be crucial for its function in LFA‐1 activa-tion. SKAP‐55 is involved in LFA‐1 activation downstream of ADAP (Wanget al., 2003). Disruption of ADAP and SKAP‐55 complex results in displace-ment of Rap1 from the plasma membrane without influencing its GTPaseactivity. Thus, ADAP/SKAP‐55 complex may control targeting of activatedRap1 to the membrane (Kliche et al., 2006). This result is consistent withthe study reporting that the localization of active Rap1‐GTP at the plasmamembrane is critical for Rap1‐dependent integrin regulation in T cells (Bivonaet al., 2004).
9. Concluding Remarks
Recent progress prompts us to think dynamic lymphocyte trafficking in termsof a spectrum of conformational states of integrins. Structural studies support aunifying model of global conformational changes from the bent to theextended‐closed to the extended‐open on activation of integrins, and furthersuggest that extended conformations by separation of the leg and cytoplasmicdomains triggered by inside‐out signals transmit allostery to activate theligand‐binding I domain. Lymphocyte integrins are also regulated spatially,which is often coupled with affinity modulation, during the processes of cellpolarization, migration, and IS formations. A wide variety of intracellularmolecules involved in integrin‐mediated adhesion are now identified by ge-netic approaches and characterized at cellular and organismic levels but needsfurther studies to clarify their regulatory mechanisms at molecular levels. Thebetter appreciation of physiological relevance of each state of conformationand valency regulation should be required to dissect integrin regulation thatoccurs in from rolling through firm adhesion to transendothelial and interstitialmigration and interactions with APC. It is important to elucidate what and howintracellular signaling processes coordinately regulate conformation and spa-tial regulation of integrins to translate external stimulation into dynamic adhe-sive responses. The answers to these questions will shed light on crucial rolesof integrin regulation in immunological surveillance and antigen response.
Acknowledgments
I would like to thank Koko Katagiri for helpful comments.

216 TATSUO KINASHI
References
Adair, B. D., Xiong, J. P., Maddock, C., Goodman, S. L., Arnaout, M. A., and Yeager, M. (2005).Three‐dimensional EM structure of the ectodomain of integrin aVb3 in a complex withfibronectin. J. Cell Biol. 168, 1109–1118.
Amsen,D., Kruisbeek, A., Bos, J. L., and Reedquist, K. (2000). Activation of the Ras‐relatedGTPaseRap1 by thymocyte TCR engagement and during selection. Eur. J. Immunol. 30, 2832–2841.
Becam, I. E., Tanentzapf, G., Lepesant, J. A., Brown, N. H., and Huynh, J. R. (2005). Integrin‐independent repression of cadherin transcription by talin during axis formation in Drosophila.Nat. Cell Biol. 7, 510–516.
Bertoni, A., Tadokoro, S., Eto, K., Pampori, N., Parise, L. V., White, G. C., and Shattil, S. J. (2002).Relationships between Rap1b, affinity modulation of integrin alpha IIbbeta 3, and the actincytoskeleton. J. Biol. Chem. 277, 25715–25721.
Bivona, T. G., Wiener, H. H., Ahearn, I. M., Silletti, J., Chiu, V. K., and Philips, M. R. (2004). Rap1up‐regulation and activation on plasma membrane regulates T cell adhesion. J. Cell Biol. 164,461–470.
Bos, J. L., de Rooij, J., and Reedquist, K. A. (2001). Rap1 signaling: Adhering to new models. Nat.Rev. Mol. Cell. Biol. 2, 369–377.
Boussiotis, V. A., Freeman, G. J., Berezovskaya, A., Barber, D. L., and Nadler, L. M. (1997).Maintenance of human T cell anergy: Blocking of IL‐2 gene transcription by activated Rap1.Science 278, 124–128.
Bousso, P., and Robey, E. (2003). Dynamics of CD8þ T cell priming by dendritic cells in intactlymph nodes. Nat. Immunol. 4, 579–585.
Burridge, K., and Connell, L. (1983). Talin: A cytoskeletal component concentrated in adhesionplaques and other sites of actin‐membrane interaction. Cell Motil. 3, 405–417.
Butcher, E. C., and Picker, L. J. (1996). Lymphocyte homing and homeostasis. Science 272, 60–66.Butcher, E. C., Williams, M., Youngman, K., Rott, L., and Briskin, M. (1999). Lymphocytetrafficking and regional immunity. Adv. Immunol. 72, 209–253.
Cairo, C. W., Mirchev, R., and Golan, D. E. (2006). Cytoskeletal regulation couples LFA‐1conformational changes to receptor lateral mobility and clustering. Immunity 25, 297–308.
Calderwood, D. A. (2004). Integrin activation. J. Cell Sci. 117, 657–666.Calderwood, D. A., Zent, R., Grant, R., Rees, D. J., Hynes, R. O., and Ginsberg, M. H. (1999). TheTalin head domain binds to integrin beta subunit cytoplasmic tails and regulates integrinactivation. J. Biol. Chem. 274, 28071–28074.
Carman, C. V., and Springer, T. A. (2003). Integrin avidity regulation: Are changes in affinity andconformation underemphasized? Curr. Opin. Cell Biol. 15, 547–556.
Carman, C. V., and Springer, T. A. (2004). A transmigratory cup in leukocyte diapedesis boththrough individual vascular endothelial cells and between them. J. Cell Biol. 167, 377–388.
Caron, E., Self, A. J., and Hall, A. (2000). The GTPase Rap1 controls functional activation ofmacrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Curr. Biol. 10,974–978.
Chan, J. R., Hyduk, S. J., and Cybulsky, M. I. (2003). Detecting rapid and transient upregulation ofleukocyte integrin affinity induced by chemokines and chemoattractants. J. Immunol. Methods273, 43–52.
Chen, J., Salas, A., and Springer, T. A. (2003). Bistable regulation of integrin adhesiveness by abipolar metal ion cluster. Nat. Struct. Biol. 10, 995–1001.
Cherry, L. K., Li, X., Schwab, P., Lim, B., and Klickstein, L. B. (2004). RhoH is required tomaintain the integrin LFA‐1 in a nonadhesive state on lymphocytes. Nat. Immunol. 5, 961–967.

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 217
Chigaev, A., Buranda, T., Dwyer, D. C., Prossnitz, E. R., and Sklar, L. A. (2003). FRET detection ofcellular alpha4‐integrin conformational activation. Biophys. J. 85, 3951–3962.
Chrzanowska‐Wodnicka, M., Smyth, S. S., Schoenwaelder, S. M., Fischer, T. H., and White, G. C.,2nd (2005). Rap1b is required for normal platelet function and hemostasis in mice. J. Clin.Invest. 115, 680–687.
Cinamon, G., Shinder, V., and Alon, R. (2001). Shear forces promote lymphocyte migration acrossvascular endothelium bearing apical chemokines. Nat. Immunol. 2, 515–522.
Cinamon, G., Matloubian, M., Lesneski, M. J., Xu, Y., Low, C., Lu, T., Proia, R. L., and Cyster, J. G.(2004). Sphingosine 1‐phosphate receptor 1 promotes B cell localization in the splenic marginalzone. Nat. Immunol. 5, 713–720.
Constantin, G., Majeed, M., Giagulli, C., Piccio, L., Kim, J. Y., Butcher, E. C., and Laudanna, C.(2000). Chemokines trigger immediate b2 integrin affinity and mobility changes: Differentialregulation and roles in lymphocyte arrest under flow. Immunity 13, 759–769.
Crittenden, J. R., Bergmeier, W., Zhang, Y., Piffath, C. L., Liang, Y., Wagner, D. D., Housman,D. E., and Graybiel, A. M. (2004). CalDAG‐GEFI integrates signaling for platelet aggregationand thrombus formation. Nat. Med. 10, 982–986.
de Bruyn, K. M., Zwartkruis, F. J., de Rooij, J., Akkerman, J. W., and Bos, J. L. (2003). The smallGTPase Rap1 is activated by turbulence and is involved in integrin aIIbb3‐mediated celladhesion in human megakaryocytes. J. Biol. Chem. 278, 22412–22417.
Dransfield, I., Cabanas, C., Craig, A., and Hogg, N. (1992). Divalent cation regulation of thefunction of the leukocyte integrin LFA‐1. J. Cell Biol. 116, 219–226.
Duchniewicz, M., Zemojtel, T., Kolanczyk, M., Grossmann, S., Scheele, J. S., and Zwartkruis,F. J. (2006). Rap1A‐deficient T and B cells show impaired integrin‐mediated cell adhesion.Mol. Cell. Biol. 26, 643–653.
Durand, C. A., Westendorf, J., Tse, K. W., and Gold, M. R. (2006). The Rap GTPases medi-ate CXCL13‐ and sphingosine1‐phosphate‐induced chemotaxis, adhesion, and Pyk2 tyrosinephosphorylation in B lymphocytes. Eur. J. Immunol. 36, 2235–2249.
Dustin, M. L., and Springer, T. A. (1989). T‐cell receptor cross‐linking transiently stimulatesadhesiveness through LFA‐1. Nature 341, 619–624.
Dustin, M. L., Bivona, T. G., and Philips, M. R. (2004). Membranes as messengers in T celladhesion signaling. Nat. Immunol. 5, 363–372.
Ebisuno, Y., Tanaka, T., Kanemitsu, N., Kanda, H., Yamaguchi, K., Kaisho, T., Akira, S., andMiyasaka, M. (2003). Cutting edge: The B cell chemokine CXC chemokine ligand 13/B lympho-cyte chemoattractant is expressed in the high endothelial venules of lymph nodes and Peyer’spatches and affects B cell trafficking across high endothelial venules. J. Immunol. 171,1642–1646.
Etzioni, A. (1996). Adhesion molecules: Their role in health and disease. Pediatr. Res. 39, 191–198.Fabbri, M., Fumagalli, L., Bossi, G., Bianchi, E., Bender, J. R., and Pardi, R. (1999). A tyrosine‐
based sorting signal in the b2 integrin cytoplasmic domain mediates its recycling to the plasmamembrane and is required for ligand‐supported migration. EMBO J. 18, 4915–4925.
Fabbri, M., Di Meglio, S., Gagliani, M. C., Consonni, E., Molteni, R., Bender, J. R., Tacchetti, C.,and Pardi, R. (2005). Dynamic partitioning into lipid rafts controls the endo‐exocytic cycle of theaL /b2 integrin, LFA‐1, during leukocyte chemotaxis. Mol. Biol. Cell 16, 5793–5803.
Fagerholm, S. C., Hilden, T. J., and Gahmberg, C. G. (2004). P marks the spot: Site‐specificintegrin phosphorylation regulates molecular interactions. Trends Biochem. Sci. 29, 504–512.
Fagerholm, S. C., Hilden, T. J., Nurmi, S. M., and Gahmberg, C. G. (2005). Specific integrinalpha and b chain phosphorylations regulate LFA‐1 activation through affinity‐dependent and‐independent mechanisms. J. Cell Biol. 171, 705–715.

218 TATSUO KINASHI
Feng, D., Nagy, J. A., Pyne, K., Dvorak, H. F., and Dvorak, A. M. (1998). Neutrophils emigratefrom venules by a transendothelial cell pathway in response to FMLP. J. Exp. Med. 187,903–915.
Fischer, A. M., Mercer, J. C., Iyer, A., Ragin, M. J., and August, A. (2004). Regulation of CXCchemokine receptor 4‐mediated migration by the Tec family tyrosine kinase ITK. J. Biol. Chem.279, 29816–29820.
Foger, N., Rangell, L., Danilenko, D. M., and Chan, A. C. (2006). Requirement for coronin 1 inT lymphocyte trafficking and cellular homeostasis. Science 313, 839–842.
Friedman, R. S., Jacobelli, J., and Krummel, M. F. (2005). Mechanisms of T cell motility and arrest:Deciphering the relationship between intra‐ and extracellular determinants. Semin. Immunol.17, 387–399.
Friedman, R. S., Jacobelli, J., and Krummel, M. F. (2006). Surface‐bound chemokines capture andprime T cells for synapse formation. Nat. Immunol. 7, 1101–1108.
Fukui, Y., Hashimoto, O., Sanui, T., Oono, T., Koga, H., Abe, M., Inayoshi, A., Noda, M., Oike,M., Shirai, T., and Sasazuki, T. (2001). Haematopoietic cell‐specific CDM family proteinDOCK2 is essential for lymphocyte migration. Nature 412, 826–831.
Gakidis, M. A., Cullere, X., Olson, T., Wilsbacher, J. L., Zhang, B., Moores, S. L., Ley, K., Swat,W., Mayadas, T., and Brugge, J. S. (2004). Vav GEFs are required for b2 integrin‐dependentfunctions of neutrophils. J. Cell Biol. 166, 273–282.
Garcia‐Bernal, D., Wright, N., Sotillo‐Mallo, E., Nombela‐Arrieta, C., Stein, J. V., Bustelo, X. R.,and Teixido, J. (2005). Vav1 and Rac control chemokine‐promoted T lymphocyte adhesionmediated by the integrin a4b1. Mol. Biol. Cell. 16, 3223–3235.
Giagulli, C., Scarpini, E., Ottoboni, L.,Narumiya, S., Butcher, E. C., Constantin, G., andLaudanna, C.(2004). RhoA and zeta PKC control distinct modalities of LFA‐1 activation by chemokines:Critical role of LFA‐1 affinity triggering in lymphocyte in vivo homing. Immunity 20, 25–35.
Girkontaite, I., Missy, K., Sakk, V., Harenberg, A., Tedford, K., Potzel, T., Pfeffer, K., and Fischer,K. D. (2001). Lsc is required for marginal zone B cells, regulation of lymphocyte motility andimmune responses. Nat. Immunol. 2, 855–862.
Gomez‐Mouton, C., Abad, J. L., Mira, E., Lacalle, R. A., Gallardo, E., Jimenez‐Baranda, S., Illa,I., Bernad, A., Manes, S., and Martinez, A. C. (2001). Segregation of leading‐edge and uropodcomponents into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. USA 98,9642–9647.
Grabovsky, V., Feigelson, S., Chen, C., Bleijs, D. A., Peled, A., Cinamon, G., Baleux, F., Arenzana,S. F., Lapidot, T., van Kooyk, Y., Lobb, R. R., and Alon, R. (2000). Subsecond induction of alpha4integrin clustering by immobilized chemokines stimulates leukocyte tethering and rolling onendothelial vascular cell adhesion molecule 1 under flow conditions. J. Exp. Med. 192, 495–506.
Grakoui, A., Bromley, S. K., Sumen, C., Davis, M. M., Shaw, A. S., Allen, P. M., and Dustin, M. L.(1999). The immunological synapse: A molecular Machine controlling T cell activation. Science285, 221–226.
Green, C. E., Schaff, U. Y., Sarantos, M. R., Lum, A. F., Staunton, D. E., and Simon, S. I. (2006).Dynamic shifts in LFA‐1 affinity regulate neutrophil rolling, arrest, and transmigration oninflamed endothelium. Blood 107, 2101–2111.
Griffiths, E. K., Krawczyk, C., Kong, Y. Y., Raab, M., Hyduk, S. J., Bouchard, D., Chan, V. S.,Kozieradzki, I., Oliveira‐Dos‐Santos, A. J., Wakeham, A., Ohashi, P. S., Cybulsky, M. I., et al.(2001). Positive regulation of T cell activation and integrin adhesion by the adapter Fyb/Slap.Science 293, 2260–2263.
Gu, Y., Filippi, M. D., Cancelas, J. A., Siefring, J. E., Williams, E. P., Jasti, A. C., Harris, C. E., Lee,A. W., Prabhakar, R., Atkinson, S. J., Kwiatkowski, D. J., and Williams, D. A. (2003). Hemato-poietic cell regulation by Rac1 and Rac2 guanosine triphosphatases. Science 302, 445–449.

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 219
Gu, Y., Jasti, A. C., Jansen, M., and Siefring, J. E. (2005). RhoH, a hematopoietic‐specific RhoGTPase, regulates proliferation, survival, migration, and engraftment of hematopoietic progeni-tor cells. Blood 105, 1467–1475.
Gunzer, M., Schafer, A., Borgmann, S., Grabbe, S., Zanker, K. S., Broker, E.‐B. E. K., and Friedl, P.(2000). Antigen presentation in extracellular matrix: Interactions of T cells with dendritic cellsare dynamic, short lived, and sequential. Immunity 13, 323–332.
Han, J., Lim, C. J., Watanabe, N., Soriani, A., Ratnikov, B., Calderwood, D. A., Puzon‐McLaughlin,W., Lafuente, E. M., Boussiotis, V. A., Shattil, S. J., and Ginsberg, M. H. (2006). Reconstructingand deconstructing agonist‐induced activation of integrin aIIbb3. Curr. Biol. 16, 1796–1806.
Hart, M. J., Jiang, X., Kozasa, T., Roscoe, W., Singer, W. D., Gilman, A. G., Sternweis, P. C., andBollag, G. (1998). Direct stimulation of the guanine nucleotide exchange activity of p115RhoGEF by Ga13. Science 280, 2112–2114.
Hemler, M. E. (1990). VLA proteins in the integrin family: Structures, functions, and their role onleukocytes. Annu. Rev. Immunol. 8, 365–400.
Hibbs, M. L., Jakes, S., Stacker, S. A., Wallace, R. W., and Springer, T. A. (1991). The cytoplasmicdomain of the integrin lymphocyte‐function‐associated antigen 1 b subunit: Sites required forbinding to intercellular adhesion molecules 1 and the phorbol ester‐stimulated phosphorylationsite. J. Exp. Med. 174, 1227–1238.
Hilpela, P., Oberbanscheidt, P., Hahne, P., Hund, M., Kalhammer, G., Small, J. V., and Bahler, M.(2003). SWAP‐70 identifies a transitional subset of actin filaments in motile cells. Mol. Biol. Cell14, 3242–3253.
Hirsch, E., Katanaev, V. L., Garlanda, C., Azzolino, O., Pirola, L., Silengo, L., Sozzani, S.,Mantovani, A., Altruda, F., and Wymann, M. P. (2000). Central role for G protein‐coupledphosphoinositide 3‐kinase gamma in inflammation. Science 287, 1049–1053.
Horwitz, A., Duggan, K., Buck, C., Beckerle, M. C., and Burridge, K. (1986). Interaction of plasmamembrane fibronectin receptor with talin: A transmembrane linkage. Nature 320, 531–533.
Hughes, P. E., Diaz‐Gonzalez, F., Leong, L., Wu, C., McDonald, J. A., Shattil, S. J., and Ginsberg,M. H. (1996). Breaking the integrin hinge. J. Biol. Chem. 271, 6571–6574.
Humphries, M. J. (2004). Monoclonal antibodies as probes of integrin priming and activation.Biochem. Soc. Trans. 32, 407–411.
Huth, J. R., Olejniczak, E. T., Mendoza, R., Liang, H., Harris, E. A., Lupher, M. L., Jr., Wilson,A. E., Fesik, S. W., and Staunton, D. E. (2000). NMR and mutagenesis evidence for an I domainallosteric site that regulates lymphocyte function‐associated antigen 1 ligand binding. Proc. Natl.Acad. Sci. USA 97, 5231–5236.
Hynes, R. O. (2002). Integrins: Bidirectional, allosteric signalling machines. Cell 110, 673–687.Ihara, S., Oka, T., and Fukui, Y. (2006). Direct binding of SWAP‐70 to non‐muscle actin is required
for membrane ruffling. J. Cell Sci. 119, 500–507.Inagaki, T., Suzuki, S., Miyamoto, T., Takeda, T., Yamashita, K., Komatsu, A., Yamauchi, K., and
Hashizume, K. (2003). The retinoic acid‐responsive proline‐rich protein is identified in promye-loleukemic HL‐60 cells. J. Biol. Chem. 278, 51685–51692.
Jenzora, A., Behrendt, B., Small, J. V., Wehland, J., and Stradal, T. E. (2005). PREL1 provides a linkfrom Ras signalling to the actin cytoskeleton via Ena / VASP proteins. FEBS Lett. 579, 455–463.
Johnson‐Leger, C., Aurrand‐Lions, M., and Imhof, B. A. (2000). The parting of the endothelium:Miracle, or simply a junctional affair? J. Cell Sci. 113(Pt. 6), 921–933.
Jones, M. C., Caswell, P. T., and Norman, J. C. (2006). Endocytic recycling pathways: Emergingregulators of cell migration. Curr. Opin. Cell Biol. 18, 549–557.
Katagiri, K., Hattori, M., Minato, N., Irie, S., Takatsu, K., and Kinashi, T. (2000). Rap1 is a potentactivation signal for leukocyte function‐associated antigen 1 distinct from protein kinase C andphosphatidylinositol‐3‐OH kinase. Mol. Cell. Biol. 20, 1956–1969.

220 TATSUO KINASHI
Katagiri, K., Hattori, M., Minato, N., and Kinashi, T. (2002). Rap1 functions as a key regulator ofT‐cell and antigen‐presenting cell interactions and modulates T‐cell responses. Mol. Cell. Biol.22, 1001–1015.
Katagiri, K., Maeda, A., Shimonaka, M., and Kinashi, T. (2003). RAPL, a Rap1‐binding moleculethat mediates Rap1‐induced adhesion through spatial regulation of LFA‐1. Nat. Immunol. 4,741–748.
Katagiri, K., Ohnishi, N., Kabashima, K., Iyoda, T., Takeda, N., Shinkai, Y., Inaba, K., and Kinashi, T.(2004a). Crucial roles of Rap1 effector molecule RAPL in lymphocyte and dendritic cell traffick-ing. Nat. Immunol. 5, 1045–1051.
Katagiri, K., Shimonaka, M., and Kinashi, T. (2004b). Rap1‐mediated LFA‐1 activation by theT cell antigen receptor is dependent on PLC‐g1. J. Biol. Chem. 279, 11875–11881.
Katagiri, K., Imamura, M., and Kinashi, T. (2006). Spatiotemporal regulation of the kinase Mst1 bybinding protein RAPL is critical for lymphocyte polarity and adhesion. Nat. Immunol. 7,919–928.
Kellermann, S. A., Dell, C. L., Hunt, S. W., 3rd, and Shimizu, Y. (2002). Genetic analysis of integrinactivation in T lymphocytes. Immunol. Rev. 186, 172–188.
Kim, M., Carman, C. V., and Springer, T. A. (2003). Bidirectional transmembrane signaling bycytoplasmic domain separation in integrins. Science 301, 1720–1725.
Kinashi, T., Escobedo, J. A., Williams, L. T., Takatsu, K., and Springer, T. A. (1995). Receptortyrosine kinase stimulates cell‐matrix adhesion by phosphatidylinositol 3 kinase and phospholi-pase C‐g1 pathways. Blood 86, 2086–2090.
Kinashi, T., Aker, M., Sokolovsky‐Eisenberg, M., Grabovsky, V., Tanaka, C., Shamri, R., Feigelson,S., Etzioni, A., and Alon, R. (2004). LAD‐III, a leukocyte adhesion deficiency syndromeassociated with defective Rap1 activation and impaired stabilization of integrin bonds. Blood103, 1033–1036.
Kliche, S., Breitling, D., Togni, M., Pusch, R., Heuer, K., Wang, X., Freund, C., Kasirer‐Friede, A.,Menasche, G., Koretzky, G. A., and Schraven, B. (2006). The ADAP/SKAP55 signaling moduleregulates T‐cell receptor‐mediated integrin activation through plasma membrane targeting ofRap1. Mol. Cell. Biol. 26, 7130–7144.
Kolanus, W., Nagel, W., Schiller, B., Zeitlmann, L., Godar, S., Stockinger, H., and Seed, B. (1996).aLb2 integrin/LFA‐1 binding to ICAM‐1 induced by cytohesin‐1, a cytoplasmic regulatorymolecule. Cell 86, 233–242.
Kozasa, T., Jiang, X., Hart, M. J., Sternweis, P. M., Singer, W. D., Gilman, A. G., Bollag, G., andSternweis, P. C. (1998). p115 RhoGEF, a GTPase activating protein for Galpha12 and Galpha13.Science 280, 2109–2111.
Krawczyk, C., Oliveira‐dos‐Santos, A., Sasaki, T., Griffiths, E., Ohashi, P. S., Snapper, S., Alt, F.,and Penninger, J. M. (2002). Vav1 controls integrin clustering and MHC/peptide‐specific celladhesion to antigen‐presenting cells. Immunity 16, 331–343.
Kucik, D. F., Dustin, M. L., Miller, J. M., and Brown, E. J. (1996). Adhesion‐activating phorbolester increases the mobility of leukocyte integrin LFA‐1 in cultured lymphocytes. J. Clin. Invest.97, 2139–2144.
Kunisaki, Y., Nishikimi, A., Tanaka, Y., Takii, R., Noda, M., Inayoshi, A., Watanabe, K., Sanematsu,F., Sasazuki, T., Sasaki, T., and Fukui, Y. (2006). DOCK2 is a Rac activator that regulates motilityand polarity during neutrophil chemotaxis. J. Cell Biol. 174, 647–652.
Labno, C. M., Lewis, C. M., You, D., Leung, D. W., Takesono, A., Kamberos, N., Seth,A., Finkelstein, L. D., Rosen, M. K., Schwartzberg, P. L., and Burkhardt, J. K. (2003). Itkfunctions to control actin polymerization at the immune synapse through localized activation ofCdc42 and WASP. Curr. Biol. 13, 1619–1624.

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 221
Lafuente, E. M., van Puijenbroek, A. A., Krause, M., Carman, C. V., Freeman, G. J., Berezovskaya,A., Constantine, E., Springer, T. A., Gertler, F. B., and Boussiotis, V. A. (2004). RIAM, an Ena/VASP and Profilin ligand, interacts with Rap1‐GTP and mediates Rap1‐induced adhesion. Dev.Cell 7, 585–595.
Larson, R. S., Davis, T., Bologa, C., Semenuk, G., Vijayan, S., Li, Y., Oprea, T., Chigaev, A.,Buranda, T., Wagner, C. R., and Sklar, L. A. (2005). Dissociation of I domain and globalconformational changes in LFA‐1: Refinement of small molecule‐I domain structure‐activityrelationships. Biochemistry 44, 4322–4331.
Laudanna, C., Campbell, J. J., and Butcher, E. C. (1996). Role of Rho in chemoattractant‐activatedleukocyte adhesion through integrins. Science 271, 981–983.
Lawson, M. A., and Maxfield, R. R. (1995). Ca2þ‐ and calcineurin‐dependent recycling of anintegrin to the front of migrating neutrophils. Nature 377, 75–79.
Legg, J. A., and Machesky, L. M. (2004). MRL proteins: Leading Ena/VASP to Ras GTPases. Nat.Cell Biol. 6, 1015–1017.
Li, S., Kim, M., Hu, Y. L., Jalali, S., Schlaepfer, D. D., Hunter, T., Chien, S., and Shyy, J. Y. (1997).Fluid shear stress activation of focal adhesion kinase: Linking to mitogen‐activated proteinkinases. J. Biol. Chem. 272, 30455–30462.
Li, X., Bu, X., Lu, B., Avraham, H., Flavell, R. A., and Lim, B. (2002). The hematopoiesis‐specificGTP‐binding protein RhoH is GTPase deficient and modulates activities of other Rho GTPasesby an inhibitory function. Mol. Cell. Biol. 22, 1158–1171.
Li, Z., Jiang, H., Xie, W., Zhang, Z., Smrcka, A. V., andWu, D. (2000). Roles of PLC‐b2 and ‐b3 andPI3Kg in chemoattractant‐mediated signal transduction. Science 287, 1046–1049.
Liu, S., Calderwood, D. A., and Ginsberg, M. H. (2000). Integrin cytoplasmic domain‐bindingproteins. J. Cell Sci. 113(Pt. 20), 3563–3571.
Lo, C. G., Lu, T. T., and Cyster, J. G. (2003). Integrin‐dependence of lymphocyte entry into thesplenic white pulp. J. Exp. Med. 197, 353–361.
Lollo, B. A., Chan, K. W., Hanson, E. M., Moy, V. T., and Brian, A. A. (1993). Direct evidence fortwo affinity states for lymphocyte function‐associated antigen 1 on activated T cells. J. Biol.Chem. 268, 21693–21700.
Lu, C., Ferzly, M., Takagi, J., and Springer, T. A. (2001a). Epitope mapping of antibodies to theC‐terminal region of the integrin beta 2 subunit reveals regions that become exposed uponreceptor activation. J. Immunol. 166, 5629–5637.
Lu, C., Shimaoka, M., Zang, Q., Takagi, J., and Springer, T. A. (2001b). Locking in alternateconformations of the integrin alphaLbeta2 I domain with disulfide bonds reveals functionalrelationships among integrin domains. Proc. Natl. Acad. Sci. USA 98, 2393–2398.
Lu, C., Shimaoka, M., Salas, A., and Springer, T. A. (2004). The binding sites for competitiveantagonistic, allosteric antagonistic, and agonistic antibodies to the I domain of integrin LFA‐1.J. Immunol. 173, 3972–3978.
Lu, C.‐H., and Springer, T. A. (1997). The a subunit cytoplasmic domain regulates the assembly andadhesiveness of integrin lymphocyte‐function‐associated antigen‐1. J. Immunol. 159, 268–278.
Lucas, J. A., Miller, A. T., Atherly, L. O., and Berg, L. J. (2003). The role of Tec family kinases inT cell development and function. Immunol. Rev. 191, 119–138.
Luo, B. H., Takagi, J., and Springer, T. A. (2004). Locking the beta3 integrin I‐like domain into highand low affinity conformations with disulfides. J. Biol. Chem. 279, 10215–10221.
McLeod, S. J., and Gold, M. R. (2001). Activation and function of the Rap1 GTPase inB lymphocytes. Int. Rev. Immunol. 20, 763–789.
McLeod, S. J., Li, A. H., Lee, R. L., Burgess, A. E., and Gold, M. R. (2002). The RapGTPases regulate B cell migration toward the chemokine stromal cell‐derived factor‐1(CXCL12): Potential role for Rap2 in promoting B cell migration. J. Immunol. 169, 1365–1371.

222 TATSUO KINASHI
Meacci, E., Tsai, S. C., Adamik, R., Moss, J., and Vaughan, M. (1997). Cytohesin‐1, a cytosolicguanine nucleotide‐exchange protein for ADP‐ribosylation factor. Proc. Natl. Acad. Sci. USA 94,1745–1748.
Medeiros, R. B., Dickey, D. M., Chung, H., Quale, A. C., Nagarajan, L. R., Billadeau, D. D., andShimizu, Y. (2005). Protein kinase D1 and the beta 1 integrin cytoplasmic domain control beta 1integrin function via regulation of Rap1 activation. Immunity 23, 213–226.
Mempel, T. R., Henrickson, S. E., and Von Andrian, U. H. (2004). T‐cell priming by dendritic cellsin lymph nodes occurs in three distinct phases. Nature 427, 154–159.
Miller, M. J., Wei, S. H., Parker, I., and Cahalan, M. D. (2002). Two‐photon imaging of lymphocytemotility and antigen response in intact lymph node. Science 296, 1869–1873.
Miller, M. J., Safrina, O., Parker, I., and Cahalan, M. D. (2004). Imaging the single cell dynamics ofCD4þ T cell activation by dendritic cells in lymph nodes. J. Exp. Med. 200, 847–856.
Miyasaka, M., and Tanaka, T. (2004). Lymphocyte trafficking across high endothelial venules:Dogmas and enigmas. Nat. Rev. Immunol. 4, 360–370.
Monks, C. R., Freiberg, B. A., Kupfer, H., Sciaky, N., and Kufper, A. (1998). Three‐dimensionalsegregation of supramolecular activation clusters in T cells. Nature 395, 82–86.
Moser, B., Wolf, M., Walz, A., and Loetscher, P. (2004). Chemokines: Multiple levels of leukocytemigration control. Trends Immunol. 25, 75–84.
Mould, A. P., Askari, J. A., Barton, S., Kline, A. D., McEwan, P. A., Craig, S. E., and Humphries,M. J. (2002). Integrin activation involves a conformational change in the alpha 1 helix of the betasubunit A‐domain. J. Biol. Chem. 277, 19800–19805.
Mould, A. P., Barton, S. J., Askari, J. A., Craig, S. E., and Humphries, M. J. (2003a). Role ofADMIDAS cation‐binding site in ligand recognition by integrin a5b1. J. Biol. Chem. 278,51622–51629.
Mould, A. P., Barton, S. J., Askari, J. A., McEwan, P. A., Buckley, P. A., Craig, S. E., andHumphries, M. J. (2003b). Conformational changes in the integrin beta A domain provide amechanism for signal transduction via hybrid domain movement. J. Biol. Chem. 278, 17028–17035.
Muller, W. A. (2003). Leukocyte‐endothelial‐cell interactions in leukocyte transmigration and theinflammatory response. Trends Immunol. 24, 327–334.
Nieminen, M., Henttinen, T., Merinen, M., Marttila‐Ichihara, F., Eriksson, J. E., and Jalkanen, S.(2006). Vimentin function in lymphocyte adhesion and transcellular migration. Nat. Cell Biol. 8,156–162.
Nishida, N., Xie, C., Shimaoka, M., Cheng, Y., Walz, T., and Springer, T. A. (2006). Threedistinctive ectodomain conformations and their interconversion in leukocyte b2 integrins.Immunity. 25, 583–594.
Nombela‐Arrieta, C., Lacalle, R. A., Montoya, M. C., Kunisaki, Y., Megias, D., Marques, M.,Carrera, A. C., Manes, S., Fukui, Y., Martinez, A. C., and Stein, J. V. (2004). Differentialrequirements for DOCK2 and phosphoinositide‐3‐kinase gamma during T and B lymphocytehoming. Immunity 21, 429–441.
Okada, T., Ngo, V. N., Ekland, E. H., Forster, R., Lipp, M., Littman, D. R., and Cyster, J. G. (2002).Chemokine requirements for B cell entry to lymph nodes and Peyer’s patches. J. Exp. Med. 196,65–75.
Okkenhaug, K., and Vanhaesebroeck, B. (2003). PI3K in lymphocyte development, differentiationand activation. Nat. Rev. Immunol. 3, 317–330.
Pearce, G., Angeli, V., Randolph, G. J., Junt, T., von Andrian, U., Schnittler, H. J., and Jessberger, R.(2006). Signaling protein SWAP‐70 is required for efficient B cell homing to lymphoid organs.Nat. Immunol. 7, 827–834.

