4;11 translocation in acute lymphoblastic leukemia: a specific syndrome
TRANSCRIPT

4;11 Translocation in Acute Lymphoblastic Leukemia: A Specific Syndrome
Helvise G. Morse, Richard Heideman, Taru Hays, and Arthur Robinson
ABSTRACT: Three patients with acute lymphoblastic leukemia (ALL) having t(4;11) (q21;q23) are described. Their clinical characteristics are compared with ten other published cases all involving similar histories and poor prognoses.
INTRODUCTION
Van den Berghe et al. [1] first reported a new chromosome anomaly, a (4;11) (q13;q22) reciprocal translocation, which they believed was nonrandom and occurred only in acute lymphoblastic leukemia (ALL). Their search of the literature showed a case reported by Oshimura et al. [2] involving the same if not identical translocation. Prigogina et al. [3] reported four cases involving a similar translocation, two showing it as the only aberration and two showing possible evolution to hyperdiploidy. Prigogina brought to our attention case 3 reported by Goh et al. [4], which appears to be this type of ALL.
In the following report we describe three additional patients having translocation 4;11. We have reviewed the laboratory and clinical findings for our patients and have compared them to those for the patients described in previous publications.
OBSERVATIONS
Since July 1979, three of 54 ALL patients examined in this laboratory have had translocation 4;11. This small group is remarkable for its initial high WBe count and poor prognosis. These patients are compared in Table 1 with patients previously reported.
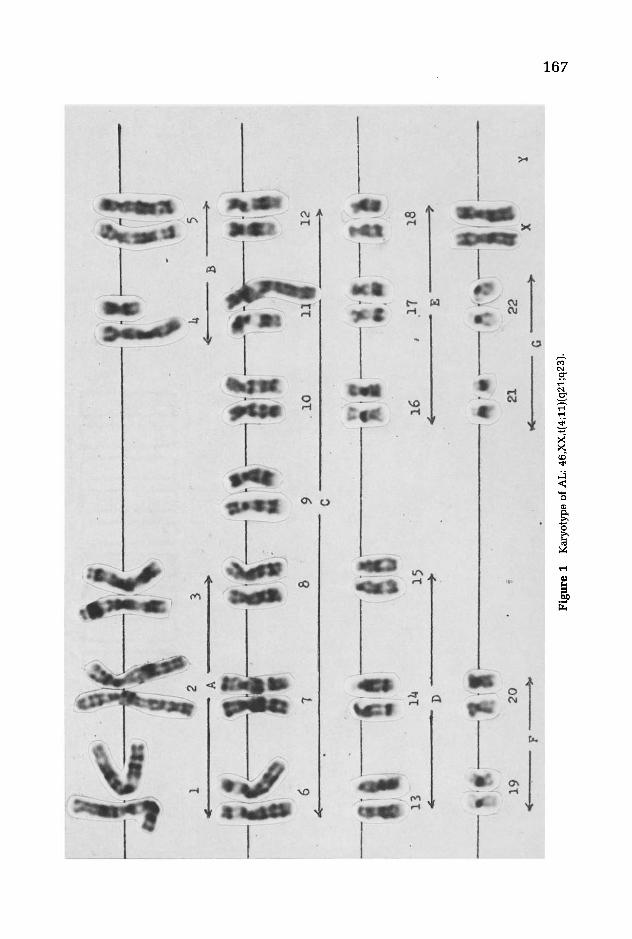
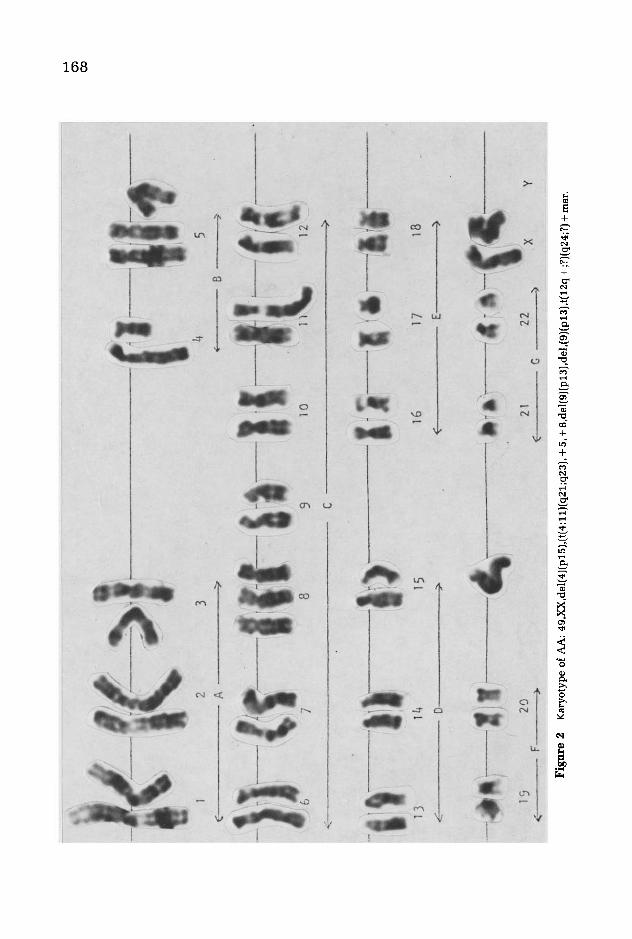
A karyotype of patient AL is presented in Figure 1. The only abnormality is the 4;11 translocation, and 25 of 25 cells examined showed this aberration. In Figure 2 is a karyotype of AA. In addition to the translocation other abnormalities are present. Her modal count is 49, with two trisomies 5 and 8 and a marker chromosome.
