a method for simultaneous determination of h2s and so2 in flue gases
TRANSCRIPT

Atmospheric Environment Pergamon Press 1969. Vol. 3, pp. 407416. Printed in Great Britain.
A METHOD FOR SIMULTANEOUS DETERMINATION OF H,S AND SO, IN FLUE GASES
CARL-ELIS BOXRGM and CYRILL BROSSET*
Department of Inorganic Chemistry, Chalmers Institute of Technology and University of Gothenburg, Sweden
(First received 21 October 1968 and in final form 18 December 1968)
Abstmct-A discontinuous method has hcen developed for the simultaneous determination of H2S and SOa in flue gases. The method is based on a pure gas phase separation whereby HIS is adsorbed on a silica gel column impregnated with silver-sulphate (Ag#L) and potassium- hydrogensulphate (KHSO.), while SO1 is subsequently absorbed in a wash-bottle contain@ a slightly acidic hydrogen peroxide solution. Owing to the acid properties of the impregnated gel, SO2 passes through the column. The experiments show that the penetration of SOI is quantita- tive while HIS is adsorbed quantitatively on the column. The method permits the determin- ation of amounts of H*S in the range of l-1500 mgNm-’ with a sampling time of 5 min and a gas flow of 024.0 1. min- *. The lower limit for the determination of SOI under the above conditions liea at about 1 mgNm-‘. The separation of H,S and SO, is not influenced by the presence of moderate amounts of water but the interferenm of ammonia is more serious. Ammonia is adsorbed on the column and tbe properties of the gel are changed SO that SOa is also adsorbed. The method is still however, valid for H,S. The interferena from organic sul- phur compounds is negligible with the procedure. Hitherto the method has successfully been used lo analyse HIS in ambient air in the ppb-range as 24&r mean values.
1. INTRODUCTION
THE METHOD was initially intended to be used for the determination of H2S and SO2 in flue gases from cellulose plants and primarily from black liquor recovery boilers but should be applicable to other types of flue gases containing HIS in the presence of other sulphur compounds. The circumstances are often such, e.g. with a recovery boiler, that the concentration of H2S is low but that of SO2 high.
Investigation showed that there were neither methods nor instruments available which were suitable for the determination of HIS in the presence of greater amounts of SOS. The methods which have hitherto been used to determine HIS can be roughly divided into the following groups.
I.
I. Methods for the analysis of aqueous solutions of H2S (a) Conductometric methods (b) Coulometric methods (c) Calorimetric methods II. Adsorption on impregnated papers III. Gas chromatography
Analysis of aqueous solutions of H,S It is necessary to take into consideration possible reaction between HIS and SOz:
8H2S +4S02~Ss +4H,O and the formation of polythionic acids: 3H,O +H,S +~SO,Z~S~O~~- +4H+, e.g. formation of S,O,‘- where n>, 3.
l this work has been performed in cooperation with the Swedish Water and Air Pollution Re- search Laboratory, Stockholm.
407

408 CARL-ELISBOSTR~M and CYRILLBROSSR
I(a) Conductomezric methods. In general the methods are rather sensitive to dis- turbances from acid or alkaline components in the gas-phase. The most common absorption medium is slightly acidic copper sulphate solution.
I(b) Coulometry. The method has, up to now, only found application in one instru- ment although the basic principle seems to be suitable for the determination of H,S. In this instrument the sum of oxidizable sulphur compounds is determined. By the use of selective filters i.e. a filter which absorbs one of the two components, e.g. adsorbs H,S but passes SO2 the instrument can be made specific for certain sulphur com- pounds. It is possible to eliminate H,S and to measure SO, but not vice versa It would therefore be necessary to first measure the sum of the two components and then to measure SO2 by eliminating H2S. The method would not thus give results of the required accuracy if the H2S concentration was low in comparison with that of soz.