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 223
Perez, O. D., Mitchell, D., Jager, G. C., South, S., Murriel, C., McBride, J., Herzenberg, L. A.,Kinoshita, S., and Nolan, G. P. (2003). Leukocyte functional antigen 1 lowers T cell activationthresholds and signaling through cytohesin‐1 and Jun‐activating binding protein 1.Nat. Immunol.4, 1083–1092.
Peterson, E. J. (2003). The TCR ADAPts to integrin‐mediated cell adhesion. Immunol. Rev. 192,113–121.
Peterson, E. J., Woods, M. L., Dmowski, S. A., Derimanov, G., Jordan, M. S., Wu, J. N., Myung,P. S., Liu, Q. H., Pribila, J. T., Freedman, B. D., Shimizu, Y., and Koretzky, G. A. (2001).Coupling of the TCR to integrin activation by Slap‐130/Fyb. Science 293, 2263–2265.
Petruzzelli, L., Maduzia, L., and Springer, T. A. (1995). Activation of lymphocyte function‐associated molecule‐1 (CD11a/CD18) and Mac‐1 (CD11b/CD18) mimicked by an antibodydirected against CD18. J. Immunol. 155, 854–866.
Reedquist, K. A., Ross, E., Koop, E. A., Wolthuis, R. M., Zwartkruis, F. J., van Kooyk, Y., Salmon,M., Buckley, C. D., and Bos, J. L. (2000). The small GTPase, Rap1, mediates CD31‐inducedintegrin adhesion. J. Cell Biol. 148, 1151–1158.
Regelmann, G. A., Danzi, N. M., Wanjalla, C., and Alexandropoulos, K. (2006). Chat‐H regulatesT lymphocyte trafficking by acting upstream of Rap1 in chemokine‐induced inside‐out signaling.Immunity. 25, 907–918.
Reif, K., Okkenhaug, K., Sasaki, T., Penninger, J. M., Vanhaesebroeck, B., and Cyster, J. G. (2004).Cutting edge: Differential roles for phosphoinositide 3‐kinases, p110gamma and p110delta, inlymphocyte chemotaxis and homing. J. Immunol. 173, 2236–2240.
Reynolds, L. F., Smyth, L. A., Norton, T., Freshney, N., Downward, J., Kioussis, D., and Tybule-wicz, V. L. (2002). Vav1 transduces T cell receptor signals to the activation of phospholipase C‐g1via phosphoinositide 3‐kinase‐dependent and ‐independent pathways. J. Exp. Med. 195,1103–1114.
Roberts, A. W., Kim, C., Zhen, L., Lowe, J. B., Kapur, R., Petryniak, B., Spaetti, A., Pollock, J. D.,Borneo, J. B., Bradford, G. B., Atkinson, S. J., Dinauer, M. C., et al. (1999). Deficiency of thehematopoietic cell‐specific Rho family GTPase Rac2 is characterized by abnormalities in neu-trophil function and host defense. Immunity 10, 183–196.
Robinson, M. K., Andrew, D., Rosen, H., Brown, D., Ortlepp, S., Stephens, P., and Butcher, E. C.(1992). Antibody against the Leu‐CAM beta‐chain (CD18) promotes both LFA‐1‐ and CR3‐dependent adhesion events. J. Immunol. 148, 1080–1085.
Rodriguez‐Fernandez, J. L., Sanchez‐Martin, L., Rey, M., Vicente‐Manzanares, M., Narumiya, S.,Teixido, J., Sanchez‐Madrid, F., and Cabanas, C. (2001). Rho and Rho‐associated kinase modu-late the tyrosine kinase PYK2 in T‐cells through regulation of the activity of the integrin LFA‐1.J. Biol. Chem. 276, 40518–40527.
Romzek, N. C., Harris, E. S., Dell, C. L., Skronek, J., Hasse, E., Reynolds, P. J., Hunt, S. W., 3rd,and Shimizu, Y. (1998). Use of a beta1 integrin‐deficient human T cell to identify beta1 integrincytoplasmic domain sequences critical for integrin function. Mol. Biol. Cell 9, 2715–2727.
Rosen, H., and Goetzl, E. J. (2005). Sphingosine 1‐phosphate and its receptors: An autocrine andparacrine network. Nat. Rev. Immunol. 5, 560–570.
Rozengurt, E., Rey, O., and Waldron, R. T. (2005). Protein kinase D signaling. J. Biol. Chem. 280,13205–13208.
Rubtsov, A., Strauch, P., Digiacomo, A., Hu, J., Pelanda, R., and Torres, R. M. (2005). Lsc regulatesmarginal‐zone B cell migration and adhesion and is required for the IgM T‐dependent antibodyresponse. Immunity 23, 527–538.
Sah, V. P., Seasholtz, T. M., Sagi, S. A., and Brown, J. H. (2000). The role of Rho in G protein‐coupled receptor signal transduction. Annu. Rev. Pharmacol. Toxicol. 40, 459–489.

224 TATSUO KINASHI
Sakakibara, A., Hattori, S., Nakamura, S., and Katagiri, T. (2003). A novel hematopoietic adaptorprotein, Chat‐H, positively regulates T cell receptor‐mediated interleukin‐2 production byJurkat cells. J. Biol. Chem. 278, 6012–6017.
Salas, A., Shimaoka, M., Chen, S., Carman, C. V., and Springer, T. (2002). Transition from rolling tofirm adhesion is regulated by the conformation of the I domain of the integrin lymphocytefunction‐associated antigen‐1. J. Biol. Chem. 277, 50255–50262.
Salas, A., Shimaoka, M., Kogan, A. N., Harwood, C., von Andrian, U. H., and Springer, T. A.(2004). Rolling adhesion through an extended conformation of integrin aLb2 and relation toalpha I and beta I‐like domain interaction. Immunity 20, 393–406.
Salas, A., Shimaoka, M., Phan, U., Kim, M., and Springer, T. A. (2006). Transition from rolling tofirm adhesion can be mimicked by extension of integrin aLb2 in an intermediate affinity state.J. Biol. Chem. 281, 10876–10882.
Samelson, L. E. (2002). Signal transduction mediated by the T cell antigen receptor: The role ofadapter proteins. Annu. Rev. Immunol. 20, 371–394.
Sampath, R., Gallagher, P. J., and Pavalko, F. M. (1998). Cytoskeletal interactions with theleukocyte integrin beta2 cytoplasmic tail: Activation‐dependent regulation of associations withtalin and a‐actinin. J. Biol. Chem. 273, 33588–33594.
Sanchez‐Madrid, F., and del Pozo, M. A. (1999). Leukocyte polarization in cell migration andimmune interactions. EMBO J. 18, 501–511.
Sanui, T., Inayoshi, A., Noda, M., Iwata, E., Oike, M., Sasazuki, T., and Fukui, Y. (2003). DOCK2 isessential for antigen‐induced translocation of TCR and lipid rafts, but not PKC‐theta and LFA‐1,in T cells. Immunity 19, 119–129.
Sastry, S. K., and Horwitz, A. F. (1993). Integrin cytoplasmic domains: Mediators of cytoskeletallinkages and extra‐ and intracellular initiated transmembrane signaling. Curr. Biol. 5, 819–831.
Sebzda, E., Bracke, M., Tugal, T., Hogg, N., and Cantrell, D. A. (2002). Rap1A positively regulatesT cells via integrin activation rather than inhibiting lymphocyte signaling. Nat. Immunol. 3,251–258.
Semmrich, M., Smith, A., Feterowski, C., Beer, S., Engelhardt, B., Busch, D. H., Bartsch, B.,Laschinger, M., Hogg, N., Pfeffer, K., and Holzmann, B. (2005). Importance of integrin LFA‐1deactivation for the generation of immune responses. J. Exp. Med. 201, 1987–1998.
Shamri, R., Grabovsky, V., Gauguet, J. M., Feigelson, S., Manevich, E., Kolanus, W., Robinson,M. K., Staunton, D. E., von Andrian, U. H., and Alon, R. (2005). Lymphocyte arrest requiresinstantaneous induction of an extended LFA‐1 conformation mediated by endothelium‐boundchemokines. Nat. Immunol. 6, 497–506.
Shimaoka, M., Takagi, J., and Springer, T. A. (2002). Conformational regulation of integrinstructure and function. Annu. Rev. Biophys. Biomol. Struct. 485, 485–516.
Shimonaka, M., Katagiri, K., Nakayama, T., Fujita, N., Tsuruo, T., Yoshie, O., and Kinashi, T.(2003). Rap1 translates chemokine signals to integrin activation, cell polarization, and motilityacross vascular endothelium under flow. J. Cell Biol. 161, 417–427.
Shimaoka, M., Salas, A., Yang, W., Weitz‐Schmidt, G., and Springer, T. A. (2003a). Small moleculeintegrin antagonists that bind to the b2 subunit I‐like domain and activate signals in onedirection and block them in the other. Immunity 19, 391–402.
Shimaoka, M., Xiao, T., Liu, J. H., Yang, Y., Dong, Y., Jun, C. D., McCormack, A., Zhang, R.,Joachimiak, A., Takagi, J., Wang, J. H., and Springer, T. A. (2003b). Structures of the alpha L Idomain and its complex with ICAM‐1 reveal a shape‐shifting pathway for integrin regulation.Cell 112, 99–111.
Shimaoka, M., Kim, M., Cohen, E. H., Yang, W., Astrof, N., Peer, D., Salas, A., Ferrand, A., andSpringer, T. A. (2006). AL‐57, a ligand‐mimetic antibody to integrin LFA‐1, reveals chemokine‐induced affinity up‐regulation in lymphocytes. Proc. Natl. Acad. Sci. USA 103, 13991–13996.

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 225
Shimizu, Y., and Mobley, J. L. (1993). Distinct divalent cation requirements for integrin‐mediatedCD4þ T lymphocyte adhesion to ICAM‐1, fibronectin, VCAM‐1, and invasin. J. Immunol. 151,4106–4115.
Shimizu, Y., Mobley, J. L., Finkelstein, L. D., and Chan, A. S. (1995). A role for phosphatidylino-sitol 3‐kinase in the regulation of beta 1 integrin activity by the CD2 antigen. J. Cell Biol. 131,1867–1880.
Shinohara, M., Terada, Y., Iwamatsu, A., Shinohara, A., Mochizuki, N., Higuchi, M., Gotoh, Y.,Ihara, S., Nagata, S., Itoh, H., Fukui, Y., and Jessberger, R. (2002). SWAP‐70 is a guanine‐nucleotide‐exchange factor that mediates signalling of membrane ruffling. Nature 416, 759–763.
Shulman, Z., Pasvolsky, R., Woolf, E., Grabovsky, V., Feigelson, S. W., Erez, N., Fukui, Y., andAlon, R. (2006). DOCK2 regulates chemokine‐triggered lateral lymphocyte motility but nottransendothelial migration. Blood 108, 2150–2158.
Sims, T. N., and Dustin, M. L. (2002). The immunological synapse: Integrins take the stage.Immunol. Rev. 186, 100–117.
Smith, A., Bracke, M., Leitinger, B., Porter, J. C., and Hogg, N. (2003). LFA‐1‐induced T cellmigration on ICAM‐1 involves regulation of MLCK‐mediated attachment and ROCK‐depen-dent detachment. J. Cell Sci. 116, 3123–3133.
Smith, A., Carrasco, Y. R., Stanley, P., Kieffer, N., Batista, F. D., and Hogg, N. (2005). A talin‐dependent LFA‐1 focal zone is formed by rapidly migrating T lymphocytes. J. Cell Biol. 170,141–151.
Springer, T. A. (1990). Adhesion receptors of the immune system. Nature 346, 425–434.Springer, T. A. (1995). Traffic signals on endothelium for lymphocyte recirculation and leukocyte
emigration. Annu. Rev. Physiol. 57, 827–872.Staunton, D. E., Lupher, M. L., Liddington, R., and Gallatin, W. M. (2006). Targeting integrin
structure and function in disease. Adv. Immunol. 91, 111–157.Stein, J. V., Rot, A., Luo, Y., Narasimhaswamy, M., Nakano, H., Gunn, M. D., Matsuzawa, A.,
Quackenbush, E. J., Dorf, M. E., and von Andrian, U. H. (2000). The CC chemokine thymus‐derived chemotactic agent 4 (TCA‐4, secondary lymphoid tissue chemokine, 6Ckine, exodus‐2)triggers lymphocyte function‐associated antigen 1‐mediated arrest of rolling T lymphocytes inperipheral lymph node high endothelial venules. J. Exp. Med. 191, 61–76.
Stewart, M. P., McDowall, A., and Hogg, N. (1998). LFA‐1‐mediated adhesion is regulated bycytoskeletal restraint and by a Ca2þ‐dependent protease, calpain. J. Cell Biol. 140,699–707.
Tadokoro, S., Shattil, S. J., Eto, K., Tai, V., Liddington, R. C., de Pereda, J. M., Ginsberg, M. H.,and Calderwood, D. A. (2003). Talin binding to integrin beta tails: A final common step inintegrin activation. Science 302, 103–106.
Takagi, J., and Springer, T. A. (2002). Integrin activation and structural rearrangement. Immunol.Rev. 186, 141–163.
Takagi, J., Petre, B. M., Walz, T., and Springer, T. A. (2002). Global conformational rearrange-ments in integrin extracellular domains in outside‐in and inside‐out signaling. Cell 110,599–611.
Takesono, A., Horai, R., Mandai, M., Dombroski, D., and Schwartzberg, P. L. (2004). Requirementfor Tec kinases in chemokine‐induced migration and activation of Cdc42 and Rac. Curr. Biol.14, 917–922.
Tamada, M., Sheetz, M. P., and Sawada, Y. (2004). Activation of a signaling cascade by cytoskeletonstretch. Dev. Cell 7, 709–718.
Tohyama, Y., Katagiri, K., Pardi, R., Lu, C., Springer, T. A., and Kinashi, T. (2003). The criticalcytoplasmic regions of the aL/b2 integrin in Rap1‐induced adhesion and migration. Mol. Biol.Cell 14, 2570–2582.

226 TATSUO KINASHI
Tommasi, S., Dammann, R., Jin, S. G., Zhang, X. F., Avruch, J., and Pfeifer, G. P. (2002). RASSF3and NORE1: Identification and cloning of two human homologues of the putative tumorsuppressor gene RASSF1. Oncogene 21, 2713–2720.
Tybulewicz, V. L., Ardouin, L., Prisco, A., and Reynolds, L. F. (2003). Vav1: A key signal transducerdownstream of the TCR. Immunol. Rev. 192, 42–52.
van Kooyk, Y., and Figdor, C. G. (2000). Avidity regulation of integrins: The driving force inleukocyte adhesion. Curr. Opin. Cell Biol. 12, 542–547.
Vicente‐Manzanares, M., Cruz‐Adalia, A., Martin‐Cofreces, N. B., Cabrero, J. R., Dosil, M.,Alvarado‐Sanchez, B., Bustelo, X. R., and Sanchez‐Madrid, F. (2005). Control of lymphocyteshape and the chemotactic response by the GTP exchange factor Vav. Blood 105, 3026–3034.
Vielkind, S., Gallagher‐Gambarelli, M., Gomez, M., Hinton, H. J., and Cantrell, D. A. (2005).Integrin regulation by RhoA in thymocytes. J. Immunol. 175, 350–357.
Vogel, V., and Sheetz, M. (2006). Local force and geometry sensing regulate cell functions. Nat.Rev. Mol. Cell. Biol. 7, 265–275.
Wang, H., Moon, E. Y., Azouz, A., Wu, X., Smith, A., Schneider, H., Hogg, N., and Rudd, C. E.(2003). SKAP‐55 regulates integrin adhesion and formation of T cell‐APC conjugates. Nat.Immunol. 4, 366–374.
Wang, H., McCann, F. E., Gordan, J. D., Wu, X., Raab, M., Malik, T. H., Davis, D. M., and Rudd,C. E. (2004). ADAP‐SLP‐76 binding differentially regulates supramolecular activation cluster(SMAC) formation relative to T cell‐APC conjugation. J. Exp. Med. 200, 1063–1074.
Ward, S. G. (2004). Do phosphoinositide 3‐kinases direct lymphocyte navigation? Trends Immunol.25, 67–74.
Warnock, R. A., Campbell, J. J., Dorf, M. E., Matsuzawa, A., McEvoy, L. M., and Butcher, E. C.(2000). The role of chemokines in the microenvironmental control of T versus B cell arrest inPeyer’s patch high endothelial venules. J. Exp. Med. 191, 77–88.
Weber, K. S.,Weber, C., Ostermann, G., Dierks, H., Nagel,W., and Kolanus,W. (2001). Cytohesin‐1is a dynamic regulator of distinct LFA‐1 functions in leukocyte arrest and transmigrationtriggered by chemokines. Curr. Biol. 11, 1969–1974.
Windh, R. T., Lee, M. J., Hla, T., An, S., Barr, A. J., and Manning, D. R. (1999). Differentialcoupling of the sphingosine 1‐phosphate receptors Edg‐1, Edg‐3, and H218/Edg‐5 to the G(i),G(q), and G(12) families of heterotrimeric G proteins. J. Biol. Chem. 274, 27351–27358.
Woods, A. J., White, D. P., Caswell, P. T., and Norman, J. C. (2004). PKD1/PKCmu promotes avb3integrin recycling and delivery to nascent focal adhesions. EMBO J. 23, 2531–2543.
Woods, M. L., Kivens, W. J., Adelsman, M. A., Qiu, Y., August, A., and Shimizu, Y. (2001). A novelfunction for the Tec family tyrosine kinase Itk in activation of beta 1 integrins by the T‐cellreceptor. EMBO J. 20, 1232–1244.
Worthylake, R. A., Lemoine, S., Watson, J. M., and Burridge, K. (2001). RhoA is required formonocyte tail retraction during transendothelial migration. J. Cell Biol. 154, 147–160.
Xie, C., Shimaoka, M., Xiao, T., Schwab, P., Klickstein, L. B., and Springer, T. A. (2004). Theintegrin a‐subunit leg extends at a Ca2þ‐dependent epitope in the thigh/genu interface uponactivation. Proc. Natl. Acad. Sci. USA 101, 15422–15427.
Xiong, J. P., Stehle, T., Diefenbach, B., Zhang, R., Dunker, R., Scott, D. L., Joachimiak, A.,Goodman, S. L., and Arnaout, M. A. (2001). Crystal structure of the extracellular segment ofintegrin aVb3. Science 294, 339–345.
Xiong, J. P., Stehle, T., Zhang, R., Joachimiak, A., Frech, M., Goodman, S. L., and Arnaout, M. A.(2002). Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex withan Arg‐Gly‐Asp ligand. Science 296, 151–155.
Yan, B., Calderwood, D. A., Yaspan, B., and Ginsberg, M. H. (2001). Calpain cleavage promotestalin binding to the beta 3 integrin cytoplasmic domain. J. Biol. Chem. 276, 28164–28170.

INTEGRIN REGULATION OF LYMPHOCYTE TRAFFICKING 227
Yang, W., Shimaoka, M., Chen, J., and Springer, T. A. (2004a). Activation of integrin beta‐subunitI‐like domains by one‐turn C‐terminal alpha‐helix deletions. Proc. Natl. Acad. Sci. USA 101,2333–2338.
Yang, W., Shimaoka, M., Salas, A., Takagi, J., and Springer, T. A. (2004b). Intersubunit signaltransmission in integrins by a receptor‐like interaction with a pull spring. Proc. Natl. Acad. Sci.USA 101, 2906–2911.
Zell, T., Hunt, S. W. R., Mobley, J. L., Finkelstein, L. D., and Shimizu, Y. (1996). CD28‐mediatedup‐regulation of beta 1‐integrin adhesion involves phosphatidylinositol 3‐kinase. J. Immunol.156, 883–886.
Zhang, W., Shao, Y., Fang, D., Huang, J., Jeon, M. S., and Liu, Y. C. (2003). Negative regulation ofT cell antigen receptor‐mediated Crk‐L‐C3G signaling and cell adhesion by Cbl‐b. J. Biol.Chem. 278, 23978–23983.

a#
Regulation of Immune Responses and Hematopoiesis by theRap1 Signal
Nagahiro Minato, Kohei Kometani, and Masakazu Hattori
Department of Immunology and Cell Biology, Graduate School of Biostudies,Kyoto University, Kyoto, Japan
A
dv2
bstract............................................................................................................. 2
229ances in immunology, vol. 93 0065-2776/07 $
007 Elsevier Inc. All rights reserved. DOI: 10.1016/S0065-2776(06)93
29
1. I ntroduction ....................................................................................................... 2 29 2. G eneral Biology of the Rap1 Signal ........................................................................ 2 30 3. R ap1 Signal in Lymphocyte Development and Immune Responses............................... 2 37 4. R ap1 Signal in Hematopoiesis and Leukemia............................................................ 2 48 5. R ap1 Signal in Malignancy: New Aspects in Cancer................................................... 2 53 6. C onclusions and Perspectives ................................................................................ 2 55R
eferences ......................................................................................................... 2 56Abstract
Rap1 (Ras‐proximity 1), a member of the Ras family of small guanine tripho-sphatases (GTPases), is activated by diverse extracellular stimuli. While Rap1 hasbeen discovered originally as a potential Ras antagonist, accumulating evidenceindicates that Rap1 per se mediates unique signals and exerts biological functionsdistinctly different from Ras. Rap1 plays a dominant role in the control of cell–celland cell–matrix interactions by regulating the function of integrins and otheradhesionmolecules in various cell types. Rap1 also regulatesMAP kinase (MAPK)activity in a manner highly dependent on the context of cell types. Recent studies(including gene‐targeting analysis) have uncovered that the Rap1 signal isintegrated crucially and unpredictably in the diverse aspects of comprehensivebiological systems. This review summarizes the role of the Rap1 signal in develop-ments and functions of the immune and hematopoietic systems as well as inmalignancy. Importantly, Rap1 activation is tightly regulated in tissue cells, anddysregulations of the Rap1 signal in specific tissues result in certain disorders,including myeloproliferative disorders and leukemia, platelet dysfunction withdefective hemostasis, leukocyte adhesion‐deficiency syndrome, lupus‐like system-ic autoimmune disease, and T cell anergy. Many of these disorders resemblehuman diseases, and the Rap1 signal with its regulators may provide rationalmolecular targets for controlling certain human diseases including malignancy.
1. Introduction
Rap1 (Ras‐proximity 1), a member of the Ras family of small guanine tripho-sphatases (GTPases), displays high overall homology (�50%) to the classicalK‐, H‐, and N‐Ras with an identical effector domain (Pizon et al., 1988). Rap1
35.00
006-5

230 NAGAHIRO MINATO ET AL .
is conserved in eukaryocytes from yeasts through mammals. In budding yeasts,Rap1 (Rsr1) is essential for the proper determination of budding sites formating(Bender and Pringle, 1989). Rap1 in Drosophila melanogaster (DRap1) is alsoan essential gene that plays crucial roles in various aspects of morphogenesis(Asha et al., 1999). The prototype, or the so‐called ‘‘roughened eyes,’’ is causedby a gain‐of‐function mutation of the Rap1 gene (Hariharan et al., 1991). Inmammals, there are two Rap1 isoforms coded by distinct genes (Rap1A andRap1B) with limited differences in constitutional amino acids and redundantactivities (Bos et al., 2001). It was first reported that overexpression of Rap1(originally called K‐rev) could revert the characteristic ‘‘malignant’’ contours ofthe fibroblasts transformed by oncogenic K‐Ras to a flat shape similar to normalfibroblasts (Kitayama et al., 1989). This raised the initial idea that Rap1 mightact as a functional antagonist of oncogenic Ras. The exact roles of Rap1 inmammalian cells, however, have remained rather enigmatic for nearly a decade.In the late 1990s, two distinct biological activities mediated by the Rap1 signal(independent of Ras) emerged, viz., the activation of MAP kinases (MAPKs)and control of cell adhesion via integrins. Since then, numerous findings on theroles of Rap1 in many cell types of various tissues have accumulated, and it hasbecome evident that the Rap1 signal mediates highly diverse cellular activitiesdepending on the cellular contexts. In the present chapter, we first summarizerecent advances on the general biology of Rap1, including regulation andfunction of the Rap1 signal, before proceeding to discuss how a ubiquitousmolecular switch (Rap1) is integrated into the signaling pathways to controlhighly sophisticated and specified cellular functions, with particular emphasison the immune/hematopoietic systems and malignancy.
2. General Biology of the Rap1 Signal
One of the most intriguing features of Rap1 is that it is activated by anextensive variety of external stimuli delivered to the cell, including numerousgrowth factors, peptide hormones, neurotransmitters, cytokines, chemokines,antigens, cell‐adhesion molecules, and physical stimuli such as cell stretch/contraction.
2.1. Regulation of Rap1 Activation
Similar to many other small G‐proteins, Rap1 binds with guanine nucleotides toform Rap1GTP (an active form) or Rap1GDP (an inactive form) that interactsor dissociates with a number of downstream effector molecules, respectively.Due to its intrinsic GTPase activity, GTP bound to Rap1 is autonomouslyhydrolyzed to GDP. Therefore, activation of Rap1 requires specific enzymes

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 231
that dissociate GDP from Rap1 to facilitate repetitive GTP loading, that is, theguanine nucleotide exchange factors (GEFs). A number of distinct Rap1 GEFssharing a catalytic GEF domain have been identified, and they are coupled withvarious receptors or intracellular second messengers (Fig. 1; Bos et al., 2001;Hattori andMinato, 2003). C3G, which is a Rap1GEFubiquitously expressed inmost cell types, binds to the SH3 domain of Crk adaptor proteins, and is re-cruited to the plasma membrane and phosphorylated on activation of receptor‐associated protein tyrosine kinases (PTKs; Gotoh et al., 1995; Ichiba et al.,1999). Phosphorylated C3G is a major Rap1 activator in the plasma membrane.CalDAG‐GEF harbor the Ca2þ ion‐ and diasylglycerol (DAG)‐binding sites.Activation of CalDAG‐GEF I is regulated by the Ca2þ ion, while CalDAG‐GEF III is translocated to the membrane by binding DAG, and thus theseGEFs may mediate Rap1 activation downstream of PLC activation (Kawasakiet al., 1998). On the other hand, the Epac family of Rap1GEFs has specificcyclic AMP (cAMP)‐binding domains at the N‐terminal region. Binding cAMPinduces conformational changes in Epacs to release the inhibitory constraintcovering the catalytic GEF domain, thus allowing interaction with the substrate
Proliferation, survivalgene activation
Cell adhesion, migrationpolarity
Smgcross-talk
GTP
GDP
Pi
C3G
Epac (1,2)
CD-GEF (I,III)
PDZ-GEF
DOCK-4
Crk (L)PTK
A-cyclase cAMP
PLC Ca2+ DAG
GEFsGAPs
SPA-1 family(SPA-1, E6TP1,SPA-L2,3)
RapGAs (1,2)
Rap1-GDP
Rap1-GTP
Extracellularstimuli
Ras-GTP
c-Raf-1
MEK1,2
ERK
B-Raf
MEK3,6
p38MAPK
RapL
Integrins
RIAM
ProfillinEna/VASP
F-actin
RalGDS
Ral, Rac
AF-6
Cadherins
Figure 1 Regulation and functions of the Rap1 signal. Refer to the text for the details.

232 NAGAHIRO MINATO ET AL .
Rap1 (Bos, 2003; de Rooij et al., 1998). It has been demonstrated that certaincAMP‐induced biological activities, such as cell adhesion and insulin secretion, aremediated by the Epac/Rap1 rather than by cAMP‐dependent protein kinasepathway (Bos, 2003). Epacs may play a major role in the cytosolic Rap1 activationdownstream of trimeric G‐protein–coupled receptors (GPCRs). Thus, distincttypes of Rap1 GEFs are tightly coupled with the major signaling pathways toinduce Rap1 activation at the different intracellular compartments via diverseextracellular stimuli (Fig. 1).Rap1 does not share a conserved catalytic residue with other small
G‐proteins, such as Ras, Rho, or Cdc42, and displays much lower intrinsicGTPase activity. The swift inactivation of Rap1GTP to terminate the signal,therefore, is crucially dependent on Rap1 GTPase‐activating proteins (GAPs).Rap1GAPs specifically bind to GTP‐bound Rap1 to provide a catalytic residue(asparagine) for Rap1 thereby enhancing the GTPase activity by multipleorders (Daumke et al., 2004). In contrast to the diverse types of Rap1GEFs,there are only two groups of Rap1GAPs (i.e., RapGAs and SPA‐1 family)that share a catalytic domain called the GAP‐related domain (GRD). WhileRapGA1 is expressed rather ubiquitously (most prominently expressed in thebrain), RapGA2 is distributed exclusively in platelets (Kurachi et al., 1997;Schultess et al., 2005). An isoform of RapGA1 binds to Ga via the N‐terminalregion and may be translocated to the plasma membrane following the activa-tion of GPCRs, thus attenuating Rap1 activation at the membrane (Mochizukiet al., 1999). The SPA‐1 family of Rap1GAPs consists of SPA‐1, SPA‐1‐like(SPA‐L) 1 (also called E6TP1 or SPAR), SPA‐L 2, and SPA‐L 3, all of whichshare a PDZ domain in addition to a GRD. SPA‐1 is most abundantly exp-ressed in lymphohematopoietic tissues and certain cancer cells, while SPAR isdistributed in epithelial tissues and the brain (Gao et al., 1999; Hattori et al.,1995; Pak et al., 2001). They are located in various intracellular compartments,such as the synaptic vesicles, actin cytoskeleton, plasma membranes, andpossibly nuclei, depending on the cell type and specific protein interactionvia the PDZ domain (Farina et al., 2004; Roy et al., 2002; Tsukamoto et al.,1999). All the Rap1GAPs are constitutively active without any protein modi-fication. As such, the expression levels of Rap1GAPs per se may determinethe threshold and degree of Rap1 activation at any given compartment (seebelow). Notably, E6TP1 is specifically degraded by human papillomavirus E6oncoprotein via E6AP ubiquitin ligase similarly to p53 in a fashion closelycorrelated with E6‐mediated epithelial cell transformation (Gao et al., 2001,2002). Rap1GAPs have no structural similarities to GAPs for other smallG‐proteins, such as Ras, Rho, Arf, or Rab, and a study has indicated that themode of GAP activity for Rap1 is also different from that of GAPs for otherG‐proteins (Daumke et al., 2004).

