1+1 - mcgill universitydigitool.library.mcgill.ca/thesisfile39994.pdf · 1 ~ 1 1 1 1 1 1 1 1 1 1 1...
TRANSCRIPT

1+1 National Libraryof Canada
Bibliothèque nat;onaledu Canada
Acquisitions and Direction des acquisitions elBibliographic serviCE'S Brcnc'> des services bibliographiques
395 WelllOglOtl Srcc! 395. rue WentnglonQnawa. Onlano Onawa {Orllano)K1AON-1 K1AON4
NOTICE AVIS
The quality of this microform isheavily dependent upon thequality of the original thesissubmitted for microfilming.Every effort has been made toensure the highest quality ofreproduction possible.
If pages are missing, contact theuniversity which granted thedegree.
Sorne pages may have indistinctprint especially if the originalpages were typed with a poortypewriter ribbon or if theuniversity sent us an inferiorphotocopy.
Reproduction in full or in part ofthis microform is governed bythe Canadian Copyright Act,R.S.C. 1970, c. C-30, andsubsequent amendments.
Omada
La qualité de cette microformedépend grandement de la qualitéde la thèse soumise aumicrofilmage. Nous avons toutfait pour assurer une qualitésupérieure de reproduction.
S'il marlque des pages, veuillezcommuniquer avec l'universitéqui a conféré le grade.
La qualité d'impression decertaines pages peut laisser àdésirer, surtout si les pagesoriginales ont étédactylographiées à l'aide d'unruban usé ou si l'université nousa fait parvenir une photocopie dequalité inférieure.
La reproduction, même partielle,de cette microforme est soumiseà la Loi canadienne sur le droitd'auteur, SRC 1970, c. C-30, etses amendements subséquents.

1
~111111111111111{'1
SPECTROSCOPIC METHODS FOR THECHARACTERIZATION OF THIN FILMS OF
POLYMER-DERIVED CERAMICS
By
Mihai Scarlete
Department of Chemisrry
McGiLl University, Montreal, Quebec, Canada
1995
A Thesis Submined to the
Faculty of Graduate Studies and Research
of McGill University in Partial Fulfilment of the Requirements fOi
the Degree of Doctor of Philosophy
Copyright © Mihai Scarlete, 1995, Montreal, Quebec, Canada

1+1 National libraryof Canada
Biblio~hèque nationaledu Canada
Acquisitions and Direction des acquisitions etBibliographie services Branch des services bibliographiques
39S Welli!"l9lon Streel 395. rue WelhnglooOttawa.Ontono Ottawa (Ottlano)K'AON4 K,AON4
The author has granted anirrevocable non-exclusive licenceallowing the National Library ofCanada to reproduce, loan,distribute or sell copies ofhisjher thesis by any means andin any form or format, makingthis thesis available to interestedpersons.
The author retains ownership ofthe copyright in hisjher thesis.Neither the thesis nor substantialextracts from it may be printed orotherwise reproduced withouthisjher permission.
L'auteur a accordé une licenceirrévocable et non exclusivepermettant à la Bibliothèquenationale du Canada dereproduire, prêter, distribuer ouvendre des copies de sa thèsede quelque manière et sousquelque forme que ce soit pourmettre des exemplaires de cettethèse à la disposition despersonnes intéressées.
L'auteur conserve la propriété dudroit d'auteur qui protège sathèse. Ni la thèse ni des extraitssubstantiels de celle-ci nedoivent être imprimés ouautrement reproduits sans sonautorisation.
ISBN 0-612-12475-4
Canada

1
\t111111111111111f1
ro my grandmorher

1
'-111111111111111f'1
PREFACE
"Many hundreds years ago. the conversion ofmolded. narurally occurring clay into fired. insoluhle
ceramic porrery began mankind's rechnical masrery over narure. These early developmenrs led ra
rhe refracrories essenrialfor rhe producrion ofroday's meral rools.....
Anonymous, Early Iron Age (750 BC - 400BC)
A few thousands years later...
"rhe supremacy of iron as a marerial for hear engines is chaIlenged by rhe "srone-like" silicon
nirride. ln 1983. rhe needfor progressively higher performance engines led ro rhe launch. in rhe
USA. ofrhe Ceramic Technologyfor Advanced Hear Engines Projecr; irs objective was ro replace
rhe old, heavy sreel engines wirh newer ceramic morors. already under advanced resting in JapanJ
Even more disrurbing, rhe major componenrs ofroday 's mosr advanced engines are based on
shaped-Si3N4 parrs and on sensitive. sman elecrronic SiC devices. Bur rhese rwo marerials.
rogerher with r/ze closely relared "silex" marerial. are weIl knownfor being the main marerials of
the "srone age" civilisalion acmally basedon their high thermal and chemicalsrabiliry. hardness and
srrengrh. Therefore, a major question arises: is rhis real progress. or are we rerurning ro rhe
"srone age" ?
A worried philosopher from the 2nd iron age, known also as the "Industrial
Revolution" (1869-19•.)
1 Sponscm:d by lhe companion DOE - Advanood Gas Turbine - Hcavy D~ty TlllIISJlOn (Diesel). The multibilUondoUar project is still in place.

1
~111111111111111fi1
ABSTRACT
Poly(methylsilane) (PMS) has bcen used as a precursor to forro a variety of ceramic
materials within the Si-CoN system. Special emphasis was placed on the synthesis of SiC.
Si3N4 and derived tertiary ceramic materials for use in semiconductor applicaèons. mainly
as thin filins coated onto various substrates.
Detailed insight into the chemical transformations occurring during pyrolysis under
inert (N2. Ar) or rea.ctive (NH3) atmOSpheres was achicved by analysis of the layers coated
on silicon single-crystal wafers. The oxidation of PMS and its transformation during
pyrolysis into SiC were monitored by IR reflectance and transmission spectIOscopy. The
degree of the oxidation of PMS is not only a critical factor in determining the elcctronic
properties of the final resulting materials. but is also a key factor in determining the
pyrolysis mechanism. An important observation was the 10w-temperature (-200°C)
Kumada rearrangement of the PMS 10 poly(carbosilane) (PCS). This reaction was evident
at 200°C on silicon substrates. when the oxygen concentration in PMS was below 40 parts
per million (ppm). Ultrathin layers of amorphous SiC (a-SiC) with thicknesses of -100
nID were obtained by deposition of the volatile species resulti.!lg frot!! the thermal cracking
of the precursor. The cracked-polymer vapor deposition (CP-VO) method allowed the
synthesis of smooth (mïrror-like) ceramic layers. The layers obtained by both CP-VD and
spin-coating procedures exhibited resistivities in the range of lQ3-106 n cm and good
adhesion properties onto the silicon substrates.
n-type SiC layers doped with nitrogen having a resistivity range suitable for Si/SiC
heterojunctions in solar cells werc also synthesized on silicon single-crystal wafers. A
homogeneous doping procedure was de':cloped that consists of reacting the Wurtz PMS
prepolymer with NH3 to form a "doping polymer'". Partial pressure of NH3 cao enhance
the rate of deposition of ceramics on cold substrates. In this case, CP-VD appears to
combine some advantages of the two currently used procedures for growing thin films -

1
~111111111111111ft1
ii
i.e., a high deposition rate characteristic of pyrolysis of spin-coated fùms of precursors and
good texture of the Iayers, characteristic of the chemical vapor deposition process (CVD).
The inclusion of higher concentrations of nitrogen into the ceramic material was
studied during the synthesis of Si3N4 by pyrolysis of PMS under pure NH3. The analysis
of the intermediate prodUClS resulting from the reaction of the precursor with NH3 ;evealed
that carbon loss occurred at temperatures below 600°C. and involves poly(carbosi1azane)
species. The stoichiometric 4:3 atomic ratio of N to Si is achieved below soooC. A
reaction pathway is proposed in which o.lly belerodebydrocoupling between Si·H and N·H
groups occurs. This pathway is sufficient to explain the incorporation of excess nitrogen
(compared 10 Si3N4) usually observed in the intermediate pyrolysis produClS. Carbon loss
is not directly related to nitrogen incorporation. thus the two processes are. al least
partially. independenL
The effcct of the thermal decomposition of NH3. as an independem variable. on the
carbon/nitrogen exchange process was slUdied. Deviations from thermodynamic
equilibrium were related 10 dynamic conditions charaeterizing the flow regime.

1
~111111111111111t'1
iii
RESUME
Poly(méthylsilane) (PMS) a été utilisé comme précurseur dans la synthèse de divers
matériaux céramiques dans le système Si-CoN. Une attention particulière a été dédiquée à
la sinthèse de SiC, Si3N4 et d'autres matériaux ternaires utilisés dans l'industrie des
sem:::onducteurs, surtout comme couches minces sur de différents suppons.
Une recherche approfondée sur les réactions chimiques agissant pendant la pyrolyse
dans une atmosphère inerte (N2, Ar) ou réactive (NH3) a été faite en utilisant des couches
minces céramiques déposées sur des gaufrenes de silicium monocristallin. L'oxidation de
PMS et sa transfonnation en SiC a été analisée par IR-TF en mode reflectence ou
aansmission. Le degré d'oxidation de PMS n'est pas seulement un paramètre CI'.lcial pour
les propriétés des produits céramiques, mais aussi très important dans le méchanisme de la
pyrolyse. Une conclusion importante a été l'observation d'une transposition de type
Kumada à basse température. Cette réaction a été observée à 200°C sur des suppons de
silicium monocristallin quant le contenu d'oxygène du précurseur a été de moins de 40
ppma. Couches ultraminces de SiC amorphe (a-SiC) avec des épaisseurs de -100 nm ont
été obtenues par la déposition des espèces volatiles produites à la suite de la dissociation
thermique du précurseur. La methôde de la déposition de la phase vapeur des polymères
cracqués (CP-VD) a permis la synthèse de couches minces avec une texture supérieure
(ref1ectives). Les couchec minces obtenues par CP-VD, ainsi que par spin-coating ont étalé
des résistivités de 1()3-1()6 nem et une bonne adhésion.
Couches minces ..le a-SiC de type n dopées avec azote, possédant des résistivités
conformes aux exigences imposées par l'utilisation dans les banéries solaires ont été
synthétisées sur des gaufrenes de silicium monocristallin. Une procédure pour le dopage
homogène du SiC est présentée, qui consiste en réagir le PMS précurseur obtenu par la
déhydrohalogénation Wurtz, avec NH3, afin d'obtenir un "polymère dopant". La préssion
partielle de NH3 résulte dans une croissance de la rate de déposition des célamiques sur des

1
~111111111111111,1
iv
supports froids. dans ces conditions, CP-VD combine les avantages des deux procédés
utilisés à présent pour obtenir des couches minces, i.e., une vitesse accrue àe déposition
qui est caractéristique pour la pyrolyse des films précurseurs, et une texture supérieure des
couches, caractéristique pour CVD.
L'accumulation d'azote dans les produits céramiques a été étudiée pendant la synthèse
de Si3N4 par la pyrolyse de PMS en NH3 pure. L'analyse des espèces intermédiaires a
révélé que la pene du carbon est complète à moins de 600°C et incumbe
poly(carbosilazanes). Le rappon stoichiometrique N:Si de 4:3 est acquis à moins de
SOO°c. Une route incluant seulement le hétérodéhydrocouplage entre des groups Si-H et
N-Hx est proposée, qui peut être responsable l'exces d'aote dans les produits
intermédiaires de la pyrolyse.
L'effet de la décomposition thermique indépendante de NH3 sur l'echange carbon
lazote a été étudié. Les déviations de l'équilibre thermodynamique qui Ont été observées
ont été liées aux conditions dynamiques imposées par le régime d'écoulement de gas.

1
-111111111111111t'1
v
ACKNOWLEDGMENTS
Graduate studies constitute a speciallearning process that allows the expansion of
fundarnental knowledge and the development of professional technical skills. The patient
guidance and enthusiastic encouragement of my research ~"1Ipervisers, Ors. I.S. Butler and
J.F. Harrod, have been in hannony with this definition, and have been greatly
appreciated. Their considerable complementary knowledge of chemistry is in panicular
much appreciated.
1 would like to express my sincere gratitude tO my colleagues in the Iaboratory,
namely J. Baldwin, H. L. Boily, V. K Dioumaev, M. E1-Khateeb, D. Hall, J. He, D.
Trojansek, A. Vreugdenhil, S. Xin, C. Dhannandat, for their friendship and the
enjoyable working environment. Special thanks are addressed to Ors. J. Ng, S. Brienne,
H. Li, R.D. Markwell, L. Tarazano, M. Spescha and H-G. Woo, for their helpful
discussions and suggestions.
1 wish to thank Dr. Fred Morin for the MAS NMR spectra, Ms. Helen Campbell
for her help in sample preparation and SEMIEOX measurements, Dr. Graham McKinnon
at the Albena Microelectronic Center for the continuous supply of silicon single-crystal
wafers during the preparation of this thesis, Dr. J. HuIse at the Institute for
Microstructural Science in Ottawa for the ellipsometric determinations and Dr. Georges
Villeneuve at INRS-Energie Varennes for the XPS measurements.
1would alse like to acknowledge all me:nbers of the support staff in the Department
of Chemistry, in particularMs. Renée Charron for her considerable help in dealing with the
bureaucratie red tape during the past years.
Fmally, 1want to express my deepest gratitude to my family members for their
unwaverïng support and constant encouragement over many years of study, and to my
friencis in Monaeal for their understanding and unconditional help.

List of abbreviations
1
~111111111111111,.1
a-SiC
a·Si3N4
AFM
CP-VD
CVD
Cz
DMZ
DRIFTS
DTA
EBCVD
e.g. A12Û3
EDX
EPR
FWHM
HBT
HOMS
i-Pr
un
LOCOS
LPCVD
MBE
·.~MNOS
ô MOSFETiL NMOS
PBL
vi
amorphous silicon carbide
amorphous silicon nitride
atomic force microscopy
cracked-polymer vapor deposition
chemical "apor deposition
Czochralski (method for growing single-crystals)
dimethylzirconocene, C1J2Zr(CH3n
diffuse reflcctance infrared Fourier transform spectrOscopy
differentia! thermal ana\ysis
electrOn-bearn chemica\ vapor deposition
electtonic-grade A\203
elcctrOn dispersive X-ray spectrOScopy
electrOn paramagnetic resonance
full width at half·maximum
HeterO BipolarTransistor
hexamethyldisilane
isopropyl
laser ablation deposition
local oxidation of silicon
low pressure chemica\ vapor deposition
molecular beam epitaXy
metal nitride oxide semiconduetol'
metal oxide semiconduetor field effect tranSistor
nitride metal oxide semiconduetor
poly-buffered local oxidation

1 vii
~PCS poly(carbosilane)
PCSZ polycarbosilazane
1 PECVD plasma-enhanced chemical vapor deposition
1 PMS poly(methylsilane)
RAM random access memory
1 sccm standard cubic centimeters pa minute
SOD spin-on-dopant
1 TFT thin film transistor
1 l'GA thenno-gravimeuic analysis
UHP ultra high purity
1 ULSI ultra large scale of integration
U1W ultrathin window
1 VPE vapor phase epitaxy
1VlSI very large scale of integration
v streClùng mode
1 li defomwion mode
CI) wagging mode
1 y scissoting mode
11111,1

1
~111111111111111('1
vüi
Table of Conte'lts
AbstraCt............•................................................................................ i
Résumé iii
AekJlovvlCligetoents v
List of Abreviations vi
Table of Contents viii
List ofFigures and SehetoeS xi
List ofTables xix
Cbapter 1. General Inaoduction 1
1.1. Advanced Structural Silicon-Based Ceramics •...••...•.••••.•.••••..•....•..•...•... 1
1.2 The Polymerie ROUle ta Ceramie Materials ..............•........................... .4
1.2.1. Polymerie Precursors ta SiC..•......••..............•...........•..............6
1.2.2. Polymerie Precursors ta Si3N4.•..............•...............................•.........•.......8
1.3. Scope of the Thesis ........•..•..•..............•... '" ..... ,..•......••............... 11
Cbapter 2. Poly(methylsilane) - A Precursor for Thin Layers of Silicon Carbide
Deposited on the Surface ofSilicon Single-<::rystal Wafers.•...•..•..•••..•................ 12
2.1. Inaoduction•..••....•.••..•.••..•..•.•..•..........•.••....................••...•....•.. 12
2.1.1. Pyrolysis of PMS and Implicalions of the Oxygen Content in Polymerie .•
Prec~rs ...•..•..•..•..•..•..•..••...•..•.......•.••............•...•......•...••...•.. 13
2.1.2. Kumada Rearrangement••..••..••..•...•.•......•......••..•.....••..•...•....• 16
2.2. Experimental Section ..•.••••.•..••..••.••..••..••.•...•.....•...•..•...•..••.......•.. 17
2.2.1. Poly(methylsilane) Synthesis, General Pyrolysis Conditions and
JIJIalJr.5is •••••..••••••••••••••••••••••••••••••••••••..•••••.•••••••••••••.••.•••••••••••••• 17

1
~111111111111111
"1
ix
2.2.2. Preparation of the Substrates " '" 20
2.2.3. Deposition of SiC Thin Films 22
2.3. Results and Discussion 2S
2.3.1. Infrared Spectroscopie Study of the Oxidation of Thin Films of
PMS on the Surface of Silicon Single-Crystal Wafers 2S
2.3.1.1. Estimation of the Oxygen Content in Poly(methylsilane) 2S
2.3.1.2. Effects of the Degree of Oxidation of the Green Poly(metilylsïlane) .. 27
2.3.1.3. Effect of Room-Temperature Oxidation on Annealed Poly(methylsilane)
between 150-45O°C. Low-temperature Kumada Rearrangement and High
Mobility of the Adsorbed Precursor on the Silicon Surface 37
2.3.2. Synthesis of Thin SiC-Layers Coated on Silicon Single-Crystal
Wafers .....•.............. ,..........................•.......... '" 42
2.4. Conclusions•.......................•....•.......................•...............•....... 50
Chapter 3. Nittogenation ofSiC Layers Deposited on Silicon Single-Crystal Wafers
via Pyrolysis of Poly(methylsilane)....•......••....•...........••....••.......•..•........... 51
3.1. Introduction..•. '" '" 51
3.2. Experimental Section 53
3.3. Results and Discussion..•....••••....••....••...••...••........•.•....•.•....•..•.•... 58
3.3.1. ln-Situ, Gas Phase Nittogen-Doping Procedure During Pyrolysis.••.... 58
3.32 The "Doping-Polymer" Route. Amination of the Wmtz-Prepolymer.•..• 67
3.4. Conclusions...••..••....••....•••.•....•••...••....•..•••.....••...•••....•••......•••.. 71
Chapter 4. Poly(methy1silane) - Precursor to Silicon Niuide via (Reactive) Pyrolysis
under Ammonia..••.••••.•••.•••....•••.•....•••••..••.....•••.••••..••••.•••••.•••••..•...••.•..• 72
4.1. Introduction.•••••••.••....••.••.••••..•.••..•.•••.•.••.•••.•..•.••.•••....••••.......... 72
42. Experimental Section 73

1
l-I11111111111111f1
x
4.3. Results and Discussions 74
4.3.1 Fr-IR Study of the Reaction of NH3 with Poly(methylsilane) 74
4.4. Conclusions 102
Chapter S. Low-temperature Carbon/Nitrogen Exchange during Reactive Pyrolysis of
Poly(methylsilane). Effect of the Thennal Decomposition of NH3 on the Composition of
Ceramic Materials 104
5.1. Introduction 104
52. Experimental Section 107
5.3. Results and Discussion 109
5.3.1. Nitrogenation of PMS via Low-Temperature Reactions 109
5.3.2. NH3 Decomposition under Thermodynamic Equilibrium 123
5.3.3. Kinetic Control over NH3 Decomposition 127
5.4. Conclusions.....•................................•..................................... 130
Chapter 6. Overall Conclusions....•••...•...........•..............•............•........... 132
6.1. Contributions te Original Knowledge 132
6.2. Suggestions for Future Work....•..........•.•.............••....................... 133
6.3. List of Publications ..•••••.....•..••••.........••......•.•. '" ........••.............. 134

1 xi
~LIST OF FIGURES AND SCHEMES
1 Figure Page
1 2.1 Infrared celI designed for in siru analysis of thin films 19
deposited on silicon wafers.
1 2.2 EDX spectra of a Si single-erystal wafer: (a) as reccived 21
1(Mag. x400, 5 kV, U1W); (b) after HF etching (Mag.
x950,5 kV, U1W).
1 2.3 Experimental setup for the spin coating of the silicon 23
wafers with a hexane solution of PMS.
1 2.4 Experimental set up for deposition of the volatile spccies 23
1on cold substratcs.
2.5 AFM images of a SiC layer deposited on Si al different 24
1 resolutions: (a) 10, (b) 5, (c) 3, (d) 2, (e) l, (0 0.8 mD.
2.6 Infrared spectrUIl1 of PMS. 26
1 2.7 vas(Si-O-Si) absorption of interstitial oxygen used for 26
calibration
1 2.8 Infrared spectra of PMS during oxidation at room- 28
1 temperature: (a) 1; (b) 5; (c) 10; (d) 30 min.
2.9 Parabolic curve fitting indicating a diffusion-controlled 29
1 oxidation of PMS.
2.10 Sub-band structure of the Si-H stretch at different 31
1 oxidation rimes al room-temperature, Pea1dit procedure: (a)
11; (b) 2; (c) 8; (d) 60 min.
2.11 Infrared spcctrum exhibiting IWO peaks for the Si-CH3 36
1 deformation al 1248 and 1260 cm-l.
r1

~ xü
b 2.12 Infrared spectra of P:'.1.S following oxidation at room- 38
1 temperature after heating for 1 h under N2 at four different
temperatures: lSOoC - after (a) l, (h) 2, (c) 4 min; 200°C -
1 after (a) 1. (h) 5. and (c) la min; 3800C - after (a) l, (b) 2.
1and {cl 4 min; 45QOC - after (a) 1min and (h) 2 h.
2.13 EDX spectra of: (a) vapor-deposited SiC layer (Mag. XSO. 43
1 5 kV, U1W) and (h) spin-coated SiC layer (Mag. xlOOO.
5kV, U1W).
1 2.14 Fr-IR spectra of: (a) vapor-deposited SiC layer on a Si 44
1single-aystal wafer. The band al 800 cm-l is charaeteristic
for SiC. The twO bands at 1100 and 1200 cm-l are
1 indicative of adventitious oxide and carbon phases.
respectively. (h) SiC-film resulting from pyrolysis of spin-
1 coated PMS layers. The broad band centered at 800 cm·l
1overIaps the 1200-1100 cm-l region.
2.15 29Si MAS NMR spectrUm confirms formation of SiC after 45
1 pyrolysis of a bu1k PMS sample al ll000C.
2.16 XPS analysis of the vapor-deposited SiC layer. (a) Survey 46-47
1 spectrum indicating presence of 0 and F in addition 10 Si
1and C on the surface. (h) Si2p region showing SiOVSiC
environments; (c) Cls region indicates the presence of
1 graphite on the surface.
3.1 Procedure used to form a pronounced lhickness gradient in 56
1 the SiC layer. The pattern imposed on the gas flow
renders the deposition rate a function ofthe the location of
1 the deposition area with respect ID the alumina plate.
f1

1 xüi
la 3.2 n-type SiC coatings on various substrates: ceramic bc'lt, 57
1 quartz, A1203. isostatically-pressed graphite. and silicon
single-crystal wafer.
1 3.3 SEM micrograph of a nia-ogenated, vapor-deposited SiC 61
1fihn deposited in an NH3IAr aonosphere.
3.4 Spot EDX analyses of (a) the continuous film on the Si 61
1 substrate and (b) the upper shattered fihn are indicative of a
unidirectional growth from the substrate with effective
1 segregation coefficients lower than 1 for both oxygen and
1nittogen.
3.5. Fr-IR specttum of a nittogenated, vapor-deposited SiC 64
1 layer coated on a Si single-crystal wafer. The absorption at
939 cm-l is assigned tov(Si-N).
1 3.6 XPS analysis of the vapor-deposited SiC layer, after 65-67
1etehing in a HF solution: (s) SUIVey 0-1000 eV; (a) Si2p
region indicates the Si-C (102 eV) and Si-O (as the
1 shoulder at 103·104 eV); (b) Cls specttum showing the
presence of Si-C at 286 eV, together with adventitious
1 caIbon at 283 eV; and (c) Nls spectIUn1 may be assigned to
1Si-N-H or cyano groups.
3.7 Fr-IR spectra of (a) the starting polymer, (b) the 68
1 amminated polymer, and (c) the polymer after partial
pyrolysis of a spin-coated layer al 300°C. Evidence for a
1 lale stage of the Kumada reanangement is provided by the
band al 1352cm·l•
1~1

1 xiv
1- 4.1 Fr-IR spectra of a thin film of PMS after exposure 10 an 76
1 NH3 atmosphere al 150°C, during oxidation al room-
lemperarure for: (a) 2; (b) 3; (c) 5 min. Insel expanded
1 region 1500-620 cm-l, which exhibits changes in spectra
1a-c for the Ss(Si-0I3) mode al 1270-1240 cm-l, v(o-Si-C)
al 967 cm-I,v(Si-C) al 796 cm-l and v(Si-N) al 870 cm-l .
1 4.2 Fr-IR spectra of (a) PMS after reaction with NH3 al 200°C 78
for 20 h, the splil of Ôs(Si-0I3) mode al 1270 cm-l shows
1 formation of (N)Si-0I3 groups; (b) po1ymer exposed te
1oxidation in air al room-temperature.
4.3 Pyr01ysis of PMS precursor under NH3: (a) TGA; (b) 80
1 DTA data. Al:z03 pan, heating rates: 5 0C/min up te 4SOOC,
8°C/min up te 11000C.
1 4.4 TGA analysis of the precursor during pyrolysis under N2 80
1atmosphere. A1203 pan, heating rates: 5 oC/min up 10
4SOOC. SOC/min up te l1000C.
1 4.5 IR spectrum showing formation of silazane spccies by 81
reaction of PMS with NH3 al 3000C, as evidenced by the
1 v(N-H) stretch al 3397 cm-l•
4.6 Fr-IR spectra exhibiting a reaction pathway excluding the 83
1 Kumacla rearrangement; the absorption al 1270 cm- l
1disappears simmultaneously with v(C-H) in the methyl
groups and Sas(Si-CH3) al 700"C (specbUm d). In
1 spectrum ci, the absorption below 1000 cm·l is assigned te
v(Si-N) and that al 800 cm-! te v(SiC)
1 4.7 Fr-IR spectrum of a silicon niuide layer coared on a silicon 84
f wafer via spin coating ofPMS
1

1 xv
~ 4.8 29Si MAS NMR of a bulk pyrolyzed sample at 1100°C in 84
1 Ar showing resonances for Si3N4 (-50 ppm) and oxidized
material (-100 ppm).
1 4.9 Oxidation of min films of SiC at llOO"C in air for (a) 4; (b) 86
18; and (c) 24 h.
4.10 Fr-IR specuum of a CP-VD layer resulting from pyrolysis 87
1 of PMS under NH3 at 11OOOC, deposited on a silicon
sllbsttate at ca. 600"c.
1 4.11 EDX analysis of me ùtin film deposited by CP-VD. 88
14.12 Fr-IR spectrUm of me mixture of species produced during 89
the pyrolysis of t1ün fùms of PMS spin-coated onto a
1 silicon single-erystal wafer and heated 10 4000C under
ammonia.
1 4.13 (a) Fr-IR spectta of the produet of the reaction of PCS 9S
1with NH3; (b) disappearance of the Si-H band in PCS \lia
heterodehydrocoupling reaetion with N-H bonds.
1 4.14 (a,b) Infrared spectl'a illustrating consumption of Si-H 96
bonds by Si-Hf N-H coupling in polycarbosilazane
1 (PCSZ) formed by PMS pyro1ysis under NH3 at SOOOC;
1(c.d) the ongoing Kumada rearrangement of PCSZ at
SOOOC uncler Ar.
14.15 Formation ofaminosilane (IR specnum b) from a silazane 97
species (IR spectrllm a) at 950°C under Ar \lia a
1 transammination reaction.
1,1

1 xvi
~ 4.16 XPS survey of th.: surface of a spin-c03ted layer resulting 98
1 from pyrolysis of a PMS film under NH3.
(a) SÏ2p signal was used for calibration. The main 99
1 component at 104 eV (A) is indicative of Si02 and
1SiOxNy, while the peaks (c,o) at 101.8 and 101.4 eV are
characteristic for Si3N4.
1 (b) The NIs signal was assigned to a SiOxNyproduet. 100
(c) The Cls signal of the residual carbon in the layer lOI
1 indicates the presence of graphitic carbon at 284.5 eV,
1while the major peak at 286 eV is assigned to cyano
groups.
1 5.1 (a) 29Si MAS NMR spectrum of the produet resulting after 111
3 h reaction of PMS with NH3 aI 3OOOC;
1 (b) 29Si MAS NMR spectrum of the produet resulting 111
1afteroxidation in air of the maIerial used for (a).
5.2 13C MAS NMR spectrum of the produet of the reaction of 112
1 PMS with NH3 at 3000C (sample 3N).
5.3 29Si MAS NMR spectrum of samp1e 4N. The Ihree peaks 114
1 in the 29Si NMR spectrum aI -7.1, -23.0. and -42.2 ppm
1are anributed to SiN2X2 (X =H, C), SiN3X and SiN4
environments, respectively. The presence of the latter
1 species can he explained oRly by taking into accouRt
cleavage of the Si-C bonds.
1 5.4 13C MAS NMR spectrum of samp1e 4N. The low inteRsity
1of the 13C peaIcs at 4 and -6 ppm in the produet at 300"C
indicateS a complete change in the caJbon environment.
f 5.5 29Si MAS NMR spectrum of sample SN. 115
1

1 xvii
~ S.6 29Si MAS ~MR spectrum of samples (al 6N; (bl 7N; (cl 116
9~; and (dl lIN. The NMR signal charaCleristic of Si~4
1 groups shifts from -43.S ppm at 600°C (al, ta -4S.4 ppm at
1 700°C (b), ta -47.S ppm at 9SO"C (c), and finally to a
struetured peak centeted around -49.1 ppm aI llOO"C (d),
1 characteristic of SiN4 groups in a-Si3N4.
1S.7 29Si MAS NMR spectrUm of the product of the pyrolysis 118
of sample 4N at 700"C under N2 is similar ta the 29Si
1 NMR panern of sample SN
S.8 29Si MAS NMR spectrum of the product of the pyrolysis 118
1 ofsample SN at 700"C under N2
1S.9 (a) The absence of the NMR signal is due ta the fuet that 119
the produet of the pyrolysis of sample 6N at 800"C under
1 N2 is pararnagnetic
(b) EPR specaum of the produet of the pyrolysis of sample
1 6N at 800"C under N2
15.10 29Si MAS NMR spectrUm of the produet of the pyrolysis 121
of sample 7N at 800"C under N2
1 5.11 Fr-IR specttum of a layer formed by pyrolysis of a PMS 122
film dip-coated onta a silicon single etystal wafer
1 5.12 InlCtpOlate:d ClD'Ve for the dependence Kp(I) 126
5.13 Dependence of the mole fraction of NH3 (y) vtrsus 127
1 temperature in the gas phase at different flow rates: (o)
1 thennodynamic equilibrium; (-) 48; (V) 57; and (x) 70
scan
1,.1

1
~1
1
1
1
11111111111~1-
5.14
xYÎü
Kinetic control over :-'1-13 de<::omposition. The flow rate
controis the thermal decomposition of NH3 in the 440
6OJOCrange
129

1xix
~LIST OF TABLES
1PageTable
1 2.1 Vibrational assignrnents for the IR spcctrurn of PMS. 27
12.2 Relative areas of the pea1cs following deconvolution of the 30
v(Si-H) band.
1 2.3 Integrated areas of the Si-H and Si-O stretches during the 33
room tempcrarurc oxidation of PMS.
1 2.4 Ratio of the integrated intensities of the symmetric and 35
1antisymmetric C-H stretehes of the methyl groups.
3.1 Influence of 1% NH3 in the carrier Ar on the resistivities of 59
1 SiC-layers.
4.1 Assignrnents for the observed absorption bands in the Fr- 93
1 IR spcctrum of poly(carbosilazane).
4.2 Peak-fit table for Si2p region. 99
1 4.3 Peak-fit table for Cls region. The main component at 286 102
1 eV is assigned ta cyano groups.
S.I C. H, Nand Si contents in the intermediate produets of the 110
1 pyrolysis ofPMS under NH3. formed in the 300-1100"C
1range.
S.2 Volume pararneter for the autotitration of lM Ha solution 124
1 for a givcn flow rate of effluent NH3 from the pyrolysis
fumace.
1 S.3 cpm data for NH3. N2 and H2. 125
S.4 Calculated functions of state for NH3 decomposition at 125
1 various tempcratures at normal pressure.,1

1
~111111111111111f'1
1
CHAPTER 1
General Introduction
1.1 Ad"anced Structural Silicon-Based Ceramics
In 1989, the US Depanment of the Interior estimated the total production of SiC for
both US and Canada, at 115,540 tons. Most of this production was destined for classic
uses such as abrasives and refractories. The production of Si3N4 with similar applications
was not monitored, because of the significandy smaller quantities involved. The
conventional methods for the production ofboth SiC (Acheson process). and Si3N4 (direct
nitridation of powdered silicon or the reaction ofSiC4 with NH3) are weil adapted for the
ton-scale proùuction of these materials in the form ofrefraclOry bricks or grinding wheels.
As a direct consequence of the progress made in the synthesis of new materials (such as
metal oxides) with excellent surrogate qualities, the use of silicon-based ceramics in these
applications has become limited. However, the value of the silicon-based ceramics market
is expected to increase from 3 billion US $ in 1990, to 6 billion US $ in 1995 and to more
than Il billion US $ by the year 2000. This development is based on the emergence of
"advanced structural and fine ,·~ramics". They have becn used wiü'l spectacular results in
the European Economic Community (EEC) and Japan, and this has led to structural
changes in the technologies used for their production. Major components of this class are
"high-grade" SiC and Si3N4 for high-modulus, refractory, non-oxide ceramic fibers and
engine components, which are considerably different from the common refractory and
abrasive products. Currently, Japan is the leading producer in this field. For example,
high purity "green" SiC current production for advanced structural ceramics in the USA is
now primarily aimed at R & D use and is cither produced in-house, or is imported from

1
~111111111111111t'1
2
Japan and the EEC. Reliable figures for the production of advanced structural Si3l"4 in the
USA are not available.!
The manufacture of silicon-based ceramic materials, mainly from preceramic
organosilicon polymers. bas recently become increasingly important because of their highly
attractive shaping properties and low fabrication temperatures.2 Significant progress has
been made, therefore. in the R & 0 of processable methods adapted tO monolithic
applications, with major industrial impact: first, ceramic fibers are now valuable
commercial products and second, the widespread use of a commercial all-ceramic engine
appears te be only a question of lime. as the cost of Si3N4 is driven down from the present
$ 20 today to $ 5 per pound, with lower manufaeturing costS-3
Research is now focussing more and more on the development of soluble or fusible
preeursors4 to these compounds for electronic applications. mainly as thin layers. As a
direet consequence, preparative techniques for both SiC and Si3N4 forro the subjeet of
intensive research activity throughout the world. A few examples are presented below.
Among silicon-based electronic ceramic materia\s, SiC is the most widely used al this
point, since it is a serniconductor with a higher band gap !han silicon. Two major
applications arise from this fearure:
(a) Deviees composed of SiC can be used at elevated temperatures. because of the higher
transition temperature for the change from an extrinsic to an intrinsic serniconductor.
compared with that for Si-based devices.
(b) In todaY's Si-based memoIy devices. the bias across the structure has to be refreshed
every few rnilliseconds due to thermal generalion of carriers that restores the charge
equilibrium. Because of the wide band gap. however. leakage currents arising from
thermal generation in SiC are extremely low. Recovery rimes for SiC data at room
! Il is curious that no domcstic supplier of higb punty Si3N4 was regislezed in the US in 1990. althougbCanadian suppIicrs were menlioned in the 1995 Annual Deparuncnt ofEnergy Repon.2Greil. P. J. Am. Ceram. Soc. 1995.78(4). 83S3RulSU, K. 1990 Amwal Rqon ofUS Depr. ofthe Intuior. Apri1. 1992, 144Wynne, lU.; Riec, R.W.; AM. Rev. Maur. Sei. 1984. 14.1!n

1
\t111111111111111f'1
3
temperature are estimated to be 1014 s, or 3 million years. In essence, RAM computer
devices produced with SiC would be both quasi-slatic and nonvolatile from the point of
view of data storage, and the need for a hard drive would be eliminated.
Other valuable electrOnic properties are the high values for saturated electron drift
velocity, junction breakdown electric field and thermal conductivity (indicating the potentia!
for high-density integration of SiC devices). FinaIly, ~SiC is transparent tO wavelengths
below 0.5 llItl and can be used as a window rnaterial for use in solar ceUs.
Layers made of Si3N4 play a key role in microelectronics, because of a unique
combination of properties. One of the most important features is the low diffusivity of
elements and compounds through its Iattice. Associated with the refractory propetties, this
renders Si3N4 an excellent material for protective coatings. For exarnple, thick membranes
of silicon nitride for X-ray masks are used in synchrotron radiation lithography because of
the chemical and structuraI inermess of Si3N4 under radiation and its relatively high optical
transparency.S,6 Advances in VLSI and ULSI technologies of semiconduetor devices
have n:sulted in complex, multilevel interconneeted architectures. where Si3N4 is the best
choice among the known dielectric rnaterials for insulation clements. Thin dielectric layers
of silicon nitride have becn exploited in metal-nitride oxide-setniconductor devices
(MNOS), nonvolatile memory devices7, and inversion layers in solar celJsll, due to their
charge-storage capabilities. Its large pertnittivity led to the use of silicon nitride as a gate
dielectric in bulk and thin film transistors - TFrs.9 Si3N4 films have also becn used as a
constituent of MNOS and NMOS structures. The use of Si3N4 films in masking
ST. Ohlll, R. Kumar, Y. Yamashilll, H. BogaJ. Vac. ScL TechnDl. B. 12(2)1,1994.5856M. SelcimOlO. H. Yoshihara, T. Ohlcubo J. Vac. Sei. TechnDl. 21,1982, 10177Habraken, F.M.P.; Kuiper, A.E.T.; Tammïnaga, Y.; 1bcelellJ. Appl. Phys•• 1982. 53. 6996Soaind. A.K.; Ackcrmann. G.K.: Lucarini,VJ.; Brauer. R.e. Solid·S~ Science & TechnDlogy 1977.124,5999Spitzer, W.G.; K1eioman. D. Phys. Rev. 1961, 121. 1324