I(c) Calorimetry. The discontinuous methods most commonly used for the determin- ation of H2S are the methylene blue method BAYER and WAGNER (1960); BOLTZ (1958); JACOBS (1949) and the molybdenum blue method BUCK and STRATKAN (1964). In both cases H,S is absorbed in a slightly alkaline Cd(OH)2 or Zn(OH)@uspension (or a slightly acidic zinc solution), the H2S being bound in the form of CdS or ZnS. The following objections may be raised: The concentration of S2- decreases with increasing storage time of the samples, i.e. S 2- is oxidized in this alkaline environment. Oxidation probably also occurs in the alkaline aqueous phase during sampling time since the gas sample contains oxygen and this can lead to an error of up to 250/, (BUCK and STRATMAN (1964)). Since the molybdenum blue method is a redox reaction all oxidizing and reducing agents (e.g. SO2 and CH,SH) ought, in principle, to interfere.
II. Adsorption on impregnated papers The methods based on the absorption of H2S on papers impregnated with e.g. lead
acetate (JACOBS (1949)) are rough and, in principle, discontinuous. Besides which, the organic sulphur compounds cause a black colouring, which diminishes the specificity of the method. High concentrations of SO2 also afIect the colour of the HsS-stain and in extreme cases the spot may be completely masked.
III. Gas chromatography At present there are no detectors which are sufficiently accurate to indicate small
amounts of H2S and SO2 (lower limit 0.01 vol-‘A). In the future, however, j?-absorp- tion or electron capture detectors may perhaps solve this analytical problem. More- over sampling causes certain problems. If these components are enriched on a silica gel column SO, will be adsorbed considerably more strongly than H,S. This means that the break-through volume of H,S on the column determines the size of the sample volume the desorption of SO2 at higher temperatures and the length of the column which can be used. Besides which, it is to be expected that other components present in the flue-gases can change the adsorption capacity of the silica gel for HIS and SO*.
2. METHOD
H,S and SO1-mixtures were prepared by mixing suitable amounts of H2S and SOz respectively, with argon in steel tubes to a total pressure of I5 atm and a concentration corresponding to 1000 mgNm_’ for H2S and 10,000 mgNmm3 for SOs. The con-
A Method for Simultaneous Determination of HIS and SO1 in Flue Gases 409
centratioas of the SO,-mixtures remained constant for several months but the H$- concentration in the H,S mixtures decreased at a rate of about 20 mgNm3 per day. The tubes were conditioned before use with gas mixtures of approximately the same
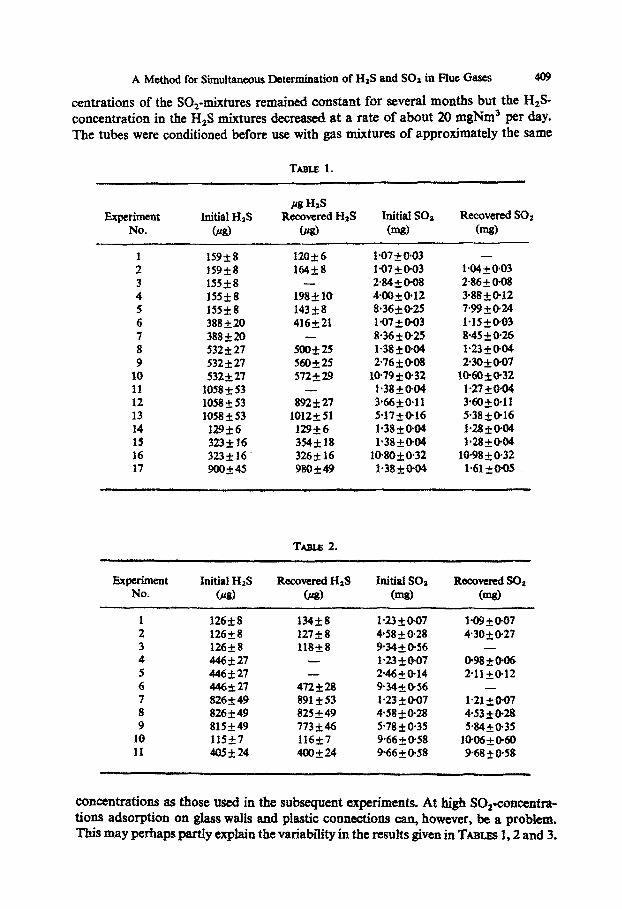
TABLE I.