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 233
2.2. Biological Function of Rap1
2.2.1. Regulation of MAPK Activation
Although it has been a matter of argument for almost a decade whether theRap1 signal activates extracellular signal‐regulated protien kinase (ERK),accumulating evidence has indicated that the Rap1 signal does activate theMAPK kinase‐1 (MEK‐1), 2–ERK pathway selectively via B‐Raf (Vossler et al.,1997), which is expressed in only selected tissue cells (Barnier et al., 1995).This finding probably explains why Rap1‐mediated ERK activation has beenobserved only in certain selected cell types. While ubiquitous c‐Raf‐1 needs tobe phosphorylated to activate MEK‐1, 2, B‐Raf is constitutively phosphory-lated at the corresponding sites (Mason et al., 1999) and activates MEK‐1, 2 bybeing recruited to the plasma membrane by Rap1GTP. This was confirmedgenetically in Drosophila melanogaster, which has only one Raf isoform (DRaf)corresponding to the mammalian B‐Raf. Thus, DRap1 that has been activateddownstream of torso receptor tyrosine kinase binds to DRaf and induces ERKactivation, which in turn incites activation of tailless and huckebein genescontrolling the terminal structures in embryos (Mishra et al., 2005). AlthoughRap1GTP binds to c‐Raf‐1 with an affinity even higher than RasGTP inmammalian cells, it may not lead to the activation of MEK‐1, 2–ERK pathway,partly because Rap1GTP is incapable of inducing c‐Raf‐1 phosphorylationrequired for the activation of the kinase activity (Mishra et al., 2005).
ERK activation by Rap1, however, shows a unique feature distinctly differentfrom Ras‐mediated ERK activation. In PC12 neuronal cell line that stronglyexpresses B‐Raf, the epithelial growth factor (EGF) and nerve growth factor(NGF) specifically induce the proliferation and differentiation, respectively(Marshall, 1995; Qui and Green, 1992; Traverse et al., 1992). NGF rapidlyelicits peak followed by sustained activation of ERK, while EGF induces onlya transient ERK activation, suggesting that a persistent ERK activation isrequired for PC12 cell differentiations. Although Ras mediates the rapid andtransient ERK activation elicited by both factors, Rap1 is responsible for theNGF‐induced sustained activation of ERK (Kao et al., 2001; York et al., 1998).Sophisticated analyses have suggested that different ERK activation kineticsmight reflect the different regulatory mechanisms of Ras and Rap1 activations(Sasagawa et al., 2005). While Ras is activated rapidly by SOS recruited to theplasma membrane via Grb2 following stimulation before recruitment of phos-phorylated RasGAP, activated ERK induces phosphorylation of SOS and thedissociation from Grb2. Because of the recruitment of RasGAP and the tightnegative feedback by ERK, the activation dynamics of Ras may primarilydepend on the temporal rate, rather than the magnitude, of stimuli. On theother hand, a negative feedback mechanism for Rap1 activation has not been

234 NAGAHIRO MINATO ET AL .
defined to date, and there is little evidence advocating that Rap1GAPs arespecifically recruited following stimulation. Thus, Rap1 activation may continueas long as the stimuli persist potently enough to overcome the basal Rap1GAPactivity, and the activation dynamics of Rap1 may directly reflect the durationand degree of stimuli. Ligand‐occupied EGF receptors (EGFRs) are inter-nalized and rapidly degraded thereafter to likely induce only transient Rasactivation with limited Rap1 activation, that is, the transient ERK activation.In contrast, NGF receptors (TrkAs) occupied with ligands are trapped andprevail in the endosomal membrane to induce sustained ERK activation viaRap1 activation. Thus, Rap1 and Ras may mediate and yield different biologicaleffects due to their distinctly different kinetics in ERK activation.Another aspect of the Rap1 signal in ERK activation is its possible effect on
Ras‐mediated ERK activation. As mentioned earlier, Rap1GTP may not con-tribute to ERK activation via ubiquitous c‐Raf‐1 but may rather competitivelyinterfere with Ras‐mediated ERK activation at the level of c‐Raf‐1 whenoverexpressed (Boussiotis et al., 1997). While ambiguity of such an effectoccurring under normal physiological conditions remains unresolved, recentreports have indicated its involvement under certain in vivo conditions (seebelow). In short, the Rap1 signal, depending on the cell contexts, may regulateERK activation in different ways.Recent evidence has demonstrated that the Rap1 signal also activates the
MEK‐3, 6–p38MAPK pathway. Rap1 is reportedly activated by cell‐stretchforce to induce MEK‐3, 6‐mediated p38MAPK activation, while Ras‐signalingis inactivated (Sawada et al., 2001; Tamada et al., 2004). On the contrary, cellcontraction force activates theRas‐mediatedMEK‐1, 2–ERKpathwayswith littleRap1 activation (Sawada et al., 2001). Similar complementary activations ofRap1‐p38MAPK and Ras–ERK pathways have also been reported in other systems.In hippocampal neurons, for instance, Rap‐1‐mediated p38MAPK activationinduces nonmetabolic glutamate receptor‐mediated removal of synaptic AMPA(alpha‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid) receptors duringlong‐term depression (Zhu et al., 2002). On the other hand, delivery of AMPAreceptors to the synaptic sites during long‐term potentiation is dependent onthe Ras‐mediated ERK activation. Under such circumstances, it appears that theRap1–MKK3, 6–p38MAPK and Ras–MKK1, 2–ERK pathways form parallel butopposing signaling modules.Since Rap1 shares the effector domain with Ras, Rap1GTP is expected
to bind other Ras effectors such as RalGDS and PI3Kp110 subunit (Boset al., 2001). The PI3K–AKT pathway is activated by receptor PTKs in aRas‐dependent or Ras‐independent manner (Shaw and Cantley, 2006). Forinstance, the regulatory effect of cAMP on cell proliferation is in part mediatedby the control of Rap1‐mediated activation of the PI3K–AKT pathway

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 235
(Tsygankova et al., 2001; Wang et al., 2001). It has been shown that the prolifer-ation and survival of hematopoietic cells induced by IL‐3 or B‐cell receptors(BCR)‐ABL oncoprotein are influenced by Rap1‐mediated activation of thePI3K–AKT pathway (Jin et al., 2006).
2.2.2. Control of Cell Adhesion
SPA‐1 overexpression (abrogating the endogenous Rap1 activation) inducedrounding and eventual detachment of inherently adherent cells from extra-cellular matrix, while overexpression of membrane‐targeted C3G (C3G‐F)enhanced cell‐spreading on extracellular matrix (Tsukamoto et al., 1999). Thiswas one of the first indications to show involvement of the Rap1 signal inregulating cell adhesion. Since then, many reports have confirmed that theRap1 signal regulates the integrin‐mediated cell adhesion induced by variousstimuli in many cell types; viz., b1 (VLA‐4) and b2 (LFA‐1) integrin activationsof lymphocytes by CD31 (Reedquist et al., 2000) and CD98 (Suga et al., 2001)stimulation; b2 (Mac‐1) integrin activation of macrophage for phagocytosis byLPS (Schmidt et al., 2001); LFA‐1 activation of lymphocytes by chemokinestimulation (Shimonaka et al., 2003); and aIIb b3 integrin activation of plateletsby thrombin or ADP (Crittenden et al., 2004). Normal resting T cells initiallyexpress a low affinity or an ‘‘inactive’’ form of LFA‐1 before conversion to the highaffinity or ‘‘active’’ form by stimulation with antigens via a process called ‘‘inside‐out’’ activation (Carman and Springer, 2003). Expressing an active form of Rap1in Tcells rapidly increases the affinity of LFA‐1, while overexpression of SPA‐1 ora dominant‐negative Rap1 mutant completely inhibits LFA‐1 activation byantigen‐receptor stimulation (Katagiri et al., 2000). These findings clearly indicatethat Rap1 is a major mediator of ‘‘inside‐out’’ activation of integrins in Tcells (seebelow). These results have clarified that Rap1 serves as amajor integrin‐activatingsignal, unveiling a novel and important functional aspect of Rap1.
The overall cell adhesiveness induced by integrins is controlled by distinctelements such as the ‘‘affinity’’ of each monomeric integrin molecule, ‘‘valency’’defined by the diffusivity or local density of integrins and ligands, and ‘‘adhesionstrengthening’’ induced following integrin interaction with ligands (Carman andSpringer, 2003). Crystal structure analyses have revealed that affinity regulationof integrins is based on their conformational changes (Xiong et al., 2001). Thus,the extracellular stork of a and b chains in a low‐affinity state is sharply bent sothat the ligand‐binding head is juxtaposed to the membrane portion of thestork (closed posture), while the binding site is free from constraints andunfurls openly (open posture) in a high‐affinity state. The conformationalchange is regulated by proteins (such as talin), which bind to the cytoplasmicdomain of integrins (Carman and Springer, 2003). In lymphoid cells, RapL (aspecific effector of Rap1) associates directly with the cytoplasmic tail of LFA‐1

236 NAGAHIRO MINATO ET AL .
a‐chain on binding to Rap1GTP to likely induce an open posture of LFA‐1(Katagiri et al., 2003). In addition, a study has indicated that activation ofintegrins by the Rap1 signal also induces redistribution and polarity of integrinexpression (Shimonaka et al., 2003). Thus, the Rap1 signal may affect theoverall integrin‐mediated cell adhesion through multiple mechanisms.In this aspect, Rap1GTP has been found to bind also with a new adaptor called
the Rap1‐interacting adaptor molecule (RIAM), which displays high homologyto lamellipodin (Lpd; Lafuente et al., 2004). Knockdown of RIAM displacesRap1GTP from the plasma membrane and reverts integrin‐mediated cell adhe-sion induced by Rap1, while RIAM overexpression enhances integrin‐dependentcell adhesion and facilitates cell spreading. Interestingly, RIAM interacts directlywith profillin and Ena/VASP family proteins to maintain the cellular content ofF‐actin (Lafuente et al., 2004). Profillin and Ena/VASP are important regulatorsof the actin cytoskeleton, and thus Rap1 not only induces integrin activation butalso may directly regulate actin dynamics required for cell spreading and migra-tion, viz., lamellipodia formation (Bailly, 2004). These results may place Rap1 in acrucial position linking cell‐signaling and actin‐cytoskeleton changes.Cumulative evidence has further advocated that Rap1 plays an important
role in the maintenance of integrity of intercellular adhesion in epithelial andendothelial cells. Convincing data on the role of the Rap1 signal in controllingadherence junctions have been derived again from a genetic study ofDrosophila(Knox and Brown, 2002). During development of the wing, where mediation byeven cell adhesion among adjacent cells via the circumferentially distributedDE‐cadherin is involved, expanding epithelial cells of the related lineages normallystay in a coherent group. The epithelial cells withmutant Rap1, however, lose thecircumferential expression of DE‐cadherin and selectively form an adherencejunction to the adjacent cells ipsilaterally, resulting in disrupted epithelial cellbehavior (Knox and Brown, 2002). This may, in part, explain the abnormalmorphogenesis in Rap1‐mutant Drosophila (Hariharan et al., 1991). The rolesof Rap1 in the formation and maintenance of E‐cadherin‐mediated adherencejunctions in epithelial cells and VE‐cadherin‐mediated endothelial barrier func-tion have been reported also in mammalian cells. For instance, the Rap1 signalplays an important role in protecting the endothelial cell barrier function againstfactors (e.g., thrombin and so on) that cause barrier dysfunctions (Cullere et al.,2005; Fukuhara et al., 2005). In epithelial cells, Rap1 regulates the endocytosis ofE‐cadherin and controls the accumulation and distribution of E‐cadherin on cellsurfaces by specific binding to a scaffold protein afadin (or AF‐6) (Hoshino et al.,2005). Interestingly, AF‐6 also binds SPA‐1 and regulates Rap1‐GAP activity atthe adherence junction (Su et al., 2003). A component of tight junction (TJ),JAM1, has been found to constitutively deliver the Rap1 signal on intercellularepithelial cell adhesion, and disruption of the Rap1 signaling results in marked

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 237
changes in epithelial cell morphology and impairment of b1‐integrin‐mediatedadhesion to extracellular matrix (Mandell et al., 2005). These results imply thatthe Rap1 signal plays an important role in the functional cross talk betweenintercellular adhesion (adherence junction) and the adhesion to extracellularmatrix in epithelial cells.
3. Rap1 Signal in Lymphocyte Development and Immune Responses
Small G‐proteins of the Ras, Rho, and Rac families play crucial roles in variousaspects of lymphocyte development and function (Cantrell, 2003). Recentstudies, including those using gene‐engineered mice, have begun to unveilunique roles of the Rap1 signal in lymphocyte development and immuneresponses (Table 1).
3.1. Thymic T Cell Development: Distinct Roles of Rap1 and Ras
Using transgenic mice expressing the RapV12 mutant gene under a humanCD2 promoter, it was shown that excessive Rap1 signals barely affected overallthymic T cell development (Sebzda et al., 2002). However, we have recentlyfound that mice conditionally expressing the RapE63 (another dominant activemutant of Rap1) transgene, driven by a more potent CAG promoter, exhibitsignificant decreases in double‐positive (DP) thymocytes (Kometani et al.,manuscript in preparation). The discrepancy may reflect the fact that V12mutation of Rap1 may not be an ideal dominant active form unlike in othersmall G‐proteins such as Ras, Rho, and Rac (Daumke et al., 2004). Theexpansion and subsequent positive selection of DP thymocytes is reportedlydependent on the Ras signal (Swan et al., 1995). Thus, it appears that Rap1activation surpassing a certain level interferes with Ras‐dependent expansionor positive selection of DP thymocytes, although the physiological significanceremains to be verified.
Amore important question of whether the endogenous Rap1 signal is requiredfor the normal thymic Tcell development warrants attention. To shed light on thisambiguity, we innovated an experimental model—the SPA‐1 transgenic mice—for further investigation. Since the mice expressing SPA‐1 transgene driven by aubiquitous CAG promoter were embryonically lethal, we generated LoxP‐franked SPA‐1 transgenic mice and then crossed them with lck‐Cre transgenicmice. The conditional transgenic mice revealed severe thymic hypoplasia inwhich thymic T cell development was arrested at the late double‐negative (DN)stage (Kometani et al., manuscript in preparation). Consistently, in fetal thymicorgan cultures (FTOC), generation of DP thymocytes from the pro‐T cells ofRag2�/� mice in the presence of anti‐CD3 antibody was suppressed by the

Table 1 Phenotypes of Gene‐Engineered Mice Related to the Rap1 Signal
Mice Phenotypes References
C3G KO Embryonic lethal (at E5) Ohba et al., 2001C3Ggt/gt mutanta Embryonic lethal (at E15) Voss et al., 2003
Vascular defectIncreased cerebral neural cells Voss et al., 2006
SPA‐1 KO Myeloproliferative disordersof late onset
Ishida et al., 2003a
Memory T cell anergy Ishida et al., 2003bLupus‐like autoimmune
disease and B1 cell leukemiaIshida et al., 2006
Diabetes insipidus Noda et al., 2004CalDAG‐GEF1 KO Impaired platelet aggregation
and adhesionCrittenden et al., 2004
Bleeding diathesisRap1A KO Reduced adhesiveness of
T and B cells(probably redundant dueto intact Rap1b)
Duchniewicz et al., 2006
RapV12 Tg (hCD2) Enhanced T cell adhesion Sebzda et al., 2002RapE63 Tg (hCD2) Reduced antibody response
to TD‐antigensLi et al., 2005b
RapGA1 Tg (hCD2) Compromised CTLA‐4‐mediatedsuppression of T cell activation
Dillon et al., 2005
SPA‐1 Tg (CAG) Embryonic lethal UnpublishedSPA‐1 Tg (LoxP/lck‐Cre)b Severe thymic hypoplasia
(impaired b‐selection)Unpublished
aMice with mutant C3Ggt allele in which pGT1.8geo has been integrated in the first intron ofC3G gene, producing less than 5% normal C3G protein.
bTransgenic mice of SPA‐1 gene franked by LoxP under a CAG promoter were crossed withlck‐Cre transgenic mice.KO, knockout; Tg, transgenic; hCD2, human CD2 promoter; CAG, CMVearly enhancer‐chicken
b‐actin hybrid promoter.
238 NAGAHIRO MINATO ET AL .
retroviral transduction with SPA‐1 or RapA17 (a potent dominant‐negativemutant of Rap1; Dupuy et al., 2005). These results strongly suggested that theRap1 signal was essential for transition from pre‐T cells to abTCR‐expressingDP‐Tcells, that is, the b‐selection.While the Ras signal does not reportedly playa major role in b‐selection (Swan et al., 1995), a report has indicated that ERKactivation is crucially involved in the process (Crompton et al., 1996; Fischeret al., 2005). Therefore, Rap1 may serve as a major signal that mediates ERKactivation required for the thymic pre‐T cell proliferation and differentiation(Fig. 2).
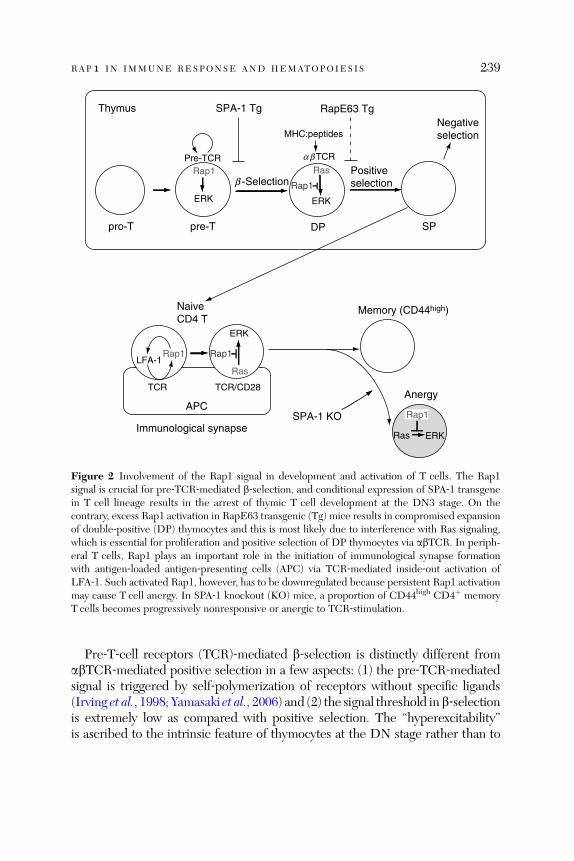
Rap1Pre-TCR
ERK
abTCR
MHC:peptides
Rap1
Ras
ERK
pro-T pre-T DP SP
b -Selection
SPA-1 Tg
Positiveselection
RapE63 TgNegativeselection
NaiveCD4 T
Rap1
Ras
ERK
APC
LFA-1Rap1
Memory (CD44high)
Anergy
Rap1
Ras ERK
SPA-1 KO
Thymus
Immunological synapse
TCR TCR/CD28
Figure 2 Involvement of the Rap1 signal in development and activation of T cells. The Rap1signal is crucial for pre‐TCR‐mediated b‐selection, and conditional expression of SPA‐1 transgenein T cell lineage results in the arrest of thymic T cell development at the DN3 stage. On thecontrary, excess Rap1 activation in RapE63 transgenic (Tg) mice results in compromised expansionof double‐positive (DP) thymocytes and this is most likely due to interference with Ras signaling,which is essential for proliferation and positive selection of DP thymocytes via abTCR. In periph-eral T cells, Rap1 plays an important role in the initiation of immunological synapse formationwith antigen‐loaded antigen‐presenting cells (APC) via TCR‐mediated inside‐out activation ofLFA‐1. Such activated Rap1, however, has to be downregulated because persistent Rap1 activationmay cause T cell anergy. In SPA‐1 knockout (KO) mice, a proportion of CD44high CD4þ memoryT cells becomes progressively nonresponsive or anergic to TCR‐stimulation.
RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 239
Pre‐T‐cell receptors (TCR)‐mediated b‐selection is distinctly different fromabTCR‐mediated positive selection in a few aspects: (1) the pre‐TCR‐mediatedsignal is triggered by self‐polymerization of receptors without specific ligands(Irving et al., 1998; Yamasaki et al., 2006) and (2) the signal threshold inb‐selectionis extremely low as compared with positive selection. The ‘‘hyperexcitability’’is ascribed to the intrinsic feature of thymocytes at the DN stage rather than to

240 NAGAHIRO MINATO ET AL .
that of the pre‐TCR (Erman et al., 2004; Haks et al., 2003). It may be possible thatthe higher excitability of pre‐T cells than DP‐T cells partly reflects the feature ofRap1‐mediated (as opposed to Ras‐mediated) ERK activation in the former(Section 1.2.1). Expression profiles of c‐Raf‐1 and B‐Raf at different developmen-tal stages may warrant further investigation. All in all, Rap1 plays significant rolesin the thymic abTcell development. In contrast, development of gdT‐lineage cellswas completely normal in the SPA‐1/lck‐Cre transgenic mice, suggesting that theRap1 signal displays a nonessential role in thymic gdT cell development if any.
3.2. Immunological Synapse and T Cell Activation
Interaction of T cells with antigen‐presenting cells (APCs) loaded by specificantigens results in the formation of three‐dimensional molecular clusters at thecontact site, that is, supramolecular activation clusters (SMAC). In SMAC,while TCR and costimulatory molecules are clustered in the central region(cSMAC), LFA‐1 is located at the periphery (pSAMC), and CD45 is excludedfrom the clusters (dSMAC; Huppa and Davis, 2003). Being maintained bycontinuous TCR signaling, this synaptic structure is quite stable and may lastmore than 10 h (Huppa and Davis, 2003). Although the exact function of sucha stable synaptic structure remains controversial, it may serve as a platform formolding architectural complexities to: (1) attain the cumulative TCR‐signalingeffect for full development of the T cell effector function and (2) accommodateregulatory mechanisms for guiding T cell activities. A hallmark of synapseformation is the ring‐cluster formation of LFA‐1 with ICAM‐1 on the APCaround TCR clusters, a process initiated by the initial contact of TCR with thespecific peptide‐loaded MHC (Huppa and Davis, 2003).It has been indicated that the Rap1 signal delivered by TCR ligation plays a
crucial role in clustering, reorganization, and activation of LFA‐1 for synapseformation. Thus, overexpression of SPA‐1 or RapN17 (another dominant‐negative mutant of Rap1) in a T cell clone strongly suppresses TCR‐mediatedLFA‐1 activation and synaptic conjugation with specific antigen‐loaded APCs,whereas that of Rap1 enhances the conjugate formation (Katagiri et al., 2002).These effects were not observed in the absence of relevant antigens, indicatingthat TCR‐mediated activation of endogenous Rap1 was crucial for clusteringand activation of LFA‐1 at the contact sites (Dustin et al., 2004; Fig. 2).A report has demonstrated that Rap1GTP is almost exclusively detected at theplasma membrane of T cells in a manner dependent on endosomal recycling,while Rap1GDP is mostly associated with the cytoplasmic vesicular membrane(Bivona et al., 2004). Interestingly, in contrast to most of the TCR‐proximalsignaling molecules, SLP‐76 rapidly dissociates from the TCR‐complex andmoves to the cytoplasm on APC interaction (Dustin et al., 2004). Thus, an

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 241
intriguing model may be proposed such that SLP‐76 associates with recyclingendosomes containing Rap1GDP, LFA‐1, and RapL to subsequently exposethem to the plasma membrane, where Rap1 is rapidly activated to Rap1GTP bymembrane‐recruited GEFs (e.g., C3G, CalDAG‐GEF I, and so on). Rap1GTPthen causes conformational activation of LFA‐1 by inducing RapL binding tothe cytoplasmic portion of a chain of LFA‐1 (Section 1.1), thus leading to tightinteraction with APCs via ICAM‐1 binding.
3.3. T Cell Nonresponsiveness and Anergy
Full activation of T cells requires additional engagement of costimulatoryreceptors such as CD28 with CD80/CD86 ligands on the professional APC(Sharpe and Freeman, 2002). Although Ras is strongly activated by the concom-itant stimulation of TCR/CD3 and CD28 receptors to induce ERK activation, itis poorly activated by TCR/CD3‐stimulation alone (Carey et al., 2000).In contrast to Ras, Rap1 is potently activated by TCR/CD3‐stimulation alone,while concomitant costimulation with anti‐CD28 markedly reduces Rap1activation (Carey et al., 2000; Reedquist and Bos, 1998). The results implythat Rap1 activation may have to be downregulated after the initiation ofsynapse formation for optimal T cell activation. In fact, persistent activation ofRap1 in T cells results in marked decreases of IL‐2 production on interactionwith antigen‐loaded APCs, albeit enhanced conjugate formation may occur(Katagiri et al., 2002). The reduced IL‐2 response is associated with compro-mised ERK activation, and this is consistent with the concept that persistentRap1 activation interferes with Ras‐mediated ERK activation downstream ofTCR (Boussiotis et al., 1997; Ishida et al., 2003b). It has further been confirmedthat T cells harboring RapE63 transgene show significantly reduced cell prolif-eration and IL‐2 production via TCR‐stimulation in vitro, and the transgenicmice exhibit compromised antibody responses to TD antigens, but not to TIantigens, in vivo (Li et al., 2005b). We have observed that SPA‐1 in T cells isspecifically recruited to synaptic sites with antigen‐loaded APCs (Harazaki et al.,2004), and thus SPA‐1most likely plays a role in restraining Rap1 activation afterestablishing efficient conjugations with APCs to yield optimal T cell activation.
CTLA‐4 has a higher affinity for CD80/86 than CD28 and exerts a negativesignal for Tcell activation, hence playing an important role in terminating Tcellresponses (Greenwald et al., 2005). Although CTLA‐4 expression is enhanced atthe late stages following T cell activation, significant CTLA‐4 expression isobserved in naive T cells (Schneider et al., 2005). A study has reported thatCTLA‐4 stimulation on naive T cells results in strong Rap1 activation to inducepotent activation of LFA‐1‐mediated cell adhesion (Schneider et al., 2005).Thus, CTLA‐4 may also contribute to synapse formation of T cells with APCs.

242 NAGAHIRO MINATO ET AL .
In contrast to CD28, however, concomitant stimulations of T cells with anti‐CD3 and anti‐CTLA‐4 antibodies potently inhibit Tcell activation by recruitingprotein tyrosine phosphatases via ITIM motif in the cytoplasmic domain(Greenwald et al., 2001). A study has demonstrated that the T cells fromRapGAP transgenic mice, which show compromised Rap1 activation, displaysignificant ERK activation by CD3/CD28/CTLA‐4 coligation, while ERK acti-vation is strongly suppressed in control T cells by the same stimuli (Dillon et al.,2005). Accordingly, T cells from the transgenic mice showed significantly lowerinhibition of IL‐2 production than control T cells by CTLA‐4 coligation,although both exhibited comparable inhibition of PLC‐g1 phosphorylation(Dillon et al., 2005). The results clearly suggest that part of the negative effectson T cell activation by CTLA‐4 ligation, especially on ERK activation and IL‐2production, is mediated by Rap1 activation.T cell anergy is a unique state, where T cells are incapable of producing IL‐2
and expanding clonally in response to antigens and is thus considered to playa role in peripheral T cell tolerance to self‐antigens (Schwartz, 1997). T cellanergy has originally been described as an in vitro phenomenon, where TCRoccupancy in the absence of costimulatory signals renders the cell nonrespon-sive, even to properly presented antigens with costimulatory signals. Theanergic state can be reversed at least partially by the addition of exogenousIL‐2, thus indicating that a major defect is in the TCR‐mediated IL‐2 produc-tion (Schwartz, 1997). Intensive analyses of anergic T cells have revealed twodominant biochemical features distinctly different from those of normalT cells. First, TCR‐mediated activation of the Ras–ERK pathway is severelyimpaired in anergic T cells, resulting in defective generation of the AP‐1complex, while other pathways (e.g., PLC‐g1 activation and so on) remainlargely intact. It is controversial whether Ras activation bypassing the TCRsignal (e.g., PMA) would restore ERK activation (Schwartz, 1997). Second,there is evidence that IL‐2 gene transcription is strongly repressed by cis‐acting elements in anergic T cells, although the exact nature of repressionremains to be elucidated.Boussiotis et al. (1997) have first reported that in vitro anergized Tcells reveal
constitutive activation of Rap1, which in fact is responsible for the defectiveRas activation and IL‐2 gene activation on CD3 and CD28 stimulations. Theyhave further suggested that constitutive phosphorylation of Cbl by Fyn and itsassociation with CrkL‐C3G may be involved in Rap1 activation of anergic T cells(Boussiotis et al., 1997). However, it has been documented that Cbl, whichpossesses an E3 ubiquitin ligase activity (Joazeiro et al., 1999), elicits ubiquitinmodification of CrkL and negatively regulates C3G recruitment and Rap1 activa-tion (Shao et al., 2003). Thus, the involvement of Cbl in constitutive Rap1activation in anergic T cells remains to be verified. Nonetheless, constitutive

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 243
Rap1 activation has also been observed in T cell populations in vivo with aner-gic features such asCD4þCD25þ andCD4þCD103þ regulatoryTcells (Li et al.,2005a,b). Although normal T cells barely express B‐Raf, T cells ectopicallyexpressing B‐Raf apparently escape anergy induction in vitro by APC stimulationof low B7 expression in association with potent Rap1 and ERK activation(Dillon et al., 2005). In addition, CTLA‐4‐deficient T cells have been shown toresist anergy induction in vivo as well (Greenwald et al., 2001). Collectively,the sustained Rap1 signal downstream of TCR stimulation plays a role in in-ducing and maintaining an anergic state in T cells, which do not express B‐Raf.Downstream effects of Rap1 in maintaining the anergic state warrant furtherinvestigations.
We have previously reported that the T cells with CD44high memory pheno-type in aged SPA‐1�/� mice selectively exhibit constitutive accumulation ofRap1GTP in vivo and manifest markedly compromised proliferation and IL‐2production via TCR stimulation (Ishida et al., 2003b; Fig. 2). We have recentlyfound that the anergic CD44high CD4þ T cells are in fact accumulated innormal mice as well with aging, albeit the extent is less than that observed inSPA‐1�/� mice (our unpublished observation). The hyporesponsiveness ofCD44high SPA‐1�/� T cells is probably attributed to impaired ERK activationby TCR‐stimulation despite the induction of normal Ras activation (Ishidaet al., 2003b). The results reinforce that downregulation of the Rap1 signal bySPA‐1 following antigen stimulation may be critical in preventing an anergicstate from occurring in primed T cells. By bypassing TCR stimulation withPMA and Ca2þ ionophore, such anergic T cells still show compromised prolif-eration and IL‐2 production, and thus, Rap1‐mediated interference with theRas–ERK pathway alone may not be able to fully account for the anergic state.A report has suggested that the specifically expressed Tob (an antiproliferativeprotein familymember gene) in anergic Tcells may be responsible for IL‐2 generepression by acting as a cofactor of Smad2/4 (Tzachanis et al., 2001). Therefore,the involvement of persistent Rap1 activation in the constitutive repression ofIL‐2 gene warrants further studies.
Importantly, the anergic Tcells in SPA‐1�/� and normal agedmice are confinedto a specific subset of CD44high T cells, and the proportions are drasticallyincreased in SPA‐1�/� mice that have developed frank leukemia (our unpub-lished observation). Studies have demonstrated that tumor‐specific T cells areanergized in hosts bearing experimental tumors with potent immunogenicity,albeit they may be efficiently generated (Willimsky and Blankenstein, 2005).Understanding the mechanisms of T cell anergy in tumor‐bearing hosts may becrucial for controlling malignancy, and SPA‐1�/� mice should provide a reli-able model to investigate the interaction between the immune system and thenaturally occurring tumor factors in vivo.