1
~111111111111111
"1
4
applications, such as local oxidation of silicon (LOCOS) and poly-buffered local oxidation
of Si (PBL) isolation, is based on their slow rate of oxidation.
While metallurgical methods such as carbothermal reduction of silica, nitridation of
metallic silicon or thermal decomposition of silicon diimide are employed in the production
of bulk Si3N4. most of the above-mentioned elcctrOnic applications are achieved via
chemical vapor deposition (CVD) or related methods. The physical, electrical, optical and
structural properties of the resulting silicon nitride species cao vary widely, depending on
the fabrication method and operating conditions employed. The f1exibility of the CVD
method with respect to the composition, permittivity, density and refractive index of the
resulting films has resulted in its widespread use in producing thin passivation layers,
barriers to a1kaIi metal diffusion and to moisrure, masks te prevent oxidation or diffusion in
the underlying material in selected panerned areas, final protection layers to fmished
devices (where the mcchanical hardness of Si3N4 is exploited), or radiation shields in
MOSFET devices.
1.2. The Polymerie Route to Ceramie Materials
The aImost universal use of silicon in semiconduClOr devices requires the synthesis of
thin ceramic films on silicon substrates. Even though CVO provides a unique way to
produce both Si3N4 and SiC films of high density and purity. high deposition temperatures
have remained a major limitation of this process. These high deposition temperatures cao
lead te substrate degradation via interlayer atomic diffusion, tG peeling of the film because
of mismatch in thermal expansion coefficients of the layer and the substrate, and to
temperature-induced changes in the shape and the crystallinity of the substrate.l0 In
lOOirolami. G.s.; Gozum. I.E. in Chemical Vapor Deposilioll ofRejrQl;tDry Metals and Ceranria;Proc. MalU. Res. Soc. Symp.; Besmann. T.Mo; Gallois, B.M~ Eds.; Materials Rescarch Society:Pittsburgh. PA, 1990; VoL 168.319

1
~111111111111111t'1
5
theory. these problems can he minimized through the use of organosilieon compounds
which already contain the bond srructure of the silicon-based eeramics (polymeric
precursors) and thus should lead to lower deposition temperatures.11 Other arawbacks of
CVD include low deposition rates and resaletions related to the size and geomeay of the
substrates. These problems limit the expansion of the CVD method to applications such as
the semiconductor silicon single-crystal growth.
A distinct sub-discipline of macromolecular science, involving the investigation of
polymeric preeursors for these layers has evolved. and the research effort into
organosilicon po!ymers has becn particularly intensive. Although the idea that inorganic
polymers might serve as precursors for ceramies was fust suggested in 1964,t2,13 active
research in me field began only in the carly 1970s, when mere was a pressing need for new
structura1 materials that could serve as replacements for metals and metallie alloys in
aircraft, spacecraft and weapons systems. Still in its infancy, the literature concemed is
spread over a wide range ofjoumals covering fields such as polymer science, materials and
ceramics science and engineering, inorganic and organometallic chemisay. electronics and
semiconductor devices. Almough ceramists have addressed me problem of shaped
ceramics wim sorne suceess, me use of preceramic polymers offers a natural route towards
monoliÙ1ÎC materials eimer direcùy or as binders for the ceramic powders from which me
shaped body is to he sintered.14
IlBaney. R.H. in UllTastruelure Processing ofCeramics. G/asses. and Co,...posiles; Hench. L.L.; Ulrich,D.R.. Eds.; John Wiley & Sons: NY. 1984.24S12Aylett, BJ. in Specüù Ceramù:s 1964; Popper, P~ Ed.; Academie: London. 1%5. lOS13Chantre11. P.G.; Popper. P. in Specüù Ceramù:s ; Popper, P~ Ed.; Academie; London. 1964. 8714seyfenh. D. in Silicon-Based Po/ymer Scient:e,199O, Zeiglec. J.M and Gordon Fearon, F. W. Eds~WashinglOll, D.c.. S96
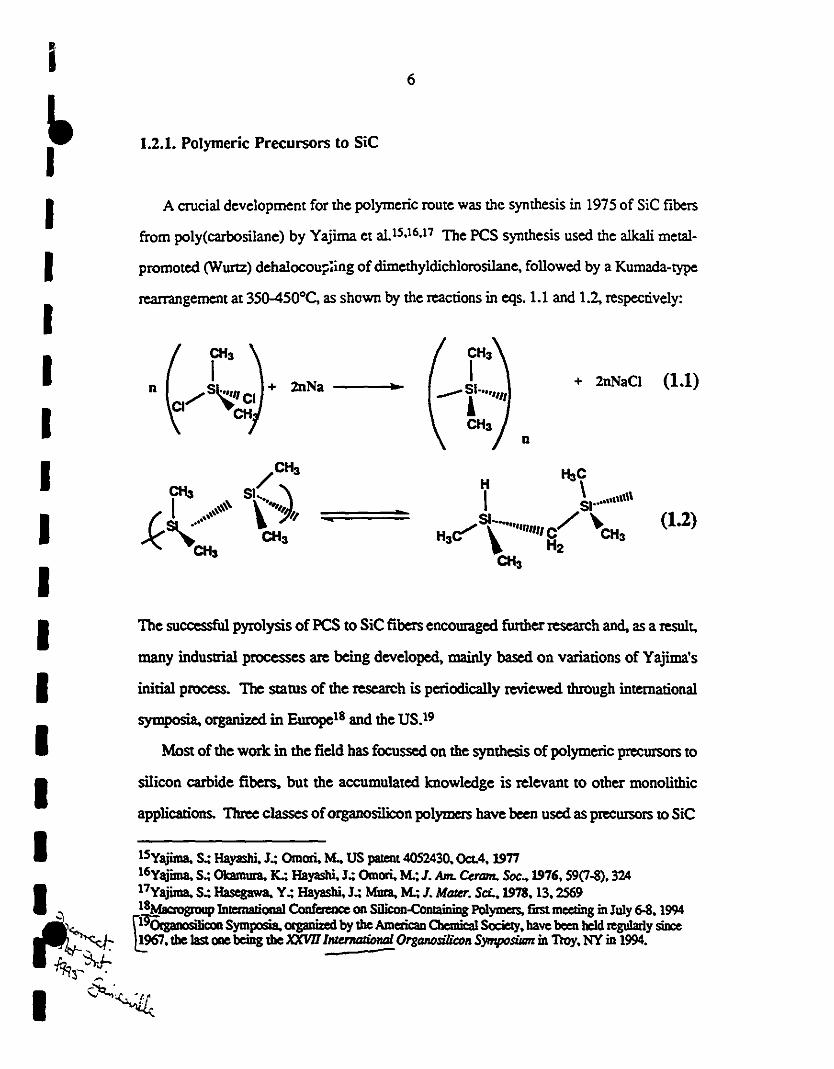
i6
rearrangement at 35Q-450°C, as shown by the reaetions in eqs. 1.1 and 1.2, respectively:
applications. 'I1uI:e classes oforganosilicon polymers have been used as precursors 10 SiC
(1.1)
(1.2)
+ 2nNaCI
n
.
..
oc
+ 2nNa
A crucial development for the polymerie route was the synthesis in 1975 of SiC fibers
MoS! of the work in the field has focusseci on the synthesis of polymerie precursors to
from poly(carbosilane) by Yajima et al)5,16,17 The PCS synthesis used the alkali metal-
1.2.1. Polymerie Precursors to SiC
promoted (Wurtz) dehalocou;;:ing ofdimethyldiehl0r0silane, followed by a Kumada-type
many industrial processes are being developed, mainly based on variations of Yajima's
silicon carbide fibers, but the aecumulated knowledge is relevant to other monolithie
initial process. The status of the research is periodically reviewed through international
symposia, organized in Europe18 and the US.19
The suecessfu1 pyrolysis of PCS to SiC fibers eneouraged further research and, as a result,
15Yajima. s.: Hayashi, J.: Omori, Mo, US paIeIIt 4052430. Oct.4, 197716Yajima, S.; Okamura. K.; Hayasbi, J.; Omori, M.; J. Am. CeraM. Soc.. 1976, 59(7-8). 32417Yajima, S.; Hascgawa. Y.; Hayasbi, J.: Mura, M.; J. MtJIU. Sci..1978. 13.256918Macrogroup IDrcmalional Confe=ce on SiJicon-ComainiDg Polymers, first meeting in July 6-8. 1994~OrgaoosiIiCOll Symposia. organized by lhe Amcric:an 0emicaI Society. bave been bc1d regularly siDce1967.lhe Jast one bcing lhe XXV111nzemation;! OrgQ1lOsiJicoll Symposillm in Troy. NY in 1994.
~111111111111111~~
fqqs-~1 ~.~

1
~111111111111111t-1
7
by pyrolysis: poly(carbosilanes).20,21,22,23.24 poly(carbosiioxanes).2S and
polysilanes.26,27,2S Despite apparent similarities in processing fibers and films via the
polymeric route, once the electronic use of the layers is considered. processing differences
arise. Requirem~nts for stoichiornetry. purity. and the consideration of a whole new dass
of properties. viz.• spinability. dopability and adhesive properties to the substrates, renders
rnost of the known precursors to SiC fibers useless. An ideal electronic-precursor design
must satisfy other specific fearures besides the requirement for a high ceramic yield. For
example, sorne remaining residual functional groups may be necessary. as they may help
the adhesion to the substrate. The presence of functionalities active in nonoxidative
crosslinking processes. such as electron beam irradiation. may also be critical since it is
generally assumed that high crosslinlàng hinders the splitting of volatile species from the
preceramic material. However. when grafted vinyl or alkynyl groups are used. an
alteration of the elemental Si:C ratio can occur. It is generally the case that carbon-rich
polymers generate carbon-rich produCts. Unlike fiber processing. subsequent removal of
excess oxygen or carbon by heating to temperarures around 1200°C, when formation of
C()29 and of Si()3o becomes thermodynatnically favored. cannot be performed for SiC
layers on silicon. At present, precursors that are stoichiometrically correct in Si:C and
which can be transformed pyrolytically to pure ceramic produets are scarce. Processability
rather than produet quality has been the primary motivation to date for many of the
precursor development studies found in the literature. As a direct consequence of these
2Overbeek, W;, Winter. G.; Ger. Ojfen2 236 078. maICh 21.197421Wmter. G.; Verbeek, W.; Mansmann.M.; US PQwu3 892 583,197522wu. H.-J.; IntemllllC. L.V.; CMm. Maler. 1989. 1. S6423Wu. H.-J.; IntemllllC, L.V. Macromolecules 1992. 25. 184024Bacqué, E.; Pillot, J.P.; Birot, M.; Dunogœs. J. Macomolecules 1988.21. 302SMutsuddy. B.e.. Ceromics /ruernational.1987. 13.4126west, R.; David, L.D.; Djurovich. PJ.; Yu. H.; Sinclair. R. Ceram. Bull. 1983.62. 89927 Schilling, CL.; Williams, T.C. Am. CMm. Soc.. Potym. Prepr. 1984. 25. 1
__28Scyfenh, D.; Yu. Y.-F. US. Paleru 4.472.59/.1988; ~oll, E.; Mocaer. D.; Villeneuve, J.F.; Pailler. R.; NasIain, R,.et al.J. Maler. Sci..1987. 26.i 1517\ 30sasaki, Y.; N"JShina. Y.; Salo. M.; Okamura. 1(., J. MtJlU. Sci..1987. 22, 443
\" '0._ \ S, \_"""'.~ •~ -eJ.o.

1
l-I11111111111111t'1
8
difficulties, the first method for producing a -SiC thin layers for electronic purposes was
reponed only in 1988.31
1.2.2. Polymerie Precursors to Si3N4
The polymeric precursor approach to Si3N4 also has proved to be an attractive
alternative, especially for bulk applications. Three major classes of organosilicon
precursors are currenùy used for the production of silicon nitride: poly(silazanes).
poly(carbosilanes) and poly(silanes). The main focus of the research for polymeric
precursors is on poly(silazanes). sinee they already contain nitrogen. as required for Si3N4
formation32. An extensive literature ex.islS on the thennal cross-linking. thermal
decornposition and pyrolysis of different poly(silazanes) under inert
aunospheres.33,34,3S,36,37,3ll,39,40,41 The structure of the poly(silazane) influences both
the ceramic yield and the composition of the final prodUCl. For example, the pyrolysis of
poly(hydridochlorosilazanes) under an inen aunosphere results in the preservation of the
nitrogen from the silazane group and leads to increased ceramic yields by facile
crosslinking \lia the Si-H function in the carbonitride.42 Intimarc mixtures of nanosized
Si3N4/SiC powders have been produced by pyrolysis of poly(silazane)
31011, c.-J•• Tmg, S.-J.; MacKenzie, J.D. SpriIIger Proc. Plrys. 1992,56,9332St:yfenh, D.; W'ISClll8lI, G.H.; Proudbomme, C. J. Am. Ceram. Soc. 1983.66,C-1333Choong Kwet Yive, N.s.; Coniu. RJ.P.; Leelerc, D.; Mutin. PJI.. Vioul. A. ChmL MtJler.1992. 4.14134Blum. Y.D.; Schwartz, 1Ul.; Laine, RoM. J. MtJlt:r. Sei. 1989.24. 17073SLegrow. G.E.; Lim. T.F.; UpowilZ, J. Reaoch, RoS. Am. Cuam. Soc. BrdJ. 19&7, 66, 363361jpowilZ, J.; FIeelI'l8II, H.A.; Chen. RoT.; Prack. ER. Adv. CutJm. Maur. 19&7. 2, 12137Mobr. D1..; Desai, p.; swr. TL. PD/]m. Prepr. 1991. 32, S6S38Monont:, A.A.; Tcxeki. W.M.; BaIicb, C.D. Malo. Leu. 1991. 11. 1939Schwart. J.M. Po/]m. Prepr. 1991.32, Sfi14Orunayama, O.;Isoda, T.;Kaya, H.; Suzuki. T.; Tasbiro. Y. PD/]m. Prepr.199l, 32, 54241BahlouI. D.; Percùa, M.; Goursat. P Key &tgint:eriIIg MtJlt:ri4/s, 1994, 89-91. 11342Sebet. M.; Bill, J.; Riedd,R.; A1diDger. F.Key &tgint:t:l'ÙIg MtJlt:ri4ls, 1994, 89-91. 101

1
1.111111111111111,1
9
Ipoly(carbosilane),43 and the initial SioN and Si-C bonds in the precursors have been
retained. As a general role, carbon removal from the organic substituents on silicon is
incomplete and SiC/Si3N4 mixtures are obtained. The chemisrry of the pyrolytic
conversion of poly(silazane) precursors, and especially carbon/nitrogen exchange. is still
poorly understood. The complexity arises from simultaneous polymerization
Idepolymerization proeesses, and decomposition and thermal cracking reactions occurring
at the operating temperanues.
To minimize the amount of residual carbon. pyrolysis can be performed under
ammonia. since silicon nitride and oxynitrides can be formed from carbon-containing
precursors.44,45.46.47 Because the SioN bond is expected tO be stronger than is the Si-C
bond (DSi-N =42 x UP J/mol and DSi-C =3.7 x UP J/mol),48 carbon/nitrogen exchange
is favoured thCIttlOdynarnically. This implies that during the niineralization step in the heat
treatment over SOO°c, insertion of nitrogen is favored over the inverse reaction with any
organic radicals present. However. the mechanism of the exchange reactions involving
clcavage of Si-C bonds is unclear. Alkyl or aryl substilUents on silicon are regarded as
nonlabile groups in typical low-temperature chemical reactions.49.so but not at the high
temperatures used for pyrolyses. Ifan ammonia stream is passed over the polymer during
the pyrolysis. a Siee cleavage reaction can occur (eq. 1.3), where exchange of the
43\loden, G.; Keutbcll, M. Key EngiMering MauriDls. 19!14, 89·91, 10744Brown-Wenstey, K.A.; Sinclair. R.A. U.s. Pat. No. 4537 942, Aug. 27. 19854SRabe, J.; Bujalslà. D. Procusfor Prepoting Cuomù: MtJleriDJs ltIÏÙlR~d Carboll Levels,Europeall Pat. Applicalion 200326, March 16, 1986460kamura. K.; Sato. Mo; Hasegawa, Y. Cuom. IlIl., 1987, 13, 5547Bums. G.T.; Cbandra. G. J. Amer. Ceram. Soc. 1989, 72(2). 33348WaIsh, R.Bond Disstx:itz:iQ1I Enugy VailleS Ùl SiliœIl.Q»luzinùtg Comporuuls lJIII1 Some ofTheirImplictJlions Ace. C/wn. Res. 1981, 14,24649weyeaberg, D.R.; Maboae, L.G.; ArweIl, W.H. AM. N.Y. AcDd. SeL 1969. 159. 38SlIMoedritzer.K. Organometallic RetJCtions; Bechz, E.I.; TSUISID, Mo, Eds.; WiJey-lntelSCÏcnce: NewYOIk, 1978,2,115

10
(1.4)
(1.3)
tnethylene groups in poly(carbosilane) with NH2 groups can result. as shown in reaction
1.4.51
Amination of poly(carbosïlanes) has becn reponed.S2 In more recent examples,
Seyfenh's groupS3 has used crosslinked poly(siIazanes) as precursors and performed the
pyrolysis under N2. N2IH2 or NH3 while the C/N ratio in the fmal materials was
monilOred. Under NH3. methylene-bridge formation is inhibited. and this results in a
decreased C/N ratio in the residues compared lO pyrolyses conducted under N2 or N2/H2.
Pyrolysis of ethylsilazane in ammonia led lO aImost complete removal of carbon in the
silicon nitride prodUCLS4 Silicon nitride containing less !han 2% carbon has been
synthesized from a vinylic polysilane.55 The efficiency ofcarbon removal in silicon-based
polymeric precursors is partly dependent on the tetDpeTatUTe of cross-linking and is
independent of the sttueture oc functional groups present in the polymer.
The reaction of the Si-H bond in PMS towards NH3 is known to lead lO
dehydrocoupling under a combined pressure of 10 atm ofammonia and methylsilane, and
the resulting poly(methyIaminosilane) gave a mixture of SiC and Si3N4 after pyrolysis
SlSeyfenh, D.; WJSeIlIllII, G.H.; Schwart, JM.; Yu,Y.F.; Pouœsse. c.A. Pmc=!ings of Ibe 193rdMccling ofIbe Amcrican CIlemic:al Society 1987. Dclner. ColOllldo, lnorgcuùc tJIId Organometa1licPo/ymers. p.l44S2T. TaIà. Mo !nui, K. OkamUSll, Mo Saro J. MalU. SeL ua. 1989. S. 918S3H.N. Hall, DA ündquisr. J.5. Haggcny. D. Seyfenh CIrDrL MalU. 1!m, 4. 70SS4Bac, Y.W.; Du, H.; Gallois, B.; GoasaIves, K.E.; WiIkeDs. BJ. C1Iem. MtzlU. 1992,4. 478SSSChmidl, W.R.; Marcbcai. P.5.; 1nIemmle. L.V.; HmIcy Jr~ WJ.; Lewis. R.H.; Doranus, R.H.;MacicI, G.E. CJrem. MtzlU. un. 4. 937
1
~111111111111111ft1

1
t.111111111111111f1
11
under nitrogen.S6 The rearrangement of PMS ioto poly(carbosilane) at 200-450°C makes
it a good precursor to Si3N4, since the laner was reponed to be a good precursor to silicon
nitride after pyrolysis under NH3.S7
1.3 Scope of the Thesis
In the flfSt part of this thesis, the effect of oxygen contamination of
poly(methylsilane) on the resu1ùng IR speCtra and the Kumada rearrangement was
investigated. Thin layers of silicon-based ceramics (SiC in Chapter 2; nitrOgen-doped SiC
in Chapter 3; Si3N4 and SiOxNy in Chapter 4) were deposited on silicon single-crystal
wafers, and analyzed by Fr-IR, EDX and XPS methods. The behavior during pyrolysis
of the spin- or dip-<:oated films of PMS parallels the behavior of the bulk samples. Thin
layers with superior texture can be produced by deposition of the volatile species resulting
from the thermal cracking of the prescursor during the pyrolysis (CP-VD). The effect of
thermal decomposition of NH3 on the composition of the residual ceramic materials was
studied in Chapter 5.
S6H.Q. Ul1, J:F. Harrod OrgQ1lO_UJ11ics. 11. 199%, 822S'Burns, G.T.; ChancIIa. G. J. Am. Coam. Soc. 1989.72(2).333

•
••
12
CHAPTER 2
Poly(methylsilane) - A Precursor for Thin Layers of Silicon
Carbide Deposited on the Surface of Single-Crystal Silicon
Wafers
2.1. Introduction
The use of preceramic polymers as a route to SiC films is being increasingly
eltplored, because of its potential for low-temperature processing, its simplicity and
versatility in synthesizing low-dimensional structures. The similarity of film synÙlesis to
the manufacture ofSiC fibers (now in Ùle industrial production stage) ensures acontinuous
supply of new precursors for films, sinee classic ones (such as PCS available from Dow
Corning - X9-6348, Aldrich - 43,749-2 or Shinetsu - PCS 380X) are readily accessible
and new precursors are continuously being developed. Two classes of organosilicon
polymers are usually convened into SiC fibers,l namely poly(carbosilanes) and
poly(carbosiloltanes). Hazards involved in Ùle synthesis and processing of polysilanes are
major drawbacks, limiting Ùleir use in ordinary applications.2 Poly(carbosilanes) produee
a -SiC at -800°C and subsequently crystalline SiC al higher temperature. On Ùle oÙler
hand, SiC formed from poly(carbosiloxanes) remains amorphous up 10 about IS000C and
is complete1y convened 10 crystalline SiC at about 1700°C.3 The polymeric precursor
method was applied 10 Ùle SYRÙlesis ofa -SiC films for the first rime in 1988 al UeLA, as a
1Hcmida, A.T.; PillOl. J.P.; Birol, Mo: Dunoguès, J.; Pailler, R.; J. Chem. Soc. Chem. Commun. 1994,23372HarwilZ, F. privait: CO/IlIIIUIIÏCaIIi3Yajima, S.; Hayashi, J.; Omori. Mo: US. PateIll40S2430. Oct. 4th, 1977

•
•
13
possible process for making windows in p-i-n amorphous silicon solar cells.
Commercially available poly(carbosïlane) was usee!, since the high pyrolysis temperatures
and high initial oxygen content would prevent the use of poly(carbosiloxane) as a precursor
for the production of SiC films on silicon substrates.
2.1.1. Pyrolysis of PMS and Implications of the Oxygen Content in the
Precursor
Large excess of Si was found in PMS pyrolyzates in the absence of DMT or DMZ,4
but not in their presence.5,6 PMS is a precursor that is not commercially available.
although it is found to produce relatively pure bulk SiC by pyrolysis at temperatures below
lOOO°C. The rearrangement of (CH3hSi-Si(CH3h to (CH3àSi(H)-CH2-Si(CH3h in the
gas phase at 600"C was first reported by Kumada.7 The rearrangement of PMS te PCS has
been observed at -400 °Cl. and this implies that PMS could be used as an in situ SOUIte of
PCS for SiC synthesis. Pyrolysis of PCS to SiC, and the subsequent formation of Ii-SiC
has already been monitored by diffuse reflectance infrared Fourier transform spectroSCOpy
(DRIFTS) of the bulk maleriaL However. the PMS used in the earlier pyrolysis slUdies
exhibited a considerable degree ofoxidation. responsible in pan for the oxygen content in
the resulting material. which had the average formula SilCo.9H<G.200.1 (about 4 wt. %
oxygen). This oxygen content was proposed as the main reason why the fibers did not
exhibit the high-temperature strength and stabi1ity expected for pure SiC.8 Consequently,
the DRIFTS data reported in the literature can be considered characteristic of a mixture of
4Seyfenh, D. in Trans/onnolion of0rgano_tallics ÙIlO Common andEzotù: MazeriDIs: Design andAcùwuion 1988, R. M. Laine El!., NATO ASI Series E, Appl. Sei. No. 141. Maninus Nijboff,Amsterdam. p.1335Seyfcnh. D.; Wood, T.G.; TI3C)'. HJ.; Robinson, JJ. J. Am. ~TQIfI. Soc. 1992. 75. 13006zhang, z.F.; Babonneau. F.; Laine, R.M.; Mu, Y.; Harrod, J.F.; RaIm, JA; J. Am.~Soc~
1991. 74(3). 6707Sbiina, K.; Klllnada, M.J. J. Org. Chem. 1958. 23. 1398Mu. Y.; LaiDe, RoM.; Harrod, J.F. Appt Organo_t. Chem. 1994. S. !l5

•
•
14
PMS and polysiloxanes, rather than of the pure precursor that might be used for electtonic
applications. The potential of PMS in the electronics induSt::y as a SiC-layer precursor is
ultimately dependent on the production ofextretnely pure SiC, with an oxygen level only in
the ppm range being acceptable. The oxygen analysis in PCS is extremely difficu1t by FT
IR spectroscopy, because both the Si-o-Si antisymmetric Stretching vibrations and the Si
CH2-Si vibrations of PCS give rise to intense bands in the 1100-1000 cm-1region. Earlier
IR srodies on the general stability toward oxidationlhydrolysis of various SiC precursors
have becn reported.9 It was observed that upon hydrolysis the IR specttum of the
poly(carbosilane) shows no significantchanges, except for a small broad band at 3650 cm
1. Therefore, the oxygen content in silicon-based precursors and ceramics derived from
!hem is usually determined by elemental analysis. For example, the elemental analysis ofa
carefully synthesized and pyrolyzed poly(silylethylene) sample still revea1ed an oxygen
content in the 0.22-6.65 wt. % range.lO From this point ofview, one major advantage of
using PMS is the complete lack ofIR absorption in the range characteristic for the oxidation
prodUCl and this distinguishes it from the pteeursors in use today.
The accurate measurement of oxygen leve1s in materials intended for e1ectronic
applications is mandatory. Since the most sensitive method for detecting the contamination
is vibrational spcctroscopy by the monitoring of the antisymmetric Si-Q-Si stretch, a clear
window in the range 1100-1000 cm-1bas 10 be available. This absolption is currently used
in routine measurements of oxygen content in silicon single etySta1s.11 Due 10 the
overlapping absorption band at 1060-1040 cm-1from the wagging mode of Si-CHz-Si
groups, monitoring contllmination of PCS with oxygen is ICStricted 10 observation of a
shoulder on the band assigned 10 m(Si-CHz-5i). Thus, the evaluation of the oxygen
content is limited by the sensitivity of the elemental analysïs, ie.. in the pen:entllge range.
This is adequate for studying the effeets of the oxygen incorporation on the
9MUISlIddy. B.e.. CD'aIIL I11t..1987. 13.41lOCcxriu, lU.P.; Leclercq. D.; Mutin, P.H.; PIaneix, l.-Mo; Vioux. A. OrgQ1l/)lIIt:lIJllia.1993. 12, 454llAMual Book ofASIM StaIIIIartlS'.199O. lo.ss. F1188-S8

•
•
15
thermomechanica1 stability of ceramic fibers. However, oxygen contamination becomes
critica1 if the semiconducting properties of the SiC layers are to be exploited. To date. this
represents the most serious constraint to the use of PCS compounds for the synthesis of
SiC layers for e1ectronics. To our knowledge, during the carlier work on the pyrolysis of
PMS the oxygen content was not considered from the point of view of the level required
for electronic processing. Moreover, the latest studies on the pyrolytic transformation of
PMS into SiC use raw materials with a significant degree of oxidation.!2 The vibrational
modes of the siloxane groups also complicate the IR spectrUm in the 21QO-2000 cm'! (vSi
H) region, in addition to the 1100-1000 cm-! region. The methylene wagging mode at
-1050 cm·1 is obscured. This band, together with a band at 1352 cm'!, are important
markers for PCS formation.13
In the present chapter, the use of PMS as a precursor for the synthesis of SiC films
on silicon substrates has been studied. Silicon single-crystal wafers with a surface
orientation [100] have been used as supports for thin layers of PMS which are pyrolyzed to
PCS. The silicon singIe-crystal wafers served as the window material for the transmission
IR measurements in the 4000-600 cm-1 range. Using silicon as a support for the thin SiC
layers may result in a buffering effect of the surface, which can accept excess silicon atoms
that usually result from the pyrolysis of PMS, or in compensating for any carbon excess
found afterthe pyrolysis ofpoly(dimethylsilane). It has previously been suggested that the
silicon substrate may react with carbon-containing species during the first step in the CVD
deposition of SiC and this he1ps to accomodate the SiC layer to the thermo-mechanica1
characteristics ofthe silicon substtate.14
12Hurwi1z,FJ.; Kacik, TA; Bu, x.-y.; MasDovi, J.; Hrimann, PJ.; Bcyeue. K.;MalD'. Ru. 5«.S1"'P' Proc.,l994, 346, 62313IbeŒ2scissotiugmodcal 1351 cm-1 ismuc:h weaJrzrlban is lbeœ2waggingmodcal I05Ocm-1•Forcxample, 011 COWpaiÏDg lbe IR speclra of"cxamelbyldjs.'a"C, (œ:fuSi-5i(œ3b. andbis(1rimcdIy1si1yl)melbane. (Œ:fuSi-CH2-Si(Œ3b.lbe fiDgapriDt vilxatioD for lbe 012 group is lbe1058 cm-1band, wbiJe lbe 1351 cm-1band is DOt obsezvedal ail.14Hasegawa, Y.; Qbmnra, KJ~J. MalD'. Sei. 1983, 18,3633

• 16
2.1.2. Kult'.ada Rearrangement
The gas-phase, thermal rearrangement of hexamethyldisilane (eq. 2.1), was
observed at 600°C.7 Although a mechanism involving radical formation was initially
proposed, later studies have revealed that the system is more complex. However, the
thermodynamics involved are still consistent with the initial observations; i.e.,
(2.1)
•
the Si-Si bond is the Most unstable bond in the system and this leads to severance of the
backbone during the thermal decomposition. An evaluation of the dissociation energy of
the bonds present in PMS and PCS from the data proposed by Walsh1S adjusted for the ex
stabilization effect of the alkyl radicals by silicon, resulted in the following values: Si-Si,
222; Si-C 318 (PMS) and 310 (PCS); Si-li. 314; and C-H. 414 kI/moL Considering that
overal1 one Si-Si bond and one C-H bond in PMS are traded for one Si-C bond and one Si
H bond in PCS, a net endothermic change of only 8 kI/mol which can be considered
negligible.
The temperature of the reaction is usually in the 350-400°C range, slightdifferences
being a fonction of the specifie system observed. In the case of poly(dimethylsilane), the
Kumada rearrangement was previously deteeted at 400"c.6 It is general1y accepted that the
rearrangement occurs around 400"c,8.12,15.16.17 instead of 6OQOC as originally reported.
1Swalsb, R. iD TIre Chemistry ofOrgallic Silicon Compounds 1; PaIai, 5.; Rappopor.. Z. ElIs.; JobnWiIcy: New Yodc, JSll'; 37116t.ame, R.M.; Balvxmc:a". F.; CIrem. Mazer•• 1993. S.26017ScbmiDg, C.L. Union Cor~ Tec1L BrdI•• 1988. Y-I2044

•
•
17
2.2. Experimental Section
2.2.1. Poly(methylsilane) Synthesis, General Pyrolysis Conditions and
Analysis
Since the oxygen content in an of the species studied is critica1, special precautions
were taken with respect to oxygen contamination during the synthesis of the precursor,
pyrolysis experiments and analytica1 procedures. Chemicals were purchased from Hüls
Petrareh Systems, and UHP compressed gases from Matheson. All solvents were dried by
conventional methods, and were stored and manipulated under nitrogen. A minimum
vacuum leve1 in the vacuum line was set to less than 5 x 10-2 torr during the operations,
and a minimum length of t1ùck-walled rubber tubing was used.
The poly(methylsilane) precursor used in the experiments was prepared by Wurtz
dehalocoupling of methyldichlorosilane (CH3SiHCI~,18 under conditions optimized in the
Iiterature:8 1 eq of the silane was refluxed with metal1ic sodium sand (2.05-210 equiv) in
6:1 hexanesIIHF (by volume) under UHP argon for 4 h. The sodium sand had previously
been washed and charged in the dry box 10 minimize oxidation. The resulting PMS was a
non-volatile, colourless oil obtained in about 80% yie1d. The sodium condensation of
(CH3)SiHCI2 is known 10 form produets of the type [(CH3SiHMCH3Si)y]n. The
composition of the resulting produets cao be sensitive 10 s1ight variations in the procedure.S
Analysis by tH NMR spectroscopy revea1ed, besides the normal chemica1 shifts for Si-H
(around 4 ppm) and Si-CH:; (0-1 ppm),19 the resonance of (O)Si-H at 5 ppm. Integration
gave an approximate formula of [(CH3)SiH]O.6[(CH3)Si]O.4, where the \ack of the
hydrogen substituent on silicon resulted from crosslinlcing of the backbones. The lower
tatio of sodium to methyldichlorosilane used in this procedure compared to that used by
18Burkhard, CA J. Am. ChmL Soc. 1949, 76, 96319Czubarow, P.; Woo, H.-G. Mn11llmItÙReseorch Reporr III, 1992

•
•
18
Seyferth et al. (25:1)20 may explain why the resulting PMS obtained in this worlc contains
residual Si-Q bonds, while that produced by Seyfenh's group did nOL21 The tetminal Q
groups were then removed by treatment of the reaction mixture with LiAlH4 at -78 oC for
30 min. The reaction could be followed by monitoring the disappearance of the Si(Cl)-H
resonance in the IH NMR spectrum at -5 ppm. Am reaction, the solvent was pumped off
and the colourless viscous liquid polymer was redissolved in toluene. Following cannula
filtration to remove inorganic components, the poly(methylsilane) was recovered by
evaporating the solvenL As mentioned a1ready, early attempts at using the PMS at this
stage in pyrolysis experiments to form SiC gave poor ceramic yields (<20 %) the produet
containing excess silicon. This polymer is ideal however for the study ofoxidation during
the rearrangement of PMS to PCS, since it has a very low oxygen contenL This bas
allowed a complete analysis of the effect ofoxidation on the Kumada remangement at low
Iemperature5 (100450" C).
On the other hand, in the synthesis of SiC, the precursor was obtained by DMZ
catalyzed dehydrocoupling of the Wurtz prepolymer.22 This precursor is known to give a
produet, not only in high ceramic yield (75-80 %), but with a stciclùometry close to that of
SiC.5,23,24
Since poly(methylsilane) rapidly oxidizes in air, in oroer to observe the influence of
different degrees of polymeroxidation, it was necessary to devise experimental conditions
in wlùch the rate ofoxidation was retaIded. Samples of the polymer were thus subjected ID
kinetically-restrieted oxidation, between two KBr windows sealed with epoxy resin. or on
20 Scyfenh, D.; Wood, T.G.; Tracy, HJ.; Robinson. JJ. J. Am. Cuam. Soc. 1992, 75, 130021UndcrcondilÏonsofexœssNa, in addition ID thedcha1ocouplingto form -MeSiH-MeSiH- units, Ihcreisan C1lC1lSive sidc teaClÏon wbicb produc:es branches of!he type -McSi(SiMeH2>-. A1Ihough Ibis sillereaclion œndcrs tbc dcIai1s ofdie poIymerstrue1IIœ IDIpœdiclable, it does not inlerlcre wim the ability ofdie~lymcr to yicld SiC on pyrolysis.ZlMu, Y.; Hamld,l.F~lnorganic and OrgQllOIMtDllic Polymus and OUgomus; Eds.l.F. HamxI and R.M.Laine; Kluwcr Academie Publishets, Dordrecht, 1991, 2323zbang, Z.F.; Baboaneau, F.; Laine, RoM.; Mu, Y.; Hamld, 1.F.; RaIm, l.A.; J. Am. Cuam. Soc.1991, 74(3), 67024Hcngge, E.; W"JCIIberger,~ J. OrgQllOIMt. Chem.1992, 433, 21

•19
a Czochralski (Cz) silicon single-erystal window mounted in a specially designed Teflon
cclI equipped for in situ etching of the window. The whole apparatus was connected to a
gas inlet for the oxidizing gas, a continuous flow of a mixrure of N2 and air above the
substtate, as shown in Figure 2.1. The thicknesses of the PMS layers were estimated by
measuring the thicknesses of the PCS films after pyrolysis at 450°C by means of a Sloan
Dektak profilometer and were in the range of 1 J.I.IIl. The percentage of the total shrinkage
was considered equal to the ceramic yield and was considered to be independent of
direction. AIl oxidations were performed at room temperarure and the observed intensity
changes were assumed to be due only to chemical reactions with negligible volatilization,
thermal or rheologica1 shrinkage.
Fig. 2.1. IR cell designed for in situ analysis of thin films deposited on
silicon wafers.
•
thlncoatlng
siliconsubstnde
1lF101ut1onand g..outIet
x
2D mlcrometrlcIIdJU8lable mnd