From the Department of Biochemistry, Biophysics, and Genetics, University of Colorado School of Medicine, Denver (H.G.M., A.R.J, the Department of Oncology and Hematology, Children's Hospital, Denver (R.H., T.H.), and National Jewish Hospital and Research Center (A.R.).
Address requests for reprints to Dr. Helvise G. Morse, Department of Biochemistry, Biophysics, and Genetics University of Colorado School of Medicine, 4200 East Ninth Avenue, Denver, CO 80262.
Received October 13, 1981; accepted February 3, 1982.
© Elsevier Science Publishing Co., Inc., 1982 52 Vanderbilt Ave., New York, NY 10017
165
Cancer Genetics and Cytogenetics 7, 165-172 (1982) 0165-4608/82/100165-08$2.75
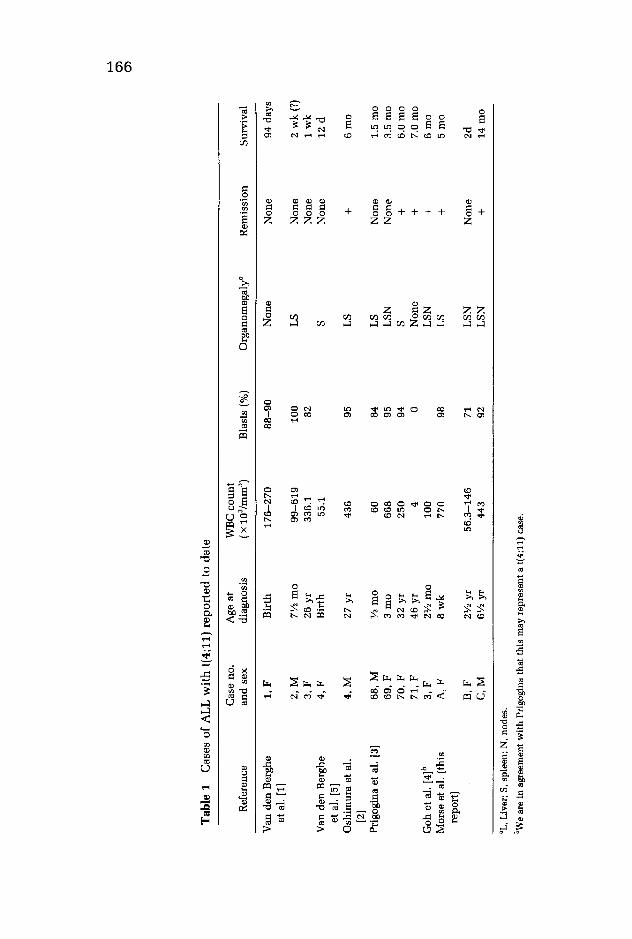
Tab
le 1
C
ases
of
AL
L w
ith
t(4
;11)
rep
orte
d to
dat
e
Cas
e no
. A
ge a
t W
BC
co
un
t R
efer
ence
an
d s
ex
diag
nosi
s (x
103 /r
nrn3
)
Van
den
Ber
ghe
1, F
B
irth
1
76
-27
0
et a
l. [1
) 2
,M
7'12
rna
99
-61
9
3,F
26
yr
336.
1 V
an d
en B
ergh
e 4
,F
Bir
th
55.1
et
al.