244 NAGAHIRO MINATO ET AL .
3.4. B Cell Development and Self‐Tolerance
Rap1 is activated by BCR stimulation in B cells as well (McLeod et al., 1998).While it has been reported that BCR‐induced Rap1 activation inhibits PI3K‐dependent AKT activation without affecting ERK activation in a B cell line(Christian et al., 2003), the physiological role of the effect remains unknown.A unique role of the Rap1 signal in development and function of B‐lineage cellsin vivo has again been uncovered using SPA‐1 knockout (KO) mice. SPA‐1 KOmice show preferential increases in peritoneal B1a cells (CD5þ Mac‐1þ B220þ
IgMhigh) with aging, accompanied by development of antinuclear antibodiessuch as anti‐dsDNA antibody (Ishida et al., 2006). While autoantibodies areof the IgM class in young SPA‐1 KO mice, significant IgG and IgA autoanti-bodies develop in elder mice to eventuate characteristic lupus‐like immunecomplex glomerulonephritis (Ishida et al., 2006). It has been documented thatB1a cells are responsible for the production of anti‐dsDNA antibodies. Perito-neal B1a cells of SPA‐1�/� mice displayed marked accumulation of Rap1GTPand were actively cycling, indicating that the cells were activated by constitu-tive self‐antigens in vivo. Rather surprisingly, however, the SPA‐1�/� peritonealB1a cells did not show enhanced proliferation via BCR stimulation, instead arather compromised response was manifested. Thus, unlike hitherto reportedmany mutant mice that developed lupus‐like autoimmune diseases, autoim-munity in SPA‐1�/� mice is not attributed to intrinsic BCR‐hyperreactivity ofB cells.B cells of SPA‐1�/� mice, however, revealed significantly altered BCR reper-
toire of the Vk genes as compared to those of control mice (Fig. 3). Studies haveindicated that unexpectedly high proportions of the newly emerged immatureB cells (>50%) in BM are autoreactive (Wardemann et al., 2003) and receptor‐editing plays a major role in negating the autoreactivity (Casellas et al., 2001).Receptor‐editing primarily involves Ig light (L)‐chain genes taking advantage ofthe fact that Vk/Jk gene rearrangements may occur repetitively unlike Ig heavychain genes because of the absence ofD gene segments.OcaB,which controls therecombination and expression of selected Vk genes as a transcriptional cofactor ofOct1,2, plays a crucial role in receptor editing (Casellas et al., 2001, 2002). It hasbeen revealed that the Rap1 signal induces transcriptional activation of OcaB viap38MAPK‐dependentCreb activation inB cells, and in fact immatureBMBcellsof SPA‐1�/� mice with excessively enhanced Rap1GTP levels exhibit augmentedOcaB gene expression (Ishida et al., 2006). The results suggest that the Rap1signal generated by the ligation of BCR with self‐antigens in self‐reactive imma-ture BMB cells may play a role in receptor editing via OcaB gene activation. Theexpected consequences of excessive Rap1 activation in SPA‐1�/� immatureB cells might be twofold: (1) it may cause skewing of the Vk gene usage toward

Pro-B
Pre-B
Immature B
Non-self-reactive
Transitional B Follicular B
Self-reactive Rap1
p38MAPKOcaB
Complete receptor editing
B1a cellsperitoneal cavitymarginal zone
IgL allelicinclusion
Genetic hit?
Pathogenicautoantibodies(Anti-dsDNA IgG)
B-CLL
IgkIgl
Apoptosis
Partialreceptor editing
Self-antigens
Self-antigens
Class switchaffinity maturation
B2
B1a
B1a
Hemolyticautoantibody
Figure 3 Rap1 signal may function as a ‘‘self‐sensing’’ signal in immature bone marrow B cells andcontrol the editing of self‐reactive BCR. Self‐reactive immature B cells in BM are tolerated byseveral distinct mechanisms including clonal deletion and receptor editing. On stimulation withself‐antigens, they may undergo repetitive rounds of Vk‐gene expression and rearrangement untilthe self‐reactivity of BCR is negated by a new Igk chain (editor Vk), viz., complete receptorediting. Rap1‐mediated p38MAPK‐dependent OcaB gene activation plays an important role in theexpression and rearrangement of selective Vk gene. With excess Rap1 activation in the absence ofSPA‐1, repetitive Vk‐gene rearrangement may proceed to Vl‐gene rearrangement, leading toallelic inclusion of Ig light chain genes. Such partial receptor editing may generate B cells withsignificantly reduced yet potential self‐reactivity. Such partially receptor‐edited B cells are deliv-ered preferentially to certain privileged sites, such as the peritoneal cavity, to become B1a (CD5þ
CD11aþ) cells. Since these B1a cells retain potential self‐reactivity, they may progress to producepathogenic autoantibodies (such as anti‐dsDNA IgG) following class switching and affinitymaturation once triggered in the periphery. Repetitive stimulations of the B1a cells by constitutiveself‐antigens may also predispose them to B1‐cell leukemia resembling human B cell chroniclymphocytic leukemia (B‐CLL) associated with autoantibody production.
RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 245
the Vk genes with higher OcaB dependency (such as the most frequently utilizedVk4 gene inmouse anti‐dsDNA antibodies; Liang et al., 2003), a finding which infact has been demonstrated in SPA‐1�/� mice (Ishida et al., 2006); and (2)excessive Rap1 signals may abnormally accelerate Vk gene recombination andexpression in SPA‐1�/� self‐reactive immature B cells. Normally, receptor editingis completed by rearrangement and expression of rare editor Vk genes to replace

246 NAGAHIRO MINATO ET AL .
the Vk genes involved in self‐reactive BCR. Excessive Rap1 signals and sustainedOcaB overexpression, however, may result in incessant Vk gene rearrangementswith ineffective editing (due to the preference for particular Vk genes), eventuallyleading to Vl gene rearrangement and expression. In fact, significant proportionsof the peritoneal B1 cells in the SPA‐1 KO mice revealed an allelic inclusion(Ishida et al., 2006), expressing both the Vk‐IgL and Vl‐IgL‐chains indicative of‘‘partial editing’’ (Fig. 3). The results suggest the important role of Rap1 inmediating the ‘‘self‐sensing’’ signal downstream of BCR in the newly derivedimmature BM B cells.The origin of B1 cells has long been a matter of argument. In mice, the
polyspecific B1 cells are supposed to have originated at the embryonic stageand subsequently segregated in the peritoneal cavity, where they can be self‐renewed (Hayakawa and Hardy, 1988). Such B1 cells play important roles ininnate immunity against bacterial infections by producing natural IgM anti-bodies broadly reactive to the various bacterial antigens (Coutinho et al.,1995). Although it has been known that pathogenic autoantibodies in systemicautoimmune diseases are also derived preferentially from B1 cells, their exactorigin remains largely unresolved. B cells expressing transgenic anti‐dsDNABCR, for instance, are segregated and distributed in marginal zones ratherthan in the follicles of spleen (Li et al., 2002). Previous report also suggestedthat VH gene usage might primarily determine the B1/B2 fates of the B celldevelopment using VH gene transgenic models (Lam and Rajewsky, 1999).Thus, the unique features of pathogenic autoreactive B cells being defined asB1 cells may be primarily attributed to BCR‐specificity per se rather than todistinct lineage. B1 cells in SPA‐1�/� mice exhibit remarkably high expressionlevels of b1‐integrin (unpublished results), and the Rap1 signal may as wellcontrol the unique distribution pattern of B1 cells having potentially pathogenicautoreactivities.Notably, a minor portion of SPA‐1�/� mice (ca. 10%) eventually developed
characteristic leukemia of CD5þ Mac‐1þ B220þ phenotypes corresponding toB1a cells with hemolytic autoantibodies (Ishida et al., 2006). Marked increasesin CD5þ B cells with high frequencies of autoimmunity (such as hemolyticanemia and autoimmune thrombocytopenia) are a hallmark of human B cellchronic lymphocytic leukemia (B‐CLL), and thus B cell leukemia in SPA‐1�/�
mice is highly reminiscent of human B‐CLL. Furthermore, leukemic B cells insome SPA‐1�/� mice revealed chromosomal translocation involving the Igl‐chain gene, t(2;6), and Igk‐chain gene, t(2;16) (Ishida et al., 2006). We there-fore propose that enhanced receptor editing and persistent stimulations byconstitutive self‐antigens of self‐reactive B1 cells may have predisposed themto the eventual leukemic transformation. Dysregulated Rap1 signals in theB‐lineage cells may provide a link between autoimmunity and B‐CLL (Fig. 3).

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 247
3.5. Lymphocyte Migration and Homing
Lymphocytes generated andmaturated in primary lymphoid organs continuouslyemigrate via the blood circulation to specific secondary lymphoid tissues.Homingof naive lymphocytes to particular areas in the secondary lymphoid tissues andmigration of primed or effector lymphocytes to local tissues where antigens existare crucial events in immunosurveillance. Attraction of lymphocytes by specificchemokines and transendothelial migration plays a major role in the lympho-cyte migration and homing to secondary lymphoid organs. The first step ofchemokine‐induced transendothelial migration is the rolling of lymphocytes viaselection followed by firm adhesion to endothelial cells against shear flow inresponse to specific chemokines (Butcher and Picker, 1996; Springer, 1995).The latter depends on integrin (LFA‐1, VLA‐4) activation and strong adhesionto their ligands (ICAM‐1, VCAM‐1) expressed on endothelial cells (Wittchenet al., 2005). Chemokine gradient induces the polarized accumulation of integrinsat the leading edge, while CD44 is mobilized to the uropod (Katagiri et al., 2003;Shimonaka et al., 2003). This polarity is vital for subsequent lymphocyte migra-tions across the endothelial cells (diapedesis). The direct interaction of LFA‐1with JAM‐1, another Ig superfamily (IgSF) protein located at the apical part of theendothelial adherence junction near the TJ, is also involved in diapedesis andmay‘‘unlock’’ the homotypic intercellular junction to guide the lymphocytes duringtransmigration (Ostermann et al., 2002).
Stimulation of the lymphocytes with specific chemokines (e.g., SLC andSDF‐1) causes rapid Rap1 activation, and all the events required for transen-dothelial migration (including adhesion, polarization, and diapedesis) arepotently inhibited by overexpression of SPA‐1 or a dominant‐negative Rap1mutant, indicating the essential role of Rap1 signals (Shimonaka et al., 2003).Recent reports have indicated that a Rap1 effector (RapL) plays a major role inlymphocyte migration (Section 1.2.2). RapL�/� mice show significant atro-phies of the secondary lymphoid organs associated with increased circulatinglymphocytes, indicating their impaired homing to lymphoid organs (Katagiriet al., 2004). Furthermore, RapL�/� mice have indicated reductions of matu-rated T cell migration from the thymus as well as impaired migration of theskin DCs to regional LNs by inflammatory stimuli (Katagiri et al., 2004). Theseresults thus clarify the essential roles of the Rap1 signal in constitutive andinflammation‐induced lymphocyte migrations and trafficking.
In a human‐inherited disease called leukocyte adhesion deficiency (LAD)syndrome, affected patients show persistent leukocytosis and life‐threateningbacterial infections due to defective leukocyte adhesion to blood vessels andtransmigration. Of the various subtype LAD patients, a majority (type‐I LAD)indicates germline mutations with impaired expression and function in the

248 NAGAHIRO MINATO ET AL .
b2‐integrin (CD18) gene (Anderson and Springer, 1987). A clinical encounterwith a rare autosomal recessive LAD syndrome (type‐III LAD), whereb2‐integrin expression and intrinsic adhesive activities of lymphocytes areapparently normal, has added an intriguing dimension to the complexity ofLAD syndrome (Alon and Etzioni, 2003). The lymphocytes from type‐III LADpatients display severely compromised Rap1‐mediated integrin activation inresponse to chemokines, although chemokine receptor signaling per se as wellas PMA‐induced Rap1 activation remains normal (Kinashi et al., 2004). Whilethe reasons for impaired Rap1 activation in response to chemokines and therelevant causative gene remain to be identified, the results strongly suggestthat functional defects in chemokine receptor‐coupled Rap1 activation areresponsible for type‐III LAD syndrome in humans.
4. Rap1 Signal in Hematopoiesis and Leukemia
SPA‐1 is most prominently expressed in the BM, in particular in the immaturehematopoietic cell population, implicating a requirement of tight control ofRap1 signal in them. Analysis of SPA‐1 KO mice disclosed unexpected yetimportant role of the Rap1 signal in regulating normal hematopoiesis (Table 1).
4.1. Hematopoietic Stem Cells and the Niche
Hematopoietic stem cells (HSCs) fulfill two opposing features: viz., per se self‐renewal without differentiation and the ability of differentiating to all lineagesof mature blood cells. Constitutive hematopoiesis depends on the homeostaticbalance of HSCs between self‐renewal and differentiation. Accumulatingevidence indicates that HSCs homeostasis is maintained by intimate interac-tions of HSCs with a specific hematopoietic microenvironment called theniche (Calvi et al., 2003; Whetton and Graham, 1999; Zhang et al., 2003).The role of Rap1 in maintaining stem cells in the niche has been wellillustrated by the male germ stem cells (GSCs) of Drosophila. In testes ofthe fruit fly, a cluster of 10–12 cells (or ‘‘hub’’) forms the niche for GSCs(Fuchs et al., 2004). When a GSC divides, one daughter cell remains anchoredto the hub cells, while the other drifts away from the hub and differentiates toform a gonialblast. Hub cells produce growth factors, such as Upd (unpaired)and Gbb (glass bottom boat), to regulate self‐renewal of GSCs, which havepreviously anchored to the hub through DE‐cadherin‐mediated cell adhesionto receive these signals (Yamashita et al., 2003). A genetic study has revealedthat the defect of Rap1 GEF (Gef26) or Rap1 causes the reduced formation ofadherens junctions at the hub–GSC interface, resulting in GSC loss due toexhaustive differentiation (Wang et al., 2006). Thus, the Rap1 signal plays amajor role in maintaining GSCs in the niche.

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 249
In mouse BM, spindle‐shaped N‐cadherinþ osteoblastic cells on the surfaceof cancellous or trabecular bone are the primary candidates of niche stroma forHSCs (Zhang et al., 2003). A previous study has demonstrated that c‐Myc‐deficient HSCs with enhanced expression of N‐cadherin and integrins aremarkedly accumulated in the niche with reduced differentiation. On thecontrary, c‐Myc overexpression accelerates the HSC release from the nicheand thereby promotes their differentiation to eventuate stem cell exhaustion(Wilson et al., 2004). Thus, the control of HSC adhesive molecules may play acrucial role in maintaining HSCs in a niche microenvironment to regulate thesignals for homeostatic balance between self‐renewal and differentiation ofHSCs (Fig. 4). SPA‐1�/� mice consistently display gradual increases of HSCcounts in BM with aging to eventually suffer overt myeloproliferative disorders(MPDs; see below). Furthermore, the diseased SPA‐1�/� mice have revealedmarked increases in the HSC population excessively expressing LFA‐1, result-ing in premature HSC mobilization out of BM to subsequently induce massiveextramedullary hematopoiesis (Kometani et al., 2006). These results indicatethat control of the Rap1 signal by SPA‐1 is crucially involved in the regulationof HSC interaction with the niche.
SDF‐1 produced by BM stroma cells is a major chemotactic factor involved inHSC migration and homing to a hematopoietic microenvironment (Nagasawaet al., 1996). The expression of SDF‐1 receptor (CXCR4) on human CD34þ
hematopoietic progenitors is reportedly enhanced by the cAMP‐induced Rap1-signal (Goichberg et al., 2006). TheRap1 signal also plays amajor role in SDF‐1‐mediated activation of b1‐integrins (Shimonaka et al., 2003), which is essentialfor migration and homing of HSCs to the hematopoietic microenvironment(Potocnik et al., 2000). We recently found that HSCs overexpressing SPA‐1 ordominant‐negative Rap17A mutant showed reduced engulfment when trans-planted into irradiated mice compared with control cells (our unpublisheddata). Therefore, Rap1 is most likely involved in HSC migration and homingto BM microenvironments or the niche, which is essential for the successfulhuman BM transplantation.
4.2. Dysregulated Rap1 Signal and Myeloproliferative Disorders
A vast majority of SPA‐1�/� mice eventually developed marked peripheral leuko-cytosis and massive splenomegaly with extensive extramedullary hematopoiesis intheir second year (Ishida et al., 2003a; Kometani et al., 2004; Fig. 4). Althoughwell‐differentiated granulocytes usually predominated in the blood of thesemice,CFU‐C assays have revealed increases in hematopoietic cells of all lineages, sug-gesting inductions of dysregulated expansion and differentiation of multipotenthematopoietic progenitors. A significant proportion of SPA‐1�/�mice additionally
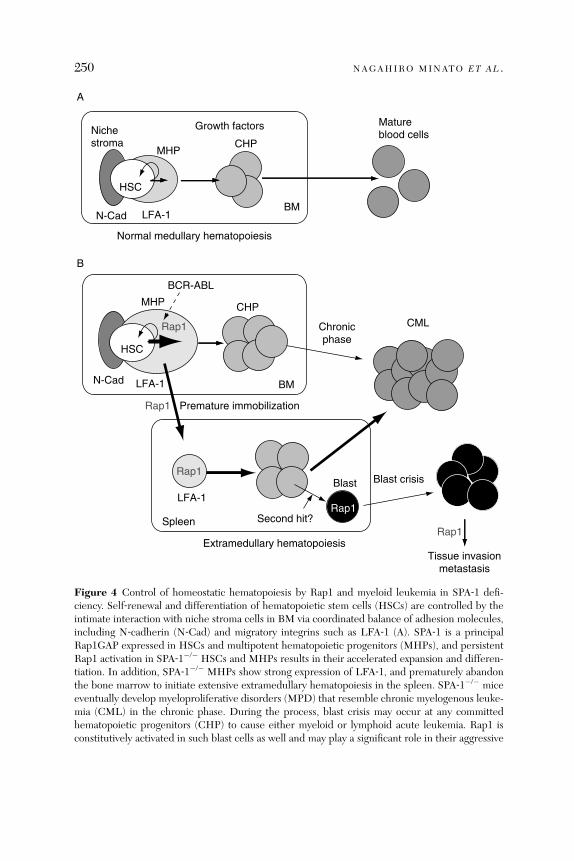
HSC
MHPCHP
Matureblood cells
HSC
MHP
Blast
CML
Blast crisis
Nichestroma
Growth factors
Premature immobilization
N-Cad LFA-1
Second hit?
Extramedullary hematopoiesisTissue invasion
metastasis
Normal medullary hematopoiesis
Rap1
Rap1
Rap1
Rap1
A
B
LFA-1N-Cad
LFA-1
CHP
BCR-ABL
Rap1
BM
BM
Spleen
Chronicphase
Figure 4 Control of homeostatic hematopoiesis by Rap1 and myeloid leukemia in SPA‐1 defi-ciency. Self‐renewal and differentiation of hematopoietic stem cells (HSCs) are controlled by theintimate interaction with niche stroma cells in BM via coordinated balance of adhesion molecules,including N‐cadherin (N‐Cad) and migratory integrins such as LFA‐1 (A). SPA‐1 is a principalRap1GAP expressed in HSCs and multipotent hematopoietic progenitors (MHPs), and persistentRap1 activation in SPA‐1�/� HSCs and MHPs results in their accelerated expansion and differen-tiation. In addition, SPA‐1�/� MHPs show strong expression of LFA‐1, and prematurely abandonthe bone marrow to initiate extensive extramedullary hematopoiesis in the spleen. SPA‐1�/� miceeventually develop myeloproliferative disorders (MPD) that resemble chronic myelogenous leuke-mia (CML) in the chronic phase. During the process, blast crisis may occur at any committedhematopoietic progenitors (CHP) to cause either myeloid or lymphoid acute leukemia. Rap1 isconstitutively activated in such blast cells as well and may play a significant role in their aggressive
250 NAGAHIRO MINATO ET AL .

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 251
accommodated variable extents of blastic cells of either myeloid or lymphoidlineages, which infiltrated in vital tissues (Ishida et al., 2003a). These featureshighly resemble human CML in the chronic phase and acute crisis. In addi-tion, a minor portion of mice developed severe anemia and pancytopenia oftenassociated with dysplastic or blastic leukocytes, and the phenotypes coincidedwell with the human myelodysplastic syndrome (MDS). The latent periods ofMPD were usually long (ca. 12 months), and secondary genetic events mightaffect the final disease phenotypes, especially in blast crisis. Despite theapparent diversity of MPD, diseased SPA‐1�/� mice share common features,including accumulation of Rap1GTP in the HSC‐enriched BM cell fraction,selective increase in HSC population with excessive LFA‐1 expression in BM,and marked HSC mobilization to the spleen (Kometani et al., 2006). Alto-gether, these findings strongly suggest that HSC disorders are the causativefactors underlying MPD (Fig. 4).
CML represents leukemia of HSCs, and leukemic stem cells generateincreasing numbers of various types of mature blood cells (Ren, 2005). Simila-rities and differences between normal and leukemic HSCs are the fundamen-tal issue in CML pathogenesis. Although it has been reported that enhancedself‐renewing capacity of HSCs in several mutant (such as Lnk�/�, c‐Myc�/�,and p18INK4C�/�) mice yields marked increases in the HSC population,these mice do not develop overt MPD (Takaki et al., 2002; Wilson et al.,2004; Yuan et al., 2004). On the other hand, a report has indicated thatconditional deletion of Pten gene in HSCs result in the development ofacute MPD (Yilmaz et al., 2006; Zhang et al., 2006a). Pten is a phosphatasethat converts PIP3 to PIP2 and negatively regulates the PI3K‐signalingpathway (Cully et al., 2006). Pten�/� mice indicate progressive reductions inself‐renewing HSCs due to their accelerated differentiation and peripheralmobilization, resulting first in massive extramedullary hematopoiesis rapidlyfollowed by blast crisis in association with frequent chromosomal transloca-tions (Yilmaz et al., 2006; Zhang et al., 2006a). The overall features of MPD inconditional Pten�/� mice resemble those of SPA‐1�/� mice, except for moreacute development in the former. Thus, the accelerated drive of HSCs indifferentiation and premature mobilization (with or without the reduced self‐renewing HSCs) seems to be the common features of CML‐like MPD, and itwould be of interest to investigate whether SPA‐1�/� and Pten�/� mice, inpart, share the dysregulation of signaling pathways in HSCs, particularly thePI3K–AKT pathway.
invasion into many vital tissues. In humans, the BCR‐ABL fusion gene from the Philadelphiachromosome is a major cause of CML and constitutive Rap1 activation downstream of BCR‐ABLoncoprotein may participate in molding phenotypes of human CML.

252 NAGAHIRO MINATO ET AL .
In humans, the vast majority of CML is caused by BCR‐ABL oncoproteingenerated by chromosomal translocation t(9;22). All lineages of maturatedperipheral leukocytes have an identical chromosomal anomaly, indicatingthat MPD is due to the leukemic transformation of an HSC clone (Wongand Witte, 2004). BCR‐ABL protein delivers a diverse array of signals viaconstitutive ABL tyrosine kinase activity, including the Ras–ERK, PI3K–AKT,and Stat5–Bcl2 pathways, as well as integrin activation (Jin et al., 2006;Sonoyama et al., 2002; Wong and Witte, 2004). Although all these signalsmay contribute to various aspects of leukemic features, the PI3K pathwayapparently plays a prominent role in terms of leukemogenesis in vivo becausethe BCR‐ABL mutant gene (lacking a domain responsible for PI3K activation)then loses the leukemogenic activity (Sattler et al., 2002). Evidence has indi-cated that Rap1 is activated constitutively by BCR‐ABL via recruitment andphosphorylation of C3G (Cho et al., 2005), or partial repression of SPA‐1 geneexpression (Kometani et al., 2006), or both. Most notably, SPA‐1 overexpressionhas significantly inhibited PI3K/AKT activation in BCR‐ABLþ cells (Jin et al.,2006). To directly examine the role of Rap1 signals in BCR‐ABL‐induced CML,we compared the leukemic phenotypes between the normal and SPA‐1�/�
progenitors transduced with BCR‐ABL oncogene in a mouse model. Thefindings demonstrated that, while both progenitors caused CML in the primaryrecipients, SPA‐1�/� leukemic progenitors persisted longer than the controlin vivo when judged by the serial transfer experiment (Kometani et al., 2006).In addition, significant proportions of the former showed blastic crisis, support-ing a role of the endogenous Rap1 signal in BCR‐ABL‐induced CML genesis inthe recipients. In some juvenile CML patients, loss of heterozygosity in NF1gene encoding a RasGAP has been observed (Shannon et al., 1994) and in factNF1þ/� mice have developed CML with a long latency of over a year (Jackset al., 1994). Due to enhanced activation of the Ras–ERK pathway, NF1�/�
cells consequently develop hyperresponsiveness to GM‐CSF (Bollag et al.,1996; Largaespada et al., 1996). In contrast, SPA‐1�/� CML cells show un-changed responsiveness to hematopoietic growth factors such as GM‐CSF(Ishida et al., 2003a), and thus deficiencies of SPA‐1 (Rap1GAP) and NF‐1(RasGAP) induce CML‐like via distinctly different mechanisms.Current literature advocates the dependence of CML‐genic potential of BCR‐
ABL on complex interactions with the intrinsic self‐renewing potential of HSCs(Huntly et al., 2004). This is of particular clinical significance because imatinibmesylate (Gleevec; a potent inhibitor of ABL kinase activity) that can rapidlyreduce the massive burden of leukemic leukocytes fails to eradicate the CMLstem cells, so‐called residual diseases (Michor et al., 2005). Under such circum-stances, most of the patients eventually develop recurrence of lethal aggressiveleukemia. SPA‐1 is among the gene set of the murine self‐renewal‐associated

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 253
signature and highly enriched in human leukemic stem cells as well (Krivtsovet al., 2006), and thus it seems likely that BCR‐ABL connects with the intrinsicRap1 signal in HSC, at least in part, to cause CML. The crucial target cells forcontrolling CML are the leukemic stem cells (Huntly and Gilliland, 2005), andRap1 may provide a rational molecular target for the eradication of CML inhumans.
4.3. Role of Rap1 in Generation and Function of Platelets
Among the various blood cells, the role of Rap1 signals in generation andfunction of platelets has been most extensively studied (Stork and Dillon,2005). Maturation of megakaryocytes depends on thrombopoietin acting onthe Mpl receptor to induce sustained ERK activation (Garcia et al., 2001).However, erythropoietin and GM‐CSF induce proliferation, but not matura-tion, of megakaryocytes, and such an action is associated with transient ERKactivation (Stork and Dillon, 2005). This represents another event where theRap1 and Ras signals mediate the distinct modes of ERK activation to inducedifferent effects (Section 1). BM stroma cells inhibit differentiation of mega-karyocyte progenitors by direct contact, an effect which has been elicited byinhibition of Rap1‐mediated persistent ERK activation (Delehanty et al.,2003). A specific type of b3‐integrin (aIIbb3), which is expressed selectivelyby platelets, plays essential roles in platelet function (e.g., aggregation andadhesion) related with homeostasis and thrombus formation. The abundantlyexpressed Rap1 in platelets is activated by many stimuli (e.g., turbulence,epinephrine, ADP, thrombin, thromboxane A2, and platelet‐activating factors)to cause platelet activation via aIIbb3 activation. CalDAG‐GEF I (RasGRP2)is responsible for Rap1 activation in platelets, and a report has revealeddefective aIIbb3‐mediated platelet aggregation in CalDAG‐GEF I�/� miceto induce markedly impaired haemostasis (Crittenden et al., 2004).
5. Rap1 Signal in Malignancy: New Aspects in Cancer
In spite of the highly convergent homology with classical Ras, there have beenlimited experimental findings implicating Rap1 to function as an oncoprotein.In 1998, however, Altschuler and Ribeiro‐Neto (1998) have revealed certainunique roles of the Rap1 signal in cell transformation; Rap1‐transfected Swiss3T3 fibroblasts are flatter and spread more with a higher saturation density thancontrol cells. Although cell growth could be strongly enhanced by cAMP or EGF,the Rap1‐transfected cells showed no anchorage‐independent growth, and thusthe cells revealed no evidence of transformation in vitro when viewed under aclassical criterion. Surprisingly, however, the cells formed tumors in nude mice

254 NAGAHIRO MINATO ET AL .
(Altschuler and Ribeiro‐Neto, 1998). This presents a very unusual phenomenon,where cells are anchorage‐dependent in vitro and yet tumorigenic in vivo. Theresults suggest that Rap1 caused tumors in vivo by constitutive interaction with,rather than bypassing, the intrinsic growth pathways of host cells. In principle, theevent may be coincidental with BCR‐ABL‐induced CML‐genesis, wherebyBCR‐ABL oncoprotein promotes the dysregulated expansion of HSCs by inter-acting with the intrinsic self‐renewal feature of HSCs (Huntly et al., 2004).According to a recent study, certain human squamous cell carcinoma cells showhigh levels of Rap1GTP. Rap1GAP transduction reportedly represses tumorigen-esis of such cancer cells in nude mice (Zhang et al., 2006b). These results suggestthat Rap1 may act as a ‘‘conditional’’ oncoprotein.Hitherto, the Rap1 signal has been documented to affect the invasiveness
and metastasis of tumors in vivo. Mutations of DOCK4 gene encoding a Rap1activator have been reported in certain human cancer cells (Yajnik et al., 2003).The mutant DOCK4 protein exhibits a dominant‐negative effect to incitedefective Rap1 activation in such cancer cells, resulting in the loss of intercel-lular adhesion among these abnormal cells. As a result, these cancers wouldmanifest highly invasive behavior in vivo. Intriguingly, introduction of a wild‐type DOCK4 gene restores the adherence junction among the cancer cells,and concomitantly represses the invasive tendency as the basal Rap1GTP isrestored (Yajnik et al., 2003). It has also been reported that the Rap1 signal incancer cells may play a critical role in metastasis. Employing the model ofspontaneous development of mammary tumors in transgenic mice of polyomamiddle T‐antigens under an MMTV promoter, Hunter and the colleagues haveindicated that genetic polymorphism of the SPA‐1 (also called SIPA‐1) gene inthe host is a major determinant for lung metastasis of primary mammarytumors (Park et al., 2005). Thus, mouse strains with SPA‐1/741A alleles displayextensive lung metastases, whereas far less lung metastases are observed inthose with SPA‐1/741T alleles. Interestingly, no significant differences ingrowth of the primary tumors are established between mice with a differentallele. The single amino acid polymorphism at position 741 in the PDZ domainof SPA‐1 protein affects Rap1GAP activity in cancer cells, with SPA‐1/741Abeing more active than SPA‐1/741T (Park et al., 2005). On the basis of thesefindings, the reduced Rap1 signal in cancer cells might favor metastasis inaddition to local invasiveness. In fact, these findings serve as the first directindication that the host genetic background can affect the metastatic behaviorof cancers (Threadgill, 2005). A report confirmed that certain SPA‐1 genehaplotypes are significantly associated with the presence of lymph nodemetastasi s and poo r prognosi s in huma n mamma ry cance rs (Crawford et al.,2006). Rec ently, we ha ve also confirme d that pro state cancer cells in primary

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 255
sites of patients with metastases exhibit expression of SPA‐1 protein signifi-cantly higher than those without metastasis (our unpublished observation). Inother words, the Rap1 signal in cancer cells may partially control malignantinvasiveness and remote metastasis by regulating intercellular adhesion amongthe cancer cells.
6. Conclusions and Perspectives
Extensive and intensive studies in recent years have unveiled unique biologicalactivities of Rap1. Twomajor activities of the Rap1 signal have been established:(1) control of cell–matrix and cell–cell adhesions via activation of integrins andother cell adhesion molecules and (2) regulation of the activation of variousMAPKs. Rap1 is activated by an extensive spectrum of extracellular stimuli viamany types of specific GEFs coupled with certain specific receptor systems, andthe activation status is tightly controlled by GAPs at different intracellularcompartments. Through such activities, Rap1 is involved in a range of diversecellular functions far more than originally anticipated. Unique and unanticipatedroles of the Rap1 signal in vivo have recently been uncovered by extensiveanalyses of gene‐targeted mice for Rap1 regulatory molecules. The Rap1 signalhas been demonstrated to play crucial roles in diverse aspects of the develop-ments and functions of immune and hematopoietic cells. Furthermore, Rap1dysregulation causes characteristically specific diseases, with highly resemblinghuman conditions. We anticipate that further analyses will reveal more as of yetundocumented important roles of the Rap1 signal in other biological systemssuch as the nervous and endocrine systems and malignant cells. While Rap1 isexpressed ubiquitously in most tissue cells in the body, predominant roles of theRap1 signal can be highly variable—depending on the contexts of specific celltypes and functions. This signaling molecule with multifaceted functional varia-bility provides a typical example, where a ubiquitous molecule may be cruciallyintegrated into the highly specified and sophisticated functions of manybiological events in a complex living system. Regulatory molecules of theRap1 signal may also serve as potentially reliable and rational molecular targetsfor controlling various human diseases including malignancy.
Acknowledgments
The authors are grateful to all personnel in the Department of Immunology and Cell Biology,Graduate School of Medicine and Graduate School of Biostudies, Kyoto University. In particular,we would like to thank Drs. D. Ishida, Li Su, Hailin Yang, Y. Hamazaki, Y. Shinozuka,M. Moriyama, M. Aoki, F. Wang, K. Shimatani, Y. Nakajima, and Y. Katayama for their kindcooperation in carrying out the study. This study was supported by Grants‐in‐Aid for ScientificResearch from the Ministry of Education, Science, Culture, Sport, and Technology of Japan.