•
•
20
The pyrolysis cycles were conducted at 5-8 torr over aonopheric pressure in a
Lindberg single-zone programmable furnace equipped with a Eurotherm PID temperature
conttoller with a rna=.:.1tIl operating temperature of 1lOO°C and providing an accuracy of
±l°C at l()()()°C. The polymer was loaded into a sealed. fused quartz, fumace tube (2.5 cm
diam, 60 cm long) under UHP Ar. The fumace tube was anached to inlet and outlet
flowmeters, which had been adapted to provide independent flow rate and pressure
adjustments for up 10 tltree components used in fotming the gas phase above the sample.
The rate ofheating was 8 0C'min and the maximum temperature (m the range 15010 4500C
in the oxidation experiments. and 1100"C for the synthesis of SiC) was maintained for 30
60 min. The samples were coated as thin films on the silicon wafers with a spin-coating
apparatus operating at ca. 100 rpm. The samples were then placed directly into the fumace
tube, under an inen gas flow. The IR spèctta were obtained by reflection from the silicon
sUIfaces. Sufficient enhancement of the signals in the deposited layers could be recotded
after multiple reflections and after 32-64 scans. The spectra (4 cm-} resolution) were
recotded on a Bruker IFS-48 specnometer equipped with an A590 IR microscope, Mer
deteetor. and a SONY Trinitron PYM 1340 co1or monitor.
2.2.2. Preparation of the Substrates
Silicon single-crystal wafers, 10 cm in diameter, 1-3° off-oriented [100], 10-20
nem P(B) and 1-10 N(P), polished on one side and etehed on the other, were used as the
coating sUIfaces. Since the silicon substtates contained a superficial native oxide layer, a
preliminaIy etching procedure was needed 10clean the substrates. Special precautions were
taken 10 ensure the creation ofa hydrophobic surface 10 pertnit initial adhesion 10 the Si-H
functions in the precursor. The substtates were cleaned initially with a H2S04/H202 (5:1)
solution at 95°C for 5 min 10 remove adsorbed residual organic materials, rinsed in
deionized water, etehed in a solution ofH201HF48% (5:1) for 4 min, washed wililacè~
•
['1
, .1•1
r1
r .11 .
1 •Lr •11 .
r· .1., .L, .L•L
21
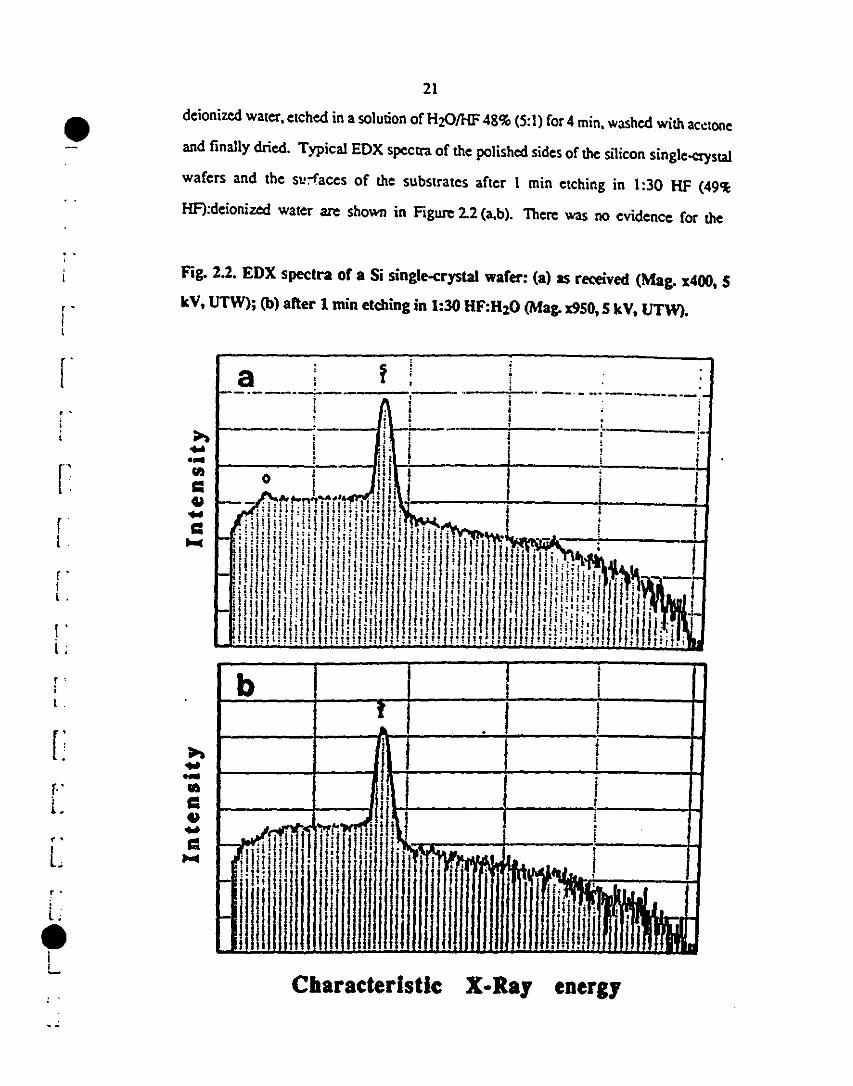
deionized water, etched in a solution of H201HF 48% (5:1) for 4 min, washed with ac~tone
and finally dried. Typical EDX spcctra of the polished sides of the silicon single-aystal
wafers and the su:-faces of the substrates after 1 min etching in 1:30 HF {49~
HF):deionized water are shown in Figure 2.2 (a,b). Therc was no evidence for the
Fig. 2.2. EDX spectra or a Si single-erystal warer: (a) as received (Mag. x400, 5
kV, UTW); (b) after 1 min etc:hing in 1:30 HF:H:O (Mag. x9S0, 5 kV. VTW).
b. 1
Characteristlc X-Ray enerey

•
•
22
presence of oxide on the surface of the cleaned sample from the EDX experimems.
However. the etched surface exhibited a variable contact angle. ranging from 84° to 74°
with dodecane. when subjected to further etching in HF for rimes ranging from 1 to 4 min.
Etching for longer rimes did not lead to a further change in contact angle. Since etching of
silicon with acid usually leaves a hydrophobie surface containing terminal hydrogen atoms.
the result obtained after 4 min was intexpreted as total removal of the oxide layer. Before
being coated, the wafers were dried and stabilized against oxidation by annealing in UHP·
Ar al 850°C for 30 min.2S
2.2.3. Deposition of SiC Thin Films
The oxygen content in the precursor was monitored by IR spectroscopy as
described in section 2.3.2. The silicon substrates were cut from 4" [100] diameter wafers.
Thin SiC films were obtained by pyrolysis of substtates spin- or dip-coated with a 5%
hexane solution of the precursor. The spin-coated SiC layers after pyrolysis were dark
brown and their roughness was estimated to be ±10%. based on profilometer data at
thicknesses of about 11UD- The resistivities of the spin-coated films were in the 1()2..1()3
nem range, an unexpected drop from the value for intrinsic SiC which will be discussed
below. During the pyrolyses, it was observed that distillation and thermal cracking of the
precursor produced a small amount of volatile species that eventually condensed on Ùle
colder walls of of the furnace. When substrates were positioned in these areas. ultraÙlÎn
films could be obtained from Ùle condensation of Ùl= Ùlermally-cracked polymer.
Simplified experimental setups for the spin coating and vapor deposition processes are
presented in Figures 2.3 and 2.4. respectively. Fr-IR and EDX data were used in the
intetpretation of the experimental data for films deposited on the silicon substrates by bath
2SHair. Ml..; Silones Swftll:e$ andllIlerfa-.es. Eds. D.E.LcydeD, Gudon and Breacb SCience Publisbels,NY. 1986. p.29
•
•
23
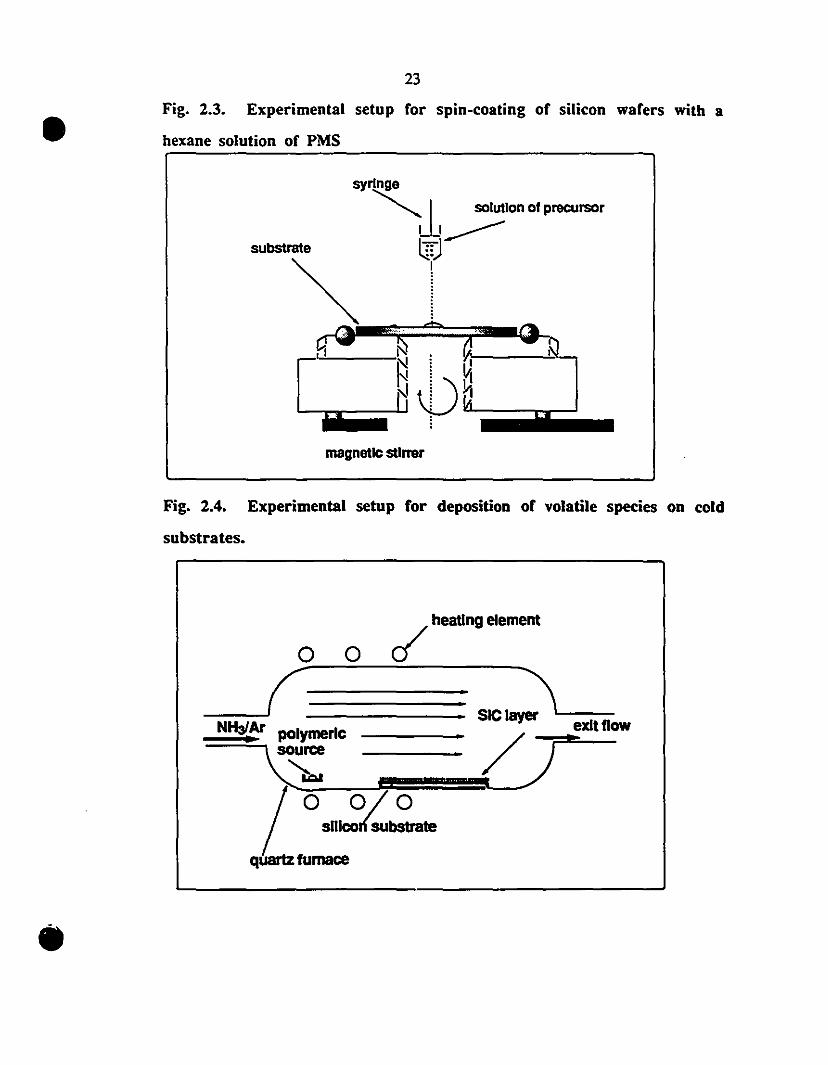
Fig. 2.3. Experimental setup for spin-coating of silicon wafers with a
hexane solution of PMS
syringe
~L solutIon of precursorLI___
subStrllte t::J
~~l~....-~,..!-.• ..~ • af----~-
IN l il" : vI
'iiiïiiiiiiiii~N 'PH'~Aiiiiiiiiiiiiiiiililiiiii~magnetle stlrrer
Fig. 2.4. Experimental setup for deposition of volatile species on cold
substrates.
o 0 ~ heatlng element
__-J(====== SICI~L--NH~Ar polymerie 7exit f10w
~~ 01O.n';;;~quartz fumace

•
r .
\,
r',
: .
l .
r .,, ..
1.~ .
· .· .
, .·.l;
•l 'W
...
24
methods. XPS measurements. used to characterize the surface of the layers. were
performed by Dr. Vei1leux atlNRS-Energie. Varennes. Quebec. Significant differences in
smoothness. texture and thickness were observed betwccn the vapor-deposited films and
the residual iayers after pyrolysis of the precursor deposited by spin-<:oating. The former
were about 150 nm thick and had a smooth. yeUow mirror-like appearance. while a rough
surface was obtained !tom spin-coating. Statistical analysis by atomic force microseopy
(AFM) of the topology of the vapor-deposited coating indicated a flat surface lO within ±
2.5 % Figure 2.S (OOl-()()3); the thickness determined by e11ipsometty was 100·200 nm.
The resistivities of the layers grown under UHP-Ar were in the range of 1()3·1()6 ncm.
Slightly smaller resistivities. with random fluctuations, but still in the 1()2.1()S ncm range
were measured for the filins produced in a Nz attnosphere. These variations in resistivity
were not subjected to a delailed analysis. since the intrinsic values were orders of
magnitude greaIer than those of inteleSt.
Fig. 1.5 AFM images of a SiC layer deposited on Si at dilTerent resolutions
(nm): (001) 10, (002) S, (003) 3, (004) 2, (00$) 1, (006) 0.8 •

•
•
25
2.3. ResuUs and Discussion
2.3.1. Infrared Spectroscopie Study of the Oxidation of Thin Films of
PMS on the Surface of Silicon Single-Crystal Wafers
2.3.1.1. Estimation of the Oxygen Content in Poly(methylsilane)
The ttansmission IR 5peCtrum of the polymer used in the present work is shown in
Figure 26. The integrated intensity of the antisymmetric Si-Q.Si stretehing absorption at
-1100 cm-1 was used to estimate the oxygen content of the polymer, using the silicon
substrate as an extemal standard. The calibration procedure was based on the absorption of
the interstitial oxygen, (Figure 2.7) in the supporting silicon wafers, which had a known
concentration of 15 pans per n.:illion atoms (ppma), equivalent to 7.5 x 1017 atomslcm3.
The calibration was based on the integrated absorption hetween 1067 and 1135 cm-l, after
baseline correction, for both the calibrant and the sample. The confidence level for the
oxygen content in the silicon wafer is high, as this is an important parameter in the
Czochralski manufaeturing process for silicon single-crystals. Based on the thickness
measurements for the wafer and the deposited layer, and assuming equal molar
absorptivities for vas(Si-û-Si) in the resulting siloxane contaminant in the polymer26 and
for the vas(Si-Q-Si) involving the interstitial oxygen in the single crystallattice, the oxygen
contentof the initial PMS polymer was found to he 25 ± 10 ppma.
26nüs assumplioD is supJlOIttd by orhcr rcsu1ls œ1aIcd ID lbe mobility of lbe polymer cbain 011 the smfaceof lbe silicon wafezs, sec sec:IÏOII2.3.2.
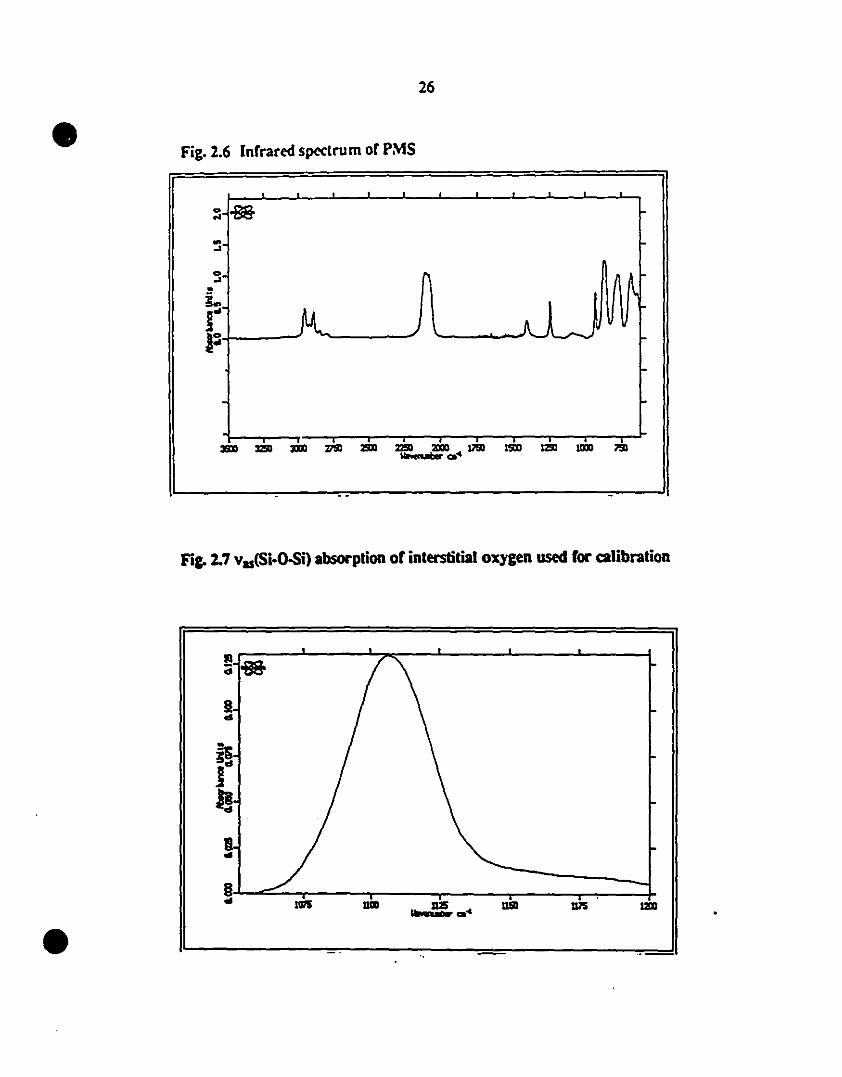
•26
Fig.2.6 lnfrared spcctrum of PMS
• fil
~-•~Dr1:::-1-----'
1-
. --.15lllI 1Z5Il IlD:I
§..
•
Fic- 2.7 vu(SI·O-Si) absorption or interstitial oxygen used ror calibration
!.!-.oe:::::=---r---~--...,.----.--- ........--,--4IlIl'I WD ~ .... lISI

•
•
27
2.3.1.2. Effect of the Degree of Oxidation on the FT-IR Spectra of
Unpyrolyzed ("Green") Poly(methylsilane)
Conflicring vibrational assignments have been proposed previously for the IR
spectrum of PMS (Table 2.1). The uncenainities arise from ambiguity in the assignment of
bands attributed 10 vs(Si-C), v(Si-H) and Ss(Si-CH3).
Table 2.1. Vibrational Assignments for the IR Spectrum of PMS
Obs. IR bands (cm-l ) Vibrational assignments
2956s v..(C-H)
2894m vs(C-H)
20755, br,27.28 210012 v(Si-H)
1406m ô..(Si-CH3)
12475,32 126()34 &(Si-CH3)
930m -y(SiHù
865vs P(CH3)
7645, br v..(Si-C)
6S5sor6S0s vs(Si-C)
The investigation of the oxidation of PMS revealed severa! interesting features in the
IR spectta. A series of IR spectra of a PMS sample supported on a silicon single-aystal
wafer during oxidation at room temperature is presented in Figure 2.8. The notable
27Mu, Y.; Hanod,J.F. inlnorganic and Org/UlOllleraJlic OlïgomersandPDlymus, Hanod, J.F. and Laine,R.M. Eds.. Kluwer Academ~ Pub\isbcrs, Nether1aDds, 1991. 23-352SHclIggc, E. in Silicon Chemistry II, SI; TDp. Curr. Chem. 1974,43
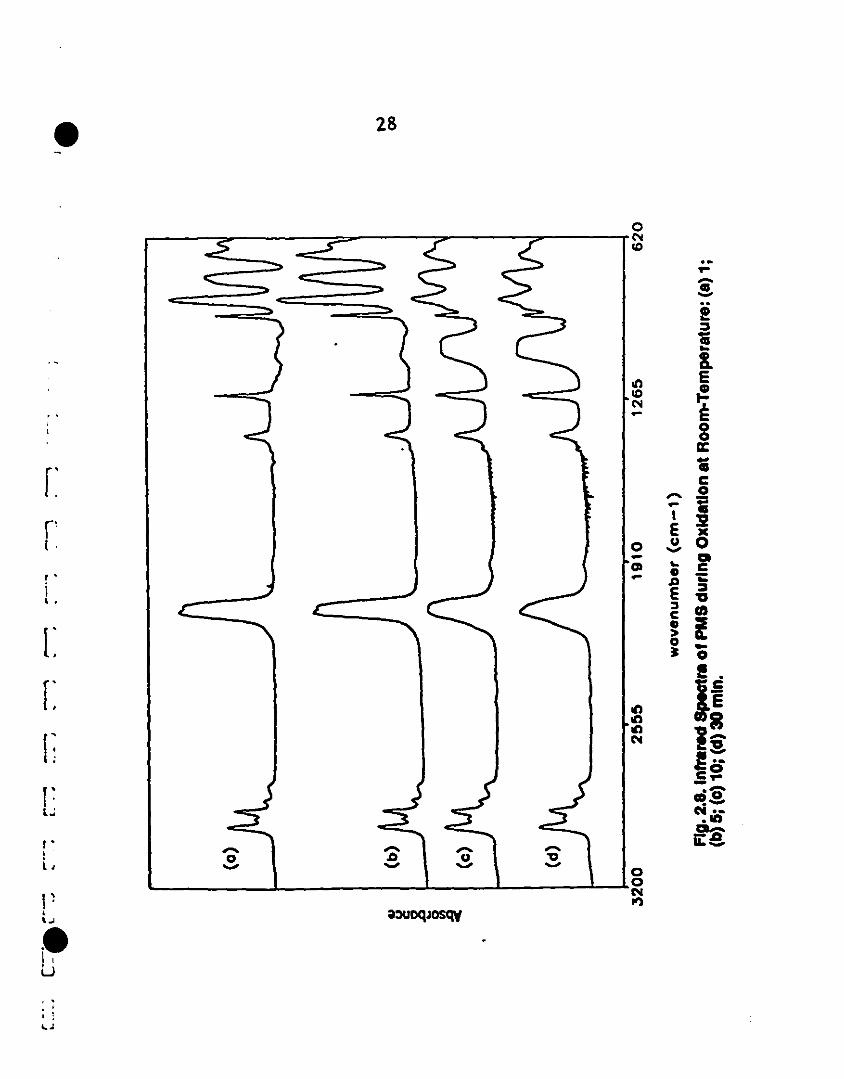
• 28
••,...-.CIl......!~
li..!.
on EID ~N- E
gli:-CIlc,.. 0- ;:1
1 1E )li
0 u 0....- I:llCIl ..- "
cS) ;:
E ~
;, "c fi)
" !>0• -0
l! .
on IlononN I~! ..
cO-,..a!$C't ••
.ID
l~,..o...,
~ ,.,.. ,...."'" ~ "0
______...JL L_=-_L_.:-...,:=-_.LJgN1')
r •
i••
r'1 .
1 •1
l ,
p•\~
••l ,LJ
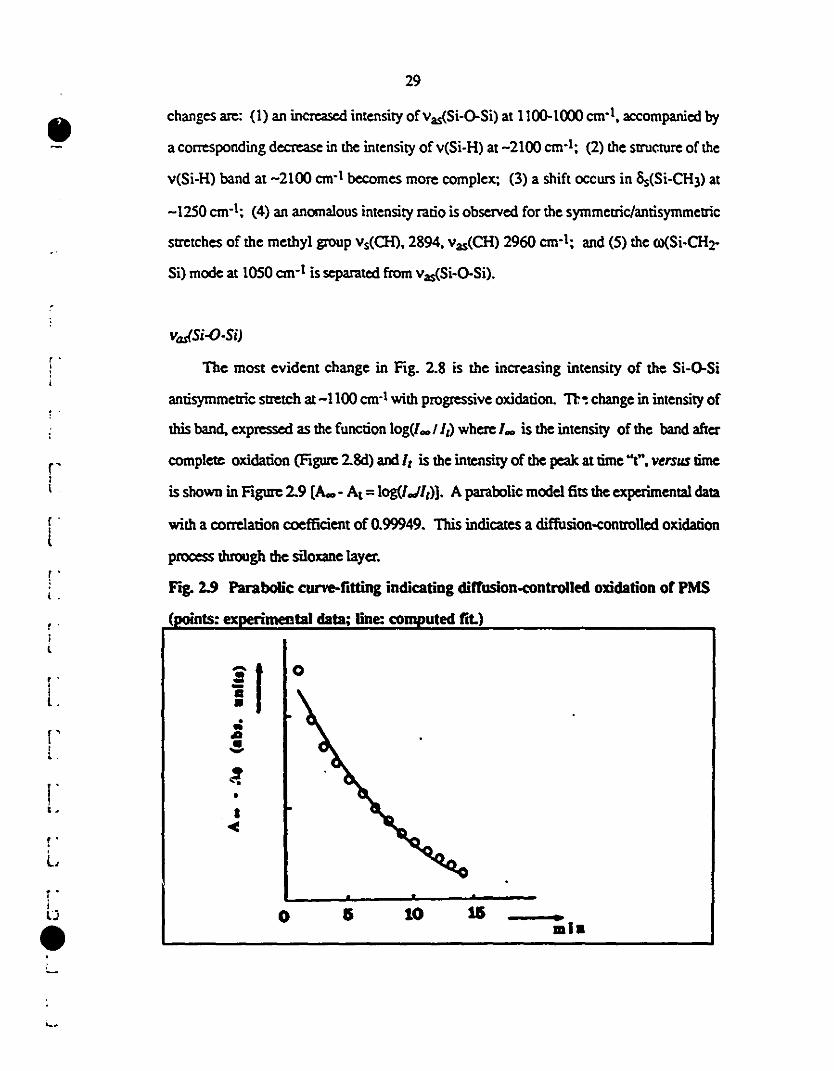
•
f'1\
r .1[ .1 .
r .11
1 .11.
r'!•
! ~
L
29
changes are: (l) an increased intensity ofvas(Si-o-Si) at 1100-1000 cm-l, accompanied by
a corresponding decrease in the intensity ofv(Si-H) at -2100 cm-l ; (2) the structure of the
v(Si-H) band at -2100 cm-I becomes more complex; (3) a shift occurs in Ss(Si-CH3) at
-1250 cm-I; (4) an anomalous intensity ratio is observed for the symmetric/antisymmetric
stretches of the methyl group vs(CH), 2894, vas(CH) 2960 cm-I; and (5) the CIl(Si-CH2
Si) mode at 1050 cm-I is separated from vas<Si-o-Si).
valSi-o-Si)
The most evident change in Fig. 2.8 is the increasing intensity of the Si-o-Si
antisymmetric stretch al -1100 cm·l with progressive oxidation. Th~ change in intensity of
this band, expressed as the function log(1..' 1iJ where 100 is the intensity of the band after
complete oxidation (Figure 28d) and l, is the intensity of the peak al lime "t", versus lime
is shown in Figure 2.9 [A.. - At =10g(1..'1,)]. A parabolïc mode! fits the experimental data
with a correlation coefficient of 0.99949_ This indicates a diffusion-controUed oxidation
process through the siloxane layer.
Fig. 2.9 Parabolic curve-fitting indicating dirrusion-controlled oxiclation of PMS
ff 0
••A.!-
~••<
r •
b•·...
o 10 •IDI •
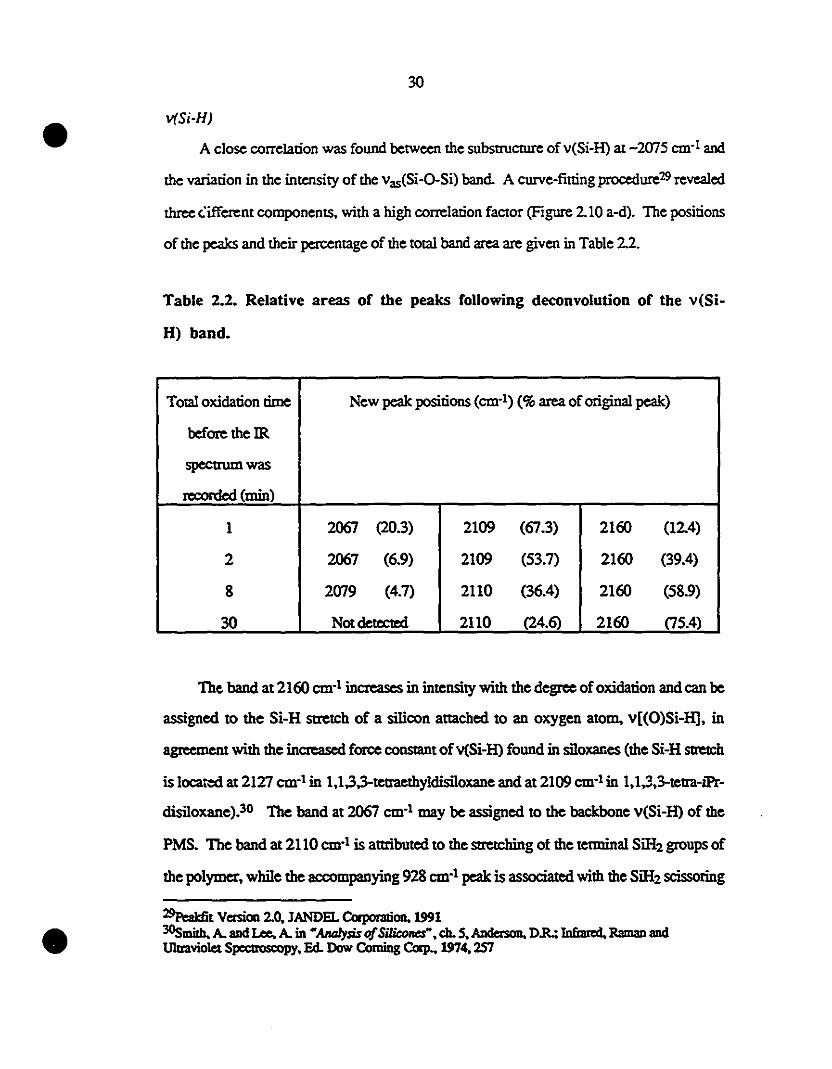
•
•
30
v(Si-H)
A close correlation was found between the substrUeture ofv(Si-H) aI -2fJ75 cm-1 and
the variation in the intensity of the vas(Si-Q-Si) band. A curve-fitting procedure29 revealed
three C:ifferent components, with a high correlation factor (Figure 2.10 a-d). The positions
of the peaks and their percentage of the total band area are given in Table 2.2.
Table 2.2. Relative areas of the peaks following deconvolution of the v(Si-
H) band.
Total oxidation time New peak positions (cm-1) (% area of original peak)
before the IR
spectIUtIl was
rccorded (min)
1 2067 (20.3) 2109 (67.3) 2160 (12.4)
2 ']J)67 (6.9) 2109 (53.7) 2160 (39.4)
8 2fJ79 (4.7) 2110 (36.4) 2160 (58.9)
30 Notdeteeted 2110 (24.6) 2160 (75.4)
The band at 2160 cm-1 increases in intensity with the degree ofoxidation and cao be
assigned 10 the Si-H stretch of a silicon anached 10 an oxygen a1Om, v[(O)Si-H], in
agreement with the increased fOtCC constant ofv(Si-H) found in sùoxanes (the Si-H stretch
is located at 2127 cm-1 in 1,1,3,3-tetraethyldisiloxane and aI 2109 cm-1 in 1,1,3,3-tetra-iPr
disiloxane).30 The band at '2JJ67 cm-1 may he assigned 10 the bacltbone v(Si-H) of the
PMS. The band at 2110 cm·1 is attributed 10 the stretching of the tetminal SïH2 groups of
the polymer, while the aceompanying 928 cm·1 peak is associated with the SïH2 scissoring
2!lFeaJ:fit VCISÏDll 2.0, JANDEL CoIporaIion, 19913Osmilh. A. and Lee. A. in •Analysis ofSilicones" , c:h. 5, Anderson, D.R.; Infmred, Raman andtntraviolet Speœoscopy, Ed. Dow Coming Corp~ 1974,2S7
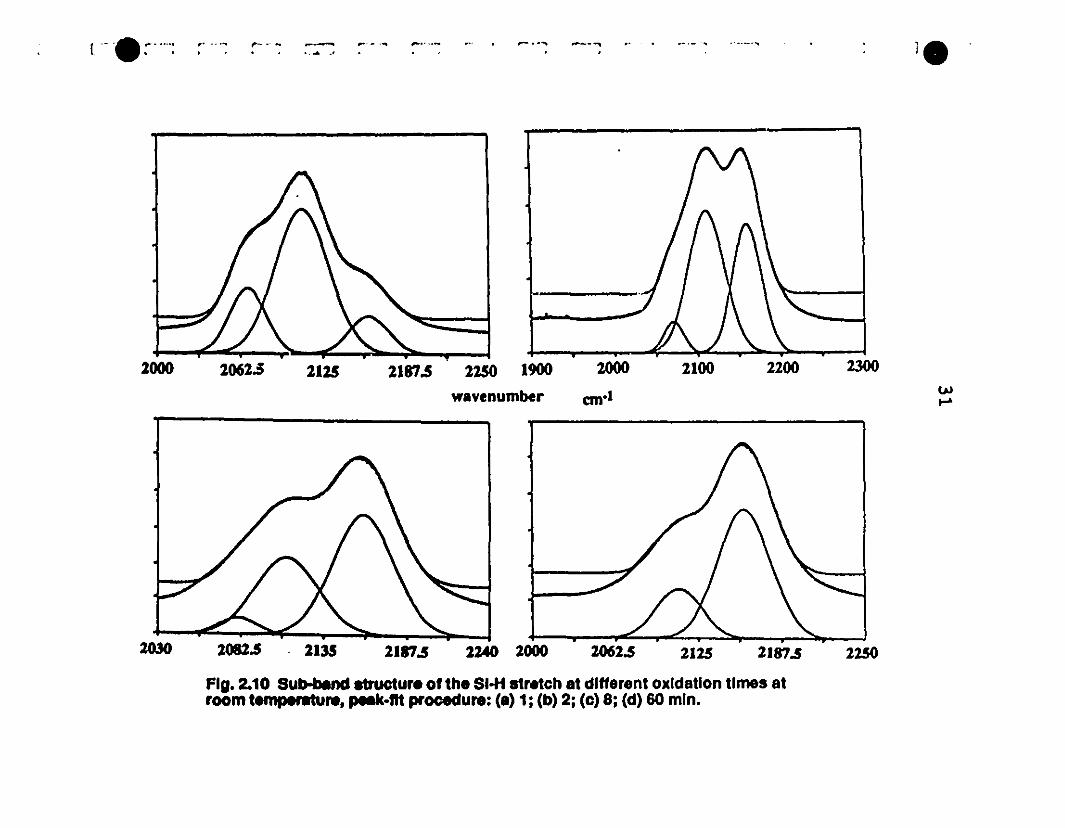
l-.~······. ., " -.... ,.-..- ... -.......'_....- _..~
--~ J.
2000
2OJO
2062.5
2082.5
2115
2135
2187.5
2187.5
2250 1900
wavenumber
2:z40 2000
2000
cm·l
2062.5
2100
:za:zs
2200
2187.5
2300
2250
W1-'
Fig. 2.10 Subobend structure o, the SloH atNtch 8t dlflerent oxldatlon limes atroom teRJDiii.tu.... peû·ftt procedure: Ca) 1; Cb) 2; Cc) 8; Cd) 60 min.