Experiment No.
w HaS Initial HIS Recovered HsS InitiaI SOa Recovered SO2
0 w (ms) (mg)
1 159i8 120*6 1.07 + 003 -
2 159i8 164+8 1*7*@03 lW+0*03 3 15528 - 2w,oo8 >86*OG8 4 155rt8 198klO 4m+o*12 3BitF12 5 155i8 14358 8+36f @25 799+@24 6 388i20 436221 1*071tw3 1*152@03 7 388i20 - 8.361025 8.45 +, 0% 8 532k27 500*25 1*38*@04 1*23+@04 9 532rt27 560+25 2.76& 0.08 23O+M)7
10 532f27 572229 lo-79+@32 1@60+_@32 11 1058 * 53 - 1*38+@04 1*27++04 12 1058 f 53 892*27 3*66*@11 3*60+@11 13 1058 f 53 1012+51 5*17+016 5.38+@16 14 129k6 12926 1*38*OoQ P28*Cm 15 323i 16 354&l% 1.38+0*04 sC!8+w4 16 323+16 326+16 1@80+0~32 10.98 5 @32 17 900,+45 98oi49 1.38 *Ow 1.61 kO-05
Turrrs 2.
Exjnximent Initial HIS No. w
Recmered HzS C!JSJ
Initial SOI Recovwed SOi (m8) (mp)
1 12648 2 126+_8 3 126f8 4 446*27 5 446f27 6 446k27 7 826i49 8 826*49 9 815k49
10 11557 11 405jz24
134kB 127k8 118+8
- -
472f23 891i53 825+49 773*46 1161-7 400224
123*097 4.58iO.28 9342@56 lQi~OG7 2.46kO.14 9.34+@56 1*23*0+7 +58+0.28 5.78 f 0.35 9*66+@58 9*66+@58
1*09+OG7 430+,@27
-
098+0+x 2*11+012
- l-21 20-07 4.53ko.28 5*84*035
1006+_@60 9*68t@58
concentrations as those used in the subsequent experiments. At high SO~-comxntra- tions adsorption on glass wails and plastic connections can, however, be a problem. This may perhaps partly explain the variability in the results given in TABLES I,2 and 3.
410 CARL-ELIS BO~TRGM and CYWL BROSS~
The H,S-mixture was analysed continually by direct absorption in an acidic ammon- ium molybdate solution according to the molybdenum blue method.
Experiments showed that from a practical point of view, taking into consideration
TABLE 3.
Experiment NO.
Initial I&S Recovered H,S
(fi@ (li&
Initial SO2
(m& Recovered SOz
b43)
1 15218 2 512-1-g 3 15258 4 149+8 S 149rt8 6 149+8 7 S42&27 8 542 + 27 9 542 + 27
10 10671ts4 11 1067f 54 12 1067+54
* * *
123+_6 1351t7 M&7 S12+26 559528 S34+ 27
122Ok21 1169fSP 1200+60
1.41 k&O6 5.28 + O-26
11*10e0’ss 1.41 fO.07 S.28*@26
11*10+055 1.41 &O-O7 2.82*0*14
11-10f0~55 1.41 &O-21 5.28 f 026 6.68 f O-33
l-33+0-07 561 f0.28
ll~S4jrO~S8 1.29 rt 0.06 485 & 0.24 9*84&0”19 144*0*07 2.77&0+14
l@lS4@Sl 1.31 *o-o7 4+8f0’24 6-36&O-32
* Means that the analysis has faikd for some reason.