256 NAGAHIRO MINATO ET AL .
References
Alon, R., and Etzioni, A. (2003). LAD‐III, a novel group of leukocyte integrin activation deficien-cies. Trends Immunol. 24, 561–566.
Altschuler, D. L., and Ribeiro‐Neto, F. (1998). Mitogenic and oncogenic properties of the smallG protein Rap1b. Proc. Natl. Acad. Sci. USA 95, 7475–7479.
Anderson, D. C., and Springer, T. A. (1987). Leukocyte adhesion deficiency: An inherited defect inthe Mac‐1, LFA‐1, and p150,95 glycoproteins. Annu. Rev. Med. 38, 175–194.
Asha, H., de Ruiter, N. D., Wang, M. G., and Hariharan, I. K. (1999). The Rap1 GTPase functionsas a regulator of morphogenesis in vivo. EMBO J. 18, 605–615.
Bailly, M. (2004). Ena/VASP family: New partners, bigger enigma. Dev. Cell 7, 462–463.Barnier, J. V., Papin, C., Eychene, A., Lecoq, O., and Calothy, G. (1995). The mouse B‐raf geneencodes multiple protein isoforms with tissue‐specific expression. J. Biol. Chem. 270,23381–23389.
Bender, A., and Pringle, J. R. (1989). Multicopy suppression of the cdc24 budding defect in yeastby CDC42 and three newly identified genes including the ras‐related gene RSR1. Proc. Natl.Acad. Sci. USA 86, 9976–9980.
Bivona, T. G., Wiener, H. H., Ahearn, I. M., Silletti, J., Chiu, V. K., and Philips, M. R. (2004). Rap1up‐regulation and activation on plasma membrane regulates T cell adhesion. J. Cell Biol. 164,461–470.
Bollag, G., Clapp, D. W., Shih, S., Adler, F., Zhang, Y. Y., Thompson, P., Lange, B. J., Freedman,M. H., McCormick, F., Jacks, T., and Shannon, K. (1996). Loss of NF1 results in activation of theRas signaling pathway and leads to aberrant growth in haematopoietic cells. Nat. Genet. 12,144–148.
Bos, J. L. (2003). Epac: A new cAMP target and new avenues in cAMP research. Nat. Rev. Mol.Cell Biol. 4, 733–738.
Bos, J. L., de Rooij, J., and Reedquist, K. A. (2001). Rap1 signalling: Adhering to new models. Nat.Rev. Mol. Cell Biol. 2, 369–377.
Boussiotis, V. A., Freeman, G. J., Berezovskaya, A., Barber, D. L., and Nadler, L. M. (1997).Maintenance of human T cell anergy: Blocking of IL‐2 gene transcription by activated Rap1.Science 278, 124–128.
Butcher, E. C., and Picker, L. J. (1996). Lymphocyte homing and homeostasis. Science 272, 60–66.Calvi, L. M., Adams, G. B., Weibrecht, K. W., Weber, J. M., Olson, D. P., Knight, M. C., Martin,R. P., Schipani, E., Divieti, P., Bringhurst, F. R., Milner, L. A., Kronenberg, H. M., et al. (2003).Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 425, 841–846.
Cantrell, D. A. (2003). GTPases and T cell activation. Immunol. Rev. 192, 122–130.Carey, K. D., Dillon, T. J., Schmitt, J. M., Baird, A. M., Holdorf, A. D., Straus, D. B., Shaw, A. S.,and Stork, P. J. (2000). CD28 and the tyrosine kinase lck stimulate mitogen‐activated proteinkinase activity in T cells via inhibition of the small G protein Rap1. Mol. Cell. Biol. 20,8409–8419.
Carman, C. V., and Springer, T. A. (2003). Integrin avidity regulation: Are changes in affinity andconformation underemphasized? Curr. Opin. Cell Biol. 15, 547–556.
Casellas, R., Shih, T. A., Kleinewietfeld, M., Rakonjac, J., Nemazee, D., Rajewsky, K., andNussenzweig, M. C. (2001). Contribution of receptor editing to the antibody repertoire. Science291, 1541–1544.
Casellas, R., Jankovic, M., Meyer, G., Gazumyan, A., Luo, Y., Roeder, R., and Nussenzweig, M.(2002). OcaB is required for normal transcription and V(D)J recombination of a subset ofimmunoglobulin kappa genes. Cell 110, 575–585.

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 257
Cho, Y. J., Hemmeryckx, B., Groffen, J., and Heisterkamp, N. (2005). Interaction of Bcr/Abl withC3G, an exchange factor for the small GTPase Rap1, through the adapter protein Crkl. Biochem.Biophys. Res. Commun. 333, 1276–1283.
Christian, S. L., Lee, R. L., McLeod, S. J., Burgess, A. E., Li, A. H., Dang‐Lawson, M., Lin, K. B.,and Gold, M. R. (2003). Activation of the Rap GTPases in B lymphocytes modulates B cellantigen receptor‐induced activation of Akt but has no effect on MAPK activation. J. Biol. Chem.278, 41756–41767.
Coutinho, A., Kazatchkine, M. D., and Avrameas, S. (1995). Natural autoantibodies. Curr. Opin.Immunol. 7, 812–818.
Crawford, N. P., Ziogas, A., Peel, D. J., Hess, J., Anton‐Culver, H., and Hunter, K. W. (2006).Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poorprognosis in breast cancer. Breast Cancer. Res. 8, R16.
Crittenden, J. R., Bergmeier, W., Zhang, Y., Piffath, C. L., Liang, Y., Wagner, D. D., Housman,D. E., and Graybiel, A. M. (2004). CalDAG‐GEFI integrates signaling for platelet aggregationand thrombus formation. Nat. Med. 10, 982–986.
Crompton, T., Gilmour, K. C., and Owen, M. J. (1996). The MAP kinase pathway controlsdifferentiation from double‐negative to double‐positive thymocyte. Cell 86, 243–251.
Cullere, X., Shaw, S. K., Andersson, L., Hirahashi, J., Luscinskas, F. W., and Mayadas, T. N. (2005).Regulation of vascular endothelial barrier function by Epac, a cAMP‐activated exchange factorfor Rap GTPase. Blood 105, 1950–1955.
Cully, M., You, H., Levine, A. J., and Mak, T. W. (2006). Beyond PTEN mutations: The PI3Kpathway as an integrator of multiple inputs during tumorigenesis. Nat. Rev. Cancer 6, 184–192.
Daumke, O., Weyand, M., Chakrabarti, P. P., Vetter, I. R., and Wittinghofer, A. (2004). TheGTPase‐activating protein Rap1GAP uses a catalytic asparagine. Nature 429, 197–201.
de Rooij, J., Zwartkruis, F. J., Verheijen, M. H., Cool, R. H., Nijman, S. M., Wittinghofer, A., andBos, J. L. (1998). Epac is a Rap1 guanine‐nucleotide‐exchange factor directly activated by cyclicAMP. Nature 396, 474–477.
Delehanty, L. L., Mogass, M., Gonias, S. L., Racke, F. K., Johnstone, B., and Goldfarb, A. N.(2003). Stromal inhibition of megakaryocytic differentiation is associated with blockade ofsustained Rap1 activation. Blood 101, 1744–1751.
Dillon, T. J., Carey, K. D., Wetzel, S. A., Parker, D. C., and Stork, P. J. (2005). Regulation of thesmall GTPase Rap1 and extracellular signal‐regulated kinases by the costimulatory moleculeCTLA‐4. Mol. Cell. Biol. 25, 4117–4128.
Duchniewicz, M., Zemojtel, T., Kolanczyk, M., Grossmann, S., Scheele, J. S., and Zwartkruis, F. J.(2006). Rap1A‐deficient T and B cells show impaired integrin‐mediated cell adhesion.Mol. Cell.Biol. 26, 643–653.
Dupuy, A. G., L’Hoste, S., Cherfils, J., Camonis, J., Gaudriault, G., and de Gunzburg, J. (2005).Novel Rap1 dominant‐negative mutants interfere selectively with C3G and Epac. Oncogene 24,4509–4520.
Dustin, M. L., Bivona, T. G., and Philips, M. R. (2004). Membranes as messengers in T celladhesion signaling. Nat. Immunol. 5, 363–372.
Erman, B., Guinter, T. I., and Singer, A. (2004). Defined alphabeta T cell receptors with distinctligand specificities do not require those ligands to signal double negative thymocyte differentia-tion. J. Exp. Med. 199, 1719–1724.
Farina, A., Hattori, M., Qin, J., Nakatani, Y., Minato, N., and Ozato, K. (2004). Bromodomainprotein Brd4 binds to GTPase‐activating SPA‐1, modulating its activity and subcellular localiza-tion. Mol. Cell. Biol. 24, 9059–9069.
Fischer, A. M., Katayama, C. D., Pages, G., Pouyssegur, J., and Hedrick, S. M. (2005). The role oferk1 and erk2 in multiple stages of T cell development. Immunity 23, 431–443.

258 NAGAHIRO MINATO ET AL .
Fuchs, E., Tumbar, T., and Guasch, G. (2004). Socializing with the neighbors: Stem cells and theirniche. Cell 116, 769–778.
Fukuhara, S., Sakurai, A., Sano, H., Yamagishi, A., Somekawa, S., Takakura, N., Saito, Y., Kangawa, K.,and Mochizuki, N. (2005). Cyclic AMP potentiates vascular endothelial cadherin‐mediated cell‐cell contact to enhance endothelial barrier function through an Epac‐Rap1 signaling pathway.Mol. Cell. Biol. 25, 136–146.
Gao, Q., Srinivasan, S., Boyer, S. N., Wazer, D. E., and Band, V. (1999). The E6 oncoproteins ofhigh‐risk papillomaviruses bind to a novel putative GAP protein, E6TP1, and target it fordegradation. Mol. Cell. Biol. 19, 733–744.
Gao, Q., Singh, L., Kumar, A., Srinivasan, S., Wazer, D. E., and Band, V. (2001). Humanpapillomavirus type 16 E6‐induced degradation of E6TP1 correlates with its ability to immor-talize human mammary epithelial cells. J. Virol. 75, 4459–4466.
Gao, Q., Kumar, A., Singh, L., Huibregtse, J. M., Beaudenon, S., Srinivasan, S., Wazer, D. E.,Band, H., and Band, V. (2002). Human papillomavirus E6‐induced degradation of E6TP1 ismediated by E6AP ubiquitin ligase. Cancer Res. 62, 3315–3321.
Garcia, J., de Gunzburg, J., Eychene, A., Gisselbrecht, S., and Porteu, F. (2001). Thrombopoietin‐mediated sustained activation of extracellular signal‐regulated kinase in UT7‐Mpl cells requiresboth Ras‐Raf‐1‐ and Rap1‐B‐Raf‐dependent pathways. Mol. Cell. Biol. 21, 2659–2670.
Goichberg, P., Kalinkovich, A., Borodovsky, N., Tesio, M., Petit, I., Nagler, A., Hardan, I., andLapidot, T. (2006). cAMP‐induced PKCzeta activation increases functional CXCR4 expressionon human CD34þ hematopoietic progenitors. Blood 107, 870–879.
Gotoh, T., Hattori, S., Nakamura, S., Kitayama, H., Noda, M., Takai, Y., Kaibuchi, K., Matsui, H.,Hatase, O., Takahashi, H., Kurata, T., and Matsuda, M. (1995). Identification of Rap1 as a targetfor the Crk SH3 domain‐binding guanine nucleotide‐releasing factor C3G. Mol. Cell. Biol. 15,6746–6753.
Greenwald, R. J., Boussiotis, V. A., Lorsbach, R. B., Abbas, A. K., and Sharpe, A. H. (2001). CTLA‐4regulates induction of anergy in vivo. Immunity 14, 145–155.
Greenwald, R. J., Freeman, G. J., and Sharpe, A. H. (2005). The B7 family revisited. Annu. Rev.Immunol. 23, 515–548.
Haks, M. C., Belkowski, S. M., Ciofani, M., Rhodes, M., Lefebvre, J. M., Trop, S., Hugo, P.,Zuniga‐Pflucker, J. C., and Wiest, D. L. (2003). Low activation threshold as a mechanism forligand‐independent signaling in pre‐T cells. J. Immunol. 170, 2853–2861.
Harazaki, M., Kawai, Y., Su, L., Hamazaki, Y., Nakahata, T., Minato, N., and Hattori, M. (2004).Specific recruitment of SPA‐1 to the immunological synapse: Involvement of actin‐bundlingprotein actinin. Immunol. Lett. 92, 221–226.
Hariharan, I. K., Carthew, R. W., and Rubin, G. M. (1991). The Drosophila roughened mutation:Activation of a rap homolog disrupts eye development and interferes with cell determination.Cell 67, 717–722.
Hattori, M., and Minato, N. (2003). Rap1 GTPase: Functions, regulation, and malignancy.J. Biochem. (Tokyo) 134, 479–484.
Hattori, M., Tsukamoto, N., Nur‐e‐Kamal, M. S., Rubinfeld, B., Iwai, K., Kubota, H., Maruta, H.,and Minato, N. (1995). Molecular cloning of a novel mitogen‐inducible nuclear protein with aRan GTPase‐activating domain that affects cell cycle progression. Mol. Cell. Biol. 15, 552–560.
Hayakawa, K., and Hardy, R. R. (1988). Normal, autoimmune, and malignant CD5 þ B cells: TheLy‐1 B lineage? Annu. Rev. Immunol. 6, 197–218.
Hoshino, T., Sakisaka, T., Baba, T., Yamada, T., Kimura, T., and Takai, Y. (2005). Regulation ofE‐cadherin endocytosis by nectin through afadin, Rap1, and p120ctn. J. Biol. Chem. 280,24095–24103.

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 259
Huntly, B. J., and Gilliland, D. G. (2005). Cancer biology: Summing up cancer stem cells. Nature435, 1169–1170.
Huntly, B. J., Shigematsu, H., Deguchi, K., Lee, B. H., Mizuno, S., Duclos, N., Rowan, R., Amaral,S., Curley, D., Williams, I. R., Akashi, K., and Gilliland, D. G. (2004). MOZ‐TIF2, but not BCR‐ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors.Cancer Cell 6, 587–596.
Huppa, J. B., and Davis, M. M. (2003). T‐cell‐antigen recognition and the immunological synapse.Nat. Rev. Immunol. 3, 973–983.
Ichiba, T., Hashimoto, Y., Nakaya, M., Kuraishi, Y., Tanaka, S., Kurata, T., Mochizuki, N., andMatsuda, M. (1999). Activation of C3G guanine nucleotide exchange factor for Rap1 byphosphorylation of tyrosine 504. J. Biol. Chem. 274, 14376–14381.
Irving, B. A., Alt, F. W., and Killeen, N. (1998). Thymocyte development in the absence of pre‐Tcell receptor extracellular immunoglobulin domains. Science 280, 905–908.
Ishida, D., Kometani, K., Yang, H., Kakugawa, K., Masuda, K., Iwai, K., Suzuki, M., Itohara, S.,Nakahata, T., Hiai, H., Kawamoto, H., Hattori, M., et al. (2003a). Myeloproliferative stem celldisorders by deregulated Rap1 activation in SPA‐1‐deficient mice. Cancer Cell 4, 55–65.
Ishida, D., Yang, H., Masuda, K., Uesugi, K., Kawamoto, H., Hattori, M., and Minato, N. (2003b).Antigen‐driven T cell anergy and defective memory T cell response via deregulated Rap1activation in SPA‐1‐deficient mice. Proc. Natl. Acad. Sci. USA 100, 10919–10924.
Ishida, D., Su, L., Tamura, A., Katayama, Y., Kawai, Y., Wang, S. F., Taniwaki, M., Hamazaki, Y.,Hattori, M., and Minato, N. (2006). Rap1 signal controls B cell receptor repertoire and genera-tion of self‐reactive B1a cells. Immunity 24, 417–427.
Jacks, T., Shih, T. S., Schmitt, E.M., Bronson, R. T., Bernards, A., andWeinberg, R. A. (1994). Tumourpredisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 7, 353–361.
Jin, A., Kurosu, T., Tsuji, K., Mizuchi, D., Arai, A., Fujita, H., Hattori, M.,Minato, N., andMiura, O.(2006). BCR/ABL and IL‐3 activate Rap1 to stimulate the B‐Raf/MEK/Erk and Akt signalingpathways and to regulate proliferation, apoptosis, and adhesion. Oncogene. 20, 4332–4340.
Joazeiro, C. A., Wing, S. S., Huang, H., Leverson, J. D., Hunter, T., and Liu, Y. C. (1999). Thetyrosine kinase negative regulator c‐Cbl as a RING‐type, E2‐dependent ubiquitin‐protein ligase.Science 286, 309–312.
Kao, S., Jaiswal, R. K., Kolch, W., and Landreth, G. E. (2001). Identification of the mechanismsregulating the differential activation of the mapk cascade by epidermal growth factor and nervegrowth factor in PC12 cells. J. Biol. Chem. 276, 18169–18177.
Katagiri, K., Hattori, M., Minato, N., Irie, S., Takatsu, K., and Kinashi, T. (2000). Rap1 is a potentactivation signal for leukocyte function‐associated antigen 1 distinct from protein kinase C andphosphatidylinositol‐3‐OH kinase. Mol. Cell. Biol. 20, 1956–1969.
Katagiri, K., Hattori, M., Minato, N., and Kinashi, T. (2002). Rap1 functions as a key regulator ofT‐cell and antigen‐presenting cell interactions and modulates T‐cell responses. Mol. Cell. Biol.22, 1001–1015.
Katagiri, K.,Maeda, A., Shimonaka,M., andKinashi, T. (2003). RAPL, a Rap1‐bindingmolecule thatmediates Rap1‐induced adhesion through spatial regulation of LFA‐1.Nat. Immunol. 4, 741–748.
Katagiri, K., Ohnishi, N., Kabashima, K., Iyoda, T., Takeda, N., Shinkai, Y., Inaba, K., and Kinashi, T.(2004). Crucial functions of the Rap1 effector molecule RAPL in lymphocyte and dendritic celltrafficking. Nat. Immunol. 5, 1045–1051.
Kawasaki, H., Spingett, G. M., Toki, S., Canales, J. J., Harlan, P., Blumenstiel, J. P., Chen, E. J.,Bany, I. A., Mochizuki, N., Ashbacher, A., Matsuda, M., Housman, D. E., et al. (1998). A Rapguanine nucleotide exchange factor enriched highly in the basal ganglia. Proc. Natl. Acad. Sci.USA 95, 13278–13283.

260 NAGAHIRO MINATO ET AL .
Kinashi, T., Aker, M., Sokolovsky‐Eisenberg, M., Grabovsky, V., Tanaka, C., Shamri, R., Feigelson, S.,Etzioni, A., andAlon, R. (2004). LAD‐III, a leukocyte adhesion deficiency syndrome associatedwithdefective Rap1 activation and impaired stabilization of integrin bonds. Blood 103, 1033–1036.
Kitayama, H., Sugimoto, Y., Matsuzaki, T., Ikawa, Y., and Noda, M. (1989). A ras‐related gene withtransformation suppressor activity. Cell 56, 77–84.
Knox, A. L., and Brown, N. H. (2002). Rap1 GTPase regulation of adherens junction positioningand cell adhesion. Science 295, 1285–1288.
Kometani, K., Ishida, D., Hattori, M., and Minato, N. (2004). Rap1 and SPA‐1 in hematologicmalignancy. Trends Mol. Med. 10, 401–408.
Kometani, K., Aoki, M., Kawamata, S., Shinozuka, Y., Era, T., Taniwaki, M., Hattori, M., andMinato, N. (2006). Role of SPA‐1 in phenotypes of CML induced by BCR‐ABL‐expressinghematopoietic progentiors in mouse model. Cancer Res. 66, 9967–9976.
Krivtsov, A. V., Twomey, D., Feng, Z., Stubbs, M. C., Wang, Y., Faber, J., Levine, J. E., Wang, J.,Hahn, W. C., Gilliland, D. G., Golub, T. R., and Armstrong, S. A. (2006). Transformation fromcommitted progenitor to leukaemia stem cell initiated by MLL‐AF9. Nature 442, 818–822.
Kurachi, H., Wada, Y., Tsukamoto, N., Maeda, M., Kubota, H., Hattori, M., Iwai, K., andMinato, N.(1997). Human SPA‐1 gene product selectively expressed in lymphoid tissues is a specificGTPase‐activating protein for Rap1 and Rap2. Segregate expression profiles from a rap1GAPgene product. J. Biol. Chem. 272, 28081–28088.
Lafuente, E. M., van Puijenbroek, A. A., Krause, M., Carman, C. V., Freeman, G. J., Berezovskaya,A., Constantine, E., Springer, T. A., Gertler, F. B., and Boussiotis, V. A. (2004). RIAM, anEna/VASP and Profilin ligand, interacts with Rap1‐GTP and mediates Rap1‐induced adhesion.Dev. Cell 7, 585–595.
Lam, K. P., and Rajewsky, K. (1999). B cell antigen receptor specificity and surface density togetherdetermine B‐1 versus B‐2 cell development. J. Exp. Med. 190, 471–477.
Largaespada, D. A., Brannan, C. I., Jenkins, N. A., and Copeland, N. G. (1996). Nf1 deficiencycauses Ras‐mediated granulocyte/macrophage colony stimulating factor hypersensitivity andchronic myeloid leukaemia. Nat. Genet. 12, 137–143.
Li, L., Godfrey, W. R., Porter, S. B., Ge, Y., June, C. H., Blazar, B. R., and Boussiotis, V. A. (2005a).CD4þCD25þ regulatory T‐cell lines from human cord blood have functional and molecularproperties of T‐cell anergy. Blood 106, 3068–3073.
Li, L., Greenwald, R. J., Lafuente, E. M., Tzachanis, D., Berezovskaya, A., Freeman, G. J., Sharpe,A. H., and Boussiotis, V. A. (2005b). Rap1‐GTP is a negative regulator of Th cell function andpromotes the generation of CD4þCD103þ regulatory Tcells in vivo. J. Immunol. 175, 3133–3139.
Li, Y., Li, H., and Weigert, M. (2002). Autoreactive B cells in the marginal zone that express dualreceptors. J. Exp. Med. 195, 181–188.
Liang, Z., Chen, C., and Mohan, C. (2003). Molecular signatures of anti‐nuclear antibodies:Contributions of specific light chain residues and a novel New Zealand Black V kappa 1 germlinegene. J. Immunol. 171, 3886–3894.
Mandell, K. J., Babbin, B. A., Nusrat, A., and Parkos, C. A. (2005). Junctional adhesion molecule 1regulates epithelial cell morphology through effects on beta1 integrins and Rap1 activity. J. Biol.Chem. 280, 11665–11674.
Marshall, C. J. (1995). Specificity of receptor tyrosine kinase signaling: Transient versus sustainedextracellular signal‐regulated kinase activation. Cell 80, 179–185.
Mason, C. S., Springer, C. J., Cooper, R. G., Superti‐Furga, G., Marshall, C. J., and Marais, R.(1999). Serine and tyrosine phosphorylations cooperate in Raf‐1, but not B‐Raf activation.EMBO J. 18, 2137–2148.
McLeod, S. J., Ingham, R. J., Bos, J. L., Kurosaki, T., and Gold, M. R. (1998). Activation of theRap1 GTPase by the B cell antigen receptor. J. Biol. Chem. 273, 29218–29223.

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 261
Michor, F., Hughes, T. P., Iwasa, Y., Branford, S., Shah, N. P., Sawyers, C. L., and Nowak, M. A.(2005). Dynamics of chronic myeloid leukaemia. Nature 435, 1267–1270.
Mishra, S., Smolik, S. M., Forte, M. A., and Stork, P. J. (2005). Ras‐independent activation ofERK signaling via the torso receptor tyrosine kinase is mediated by Rap1. Curr. Biol. 15,366–370.
Mochizuki, N., Ohba, Y., Kiyokawa, E., Kurata, T., Murakami, T., Ozaki, T., Kitabatake, A.,Nagashima, K., and Matsuda, M. (1999). Activation of the ERK/MAPK pathway by an isoformof rap1GAP associated with G alpha(i). Nature 400, 891–894.
Nagasawa, T., Hirota, S., Tachibana, K., Takakura, N., Nishikawa, S., Kitamura, Y., Yoshida, N.,Kikutani, H., and Kishimoto, T. (1996). Defects of B‐cell lymphopoiesis and bone‐marrowmyelopoiesis in mice lacking the CXC chemokine PBSF/SDF‐1. Nature 382, 635–638.
Noda, Y., Horikawa, S., Furukawa, T., Hirai, K., Katayama, Y., Asai, T., Kuwahara, M., Katagiri, K.,Kinashi, T., Hattori, M., Minato, N., and Sasaki, S. (2004). Aquaporin‐2 trafficking is regulatedby PDZ‐domain containing protein SPA‐1. FEBS Lett. 568, 139–145.
Ohba, Y., Ikuta, K., Ogura, A., Matsuda, J., Mochizuki, N., Nagashima, K., Kurokawa, K., Mayer,B. J., Maki, K., Miyazaki, J., and Matsuda, M. (2001). Requirement for C3G‐dependent Rap1activation for cell adhesion and embryogenesis. EMBO J. 20, 3333–3341.
Ostermann, G., Weber, K. S., Zernecke, A., Schroder, A., and Weber, C. (2002). JAM‐1 is a ligandof the beta(2) integrin LFA‐1 involved in transendothelial migration of leukocytes. Nat.Immunol. 3, 151–158.
Pak, D. T., Yang, S., Rudolph‐Correia, S., Kim, E., and Sheng, M. (2001). Regulation of dendriticspine morphology by SPAR, a PSD‐95‐associated RapGAP. Neuron 31, 289–303.
Park, Y. G., Zhao, X., Lesueur, F., Lowy, D. R., Lancaster, M., Pharoah, P., Qian, X., and Hunter,K. W. (2005). Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1.Nat. Genet. 37, 1055–1062.
Pizon, V., Chardin, P., Lerosey, I., Olofsson, B., and Tavitian, A. (1988). Human cDNAs rap1 andrap2 homologous to the Drosophila gene Dras3 encode proteins closely related to ras in the‘effector’ region. Oncogene 3, 201–204.
Potocnik, A. J., Brakebusch, C., and Fassler, R. (2000). Fetal and adult hematopoietic stem cellsrequire beta1 integrin function for colonizing fetal liver, spleen, and bone marrow. Immunity 12,653–663.
Qui, M. S., and Green, S. H. (1992). PC12 cell neuronal differentiation is associated withprolonged p21ras activity and consequent prolonged ERK activity. Neuron 9, 705–717.
Reedquist, K. A., and Bos, J. L. (1998). Costimulation through CD28 suppresses T cell receptor‐dependent activation of the Ras‐like small GTPase Rap1 in human T lymphocytes. J. Biol. Chem.273, 4944–4949.
Reedquist, K. A., Ross, E., Koop, E. A., Wolthuis, R. M., Zwartkruis, F. J., van Kooyk, Y., Salmon,M., Buckley, C. D., and Bos, J. L. (2000). The small GTPase, Rap1, mediates CD31‐inducedintegrin adhesion. J. Cell Biol. 148, 1151–1158.
Ren, R. (2005). Mechanisms of BCR‐ABL in the pathogenesis of chronic myelogenous leukaemia.Nat. Rev. Cancer 5, 172–183.
Roy, B. C., Kohu, K., Matsuura, K., Yanai, H., and Akiyama, T. (2002). SPAL, a Rap‐specificGTPase activating protein, is present in the NMDA receptor‐PSD‐95 complex in the hippocam-pus. Genes Cells 7, 607–617.
Sasagawa, S., Ozaki, Y., Fujita, K., and Kuroda, S. (2005). Prediction and validation of the distinctdynamics of transient and sustained ERK activation. Nat. Cell Biol. 7, 365–373.
Sattler, M., Mohi, M. G., Pride, Y. B., Quinnan, L. R., Malouf, N. A., Podar, K., Gesbert, F.,Iwasaki, H., Li, S., Van Etten, R. A., Gu, H., Griffin, J. D., et al. (2002). Critical role for Gab2 intransformation by BCR/ABL. Cancer Cell 1, 479–492.

262 NAGAHIRO MINATO ET AL .
Sawada, Y., Nakamura, K., Doi, K., Takeda, K., Tobiume, K., Saitoh, M., Morita, K., Komuro, I.,De Vos, K., Sheetz, M., and Ichijo, H. (2001). Rap1 is involved in cell stretching modulation ofp38 but not ERK or JNK MAP kinase. J. Cell Sci. 114, 1221–1227.
Schmidt, A., Caron, E., and Hall, A. (2001). Lipopolysaccharide‐induced activation of beta2‐integrin function in macrophages requires Irak kinase activity, p38 mitogen‐activated proteinkinase, and the Rap1 GTPase. Mol. Cell. Biol. 21, 438–448.
Schneider, H., Valk, E., da Rocha Dias, S., Wei, B., and Rudd, C. E. (2005). CTLA‐4 up‐regulationof lymphocyte function‐associated antigen 1 adhesion and clustering as an alternate basis forcoreceptor function. Proc. Natl. Acad. Sci. USA 102, 12861–12866.
Schultess, J., Danielewski, O., and Smolenski, A. P. (2005). Rap1GAP2 is a new GTPase‐activatingprotein of Rap1 expressed in human platelets. Blood 105, 3185–3192.
Schwartz, R. H. (1997). T cell clonal anergy. Curr. Opin. Immunol. 9, 351–357.Sebzda, E., Bracke, M., Tugal, T., Hogg, N., and Cantrell, D. A. (2002). Rap1A positively regulatesTcells via integrin activation rather than inhibiting lymphocyte signaling.Nat. Immunol.3, 251–258.
Shannon, K. M., O’Connell, P., Martin, G. A., Paderanga, D., Olson, K., Dinndorf, P., andMcCormick, F. (1994). Loss of the normal NF1 allele from the bone marrow of children withtype 1 neurofibromatosis and malignant myeloid disorders. N. Engl. J. Med. 330, 597–601.
Shao, Y., Elly, C., and Liu, Y. C. (2003). Negative regulation of Rap1 activation by the Cbl E3ubiquitin ligase. EMBO Rep. 4, 425–431.
Sharpe, A. H., and Freeman, G. J. (2002). The B7‐CD28 superfamily. Nat. Rev. Immunol. 2,116–126.
Shaw, R. J., and Cantley, L. C. (2006). Ras, PI(3)K and mTOR signalling controls tumour cellgrowth. Nature 441, 424–430.
Shimonaka, M., Katagiri, K., Nakayama, T., Fujita, N., Tsuruo, T., Yoshie, O., and Kinashi, T.(2003). Rap1 translates chemokine signals to integrin activation, cell polarization, and motilityacross vascular endothelium under flow. J. Cell Biol. 161, 417–427.
Sonoyama, J., Matsumura, I., Ezoe, S., Satoh, Y., Zhang, X., Kataoka, Y., Takai, E., Mizuki, M.,Machii, T., Wakao, H., and Kanakura, Y. (2002). Functional cooperation among Ras, STAT5, andphosphatidylinositol 3‐kinase is required for full oncogenic activities of BCR/ABL in K562 cells.J. Biol. Chem. 277, 8076–8082.
Springer, T. A. (1995). Traffic signals on endothelium for lymphocyte recirculation and leukocyteemigration. Annu. Rev. Physiol. 57, 827–872.
Stork, P. J., and Dillon, T. J. (2005). Multiple roles of Rap1 in hematopoietic cells: Complementaryversus antagonistic functions. Blood 106, 2952–2961.
Su, L., Hattori, M., Moriyama, M., Murata, N., Harazaki, M., Kaibuchi, K., and Minato, N. (2003).AF‐6 controls integrin‐mediated cell adhesion by regulating Rap1 activation through the specificrecruitment of Rap1GTP and SPA‐1. J. Biol. Chem. 278, 15232–15238.
Suga, K., Katagiri, K., Kinashi, T., Harazaki, M., Iizuka, T., Hattori, M., and Minato, N. (2001).CD98 induces LFA‐1‐mediated cell adhesion in lymphoid cells via activation of Rap1. FEBSLett. 489, 249–253.
Swan, K. A., Alberola‐Ila, J., Gross, J. A., Appleby, M. W., Forbush, K. A., Thomas, J. F., andPerlmutter, R. M. (1995). Involvement of p21ras distinguishes positive and negative selection inthymocytes. EMBO J. 14, 276–285.
Takaki, S., Morita, H., Tezuka, Y., and Takatsu, K. (2002). Enhanced hematopoiesis by hemato-poietic progenitor cells lacking intracellular adaptor protein, Lnk. J. Exp. Med. 195, 151–160.
Tamada, M., Sheetz, M. P., and Sawada, Y. (2004). Activation of a signaling cascade by cytoskeletonstretch. Dev. Cell 7, 709–718.
Threadgill, D. W. (2005). Metastatic potential as a heritable trait. Nat. Genet. 37, 1026–1027.