•
•
32
mode.
The band at 2f>67 cm-! disappears completely after the fourth deconvolution cycle.
This is accompanied by maximum intensity in the band assigned to the antisynuneaic Si-Q
Si stretch. At this stage, ail the Si-Si-H vibrations have been traDsformed into Q-Si-H
vibrations. The totald= in the intensity of the Si-H peak at 2067 cm-l, and the fact
that the terminal SiH2 groups seem to be unaffected (from the constant intensities of the
peaks at 928 and 2110 cm-l), can be ex.plained by the smaller molar absorptivity of
v[(O)Si-H] at 2160 cm-! relative to that of v(Si)Si-H at 2067 cm-Jo The ratio of the
absorptivities of the IWO vibrational modes cao be calculated from the ratio of the integrated
areas of the v(Si)Si-H band in Fig. 2.1Oa and the v[(O)Si-H] band in Fig. 2.1Od. The final
ratio of the absorbance values obtained this way was 0.78.
It is expected that groups attached to a silicon atom will tend to show more
characteristic group frequencies than do organic compounds. This is due in part to the faet
that the increased size and mass of the silicon atom compared to carbon cao provide better
vibrational insulation in a molecule. However, we observed significaot shifts in the
position and changes in the intensity of v(Si-H) during the oxidation of PMS. The
intensity ofv(Si-H) when the Si atom is attaehed 10 another Si (as in PMS). compared 10 0
(as in the oxidized polymer), 10 H (as in a terminal SiH2 group), or to C (as in PCS),
depends on tbe magnitude of the change in the dipole moment of the bond. Such
interferences usually arise from inductive effects of neighboring atoms, coupling or
interaction of vibrations or steric and other geometrical effects. The results of our attempt
to relate the electron-withdrawing properties of the substituents on silicon are presented
below.
Advancing oxidation is reflected in the area of the vas(Si-O-Si) band (percentage of
the final area of the peak while the degree ofoxidation can be obtained from the ratio of the
adjusted areas of the peaks at "2f)67 and 2160 cm-l. However, equal weighing factors in
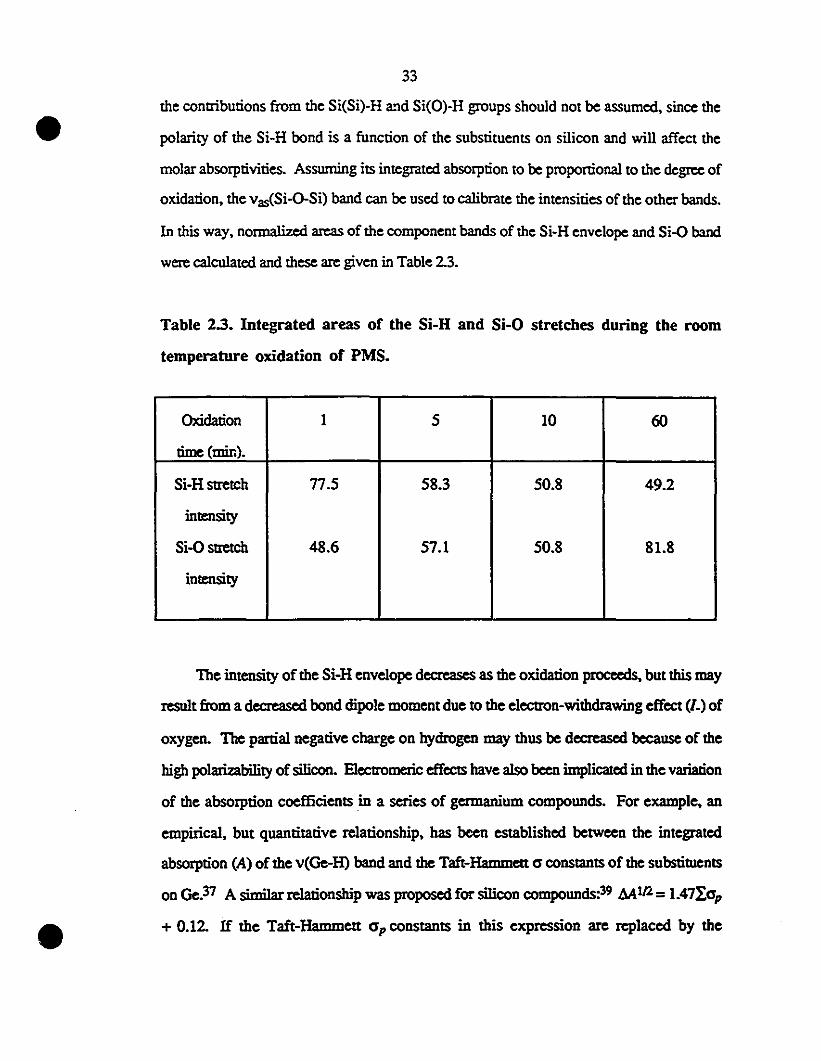
•
•
33
the contributions from the Si(Si)-H a..'1d Si(Q)-H groups should not he assumed, sincc the
polarity of the Si-H bond is a function of the substituents on silicon and will affect the
molar absorptivities. Assurning its integrated absorption to be proportional to the degree of
oxidation, the vas(Si-o-Si) band can be used to calibrate the intensities of the other bands.
In this way. normalized areas of the component bands of the Si-H envelope and Si-o band
were calculated and these are given in Table 2.3.
Table 2.3. Integrated areas of the Si-H and Si-Q stretches during the room
temperature oxidation of PMS.
Oxidation 1 5 10 60
rime (min).
Si-H stretch 77.5 58.3 50.8 49.2
intensity
Si-o stretch 48.6 57.1 50.8 81.8
intensity
The intensity of the Si-H envelope deaeases as the oxidation ploceeds, but this may
result from a decreased bond dipole moment due to the electrOn-withdrawing effect (1_) of
oxygen. TIte partial negative charge on hydrogen may thus be decreased because of the
high polarizability of silicon. ElCClI'Omeric effects have also been implieated in the variation
of the absorption coefficients in a series of germanium compounds. For example, an
empirical, but quantitative relationship, has been established between the integrated
absorption (A) of the v(Ge-H) band and the Taft-Hammett C1 constants of the substituents
on Qe.37 A similar relationship was proposed for silicon compounds:39 M 112 = 1.47IC1p
+ 0.12. If the Taft-Hammett C1p constants in this expression are replaced by the

•
•
34
electronegativities of oxygen and silicon, the predieted decrease in intensity at the end of the
oxidation process is 0.84, a value which is in reasonable agreement with that of 0.78
obtained from the IR spectra. In this case, the contributions from polymeric effects are
small, and the application of the concept of group frequencies is still valid, despite the
electronic effects along the silicon backbone.
The progression of....e vs(C-H) band during oxidation can be analyzed similarly. The
ratio of the intensities of the symmetric and antisymmetric C-H stretches of the methyl
groups is strongly influenced by the oxidation reactïon, although the integrated areas of the
antisymmetric C-H stretch for the PMS sample remained approximately constant: 7.84,
8.15, 8.29 and 7.87 in Figs. 2.8 a, b, C, and d, respectively. Therefore, t1ùs absorption
can also be used as an internal standard for normalization when oxidation has to be
avoided. The integrated areas of the bands listed in Table 2.4 have been normalized with
respect to the vas(C-H) band, whereas normaIized integrated areas for vasSi-Q-Si are
expressed as a pettentage of the integrated area after saturation.
Ifsaturation of the Si-O stretch band is associated with a total tranSformation of PMS
to polysiloxane, then the lowest obsetved ratio of the intensities of the IWO C-H stretching
vibrations of the methyl group (0.56) should correspond to that observed in
poly(methylsiloxane). The value obsetVed for the same ratio in poly(methylhydrosiloxane)
was 0.41.
The significantly decreased ratio of the intensities vs(C-H)/Vas(C-H) can also be
related to electronic effects. A change in the effective charge on the Ge atom due to the
effect of alkyl substituents has been mentioned in relation to the intensity of the Ge-H
absorption}1 Methyl groups have an (1+) effcet and seem to participate in the G-<1 conju-
31Egcrochl:in, A.N.; Khorsbev, S.Y.; Oslasheva, N.s.; Sevasyanova, E.L; S8rge, J.; Riviere, p.; Banau,J.; J. Organomt:l. ChmL, 1976, lOS, 311
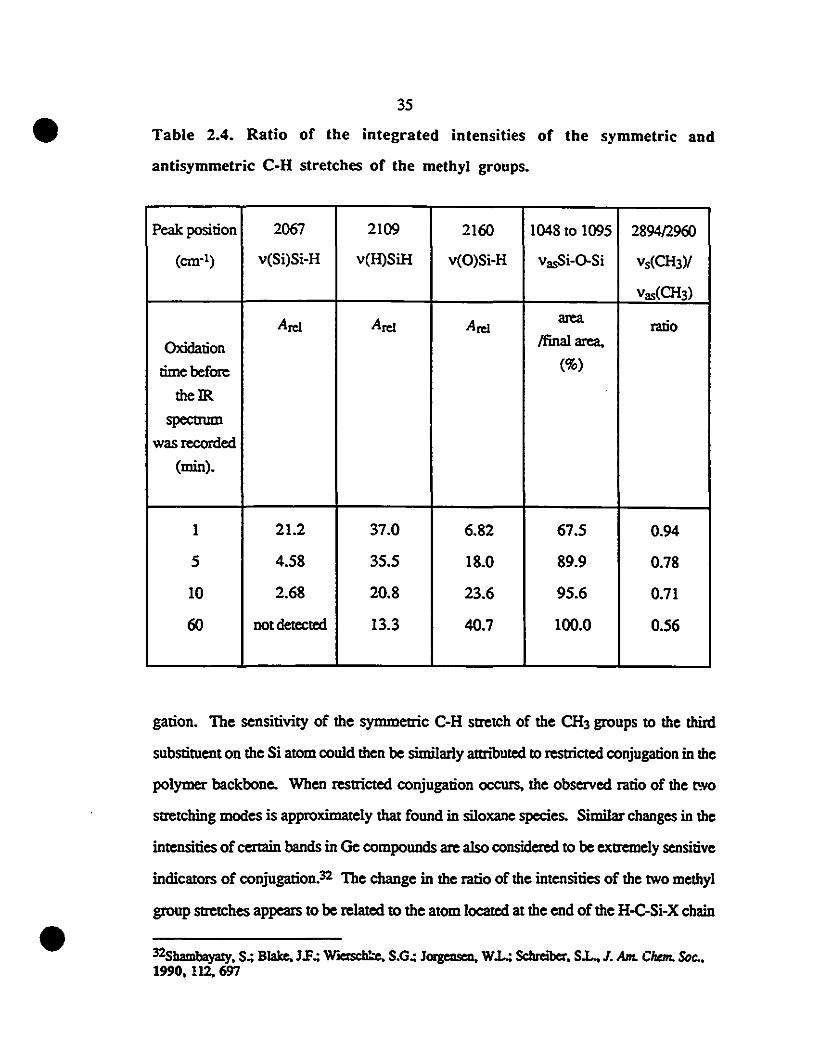
•
•
35
Table 2.4. Ratio of the integrated intensities of the symmetric and
antisymmetric C-H stretches of the methyl groups.
Peak position 2067 2109 2160 1048 to 1095 2894/2960
(cm-t) v(Si)Si-H v(H)SiH v(Q)Si-H va$i-Q-Si Vs(CH3)1
Vas(CH3)
Are! Are! Are!area ratio
Oxidation {final area,
timebeforc (%)
the IR
specttum
was recorded
(min).
1 21.2 37.0 6.82 67.5 0.94
5 4.58 35.5 18.0 89.9 0.78
10 2.68 20.8 23.6 95.6 0.71
60 not deteeted 13.3 40.7 100.0 0.56
gation. The sensitivity of the symmeaic C-H stretch of the CH3 groups 10 the third
substituent on the Si atom could then be similarly attributed to restrieted conjugation in the
polymer backbone. When resaieted conjugation occurs, the observed ratio of the ~o
stretching modes is approximately that found in siloxane species. Similar changes in the
intensities of certain bands in Ge compounds aIe also considered to be extremely sensitive
indicators of conjugation.32 The change in the ratio of the intensities of the IWO methyl
group stretches appears to be reiated to the atom located at the end of the H-e-Si-X chain
325bambayaty, 5.; Blake, J.F.; WJCrSCh:;c, 5.G.; JorgeascD, W.L.; 5c:brcibcr, 5.L., J. Am. Chem. Soc..1990, 112, 697
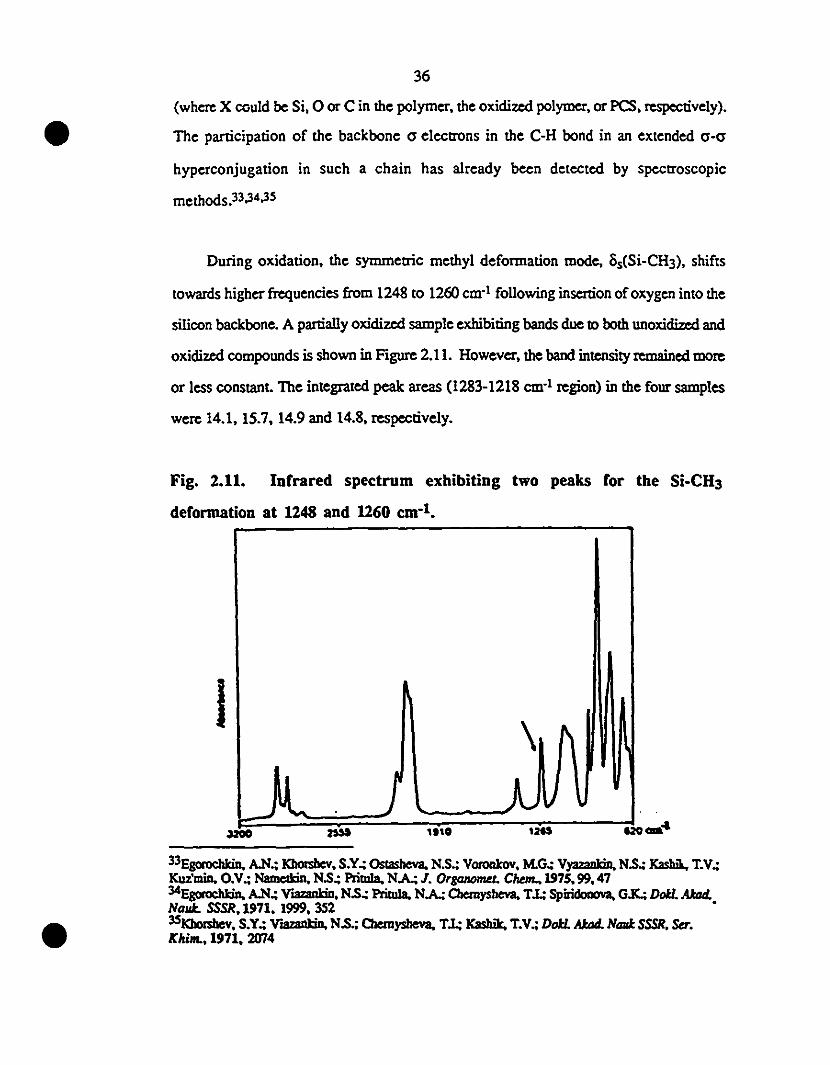
•36
(where X could be Si. 0 or C in the polymer, the oxidiz.ed polymer, or PCS, respectively).
The participation of the backbone a elcctrons in the C-H bond in an eXlended a-a
hyperconjugation in such a chain has already been delccled by spectroscopic
methods.33,34,3S
During oxidation, the symmetric methyl deformation mode, Ss(Si-CH3), shifts
lOwards higher frequencies from 1248 to 1260 cm-! following insertion of oxygen inlO the
silicon backbone. A partia1ly oxidized sample exltibiùng bands due 10 both unoxidiz.ed and
oxidiz.ed compounds is shown in Figure 2.11. However, the band intensity remained more
or less constant. The integraled peak areas (1283-1218 cm-t region) in the four samples
were 14.1, 15.7, 14.9 and 14.8, respectively.
Fig. 2.11. Infrared spectrum exhibiting two peaks for the Si-CH3
deformation at 1248 and 1260 cm-l •
\ "-.--JU...... J JL.v
1
"'0 '2U
•33Egorochkin, A.N.; K!lotsIlcv, S.Y.; 0sIasbeva. N.S.; Voroakov, M.G.; Vyannkin, NS.; Kashù., T.V.;Kuz'min, O.V.; Namelkin, NS.; Prilllla, N.A.; J. Org~t. Chem..l!l75, 99, 4734Egorochkin, A.N.; ViuMkin, NS.; Prilllla, N.A.; Olemysheva, Tl.; Spiridooova, G.K.: DoI:1. AkdtLNall1. SSSR,lll71. lm. 352 •3SKborshev, S.Y.; VI871llltin, NS.; 0Icmysheva, Tl.; KashiIt, T.V.; Dokl. AkdtL NaJll:SSSR. Su•KIùm., 11171, 2074

•
•
37
2.3.1.3. Effect of Room-Temperature Oxidation on
Poly(methylsilane) Annealed at ISO-450°C. Low-Temperature
Kumada Rearrangement and High Mobility of the Adsorbed
Precursor on the Silicon Surface.
The pyrolytically induced lI'ansfonnation of PMS into PCS involves insertion of a
silylene group into a C-H bond. The final product bas methylene groups located belWeen
the silicon atoms fonning the backbone of the polymer. During oxidation, the direct
insertion of an oxygen atom into the Si-Si bond could be detected spectroscopically
(Cbapter 2.3.1). Earlier pyrolysis srudies employed starting polysilane spccies with a
considerably lùgher degree of oxidation. Oxygen presence was evident in the IR spectra of
the starting materials.6•18 The IR spcctra of the pyrolyzed material alse exhibited features
due ta both the polysilane and the polysiloxane. In our expcriments, the vibrational bands
particularly affected were the symmetrie C-H stretch of the methyl group at 2894 cm-l, the
symmetrie deformation of the Si-CH3 group at 1248 cm-l, and the wagging vibration of the
CH2 group at 1050 cm-l . AIl three IR bands are shifted or overlapped with polysiloxane
species in the spectra of mixtures.
The room-temperature reactivity towards oxidation ofsamples pteviously heated in an
inert annosphere (Ar or N2> at different temperatures in the 150-45O"C range is illusttated in
Figs. 2.12 a-d. Thin films were coated on silicon wafers, prebeated at the desired
temperatttre and then subjected to oxidation at room temperarure. The saute spectral
fearures appear as in the case ofoxidation of unheated PMS, but on a different time scale.
As a general rule, the higher the preheat tempe:2ture in an inert atmosphere, the slower is
the rate of oxidation at room temperarure. Specifie behavior ta annea\ed PMS is also
exhibited. After 1h at 200°C, the intensity ratio of the two methyl
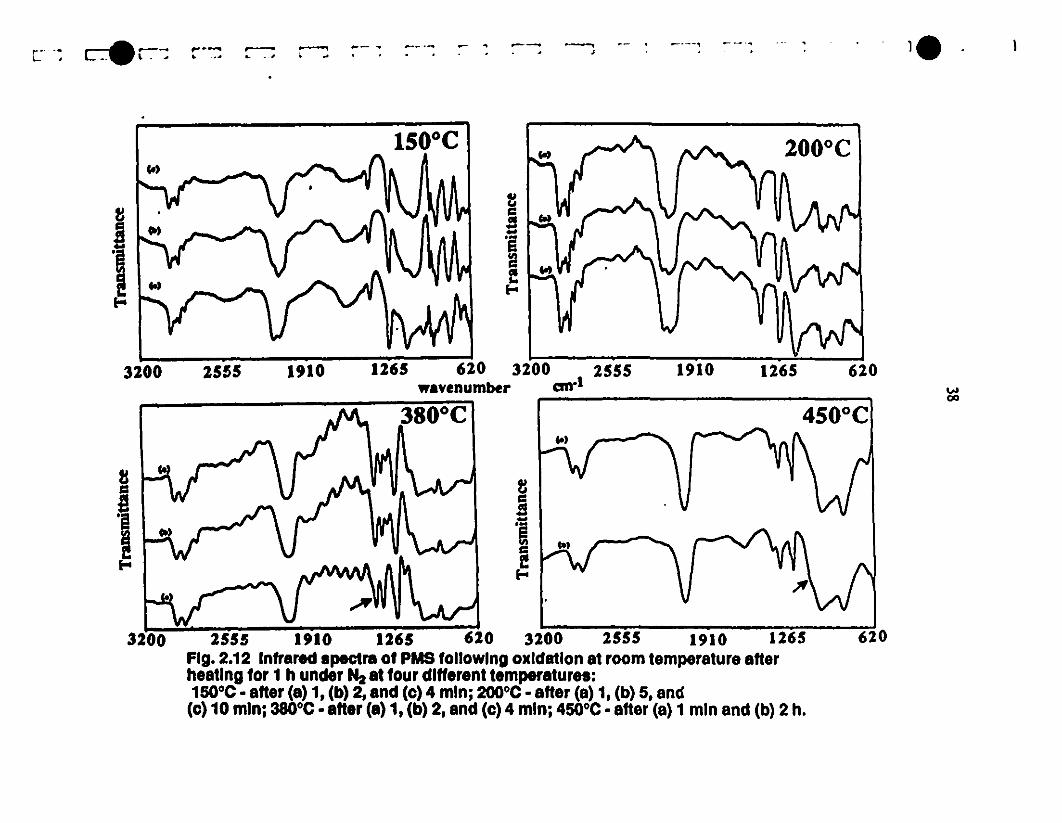
[ '. -. -..A~~ L-.'W''' .. -i
...~. -.~
....~ --~
--... ~- ... ~
- . '--', -----, 1. .
w00
620
450°C
200°C
U6519101
Z555cm'
j
J
J'§e
1-0
6Z0 3%00".venumber
380°C
150°C
UU1910Z555
3Z'00- Z555 1910 U65 6Z0 3%00 Z555 1910 U65 610Fig. 2.12 InfraNd spectre o' PMS .ollowlng oxldatlon at room temperature alterheatlng 'or 1 h under~ at four dlflerent temperatures:150°C· alter (a) 1, (b) 2, and (c) 4 mIni 200°C· alter (a) 1, (b) 5, and(c) 10 mIni 380°C· alter (a) 1, (b) 2, and (c) 4 mIni 450°C· alter (a) 1 min and (b) 2 h,
3%00
fi
t1-0
Jl1-0

•
•
39
stretches becomes less sensitive to oxidation. A new and unexpected feature is the
sharpening of the Si-O band at 1104 cm- I . The corresponding vas(Si-O-Si) band in
methyl-substituted siloxanes is broad with a characteristic half-width of -100 cm- I and has
an upper limit of just below 1100 cm-I . The above mentioned shift and sharpening effects
have !Wo important consequences.
First, the band shift allowed observation of an additional absorption at 1050 cm-l,
which is assigned to the ro(Si-CH2-Si) mode of PCS.36 This is an indication that the
Kumada rearrangement might be triggered at a much lower temperature !han earlier reponed
based on the IRdetection of the CH2 scissoring mode at 1350cm-l. The scissoring mode
of the Si-CH2-Si group at -1350 cm-l, may not always be observed due to its low molar
absorptivity. For example, on comparing the IR spectra of hexamethyldisilane, (CH3hSi
Si(CH3h, and bis(trimethylsilyl)methane, (CH3hSi-CH2-Si(CH3h, the characteristic
vibration for the Si-CH2-Si group is the ro(CHù al 1058 cm-I band, while the 1350 cm-I
band is not observed at al1.37 Onset of the Kumada rearrangement was indirect1y implied in
Schilling's work when it was noted that the intensity of the Si-H band in a vinylic
polysilane increased as the pyrolysis temperature rose from 250"C to 400"c, an indication
ofan ongoing rearrangement in that temperature range.38 The interpretation of the Fr-IR
spectra of the intermediate products of pyrolysis below 400"C ofanother vinylic polysilane
could be explained similarly.39 This result is also consistent with other IR studies and
mass balance indicative of the formation ofPCS at 18G-19O"C in polysilane systems.40
Second, the sharpening effect of the vas(Si-O-Si) band can be associated with an
increased order in the environment ofoxygen atoms. The position of the v(Si-O) mode al
1104 cm-1 is very close to the characteristic absorption for interstitial oxygen atoms
36gcarlete, M.: Brienne, S.; Hamld, JoF.; Butler, lS.; Chem. Mazu., 1994, 6, 97737Aldrich Library ofFr-IR SpeclIll II, 1991 21706-9 CAS [l450-14-2Il111d 28213-8 CAS [2117-28-41.iespectively38Schi1ling, CL., Jr.; Bri:. Po/ym. J.; 1986, 18(6). 3SS39Sclunidt, W.R.; InienaDte, L.V.; Doremus, R.H.; Trout, T.K.; Man:hctti, P.5.; Macic1, GA; Chem.Maze~,1991,3,2S7
4OMartin, H.-p.; M01ler, E.; Richter, R.; Roewer, G.; Brendlcr, E.; 1995,personal œmmunicazioll

•
•
40
captured in the lanice of silicon single crystals. observed at 1107 cm-l. The upward shift
of the Si-O stretch is characteristic of a highly ordered environment. During oxidation of
SiC single crystals. the Si-O stretch shifts to 1100-1098 cm-l, while for the polycrystalline
material it is observed at 1094-1086 cm-I .4l These observations can be explained by a
high mobility of PMS on the surface of the silicon substrate, which results in a preferential
absorption of the oxygen bridges of the siloxane backbone formed by oxidation in the
interstitial sites present in the silicon substrate just below the surface. Such a process.
which is thermodynamica11y driven, is expected te have a very 10w activation energy, as is
evident from the extremely high mobility of interstitial oxygen in the bulle of the silicon
lanice.42 The net result is a stimulated ordered conformation of the po1ymer induced by
interaction of the oxygen alOms in the backbone with the substrate. Funher ordering could
be enhanced by the interaction of labile Si-Si bonds in the polymer thereby allowing
interaction with the silicon dangling bonds on the silicon surface. The good adhesion of
the film suggests that such a sttong interaction between the film and the substraIe is
present.
The IR spectra ofPMS samp1es heated at 30ll-4SOoC in N2 and subsequently oxidized
at room temperature exhibit bands due to both vas<Si-o-Si) at 1107 cm·l and the wagging
mode of the inserted CH2 group at 1046 cm-l• The appearance of the scissoring mode
')(Si-CH2-Si) at 1351 cm-l in the sample preheated at 380°C (Figure 2.12c), confirms!bat a
Kumada rearrangement occumd at !bat temperature, but it is only evident during the final
stages of the reaction. On the other band, the ')(Si-CH2-Si) band at 1030-1050 cm-l
(usually stronger than the scissoring band) may be overlapped and even obscured by the
vas(Si-o-Si) band. Therefore, for high reaction coordinates the scissoring mode is a more
reliab1e indicator. If the object is to establish the thresho1d temperature for the
41EgorocblàD, AN.; VV!?Imkin N.S.; Kborsbcv. S.Y~ /:zv. AJ:JJtI. NauI:. SSSR. Su. Khim..1971, 207442oiumazd.A.: ~cbrlllCl', W.; Bourret, A. J. Appl. PIrys.1984. 56. 1670

•
•
41
rearrangement. then the weak peak at 1351 cm-! is not reHable - the more intense CH2
wagging mode should be used instead. For silicon single-crystal wafer substrates. this
mode is observed after heating for lh at ISO oC (Figure 2.l2a), while the CH2 waggir.g is
only deteeted after heating the sample (as deposited thin film) for 1 h at 380 oC in an inert
(Nv atmosphere (indicated by the arrow in Figure 2.l2c).
Significant variations were observed in the ratios of the integrated intensities of the
antisymmetric and symmetric C-H slretching modes during pyrolysis of the layer deposited
on silicon substrates. The ratio of the intensities. vs(C-H)/vas(C-H), reach::s a maximum at
higher values than in the unpyrolyzed polymer. and a possible explanation is that the degree
ofconjugation in the (J bonds ofthe polymer backbone is presumably affected by the nature
of the inserted group. In this case, there is apparenùy a long-range effect through a Si-C
bond from the conjugated chain. The polarizability of the silicon electronic shell facilitates
transmission of the electronic demands of the insened group towards the C-H bond. The
effect is detectable. not in terms of band positions. but in the ratio of the integrated
intensities of the symmetric and antisymmetric C-H stretching modes. Fmally, the band al
967 cm-! which appears at late stages of the oxidation can be associated with the 964 cm-!
peak in crystalline SiC, and it bas previously been attributed to the formation of a Si-Q-C
phase.43
Even after pyrolysis for 1 h at 450 oC under inen (Nv atmosphere, the polymer is
still reactive towards oxygen (Figure 2.12d). Sensitivity of the PCS towards oxidation is
also observed in the commercial produets from Dow-Corning.44 The presence ofoxygen
could not be completely eliminated but the low intensity of the vas(Si-Q-Si) band (mdicated
by the arrow in Figure 2.12d) still allowed observation of the CH2 wagging mode al 1042
cm-l • At 380 oc, a new band appeared al 1311 cm-l, accompanied by a shoulder on the
43Lee, S.Y. HcIIch, L.L. in Ul1TtzslnlclllTe Processùlg ofAdvanudSlTUCIUTal and E/eClTOnic Mazerials, PadcRidge Ed. Noyes, NJ~ 1983, 17144Malsudi, B. Ceram. 111I. 1987. 3, 286

•
•
42
antisymmetric C-H bending mode of the CH3 group at 1409 cm'! (Figure 212c). These
two new bands were not detected for PMS pyrolyzed at 300°C for up to 8 h or upon
prolonged exposure of the polymer 10 air. The position of the former is in a region where a
Si=C double bond is expected.4S and its presence at -400oC would assure an
organic/mineral transformation involving an ethylenic pathway. The intensity of the
antisymmetric deformation of the methyl group is decreased if insertion is not complete
and the polymer is subject 10 oxidation.
2.3.2. Synthesis of Thin SiC-Layers Coated on Silicon
Single-Crystal Wafers
The elemental analyses of the vapor-deposited and spin-coated layers on silicon wafers
produced at llOOOC are il!dicated by the EDX spcctra presented in Figure 2.13. The
presence of Si and C was normally detected by EDX in both films; however. the spin
coated films exhibited a slightly stronger tendency towards cODtamination with oxygen.
This feature can he attributed to the tendency of the adventitious siloxane species 10
concentrate in the ceramic residue. rather than in the volatile species. The IR spectra of
both the spin-coated and the vapor-deposited layer after pyrolysis exhibited the
characteristic absorption of SiC at 800 cm-1• as shown in Figures 2.14 a and b.
respectively. A relatively sharp absorption is characteristic for the u1Irathin layers deposited
by the condensation froID the vapor phase. A bulk sample of the precursor. pyrolyzed
simultaneously with the spin-coated wafer. was examined by 29Si MAS NMR
spectroscopy. The resonances at about -20 ppm confumed SiC formation (Figure 215).46
The formation of SiC layers from the spin-coated precursor thus parallels the known
4SBaskir. E.G.: MailSCV. A.K.; Nefedov. O.M.l::v. A1:JJd. NIllI1c. SSSR. Su. Khim.1983. 131446Rcpons ofaœnain 3IIIOUIIt of silicon iD excess oftbe SiC composition aftcrpyrolysis ofPMS iD tbep=œeofCp:zTiMC2 may bc associared wiIb appeciabledegzee ofoxicfation ortbe preç1IISOr.1eading IDpyrolysis padlwaysproper for siIoxane species.
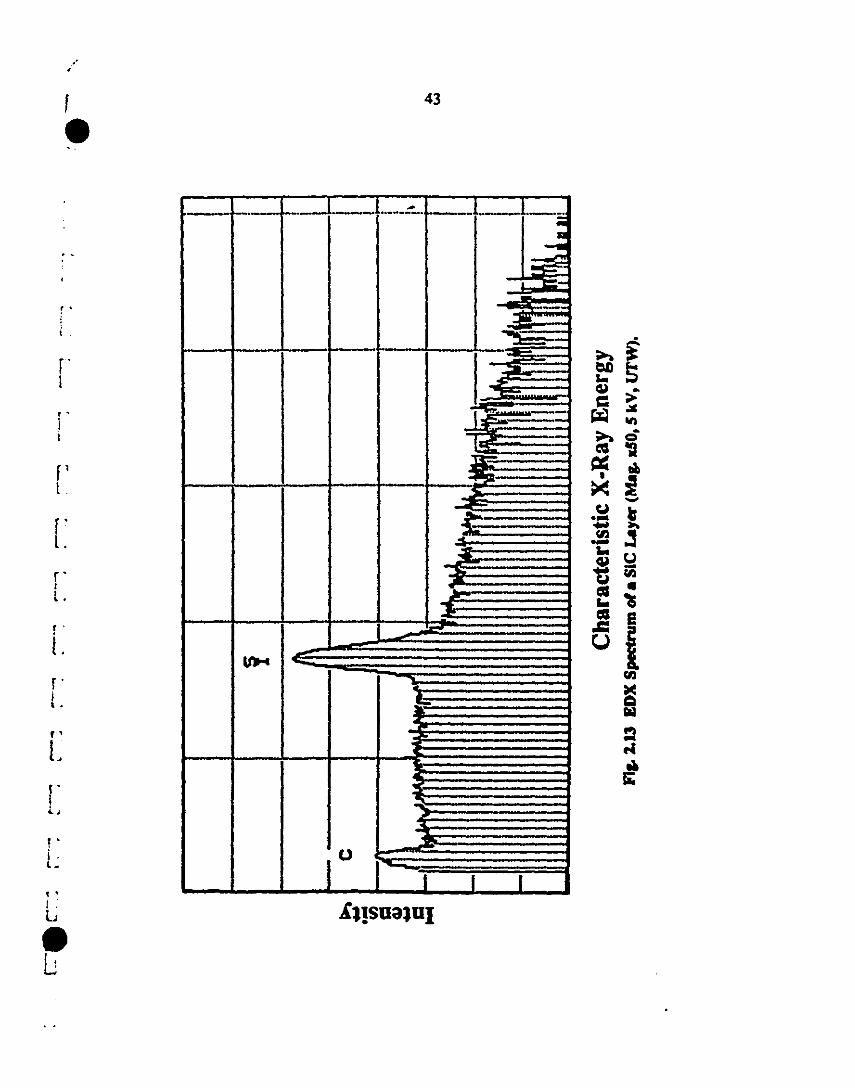
.'
1•
, -11
f 1
t
rl.
r-Il.
,'
L!-L
! .i .l .
43
.'1 .l ••L
1
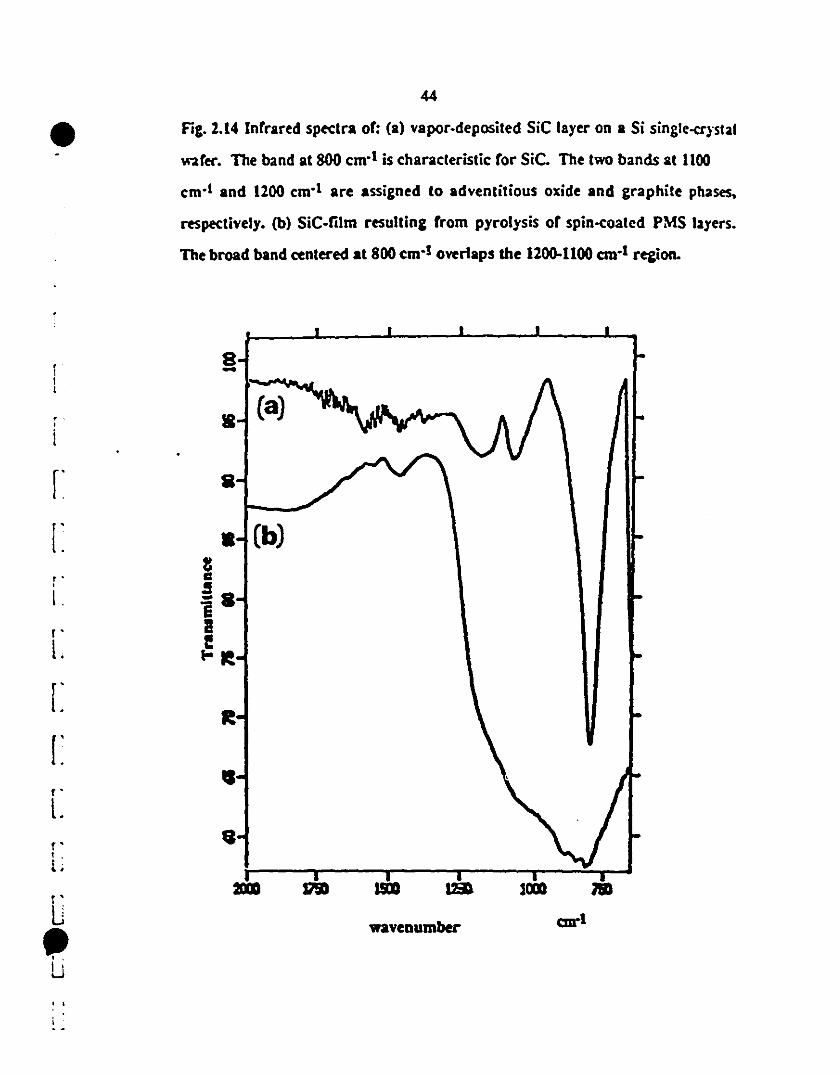
• Fig. 2.14 Infrared spectra of: (a) vapor-deposited SiC layer on a Si singleoOlstal
wafer. The band at 800 cmol is characleristic for SiC The two bands at 1100
cm-t and 1200 cm-t are assigned to adventitious oxide and graphite phases.
respectively. (b) SiC-film resulting rrom pyrolysis of spin-coalcd PMS layers_
The broad band centered at 800 cm-Soverlaps the 1200-1100 cm-l re&ion.
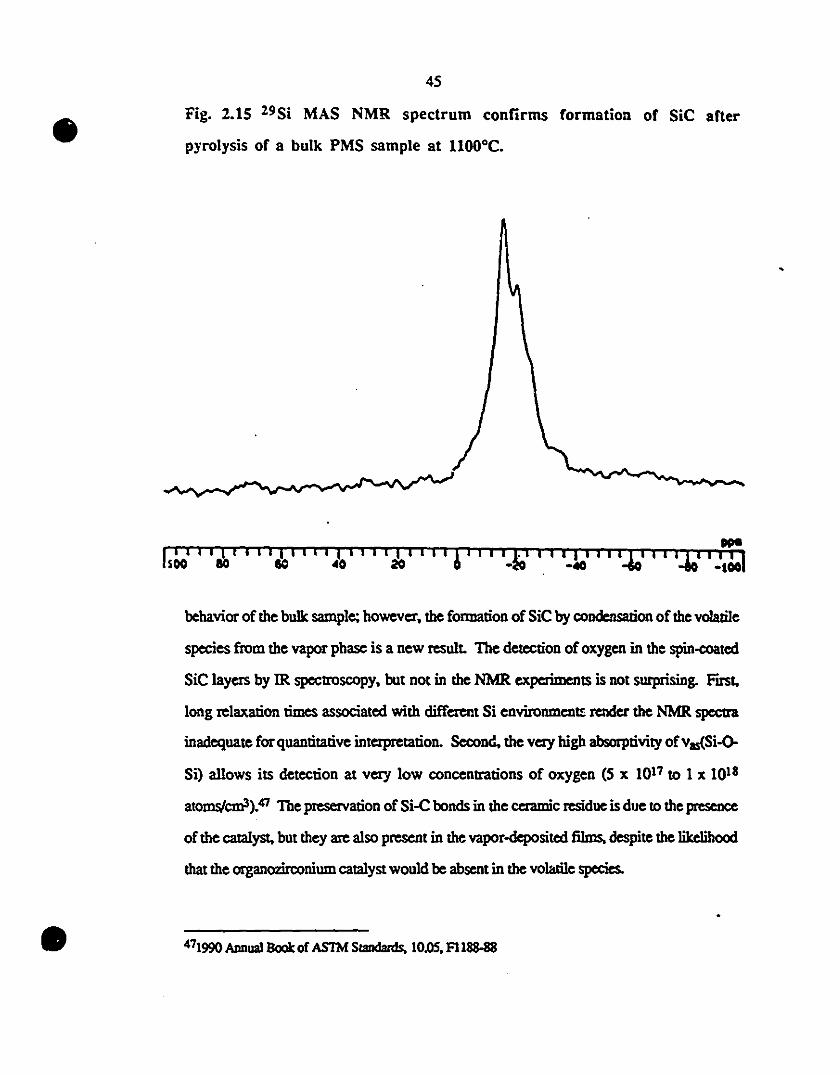
•
•
45
Fig. 2.15 29Si MAS NMR spectrum c:onfirms formation of SiC arter
pyrolysis of a bulk PMS sample at noo°c.
".1 i 1 i I~ f i i i 1 i i i il' i i i 1 i i i i 1i 1 i i lO' i , Iii i i ~ i 11=10' 1i i 1100 ISO 40 20 -. -40 -100
behavior of me bulk sample; however, ÛlC fonnation of SiC by condensation of me volatile
species from me vapor phase is a new result. The deteCtion ofoxygen in me spin-c:oated
SiC tayers by IR spectroscopy, but not in the NMR experiments is not surprising. FIJ'St,
long relaxation limes associated wim different Si environmen~ render me NMR specaa
inadequate for quantitative inteIpretation. Second, me very high absorptivity ofvas(Si-o
Si) allows its detcction at very low concentrations of oxygen (5 x 1017 to 1 x 1018
atomslcrrhC The presetvation of Si-C bonds in me ceramic residue is due to me presence
ofme eata1yst, but mey are also present in Ûle vapor-deposited films, despite the like1ihood
mat me organozirconium catalyst would be absent in ÛlC volatile species.
471990 Annual Book of ASTM SIlIDdards, 10.05. FlI88-88
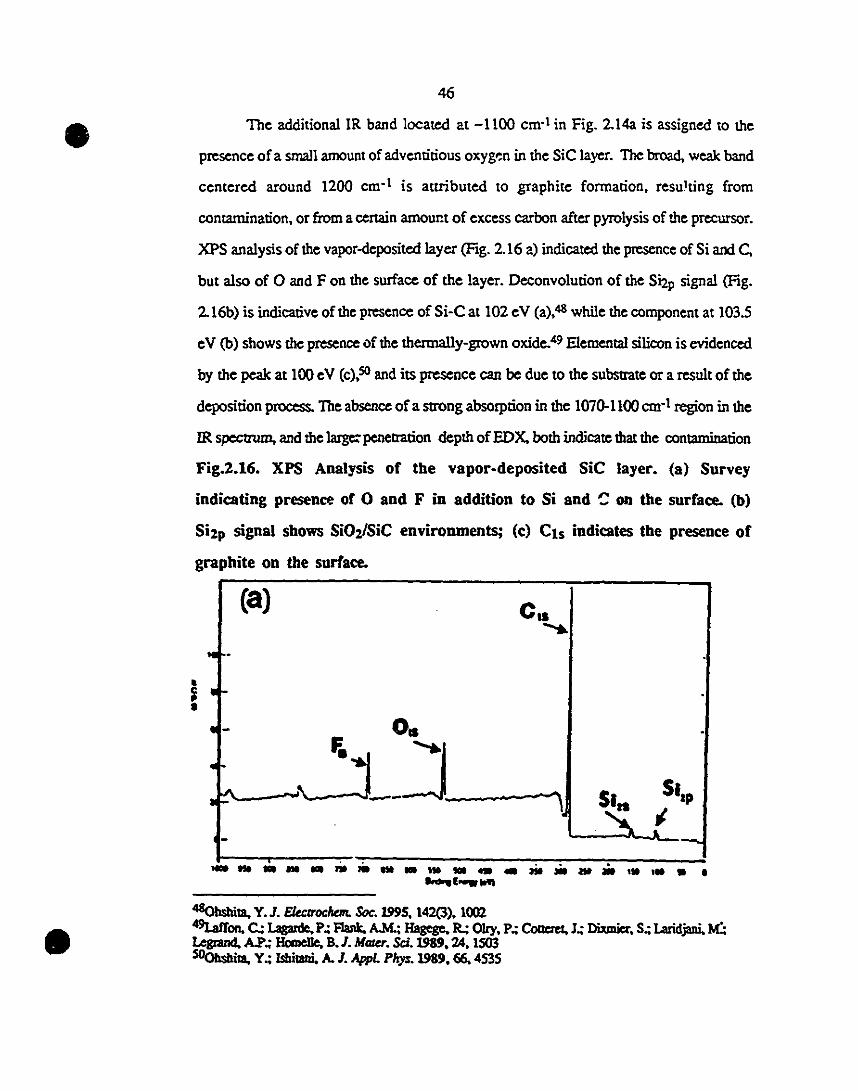
•46
The additionallR band located at -1100 cm'! in Fig. 2.l4a is assigned to the
presence ofa slllall amount ofadventitious oxyg~n in the SiC layer. The broad, weak band
centered around 1200 cm-! is attributed to graphite formation, resu1ting from
contamination. or from a cenain amour.t of excess carbon after pyrolysis of the precursor.
XPS analysis of the vapor-deposited layer (Fig. 2.16 a) indicated the presence of Si and C.
but also of 0 and F on the surface of the layer. Deconvolution of the Si2p signal (Fig.
2.l6b) is indicative of the presence of Si-C at 102 eV (a),48 while the component at 103.5
eV (b) shows the presence of the thermally-grown oxide.49 Eemental silicon is evidenced
by the peak at 100 eV (c),5O and its presence cao be due to the substrate or a result of the
deposition proeess. The absence ofa strong absolption in the 1070-1100 cm-1region in the
IR speclIllm, and me 1aIge:penetration depth of BDX, bath indicate thaI me contamination
Fig.2.16. XPS Analysis of the vapor.deposited SiC layer. (a) Survey
indicating presence of 0 and F in addition to Si and ~ on the surface. (b)
Si2p signal shows Si02/SiC environments; (c) Cls indicates the presence of
graphite on the surface.
(a) Cil......'" .If- .
1- O. .
.. - -~__Ja~ "\ SI.. 5.,p
." ~•-
. .~-.-.·· •• __ ~ a •••••
......'-""480bsbil3, Y. J. Ekcrrochort. Soc. ms, 142(3), 100249tafr0ll, c.: l.aprdc, P.; FIant, A.M.; Hagege, R.; Oiry, p.; Coaerel, J.; Dixmier, S.; Laridjani, M::Lcgzand, Al'.; HomclJe, B. J. M_. Sd. 1989,24, 1S03SOObsml3, Y.; lsIùl8llÎ, A. J. Appt Phy$.1989, 66, 4S3S
•~•
•
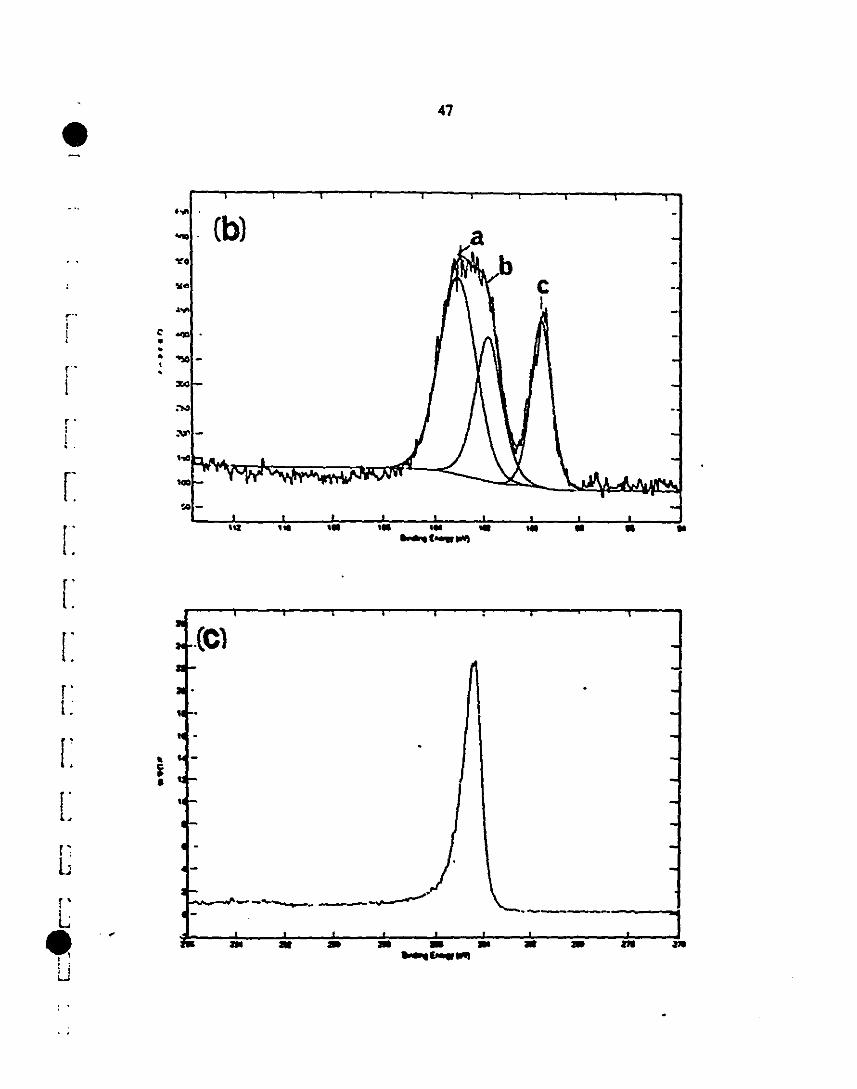
• 47
....._. Cb)-.<. b'''' c.... 1
r'i
"1 -••• -.0 -r • ,•
1 :x.o
"'"r .1
~ -! . ....r: -
>o-
r:..a ... •• •• ... .. .. .. .. ..
_1_11"I
r:L .(C)
r'L
r: •~•r.
l.
r~
Lr• --~- -----L .._---
• . ~
• - - - .. • - ...II
_e-WIlu