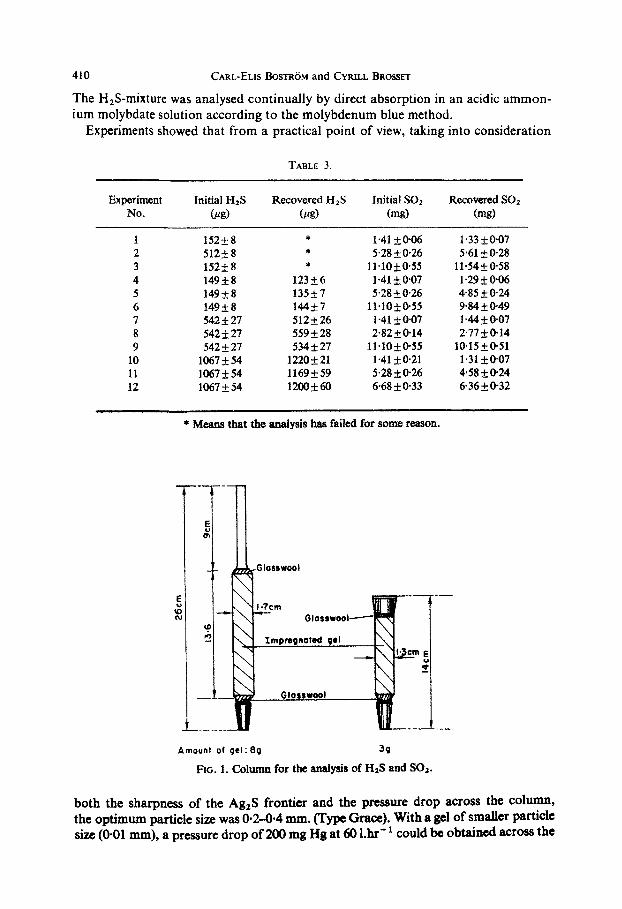
Amount of 9~1: 89 39
FIG. 1. Column for the analysis of HIS and Sot.
both the sharpness of the Ag,S frontier and the pressure drop across the column, the optimum particle size was O-2-0-4 mm. (Type Grace), With a gel of smaller particle size (O-01 mm), a pressure drop of 200 mg Hg at 60 l.hr” could be obtained across the

A Method for Simultaneous Determination of HzS and SO1 in Flue Gases 411
cotwnn wltich is hardly desirable from a practical point of view. After experiments we
arrived at the foltowiug simple method for impregnating the gel: A solution of b ml 5% KHSQa solution and b ml silver sulphate solution, saturated at room temperature, was added under stirring, to a beaker containing b g gel. After thorough mixing, the gel was dried (at 1 ICW) for 12 hr. The impregnated gel was then packed in columns of the type illustrated in FIG. 1. The different columns contain about 3 and 8 g gel
P -R
Fx?. 2, Apparatus &x s&&&g absorption and separation of HIS and SO,; D 3 dosing arrangement for air of atmospheric presswe; Vi and Vs : OHS pipettes of known volume; K = c&mm with impregnated gel; TV J Wash-b&e containing 1 per ant &Or and HCiO~ fpI3 = 4.5) P = Pump with rotary transformer; R = Rotameter.
Flio. 3. ant for dewrption of I&S frum the impregnated gei; D = dosing arrangement for Ns of atmospheric pressure; A 3 Reverschd wash-bottle containing 25 ml 100 g S~Cl~/looO ml. CO~C. HCI; B = dcsorpticm bottle; C and D = Wash bottles ContainEng &if+ and diluted I%CI @3X! ml cone. ‘i;lct+rloo rnI H&l>;
E = “Drop catcher”; F = Impinger with sintered glass disc containing 50 ml absorption solution; 0 = a&a&q P = Pump with rotary transformer; R = Rotameter.
respectively. ft is not advisable to use “acid-wasb;hed” gel and the gel sbaufd be dried before impregnation.
To attain a material balance in the adsorption experiments the apparatus shown in FIG. 2 was used. Gas pipettes of different volumes were f&ed after evacuation with known concentrations of the SOz and H,S-mixtures, respectively. The pipettes were connected in series and the gas mixtures were driven through the column with purified

412 CARL-ELIS E#OSTR~~M and CYRILL BROSSET
air as carrier gas. SO, was absorbed in a wash bottle containing a slightly acidic hydrogen peroxide solution. (PM = 4.5 with HCIO,). This procedure with separate pipettes was used in all subsequent experiments since the preparation of gas mixtures of H$ and SO, of known concentrations proved to be extremely difficult. The SO2 which passed through the column was determined as sulphate in the H,O,-solution according to the torin-method PERSWN (1966).