RAP1 IN IMMUNE RESPONSE AND HEMATOPOIESIS 263
Traverse, S., Gomez, N., Paterson, H., Marshall, C., and Cohen, P. (1992). Sustained activation ofthe mitogen‐activated protein (MAP) kinase cascade may be required for differentiation of PC12cells. Comparison of the effects of nerve growth factor and epidermal growth factor. Biochem. J.288(Pt. 2), 351–355.
Tsukamoto, N., Hattori, M., Yang, H., Bos, J. L., and Minato, N. (1999). Rap1 GTPase‐activatingprotein SPA‐1 negatively regulates cell adhesion. J. Biol. Chem. 274, 18463–18469.
Tsygankova, O. M., Saavedra, A., Rebhun, J. F., Quilliam, L. A., and Meinkoth, J. L. (2001).Coordinated regulation of Rap1 and thyroid differentiation by cyclic AMP and protein kinase A.Mol. Cell. Biol. 21, 1921–1929.
Tzachanis, D., Freeman, G. J., Hirano, N., van Puijenbroek, A. A., Delfs, M. W., Berezovskaya,A., Nadler, L. M., and Boussiotis, V. A. (2001). Tob is a negative regulator of activation that isexpressed in anergic and quiescent T cells. Nat. Immunol. 2, 1174–1182.
Voss, A. K., Gruss, P., and Thomas, T. (2003). The guanine nucleotide exchange factor C3G isnecessary for the formation of focal adhesions and vascular maturation. Development 130,355–367.
Voss, A. K., Krebs, D. L., and Thomas, T. (2006). C3G regulates the size of the cerebral cortexneural precursor population. EMBO J. 25, 3652–3663.
Vossler, M. R., Yao, H., York, R. D., Pan, M. G., Rim, C. S., and Stork, P. J. (1997). cAMP activatesMAP kinase and Elk‐1 through a B‐Raf‐ and Rap1‐dependent pathway. Cell 89, 73–82.
Wang, H., Singh, S. R., Zheng, Z., Oh, S. W., Chen, X., Edwards, K., and Hou, S. X. (2006). Rap‐GEF signaling controls stem cell anchoring to their niche through regulating DE‐cadherin‐mediated cell adhesion in the Drosophila testis. Dev. Cell 10, 117–126.
Wang, L., Liu, F., and Adamo, M. L. (2001). Cyclic AMP inhibits extracellular signal‐regulatedkinase and phosphatidylinositol 3‐kinase/Akt pathways by inhibiting Rap1. J. Biol. Chem. 276,37242–37249.
Wardemann, H., Yurasov, S., Schaefer, A., Young, J. W., Meffre, E., and Nussenzweig, M. C.(2003). Predominant autoantibody production by early human B cell precursors. Science 301,1374–1377.
Whetton, A. D., and Graham, G. J. (1999). Homing and mobilization in the stem cell niche. TrendsCell Biol. 9, 233–238.
Willimsky, G., and Blankenstein, T. (2005). Sporadic immunogenic tumours avoid destruction byinducing T‐cell tolerance. Nature 437, 141–146.
Wilson, A., Murphy, M. J., Oskarsson, T., Kaloulis, K., Bettess, M. D., Oser, G. M., Pasche, A. C.,Knabenhans, C., Macdonald, H. R., and Trumpp, A. (2004). c‐Myc controls the balance betweenhematopoietic stem cell self‐renewal and differentiation. Genes Dev. 18, 2747–2763.
Wittchen, E. S., van Buul, J. D., Burridge, K., and Worthylake, R. A. (2005). Trading spaces: Rap,Rac, and Rho as architects of transendothelial migration. Curr. Opin. Hematol. 12, 14–21.
Wong, S., and Witte, O. N. (2004). The BCR‐ABL story: Bench to bedside and back. Annu. Rev.Immunol. 22, 247–306.
Xiong, J. P., Stehle, T., Diefenbach, B., Zhang, R., Dunker, R., Scott, D. L., Joachimiak, A.,Goodman, S. L., and Arnaout, M. A. (2001). Crystal structure of the extracellular segment ofintegrin alpha Vbeta3. Science 294, 339–345.
Yajnik, V., Paulding, C., Sordella, R., McClatchey, A. I., Saito, M., Wahrer, D. C., Reynolds, P., Bell,D. W., Lake, R., van den Heuvel, S., Settleman, J., and Haber, D. A. (2003). DOCK4, a GTPaseactivator, is disrupted during tumorigenesis. Cell 112, 673–684.
Yamasaki, S., Ishikawa, E., Sakuma, M., Ogata, K., Sakata‐Sogawa, K., Hiroshima, M., Wiest, D. L.,Tokunaga, M., and Saito, T. (2006). Mechanistic basis of pre‐T cell receptor‐mediated autono-mous signaling critical for thymocyte development. Nat. Immunol. 7, 67–75.

264 NAGAHIRO MINATO ET AL .
Yamashita, Y. M., Jones, D. L., and Fuller, M. T. (2003). Orientation of asymmetric stem celldivision by the APC tumor suppressor and centrosome. Science 301, 1547–1550.
Yilmaz, O. H., Valdez, R., Theisen, B. K., Guo, W., Ferguson, D. O., Wu, H., and Morrison, S. J.(2006). Pten dependence distinguishes haematopoietic stem cells from leukaemia‐initiatingcells. Nature 441, 475–482.
York, R. D., Yao, H., Dillon, T., Ellig, C. L., Eckert, S. P., McCleskey, E. W., and Stork, P. J. (1998).Rap1 mediates sustained MAP kinase activation induced by nerve growth factor. Nature 392,622–626.
Yuan, Y., Shen, H., Franklin, D. S., Scadden, D. T., and Cheng, T. (2004). In vivo self‐renewingdivisions of haematopoietic stem cells are increased in the absence of the early G1‐phaseinhibitor, p18INK4C. Nat. Cell Biol. 6, 436–442.
Zhang, J., Niu, C., Ye, L., Huang, H., He, X., Tong, W. G., Ross, J., Haug, J., Johnson, T., Feng,J. Q., Harris, S., Wiedemann, L. M., et al. (2003). Identification of the haematopoietic stem cellniche and control of the niche size. Nature 425, 836–841.
Zhang, J., Grindley, J. C., Yin, T., Jayasinghe, S., He, X. C., Ross, J. T., Haug, J. S., Rupp, D.,Porter‐Westpfahl, K. S., Wiedemann, L. M., Wu, H., and Li, L. (2006a). PTEN maintainshaematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 441,518–522.
Zhang, Z., Mitra, R. S., Henson, B. S., Datta, N. S., McCauley, L. K., Kumar, P., Lee, J. S., Carey,T. E., and D’Silva, N. J. (2006b). Rap1GAP inhibits tumor growth in oropharyngeal squamouscell carcinoma. Am. J. Pathol. 168, 585–596.
Zhu, J. J., Qin, Y., Zhao, M., Van Aelst, L., and Malinow, R. (2002). Ras and Rap control AMPAreceptor trafficking during synaptic plasticity. Cell 110, 443–455.

a#
Lung Dendritic Cell Migration
Hamida Hammad and Bart N. Lambrecht
Department of Pulmonary Medicine, Erasmus Medical Center, Dr Molewaterplein50, 3015 GE Rotterdam, The Netherlands
A
dv2
bstract............................................................................................................. 2
265ances in immunology, vol. 93 0065-2776/07 $
007 Elsevier Inc. All rights reserved. DOI: 10.1016/S0065-2776(06)93
65
1. I ntroduction ....................................................................................................... 2 65 2. A irway DC Subsets: Localization and Phenotype....................................................... 2 66 3. R ecruitment of DCs to the Lung............................................................................ 2 67 4. M igration of Airway DCs to the LNs ...................................................................... 2 69 5. R ecruitment of pDCs to the Sites of Inflammation .................................................... 2 72 6. C onclusions........................................................................................................ 2 72R
eferences ......................................................................................................... 2 73Abstract
Dendritic cells (DCs) are crucial in regulating the immune response by bridg-ing innate and adaptive immunity. DCs are constantly migrating from theblood to the lungs and from the lungs to the draining lymph nodes. How DCspopulate the lung in the absence of inflammation and how they are recruitedthere during inflammation remain unclear. Since DCs play a central role inimmune responses, both under steady‐state and inflammatory conditions,detailed characterization of their migratory behavior may be essential for thedevelopment of future therapeutic strategies.
1. Introduction
Numerous environmental pathogens, particulate matter, allergens, and harmlessantigens are present in the air we breathe. Althoughmost of these particles will beheld up in the upper airways, the lung is one of the most challenged organs of thebody. The usual functional outcome of harmless antigen encounter in the lung isignorance or tolerance. Yet, when faced with pathogens, the immune defensemechanisms of the lung can generate a protective immune response.
Many cells of the innate and adaptive immune system play an important rolein the induction of inhalation tolerance or immunity. Dendritic cells (DCs) arecrucial in regulating the immune response by bridging innate and adaptiveimmunity. At least two subsets of DCs have been described in both human andmice, namely myeloid and plasmacytoid DCs (pDCs). Whereas mDCs are theclassical T cell priming subset (Lambrecht et al., 2000), the function of pDCs isless clear, although they might play an important role in the maintenance oftolerance to inhaled antigens (de Heer et al., 2005).
35.00
007-7

266 HAMIDA HAMMAD AND BART N. LAMBRECHT
In the airways, DCs are strategically located to sample inhaled antigens andexist in an immature form, efficient at taking up and processing antigens(Mellman and Steinman, 2001). Once activated by ‘‘danger’’ signals, such ascontact with microbial products or proinflammatory cytokines, DCs undergomaturation into competent antigen‐presenting cells, expressing high levels ofMHCII and costimulatory molecules (Banchereau and Steinman, 1998).An important question for the understanding of airway DC biology is how
DCs populate the lung in the absence of inflammation, and how they arerecruited there during inflammation. It is known that myeloid DCs enter thebloodstream from bone marrow and circulate as precursor/immature DC(Nikolic et al., 2003). Under inflammatory conditions, adhesion molecules atthe surface of endothelial cells as well as chemokines are upregulated, facil-itating efficient recruitment of circulating DC to the inflamed site (Robertet al., 1999). On maturation, DCs reprogram their repertoire of chemokinereceptors (Sallusto et al., 1999; Sozzani et al., 1999) and migrate to lymphnodes (LNs), where they will activate naive T cells (Steinman, 1991). Themigratory pathways of pDC are less well understood. pDCs circulate inperipheral blood in precursor form and represent a significant population inmost secondary lymphoid organs (Asselin‐Paturel et al., 2001, 2003; Colonnaet al., 2002).Since DCs play a central role in immune responses, both under steady‐state
and inflammatory conditions, detailed characterization of their migratoryproperties may be essential for the development of future therapeutic strate-gies. Here, we discuss the different mechanisms used in the migration ofairway DCs.
2. Airway DC Subsets: Localization and Phenotype
Immature DCs are distributed throughout the lung and are at the focal controlpoint determining the induction of pulmonary immunity or tolerance (Akbariet al., 2001, 2002; Lambrecht and Hammad, 2003). Airway DCs form a densenetwork in the lung ideally placed to sample inhaled antigens, and these cellsmigrate to draining mediastinal LNs to stimulate naive T cells (Lambrechtet al., 1998; Vermaelen et al., 2001).Several populations of DCs can be found in every compartment, including
the conducting airways, lung parenchyma, alveolar space, visceral pleura, andthe pulmonary vascular bed (Gong et al., 1992; Holt et al., 1994; Pollard andLipscomb, 1990; Sertl et al., 1986; Suda et al., 1998). However, the differentsubsets of lung DCs are differentially distributed.In the conducting airways, CD11chigh DCs form a dense network under-
neath and within the epithelium with dendrite projections toward the lumen to

LUNG DENDRITIC CELL MIGRATION 267
sample for environmental antigens. In mice and rats, these cells are mainly ofthe myeloid origin and show a high turnover of about 2–3 days under steady‐state conditions (Holt et al., 1994; Lambrecht et al., 1998). DCs in the lunginterstitium are predominantly CD11cþ myeloid and are immature as assessedby the low expression of the costimulatory molecules CD40, CD80, CD86 (deHeer et al., 2005; Huh et al., 2003; Stumbles et al., 1998; van Rijt et al., 2005;Vermaelen and Pauwels, 2003) and by the high expression of several receptorsfor inflammatory chemokines and endocytic receptors (Cochand et al., 1999).We and others have been able to identify a population of pDCs in enzymaticlung digests of mice and humans, respectively (Demedts et al., 2005). Theanatomical location of human pDCs in the lung has not yet been characterized.In the mouse, pDCs have been identified in the interalveolar interstitium ofthe lung (De Heer et al., 2004) and within the alveolar lavage fluid of mice withallergy (unpublished observations).
3. Recruitment of DCs to the Lung
DCs are often referred to as ‘‘sentinels’’ of the immune system. The roleof DCs is the continuous surveillance of peripheral sites highly exposed toantigens, such as the lung. In the absence of inflammatory signals, DCs andtheir precursors are recruited from the bloodstream into the lung where theyhave a very rapid turnover of about 2–3 days compared to skin DCs (Holt et al.,1994). In the presence of inflammatory signals, DCs can be recruited veryrapidly to the lungs as a response to the increased requirement for surveillanceat the local site. McWilliams et al. (1994) showed that the earliest detectablecellular response after inhalation of bacteria by naive mice is the recruitmentof MHCIIþ DC precursors into the airway epithelium. Because the wave ofDCs arrives in the airways before the neutrophils and other mononuclear cells,DCs contribute to the very early phase of the immune responses in theairways. DCs are also found in increased numbers in the lung during second-ary immune responses. Indeed, the number of airway CD11cþ CD11bþ
myeloid DCs is strongly increased within the airway epithelium followingallergen challenge in sensitized animals (van Rijt et al., 2005). This increasein the number of airway DCs is accompanied with an increase in CD11c–CD11bþ MHCIIþ monocytes that could be recruited to the airways andprobably further differentiate to DCs (van Rijt et al., 2002).
Like for other leukocytes, the recruitment of DCs and their precursors fromthe bloodstream to peripheral tissues first involves a cascade of cellular inter-actions between the circulating cells and the endothelium (Springer, 1994)Molecules such as E‐ and P‐selectin, VCAM‐1, and L‐selectin ligands canmediate leukocyte tethering and rolling (Carlos and Harlan, 1994) and are

268 HAMIDA HAMMAD AND BART N. LAMBRECHT
upregulated on endothelial cells on inflammation. However, whereas thesemolecules have been extensively described in the transendothelial migration ofskin DCs, their involvement in the recruitment of precursors or DCs to thelung has never been proven.Once in the lung, chemokine expression within tissue may direct DC
localization after extravasation. Chemokine concentration gradients elicit adirected movement called chemotaxis. In vitro studies show that many cellsin the lung produce many chemokines known to have an effect on DCs(Godiska et al., 1997; Gonzalo et al., 2000; Hieshima et al., 1997; Schall,1997; Thorley et al., 2005). However, only a few have been studied in detailwith respect to pulmonary DCs. In vivo, different chemokines orchestrate therecruitment of DCs into the lung depending on the inflammatory stimuluspresent. Isolated lung DCs express several chemokine receptors, includingCCR1, CCR2, CCR5, CXCR4, and CCR6 (Chiu et al., 2004; Power et al.,1997). In a mouse model of infection with Mycobacterium tuberculosis, it wasshown that the recruitment of DCs and T cells to the lungs of CCR2�/� micewas reduced compared to wild‐type animals, and resulted in the prematuredeath of the animals (Peters et al., 2001, 2004). One crucial receptor in therecruitment of DCs to the lung is CCR6. CCR6 is the receptor for MIP‐3a orCCL20, a chemokine abundantly released by bronchial epithelial cells andprimary alveolar type 2 cells (Reibman et al., 2003; Starner et al., 2003).Pichavant et al. (2005) have shown that bronchial epithelial cells of asthmaticpatients stimulated with the house dust mite allergen Der p 1 showed anincreased production of CCL20. CCR6 involvement in the recruitment ofDCs to the lung has been shown using CCR6�/� mice in which the accumu-lation of DCs in the airways was impaired (Osterholzer et al., 2005). Interest-ingly, several features of asthma, a disease mediated by airway DCs (ourreferences), were reduced in mice lacking CCR6 expression (Lukacs et al.,2001; Lundy et al., 2005). DC and monocyte chemokine‐like protein (DMC), anew chemokine constitutively expressed in the lung has been characterized(Pisabarro et al., 2006). This chemokine specifically attracts monocytes andDCs in vitro; however, whether it has the same role in vivo remains to beelucidated.In addition to chemokines, the airway epithelium can attract DCs by means
of defensins, cationic peptides with bactericidal activity engaging CCR6 onimmature DCs (Cole and Waring, 2002; Yang et al., 1999). In this way,defensins may promote adaptive immune responses by recruiting DCs to thesite of microbial invasion. Besides chemokines, other molecules can also favorthe migration of DCs into the lungs. During inflammation, matrix metallopro-teinase (MMP)‐9�/� mice have been shown to have an impaired recruitment

LUNG DENDRITIC CELL MIGRATION 269
of DCs into the airways, whereas the migration of DCs to the draining LNswas unaffected (Vermaelen et al., 2003).
In summary, DCs are recruited to the lungs via different mechanisms, and itseems very likely that these chemotactic agents may act sequentially to attractrecently transmigrated DC, and position them at the inflamed site. Oneunclear point remains, however, whether lung DCs are recruited from theblood in a differentiated form or as early precursors. It is possible that DCprecursors, possibly monocytes, could first be attracted from the bloodstreaminto the lung and subsequently differentiate into DCs (van Rijt et al., 2005).
4. Migration of Airway DCs to the LNs
4.1. Migration of DCs Under Steady‐State Conditions
DCs constantly migrate from peripheral tissues to the draining LNs. It hasbeen proposed that under steady‐state conditions, mDCs continuously migrateto draining LNs and present either (self)‐auto antigens or harmless antigen in atolerogenic form (Steinman and Nussenzweig, 2002). Airway DCs extend longdendrites to the lumen of the airways, forming bud‐like extensions at theborder of the air interface (Brokaw et al., 1998). Most of our knowledge onhow DCs migrate from the lung to the mediastinal draining lymph nodes(MLNs) involves the intratracheal inoculation of fluorescently labeled antigen.Within a few hours after inhalation of FITC‐labeled ovalbumin (OVA), airwaymyeloid DCs and plasmacytoid DCs loaded with fluorescently labeled antigencould be detected within the draining mediastinal LNs (De Heer et al., 2004;Hammad et al., 2003; Vermaelen et al., 2001). After 24 h, both mDCs andpDCs in the mediastinal LNs contain antigen inside vesicles of the cytoplasm.What is unclear at present is whether pDCs take up antigen in the periphery ofthe lung and subsequently migrate to the nodes, or whether antigen is beingtransported to them by migratory mDCs. The phenotype of antigen‐transporting DCs is still uncertain since Holt’s group showed that these cellswere expressing low levels of CD8a (Wikstrom et al., 2006), whereas Belz et al.(2004a,b) have found that Ag‐loaded DCs were CD8a� CD11b�. Transport ofantigenic material from one nonmigratory DC to another is certainly a possi-bility, as we and others saw that CD8aþ DCs injected into the lung or skininduce an immune response in the draining node without migrating into it(Hammad et al., 2004; Smith and Fazekas De St Groth, 1999).
In another system, we also found that the intratracheal administration ofbone marrow‐derived DCs results in the accumulation of the transferred cellsin the MLNs of the mice (Havenith et al., 1993; Lambrecht et al., 2000). Themechanisms involved in the steady‐state migration of airway DCs to the MLNs

270 HAMIDA HAMMAD AND BART N. LAMBRECHT
are unclear. It has been suggested that semimature DCs, which are immatureDCs with an intermediate phenotype, might use CCR7 to continuouslymigrate to the LNs in the absence of inflammatory signals (Ohl et al., 2004;Sanchez‐Sanchez et al., 2006); however, these studies have been performed onskin DCs. It is not clear whether this would also happen in the lung as somestudies have reported the absence of functional CCR7 expression by DCs atmucosal surfaces (Kobayashi et al., 2004).
4.2. Migration of DCs Under Inflammatory Conditions
The presence of ‘‘danger’’ signals in organs exposed to antigens is a strongstimulus for the migration of antigen‐bearing DCs toward LNs. In a mousemodel of allergic inflammation, ongoing airway inflammation was shown tocause a massive and accelerated flux of allergen‐loaded DCs from the airwaymucosa to the MLNs (Huh et al., 2003; Vermaelen and Pauwels, 2003). Thesame observations were made after the intranasal administration of influenzavirus (Legge and Braciale, 2003). The mechanisms behind this increasedmigration to the LNs are unknown, but some likely mechanisms are describedin Section 4.2.1 to 4.2.5.
4.2.1. Involvement of Chemokine Receptors
The most important factor driving DC migration from peripheral tissues to theT cell area of LNs under inflammatory conditions is CCR7 (Saeki et al., 1999).Although the involvement of CCR7 is well documented in skin DC migration,very little is known regarding the involvement of this molecule in the traffick-ing of lung DCs. CCR7 has been described on a subset of lung DCs (Swansonet al., 2004) and is regulated by the transcription factor RunX3 (Fainaru et al.,2005). In RunX3‐deficient animals, CCR7 expression was upregulated and themigration of DCs to the MLNs was increased (Fainaru et al., 2005). We havepreviously shown that in vivo neutralization of CCL21 could prevent humanDC migration to MLNs of humanized severe combined immunodeficient(SCID) mice and the subsequent development of asthma features (Hammadet al., 2002). Moreover, the intranasal injection of latex beads into mice led to aCCR7‐dependent accumulation of DCs in the MLNs (Jakubzick et al., 2006).Interestingly, this phenomenon was not only CCR7‐dependent but alsoinvolved another chemokine receptor, namely CCR8. The authors have de-monstrated that CCR8‐deficient mice manifested a decreased accumulation ofbeadþ DCs to MLNs, reminiscent of the reduced emigration of DCs from skin(Qu et al., 2004), suggesting that lung and skin DCs use similar mechanismsfor their migration to the LNs.

LUNG DENDRITIC CELL MIGRATION 271
4.2.2. Involvement of Cysteiny l Leukotrienes
In addition to chemokines, lipid metabolites such as leukotrienes and prosta-glandins are emerging as important upstream controllers of DC migrationtoward LNs. Cysteinyl leukotriens are important inflammatory mediators inasthma (Busse, 1998) and can affect DC functions (Okunishi et al., 2004).Molecules like LTC4 and its transporter, multidrug resistance‐related protein 1(MRP1, also called ABCC1) participate in DC migration from skin to LNsbecause they contribute to the sensitization of CCR7 to its ligands (Hopkenand Lipp, 2004; Robbiani et al., 2000). However, it appears that LTC4 is notneeded for lung DCs accumulation in the MLNs (Jakubzick et al., 2006), mostlikely because in the lung other lipid mediators might be able to take over thefunction of LTC4. Future studies will be required to address this possibility,as additional mediators of DC migration from lung to MLNs are described.
4.2.3. Involvement of Prostaglandins
Several studies have implicated PGE2 in the chemotactic response of DCs toCCR7 ligands, the latter response being dependent on the EP4 receptor (Berthoet al., 2005; Kabashima et al., 2003; Luft et al., 2002; Scandella et al., 2004).In contrast, PGD2 exerts an opposite effect: we showed that PGD2, through theligation of DP1, one of its two receptors, could inhibit the emigration of airwayDCs towardMLNs and consequently prevent the induction of a primary immuneresponse and of eosinophilic airway inflammation (Hammad et al. (2003); ourunpublished observations). More recently, we have looked at the role of PGI2 onDCmigration to the MLNs, and showed that a stable analogue of PGI2, Iloprost,could inhibit the migration of lung DCs to the MLNs, thereby abolishingthe induction of an allergen specific Th2 response in these nodes (Idzko, 2007).The same effect was obtained with pharmacological agonists of the peroxisomeproliferator‐activated receptor‐g (PPAR‐g), an important intracellular mediatorof prostaglandin signaling (Angeli et al., 2003).
4.2.4. Involvement of Sphingosine‐1‐Phosphate
Sphingosine‐1‐phosphate (S1P) is predominantly generated by stimulatedplatelets and leukocytes. In immune cells, S1P can modulate many differentfunctions including migration, cytokine, and chemokine release (Graeler andGoetzl, 2002; Spiegel and Milstien, 2003). We have recently shown that theinhalation of FTY720, a structural homologue of S1P, reduced the number ofmigrating mDCs in MLNs of naive and allergen challenged mice and reducedasthma featu res (Idzko et al., 2006).

272 HAMIDA HAMMAD AND BART N. LAMBRECHT
4.2.5. Involvement of CD38
Generation of cyclic adensine diphosphate ribose from NADþ by the action ofCD38 initiates calcium mobilization that is required for chemotaxis of DCs to avariety of chemokines, including CCR7 ligands (Partida‐Sanchez et al., 2004).CD38 has been shown to regulate themigration ofDCprecursors from the bloodto peripheral sites and to control themigration ofmatureDCs from inflamed skintoLNs (Partida‐Sanchez et al., 2004).We also have evidence thatCD38 is used bylung DCs to migrate to the MLNs. The intratracheal administration of thecADPR antagonist 8‐Br‐cADPR, which renders DCs unable to flux Ca2þ inresponse to chemokine stimulation, strongly decreased the number of migratinglung DCs to the MLNs (Hammad H., unpublished observations). Since theextracellular NADþ levels have been shown to rise significantly in serum and atlocal sites of tissue damage due to release of NADþ from necrotic cells (Okamotoet al., 1998), extracellular NADþ could be thought of as an inflammatory mod-ulator, similar to other extracellular nucleotides such as adenosine (Ohta andSitkovsky, 2001) and ATP (Di Virgilio et al., 2001), and compounds that alterNADþ levels or CD38 enzyme activity could potentially be used to block DC‐mediated inflammation by altering their migration to the MLNs and thesubsequent T cell activation.
5. Recruitment of pDCs to the Sites of Inflammation
pDCs can be recruited to nonlymphoid organs in inflammatory conditions.Indeed, for instance, the number of pDCs was increased in the nasal mucosalof patients suffering from allergic rhinitis (Jahnsen et al., 2000). We have alsofound an increased number of pDCs in the lung and in the bronchoalveolarlavage of asthmatic mice (M. Kool, unpublished observations). In nasal allergyand in asthma, there is an increased expression of PNAd, ICAM‐1, and VCAMon the vessels of affected tissues. Interactions with these adhesion moleculesare likely to be involved in DC migration into this site, as circulating pDCsexpress the appropriate ligands, namely CD62L, b2, and a4 integrins, respec-tively (Cella et al., 1999; Olweus et al., 1997). Moreover, pDCs expressCXCR3, the receptor for CXCL10, which is upregulated under inflammatoryconditions in the lung (Medoff et al., 2002).
6. Conclusions
DCs play a central role in the induction of immune responses against foreignantigens, but DCs are also involved in the maintenance of inflammation andtolerance in the periphery. A critical factor for the DC role in both immunityand tolerance may be their migratory capacity. As inflammation induces DC

LUNG DENDRITIC CELL MIGRATION 273
maturation and migration, DCs reach the LNs fully activated so that T cellpriming can occur. In the absence of inflammation, smaller numbers of DCmigrate to LNs carrying self‐antigens, and therefore might induce T celltolerance. Although a lot is known about DC migration in the skin, manyopen questions still remain especially regarding lung DCs. For example, DCmigrate to many different peripheral tissues; however, little is known about thespecific molecular mechanisms involved. Moreover, the constant migration ofDC from various tissues such as the lung to LNs may be important fortolerance induction. Understanding the mechanisms by which this occurs,and the exact subset of DC that migrates, may be critical for the design offuture therapeutic strategies.
References
Akbari, O., DeKruyff, R. H., and Umetsu, D. T. (2001). Pulmonary dendritic cells producing IL‐10mediate tolerance induced by respiratory exposure to antigen. Nat. Immunol. 2, 725–731.
Akbari, O., Freeman, G. J., Meyer, E. H., Greenfield, E. A., Chang, T. T., Sharpe, A. H., Berry, G.,DeKruyff, R. H., and Umetsu, D. T. (2002). Antigen‐specific regulatory T cells develop via theICOS‐ICOS‐ligand pathway and inhibit allergen‐induced airway hyperreactivity. Nat. Med. 8,1024–1032.
Angeli, V., Hammad, H., Staels, B., Capron, M., Lambrecht, B. N., and Trottein, F. (2003).Peroxisome proliferator‐activated receptor gamma inhibits the migration of dendritic cells:Consequences for the immune response. J. Immunol. 170, 5295–5301.
Asselin‐Paturel, C., Boonstra, A., Dalod, M., Durand, I., Yessaad, N., Dezutter‐Dambuyant, C.,Vicari, A., O’Garra, A., Biron, C., Briere, F., and Trinchieri, G. (2001). Mouse type I IFN‐producing cells are immature APCs with plasmacytoid morphology.Nat. Immunol. 2, 1144–1150.
Asselin‐Paturel, C., Brizard, G., Pin, J. J., Briere, F., and Trinchieri, G. (2003). Mouse straindifferences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclo-nal antibody. J. Immunol. 171, 6466–6477.
Banchereau, J., and Steinman, R. M. (1998). Dendritic cells and the control of immunity. Nature392, 245–252.
Belz, G. T., Smith, C.M., Eichner, D., Shortman, K., Karupiah, G., Carbone, F. R., andHeath,W. R.(2004a). Cutting edge: Conventional CD8 alphaþ dendritic cells are generally involved inpriming CTL immunity to viruses. J. Immunol. 172, 1996–2000.
Belz, G. T., Smith, C. M., Kleinert, L., Reading, P., Brooks, A., Shortman, K., Carbone, F. R., andHeath, W. R. (2004b). Distinct migrating and nonmigrating dendritic cell populations areinvolved in MHC class I‐restricted antigen presentation after lung infection with virus. Proc.Natl. Acad. Sci. USA 101, 8670–8675.
Bertho, N., Adamski, H., Toujas, L., Debove, M., Davoust, J., and Quillien, V. (2005). Efficientmigration of dendritic cells toward lymph node chemokines and induction of T(H)1 responsesrequire maturation stimulus and apoptotic cell interaction. Blood 106, 1734–1741.
Brokaw, J. J., White, G. W., Baluk, P., Anderson, G. P., Umemoto, E. Y., and McDonald, D. M.(1998). Glucocorticoid‐induced apoptosis of dendritic cells in the rat tracheal mucosa.Am. J. Respir. Cell Mol. Biol. 19, 598–605.
Busse, W. W. (1998). Leukotrienes and inflammation. Am. J. Respir. Crit. Care Med. 157,S210–S213; discussion S247–S218.