•
•
with oxygen is mosüy in a surface layer. Similarly to the SiC films obtained from PeS,5!
or tO the Nicalon NML 202 SiC fibers,52 it appears that the surface of the fùm may Oc
oxidized under ambient conditions after being exposed to air for a long period of time. The
Cls peak (Fig. 2.l6c) is indicative of significant grar>hite on the surface. This is consistent
with the information supplied by a depth-profiling analysis of an identical sample using
Auger spectroscopy, which has shown that both the silicon substrate and the SiC layer are
covered with adventitious carlxm.53 The formation of the carbon layers may be the resclt
ofcontamination of the sUIface during preparation of the subStrate before coating and ofthe
SiC layer during handling. Its formation may be in part responsible for the observed
decrease in the resistivity of the SiC films from the theoretical value (sec Chapter 3).
At the onset ofoxidation, the oxygen content cannot be measured accuratcly,S4 due
to the extreme broadness of the main IR band, which sometimes e.'ttends up to 1200 r.m.
1.55 DRIFTS analysis allows a qualitative estimation of the degree of oxidation by
comparison with the spectrUtI1 of silicon oxycarbide. since a shoclder can be observed at
high frequencies overlapping the main SiC absorption.56 However, DRIFTS spectra can
be deceptive.51 Advanced oxidation is responsible for upward frequen..'j' shifts to 900, or
even 1000 cm-l, while samples with 10w oxygen content exhibit an absorption at 800 cm-l.
Usually, the broadness of the absorption prevents the detection of oxidation or graphite
incorporation. The relative sharpness of the SiC band appears to be characteristic of a
higher degree oforoer in the SiC layer. For example, polycrystal.line SiC films obtained
from polymeric precursors. or by laser ablation deposition (LAD), after annealing, usually
exhibit an IR band roughly three times broader than that observed in Fig.2.14a, while
51Mucalo. M.R.; Milcslooe, N.B.; Viclaidge,lC.; Swaïn. M.V. J. Am. Cuant. Soc. 1994.448752Scbm:k, P.; Vix-GUItrl, C.; Ebrburger, P.; Lamaye, J. J. MIJlU. Scï.1992, 27. 4243S3HuIse, J. penolUZl CtJIrlmJlIÙCtJt NRC-1IlslibJte for Mic:rosauctural ScieIlce, 1993S4t.aine. R.M.; BabonMiu. F.; CheIn. MaleT'. 1993. 5, 260SSscbmidt, W.; lnlemImc, L.V~ CheIn. MIJlU.1991, 3, 2S1S6LaiDe, R.M.; BaboDMilI. F.; RaIIII, JA; ZhaDg, Z.-F.; Youngdah!, K.A.; 37-th SogamoTe Army Mal.Ru. Conf. hoc.; Vll'àmiclci. DJ~ Ed.; PubL IlcpL of the Army, 1991, 15951Tsuge, A.; UW8lIÙllO, y.; Isbizub, T.; Appl. Spec. 198(i, 40. 310

•
•
49
similar bandwidths to that observed here are obtained only in CVD deposition procedures
of highly orclered SiC films. This observation is indicative of a higher degreee of orcier in
the films obtained in our experiments and suggests that this polymerie precursor may be
considered from the point of view of a potential single-source for SiC layers with superior
texrural properties. This feature of the precursor has been exploited in processing thin
layers of SiC doped with nitrogen from NH3, and will be presemed in Chapter 3.
The crystallinity of the SiC layers was not of particular concem in this study, although
the unusual mobility of the precursor on the silicon surface rnight lead, under appropriate
conditions, 10 an increased degree ofcrystallinity. However, AFM measurements revealed
that most of the material is amorphous (Fig 25 004-006). Increased crystallinity should be
possible in principle, because crystalline SiC fùms can be grown below lOOO·C from
compounds that already comain the Si-C bonds in single-source CVD proeesses.SS
SSLadi:in. DJ.; IntemlDlc, L.V.; Springer Proceedings in Physics, Amorphousœul CrySlDlUne Süü:D"Carbitk, Eds. C.Y. Yang, MM. RabmaJI and G.L. HaIris, Berlin 1989. 239

•
•
50
2.4. Conclusions
The use of PMS as a polymeric precursor in the synthesis of a-SiC layers has been
srudied. Thin layers of semiconducting a-SiC were deposited by spin-eoating and via
vapor deposition on silicon wafers by pyrolysis of PMS. The oxygen content of the
precursor has a pronounced effect on the resulting vibrational spectra. A careful
interpretation of the vibrational spectroscopic data permits separation of the changes due ta
pyrolysis from those due :0 oxidation during the PMS to PCS transformation. The
development of the vas(Si-Q-Si) absorption band because of oxidation of the polymer
results in an overlap in the fingerprint region during the initial stages of the Kumada
rearrangement with the wagging mode of Si-CH2-Si groups, oo(Si-eH2-Si). The clean
1100-1000 cm-l region in the Fr-IR spectrum makes PMS arguably the best precursorta
date, if control of the oxygen content in the precursor during the synthesis ofSiC layers is
required. The quantitative analysis of the oxygen content in the precursor can be achieved
by means of IR spectroscopy, by using a calibration procedure involving the content of
interstitial oxygen in the silicon substrate.
IR specaoscopy can be conveniently used to monitor the transformation of PMS into
PCS as thin layers deposited on silicon single-erystal wafers. On the basis of the molar
absorptivities, the Ïmal stages of the Kumada rearrangement are best followed by
monitoring the behavior of the Oiàgroup scissors vibration at 1351 cm-l, wbi1e the CH2
wagging mode is a better probe for the initial stages of the rearrangement. The polymer
containing 25 ± 10 ppma ofoxygen undergoes the Knmada reammgement at temperatures
be10w 200°C, and the cbains remain mobile on the substtate.

•
•
51
CHAPTER 3
Nitrogenation of Silicon Carbide Layers Deposited on
Silicon Single-Crystal Wafers via Pyrolysis of
Poly(methylsilane)
3.1. Introduction
The semiconducting properties of SiC layers cao only be fully exploiled if reliable
doping procedures cao be developed. Doped semiconduclor-grade SiC layers are currently
produced primarily by chemical vapor deposition (CVD),I vapor phase epitaxy {VPE),2
molecular beam epitaxy (MBE),3 glow discharge4 or spunering processes.s A single
source LPCVD method, using CH3SiH3 as a source gas has been reported 10 10wer the
deposition temperature necessary to values useful for fabricating Si HBTs.6 Recently,
results based on the last methodhave been reported for the sYDthesis ofn-type SiC layers
doped with phosphorus.7 Still, there is considerable interest in the development of new
synthetic routes to thin ÏÙInS of SiC. Routes that involve polymeric precursors are
especially being considered, because of their succesful application in the formation of bulk
a-SiC. Consequently, the layers obtained from polymeric precursors are also largely
lHauory, Y.; Kruangam. D.; KaIoh. K.; Nitra, Y.; OIcamOIO, H.; Hamakawa. Y.; hoc. 191hlEEEPholOvollt1ic Specialists Conf•• New Orleans, 1987.6892KimolO. T.; N"15hino. H.: Yamashilll, A.; Woo. W.S.; Malsunami, H.: Springer Procecdings in Physics71. Proc. Amorphous and Crysuzl/ine Silicon Carbide IV. ElIs. C.Y•Yang. M.M. Rahman and G.L. Harris.Berlin, 1989.313Rowland, L.B.; Tanaka. S.; Kem. R.S.; na-.15. R.F.; ibid.. 844Tran. A. Springer PI'OC"",lings in Physic:s 34. Proc. Amorphous and CryslD1/ine Silicon Carbide J. ElIs.G.L. Ranis and C.Y.-W. Yang. Ber1in 1989. 167SManning. B.M.; Hewiu. S.B.; Tm. N.G.; MacEJwee. T.W.; Springer Pux:eediDgs in Physics 71. Proc.Amorphous and Crysuzl/ine Silicon Carbide IV. ElIs. C.Y•Yang. M.M. Rahman and G.L. Harris. Berlin1989.2526oo1ec1â. 1.; ReidiDger, F.; Mani. J.; Appt PIry.~. Len•• 1992, 57, 60S70hsiœ. Y.; J. EkclrOChem. Soc., 1995. 142(3). 1002

1
~111111111111111,.1
S2
a-SiC. Consequently. the layers obtained from polymeric precursors are also largely
amorphous. as opposed to the strUctured CVD films. As a consequence, me main researeh
focus is in applications for me manufacture of solar cells. The major expected advantages
are the relatively simpler procedure compare<! to the alternative memods. and c\osely
related. the reduced lime and COSt of coating large areas of complicated shapes. An
important limitation for the precursor-derived SiC films is the dopant profile. Resistivity
control in polymer-derived fùms can be achieved by diffusing the dopant into the SiC
layers by using the spin-on-dopant (500) technique. However. the SOD approach is DOt
always applicable.. When a SiC/Si heterojunction for solar cens is required. in situ
doping ofCVD-produced fùms is essential for reducing resistive losses and increasing the
fill faclOr9. An alternative in situ route, which presumably could be applied to polymeric
precursors. is the reaction of these precursors with dopants (boron or phosphorus
compounds) priorto pyrolysis.lO Unfottunately. the details ofthesedoping procedures are
not fully described in the open literature.
To our knowledge, there have been no procedures reported for the Ùl situ doping of
SiC layers via the polymeric route. We shall present below such a doping procedure with
nitrogen. whicb cao he performed at the same time as the pyrolysis. Nitrogen is
electronica1ly active in SiC and it is pteferred as a donor because ofits higb solubility and
reIa!ively low ionizalion energy (-70 meV).11 The one-step approach is based on an carlier
reported observation that annealing organosùanes, wbich initially coatain no nitrogeu,
onder NH3 resu1ts in the elimination of the organic groups anached ID the silicon precursor
·Solangi, A.: Qaudllly. Mo; J. MtJleT. Res. 1992,7. 247~awada, y.; XoadD, Mo; Otamoco, H.; Hamakawa, Y.; SolDr EMrgyMalO•• DI2. 6, 299lCOta, CJ.; TJDI, SJ.; Mackenzie, J.o.; Sprin&er Pmœedings in Pbysics S6, hoc. AmorphoKS tw1Cry$lGl1iM SiIicoft CDTbiIJe. Eds. G.L. Hatris, MG. Speoœr aDd C.Y. Yq. 1992,87l1TIIII. C.J.;~ S.J.; Cha, J.o. ibëd., 90

1
~111111111111111,1
53
and incorporation of nitrogen.12.13.14.1S This process. which rakes place al 400-600oC.
has already been used successfully in the production of pure silicon ninide (Si3N4).16
The presence of functional groups in the precursor material is vital for the crosslinking
and doping sleps. as well as for promoting adhesiOll to the substrate. Poly(methylsilane) is
a promising candidale from these points of view. The initial presence of Si-H bonds leads
to specifie features in the thermally-induced crosslinlcing process ocCUIring during the
pyrolysis of PMS. compared. for example, to the pyrolysis of l'CS. The bifunctionality of
silicon in terms of its erosslinking properties has already been noted.17•18 Other studies
have shown that SiC fonnation is controlled by the formation of HSi-SiH bonds leading to
the crosslinking of polymer ehains.19 The presence of catalytic amounts ofQl2Zr(CH3n
in PMS is particularly effective in favoring the formation of Si-C bonds rather than Si-Si
bonds during pyrolysis. thereby reducing the Si exeess down to stoichiomenically pure
SiC.20,21,22
3.2. Experimental
Poly(methylsïlane) was prepared according to the previously described procedure
(Chap. 2). Crosslinked samples were obtained by anionie crosslinJàag of !he Wurtz-
12BlIlWlI-wens1cy. !C.A.; Sinclair. RA; US PIIIDIl No. 4537942. DIS13Bujalslà. RJ.D.; EIITO~Ptllelfl ApplicodDII 200326. DI'14Bums, G.T.; Cbandra, G.; 1. Am. eu-. Soc•• 1919,72(2).333ISVarsbney. S.K.; BeaIry. CL.; Proc. 6111 AMual Corrfemu:e 011 Compomes tIIId AdwJIIœd CeramicMalUids. Cocoa Beach. FIorida, 1989. 55516okamura, K.; Salo. M,; Hasegawa, Y.; Ceam.llfl. UI1. 13.55l'Han, RN.; LiDdquisz, DA; HasgC/t)'. J.5.; Seyfenh, D.; Chem. Maur•• 199204. 70S18Schmidt. W.R.; Man:betti. P.5.; IntemlJlle, LV.; HurIcy. WJ. Jr.; Lewis, R.H.; DoRmIlS, lUl;MacieI. G.E.; CIrDrL MalU. 19920 4. 93719Bouzy. B.; CalpeDter. L.; Ccxriu. R.; Allgew. Chem.llfl. Ed. Ellgl.. 199O. 29(7). 78S2OMu.. Y.; Hmod.J.F•• /lIOrgaIIic tw1 OrgtlllOmelll11û: PolymenaNl Olipnen, J.F. HInod lIld RoM.ùine Eds., KJuwre Açadcmic: PubIisbeIS, Docdn:cIu. U9L 2321Seyfenh, D.; Wood, T.G.; TIxy. HJ.; Robinsoa, JJ.I. Am. Ceram. Soc. lm, 75.130022Hengge, E.; W'1Cllbc:rJp::r. M./.O~C1rDrL lm,433. 21

1
~111111111111111,1
54
coupled prepolymer. then elimination of chlorine end-capping promoled by LiAléil.
Soluble polymers were prepared by further polymcrization of the Wurtz prepolymer with
CP2znCH3h. They were then subjecled tO pyrolysis. A typical pyrolysis cycle consisled
of [WO stages: in the fU'St Step the heating rate was 2-5 oC/min up 10 450°C, followed by
maintaining the temperature at 45O"C for 1 h in order 10 produce PCS; in the second step,
the heating rate was SOC/min up to 1100°C and the samples were maintained at the final
temperature for 90 min. The pyrolyses were performed under various gas atmospheres
such as UHP Ar, NH3, 7% H2IAr and different mixtures of N2fNH3 and ArINH3.
Prepurified NH3 from Matheson was passed immediately prior to use through a column
(l.5 m length, 2" diameter) of KOH and 4Â molecular sieves at rates ofl-1OO mUmin.
A significanùy different behaviour of the precursor occurred under NH3. or NH3IAr:
annospheres compared with that observed during pyrolysis in Ar. with respect to the mass
transpott through the gas phase. During the doping experiment performed under a reactive
NH3 annosphere, a significantly increased mass-transpott of a volatile species onto
subslrates located in the colder regions of the fumace was observed. This enhanced mass
transpott led to metallica1ly bright. reflective coatings on any object placed in the cooler
zone. Therefore, in addition to the usual dip- or spin-coating procedures, the technique
involving the transpon of the volatile species to a substrare located al the colder end of the
horizontal furnace was also adapted to the pyrolysis cycle. Superior topological
characteristics of the layers compared to the films obtained by the classic methods were
obtained for the films deposited using !bis procedure, referred to below as the cracked
polymer vapor dcpositiOD (CP-VD) technique, due to the involvement of the thermally
cracked precursor in the vapor deposition process. Various substrates were used,
including electItlllic grade alumina. quanz plateS, isostaIica1ly pressed graphite, and ceramic
boats. The most tholOUghly studied substrate was silicon single-crysta1 wafer (4", 12-20
Q cm n-type doped with phosphorus, orientation [100], polished on one side and Iapped

1
'-111111111111111,1
55
on the other). The silicon substrales were prepared follo....ing the procedure outlined in
Chapter 2.3.1. Spin- and dip-<:oating procedures were used tO prepare the layers thal were
doped by lIÙxing the DMZ-<:atalyzed PMS with a doping-polymer. prepared by reaction of
the Wunz-dehalocoupled prepolymer with NH3.
To facilitate the measurement of nitrogen in samples. a special deposition procedure
was developed in this work. based on the observed sharpness of the SiC IR band for very
thin films. In this procedure, an alIllOSphere of Ar and NH3 (10:1) was used and gas flow
over the substrate was blocked by a small alumina plate. placed perpendicular to the
substrate. as shown in Figure 3.1. In this way. different areas of the substrate were
shielded from the gas flow.leading to very slow deposition rates. This experimcnt led to a
pronounced gradient in the thickness of the deposited SiC la)'C' with sorne areas only being
coated up to a thickness of-20 nm.
The purity of the starting polymers was determined by comparison of their IR spectta
with those previously reported,23 and the degree of initial aosslinking was determined by
1H NMR specnoscopy from the integrated intensities or the Si-H and CH3 groups. The
pyrolyses were perfonned in a Lindbetg single-zone programmable fumace equipped with
a Eurotherm PlO temperature controller which could he operated up to llOO"C lIId
provided an accuracy of±lo at llOO"C. EDX and XPS lIIla1yses were used todetermine
the e1emental composition of the layers obtained. Identical samples ofbulk material were
pyrolysed al the same lime as the coated wafers and 29Si MAS NMR specuoscopy wu
used 10 deteet the presence of different Si enviroaments (such as SiC, SiJN4. Sic.: lIId
e1emental Si) in the IeSU1ting powders. The texture of the layers wu examined by SEM.
The thicknesses of the minor-like layers were determined by eIlipsomeb'y for tbicknesses
less !han SOO nID and a Sloan-DeJctak profilometer wu used for measuring the micrometcr
thickness range of the spin coated layers. The resislivities or the coatings were measurcd
with a four-point probe and the conductivity type wu determined by EFM (hot probe)
23Sear1c1c.. Mo; Brieanc, S.R.: Hanod, JoF.; ButJcr. LS.; C1IDIr. IIt111:r. llIM. 6, m

..1
1
S6
Fig. 3.1 Procedure used to fonn a pronounced thickness gradient in the SiC layer.
The pattern imposed on the gas now renders the deposition rate a function of
the location of the deposition area with respect to the alumina plate.
measurements. The Fr-IR specaa (4 cm-! IeSOlution) were recoIded on a Bruker lFS-48
spectrometer equipped with a microscope, MCT deteetor. and a SONY-Trinitron PYM
1340 color monitor for specaal display. Some ofthe coated samples prepared in our wodc
are illustra1ed on the front page from the Canadian Ceramics Quanerly (Figure 3. 2.)
( . ~;;;;:::::.- \~-N-H_-,Ar---J - If'.... - _SiCJayerex"itflow
.... ~,:riç - if:'.....--- -/\~~ : J~--~..... . .
/ O-.O~~...qual1Z fumace
1
1
1
1
1
1
1
1
1
1
1
1
1
~1
o ocf healing element

Si
JOURNAL OF THE
CANADIAN CERAMIC SOCIETYVol 63, No. 4
November 1994
," . ': - CANADIAN ' ' .:. .,'
1
lJPiC'E'R~À\Mœ-C'S' N,w', S"'"" andni, l "'-.i QeJU.......A~ R..... "'aT"E+<i.R~L' :;- ,;' Technology for thel...- 1 _...... Canadian Ceramic Industry
1 il N'-TvPE 51'( ,,'-==~~"~'• 1 ):\1-:,-, ~
1 ON VARIOeS :-;:, S:' 'Z \ ' ::.
111111111111,1

1
'-111111111111111,1
58
3.3. Results and Discussion
3.3.1. ln Situ, Gas-Phase Nitrogen-Doping Procedure During Pyrol~'Sis
The SiC layers that were deposited on silicon wafers (sec Chapter 2.4.) exhibited
resistivities of the order of 1()2-1()6 n cm. This shows the combined effects of all the
adventitious impurities in producing charge carriers. From this level ofelectrical purity, the
effectiveness ofany doping proccss !hat car.: be considered is limited to resistivities lower
than 1n cm. The resistivties required for SiC layers in SiC/Si heterojunctions are in the
range of 1 n cm-Q.Ol n cm. Considered together with the texturaI characteristics of the
SiC layers deposited from polymeric precursors, the resistivity factor suggests the use of
the resulting thin films for solar cells.
As mentioned in section 3.1. the dopant-profile requîrements are critical. An in situ
doping process with nitrogen was explored. by using the reactivity of the Si-C bond with
NH3 under pyrolysis conditions. Attempts were made to fonn homogeneously distributed
SioN bonds !hat can be presetVed in the pyro1ysis prodUCL The reactive atmosphere of NH3
aets not ooly as an amination reagent, but also as a reagent for' the traDsaminatioo reactions,
resulting in the smoothing of the dopant profile by redistribution ofthe SioN bœds. 'Ibus,
partial conversion of the Si-C bonds into SioN bonds under carefully contro1led partial
pressure of NH3 in the gas phase was used to introducc amounts of nitrogen into the
growing SiC layer, in th~ range of interest for the doping process. By usiog partial
pressures of NH3 in an Ar carrier, silioon single-<:r'},stal wafers coated with heavily doped
SiC layers COuld be produced. When the mole ratio NH31'carrier was in the 0.1 to 0.01
range, the resistivities dropped dramatically froID those measured for SiC layers obtained in
either Ar, or N2 atmospheres (sec Chapter 2.4) to 10-0.03 Q cm, corresponding to
concentratioDS of 101S-1018 donorslcm3• respectively, after compensation (Table3.1).

1
~111111111111111f'1
59
Table 3.1 Influence of 1% :"113 in the carrier Ar on the resistivities of SiC-
layers.
Substrate Resistivity of the Layers [Qcm]
Inen AlIIlOsohere (Ar) Ar:NH~(-l%)
SiÜ2 > lOS 0.055
Al2Û3 >9 x lOS 0.030
Si > 3 x lOS 3.3
The resulting product!: exhibited -100% spati'.t.1 di;persion in the resistivity. for substrates
with areas of2 cm2. The large observed variation is probably due to the variable gas-flow
regirne leading to inhomogeneities on the substrate surface. This assumption is based on
close similarities with the previously reponed variations in dopant profiles obtained in
silicon crystals because of unstable melt-flow panerns of the molten silicol'i during the
growth process. Since standard flow meters were used to control the pressure and flow
rates, and since no special precautions were taken t:l insure the mixing of components in
the gas phase, improved results could be expected ü mass flow controllers (MFC) were
used and the design of the geometry of the furnace was compensated for the resuiction of
the flow.
When the mole ratio ofNH3 that was employed during pyrolysis was above 0.15. the
produets were different from those obtained at Iower NH3 ratios. A more complieated
situation arose, as indiCated by the SEM image (Figure 33) and EDX spectra (Figure 3.4
a,b) of a typical coating for these conditions. Two separate layers were formed on Ihe
substrate which were wgetted in the EDX experiments. The IWO highlighted 3IeaS a and b
in Figure 3.4 were probed by EDX for (a) the continuous layer and (b) the shanered film.
The concentrations of both nitrogen and oxygen were significantly greater in the top layer
(b). The presence of both elements is significanr. as surface contamination during

1
~11111111111111l'1
60
manipulation and analysis would only result in enhanced oxygen contamination being
observed. Moreover, since tlle thennal decomposition of NH3 al higher temperature
should lead to a decreased amount of nitrogen in the growi.'lg SiC film, a reasonable
explanation would be that both the excess nitrogen (i.e.• that beyond the solubility Iimit in
SiC) and adventitious oxygen present in the pyrolysis atmOsphere during the experiment
were segregated into the emerging phase upon cooling. or were continuously segregated
during the growth. The pronounced segregation of both major impurities (nitrogen and
oxygen) in the top layer suggests atl unidirectional solidification process which stanS from
the substrate and involves distribution coefficients lower thall 1. The partition coefficients
for both oxygen and nitrogen are known to be smaller thall 1 in all the unidirectional
solidifying processes observed for SiC (Acheson process, epitaxial growth or melt
solidification).24 nie clear delimitation of the !Wo layers Catl be attributed to IWO
immiscible phases.. a "carbide-like" lower layer and atl emerging. cracked "niaide-like"
phase. since Si3N4 and SiC are immiscible. The homogeneous texture of the underlying
phase indicates that a signifiCatlt amount of nitrogen Catl be bomogeneously incorporated
(on the SEM scale) into the SiC lattice, in accordance with the higb levels of doping
observed during the experiments.
The use of IR spec!:roSCopy did IlOt provide atly additional information on the presence
and type of bonding for nitrogen. most probably because the cbaraeteristic vibrational
modes ofSi-N.expectedal-9SO. 840 and 8OOcm-1•2S were buried UDderthemucb lIlOIe
intense and broat! absotplion al -800 cm-1 due to the amorphous SiC layer. Several bulk
SatDples, whicb were pyrolysed simul.'"meously with the layers produced on the silicon
substrale, were examined by 29Si MAS NMR. A varlet)' of SiCxN)' materials, ranging
from pure SiC to pure Si3N4. were produced depending DOt only on the NH3 camer gas
mole ratio. but also on other conditions such as flow rate, the annealing program and the
24yen, CT.; Tiller. W.A. J. Cry#. Growt1l 1991, 109. 1422SNaimaD, Ml..; Kilt, CT.; Auc:oiD. RJ.; Tar)'. F.L.: Senturia. S.D.; J. Ekctrodrem. Soc. 1984. 131.637
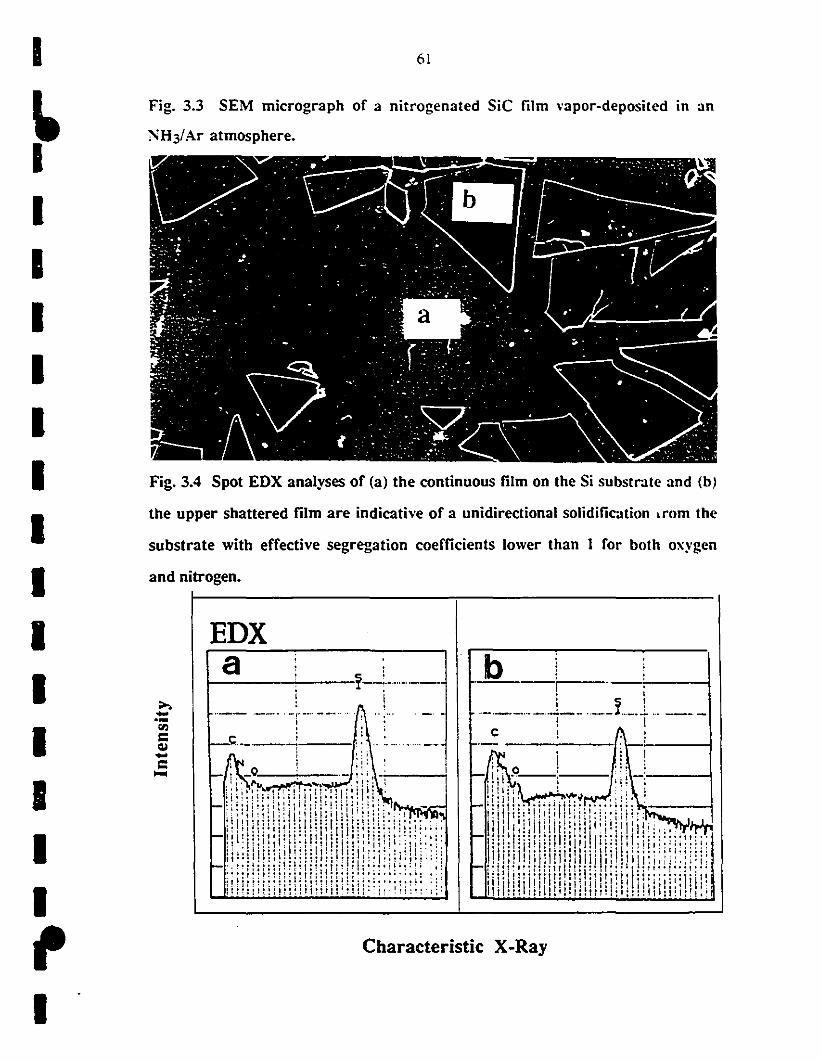
Characteristic X-Ray
Fig. 3.4 Spot EDX analyses of (a) the continuous film on the Si substrate and (b)
b
61
EDX
;..,-.-<IlC~-c....
the upper shattered film are indicative of a unidirectional solidification •rom the
substrate with effective segregation coefficients lower than 1 for both oxygen
and nitrogen.
Fig. 3.3 SEM micrograph of a nitrogenated SiC film vapor-deposited in an
NH3/Ar atmosphere.
1
l-I11111111111111,1

1
~111111111111111f1
62
sizc of the samples. Characteristic 29Si-NMR signais for SiC (a SlIUCtured band aI -20
ppm)26.27,28 and Si3N4 (a featureless. broad band at -50 ppm or a band envelope with
subsnuctures at -47 and -49 ppm)29,30 were observed for the limiting cases. There was no
NMR evidence for the presence ofany e1emental silicon (-80 ppm) or SiÛ2 (-100 ppm) in
any of the pyrolysis products. Results related to pyrolysis produets with Iùgher niuogen
content, such as silicon niaide and the intermediate species formed during il'le
carbonlniuogen exchange towards Si3N4 will f<ll'IIl the subjects of Chapters 4 and 5.
The detection of nitrogen in a SiC lanice (e.g.• shallow donors in SiC-based
semiconductor devices) is usually quile a difficult task. At about llOO"C. the materials are
essential1y amorphous and no useful information can be obtained by diffraction merlJods.
NMR methods have been extremely useful for the characterization of bu1k samples, but 50
far techniques for srodying the chemistIy of thin f.ilms have not been widely developed. A
serious problem with silicon-containing materials is the very slow relaxation of 29Si in the
rigid ceramic matrix. This gives rise to broad resonances in which much of the chcmical
information is obscured and signal amplitude is not Iùgl! enough to aI10w detection of
signals from microscopic samples31• For example. although 13C and 29Si spin-lanice
relaxation measurements have becn used to deteet ciuogen locaJ:ion in bulk 6H-polytype
silicon carbide.26 indirect methods such as the Hall effect,32,33:14,35 luminescence36. or
26Rocbe1eau, R.E.: Zhana. z.; J. Appl. PIrys~ 1992, 72(1). 28227Marcband, A.; FoId. M.T.; MetIlIS, F.; Valade, I.;J. CIùm. Plrys•• 196C. 61(3). 3432SRichaRIsan. Ml'.; HartmaII, J.5.: Guo, D.; CGIL J. CItar&.. 1992, 70, 70029onvicri, A.C.; HalfieId, GoR. J. ofMoglI. ResoIL, 1991. 94. S3S30rb0mpson, D.P. J. Am. Ceram. Soc•• 1991. 74. m3111bas bc:en lIlCCIIlIy demonsll'lllCd Ihalmore inforrnaIioo c:an tic extraClt.d by aDowiDg a very long lime fcrdie Duclei ID relax (HartmaD, 1.5.; Narayanao, A.; W8llg. y.x.; J. Am. CItar&. Soc., 1994. 116,4019).~.lbcse moreeJabcn'C tedmiques gteIlIIy iDa acquisilion time.32yacbiballl!, T.; lCœa. H.S.; Waoa, Y.C.; Davis, ItF.; J. Appl. PIrys., 199O. 67.637S33A'elcseeako. M.V.; zatxodskii. A.G.; Tlll'lofeev. M.P.; Sov. Plrys. Semico1ld..19S7. 21.49434
3SzJwIg. H.; PensI, o.; Glasow. p.; T r"bcDzeder. S.; TN: Ekcrroehemical S«kty Ezwtded Abszracu.l!JI9. 89-2, 7143Gcboyte, W.J.; Pmict, L.; Pity$. RD~ 1962. 127. 1868