[5)
Osh
irnu
ra e
t al
. 4
,M
27 y
r 43
6 [2
) P
rigo
gina
et
al.
[3)
68,
M
'12 r
na
60
69
,F
3 rn
a 66
8 7
0,F
32
yr
250
71,
F 46
yr
4 G
oh e
t al
. [4
)b
3,F
2'1
2 rn
a 10
0 M
orse
et
al.
(thi
s A
, F
8w
k
770
repo
rt)
B,F
2'1
2 y
r 5
6.3
-14
6
C,M
6'1
2 y
r 44
3
"L, L
iver
; S,
spl
een;
N,
node
s.
bWe
are
in a
gree
men
t w
ith
Prig
ogin
a th
at t
his
may
rep
rese
nt a
t(4;
11)
case
.
Bla
sts
(%)
Org
anor
nega
lya
88
-90
N
on
e
100
LS
82
S
95
LS
84
LS
95
LS
N
94
S 0
Non
e L
SN
98
L
S
71
LS
N
92
LS
N
Rem
issi
on
No
ne
No
ne
Non
e N
one
+
No
ne
Non
e +
+
+
+
No
ne
+
Sur
viva
l
94 d
ays
2 w
k (
?)
1 w
k
12 d
6 rn
a
1.5
rna
3.5
rna
6.0
rna
7.0
rna
6 rn
a 5
rna
2d
14 r
na
.....
C1'l
C1'l
l J1
IIIItI
) \.
. .~
~ \ " 't' ,11
; \.('
11// .~
\'
.,
1 ,
, '
" ~,
'
1 4
5 .L
...
a ~
-II
. \ r
7 6
il' .. (1'
'.
f ~~
\ \
i
\ ,~
'\'
/ \
'.
"
8 9
.10
1
2
.J
c
-Iii!
It
lil,
11
1
~lrl
' ~Ir
\,\
~ \"
""
,-,.
13
l~
15
16
.17
IB
1>
-'--
----
---
E
'
1:'l
i l".).'
lll'..--
-:I.fl··
'. '--1,
,
\"
\ "
\. ",
'{';.
,-'~
~ -,
,' ,
~I
19
2
0
'j
21
22
\.
}'
r
----
+0
tII('"
X
'! G
F
Fig
ure
1
Kar
yoty
pe o
f A
L:
46,X
X,t(
4;11
)(q2
1;q2
3).
.....
O"l
'1
--
--------
i.if: .. "I!
'I. i.:l
r'·····
··~.i: t
'~'\
I ~
, "
; 1
/ ..
.•
\ I." /
2 3
4 5
:------A
)r
( 8
~
;1.:
~t-'
.l~I
;1
,II!
...
' /
8 9
10
i ~
C
>
I~i'
. .A
·
i-Ill:
,11
[·1''(
-'\
, '.'
\, ',,'
,-00 ,,
';
'13
14
15
16
17
18
<£
0
i?
<7 E
~
i .
i .'
If \
I.
-.
i dl'
\ 't
' ')\1';
.~
.. , \
: .\.'
\ 1,'
;/1"
...
'.' .
} ",<
)
\ ...
... ):a1i~~/
I" .)\,
J
19
f £
-.
20
21
22
('
G
') y
. -~------------.
Fig
ure
2 K
aryo
type
of
AA
: 49
,XX
.del
(4J(
p15)
.(t(
4;11
)(q2
1;q2
3). +
5, +
8.de
l(9J
(p13
).de
l.(9
)(p1
3).t
(12q
+ ;?
J(q2
4;?)
+ m
ar.
.... O'l co

4;11 Translocation in ALL 169
Chromosome No.4, which has donated most of its long arm in the translocation, also has a deletion of its short arm. All 33 cells examined showed the abnormal karyotype.
The karyotype in Figure 3 is from a relapse specimen of DK showing an isochromosome 7q in addition to the translocation. Initially chromosomes No.7 were normal, and at least 11 of 20 cells showed the translocation. In other cells it was impossible to determine its presence because of blurred and poor bands. During remission phytohemagglutinin (PHA)-stimulated blood showed a normal male karyotype for DK.