274 HAMIDA HAMMAD AND BART N. LAMBRECHT
Carlos, T. M., and Harlan, J. M. (1994). Leukocyte‐endothelial adhesion molecules. Blood 84,2068–2101.
Cella,M., Jarrossay, D., Facchetti, F., Alebardi, O., Nakajima, H, Lanzavecchia, A., and Colonna,M.(1999). Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amountsof type I interferon. Nat. Med. 5, 919–923.
Chiu, B. C., Freeman, C. M., Stolberg, V. R., Hu, J. S., Zeibecoglou, K., Lu, B., Gerard, C., Charo,I. F., Lira, S. A., and Chensue, S. W. (2004). Impaired lung dendritic cell activation in CCR2knockout mice. Am. J. Pathol. 165, 1199–1209.
Cochand, L., Isler, P., Songeon, F., and Nicod, L. P. (1999). Human lung dendritic cells have animmature phenotype with efficientmannose receptors.Am. J. Respir. Cell Mol. Biol. 21, 547–554.
Cole, A. M., and Waring, A. J. (2002). The role of defensins in lung biology and therapy.Am. J. Respir. Med. 1, 249–259.
Colonna, M., Krug, A., and Cella, M. (2002). Interferon‐producing cells: On the front line inimmune responses against pathogens. Curr. Opin. Immunol. 14, 373–379.
De Heer, H. J., Hammad, H., Soullie, T., Hijdra, D., Vos, N., Willart, M. A., Hoogsteden, H. C.,and Lambrecht, B. N. (2004). Essential role of lung plasmacytoid dendritic cells in preventingasthmatic reactions to harmless inhaled antigen. J. Exp. Med. 200, 89–98.
De Heer, H. J., Hammad, H., Kool, M., and Lambrecht, B. N. (2005). Dendritic cell subsets andimmune regulation in the lung. Semin. Immunol. 17, 295–303.
Demedts, I. K., Brusselle, G. G., Vermaelen, K. Y., and Pauwels, R. A. (2005). Identification andcharacterization of human pulmonary dendritic cells. Am. J. Respir. Cell Mol. Biol. 32, 177–184.
Di Virgilio, F., Chiozzi, P., Ferrari, D., Falzoni, S., Sanz, J. M.,Morelli, A, Torboli, M., Bolognesi, G.,and Baricordi, O. R. (2001). Nucleotide receptors: An emerging family of regulatory molecules inblood cells. Blood 97, 587–600.
Fainaru, O., Shseyov, D., Hantisteanu, S., and Groner, Y. (2005). Accelerated chemokine receptor7‐mediated dendritic cell migration in Runx3 knockout mice and the spontaneous developmentof asthma‐like disease. Proc. Natl. Acad. Sci. USA 102, 10598–10603.
Godiska, R., Chantry, D., Raport, C. J., Schweickart, V. L., Trong, H. L., and Gray, P. W. (1997).Monocyte chemotactic protein‐4: Tissue‐specific expression and signaling through CC chemo-kine receptor‐2. J. Leukoc. Biol. 61, 353–360.
Gong, J. L., Mc Carthy, K. M., Telford, J., Tamatani, T., Miyasaka, M., and Schneeberger, E.(1992). Intraepithelial airway dendritic cells: A distinct subset of pulmonary dendritic cellsobtained by microdissection. J. Exp. Med. 175, 797–807.
Gonzalo, J. A., Lloyd, C. M., Peled, A., Delaney, T., Coyle, A. J., and Gutierrez‐Ramos, J. C. (2000).Critical involvement of the chemotactic axis CXCR4/stromal cell‐derived factor‐1 alpha in theinflammatory component of allergic airway disease. J. Immunol. 165, 499–508.
Graeler, M., and Goetzl, E. J. (2002). Activation‐regulated expression and chemotactic function ofsphingosine 1‐phosphate receptors in mouse splenic T cells. FASEB J. 16, 1874–1878.
Hammad, H., Lambrecht, B. N., Pochard, P., Gosset, P., Marquillies, P., Tonnel, A. B., and Pestel, J.(2002). Monocyte‐derived dendritic cells induce a house dust mite‐specific Th2 allergicinflammation in the lung of humanized SCID mice: Involvement of CCR7. J. Immunol. 169,1524–1534.
Hammad, H., de Heer, H. J., Souillie, T., Hoogsteden, H. C., Trottein, F., and Lambrecht, B. N.(2003). Prostaglandin D2 modifies airway dendritic cell migration and function in steady stateconditions by selective activation of the DP‐receptor. J. Immunol. 171, 3936–3940.
Hammad, H., de Vries, V. C., Maldonado‐Lopez, R., Moser, M., Maliszewski, C., Hoogsteden,H. C., and Lambrecht, B. N. (2004). Differential capacity of CD8 þ alpha or CD8‐ alphadendritic cell subsets to prime for eosinophilic airway inflammation in the T‐helper type 2‐pronemilieu of the lung. Clin. Exp. Allergy 34, 1834–1840.

LUNG DENDRITIC CELL MIGRATION 275
Havenith, C. E. G., Van Miert, P. P. M. C., Breedijk, A. J., Beelen, R. H. J., and Hoefsmit, E. C. M.(1993). Migration of dendritic cells into the draining lymph nodes of the lung after intratrachealinstillation. Am. J. Respir. Cell Mol. Biol. 9, 484–488.
Hieshima, K., Imai, T., Baba, M., Shoudai, K., Ishizuka, K., Nakagawa, T., Tsuruta, J., Takeya, M.,Sakaki, Y., Takatsuki, K., Miura, R., Opdenakker, G., et al. (1997). A novel human CC chemokinePARC that is most homologous to macrophage‐inflammatory protein‐1 alpha/LD78 alpha andchemotactic for T lymphocytes, but not for monocytes. J. Immunol. 159, 1140–1149.
Holt, P. G., Haining, S., Nelson, D. J., and Sedgwick, J. D. (1994). Origin and steady‐state turnoverof class II MHC‐bearing dendritic cells in the epithelium of the conducting airways. J. Immunol.153, 256–261.
Hopken, U. E., and Lipp, M. (2004). All roads lead to Rome: Triggering dendritic cell migration.Immunity 20, 244–246.
Huh, J. C., Strickland, D. H., Jahnsen, F. L., Turner, D. J., Thomas, J. A., Napoli, S., Tobagus, I.,Stumbles, P. A., Sly, P. D., and Holt, P. G. (2003). Bidirectional interactions between antigen‐bearing respiratory tract dendritic cells (DCs) and T cells precede the late phase reaction inexperimental Asthma: DC activation occurs in the airway mucosa but not in the lung paren-chyma. J. Exp. Med. 198, 19–30.
Idzko, M., Hammad, H., van Nimwegen, M., Kool, M., Muller, T., Soullie, T., Willart, A. M.,Hijdra, D., Hoogsteden, H. C., and Lambrecht, B. N. (2006). Local application of FTY720 to thelung abrogates experimental asthma by altering dendritic cell function. J. Clin. Invest. 116,2935–2944.
Idzko, M., Hammad, H., van Nimwegen, M., Kool, M., Hoogsteden, H. C., and Lambrecht, B. N.(2007). Inhaled iloprost suppresses the cardinal features of asthma via inhibition of airwaydendritic cell function. J. Clin. Invest. (in press).
Jahnsen, F. L., Lund‐Johansen, F., Dunne, J. F., Farkas, L., Haye, R., and Brandtzaeg, P. (2000).Experimentally induced recruitment of plasmacytoid (CD123high) dendritic cells in humannasal allergy. J. Immunol. 165, 4062–4068.
Jakubzick, C., Tacke, F., Llodra, J., van Rooijen, N., and Randolph, G. J. (2006). Modulation ofdendritic cell trafficking to and from the airways. J. Immunol. 176, 3578–3584.
Kabashima, K., Sakata, D., Nagamachi, M., Miyachi, Y., Inaba, K., and Narumiya, S. (2003).Prostaglandin E2‐EP4 signaling initiates skin immune responses by promoting migration andmaturation of Langerhans cells. Nat. Med. 9, 744–749.
Kobayashi, H., Miura, S., Nagata, H., Tsuzuki, Y., Hokari, R., Ogino, T., Watanabe, C., Azuma, T.,and Ishii, H. (2004). In situ demonstration of dendritic cell migration from rat intestine tomesenteric lymph nodes: Relationships to maturation and role of chemokines. J. Leukoc. Biol.75, 434–442.
Lambrecht, B. N., and Hammad, H. (2003). Taking our breath away: Dendritic cells in thepathogenesis of asthma. Nat. Rev. Immunol. 3, 994–1003.
Lambrecht, B. N., Salomon, B., Klatzmann, D., and Pauwels, R. A. (1998). Dendritic cells arerequired for the development of chronic eosinophilic airway inflammation in response to inhaledantigen in sensitized mice. J. Immunol. 160, 4090–4097.
Lambrecht, B. N., De Veerman, M., Coyle, A. J., Gutierrez‐Ramos, J. C., Thielemans, K., andPauwels, R. A. (2000). Myeloid dendritic cells induce Th2 responses to inhaled antigen, leadingto eosinophilic airway inflammation. J. Clin. Invest. 106, 551–559.
Legge, K. L., and Braciale, T. J. (2003). Accelerated migration of respiratory dendritic cells tothe regional lymph nodes is limited to the early phase of pulmonary infection. Immunity 18,265–277.
Luft, T., Jefford, M., Luetjens, P., Toy, T., Hochrein, H., Masterman, K. A., Maliszewski, C.,Shortman, K., Cebon, J., and Maraskovsky, E. (2002). Functionally distinct dendritic cell (DC)

276 HAMIDA HAMMAD AND BART N. LAMBRECHT
populations induced by physiologic stimuli: Prostaglandin E(2) regulates the migratory capacityof specific DC subsets. Blood 100, 1362–1372.
Lukacs, N. W., Tekkanat, K. K., Berlin, A., Hogaboam, C. M., Miller, A., Evanoff, H., Lincoln, P.,and Maassab, H. (2001). Respiratory syncytial virus predisposes mice to augmented allergicairway responses via IL‐13‐mediated mechanisms. J. Immunol. 167, 1060–1065.
Lundy, S. K., Lira, S. A., Smit, J. J., Cook, D. N., Berlin, A. A., and Lukacs, N. W. (2005).Attenuation of allergen‐induced responses in CCR6‐/‐mice is dependent upon altered pulmonaryT lymphocyte activation. J. Immunol. 174, 2054–2060.
McWilliam, A. S., Nelson, D. J., Thomas, J. A., and Holt, P. G. (1994). Rapid dendritic cellrecruitment is a hallmark of the acute inflammatory response at mucosal surfaces. J. Exp.Med. 179, 1331–1336.
Medoff, B. D., Sauty, A., Tager, A. M., Maclean, J. A., Smith, R. N., Mathew, A., Dufour, J. H.,and Luster, A. D. (2002). IFN‐gamma‐inducible protein 10 (CXCL10) contributes to airwayhyperreactivity and airway inflammation in a mouse model of asthma. J. Immunol. 168,5278–5286.
Mellman, I., and Steinman, R. M. (2001). Dendritic cells: Specialized and regulated antigenprocessing machines. Cell 106, 255–258.
Nikolic, T., de Bruijn, M. F., Lutz, M. B., and Leenen, P. J. (2003). Developmental stages ofmyeloid dendritic cells in mouse bone marrow. Int. Immunol. 15, 515–524.
Ohl, L.,Mohaupt,M., Czeloth, N.,Hintzen, G., Kiafard, Z., Zwirner, J., Blankenstein, T.,Henning,G.,and Forster, R. (2004). CCR7 governs skin dendritic cell migration under inflammatory andsteady‐state conditions. Immunity 21, 279–288.
Ohta, A., and Sitkovsky, M. (2001). Role of G‐protein‐coupled adenosine receptors in down-regulation of inflammation and protection from tissue damage. Nature 414, 916–920.
Okamoto, S., Azhipa, O., Yu, Y., Russo, E., and Dennert, G. (1998). Expression of ADP‐ribosyl-transferase on normal T lymphocytes and effects of nicotinamide adenine dinucleotide on theirfunction. J. Immunol. 160, 4190–4198.
Okunishi, K., Dohi, M., Nakagome, K., Tanaka, R., and Yamamoto, K. (2004). A novel role ofcysteinyl leukotrienes to promote dendritic cell activation in the antigen‐induced immuneresponses in the lung. J. Immunol. 173, 6393–6402.
Olweus, J., BitMansour, A., Warnke, R., Thompson, P. A., Carballido, J., Picker, L. J., and Lund‐Johansen, F. (1997). Dendritic cell ontogeny: A human dendritic cell lineage of myeloid origin.Proc. Natl. Acad. Sci. USA 94, 12551–12556.
Osterholzer, J. J., Ames, T., Polak, T., Sonstein, J., Moore, B. B., Chensue, S. W., Toews, G. B., andCurtis, J. L. (2005). CCR2 and CCR6, but not endothelial selectins, mediate the accumulation ofimmature dendritic cells within the lungs of mice in response to particulate antigen. J. Immunol.175, 874–883.
Partida‐Sanchez, S., Goodrich, S., Kusser, K., Oppenheimer, N., Randall, T. D., and Lund, F. E.(2004). Regulation of dendritic cell trafficking by the ADP‐ribosyl cyclase CD38: Impact on thedevelopment of humoral immunity. Immunity 20, 279–291.
Peters, W., Scott, H. M., Chambers, H. F., Flynn, J. L., Charo, I. F., and Ernst, J. D. (2001).Chemokine receptor 2 serves an early and essential role in resistance to Mycobacteriumtuberculosis. Proc. Natl. Acad. Sci. USA 98, 7958–7963.
Peters, W., Cyster, J. G., Mack, M., Schlondorff, D., Wolf, A. J., Ernst, J. D., and Charo, I. F.(2004). CCR2‐dependent trafficking of F4/80dim macrophages and CD11cdim/intermediatedendritic cells is crucial for Tcell recruitment to lungs infected withMycobacterium tuberculosis.J. Immunol. 172, 7647–7653.
Pichavant, M., Charbonnier, A. S., Taront, S., Brichet, A., Wallaert, B., Pestel, J., Tonnel, A. B.,and Gosset, P. (2005). Asthmatic bronchial epithelium activated by the proteolytic allergen

LUNG DENDRITIC CELL MIGRATION 277
Der p 1 increases selective dendritic cell recruitment. J. Allergy Clin. Immunol. 115,771–778.
Pisabarro, M. T., Leung, B., Kwong, M., Corpuz, R., Frantz, G. D., Chiang, N., Vandlen, R.,Diehl, L. J., Skelton, N., Kim, H. S., Eaton, D, and Schmidt, K. N. (2006). Cutting edge:Novel human dendritic cell‐ and monocyte‐attracting chemokine‐like protein identified byfold recognition methods. J. Immunol. 176, 2069–2073.
Pollard, A. M., and Lipscomb, M. F. (1990). Characterisation of murine lung dendritic cells:Similarities to Langerhans cells and thymic dendritic cells. J. Exp. Med. 172, 159.
Power, C. A., Church, D. J., Meyer, A., Alouani, S., Proudfoot, A. E. I., ClarkLewis, I., Sozzani, S.,Mantovani, A., and Wells, T. N. C. (1997). Cloning and characterization of a specific receptor forthe novel CC chemokine MIP‐3 alpha from lung dendritic cells. J. Exp. Med. 186, 825–835.
Qu, C., Edwards, E. W., Tacke, F., Angeli, V., Llodra, J., Sanchez‐Schmitz, G., Garin, A., Haque,N. S., Peters, W., van Rooijen, N., Sanchez‐Torres, C., Bromberg, J., et al. (2004). Role of CCR8and other chemokine pathways in the migration of monocyte‐derived dendritic cells to lymphnodes. J. Exp. Med. 200, 1231–1241.
Reibman, J., Hsu, Y., Chen, L. C., Bleck, B., and Gordon, T. (2003). Airway epithelial cells releaseMIP‐3alpha/CCL20 in response to cytokines and ambient particulate matter. Am. J. Respir. CellMol. Biol. 28, 648–654.
Robbiani, F., Finch, R. A., Jager, D., Muller, W. A., Sartorelli, A. C., and Randolph, G. J. (2000).The leukotriene C4 transporterMRP1 regulates CCL19 (MIP‐3b, ELC)‐dependentmobilizationof dendritic cells to lymph nodes. Cell 103, 757–768.
Robert, C., Fuhlbrigge, R. C., Kieffer, J. D., Ayehunie, S., Hynes, R. O., Cheng, G., Grabbe, S.,von Andrian, U. H., and Kupper, T. S. (1999). Interaction of dendritic cells with skin endothelium:A new perspective on immunosurveillance. J. Exp. Med. 189, 627–636.
Saeki, H., Moore, A. M., Brown, M. J., and Hwang, S. T. (1999). Cutting edge: Secondarylymphoid‐tissue chemokine (SLC) and CC chemokine receptor 7 (CCR7) participate in theemigration pathway of mature dendritic cells from the skin to regional lymph nodes. J. Immunol.162, 2472–2475.
Sallusto, F., Kremmer, E., Palermo, B., Hoy, A., Ponath, P., Qin, S., Forster, R., Lipp, M., andLanzavecchia, A. (1999). Switch in chemokine receptor expression upon TCR stimulationreveals novel homing potential for recently activated T cells. Eur. J. Immunol. 29, 2037–2045.
Sanchez‐Sanchez, N., Riol‐Blanco, L., and Rodriguez‐Fernandez, J. L. (2006). The multiplepersonalities of the chemokine receptor CCR7 in dendritic cells. J. Immunol. 176,5153–5159.
Scandella, E., Men, Y., Legler, D. F., Gillessen, S., Prikler, L., Ludewig, B., and Groettrup, M.(2004). CCL19/CCL21‐triggered signal transduction and migration of dendritic cells requiresprostaglandin E2. Blood 103, 1595–1601.
Schall, T. (1997). Fractalkine: A strange attractor in the chemokine landscape. Immunol. Today 18,147.
Sertl, K., Takemura, T., Tschachler, E., Ferrans, V. J., Kaliner, M. A., and Shevach, E. M. (1986).Dendritic cells with antigen‐presenting capability reside in airway epithelium, lung parenchyma,and visceral pleura. J. Exp. Med. 163, 436–451.
Smith, A. L., and Fazekas De St Groth, B. (1999). Antigen‐pulsed CD8aþ dendritic cells generatean immune response after subcutaneous injection without homing to the draining lymph node.J. Exp. Med. 189, 593–598.
Sozzani, S., Allavena, P., Vecchi, A., and Mantovani, A. (1999). The role of chemokines in theregulation of dendritic cell trafficking. J. Leukoc. Biol. 66, 1–9.
Spiegel, S., and Milstien, S. (2003). Sphingosine‐1‐phosphate: An enigmatic signalling lipid. Nat.Rev. Mol. Cell Biol. 4, 397–407.

278 HAMIDA HAMMAD AND BART N. LAMBRECHT
Springer, T. A. (1994). Traffic signals for lymphocyte recirculation and leukocyte emigration: Themultistep paradigm. Cell 76, 301–314.
Starner, T. D., Barker, C. K., Jia, H. P., Kang, Y., and McCray, P. B., Jr. (2003). CCL20 is aninducible product of human airway epithelia with innate immune properties. Am. J. Respir. CellMol. Biol. 29, 627–633.
Steinman, R. M. (1991). The dendritic cell system and its role in immunogenicity. Annu. Rev.Immunol. 9, 271–296.
Steinman, R. M., and Nussenzweig, M. C. (2002). Avoiding horror autotoxicus: The importance ofdendritic cells in peripheral T cell tolerance. Proc. Natl. Acad. Sci. USA 99, 351–358.
Stumbles, P. A., Thomas, J. A., Pimm, C. L., Lee, P. T., Venaille, T. J., Proksch, S., and Holt, P. G.(1998). Resting respiratory tract dendritic cells preferentially stimulate T helper cell type 2(Th2) responses and require obligatory cytokine signals for induction of Th1 immunity. J. Exp.Med. 188, 2019–2031.
Suda, T., McCarthy, K., Vu, Q., McCormack, J., and Schneeberger, E. E. (1998). Dendritic cellprecursors are enriched in the vascular compartment of the lung. Am. J. Respir. Cell Mol. Biol.19, 728–737.
Swanson, K. A., Zheng, Y., Heidler, K. M., Zhang, Z. D., Webb, T. J., and Wilkes, D. S. (2004). Flt3‐ligand, IL‐4,GM‐CSF, and adherence‐mediated isolation ofmurine lung dendritic cells: Assessmentof isolation technique on phenotype and function. J. Immunol. 173, 4875–4881.
Thorley, A. J., Goldstraw, P., Young, A., and Tetley, T. D. (2005). Primary human alveolar type IIepithelial cell CCL20 (macrophage inflammatory protein‐3alpha)‐induced dendritic cell migration.Am. J. Respir. Cell Mol. Biol. 32, 262–267.
van Rijt, L. S., Prins, J. B., deVries, V. C., Leenen, P. J., Thielemans, K., Hoogsteden, H. C., andLambrecht, B. N. (2002). Allergen‐induced accumulation of airway dendritic cells is supportedby an increase in CD31hi Ly‐6Cneg hematopoietic precursors. Blood 100, 3663–3671.
van Rijt, L. S., Jung, S., Kleinjan, A., Vos, N., Willart, M., Duez, C., Hoogsteden, H. C., andLambrecht, B. N. (2005). In vivo depletion of lung CD11c þ dendritic cells during allergenchallenge abrogates the characteristic features of asthma. J. Exp. Med. 201, 981–991.
Vermaelen, K., and Pauwels, R. (2003). Accelerated airway dendritic cell maturation, trafficking,and elimination in a mouse model of asthma. Am. J. Respir. Cell Mol. Biol. 29, 405–409.
Vermaelen, K. Y., Carro‐Muino, I., Lambrecht, B. N., and Pauwels, R. A. (2001). Specificmigratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes.J. Exp. Med. 193, 51–60.
Vermaelen, K. Y., Cataldo, D., Tournoy, K., Maes, T., Dhulst, A., Louis, R., Foidart, J. M., Noel, A.,and Pauwels, R. (2003). Matrix metalloproteinase‐9‐mediated dendritic cell recruitment into theairways is a critical step in a mouse model of asthma. J. Immunol. 171, 1016–1022.
Wikstrom, M. E., Batanero, E., Smith, M., Thomas, J. A., von Garnier, C., Holt, P. G., andStumbles, P. A. (2006). Influence of mucosal adjuvants on antigen passage and CD4 þ T cellactivation during the primary response to airborne allergen. J. Immunol. 177, 913–924.
Yang, D., Chertov, O., Bykovskaia, S. N., Chen, Q., Buffo, M. J., Shogan, J., Anderson, M.,Schroder, J. M., Wang, J. M., Howard, O. M., and Oppenheim, J. J. (1999). Beta‐defensins:Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 286,525–528.

INDEX
AA proliferation-inducing ligand
(APRIL), 14–15, 32–33A-T. See Ataxia-telangiectasiaABC. See Activated B cellsActin cytoskeleton, 134–135, 156–157,
194, 201–202, 208, 210–213, 215, 232Actin regulating enabled (Ena), 207Activated B cells (ABC), 2, 5–6, 13Activation-induced deaminase (AID), 3
CD40 signaling, 25expression, regulation in CSR
AICDA gene, 13B cell-specific enhancer, 12NF-kB p50, 13Pax5, 12–13PKA, 13Smad proteins, 13Sp1, 12–13Sp3, 12–13STAT6, 13in human and mouse, 25–26IL-4, 25intronic enhancer, 25LPS, 25NF-kB p50, 25PKA phosphorylation sites, 25S38, 25–26STAT6, 25T27, 25–26
Activator protein 1 (AP-1), 14, 152transcription factor of, 128, 152
ADAP/SKAP-55 complex, 215ADP ribosylation factor (ARF), 204AF-6. See AfadinAfadin (AF-6), 236AI domain, 187–188, 190–192AICDA gene, 13, 25
279
AID. See Activation-induced deaminaseAirway DC migration, to LN
under inflammatory conditions
CD38 involvement, 272chemokine receptor involvement, 270cysteinyl leukotriene involvement, 271prostaglandin involvement, 271S1P involvement, 271under steady-state conditionsbone marrow-derived DCs, 269–270CCR7, 270CCR8, 270CD8a, 269in MLN, 269mDCs, 269pDCs, 269
Airway DC subsets, localization andphenotype of
CD11cþ myeloid, 267CD11chigh DCs, role of, 266–267pDCs, role of, 267
ALb2 unclasping, 191Allergen-based immunotherapy, 83
anti-IgE and RIT, 84–85anti-IgE and SIT, combination of, 84
Allergen-specific IgE-occupiedreceptors, 92–93
‘‘Allergic asthma,’’ 74–75Allergic diseases, 78
allergic rhinitis treatment, 80
CGP51901, 79omalizumab, 79in case studies, 81anaphylactic reactions, 83atopic dermatitis, 82omalizumab, 82SIT, 83

280 index
Allergic diseases (continued)definition of, 66latex sensitivity treatment, with
omalizumab, 81peanut sensitivity treatment, anti-IgE
studies on
with epinephrine, 80with omalizumab, 81with TNX-901, 80Allergy, as medical specialty, 83Alpha-amino-3-hydroxy-5-methyl-4-isoxazole
propionic acid (AMPA), 234AMb2, 187, 209AMPA. See Alpha-amino-3-hydroxy-5-
methyl-4-isoxazole propionic acidAnti-CD72 mAb, 123Anti-IgE, 73adverse reactions, 103‘‘allergic asthma’’
b2 adrenergic receptor agonists, 74corticosteroids, 74omalizumab medication, 75
antibodies, rationale/scope/pharmacologicalbasis of, 67
binding specificities, 68hybridoma clones, 69MAbs, 69mIgE, as immunological site, 69unique set of, 70
with CGP51901, 73–74clinical parameters of, 77
corticosteroid, use of, 75omalizumab treatment, 75–76
confirmation in asthma pathogenesisin allergic rhinitis, 77inhibitory effects of, 77
development of, major eventsCGP51901, 64CGP56901, 64omalizumab (E25), 64–65TNX-901, 64
disease-affected tissuesin allergen molecules, 100in mast cells, 99–100
good responders in patients,analyses of, 78
IgE cytokinergic propertiesneutralization, 98
IgE isotype-specific control/IgE targetingB cells, 68
IgE suppression, 67OKT3 antibody, 68T cell receptors, 68
immune defense functionIgE antibody class, 101–103in parasites, 102role of IgE, 102
immune reactivity attenuationCD23, role of, 99
long-term remission state, 100–101modulation of, IgE-committed B
lymphoblasts and memory B cell, 96in vivo mouse model, 97–98
omalizumab, clinical development of, 74prevailing concepts during invention, 71, 73therapeutic, chemical features of
CDR, 65CHO cell line, 66omalizumab, 65–66TNX-901, 66
unique binding specificities, structuralbasis of, 69–71
Antigen receptor signaling pathways inB cells, 151–152mast cells, 151–152T cells, 151–152
Antigen-peptide-MHC complex, 200Antigen-presenting cells (APC), 126–127,
186–187, 195–196, 200–201, 211,215, 239–241, 243, 266
AP-1. See Activator protein 1APC. See Antigen-presenting cellsAPE. See Apurinic/apyrimidic endonucleaseAPEX. See Apurinic/apyrimidic endonucleaseApolipoprotein B mRNA editing catalytic
polypeptide 1, 4APRIL. See A proliferation-inducing ligandApurinic/apyrimidic endonuclease (APE or
APEX), 5, 37–38ARF. See ADP ribosylation factorArtemis, 10–11, 36Ataxia-telangiectasia (A-T), 6, 11, 35, 38–40Ataxia-telangiectasia and RAD3-related
(ATR) protein, 6, 10–11, 34–35, 39ATM deficiency, in human and mousein A-T, 38–40CSR junctions, 39–40interswitch recombination, 39intraswitch recombination, 39mouse knockout models, 39

index 281
Sm�Sa, 39Sm�Sg, 39Sm�Sg1 junctions, 39
ATR. See Ataxia telangiectasia and RAD3AXb2, 187, 190–191
BB cell activation factor (BAFF), 14–15, 32–34B cell maturation antigen (BCMA), 14–15, 32B cell receptor (BCR), 32, 34, 68–69, 97, 104,
123–126, 148, 151, 155, 159, 165, 168,213, 244–246
B-cell chronic lymphocytic leukemia(B-CLL), 245–246
B-cell development and self-tolerance, roleof Rap1
autoreactivity, 244B1 cells, 246B1a cells, 244in B-CLL, 246in CD5þ B cells, 246IgA, 244IgG, 244OcaB, 244Oct1,2, 244‘‘self-sensing’’ signal, 245–246SPA-1�/�, 244 –246in SPA-1 KO mice, 244–246VH gene, 246Vk genes, 244–246Vl gene, 246
B-CLL. See B-cell chronic lymphocyticleukemia
BAFF. See B cell activation factorBAFF–APRIL–TACI pathway, in human
and mouseIgG, IgA, and IgE switching, 32TACI, 32TACI-deficient phenotype, 33TNFRSF13B, 32
BAP/TFII-I Btk phosphorylation, 151Base excision repair (BER), 3, 7, 8BCMA. See B cell maturation antigenBCR. See B cell receptorBCR signaling, 123–125, 155, 165BER. See Base excision repairBH. See Btk homologyBmx, 146, 156, 158–161BRDG1, 159Btk deficiency in mast cell, 169
Btk homology (BH), 146Btk in mast cell, function and signaling, 169Btk receptor signaling, 157
Cc-cb1, 159–160Calf-1, 188–190Cas–Crk complex, 156Caveolin-1, 160CCR7, 270–272CCR8, 270CD2 signaling protein, 157–158CD28 signaling protein, 157–158CD38 involvement, in airway DC migration,
to LN8-Br-cADPR, 272cADPR, 272NADþ, 272
CD40 signaling, 14, 25, 31CD40–CD40L interaction, 14CD40–CD40L pathway, in human and mouse
anti-CD40 antibodies, 32CSR pathways, 31HIGM, 31IgM serum levels, 31IL-10, 32
CD72 tyrosine-phosphorylation, 124CD8 T cell, development and function, 163CD94/NKG2C, 133CDC42/Rac-dependent serine/threonine
kinase, 158CemX domain in mIgE, 105CemX sequence, 104–105CGP51901, 64–65, 73, 79, 93, 97Chat-H, 206Chemokine, 87, 99, 135, 166, 186, 195–196,
198–200, 202–212, 230, 235, 247–248,266–268, 270–271
with APC, 201C-C-chemokine ligand 21, 197CXC chemokine ligand 12, 197CXCL13, 197receptor involvement, in airway DC
migration, to LN
CCL21, 272CCR7, 272in RunX3-deficient animals, 272Chemotaxis, 136, 200, 203–204, 208–210,268, 272
Chinese hamster ovary (CHO), 66

282 index
CHO. See Chinese hamster ovaryClasped aLb2, 191Clasped aXb2, 191Class switch recombination (CSR), 1in ABC, 3
IgM class, 2isotype switching, 2region-specific process, 2switch (S) regions, 2
AID, function ofAPOBEC-1, 4cytidine deamination, 4deamination of ssDNA, 4ectopic expression, 4editing of mRNA, 4in gene conversion, 4RGYW motifs, 5RPA, 4–5
APRIL, 14–15BAFF, 14–15CD40–CD40L interaction, 14cytokine differential regulation, in human
and mouseCg3 expression, 28IFN-g, 28IgG2, 28–29IL-10, 28IL-27, 29IL-4, 28–29LPS, 29‘‘switch factor,’’ 28
DNA damage response/repair pathways inIg gene diversification, 8–9
Artemis, 10MLH3, 10MMR proteins, 7polm role, 11TDT role, 11in XP-V patients, 11
DNA DSB resolutions inATM/ATR signaling, 6HR, 6–7NHEJ, 6–7
DNA repair factors andAPEX, in human and mouse, 37–38ATM deficiency, in human and
mouse, 38–40immunodeficiency, 37NBS1 deficiency, in human and
mouse, 40–42
pleiotropic phenotypes, 37UNG deficiency, in human and
mouse, 34, 37dU:dG mismatches in
APE1 (APEX1), 5APEX2, 5Mre11/Rad 50 pathway, 5Mre11/Rad50/NBS1 complex, 6MSH2-dependent pathway, 5
functional properties of IgG subclasses, inhuman and mouse
FcgPI receptors, 28IgG1, 27–28IgG2a, 28IgG2b, 28IgG3, 27–28
GL promoters, in human and mouse, 30ECS-Ig, 29NF-kB-binding sites, 29STAT6-binding site, 29
IgA1/IgA2, in human and mouse, 26IgH 30 enhancers, 12–13regulation of, 11
accessibility model, 12AID expression, 12–13
regulation to IgA, in human and mouse, 26IgA production in mMT mice and
Cm-deficient patients, 27SHM and
point mutations, 3SSB, 4
TGF-b1, in human and mouse, 26toll and toll-like receptor, 15V(D)J recombination and
RAG1, 3RAG2, 3site-specific process, 3
Common variable immunodeficiency(CVID), 31–32, 34
Complement receptor (CR), 187Complementarity-determining regions
(CDR), 65Constant region gene locus, in human
and mouseCg2a, 16Cg2b, 16IGHC gene, 15–16pseudo-g-genes, 16
Coronin1, 210Corticosteroids, 74–75, 78, 80

index 283
CR. See Complement receptorCromones, 92CSR. See Class switch recombinationCVID. See Common variable
immunodeficiencyCyclophilin, peptidyl-prolyl isomerase, 155Cysteine-string motif, 146Cysteinyl leukotriene involvement, in airway
DC migration, to LNABCC1, 271CCR7, 271LTC4, 271MRP1, 271
Cytokines, 2, 11–14, 28, 32–33, 40, 87, 98, 126,128, 135–138, 166, 230, 271
DD1PTPD1. See Protein-tyrosine phosphataseDAG. See DiasylglycerolDAP12, 132–134DC. See Dendritic cellsDC and monocyte chemokine-like protein
(DMC), 268DC recruitment, in lung, 269
airway epithelium, 268alveolar type 2 cells, 268in asthma, 268CCL20, 268CCR2�/�, 268CCR6�/�, 268chemokine, role in, 268chemotaxis, 268defensins, 268Der p 1, 268DMC, 268E-selectin, 267L-selectin, 267MHCIIþ DC precursors, 267MMP9�/�, 268in mouse model, 268P-selectin, 267role of, 267‘‘sentinels’’ of, immune system, 267VCAM-1, 267
Defensins, 268Dendritic cells (DC), 185–186, 265
in airways, 266in immune responses, 266inflammation, role of, 272–273migratory capacity of, 272
Diacylglycerol (DAG), 152, 213, 231Disodium cromoglycogate, 92DMC. See DC and monocyte
chemokine-like proteinDNA damage response/repair
pathways, 7–11DNA ligase IV, 6–7, 34, 36DNA-PKcs, 6bI domain, 188–193Double strand breaks (DSB), 3–5
repair mechanisms
HR, 6–7NHEJ, 6–7Double-positive (DP) thymocytes, 237DP. See Double-positiveDRap1. See Rap1 in Drosophila
melanogasterDSB. See DNA double strand breaks
EEBV. See Epstein-Barr virusECM. See Extracellular matrixECS-Ig. See Evolutionarily conserved
sequenceEGF. See Epithelial growth factorEna. See Actin regulating enabled30 Enhancers, in human and mouse
GLe and g2b promoters, 24HS3 enhancers, 24HS4 enhancers, 24a1HS1, 2, 24a2HS1, 2, 24
Epinephrine, 80, 253Epithelial growth factor (EGF), 189–190,
233–234, 253Epstein-Barr virus (EBV), 206ERK1/ERK2, 152Evolutionarily conserved sequence
(ECS-Ig), 29–31Extracellular matrix (ECM), 156,
200, 235–237
FFAK. See Focal adhesion kinaseFas, 158–159FceRI and IgE in Type I hypersensitivity.
See IgE and FceRI in Type Ihypersensitivity, roles of
FceRI activation-induced signaling, 167