1
~111111111111111
"1
63
EPR rneasuremems37 normally have had to be useci for the analysis of thin films. AuemplS
have been made to relate the IR bands appearing at very low temperatures (7-80 K) to
different nitrogen locations in the SiC lanice)! In our work. we applied Fr-IR
spectroscopy in the characterization of thin fùms. due to ilS adaptability to the SiC/Si
system, since the Si substrate is transparenL
The combined effects of (a) considerable nitrogen insertion because of the higher
mole fraction of NH3 (O.IO) present in the gas phase d!IIÏng the pyrolysis and (b) the very
small thickness of the obtained SiC laye:rs, resulted in the strUetured IR-absorption band
shown in Figure 3.5_ The shift in the v(Si-C) band from 800 cm-. in pure SiC, to 822 cm
• suggests the formation of a silicon carbonitride species and is consistent with the expeclCd
increase in the SiC force constant in the series Si-C-Si < C-Si-N. This is aise strong
evidence for the existence ofa homogen~ silicon carbonitride phase, where the niuogen
concentration is quite low. The relatively large shift argues strongly ~gainst a two-phase
system consisting ma:nly of amorphous SiC. Together with SEM measuremellts
anaIogous to those mentioned above, this is an indication that more than a single
carbonitride phase may exist in this range. In addition. a new band was deteeted al 939 cm
1 (Figure 3.5). This additional band is charaeteristic for the SioN stretch and, tO our
knowledge, is me fust time that nitrogen b3s been detected in SiC laye:rs by IR
SptCtroscopy at room temperatme. The planar geometry of niaogen, as suggesled by its
band posItion, may he a remnant state froID the precursor (sec QapIer 4 and referenœ 39).
37Carlos, w.E.; Moore, WJ.; Siebeamann, P.G.; FteiIaS, Jr~ JA; lCaplaD, R.; Bisbap, S.G.; NonIquislJr~ P.E.R.; Kong, M.; davis, R.F.; l'Toc. Mar. Res. Soc. Symp~ 1917, Ambejm, ca, 27638SUIIIOP, w.; P=sl, G.; CboyIce, WJ.; Dllmen, A.; LeibenzrdeT, S.; SIeÙI, R.; Sprïnaer Pnxecdinp iDPhysics 71, Amorp/lDllS cwl Crystal1üle Silicoll Corbïde IV, Eds. C.V. Vq. M.M. Rabmaa lIIld G.L.Harris, lm, llerIin. 12939He, J.; SCarIele, M.; Harrod, J.F. J. Am. ~TQIII. Soc., 1995, iD pRSS
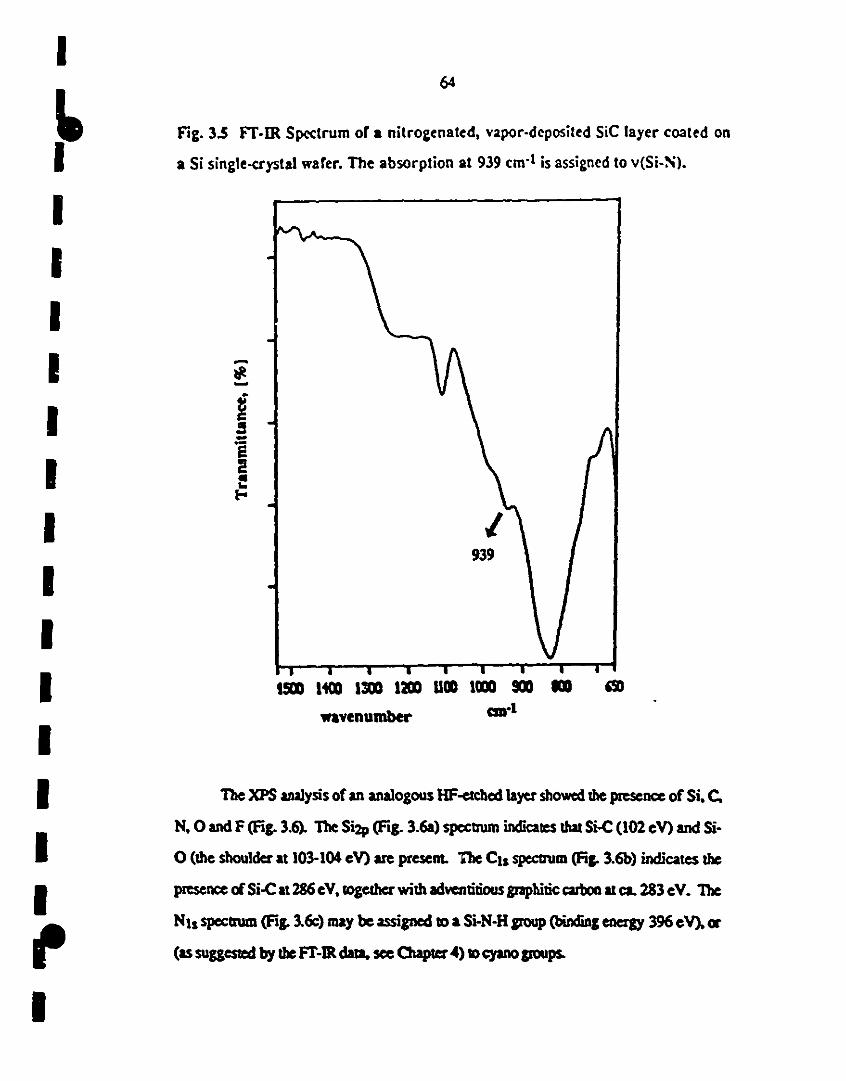
Wlvenumber
1
~11111111111111f'1
64
Fig. 3.5 FT·IR Spcclrum or a nilrogenaled, vapor·dcposiled SiC layer coaled on
a Si single·crystal wafer. The absorplion al 939 cm-l is assigned 10 v(Si·S).
939
I!D) 11CXl lB lB UlD lem 9CXI IIIœrl
The XPS analysis of an analogous HF-elthed layer showed the presence of Si, C.
N, 0 and F (Fig. 3.6). The Si2p (Fig. 3.6&) spcctrum indicalCS tba Si-<: (102 cV) and Si·
o (die shoulder al 103-104 cV) &le prcsenL The Cil spcclI'Um (Fil- 3.6b) indicalcs the
presence ofSi-<: al 286 cV, rogclhcr widl adYCntilÏOuS gnphitiç c:arboo al ca. 283 cV. The
Nb spc:ctrum (Fil- 3.6c) may bc assigned ID a Si-N-H JIOUP (biIlding energy 396 eV),«
(as suggcsœd by the Fr-IR. data. sec 0Iaplcr 4) 10 ÇYIIlO groups.
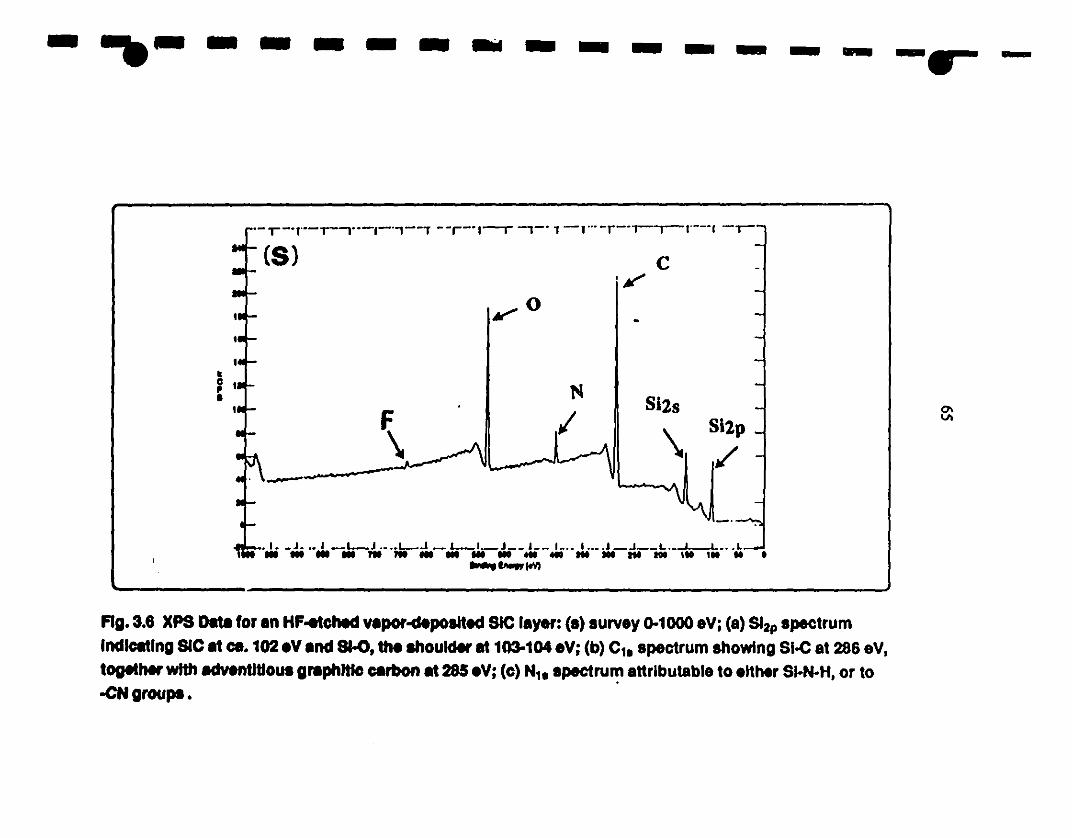
- --- - - - - - .. - - - - - - .. -r- -
0_,-0"-_'--'00-'_0'_' --r-o,-r -,_o,-,"--r--' 1 ,,---'-r-
.1 .. _J I_L-J L-..J_L--._J__ l __ I_ J_J__L. l ' L•••• ~~ •••• 'MIMM •
....~1eYl
c'~
...... 0
(S)
•0,• 1
F..
Si2sSi2p ï
\ \,1 Q\
VI
. .......".,....-- - - - - ,/
Flg.3.6 XPS Data for en HF4tched vepor..poaIted SIC leyer: (a) aurvey 0-1000 eV; (e) Sl2p apectrum
Indlcetlng SIC et ca. 102 eV end 81-0, the shoulder et 1Q3.104 eV; (b) Cl' apectrum showlng SloC et 286 eV,
togelher wtth edventJtIous grephltlC carbon et 285 eV; (c) N,. apectru'!' ettrlbutable to _"Mr S~H,or to-cNgroupe.
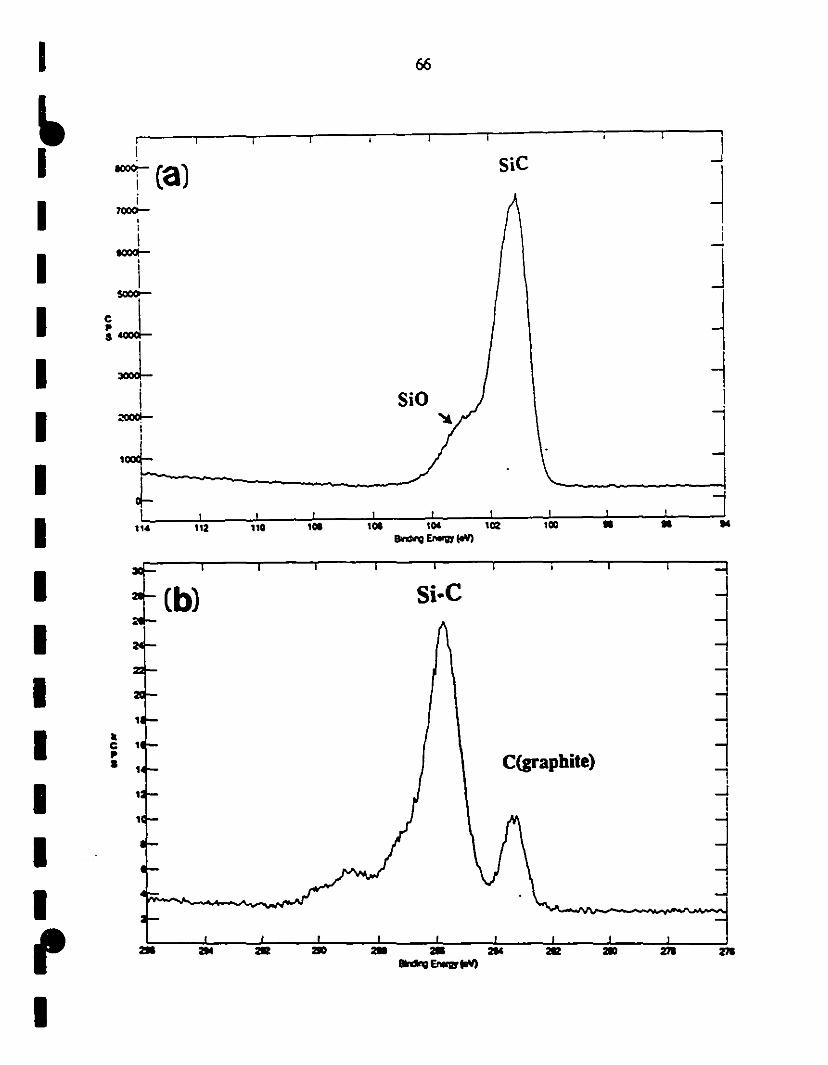
1 66
'- ]1
1 1
SiCIca)~1
i7_,1 ,1 1-
1100»-
J,
1--~1 :.1
1 1- -11
1 1
1 ~ ]11
1j
1 1 , , 1 1 1 1 , ,1 "4 112 110 '01 '01 'OC 102 ,ClO • • ..
llInl"'O E-w (eV)
1 ±(b)'=1
Siee
~11
1 ,1
,CO ,~ C(grapbite)• , ...,
1,, ...J1,
j11, 211 ZIG :III - ZM ZID 271
llOdrv E-wloVl
1
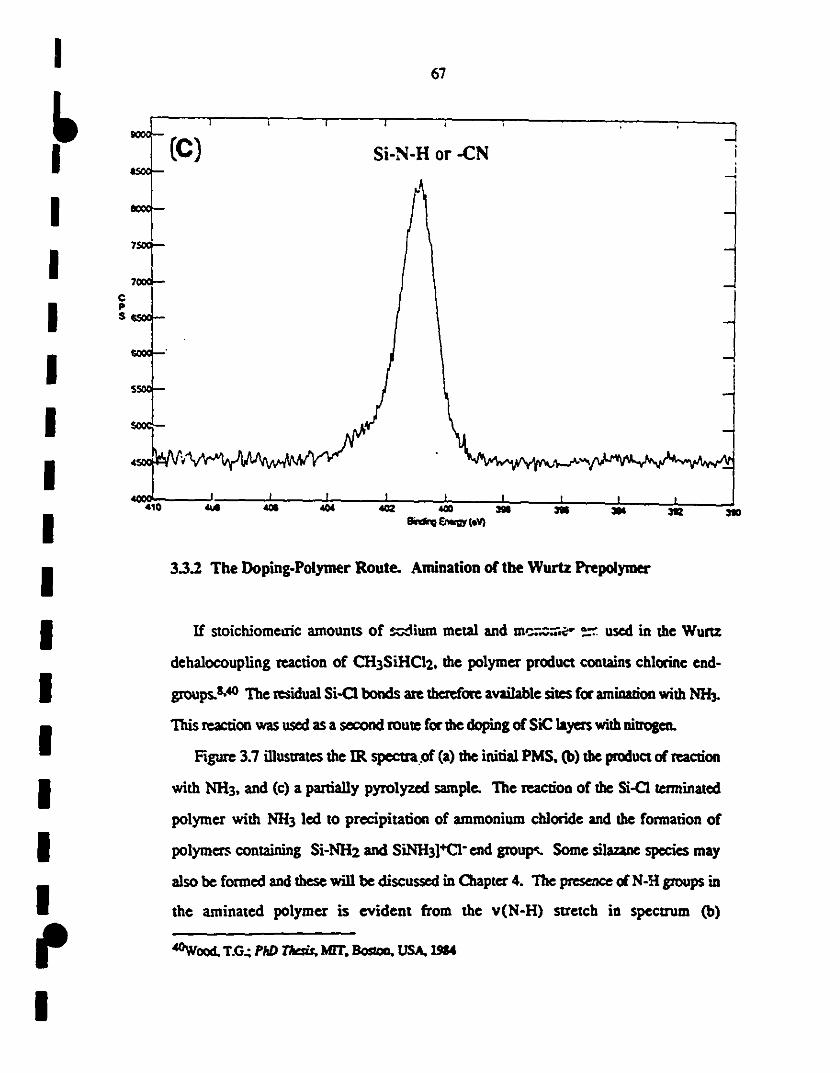
i-
67
Si-N-H or -CN
If stoichiomeaic amounts of ~ium metal and m'::;".;;;;;~ ~ used in the Wurtz
4Owood. T.G.; PIrD T1Iesis, MIT. flos!oD. USA. 1984
dehalocoupling reaetion of CH3SiHCI2. the polymer prodUCl coatains chlorine end
groups.a,.c0 The l'CSidual Si-Cl bonds are therl:fore available sites for aminaàon with NH,.This reaction was uscd as a second route for lbe doping orSiC layers with nitrogen.
Figure 3.7 illustrates the IR sptel1a9f (a) the initial PMS. (b) the produet or reaction
with NH3. and {cl a partially pyrolyzcd sample. The reaetïon of the Si-Cl rcnninated
polymer with NH3 led to precipitation of ammonium chloride and the formation of
polymm: containing Si-NH2 and SiNH3]+cI- end grouJl'l. Some silazane species may
also be fonned and lhese will be discussed in Chapter 4. The presence or N-H groups in
the aminated polymer is evident (rom the v{N-H) stretch in spectrum (b)
3.3.2 The Doping-Polymer Route. Amination of the Wurtz Prepolymer
CC)
400Ç4""'O---'.....--.:..--.i;o--"';jj_~----:;;600;;---_;---:=-----;!=---::!::---~lIrdrv EnoovJ (ov)
1
~111111111111111ft1
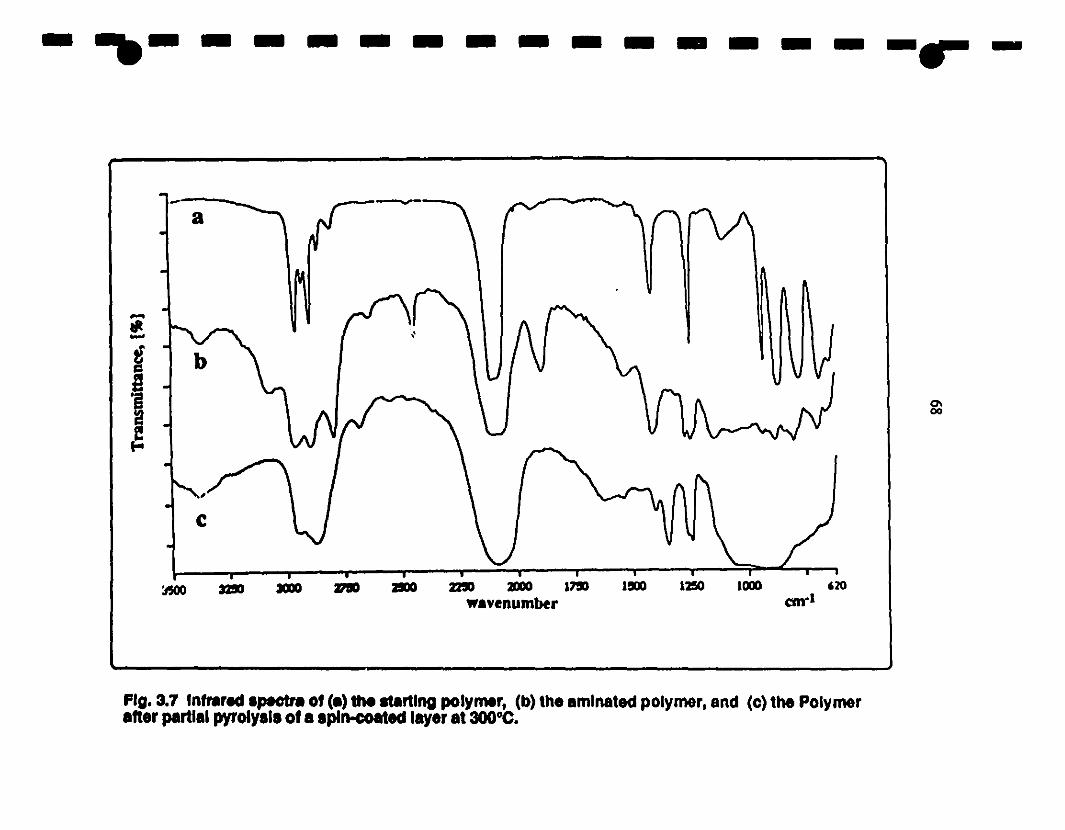
- .,.- -
- -a
-~....
- - --
-~
- - - - -- - - -,,-
g:
-
~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ~ ,~
wavenumbtr cnr1
FIg. 3.7 In"'r. spectre of (e) the atartlng polymer, (b) the amlnatGd polymer, and (c) the Polymerafter partl.' pyrolYlla of a apln-coated layer at 3OO"c.

1
~111111111111111,.1
69
at 3380 cm-1ft A shift of the Ôs(Si-CH3) band from 1247 cm·! to 1270 cm-! (Figure 3.7
b) accompanies the nitrogenation reaction_ This resu1t is analogous 10 the shift to 1260
cm-! which was observed during oxidaùon of PMS (sec Chapter 2.3.2.) and is a direct
effect of the e1ectron-withelrawing properties ofoxygen and nitrogen.
The effect of pyro1ysis was also investigated. A mixture of the aminated po1ymer and
the po1ymer produced by the Cp:Zr(CH3h-catalyzed po1ymerization of the hydrogcnated
Wunz prepo1ymer was spin-coated onto a Si wafer. The pyro1ysis was performed under
an inen annosphere (UHP Ar) and the IR spectrum of a typical samplc which had bcen
hcated up to 300"C is shown in Figure 3.7c. The split in the Ôs(Si-ŒI3) band at 1270 cm-!
paralle1s that observed when PMS undergoes oxidation. In the case of oxidation. the
appearance of a second band was attributed to the replacement of Si-Si(Œ3)- groups by
Si-O-Si(CH3) groups (sec Chapter 2.3.2. and ref. 20). In the case of the aminated
prepo1ymer. this suggests a reanangement of Si-Si(CH3)(H)NH2 groups to Si-NH
Si(Œ3)H2. The N-H stretehing band was deteeted up to 4SO"C. This result is impoll:ant
as the KlImada rearrangement.42 which takes place bclow 400"C during pyrolysis of PMS.
involves mainly uansfonnations in the chain and may result in the possible loss of the
nitrogen-containing groups during the rean:angemenL The final stages of the formation of
PCS are readi1y deœcted by the appearance orthe Œ2 scissoring mode iQ the IR spccttum
at 1352 cm-l . Figure 3.7c reveals that the KlImada rean:angment bas indeed taken place
and IDŒCOver the persistence of the v(N-H) band at 3400 cm-1indicates that the nitrogCll in
the precursor bas survived the rearrangemenL The eventual disappearance of the v(N-H)
band at higher tempQatures (such as 4SO"C in Ibis experiment) is most probably the result
41[R3Si.NH~-groups iIIlbcsampleandJeSiduallOlucne(used as a bydraulic valve iIIlbcps Dowcircuit) are 1eS(lDDSI1lIc fer lbc addilional bends.24OO cm-l,and. 1900and 3100 cm-IItSPCClhdy, ialbc spccuum Sb. Bolh bands are ItlIlOved during pyroIysis.42SbiiJla. Je.: Kumada. Mo. J. Org. C1Iem.. 1951.23. 139

1
~1
1
1
111111111111ft1
70
of dehydrocoupling between Si-H and N-H bonds. This is supported by IR investigations.
in which the sirnultaneous disappearance of the v(N-H) and v(Si-H) bands for Si3N4 thin
films formed by PECVD from SïI-l4 and NH3 was observed.43 It is funher corroborated
at higher temperatures. and in similar environments with EBCVD of Si3N4. if samples
more cor.centrated in N-H bonds are used.44 It has been observed previously that a large
excess of Si-H bonds willlead to complete reaction of the K-H bonds, as in the case of
poly(carbosilazanes), where three-dimensional backbones are known tO be formed by
facile coupling of Si-H and N-H bonds. with the subsequent formation of H2 and the
creation of Si3N knots.4S From the present IR spectra, there was no direct evidence of
such a process taking place because the v(SiC) band could not be resolved sufficienùy to
allow the detection of nitrogen after the pyrolysis. However, n-type resistivities in the
range 50 ma cm to 5000 n cm were obtained by varying the amounts (1-10%) of the
Wurtz·prepolymer to generate donor concentrations greater than 1016 donorslcm3. The
ceramic yield with respect only to the doping prepolymer was 5-10% (assuming thaI the
concentration of the adventitious impurities-related carners is negligible for resistivities
below 100 mncm). This value is increased compared to the 2-5% ceramic yield achieved
for pyrolysis of the prepolymcr alone and may be due to the increased concentration ofSi
H bonds available for crosslinking.
Despite sorne limitations. the material resulting from the above process cm be
considered ofpotential intere:>( for doped-SiC for use in solar ceI1s. where the ~uirements
related to the dispelsion ofthe resistivity are Jess strïng"'..Dt.
43Ricbardsoo, M.F.; Hanman, J's.; Guo, D.; CIZIL J. ChmL. 199Z, 70, 70044Zhang, B.-R.; Yu, Z.; CoUins, GJ.; HWlIIIg, T.; Ritclùe, W.H.; J. Vac. Sei. Technol. 1989, A7(2). 1764SPi1ol, J.P.; ProuediJlgs ofdie It1lel'llDlioMl SymposùIm 011 Organosiliœll Chemïstry. 1993. PomaD. 16

1
~111111111111111,.1
71
Conclusions
Nitrogen concentrations in a range suitable for the production of solar grade n++ SiC/Si
heterojunctions have becn obtained in a one-step process that involves the direCt pyrolysis
of PMS into n-type SiC layers deposited on silicon single-cryslal wafers. Two
procedures for doping the SiC layers -,vith nitrogen have becn developed. A good degree
of reproducibility has becn c.btained by contro11ing the partial pressure of NH3 in the gas
phase during the pyrolyses. Amination of a Wunz prepolymer can also be used in
synthesizing doping-polymers. Pyrolysis of mixrures of these polymers willt the precursor
under argon led 10 retention of nitrogen in the SiC layers. after Kumada rearrangement
The resistivities were typically 30 ma cm to ID n cm for the n-type SiC layers of
thicknesses varying from 100 nm to 2.S J.I.II1.

1
l-I11111111111111
"1
72
CHAPTER4
Investigation into the Synthesis of Amorphous Silicon Nitride and
Silicon Oxynitride During the Reactive Pyrolysis of
Poly(methylsïlane) under Ammonia
4.1. Introduction
When NH3 was used during pyrolysis of PMS. as described in chapler 3. silicon
based ceramic malerïals incorporating nilI'Ogen were oblained. NilI'Ogen concentrations in
the doping-range of lOIS _1018 atomslcm3 were achieved in SiC thin fùms by pyrolysis
under a partial pressure of NH3.1.2 which resulled in partial cleavage of the Si-C bonds as
a result of the reaction of PMS with NB3 catalyzed by Cp2Zr(CH3n. (Cp = TlS-CsHs.
DMZ). The reactive pyrolysis of various polymerie precursors under NH3 is a well
established route for the synthesis of bulk produets in the Si-C-N system. However. a
specific hehavior might he expected in the tranSformation of PMS into Si3N4. The Si-H
functionality in PMS not only suppteSSCS the independence of the crosslinking ptOCesss
versus the ammonolysis IellCtion, but may also permit a specifie palhway for the Kumada
rearrangemenL For example, the silylene icsenion mechanism may he favored over the
rndical mechanism proposed for poly(dimethylsilane).3 Such a situation may be compared
to other cases, where the design of the precursor involves independent crossIinking
mechanisms. e.g.. througb pendant vinyl groups grafted on the silicoo backbone. The
IScarIeœ, M; LS. Buller; J.F. Hanod; C"-- Maler. 1995.7(6), 12142 AppliCQtiotlS o/Orgtl1llJlMlQ/lic Chemistr] ÙI lM Syrrlhesis tJNJProc=ütg of~mtJIeriD1s. Eds.Laine. RoM. and Harrod,J.F~ K1uwer AI .....mie Publisbels. DanlœcbI.l995 Cm pn::ss)3Laine. RoM.; BaboallC'!ln, F. Chem. MQlU.1993. S. 260

1
~111111111111111
"1
73
reaction of the Si-H bond in PMS wiÙl )I,'H3 is known 10 lead to dehydrocoupling under
10 atm combined pressure of ammonia and methylsilane, and the resulting
poly(methylaminosilane) gives a mixture of SiC and Si3N4 arter pyrolysis under nitrogen.4
The rearrangement of PMS into polycarbosilane (PCS) at 20Q-4S0·C makes it a good
precursor te Si3N4. since PCS was reported earlier te be a good precUISor te silicon niaide
by pyrolysis under NH3.S The efficiency of carbon removal in silicon-based polymerie
precursors is paroy dependent on Ùle temperature of cross-linking and relatively
independent of the structure or functionality of the polymer.
Obviously, the process is extremely complex. In addition to the as yet unsolved
pyrolysis pathway of polysilazanes. for example, there are two main reactions: the reaction
of PMS wiÙl ammonia and the Kumada rearrangement of polysilane leading to
polycarbosilane. These processes render the goal of providing a mechanistic model
difficulL Nevertheless, some critical steps in Ùle transformation of PMS in the presence of
NH3 inte Si3N4 were uncovered in the work described below.
4.2. Experimental Section
Synthesis of PMS Wunz-prepolymer. polymerization of the Wunz-coupled
prepolymer with DMZ, substrate preparation. dip- and spin-coating film deposition,
pyrolyses and produet charaeterlzation were carrïed out as described in Chapter 2.
Prepurified NH3 from Matheson was passed immediately prior to reaction with the PMS
thin films througb a column (l.5 m in length, 7.6 cm in diameter) of KOH and a mixture of
molecular sieves 3-4 Aat flow rates of IG-200 mUmin. Because of the large amount of
4H.Q. Liu, J.F. Hanod OrgaNlmeuzUïcs. 11, m2, 822SslUll$, G.T.; 0IaDdIa, G.J. Am. eu-. Soc. lm. 72(2). 333

1
~111111111111111f'1
74
NH3 used in tffis reaction, the column was regenerated before each experiment by heating
the contents of the column at 280°C in the presence of UHP Ar for 6 h. Pure NH3 or
mixtures of UHP Ar/NH3 were used as flowing gas phases during the pyrolyses. DTA
and TGA analyses were performed on a Seiko 220 instrument; Al203 pans, PIN50-026
were used and similar heating cycles to those followed during the pyrolyses were used.
4.3. Results and Discussion
4.3.1. FT-IR Study of the Reaction of NH3 with PoIy(methylsilane)
Reacrions in the range ofroom temperature 10 300"C. At room temperature. no
reaction was obsetVed between NH3 and spin-coated films of PMS on any of the tested
substrates. Sinee some eomparatively low-temperature reaetions had been previously
observed in simiIar systemS,6 funher anaIysis was performed 10 exclude any hidden effects
sueh as the formation of small amounts of Si3N knOlS. The specaa exhibit essentially the
saIne featutes as those obtained in the study of the oxidation of the starting polymer
described in Chapter 2.7 The growth in the intensity of the band around 1100 cm-l (vasSi-
o-Si) suggests that the predominant reaction involves the formation of siloxane. The
broadness and the splitting of the band are ebaracteristic for a polymerie species, while the
maximum intensity at 1106 cm-l is associated with themobility of the chain on the silicon
substrate. This results from the predominant adsorption of the oxygen bridges in the
polymer onto inlerstitial sites al the silicon surface (sec Chapter 2). The appearance of
bands at 908 cm-l [v(Si-Q)]. and especially that al 967 cm-l (Q-Si-C) whieh appem al
6ua. H.Q.; Harrod, J.F. OrgtUlOmt:ltJllks 199%, Il. 8227SCarJcte. Mo; BriClllle, S.R.; Hamld. J. F.; BUIIcr, LS. C1IDrL 1tI_~ 19!14, 6, m

1
'-111111111111111t'1
75
higher degrees of oxidaùon. confums this observaùon.
A series of IR spectra was measured on 5Q-mg samples, which were kepl al 15O"C
for 2.5 h under a flow of NH3 (100 mUmin) and subsequently subjected to oxidation by
exposure ta air al room temperalure (Fig. 4.1). Some minor changes compared 10 the
oxidation of the "green" po1ymer were deteeted in this series. in the 1500-700 cm'! range
(inset Fig. 4.1) These include the appearance of new but 10w intensity bands in the 1000
750 cm'! region. In addition. the ôas(Si-0i3) band al 1247 cm-1 shows [WO significant
differences compared to the spectrum of PMS pyro1yzed in an inen atmosphere al 15O"C
and that of the unoxidized po1ymer al room lemperature. FlIStly, instead of the expected
doublet of the Ôas0i3 band at 1260 cm-!,S a new band deve10ps at 1270 cm-!. This new
band may be assigned ta the presence of (N)Si-CH3 groups, based on a similar shift
toward higher wavenumbers usually observed in the case of substituents with e1ecaoo.
withdrawing properties (sec page 36 and refs.38-40 in Chapter 2). Secondly, the ratio of
the intensitiesof thevJvas C-H strelChesof themethy1 groups at 2897 and 296Scm'I ,
respective1y, bas an abnonnally 10w value. This ratio of intensities is lower than that
characteristic for siloxane species. Moreover, the intensity (oormalized with resp«l ta die
vasC-H band) of the SïHZdefonnation at 930 cm-! is reduced, wbile the bands al 796 aDd
870 cm-!, which appear during the reaction of PMS with NH3 disappear during the
oxidation cycle. These last effects are associated with oxidation or byclrolysis of SioN
bonds, whicb leads ID the disappearance of the vas(N-Si-N) and v(C-Si-N) modes,
Snûs vibration is scnsilive ta lbc SUbslilllClllS 0lI siIicOlI, c.g. 1247 c;m-l for (Si)Si.cH3 groupsand 1260cm-1 for (O)Si-CH3 groups
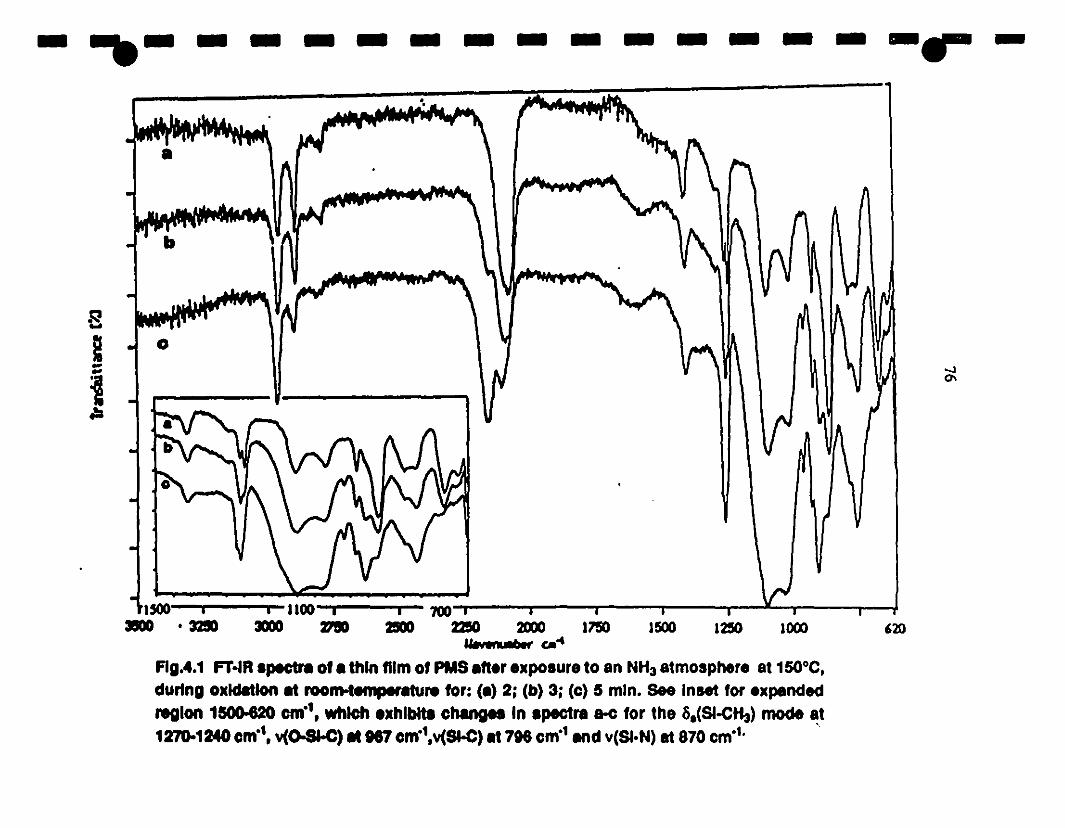
- ---- - - -- - - - - - - - --,,- -
BB-t~
"
o-.l0\
1500 i i 1100 i i 700BIO • 32S) 3llOO 2'1ID 2!100 22!lO 2000 17!lO
1Iamm...... CIl"1500 12!lO 1000 620
F1g.4.1 Fr-IR .peen O, a thln film o. PUS after .xpo.ure to an NHa atmospher. at 150°C,durlng oxldatlon et room-temperature IOr: (a) 2; (b) 3; (c) 5 min. see Inset for expendedreglon 1500-620 cm", whlch exhlblta changes ln .pect,. a-c for the 6.(S1oCHa) mode at127001240 cm-1, v(o-sI C) al t87 cm-1,v(SI-C) et 7lH5 cm" and v(S1-N) et 870 cm'" '