CASE REPORTS
Case A
Case B
AL was an 8-week-old white female found to have ALL after a brief history of irritability and poor feeding. A physical examination at the time of admission revealed the liver and spleen palpable to 5 cm below their respective costal margins. A chest x-ray was unremarkable, and an intravenous pyelogram (IVP) showed markedly enlarged kidneys with calyceal splaying. A lumbar puncture showed the presence of leukemic cells in the cerebrospinal fluid (CSF). Further laboratory determinations revealed normal BUN and creatinine levels, a uric acid level elevated to 7.8 mg%, a LDH level elevated to 608 IU, and an elevated GOT level of 44 IU. A complete blood count (CBC) revealed a hematocrit of 22%, a platelet count of 90,000/mm3
,
and a WBC count of 770,000/mm3 with 98% blasts with variable morphologic features. A bone marrow examination showed 67% blasts with coarsely granular periodic acid-Schiff (PAS)-positive cytoplasm. One percent of the marrow cells rosetted with sheep erythrocytes, 1% contained surface membrane immunoglobulin, and the remaining 98% were unlabeled. Morphologically the lymphoid cells present in the marrow and peripheral smear ranged between typical FAB-L1 leukemic cells and atypical lymphoid cells. Electron microscopy confirmed the lymphoid origin of all these cells. The patient was treated with standard leukemic induction therapy for ALL and attained remission by day 28. Cranial irradiation (1800 rads) and several doses of intrathecal methotrexate were given as treatment for central nervous system (CNS) leukemia. Within 5 months of diagnosis, the patient had a bone marrow relapse and did not respond to reinduction therapy. She died shortly thereafter of bacterial sepsis.
AA was a 2Yz-year-old female of Sioux Indian and black parents. She was found to have ALL after 1-week history of lethargy, low-grade fever, and abdominal pain. On admission a physical examination showed a moderately ill female with generalized shotty lymphadenopathy and a markedly enlarged distended abdomen. The liver edge was palpable 7 cm below the right costal margin and the spleen 10 cm below the left costal margin. A large, firm mass filling the entire anterior abdomen was later identified as massively enlarged kidneys. A chest x ray was normal. A CBC showed an hematocrit of 23%, a platelet count of 31,000/mm3
, and a WBC count of 56,300/mm3 with 71 % blasts. A bone marrow aspiration was performed which showed complete replacement of the marrow by cells that were morphologically identifiable as lymphoblasts (FAB-Ll). Special stains revealed only block PAS-positive activity in the cytoplasm of these cells. Fourteen percent of the bone marrow-derived cells rosetted with sheep erythrocytes, 2% contained surface-bound immunoglobulin, and

(ILl
~ ~,
i--f.' -------,
. !
i i
\ \
" ;
1.\
\ : ,
/
\, )
2 3
. ' '.
1;"\
~ \.
' I,
\
i--lI~If-
1'. '1M
... ~
, ,
tl I
I 1
, >
;',
!)
,I ~ \'
6 7
8
fl/~
I "
\'
". '
( '}i
\ i
\ \
".
. .....
,.)
13
14
15
« 0
'>
-,--
,
19
20
""--
-F ).
4 5
'" B
)
"1' i
i---
-;
; !
\. \
' ,
~."",i
~
9 \,
~ ,
>
c---
----
----
----
----
--
16
17
I8
( E
)
7 I ".. ,,-~
21
Fig
ure
3 R
elap
se k
aryo
type
of
DK
: 46
,XY
,t(4;
11)(
q21;
23),
i(7q
).
~~-----~
---.
-,
'I
~
'I
o

4;11 Translocation in ALL 171
CaseC
86% were unlabeled. Other laboratory determinations revealed normal immunoglobulin levels and an elevated LDH level of 1940 IU. The uric acid level was 13 mg/dl, the BUN level 64 mg/dl, and the creatinine clearance 2.3 mg/dl.
Over the initial 12 hours after admission, the patient's WBC count increased to 146,000/mm3
• Concomitant with this, there was marked and rapid deterioration of her clinical and metabolic status, and she suffered cardiorespiratory arrest. Intensive supportive care and treatment aimed at reversing metabolic acidosis and renal failure due to both organ infiltration and uric acid nephropathy were unsuccessful, and the patient died 30 hr after admission. Autopsy revealed extensive leukemic infiltration of all the reticuloendothelial organs and the kidneys as well. No evidence of primary CNS leukemia was noted.