284 index
FceRI downregulationanti-IgE, as mast ‘‘cell-stabilizing’’ agent
cromones, 92mast cells, role of, 91–92
IgE and FceRI, relationshipbetween, 91–92
insensitivity of FceRI�IgE; for amast cell
allergen concentration, 92–93FceRI density, 92–93
FceRI on CH3 domains, binding siteof a chain, 72
FceRI stimulation, 155Fetal thymic organ cultures (FTOC), 237Focal adhesion kinase, 156, 199FTOC. See Fetal thymic organ cultures
GG-protein b and g subunits, 158G-protein-coupled receptors (GPCR), 198,
203, 232Gbg. See G-protein b and g subunitsGAP. See GTPase-activating proteinsGAP-related domain (GRD), 232GEF. See Guanine exchange factor; Guanine
nucleotide exchange factorGenome sequences, 1–2Germ line (GL), 2–3, 11–14, 21, 24, 26–27,
29–31, 34, 37, 39, 41Germ stem cells (GSC), 248GFFKR motif, 188, 193–195, 206Gi-coupled signaling, 198GL. See Germ lineGL transcription, 11–13, 34, 39, 41Glutamic acid mutation, 192Glycosylphosphatidylinositol (GPI), 122GPCR. See G-protein-coupled receptorsGPCR-triggered signalsdevelopment of
leading edge, 203uropod structure, 203
integrin activation, 203GPI. See GlycosylphosphatidylinositolGrb10/Grb1R, 159GRD. See GAP-related domainGSC. See Germ stem cellsGTPase RhoH, 210GTPase-activating proteins (GAP), 232GTPases. See Guanine triphosphatasesGuanine exchange factor (GEF), 204, 212
Guanine nucleotide exchange factors(GEF), 231
Guanine triphosphatases (GTPases), 133–135,157, 199, 203–205, 208, 210–212, 215,229–230, 232
HH2AX, 6, 10–11, 34Hematopoiesis and Rap1 signal. See
Hematopoietic stem cells (HSC)Hematopoietic stem cells (HSC), 210, 250, 254deregulated Rap1 signal
BCR-ABL protein, 252–253CML, 251–253in NF1 þ/� mice, 252PI3K/AKT activation, 252Pten gene, 251–253in Pten�/� mice, 251–253in SPA-1�/� mice, 249, 251–253
regulated Rap1 signalc-Myc, 249in Drosophila, 248in GSC, 248in N-cadherinþ osteoblastic cells, 249in SPA-1�/� mice, 249niche, 248–249SDF-1, 249
Heterotrimeric G-protein, 158High endothelial venules (HEV), 186, 196,
197, 200, 204–205, 210in sequential adhesion steps, 195
Homeostatic hematopoiesis, by Rap1 andmyeloid leukemia, 250
Homologous recombination (HR), 2, 6–7, 9, 31HR. See Homologous recombinationHSC. See Hematopoietic stem cells
IIC3b-coated particles, 187ICAM. See Intercellular adhesion moleculeIFN-gpromoter region, 150transcription, 151
IgA, 27, 31–33, 36–42IgE and FceRI in Type I hypersensitivity,
roles ofanti-IgE pharmacological effects of, binding
to FceRI and FceRII, 87–88IgE-mediated allergic pathway
chemokines, 87

index 285
cytokines, 87lipid mediators, 87manifestation stage, 87prepacked mediators, 87sensitization stage, 85triggering stage, 85–86
IgE neutralization, 88IgE concentration versus IgE occupancy
of FceRI, 89–90total IgE and proportion of
allergen-specific IgE
‘‘hygiene hypothesis,’’ 89sensitivity of mast cells/basophils, 88–89IgE or IgE-expressing B cell targetingIgE-mediated allergic pathway attenuation
anti-CD23, 103IL-4, 104immune modulators, 103mIgE, epitope onB lymphoblasts, 104CemX sequence, 104–105memory B cells, 104
IgE-mediated allergyallergic diseases, 66allergic reactions, 66IgE roles, 66
IgE:anti-IgE immune complexes, beneficialeffects of
antigen trappers
antigen-binding sites, 95omalizumab treatment, 96clinical improvementcase studies, 94CGP51901, 93–94molecular/cellular pharmacological
mechanisms, 94omalizumab, 94
IL-4, 12–13, 25, 28–29, 31, 33–34,40–41, 89, 97, 99, 102–104, 127–128
Iloprost, 271Immunoglobulin (Ig), 2–3, 6–8,
13–14, 15–16, 20, 21, 29, 33, 38,244–245, 247
Immunological synapse and T-cellactivation, role of Rap1
LFA-1, 240–241Rap1GDP, 240Rap1GTP, 240–241SLP-76, 240–241in SMAC, 240
synapse formation, 240TCR signaling, 240
Immunoreceptor tyrosine-based inhibitorymotif (ITIM), 123–125, 133, 168, 242
Inositol-1,4,5-triphosphate (IP3), 152, 168Inside-out signaling, 186–187, 194, 198, 200,
203–204, 206, 209, 211–212, 214Integrin
adhesion receptor, role of, 186avidity regulation, 197conformational change
affinity regulation of bI domain, 192coherent model of, 191cytoplasmic domain, 193extensions of extracellulardomains, 190–191global changes of extracellular
domain, 189–190multiple affinity states of aI domain, 192regulation of aI domain
conformation, 192cytoplasmic domain, 187
a4 Integrins, 186–187, 196–197, 199–200,209, 272
b2 Integrin, 187, 190–191, 199, 204, 207–208,212, 214, 247–248
structure of, 188Intercellular adhesion molecule
(ICAM), 186, 196, 201Interleukin-1 receptor-associated kinase-1
(IRAK), 157IP3. See Inositol-1,4,5-triphosphateIRAK. See Interleukin-1 receptor-associated
kinase-1ITIM. See Immunoreceptor tyrosine-based
inhibitory motifItk
role in mast cells, 167SH2 domain, 155
JJAM-1. See Junctional adhesion molecule-1JUN amino-terminal kinase, 152Junctional adhesion molecule-1, 187
LLAD. See Leukocyte adhesion deficiencyLAT. See Linker for activation of T cellsLeukocyte adhesion deficiency (LAD), 187,
206, 247–248

286 index
Leukocyte integrinaffinity, 189avidity, 189conformational changes, 190–195genu, structural studies of, 190inside-out signaling, 187valency regulation, 189
LFA-1; aL/b2. See Lymphocytefunction-associated antigen
Lig4 gene, 6–7, 9, 36Ligand-binding headpiece, 187, 191, 198Linker for activation of T cells, 152, 167–172,
211–213Lymph nodes (LN), 26, 194–195, 197,
199–200, 206–207, 210, 247, 266, 268–273Lymphocyteinteraction, swarming pattern, 201role of Rap1
CD44, 247chemokine migration, 247homing of, 247in immunosurveillance, 247in LAD patients, 247–248in RapL�/� mice, 247Rap1 activation, 247–248transendothelial migration, 247
Lymphocyte b2 legpiece extension, 198Lymphocyte function-associated antigen, 186
MMAb. See Monoclonal antibodyMac-1, 187, 235MAdCAM. See Mucosal addressin cell
adhesion moleculeMammalian Ste20-like kinase MST1/
STK4, 207Matrix metalloproteinase (MMP), 268MDC. See Myeloid dendritic cellsMDS. See Myelodysplastic syndromeMediastinal draining lymph nodes
(MLN), 269–272Metal ion-dependent adhesion site, 188,
190–193MIDAS. See Metal ion-dependent
adhesion siteMismatch repair (MMR), 3, 5, 7–8, 10, 37MLN. See Mediastinal draining lymph nodesMMP. See Matrix metalloproteinaseMMR. See Mismatch repairMonoclonal antibody (MAb), 64, 68, 191
MPD. See Myeloproliferative disorderMre11, 5–6, 10, 34, 36MRP1. See Multidrug resistance-related
protein 1Mucosal addressin cell adhesion molecule, 186Multidrug resistance-related protein 1
(MRP1), 271Mus musculus, 1MyD88 adapter-like protein, 157Myelodysplastic syndrome (MDS), 251Myeloid cell-2 (TREM-2)–DAP12 complex, 132Myeloid dendritic cells (MDC), 265, 269, 271Myeloproliferative disorder (MPD), 238, 249,
250–251. See also Hematopoietic stemcells (HSC)
NNBS. See Nijmegen breakage syndromeNBS1, 6, 10–11, 34, 36, 40–42deficiency, in human and mouse
5-bp deletion hypomorphic allele, 41CSR defect, 41NBS1 mutation, 657del5, 40–41null mutation, 40
Nerve growth factor (NGF), 233–234NF-kB. See Nuclear factor-kBNFAT transcription factor, 152NGF. See Nerve growth factorNHEJ. See Nonhomologous end joiningNijmegen breakage syndrome (NBS), 7, 36,
40–41NKT cell development, 163Nonhomologous end joining (NHEJ), 3, 6–7, 9Nuclear factor-kB (NF-kB) activity, 13activation in B cells, 151p50 of, 25
OOmalizumab, 64, 65–66, 70, 74–76, 78–85, 94,
96–97, 103, 105, 10730 Phase II and III clinical trials, 106clinical utility of, 106
OVA. See OvalbuminOvalbumin (OVA), 269
PP13K. See Phosphatidylinositol 3-kinaseP21-activated kinase 1, 158Pax5, 12–13, 22, 25

index 287
pDC. See Plasmacytoid dendritic cellsPeroxisome proliferator-activated receptor-g
(PPAR-g), 271PH. See Plekstrin homologyPH/TH domain-mediated association of
Btk, 158Phosphalidylinositol (PI)-3-kinase
(PI3K), 42, 147–149, 154,167–169, 203–205, 212, 252
Phosphatidylinositol (3,4,5) triphosphate(PIP3), 147–149, 154–155, 158, 167, 168,204, 210, 251
PIP3. See Phosphatidylinositol (3,4,5)triphosphate
PKA. See Protein kinase APKC-z kinase activity, 205Plasmacytoid dendritic cells (pDC),
265–267, 269recruitment, to sites of inflammation
in asthma, 272CD62L, 272CXCR3, 272ICAM-1, 272a4 integrin, 272b2 integrin, 272in nasal allergy, 272PNAd, 272VCAM, 272Plekstrin homology (PH), 146–149, 151,154–156, 158–159, 167, 168, 204, 207,210, 214
PPAR-g. See Peroxisomeproliferator-activated receptor-g
Proline-rich region (PRR), 146, 150–151,154, 161–163
b-Propeller, 188–192Prostaglandin involvement, in airway DC
migration, to LNiloprost, 271PGD2, 271PGE2, 271PGI2, 271PPAR-g, 271
Protein kinase A (PKA), 13, 25–26Protein tyrosine kinases (PTK), 145,
161, 231, 234Protein-tyrosine phosphatase D1, 159PRR. See Proline-rich regionPRR mutant of Rch1a in Jurkat cells, 151PTK. See Protein tyrosine kinases
RRac pathways, 208–210Rap1. See Ras-proximity 1Rap1 in Drosophila melanogaster
(DRap1), 230, 233Rap1-binding protein (RAPL), 206, 213,
235, 241overexpression, 207
Rap1-interacting adaptor molecule(RIAM), 207, 236
Rap1A, 205, 213, 230, 238Rap1B, 205–206, 213, 230, 238RAPL. See Rap1-binding proteinRAS guanyl-releasing protein, 152Ras-proximity 1 (Rap1), 229
activation, regulation of
C3G, 231CalDAG-GEF I, 231CalDAG-GEF III, 231cAMP, 231E6TP1, 232Epac/Rap1, 232Epacs, role in, 232in GTPase activity, 232Rap1GAP, 232Rap1GDP (an inactive form), 230Rap1GEF, 231–232Rap1GTP (an active form), 230RapGA1, 232RapGA2, 232SPA-1, 232in budding yeasts, 230cell adhesion, control ofactin dynamics, regulation of, 236AF-6, 236DE-cadherin, 236in Drosophila, 236Ena/VASP, 236‘‘inside-out’’ activation, 235integrins, 235JAM1, 236–237LFA-1 activation, 235profillin, 236Rap1GTP, 236RapL, 235–236RIAM, 236SPA-1 overexpression, 235
in cancer, 253, 255BCR-ABL oncoprotein, 254‘‘conditional’’ oncoprotein, 254

288 index
Ras-proximity 1 (Rap1) (continued)
DOCK4 gene, mutations of, 254Rap1GAP transduction, 254SPA-1/741A alleles, 254in Drosophila melanogaster, 230exchange factor, 206gene-engineered mice, phenotypes
of, 238in hematopoiesis and leukemia
deregulated Rap1 signal andmyeloproliferative disorders, 249,251–253
HSCs, 248–249in platelet generation and function,
role of, 253in lymphocyte development and immune
responsesB-cell development and
self-tolerance, 244–246immunological synapse and T-cell
activation, 240–241lymphocyte migration and
homing, 247–248T-cell nonresponsiveness and
anergy, 241–243thymic T-cell development, 237–240
in mammals, 230MAPK activation, regulation of
B-Raf, role of, 233c-Raf-1, role of, 233–234DRap1, 233in hippocampal neurons, 234ligand-occupied EGF receptors, 234in MEK-1, 2–ERK pathway, 233in MEK-3, 6–p38MAPK
pathway, 234NGF receptors, 234in PC12 neuronal cell line, 233PI3K–AKT pathway, 234–235PI3Kp110, 234RalGDS, 234Rap1GTP, 234Ras-mediated ERK activation, 234
overexpression of, 207, 230regulation and functions of, 231roles of, 230‘‘self-sensing’’ signal in immature bone
marrow B cells, 245RASGRP. See RAS guanyl-releasing proteinRassf 5, 207
Receptor-proximal pathways in FceRIsignaling, 167
Recombinant integrins electronmicroscopic analysis, 190
Replication protein A (RPA), 4–5, 10, 13RHOH gene inactivation, 210RHOH-specific siRNA, 210RIAM. See Rap1-interacting adaptor
moleculeRIAM knockdown, 208RIT. See Rush immunotherapyRPA. See Replication protein ARush immunotherapy (RIT), 84–85, 101, 106
SS1P. See Sphingosine-1-phosphateS1P involvement, in airway DC migration,
to LNFTY720, 271in immune cells, 271
S38. See Serine 38Sab, overexpression in B cell, 155SAC. See Staphylococcus aureus Cowan ISak kinase, 159b-Sandwich hybrid domain, 189–190b-Sandwich module, 189Sema4Aexpression pattern of, 127involvement in T cell activation and
differentiation, 129receptors in immune system, 130Sema4A-Fc fusion protein, 127Th1 differentiation of, 128
Sema4DCD72 receptor for, 123CD72 interaction and, 123exogenous expression of, 123human Sema4D-Fc protein, 123in immune system, 123in T cell-mediated immunity, 126
Sema4D-CD72 interaction, 123B cell homeostasis, 124mechanism of, 124regulation of CD40, 124TLR4 signal, 124
Sema4D-Fc, 123Sema6D-Plexin-A1 interactionin cardiac development, 131in DC function, 131in osteopetrosis, 132

index 289
Sema7Aas monocyte stimulator, 135as negative regulator for T Cells, 136
Semaphorinbiological functions in immune
system, 121–138Class III semaphorin, 122, 137–138glycosylphosphatidylinositol, 122neuropilin, 122plexin-D1, 122
Serine 38 (S38), 25–2644-kDa serine/threonine kinase Pim-1, 158SH2-domain-containing leukocyte protein
of 76 kDa, 152SH3-domain binding protein, 155SHIP proteins
Dok-1, 154Dok-2, 154signaling of, 154
SHM. See Somatic hypermutationSingle-strand breaks (SSB), 3–5SIT. See Specific immunotherapySKAP-55, 214–215SLP-76. See SH2-domain-containing
leukocyte protein of 76 kDaSMAC. See Supramolecular activation
clustersSmad proteins, 13Sodium nedocromil, 92Somatic hypermutation (SHM), 3–11, 14,
37–38Specific immunotherapy (SIT), 83–84,
101, 106Sphingosine-1-phosphate (S1P), 205–206, 271SSB. See Single-strand breaksStaphylococcus aureus Cowan I (SAC), 28, 31STAT6, 13, 25, 29, 31Stem cell factor (SCF), 154, 210–211Supramolecular activation clusters
(SMAC), 200, 240SWAP-70, 210Switch (S) regions, in human and mouse, 42
characteristics of, 16, 19–20
structural, 17–18dot matrix analysis, 16, 19polymorphism of, 20–21secondary structures, 21–22Sg1, 17, 18, 20–21Sg3, 17–21Sm Sa, Se and Sg, 16, 19–20
TT27. See Threonine 27T cell receptors (TCR), 68, 146
complex, 200, 211T cell-APC conjugate, 201T-cell nonresponsiveness and anergy, role
of Rap1Cbl, 242CD44highmemory phenotype, 243CTLA-4, 241IL-2 gene, 242–243IL-2 response, 241in RapGAP transgenic mice, 242–243RapE63 transgene, 241Ras–ERK pathway, 242in SPA-1�/� mice, 243T-cell anergy, 242TCR/CD3-stimulation per se, 241
TACI. See Transmembrane activator andAML interactor
TalinaIIbb3 activation and, 208intracellular regulator, in lymphocyte
adhesion and migration, 201–202TCR. See T cell receptorTCR-mediated inside-out signals, 201TCR-stimulated lymphocytes, inside-out
signaling events, 211adaptor protein, 214PKD1, 214Rac signaling pathways, 212Tec family kinases, 212
TCR-triggered signals, 211Tec homology (TH), 146, 152, 155Tec kinase
activation, regulation of
by interdomain interactions, 161–162intramolecular domaininteractions, 161–162by tyrosine phosphorylation, 160
in antigen receptor signaling pathway, 151C-terminal kinase domain, 145cytoskeletal components interactions
with, 156–157interacting protein, 153level of, 146membrane localization of, 148negative regulators interactions
with, 153–155nuclear localization, 150–151

290 index
Tec kinase (continued)overview of, 145–146regulation of, 146roles in mast cells, 167SH3 domain, 145Src homology (SH)2 domain, 145subcellular localization, regulation of
membrane recruitment and, 147in T cell, 145–172
Tec/SHIP/Dok complex, 154Tetherto-synapse dynamic, 201TFII-I-dependent transcriptional activation
in COS7 cell, 151TH. See Tec homologyTherapeutic anti-IgE antibody, 64Threonine 27 (T27), 25–26Thymic T-cell development, roles of
Rap1 and RasCAG promoter, 237in DP thymocytes, 237in ERK activation, 238in FTOC, 237–238‘‘hyperexcitability,’’ 239–240pre-TCR-mediated signal, 239RapE63 transgene, 237in b�selection, 238–239SPA-1 transgene, 237in transgenic mice, 237–240
Timloci, 130Tim-1, 130Tim-2, 130Tim-3, 130
TKB. See Tyrosine kinase bindingTLR. See Toll-like receptorTLR4. See Toll-like receptor 4TLR4 signal transduction, 157TLR9 pathway, 15TNX-901, 64, 66, 70, 80–81, 105–107Toll and Toll-like receptor, 15in human and mouse
CpG DNAs, 33–34in CVID patients, 34TLR4 expression, 33TLR9, 33–34
Toll-like receptor (TLR), 157Toll-like receptor 4 (TLR4), 15, 33, 124, 157Transmembrane activator and CAML
interactor (TACI), 14, 32–33Tyrosine kinase binding (TKB), 159Tyrosine phosphatase SHP-1, 123Tyrosine phosphorylation, 123–124, 130,
133, 147–148, 155, 160
UUracil DNA glycosylase (UNG), 5, 7–8deficiency, in human and mouse
HIGM2, 34, 37HIGM5, 34, 37knockout model, 37MSH2 pathway, 37SMUG1, 37
noncanonical role, 42
VV(D)J recombination, 3, 7, 10–11, 34Vaccinia virus semaphorin A39R, 137Vascular cell adhesion molecule
(VCAM), 186Vasodilator-stimulated phosphoprotein
(VASP), 207–208VASP. See Vasodilator-stimulated
phosphoproteinVav, guanine nucleotide exchange factor, 157VCAM. See Vascular cell adhesion moleculeVEGFR2, 131–133Viral semaphorin, 137
XX-linked agammaglobulinemia, 157Xeroderma pigmentosum variant (XP-V), 11XRCC4, 6–7, 9

Contents of Recent Volumes
Volume 83Lineage Commitment and Developmental
Plasticity in Early LymphoidProgenitor SubsetsDavid Traver and Koichi Akashi
The CD4/CD8 Lineage Choice: New Insightsinto Epigenetic Regulation during T CellDevelopmentIchiro Taniuchi, Wilfried Ellmeier, andDan R. Littman
CD4/CD8 Coreceptors in ThymocyteDevelopment, Selection, and LineageCommitment: Analysis of the CD4/CD8Lineage DecisionAlfred Singer and Remy Bosselut
Development and Function of T Helper 1 CellsAnne O’Garra and Douglas Robinson
Th2 Cells: Orchestrating Barrier ImmunityDaniel B. Stetson, David Voehringer,Jane L. Grogan, Min Xu, R. Lee Reinhardt,Stefanie Scheu, Ben L. Kelly, andRichard M. Locksley
Generation, Maintenance, and Function ofMemory T CellsPatrick R. Burkett, Rima Koka,Marcia Chien, David L. Boone, andAveril Ma
CD8þ Effector CellsPierre A. Henkart and Marta Catalfamo
291
An Integrated Model of ImmunoregulationMediated by Regulatory T Cell SubsetsHong Jiang and Leonard Chess
Index
Volume 84Interactions Between NK Cells and
B LymphocytesDorothy Yuan
Multitasking of Helix-Loop-Helix Proteinsin LymphopoiesisXiao-Hong Sun
Customized Antigens for DesensitizingAllergic PatientsFatima Ferreira,Michael Wallner, andJosef Thalhamer
Immune Response Against DyingTumor CellsLaurence Zitvogel, Noelia Casares,Marie O. Pequignot,Nathalie Chaput,Mathew L. Albert,and Guido Kroemer
HMGB1 in the Immunologyof Sepsis (Not Septic Shock)and ArthritisChristopher J. Czura,Huan Yang,

292 contents of recent volumes
Carol Ann Amella, andKevin J. Tracey
Selection of the T-Cell Repertoire:Receptor-Controlled Checkpoints inT-Cell DevelopmentHarald Von Boehmer
The Pathogenesis of Diabetes in theNOD MouseMichelle Solomon andNora Sarvetnick
Index
Volume 85Cumulative Subject Index
Volumes 66–82
Volume 86Adenosine Deaminase Deficiency:
Metabolic Basis of ImmuneDeficiency and PulmonaryInflammationMichael R. Blackburn andRodney E. Kellems
Mechanism and Control of V(D)JRecombination Versus Class SwitchRecombination: Similarities andDifferencesDarryll D. Dudley, Jayanta Chaudhuri, CraigH. Bassing, and Frederick W. Alt
Isoforms of TerminalDeoxynucleotidyltransferase:Developmental Aspects and FunctionTo-Ha Thai and John F. Kearney
Innate AutoimmunityMichael C. Carroll and V. Michael Holers
Formation of Bradykinin: A Major Contributorto the Innate Inflammatory ResponseKusumam Joseph and Allen P. Kaplan
Interleukin-2, Interleukin-15, and Their Rolesin Human Natural Killer CellsBrian Becknell and Michael A. Caligiuri
Regulation of Antigen Presentation and Cross-Presentation in the Dendritic Cell Network:Facts, Hypothesis, and ImmunologicalImplicationsNicholas S. Wilson andJose A. Villadangos
Index
Volume 87Role of the LAT Adaptor in T-Cell Development
and Th2 DifferentiationBernard Malissen, Enrique Aguado, andMarie Malissen
The Integration of Conventional andUnconventional T Cells that CharacterizesCell-Mediated ResponsesDaniel J. Pennington, David Vermijlen,Emma L. Wise, Sarah L. Clarke,Robert E. Tigelaar, and Adrian C. Hayday
Negative Regulation of Cytokine and TLRSignalings by SOCS and OthersTetsuji Naka, Minoru Fujimoto,Hiroko Tsutsui, and Akihiko Yoshimura
Pathogenic T-Cell Clones in AutoimmuneDiabetes: More Lessons from theNOD MouseKathryn Haskins
The Biologyof Human LymphoidMalignanciesRevealed by Gene Expression ProfilingLouis M. Staudt and Sandeep Dave
New Insights into Alternative Mechanisms ofImmune Receptor DiversificationGary W. Litman, John P. Cannon, andJonathan P. Rast
The Repair of DNA Damages/ModificationsDuring the Maturation of the Immune

contents of recent volumes 293
System: Lessons from Human PrimaryImmunodeficiency Disorders andAnimal ModelsPatrick Revy, Dietke Buck,Franc,oise le Deist, andJean-Pierre de Villartay
Antibody Class Switch Recombination: Rolesfor Switch Sequences and MismatchRepair ProteinsIrene M. Min and Erik Selsing
Index
Volume 88CD22: A Multifunctional Receptor That
Regulates B Lymphocyte Survival andSignal TransductionThomas F. Tedder, Jonathan C. Poe, andKaren M. Haas
Tetramer Analysis of Human AutoreactiveCD4-Positive T CellsGerald T. Nepom
Regulation of Phospholipase C-g2 Networksin B LymphocytesMasaki Hikida and Tomohiro Kurosaki
Role of Human Mast Cells and Basophils inBronchial AsthmaGianni Marone, Massimo Triggiani,Arturo Genovese, and Amato De Paulis
A Novel Recognition System for MHC Class IMolecules Constituted by PIRToshiyuki Takai
Dendritic Cell BiologyFrancesca Granucci, Maria Foti, andPaola Ricciardi-Castagnoli
The Murine Diabetogenic Class IIHistocompatibility Molecule I-Ag7:Structural and Functional
Properties and Specificity ofPeptide SelectionAnish Suri and Emil R. Unanue
RNAi and RNA-Based Regulation of ImmuneSystem FunctionDipanjan Chowdhury andCarl D. Novina
Index
Volume 89Posttranscriptional Mechanisms Regulating
the Inflammatory ResponseGeorg Stoecklin Paul Anderson
Negative Signaling in Fc Receptor ComplexesMarc Daeron and Renaud Lesourne
The Surprising Diversity of Lipid Antigens forCD1-Restricted T CellsD. Branch Moody
Lysophospholipids as Mediators of ImmunityDebby A. Lin and Joshua A. Boyce
Systemic MastocytosisJamie Robyn and Dean D. Metcalfe
Regulation of Fibrosis by the Immune SystemMark L. Lupher, Jr. and W. Michael Gallatin
Immunity and Acquired Alterations inCognition and Emotion: Lessons from SLEBetty Diamond, Czeslawa Kowal,Patricio T. Huerta, Cynthia Aranow,Meggan Mackay, Lorraine A. DeGiorgio,Ji Lee, Antigone Triantafyllopoulou,Joel Cohen-Solal Bruce, and T. Volpe
Immunodeficiencies with AutoimmuneConsequencesLuigi D. Notarangelo, Eleonora Gambineri,and Raffaele Badolato
Index

294 contents of recent volumes
Volume 90Cancer Immunosurveillance and
Immunoediting: The Roles of Immunity inSuppressing Tumor Development andShaping Tumor ImmunogenicityMark J. Smyth, Gavin P. Dunn, andRobert D. Schreiber
Mechanisms of Immune Evasion by TumorsCharles G. Drake, Elizabeth Jaffee, andDrew M. Pardoll
Development of Antibodies and ChimericMolecules for Cancer ImmunotherapyThomas A. Waldmann and John C. Morris
Induction of Tumor Immunity FollowingAllogeneic Stem Cell TransplantationCatherine J. Wu and Jerome Ritz
Vaccination for Treatment and Prevention ofCancer in Animal ModelsFederica Cavallo, Rienk Offringa,Sjoerd H. van der Burg, Guido Forni, andCornelis J. M. Melief
Unraveling the Complex RelationshipBetween Cancer Immunity andAutoimmunity: Lessons from Melanomaand VitiligoHiroshi Uchi, Rodica Stan, Mary Jo Turk,Manuel E. Engelhorn, Gabrielle A. Rizzuto,Stacie M. Goldberg, Jedd D. Wolchok, andAlan N. Houghton
Immunity to Melanoma Antigens: FromSelf-Tolerance to ImmunotherapyCraig L. Slingluff, Jr., Kimberly A.Chianese-Bullock, Timothy N. J. Bullock,William W. Grosh, David W. Mullins,Lisa Nichols, Walter Olson, Gina Petroni,Mark Smolkin, and Victor H. Engelhard
Checkpoint Blockade in CancerImmunotherapyAlan J. Korman, Karl S. Peggs, andJames P. Allison
Combinatorial Cancer ImmunotherapyF. Stephen Hodi and Glenn Dranoff
Index
Volume 91A Reappraisal of Humoral Immunity Based on
Mechanisms of Antibody-MediatedProtection Against Intracellular PathogensArturo Casadevall and Liise-anne Pirofski
Accessibility Control of V(D)J RecombinationRobin Milley Cobb, Kenneth J. Oestreich,Oleg A. Osipovich, and Eugene M. Oltz
Targeting Integrin Structure and Functionin DiseaseDonald E. Staunton, Mark L. Lupher,Robert Liddington, and W. Michael Gallatin
Endogenous TLR Ligands and AutoimmunityHermann Wagner
Genetic Analysis of Innate ImmunityKasper Hoebe, Zhengfan Jiang,Koichi Tabeta, Xin Du, Philippe Georgel,Karine Crozat, and Bruce Beutler
TIM Family of Genes in Immunityand ToleranceVijay K. Kuchroo, Jennifer Hartt Meyers,Dale T. Umetsu, andRosemarie H. DeKruyff
Inhibition of Inflammatory Responses byLeukocyte Ig-Like ReceptorsHoward R. Katz
Index
Volume 92Systemic Lupus Erythematosus: Multiple
Immunological Phenotypes in aComplex Genetic Disease

contents of recent volumes 295
Anna-Marie Fairhurst, Amy E. Wandstrat,and Edward K. Wakeland
Avian Models with SpontaneousAutoimmune DiseasesGeorg Wick, Leif Andersson, Karel Hala,M. Eric Gershwin,Carlo Selmi, Gisela F.Erf, Susan J. Lamont, and Roswitha Sgonc
Functional Dynamics of NaturallyOccurring Regulatory T Cells in Healthand AutoimmunityMegan K. Levings, Sarah Allan,Eva d’Hennezel, and Ciriaco A. Piccirillo
BTLA and HVEM Cross Talk RegulatesInhibition and Costimulation
Maya Gavrieli, John Sedy, ChristopherA. Nelson, and Kenneth M. Murphy
The Human T Cell Response toMelanoma AntigensPedro Romero, Jean-Charles Cerottini, andDaniel E. Speiser
Antigen Presentation and theUbiquitin-Proteasome System inHost–Pathogen InteractionsJoana Loureiro and Hidde L. Ploegh
Index