1
~111111111111111{'1
77
respectively. The N-Si-N groups may result from -Si(NH21:z- groups forrned byamination
of terminal SiH2 groups in the precursor. Ali these changes may be explained by a slow
coupling reaction between the Si-Ho or SiH2. bonds and the N·H bonds. tO give (N)Si
CH2X (X =H. SiR3) groups. The change in the C-H bond polarity associated with the
presence of the electronegative nitrogen anached 10 silicon is probably responsible for the
decrease in the intensity of the symmetric C-H sttetch. This was previously observed
during oxygen insertion into the silicon backbone of PMS (sec Chapter 2). The extent of
the coupling reaetion presenl1y observed at 150°C is still very small compared to the
dehydrocoupling reaction observed at higher temperature between Si-H and N-H bonds in
vinylpolysilazane (·SiViH·NH-) or (-SiMeH-NH-)n precursors. where Vi represents a
vinyl group.9 The low-temperature reaetion of the precursor with NH3 is important for the
production ofcarlxm-free Si3N4. considering the crosslinking process and the instability of
NH3 al high tempetatures (sec Chapter S).
The absence ofabsorptions due to N-H may be due ID extensive formation of Si3N
knots. since facile SiH/NH coupling is Icnown to occur in polycarbosilazanes.lO AIl
increased rate for the reactions ofaminosilane and silazane NH groups with SiR, compared
to NH3. would explain the complete absence of N-H bands at 3400-3300 cm-} and the
modified absorption pattern in lhe 1000-900 cur1tegion.
PyrolysisofPMS for 20 h at 200"C led 10 the developmentofa strong band al 1000
cm-l instead of a SioN vibration ncar 950 cm-1 (Fig. 4.2). This band is assigned ID a
mixed Si-Q-N phase, as suggested by the Fr-IR data obtained on thin films of SiIO/N
9Cbooag Kwet Yi~ N.s.Co; Corrill, RJ.P.; Leclerc, D.; MutiD, P.H.; VIOUX, A. C1JDrL MQ16.1992,4,141lOpj}oc. J.P. Procdngs cf the llIlUMIioMl SympoSÙIM 011 OrgtWJsiIi&OII C1Iemistry.1993. Paznan, 16
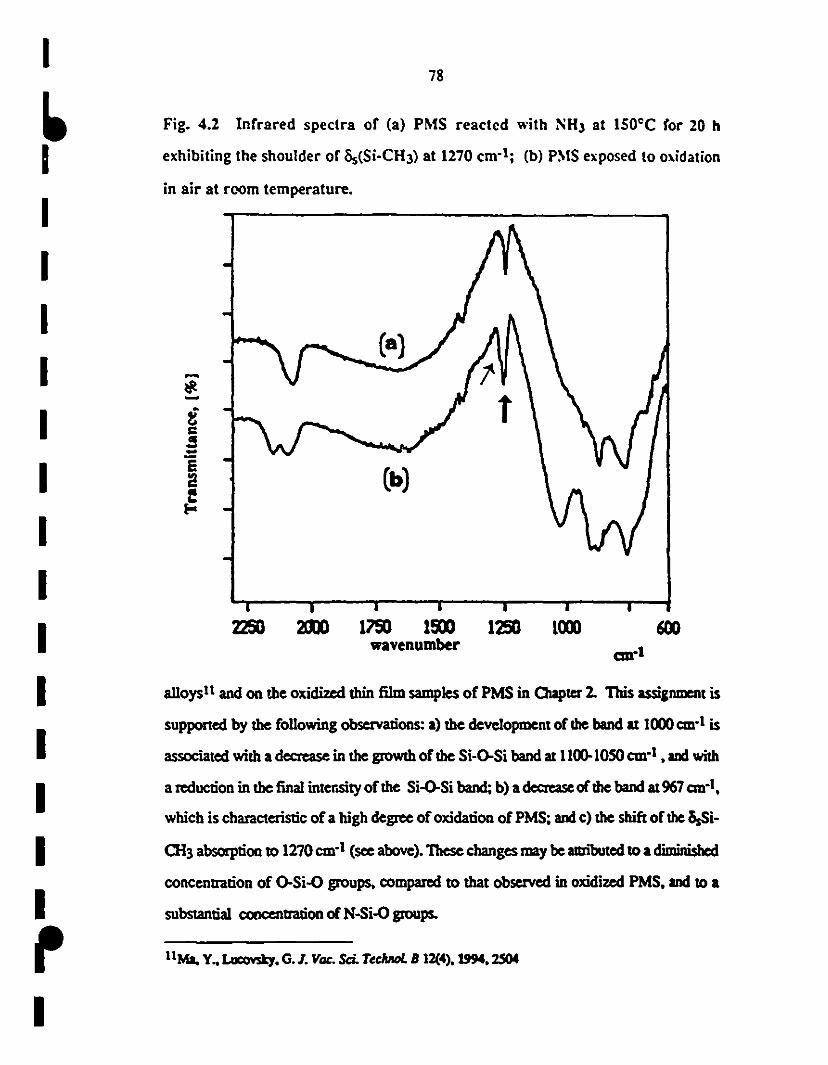
um
(b)
1750 ID 1250wavenumber
2250
78
11Ma, y ~ Lucovsty. G. J. VGC. Sei. Tcc1wlL B 12(4). 1994.2504
allOys11 and on the oxidized thin film samples of PMS in Chapter 2. This assi&nmcnt is
supponed by the following observalions: a) the devdopment of die band Il 1000 cm-l is
assoclated with a decrease in the growth of the Si-Q-Si band u 11()().10SO cm-l •and with
a reduction in the final inter.sityofthe Si-o-Si band; b) adccreaseofthe band u967 anal.
which is charaeteristic ofa high degree ofoxidation of PMS; and c) the shift of die ~i
CH3 absorptiOll to 1270 cm-1 (sec above). These changes may Ile altributed to a diminished
concentration of Q-Si-o groups, compared to mat observed in oxidized PMS. and to a
substantial COllCelltration of N-Si-o groups.
in air at rcom temperature.
Fig. 4.2 Infrared spectra of (a) PMS reactcd with NH) at 150°C for 20 h
exhibiting the shoulder of Ôs(Si-CH3) at 1270 cm-l : (b) P~1S exposed to oxidation
1
~111111111111111
"1

1
~111111111111111f1
79
The lability of the SioN bond towards oxidation and hjdrolysis during the CVD
formation of"soft-vacuum" silicon (oxy)nitride laye~ is weil docurnented in the lilCTature
and it is accepled that low-temperarure depositions usually lead ID a higher oxygen COlllent
in the coating.12 This type of contamination is panly due ta the higher activation eoergy
for the ninidation reaction compared rD oxidation aI temper'dtureS below JOOCc.13 It is also
known that the lability of the Si-N bond in polymerie prectlISO~ IOwards traces of l!IOisture
is responsible for oxygen incorporation in the polymer. and ultimately oxygen
contamination of me products of pyrolysis.14 This effect is a considerable limitation on me
use of me low-temperature reaction of PMS wim NH3 as a crosslinking process for the
polymer. The presence of adventitious oxygen in the samp\es reacted at temperaIUres
below 300"C could not be avoided. Sïmilar results were observed during CVD deposition
of silicon ninide. where the substrate temperature bas rD be increased to higher values man
3SO"C, in order rD counter me oxidation.1S
Reactions Ùl the 300 10 700"C range. A major change in the reaction of PMS with NH3
OCC\m above 300"c, as is evident!rom DTA (Fig. 4.3b), TGA(Fig. 4.4) sans and Fr-IR
(Fig. 45) results. DTA reveals an exothermic process between 300 and 400"C during the
pyrolysis under NH3, wbich is not present in the pyrolysis under inen atmosphere. while
TGA shows a different pathway UDder NH3 compaml rD the pyrolysis under Ar (Figs.
4.3a and 4.4, n:spectïvely). A major weight loss appears in the TGA curves wben the
reaction is performed under NH3 at 300-400"C, and an unusual increase iD the weight is
observed in the 600-700"C range (Fig. 4.3a). A suong absorplion due to v(NH) appears
al 3397 cm-1in the Fr-IR spectrum aI 300"C (Fig. 4.5), substaDliating the occurrence ofan
atnination process. The general shape and position of the IR absorption is more
12zJuma. B.R.: Ya, z.: (Wljns, G.I.: Hwaug, T.; Ritcbie. WJL J. Vac. Sei. T~cMol. 1989, A 7(2), 17613HéJlIdboo.t cfC1lemi.ltry andp~ S7d1 ed.,editedby Co WeastCRCEd., CIevdaDd. OH, 19'77. 126414 P""stoWeIY. G eo-. It1l. 1989, IS,21315zbang. B.R.: Ya, z.: Co1IiDs. G.I. J. Voc. Sei. T«1wIl. 1919. A{T). 176
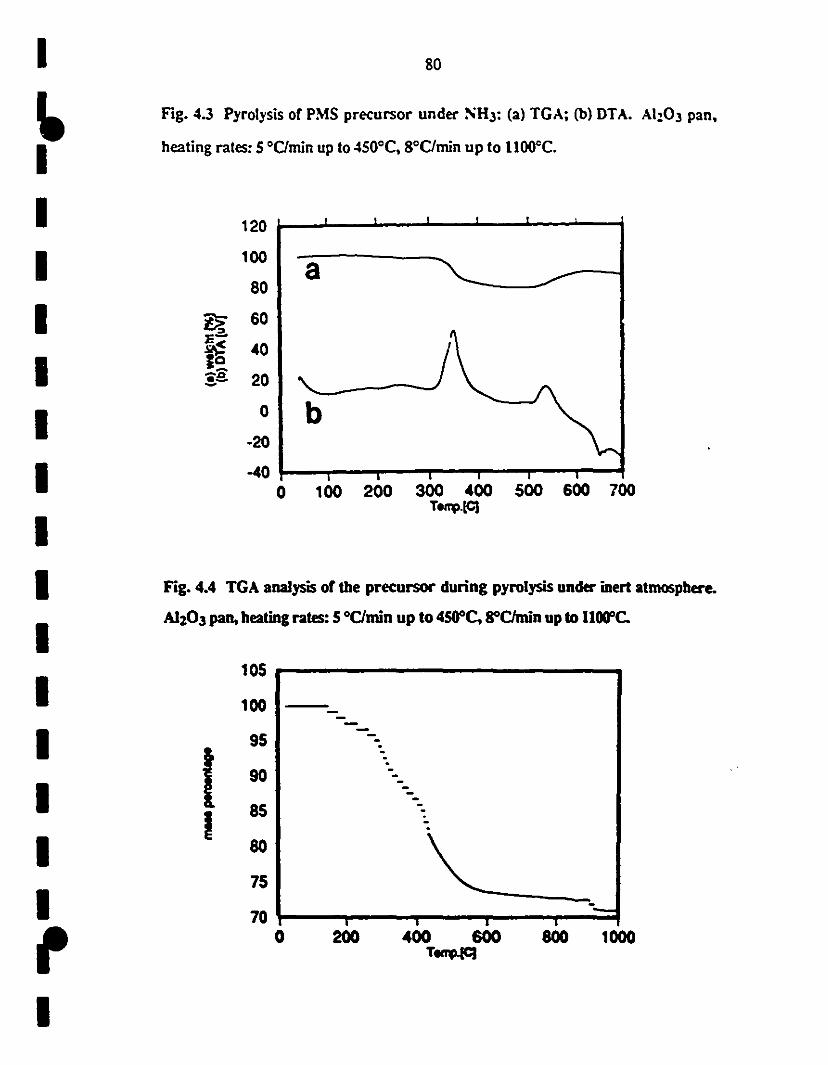
1
~111111111111111,1
80
Fig. 4.3 Pyrolysis of PMS precursor under :"OHJ: (a) TGA; (b) DTA. AI:OJ pan,
heating raIes: 5 oC/min up to 450°C, SOC/min up to llOO·C.
120
100 a80
~ 60E-c 40~~
~D 20 '\...---0 b
·20
·400 100 200 300 400 500 600 700
T."".(C]
Fig. 4.4 TGA analysis of the precursor during pyrolysis under iDen atmosphere.
Al~03 pan, heatiDg rates: 5 °C/nùn up to 4SO"C, SOClmin up CO 1l00"c.
105
100 -95 -
1.".
90 ----85 -
1 ···80
~75
70 --1 1 • 1
0 200 400 600 800 1000T-.tCI
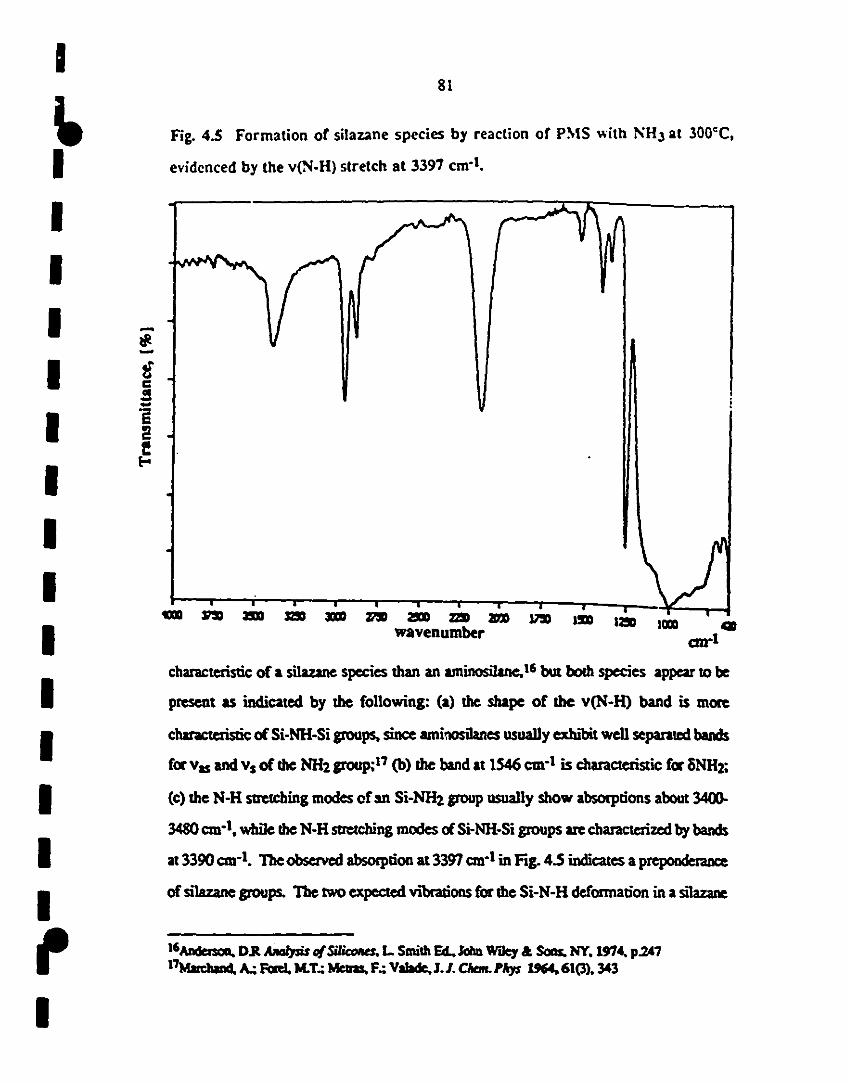
81
lSIl lai IlIlll QI
cm-1
Fig. 4.5 Formation or silazane species by reaction or p~tS with ~H3 at 300·C.
evidcnced by the v(N-H) stretch at 3397 cm·l•
cbaracleristiç or a silazane species than an aminosiJane,16 but botb species appear 10 Ile
present as indicated by the following: (a) the shape of me v(N-H) band is m~
cbaractcristic orSi-NH-Si groups, since arnillOSilanes usually Wùbit weB separalCd bands
for Vas and Vs or the NR2 group;17 (b) the band at lS46 cm-l is cbaraeteristic for llNH2;
(c) the N-R stretcbing modes cf311 Si-NH2 group usually show absotptions about 3400
3480cm-1•wtù1e the N-Rstretcbing modes ofSi-NH-Si groups arecb3JaClerized bybands
at 3390 cm-1• The observed absorption al 3397 cm-1in Fig. 4.5 ïndicales a prepooderanœ
of silazane groups. The IWO expected vibrations for the Si-N-H cleformation in a S11azane
16ADdaSOll, DJt AJtGlyàs t;fsm-. L Smilll F4.1aIlD WiIe)'.t Soas. NY. 1914, p.247t7Marcb1nd. A.: FcnI. KT.; MclnIs,F.; VIlade.J.J. Cltœ.P1Iys 19f4.61(3).343
1IllIllftBlllJZ!l3llll2i'Sla1D2ZIIZI»VSlwavenumber
-~-ic.!-Ê2~
1
~111111111111111t'1

1
~111111111111111,1
82
species are: "(N-H (out of plane) at 1179 cm-! and "(.II-H (in plane) at 778 cm'!, The latter
is usually overlapped with the SioN absorption band, and its presence in nitride layers can
olùY be detected arter deconvolution or peak fitting procedures. The anûsymmetric stretch,
vas(Si-N-Si), is expected in the 1000-900 cm'! region, while vs(Si-N) \s expected to
absorb below 600 cm'!, The laner is usually a very wea.k absorption and is outside the
range (620 cm-l) of the instrument used in these experiments. The strong absorption
below 1()()() cm·l is indicative of the presence of SioN bonds, although the formation of a
certain amount of siloxane units can still be observed as a shoulder at 1100 cm·l. The band
appearing at 1356 cm-l, assigned to)'(Si-0i2-Si), reveals that a Kumada rearrangement to
produce CH2 groups has occurred. The strong and sharp absorption at 1270 cm-l is
assigned to Ss<Si-0i3).
The formation of Si3N Icnots by NH3 curing at low temperature inhibits the
rearrangement of PMS to PCS. A sample of PMS exposed to NH3 for 10 Il at 300"C
(beating rate 2°C/min) sbowed no evide!1Ce ofSi-H bonds in its IR spectrum.18 Pyrolysis
of this sample under NH3 up to 1100°C was monitored by IR (Fig. 4.6) and the lack of
absorption al -1350 cm-lover the whole ICmperature range indieates dlat the formatiOll of
polycarbosilane via the Kumada rearrangement does not occur. The final prodUCl of the
pyrolysis exhibited a StroDg broad IR absorption between 12OG-900 cm-l (Fig. 4.7).
assigned ta the presence ofa Si-N-C-conraining maIerial. Residual hydrogen appcars ta he
bonded mainly as NHxgroups. A bulle sample proc:essed similarIy exhibited the 29Si MAS
NMR spectrum shown in Fig. 4.8. The main peak at -SO ppm is ch3racteristic of Si3N40
while the weaJc, broad signal al -100 ppm can he assigned ta both SiOxNy and Si04
groups. The dominant SioN absorption al 920 etn-l in the Fr-IR spectrum (Fig. 4.7) cao
he assigned ta a constrained planar geometry of nitrogen. charaeterized by IR absorptions
18N.B. l'be ex1ICI1l ofdie oxjc!"liaIIl3OO"C is lDwct lhaD in die procIuct reac1I:d 1l2OO'C <_die absenceofdiebaDd Il 11()().1010 cm-1).
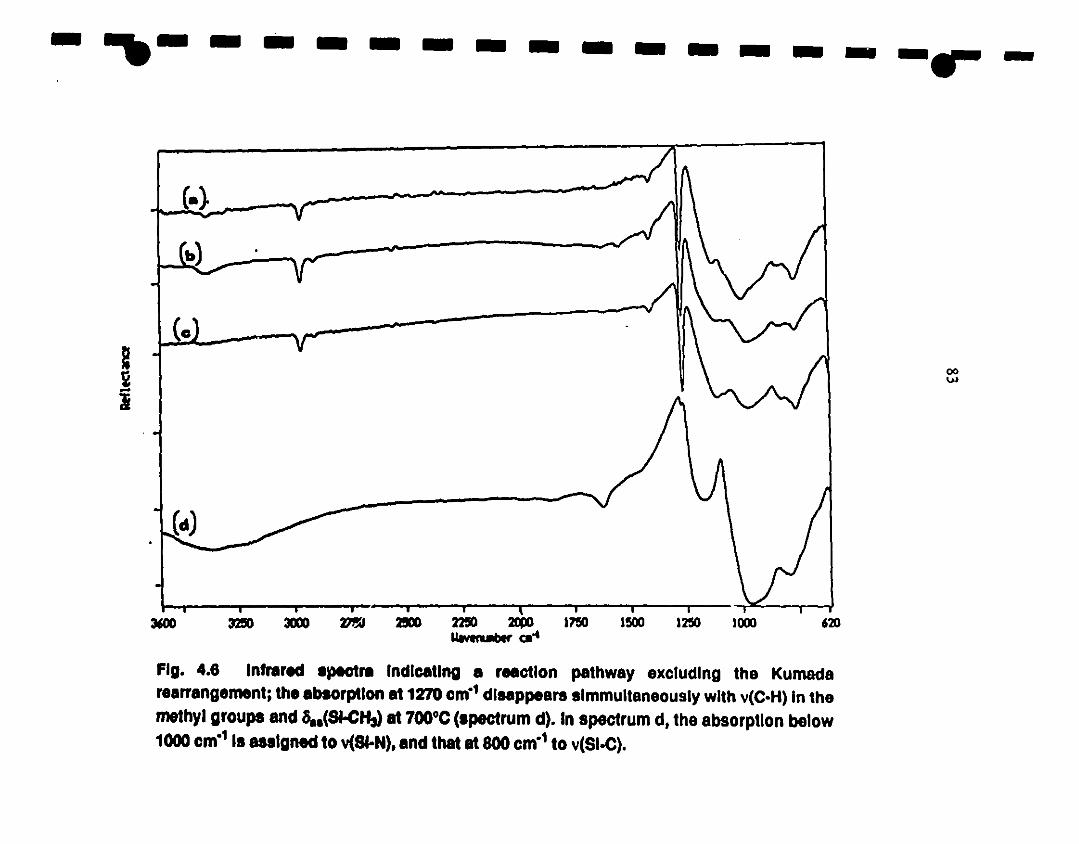
- .,.-- ------ - - -- - - -".. -
c·}b)
(cSii
(d)
3f(lO 32!lO 3000.------.--- - ., - -- -r T -- --T
27!!a 2!JOO 22!lO ~ 17SI I~ lm~e-"
1000
00V>
620
Fig. 4.6 Infnlred .~... Indlcetlng a r..ctlon pethway excludlng the Kumadarearrangement; the absorption et 1270 cm-1 dlsappear8 elmmultaneously wlth v(C-H) ln the
rnethyl groups and 3u(Sl-CH,) et 700·C (.pectrum dl. In lpectrum d, the absorption below1000 cm-1 Il asslgned to v(Sl-N), and that et 800 cm-1 to v(SI.c).
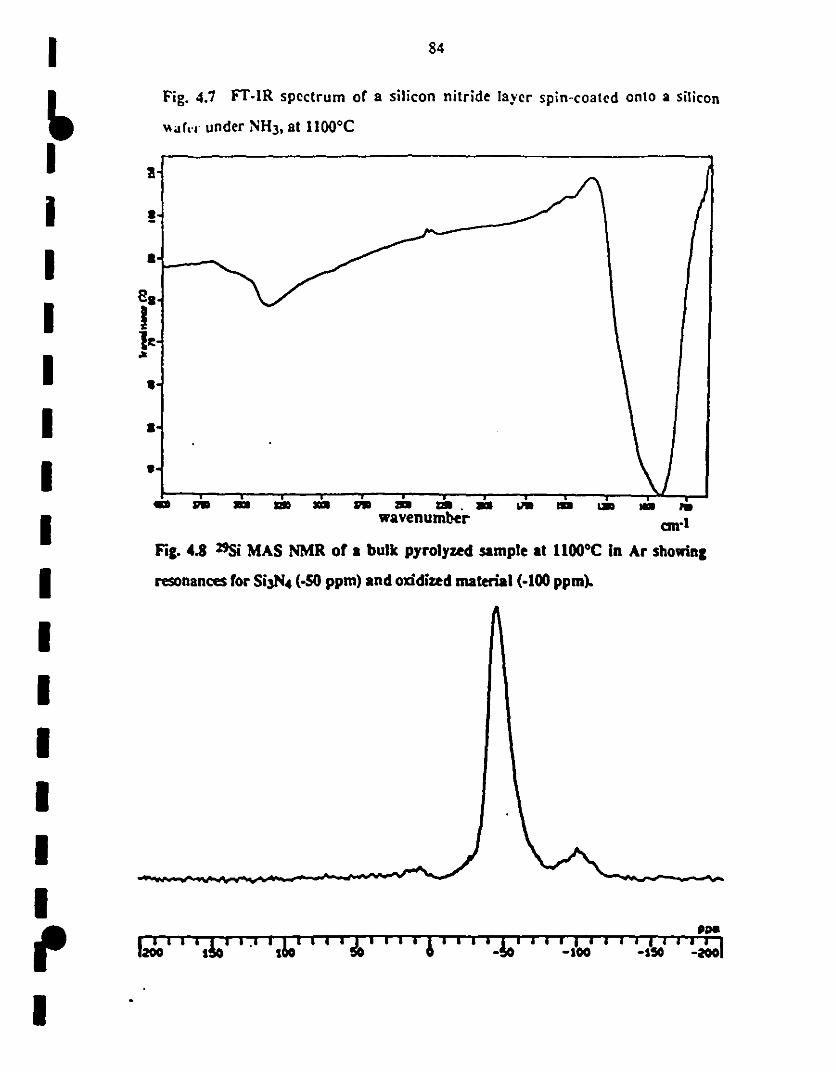
Iii Iii-150 -200
'ii-100
84
Fig. 4.7 FT·IR spectrum of a silicon nilride layer spin-coated onlo a silicon
"al.·.. under NH3. at llOO°C
- _ D a •. D ~ M
wavenumber an-.
FiC. 4.8 %9Si MAS NMR or a bulk pyrolyzed sample at noo°c ln Ar showinc
resonances ror Si)N4 (-50 ppm) and oxidized material (.100 ppm).
!
•
.'l.---.....
••
laJo i i 1,101 i .1 i ,&Ii i i JO 1 i IIi ii_Li
1
~111111111111111t'1

Pagina~ion Error
'l'ext complete
National Library of Canada
Canadian 'l'heses Service
Erreur de pagination
Le texte est complet
Bibliothèque nationale du canada
Service des thèses canadiennes
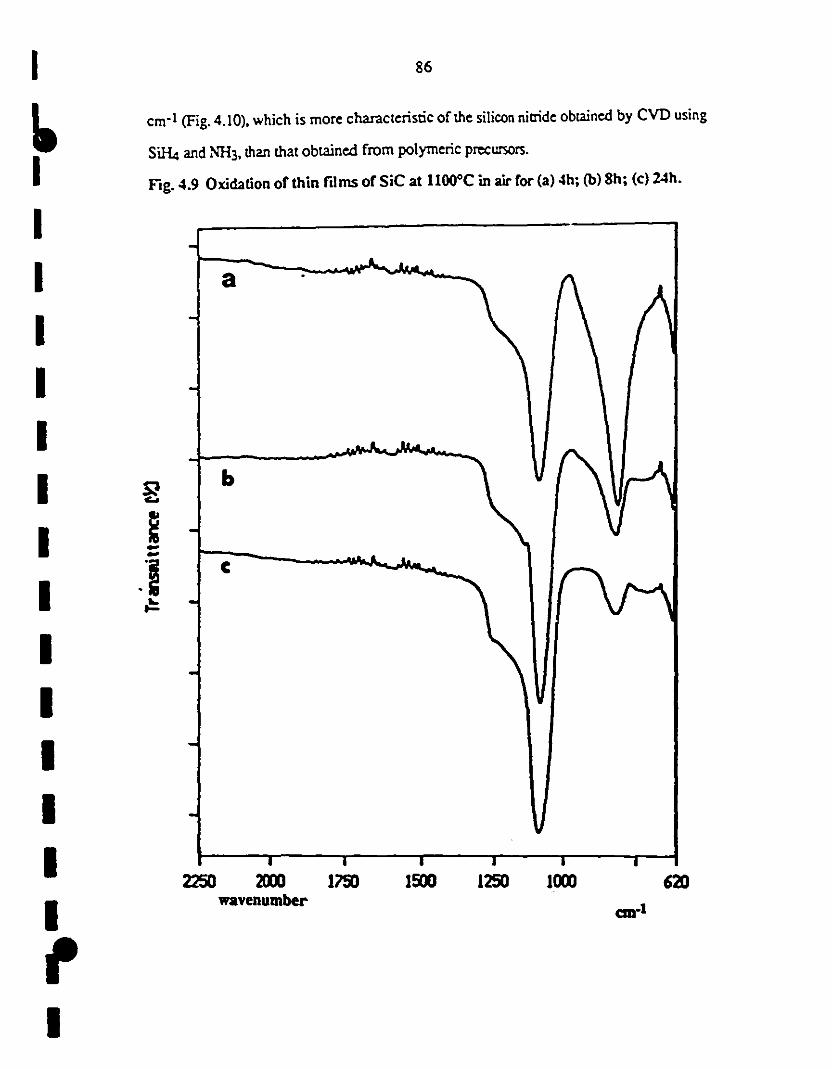
uO)12501500
a
86
b
2250 2ŒO 1750wavenumber
c
em-! (Fig. 4.10). whieh is more ehar.!eleristie of the siliron ninide obtained by CVD using
Sil-4 and NH3. than thal obtained from polymerie precursors.
Fig. 4.9 Oxidation of thin films of SiC at UOO°C in air for (a) 4h; (b) Sh; (e) 24h.
1
1.111111111111111l'1
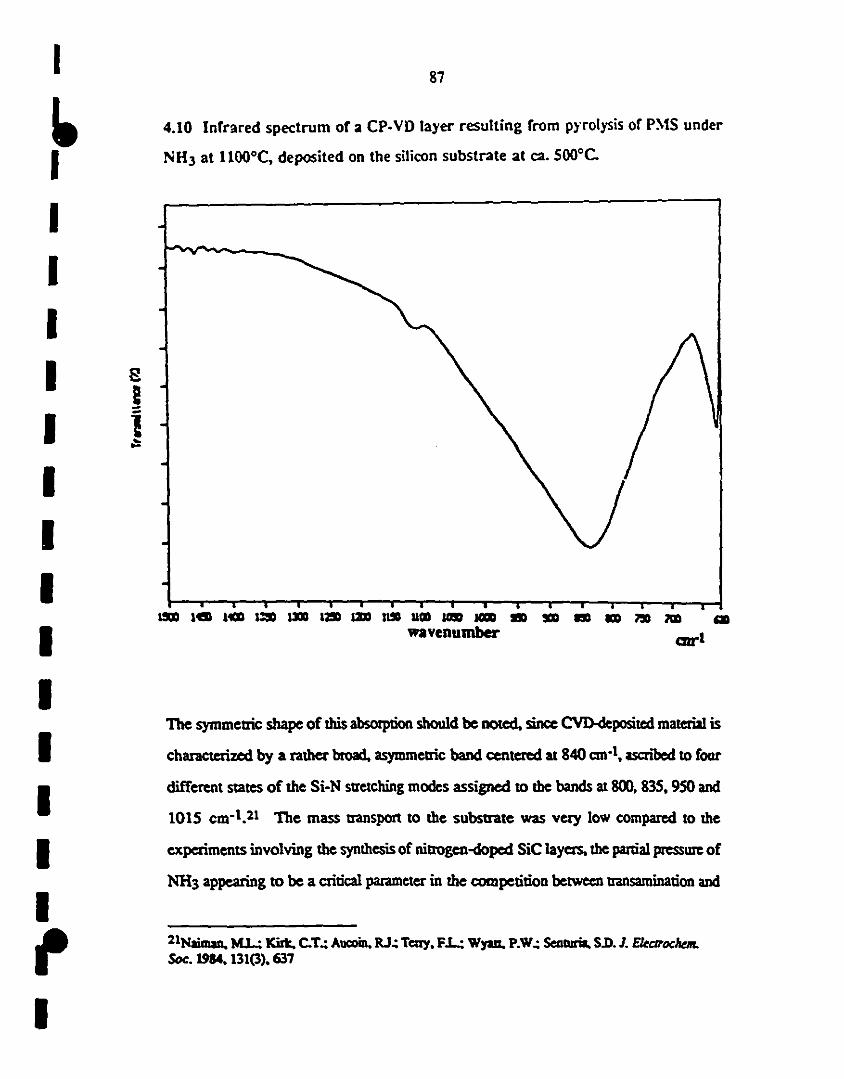
1
~111111111111111
"1
87
4.10 Infrared spectrum of a CP·VD layer resulting from p)"rolysis of P~tS under
NU) al llOO°C, deposited on the silicon substrate at ca. SOO°c.
lSlIl lel lC 1:sl ID lZll l2lIl Ils UCllI 1GIIl 1CllID III SIl ISI llXl 1Sl 1IIl QII
wavenumber œr1
The symmelric shape of Ihis absorplÎOll should be noted, sinc:e CVD-deposilCd material is
characterized by a ralher broad, asymmetric band centered al 840 cm·1, ascnOed 10 four
different states of the SioN suetching modes assigned 10 the bands al 800, 835, 950 and
1015 cm·1•21 The mass uanspon to the substrate was very low compared to the
experiments involving the synthesis of nitrogen-doped SiC layas, the partial pzessure of
NH3 appearing to be a critical parameter in the competition between uansamination and
21Naiman. ML.: Kilt., CT.; AUCOÏII, RJ.; Tcny, F.L.: Wym. P.W.; Sealllril. S.D. J. Ekczroc1leJrc.Soc. 19114. 131(3).637
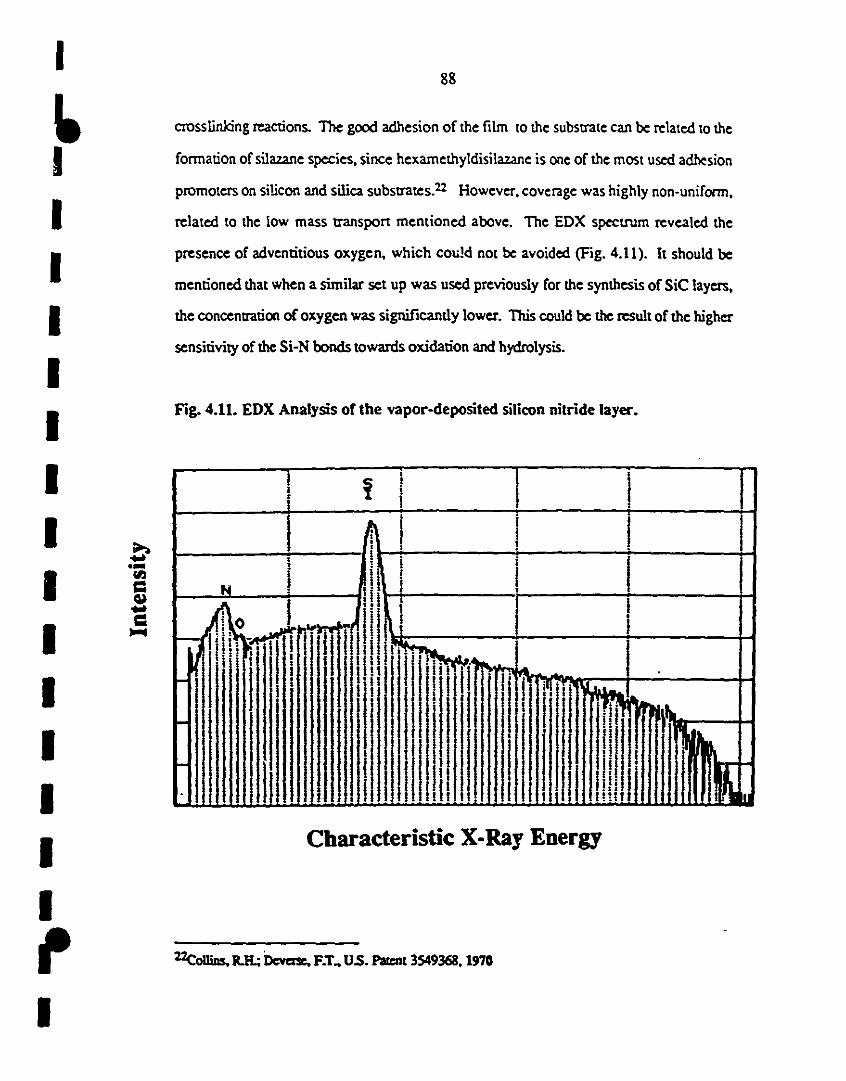
.1
1
~111111111111111,.1
88
crosslinldng reactions. 1be good adhesion of the film tO the substrate can he related to the
formation of silazane species. since hexamethyldisilazane is one of the most used adhesion
promoters on silicon and silica substrates.22 However. coverage was highly non-unifonn.
related to the iow mass transport memioned above. The EOX specuum revealed the
presence of adventitious oxygen. which cou!d not he avoided (Fig. 4.11). It should he
mentioned that when a similar set up was used previously for the synthesis of SiC layers.
the concentration ofoxygen was significantly lower. This could he the result of the higher
sensitivity of the SioN bonds towards oxidation and hydrolysis.
Fig. 4.11. EDX Analysis or the vapor-deposited silicon nilride layer.
1 i 1 1
1 ~ 1 1 1 1I-----;-!---'1 l-r-----.r-----t,------+--I
1-_...:N.1-__!~--...I\lH------li-----+_----_HJ1'\.. 1. i1 l,III \:r:!__-!!---__l~-_+-f'lI rr~I'" Il 1'1 1111 ~I· ~i'rli1rn"II"i1irrrn,·~~....-~I~.~--u
1 l'l' il. ilil 1 1 i~ "\It. .1
1 ï ï n~ l)'r.r.:-
11,1 l ,1 ,IIII! 11iN\{11
1 1111 . 1 1 I!lit .!DilI
Characteristic X-Ray Energy
27<:oIIiDs. R.H.: Devcnc. F.T~ US. Patent 3549368. 1970
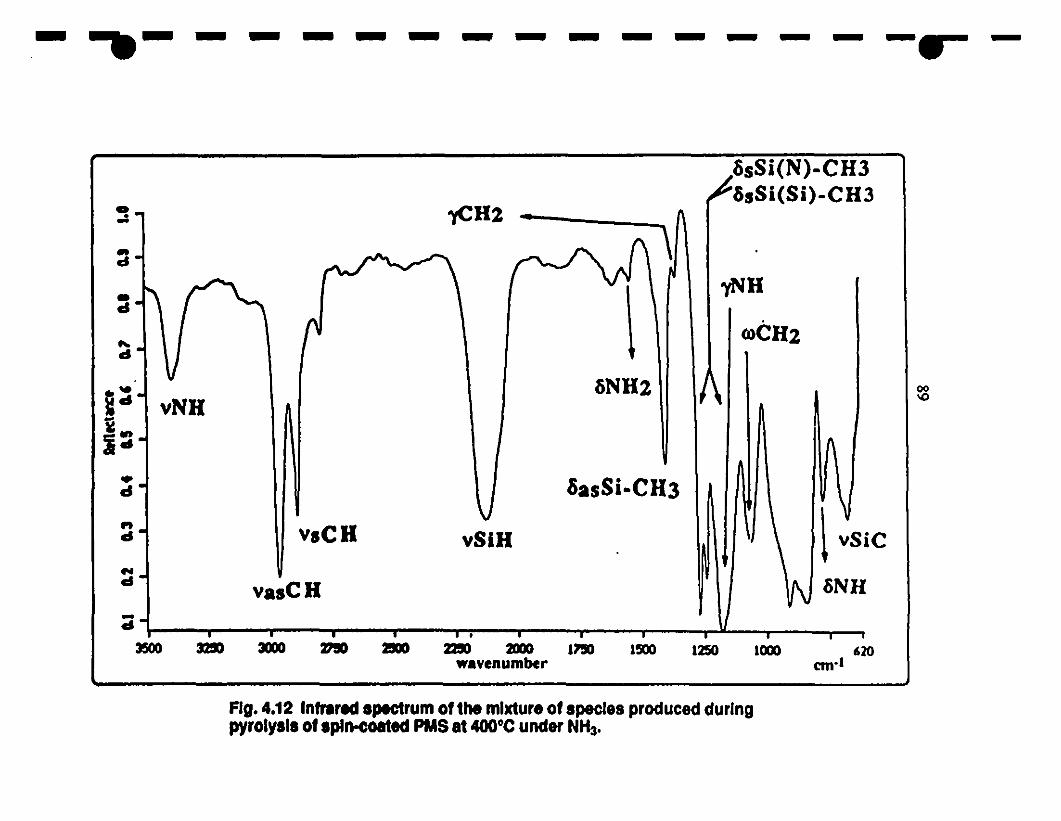
-~---------------~-
~
t vSiC
~NH
1000 '20cm-I
œCH2
,~sSi(N)-CH3
8sSâ(Si)-CH3
"fNH
12501!lIOO
vSiH
8NH2
SasSi-CH3
oyCH2 _.. _
\
vsCR
vasCH
•3500 32!lO 3000 2'1lIO 2!00 22llO 2DOO 17!lO
wavenumber
...cS
-eS
ftd
•d
....d
•d
:;
•.:
..id 1 vNH-~ ...ieS
Flg.4.12 Infrar. apectrum of the mixture of specles produced durlngpyrolysll of Ipin-cOlted PMS 8t 400°C und., N~.