KD was a 6¥2-year-old Caucasian male found to have ALL after a brief history of malaise and low-grade fever. A physical examination showed a 5-cm enlarged liver, a 9-cm enlarged spleen, and generalized shotty lymphadenopathy along with bilateral papilledema and priapism occurring soon after admission. A chest x ray was normal. His WBC count was 443,000/mm3 with 92% blasts, the platelet count was 19,000/mm3 and the hematocrit 11%. A bone marrow aspiration revealed complete replacement by leukemic cells which were morphologically identified as lymphoblasts (FAB-L1). A lumbar puncture was performed and showed no evidence of leukemic infiltration or increased intracranial pressure. Other significant laboratory evaluations included an elevated LDH level of 543 IU, an elevated GOT of 36 IU, and normal immunoglobulin levels. Further evaluation of the leukemic cells showed block PAS-positive staining in the cytoplasm and negative reactions to Sudan black, nonspecific esterase, and peroxidase stains. One-hundred percent of the patient's leukemic cells were unlabelled, showing no evidence of surface immunoglobulin and no rosetting with sheep erythrocytes.
Although the response to initial induction chemotherapy with vincristine, prednisone, and L-asparaginase was slow as judged by the fall in peripheral blasts and rate of resolution of organomegaly, complete remission was obtained by day 28, the end of the induction therapy. Eight months after diagnosis this patient had a bone marrow relapse and underwent reinduction therapy to which he had only a partial response. Further attempts at induction failed to clear his marrow of leukemic cells, and he expired 14 months after diagnosis.
DISCUSSION
The cases of ALL found to have t(4;11) (q21;q23) make up a small but specific group of this leukemia. In the group of patients reviewed here 7 of the 13 patients were less than 8 months of age, and in 3 of them onset occurred within 2 weeks after birth. The disease is characterized by extremely high WBC counts. Only 1 patient reviewed had a count less than 50,000/mm3
, and 10 patients had counts 100,000/mm3 or greater. A high percentage of blasts in the blood is a frequent finding.
The disease in the patients we have described has had a sudden onset and has had grave complications including renal involvement and cardiorespiratory arrest. The longest survival time has been case C of this report who survived 14 months.
The 4;11 translocation is remarkable in that it apparently results from very consistent breaks (q21;q23) in the respective chromosomes. To our knowledge, no balanced translocation with this description has been reported in routine cytogenetic

172 H. G. Morse et al.
studies. Further chromosomal aberrations arising in these cells are usually of a trisomy nature producing hyperdiploidy. Such hyperdiploidy is characteristic of ALL cytogenetics. A large percentage of metaphases examined are often abnormal, a factor that may contribute to the poor prognosis. It is our contention that the 4;11 translocation is a primary event and that additional aberrations are secondary events in the progression of the disease.
This study has been supported by the Helen S. Fisher Cancer Research Fund of Children's Hospital, Denver, Colorado.
The authors wish to thank Drs. Avery A. Sandberg, E. 1. Prigogina and Kong-oo Goh for patient information not included in previous reports.
ADDENDUM
While this article was being reviewed, the Third International Workshop on Chromosomes in Leukemia was published in Cancer Genetics and Cytogenetics, Vol. 4, pp. 96-137. Nineteen leukemia patients having t(4;11) were considered by the members of this group. Because the total cases of ALL studied included both published and unpublished cases, no attempt will be made to incorporate into our study the patients included there. Briefly, the Lund groups t(4;11), t(8;14), and PhI-positive were thought to be specific clinical syndromes. Patients with t(4;11) or t(8;14) ALL were found to respond poorly to treatment and were found to have the shortest survivals. In the Lund group t(4;11) there were nine children and nine adults. The median age at diagnosis for children was 1 year, whereas for adults it was 28. For further details the reader is referred to the comprehensive report of the workshop.
REFERENCES
1. Van Den Berghe H, David G, Broeckaert-Van Drshoven A, Louwagie A, Verwilghen R, Casteels-Van Daele M, Eggermont E, Eeckels R (1979): A new chromosome anomaly in acute lymphoblastic leukemia (ALL). Hum Genet 46, 173-180.
2. Dshimura M, Freeman AI, Sandberg AA (1977): Chromosomes and causation of human cancer and leukemia. XXVI. Banding studies in acute lymphoblastic leukemia (ALL). Cancer 40, 1161-1172.
3. Prigogina EL, Fleischman EW, Puchkova GP, Kulagina DE, Majakova SA, Balakirev SA, Frenkel MA, Khvatova NV, Peterson IS (1979): Chromosomes in acute leukemia. Hum Genet 53, 5-16.
4. Goh K, Lee H, Klemperer M (1980): Evidence of clastogens in acute leukemia: Chromosomal p.bnormalities in healthy parents of congenital leukemic patients. Cancer 46, 109-117.
5. Van Den Berghe H, Fryns JP, Verresen H (1972): Congenital leukaemia with 46,XX, t(Bq + ,Cq - ) cells. J Med Genet 9, 468-470.