H,S was desorbed from the column according to a method described by BUCK and GIES (1966) using Sn2 + and cont. HCI and the apparatus illustrated in FIG. 3. The gel is first poured into bottle B and the solution in A is then transferred to B by starting the pump. The H,S liberated is finally absorbed in F; C and D are used to absorb the hydrogen chloride which escapes from A. The desorption must be carried out under carefully controlled conditions and with a low gas flow (10 l.hr-‘) in order not to change the pH in the absorption solution F. A teflon or glass stirrer is necessary in B to insure that all the gel comes in contact with the desorption solution. The desorption time is 15 min and after this the pH in F should be checked (most simply by means of an indicator paper).
TABLE 4. THE SAME GAS MMTURE IS ANALYSED BY TWO D- MRTHODS: (l) THE MoLyBDENuM BLUE MElHOD m THE MODIFIED HYDROOJJN PERO= WlliOD
Sample No.
(1) ~18 &S/ml-’
1 2 3 4 5 6 7 8
1.16 1.13 1.15 0.93 0.70 0.95 0.87 1.01
(II) Leg W/ml- 1 1.28 1.16 1.17 0.97 0.76 1.07 0.98 1.06
H2S was analysed by the following two different methods: (1) the molybdenum blue method and (2) a modification of the hydrogen peroxide method.
(1) The molybdenum blue method was carried out according to the procedure described by BUCK and Gm.q (1966), i.e. H,S was absorbed in an acidic solution of ammonium molybdate.
(2) By increasing the pH of the hydrogen peroxide solution to 9.5 with ammonia it was possible to obtain a 98% absorption in an impinger with a sir&red glass disc and a quantitative oxidation of E&S to HzS04 in the absorption solution. By this pro- cedure both SO, and HIS could be analysed by the same method which is an advant- age. TABLE 4 shows the results of the analysis of some gas mixtures by the two d&rent methods (1) and (2). A gas pipette with a mixture of H2S and Ar was connected in series with an absorber containing a smtered glass disc. The gas mixtures were driven through the absorption solution with air at a flow rate of 20 l.hr-‘.
3. EXPERIMENTAL
All experiments have been carried out at room temperature (22°C) and under an estimated pressure of 760 mm Hg. The gas mixtures were drawn through the columns

A Method for Simultaneous Determination of HIS and SOz in Flue G~.SCS 413
with a reciprocator pump and their rates of flow determined with calibrated rota- meters.
4. RESULTS
The results of experiments in which the gas flow was 20 Lhr” and the ex&mentaI time 15 min are given in TABLE 1. As we found that the length of the column darkened due to Ag,S-formation did not vary appreciably for flow-rates between 10 and 240 l.hr- 1 we chose a flow rate of 15 or 20 l.hr - ’ for the experiments.
The molybdenum blue method was used for the analysis of H2S and the to&- method for SOz. Estimated errors were f 5% for the former method and f 3% for the latter. The blank value for the desorption of pure gel using the molybdenum blue method was 2.5 i-0.7 pg H&g gel.
To study the interference of humidity on the separation of HaS and SO2 the carrier gas was allowed to bubble through water to achieve saturation with water vapour at the relevant temperature (22”C, e.g. 20 g HzO/m3). The water content of the gas mixture was determined by weighing the column containing the gel before and after the experiment. The gas flow was 15 lbr” and the experimental time 15 min. The results are shown in TABLE 2.
The results for the modified toxin-method for the analysis of HaS are given in TABLE 3. Gas flow and experimental time as before.
The interference of organic sulphides was studied by simultaneous adsorption of H,S and methylmercaptan (CH,SH) on the column. It was found that H2S and CH3SH separated on the c&m.n as a black layer of Ag,S followed by a yellow layer of Ag(CH,S). This is in accordance with what could be expected from a consideration of the solubility products of Ag,S and Ag(CH,S). Desorption showed that CH,SH only raised the blank value from 7 f 2 m H$/ g pure gel to 28 f 12 pg HzS/ g gel when the impregnated gel was saturated with CH3SH (920 pg g-l gel).
It is to be expected from the properties of the column that ammonia will be ad- sorbed. This was shown to be the case and the adsorption appeared to be quantitative. The gel was impregnated with an indicator (I’bymol blue) in order to study the change in the acid properties of the gel. Three gas pipettes containing H$-SO,-mixtures and pure 3260 c(g NH3, were coupled in series and the amounts of HI,S and SO, were systematically varied between 100 and 1000 pg. The gas-flow was 15 l.hr” and the experimental time 15 min. On adsorption of NH, by the gel to form Ag(NH3)a+, the indicator assumed its alkaline colour and a sharp boundary was observed between this layer and the preceding layer of AgaS, which is a weaker complex than Ag,(NI-I,),+. It was further evident from the experiments that the method still worked for HzS although SO, was bound nearly quantitatively to the column and desorption from the gel proved to be impossible. This indicates that SO? is oxidized to SOb2- as it is not possible to reduce SO,‘- to H,S with Sn2+ and cont. HCl.
The relation between the length of the column which was blackened and the amount of H2S has been studied under different conditions. It was shown that the way in which the column was packed and the method of impregnation were of great import- an=. Although it is possible to obtain linear calibration curves for a specific batch of gel and a certain gas flow, it is difficult to obtain accurate general curves. Our experi- en= is that it is possible to determine the H&concentration in a gas mixture only to an accuracy of + 10% by measuring the length of the Ag,S-layer on the column.
414 CARL-ELIS Bosrai% and CYRLL Bxoss~r
6. DISCUSSION AND RECOMMENDED METHOD FOR FIELD MEASUREMENTS
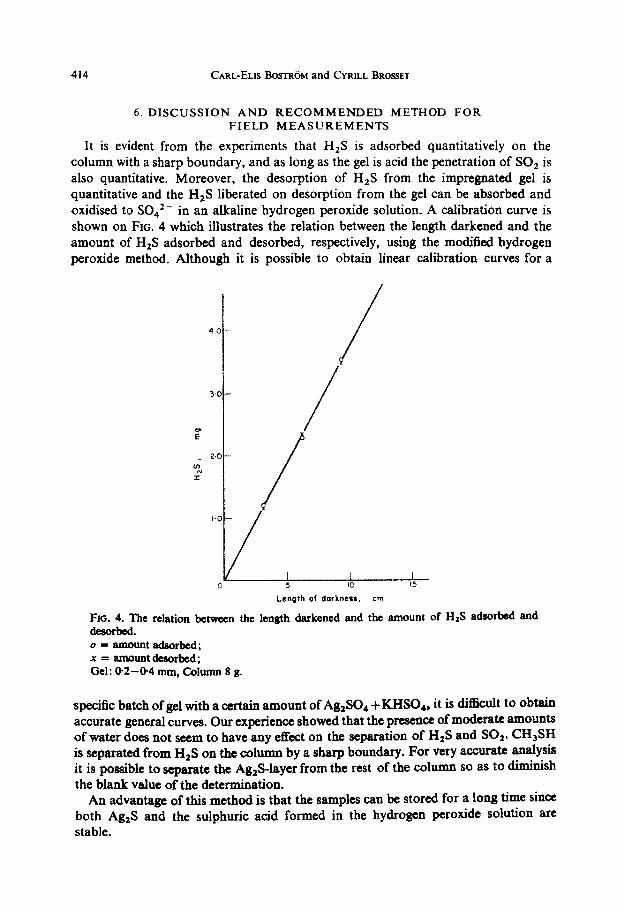
It is evident from the experiments that H2S is adsorbed quantitatively on the column with a sharp boundary, and as long as the gel is acid the penetration of SOz is also quantitative. Moreover, the desorption of H2S from the impregnated gel is quantitative and the H2S liberated on desorption from the gel can be absorbed and oxidised to SO,’ - . m an alkaline hydrogen peroxide solution. A calibration curve is shown on FIG. 4 which illustrates the relation between the length darkened and the amount of HIS adsorbed and desorbed, respectively, using the modified hydrogen peroxide method. Although it is possible to obtain linear calibration curves for a
Length of dorknetr.
FICL 4. The relation between the length darkened and deaorbed. 0 = amount adaorlJcd; x=amountdeEiorbed; Gel: @2-04 mm, Cdunn 8 g.
cm
the amount of H2S adsorbed and
specific batch of gel with a certain amount of Ag,SO., + KHS04, it is difficult to obtain accurate general curves. Our experience showed that the presence of moderate amounts of water does not seem to have any effect on the separation of H$ and SO1. CH,SH is separated from H2S on the oolumn by a sharp boundary. For very accurate analysis it is possible to separate the Ag,S-layer from the rest of the column so as to diminish the blank value of the determination.
An advantage of this method is that the samples can be stored for a long time since both Ag,S and the sulphuric acid formed in the hydrogen peroxide solution are stable.

A Method for Simultaneous Determination of HIS and SOS in Flue Gases 415
In some flue gases the water control provides a serious problem in sampling. It is, however, known that H$ and SO2 neither interact with each other nor are adsorbed on CaC12 as long as the CaC12:H20 molar ratio is greater than unity. This procedure is suitable when taking gas chromatographic samples, e.g. CO, COz, O2 in the same gas stream used for the H,S and SOz analysis. Otherwise, the water vapour can be condensed before analysis of H2S and SOz. The difficulty with this procedure is that there is considerable absorption of SO1 and H,S in the condensed water.
The apparatus recommended for use in field measurements is shown in FIG. 5. It consists of a column of CaC12 to remove the greater part of water vapour from the flue gas, a column with impregnated gel, a wash-bottle containing 50 ml 3% slightly acidic hydrogen peroxide solution, a reciprocator pump, a “dry meter” with thermo- meter and a rotameter. The speed of the reciprocator pump is regulated by means of a rotary transformer making it possible to vary the gas flow from O-2 to 4.0 l.min- ‘. It is thus possible to analyse H2S and SOz in the interval l-1500 mgNmB3 at a sample time of 5 min and to vary the gas flow and the sample time in order to obtain optimum conditions. For larger amounts of H2S and SOz it is possible to expand the range of measurement by dilution with e.g. nitrogen before entrance to the column.
Fro. 5. Apparatus for determination of HIS and SOS in flue gases; A = Probe containing CaClr and connection to sample probe; K = C&mn containing impregnated gel; TV = wash bottle containing 3 per cent H,Ot and HCIO‘ @ii = 4-5); T=d@lgtiIXXWith8ililXgCl; R1 and Rt = rotameters; P = pump with rotary transformer; Nz = tube containing nitrogen.
When packing the columns a small layer of gel impregnated only with KHSOb and a pH-indicator may be included. By this method sharper Ag,S-boundaries are obtained and it is possible to check that the acidity of the column is unchanged.
The method can be applied to environmental measurements by prolonging the sample time. With a sample time of 24 hr and a gas flow of 1.2 1. min-’ it is possible to analyse 2 ppb H,S.
This method has been tested under practical conditions in Sweden (for instance in process study of the sulphate process with black liquor recovery boilers) with good

416 CARL-ELIS BOSTR~M and CYRILL BROSSET
results. Concentrations of H,S greater than O*OS”/O were checked by means of gas chromatography.
The method would appear to be useful because of its wide generality and the columns could also be used in connection with continuous instruments for registering SO, in which one wishes to eliminate the interference of H2S.
Acknowledgements-The authors wish to thank ING K. ANDREAWN and Mrs E. M. LORD for invaluable assistance with the experimental work, the Swedish Water and Air Pollution Research Laboratory for making it possible to test the method under practical conditions and finally “1964 Brs Naturresursutredning” for financial assistance.
They are grateful to SVSAN JAGNER, M.A. (Cantab), 6Llic. for revising the English text of this paper.
REFERENCES
BAYER F. and WAGS J. (1960) Gasanalyse, Me&den akr Arbeitsprarir, Ferdinand Enke, Stuttgart. BoLTZ D. F. (1958) Calorimetric determination of non-metals, hteracience, NCW York. JACOBS M. B. (1949) 77te adytical chemistry of industrialpoisons, hazards and solvents, Interscience,
New York. BUCK M. and STRATMAN H. (1964) Bestimmung von Schweeflwasserstoff in der Atmosphilre. Staub
24,241-50. hItssoN G. A. (1%6) Automatic calorimetric determination of low concentrations of sulphate for
measuring SO2 in ambient air. Inf. J. Air War. Polht. 10,845-852. BUCK M. and GIES H. (1966) Die Messung von HIS in der Atmosptire. Kombinierta HzS und
SOZ-MCWUQL Staub 26.379-384.