1
~111111111111111,1
90
The complex nature of the chemical processes involved in the pyrolysis of PMS
under anunonia is revealcd by the large number of vibrational bands observed in the FT-IR
spectnlm of a spin-<:oated layer that was annealed at 400"C (Fig. 4.12). The simultaneous
presence of specific features for silane, silazane and carbosilane species is observed.
Assigning specific bands to these species is complicated due 10 the presence of overlapping
bands and the possibility of contamination wit!l oxygen. The frequency range 1100-1000
cm-1 would be the most affected since this region may contain overlapping bands due 10
vas(Si-Q-Si), vas(N-Si-N) and CJ>(Si-CH2-Si). Therefore, special precautions have to be
taken in order 10 minimise oxidation problems, such as dry box manipulations, UHP
gases, and the use of an inen atmosphere during physical measurements.
Some uncertainty existS in the literature concerning the assignment of the bands in
the l4OQ-l(XlO cm-1 region. For instance, the band at lOSOcm·1 bas been atnibu1ed 10 both
the wagging mode of a ·.:H2 group in a carbosilane unit,23,24,2S,26. and 10 vas<Si-Q-Si).27
The position of die -y(CHz) mode in this region is based OQ die assignment of a similar
mode in H3SiŒ2SïH3 aI 1370 cm-1•2S
1be spectrum in Fig. 4.12 shows bands suggesting that the intermediate produet of
the pyrolysis aI 400"C is acarbosilazane. The simultaneOUS ptesCDCe of the bands at 3396.
1179 and 778 cm-1 strongly supports the p~minanceof a silazane species over an
aminosilane; die presence of a small amount of die laaer is œsponsible for the weak
absorption aI 1544 cm-1 (~NHz)- In the 2000 cm-1 regioo, the v(Si-H) band appears al
2131 cm-l, shifted from 2075 cm-1 in the starting material. This sbift signais the
23colthup. N.B.; DaI)', Ut: Wibericy, S.E./NTodvrlj". ID Infrand twlR_ SpeCUD=PY;Academie~New Yœk, 1!l6424Wu, HJ.; Intemlllte, L.V. Chut. Maur. 19S9, l, S642SBu1IoI, J.; SchmidJ. MP. Pirys. S/QlIU SolùJi 8,1987,143, 34S26Schmidt, W.R.; InImante, L.V.; Domnus, JUL; Trœt. TJC.; Martbcui, P,S.; MacieI G.E.; Chut.Maler. 1991, 3,2lI627Ma1suddy, B. Cmun./Ilt.19S7, 13,4128McKean, D.C.; Davidsoa, G.;.Woodw..d LA. Spearoclùm. AcID lm. 26(A), 181S

1
~111111111111111ft1
91
conve~ion of mast of the Si-SiH-Si groups in the baclcbone inlo S-SiH-N unilS. The high
intensity of the Si-H band ailer amination may be explained in r,r."O ways: (a) as Si-H
groups are undergoing amination, the Kumada rearrangemenl generales new Si-H groups,
and (b) NH3 reaets mainly with the Si-Si bonds and not with the Si-H bonds. The last
process would explain the scission of the polymer baclcbone and may explain the inaeased
weight loss observed in l'GA under NH3· The increased mass transport through the gas
phase may be responsible for the high deposition rates of ceramics on colder substtates
during the CP-VD process under these conditions.
The presence of methylene bridges, confinning an ongoing Kumada rearrangement
is indicated by the scissoring mode ()CP.ù at 1356 cm- l _ The rather sharp absorption at
1048 cm-l is assigned ta the methylene wagging mode (CIlCHù ronfinning an advanced
stage of the Kumada reammgement. Siloxancs have strong, broad absorptions in this
region, but their bands are generally not as sharp as Cl>(Si-CHZ-Si) and cao be easily
recognized. Analyses of mixtures of carbosilane and siloxane specics is usually
complicated, since the vasCSi-o-Si) band is much suonger than CIl(Si-eH2-Si), in which
case the presence of Si.Qi2-Si groups can be easily overlooked. The lower inlcnsity fer
the ){0i2l vibration compared to that observed in the FT-IR spectrum of PMS-derlved
PCS at the satne !i::mperature indicates that the Kumada rearrangement occurs ta a lesser
exteDt in the presence of NH3- This is most likely due ta the competition betweea the
fonnation ofSi-CHz-Si and Si-NH-Si and Si]N groups.
The symmetric defonnations of the (Si)Si-0i3 and (N)Si-CH3 groups are
observed at 1244 cm-1 and 1275 cm-l, respeclively. It should be mentioned mat the
absence of absorption bands at 2160, 1260 and al 1100 cm-1 associated with (O)Si-H,
(O)Si-CH3, and Si-0-5i groups respectively, indicates negligible œidation ofdie backbœe
in this sample. The bands at 916 and 835 cm-1are assigned ta the trigonal planar and
pyramidal N geomeuies. Bulk satnples ofcomposite SiCxNy powdeJs with low Ncontent

1
~111111111111111('1
92
have been shown tO contain N atoms dissolved in the SiC latùce as both the teuahedra1
fonn. bonded to four Si atoms and the trigonal form. bonded to three Si atoms.29 The
main SioN band for Si3N4 obtained by CVD is asymmeaic and cao be deconvoluted into
four best-fit Si·N Gaussian components, the IWO most intense bands being located around
840 and 940 cm-1•1S,30 Force constant calculations suggest that N-Si·C groups should
give absorption bands in the 800-850 cm-1 n:gion. Finally. the band at 678 cm-I is
associated with Si-C bonds. Ali of the proposcd assignments are listed in Table 4.1.
Given the conclusions of the analysis of the speclIUm in Fig. 4.12, pyrolysis at
higher temperatureS will evidently involve n:actions charaeteristic of poly(silazane).
poly(aminosilane) and poly(carbosilazane) species. Each species has its own complex
,behaviour as a result of simultaneous polymerizaùon. decomposition and degradation
n:actions. The Si-H bond is al50 known to participate in the thermal crosslinkiDg of
polY(S11anes). poly(carbosilanes) and poly(silazanes).31 In addition. transamination
n:actions are common for the group 14 elements.32 A further complication arises from the
thermal cfecomposition ofammonia, which may become significant al temperatures in the
range of500-SOO"c' 50 possible transformations under N2. H2 and NH3 almosphere of all
of these species must be taken into account. Even under pwe NH3. n:actions involving
hetero-dehydrocoupling of SiHINH. homo-dehydrocoupling of SiHlSiH and
transamination must be considen:d. as weil the Kumada rearrangement and NIC bond
interchanges.33
Nilridarion 01polycaroosilane. A PCS film on a silicon substrate was prepan:d
by the thermal n:arrangement of PMS undcr Ar (heating rate «)OC/min) up 10 4SO"C, with
29Suzuti. Mo; HastBawa. y.; Aizawa. Mo; Natall, y.; OIallallÏ. T. J. AM. CcrGllL Soc. 1995.78(1), 833OI..evmc.LN. iD MokclI14r SpeCVO#O/11. WiJer. New YOIt. U75. p.13231PiJoc,J.P. Proc:«dillls tl/Ille X-t1lI_T1IDIioNJ1 ()rposiIWm S)mpOJiMM, l'I:lznan, 1993. p.163~ JA; Oja... CM.; Dubois, LA J. E1«vocJIeJIL Soc. 1993. 140(9), 2695~dtbeexpeaÙllallS iDvohe maiPl'i • ofaSlrictCClllllll~such dyDamics--asas tbe laIiD& nIC lIId tbe ps Dow. as discus3cd iDOapcr5.

1
~111111111111111,.1
93
Table 4.1. Assignments ror the observed absorption bands in the FT-IR
spectrum (cm-t) or poIycarbosilazane.
f=luencv fern-II assimment
3396 vïp{NH)
2966 vas<CH)
2888 vs<œ)
2131 v(SiH)
15S4 S<NHù·
1410 8as<Si-CH3)
1351 'lCHz bridges
1270 SsSi('rl}-CH3··
1244 .. S"("i)-CH ••<>;1,'13
1179 yop(NH)
1048 Cll(Si-Olz-Si)
916 Si·Npi-Si (N planar)
836 Si·NII"'Si (N-lrigonal)
778 a(NH)
678 v(Si-C)
·Fourdeformation modes are expected for NH2. S(NHZ) represenlS the deformation of the
angle H·N-H; Q),'t and Pare related lO the angle SiNH.
••AssignmenlS based 011 comparison wim the specttum ofoxidizcd or PMS presented in7
a residencc lime of90 min al 4SO"C. Ammonia was tben inaoduced in the system and the
reactioD wim NH3 Il 4SO"C was canied out uotil the IR showed the absence of the Si-H

1
~111111111111111,1
94
band at 2100 cm-!. The spectra of the initial PCS and ammonolys~s product are sho.....n in
Fig. 4.13. The presence of the antisymmetric S-H stretch, the lower frequency of the
vs<N-H) band and the presence of the band at 1549 cm-! due to O(NHù in the prodUCl, all
indica!e formation of aminosilane in large amounts. Funher pyrolysis of this produet led
to the spectra presented in Fig. 4.14. A decrease in the intensity of the Cll(CHù mode at
1350 cm-! is initially accompanied by an increase in the intensity of the broad absorption al
-940 cm-l • followed at higher temperature by a redistribution of intensity between the IWO
absorptions at 940 and 840 cm- l . Another interesting fcarure is the merging of the
Ôs(Si(N)-CH31 at 1270 cm-l with the broad absorption shifting from 1200 to 1250 cm- l ;
this convergence ilIustrates the difficulty in assigning the different vibrationa1 modes in this
region.
Another pyrolysis cycle was performed by heating PMS undcr NH3 up to 800"C al
2°Clmin. followed by a fast heating rate (l5°C/min) under Ar up to 950"C. Formation of
NH2 groups from the silazane species under these conditions is iDdicated by the appearance
of both vs<N-H) and vas(N-H) modes, as weil as the appearance of the li(NHù absorption
al 1544 cm-l (Fig. 4.15). Formation of an NH2 group from the silazane species is the
result ofa transamination Ie3ClÎOII of the type pn::sented in Equation 3:
NH21
....S~
(3)
Such redistribution IeaCtions are unavoidable during the reaction of the gIOUp 14 clements
with NH3,33 This reaction has becn prevïously cited as a cause of deaused ceœnic
yields, by favouring the formation of volatile species which CID distill out during
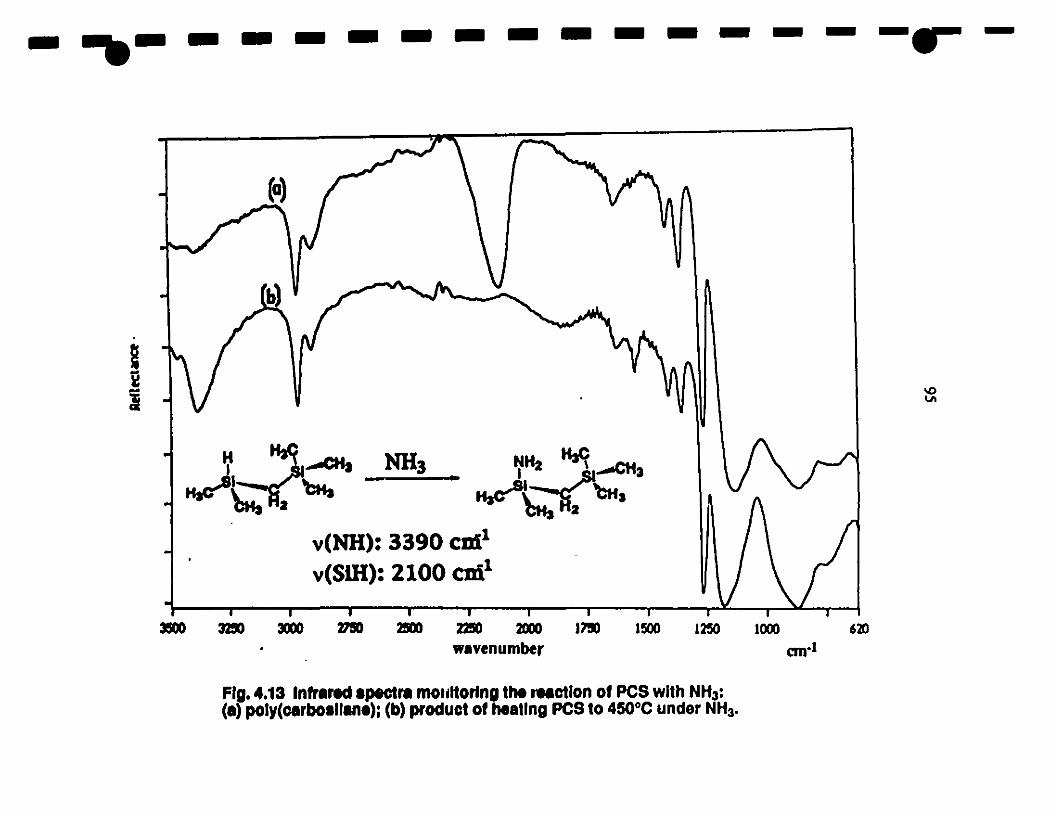
-~---------------~-
e2i
(a)
~~Haq -cHa NH3 ~H2~~-cHa~I "cHa - • ~ ,~ z ~ z
v(NH): 3390 cDi1
v(SIH): 2100 cm1
31500 32lIO 3000 21!111 2!lOO 22!10 2000 17Sl 1500 1250 1000 620wavenumber cm'}
Flg.4.13 Inrrared lpect... mo'i"orlng the reaetlon of PCS wlth NH3:(a) poly(carboillan.); (b) product of heatlng PCS to 450°C under NH3•
'DVI
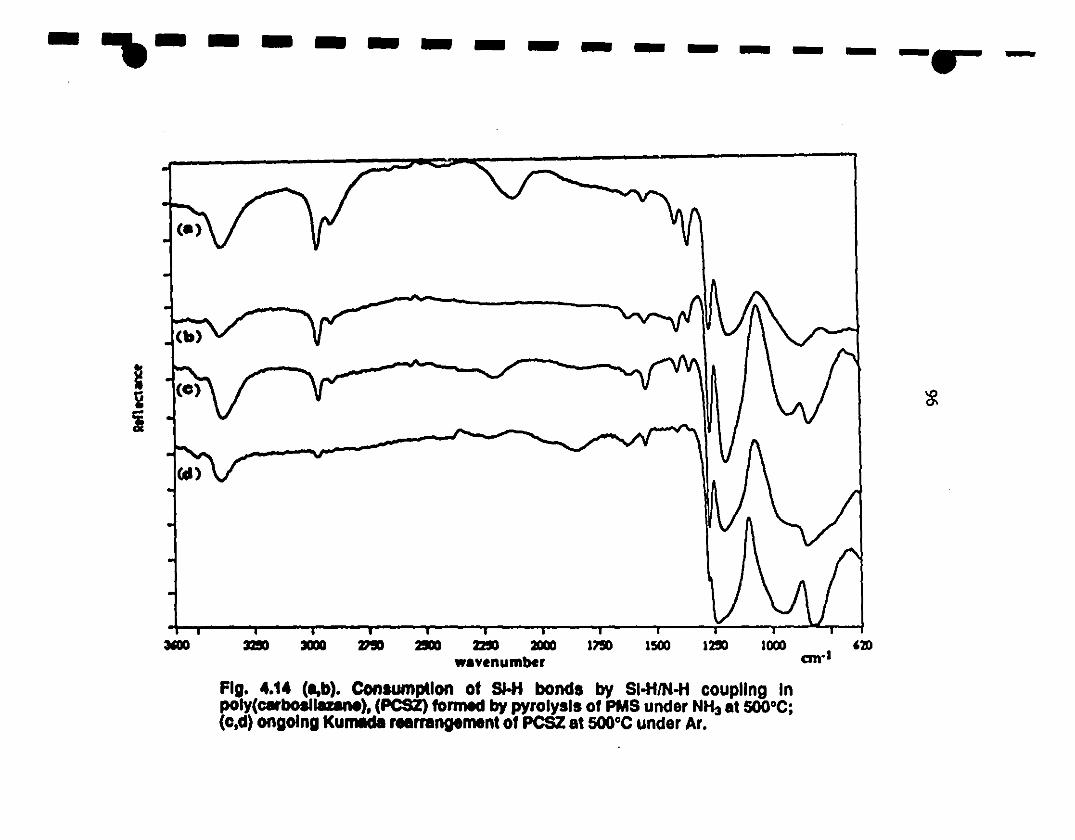
- ...------------- --.- -
(b)
eii
~
3QlO 3Z!lO 3IlOO P!IO 2!IOlI 22S) 2OQO 17Sl 1500 1251 1000 ''IDwavenumber arrl
Fig. 4.14 (I,b). Consumptlon of SI-H bonds by SI-HIN-H coupllng lnpoly(eatbosllarane), (PCSZ) formed by pyrolysls of PMS under N~ at soooC;(O,d) ongolng Kumllda reerrengement of PCSZ at soooC under Ar.
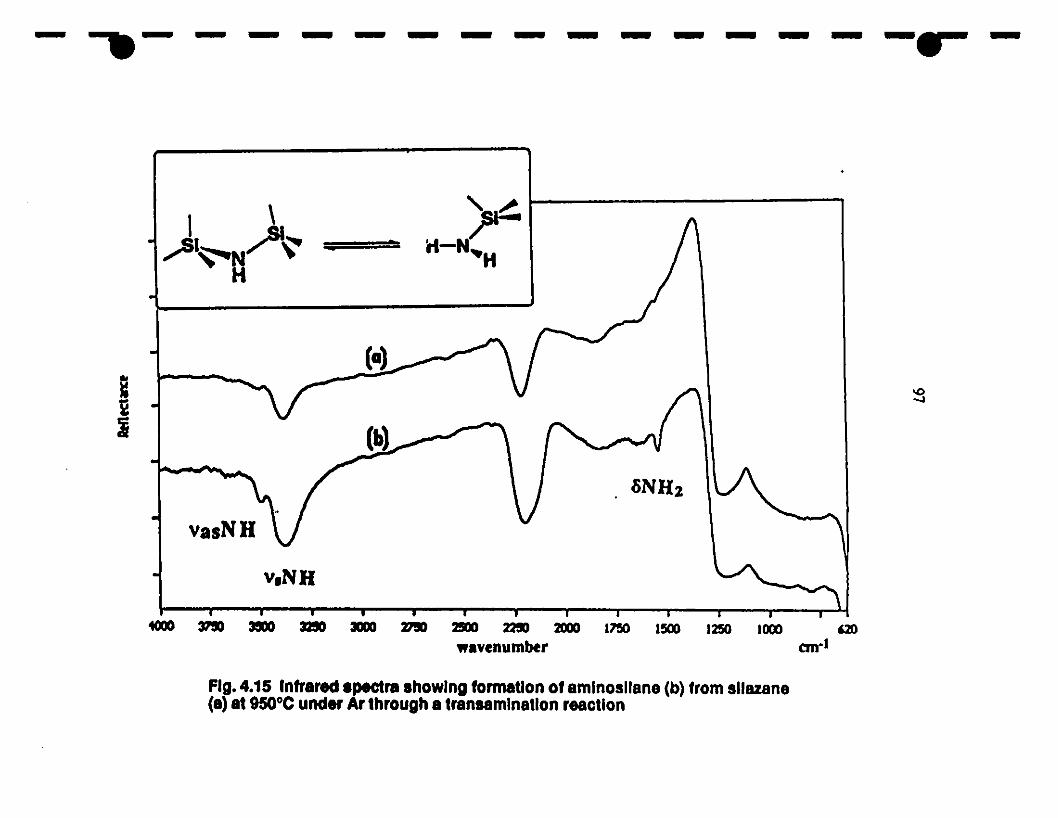
- -.- - - - -- - - - - - - - - -.- -
~
~NH2
,,;H-N~
~H
(a)
(b).......
v.NB
vasNH
1 \,..s1~N/'~
H
ii
1000 37BO 3!lOO :rao 3000 %1lIO 2!lOO 22!10 2IlOO I~ 1500 1:l5O 1000 QI)
wavenumbtr cm-I
Flg.4.15 Infrareel spectre showlng formation of amlnosllane (b) from sllazane(8) at 95Q°C und., Ar through 8 trensamln8110n reactlon
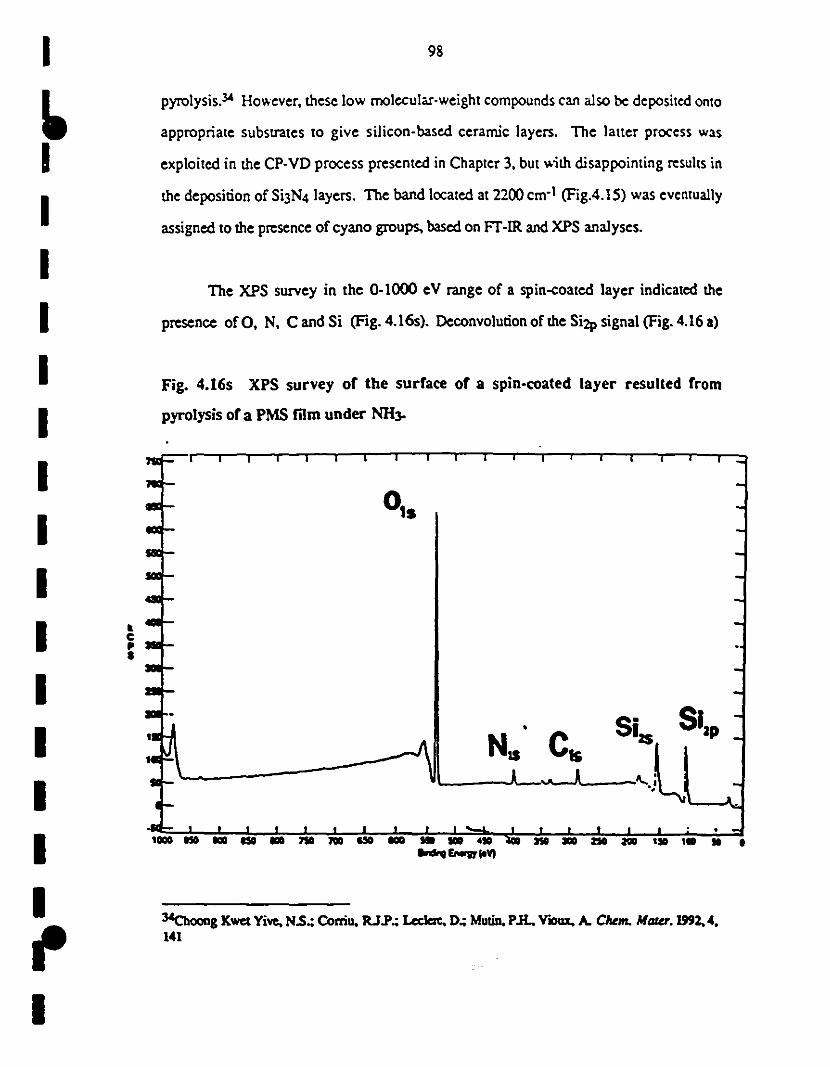
98
34choong Kwet YiYe, N.s.; Corriu, RJ.P.;~ D.; MulÎll, P.H.. YIDlŒ, A. CMm. Maw. llI92, 4.141
JIll lIllO ZM aoo '10 •• • •
0,.
Fig. 4.16s XPS survey of the surface of a spin·coated layer resulted from
pyrolysis of a PMS film under NH).
The XPS survey in the 0-1000 eV range of a spin-coated layer indicated the
presence of O. N. C and Si (Fig. 4.16s)_ Deconvolution of the Si2p signal (Fig. 4.16 a)
pyrolysis.304 However. these low molecular·weight compounds can also be depositcd onto
appropnale substrates to give silicon-bascd ceramic layers. The latter process was
exploitcd in the CP-VD process presented in Chapler 3. but ",ith disappointing results in
the deposition of Si3N4 layers. The band located at 2200 cm-! (Fig.4.l 5) was evenlua1ly
assigned to the presence of cyano grouPS, based on FT-IR and XPS analyses.
'000 150 lllII 150 lllII no JllO 150 _ _ lIIlI <10
~E-w"V1
,cp
•
1
~111111111111111t'1
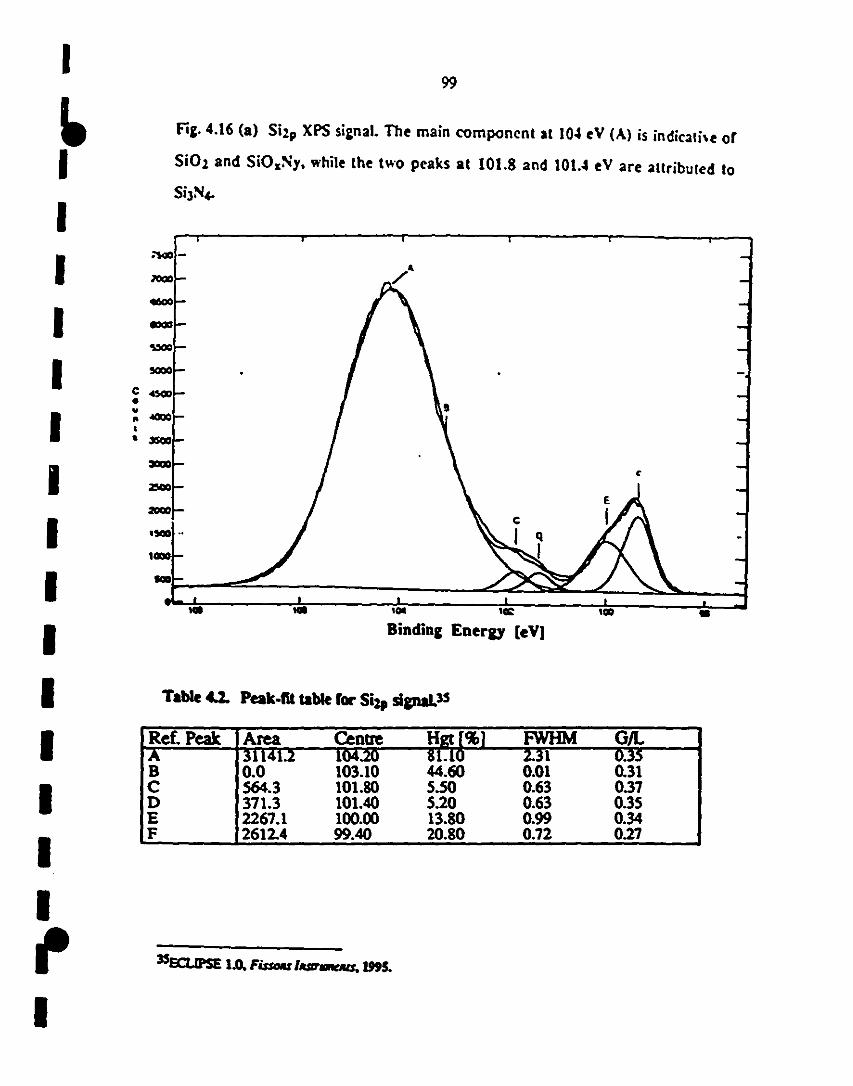
99
Bindin& Enerl)' [eV)
Fie· 4.16 (a) Si2p XPS siena!. The main componenl al IO~ eV (A) is indicalÏ\e or
Si02 and SiOxSy, while the IwO peaks al 101.8 and IOU eV are allribuled to
SiJS4-
3SECLIPSE 1.0. FÙSl1WII-.-., ms.
Table 4.2. Peak.fIt table 'or Si1p sipL35
Ref. Peak Area Centre FWHM. 1
B 0.0 103.10 44.60 0.01 0.31C 564.3 101.80 5.SO 0.63 0.37D 371.3 101.40 5.20 0.63 0.35E 2267.1 100.00 13.80 0.99 0.34F 2612.4 99.40 20.80 0.72 0.27
:wo --4IlOO
.,.,.~
~
c .-•v .--•• -)000
ZIOO-.- ..,-- .. .. '01 'C 'GO •
1
~111111111111111,1
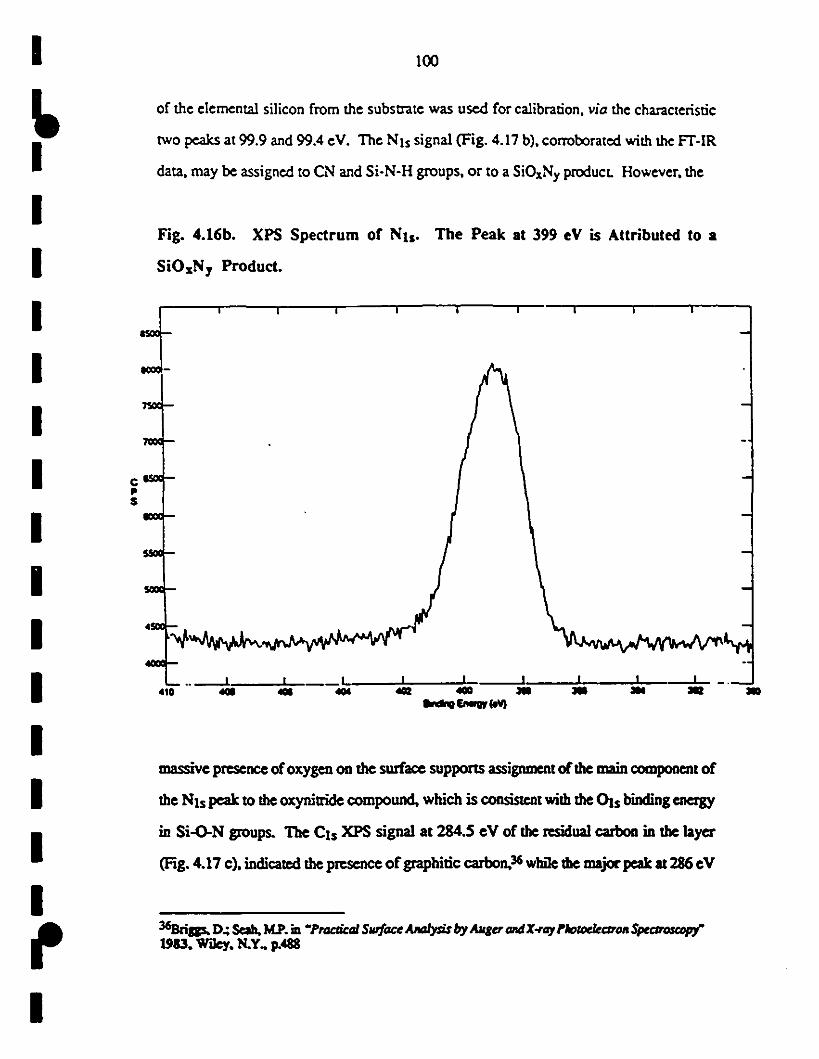
:1
•..---1.-_-:'~_-L_--+-J =--_-:':-__....,:'!:-_-='::-_-='::-_-='=_~ - - - - - - - - -.r-.(oV)
100
massive presence ofoxygen on the surface suppons assignmcnt orthe main component of
the Nls peak to the oxynitrïde compound, which is consistent wilb the Ols binding energy
in Si.().N groups. The Cis XPS signal at 284.5 eV of the residual carbon in the layer
(Fig. 4.17 cl. indicated Ibe presence ofgraphitic carbon,36 wbile the major' peaIt at 286 eV
IWO peaks at 99.9 and 99.4 eV. The NIs signal (Fig. 4.17 bl, corroborated with the FT·IR
data. may he assigned 10 CN and Si·N-H groups. or to a SiOxNy prodUCL However. the
of the elemental silicon from the substrate was used for calibration, via the characteristic
Fig. 4.16b. XPS Spectrum of Nb. The Peak at 399 eV is Attributed to a
SiOxN" Product.
cp$
1
~111111111111111f1
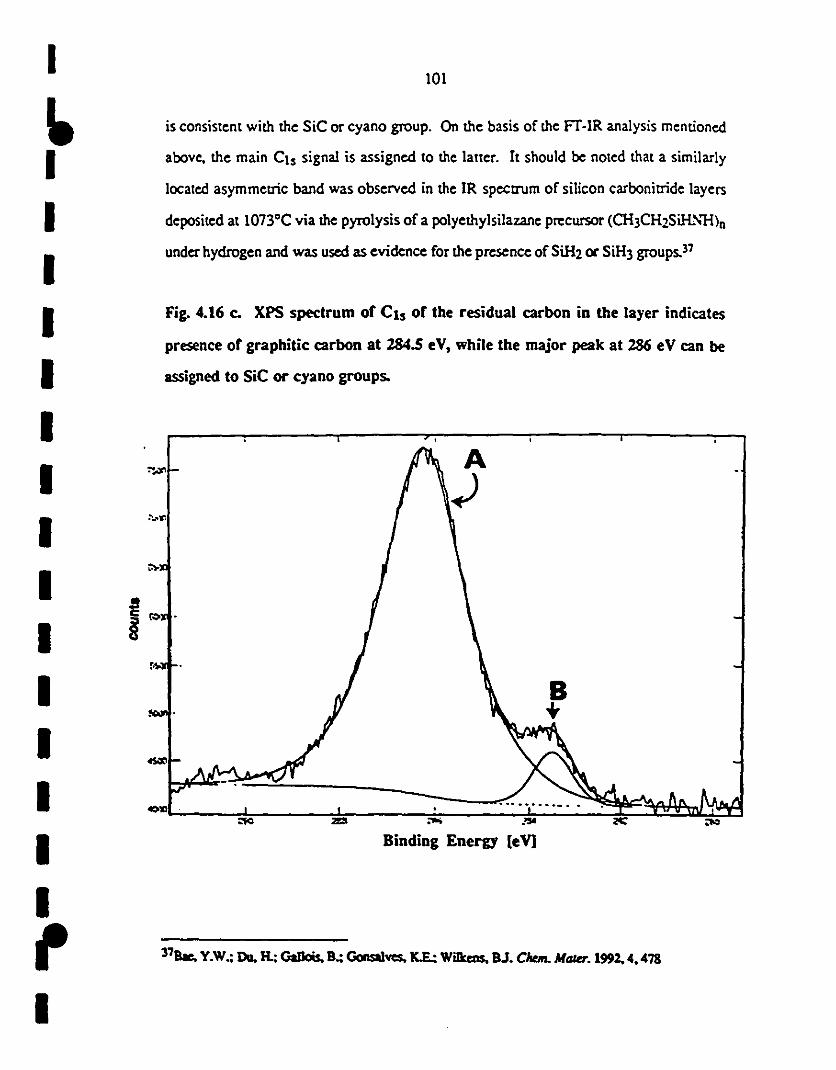
1
~111111111111111,1
lOI
is consistent with the SiC or cyano group. On the basis of the Fr-IR analysis menùoned
above, the main Ch signal is assigned to the laller. It should be noted that a similarly
localed asymmetric band was observed in the IR spectrum of silicon carbonitride layers
deposited at 1073°C via the pyrolysis of a polyethylsilazane precursor (CH3CH2SiH.'Œ)n
under hydrogen and was used as evidence for the presence of Sili2 or SiH3 groups.37
Fig. 4.16 Co XPS spedrum of Cls of the residual carbon in the layer indicates
presence of graphitic carbon at 284.5 eV, while the major peak at 286 eV can be
assigned to SiC or cyano groups.
«>'IIL---:;:,oi;;----...,:~---~:oo;~.==::::~....;:..:..:.:.:-=~~2C~~ei.\&~:oo~.~Binding Energy [eV]
3'BIe, Y.W.; Du. H.; GaDois, B.; Gonsalves. K.E.; Wilkens, BJ. CIruL Makr. 1992. 4, 478

1
~111111111111111,1
102
Table 4.3 Peak·fit table for Cls region.
Ref. Peak Alea Centre Hei~t [%] FWHM GausslLon:ntz
A 9054.5 286.20 91.50 2.21 0.78
B 612.6 283.50 14.10 1.06 0.38
4.4. Conclusions
Poly(methylsilane) (PMS) has been used as a precursor in the preparation of
amorphous silicon niuide (a·Si3N4). Thin layers of a·Si3N4 wcre synthesized by the
pyrolysis of thin films of PMS deposited on siliCO'l single crystal wafers and via deposition
of the volatile spccies resulting from the thermal cracking of the bulk precursor in the
presence of ammonia. The chemical processes involved in the pyrolytic steps wcre
monitored by FT-IR specaoscopy. thermogravimeuic and differential thermal analysis. In
the presence of catalytic amounts of CP2Zr(CH3)z (DMZ; Cp-...."S·CsHS). the reaction
between PMS and NH3 stans at ca. 200°C with the slow production of a slightly
crosslinked product most like1y involving Si3N knots. At this temperamre. the silazane
links are extreme1y sensitive to hydrolysis and oxidation. and oxygen contamination of the
product could not be avoided. Extensive amination of PMS, with little oxygen
incorporation. occurred on pyrolysis at 300"C. under 5-10 torr NH3 overpressure. The
product exhibits IR bands charaeteristic of both a silazane and an aminosi1ane species.
which are presumably formed by Si-H and N-H heterodehydrocoupling. Between 200 and
4500C. this cross dehydrocoupling reaction compete5 very effectively with the Kumada
rearrangement. Vibrational assignments are proposed for the observed bands. Significant
10ss of carbon occurs from the resulting poly(carbosilazane) between 500 and 600"c.
Prolonged curing under NH3 at 300"C. to remove aIl Si-H groups and to give a densely

1
~111111111111111{'1
103
crosslinked polysilazane suppresses the Kumada rearrangement which is not observed even
after raising the pyrolysis temperarure to 7000C. In this case, although the main product is
still a-Si3N4. there is an increased amount of residual carbon. Pyrolysis of a PMS sample
under NH3 up to SOO°C, followed by annealing under N2 up to 950°C leads to the
formation of an aminosiiane species from the silazane. possibly through a ttansamination
reaction.

1
~111111111111111('1
104
CHAPTER5Low-Temperature Nitrogen-Carbon Exchange
During Reactive Pyrolysis of Polymethylsilane under Ammonia.
The Influence of l\'H3 Decomposition
5.1. Introduction
Economical1y important structural and microclcctronics applications exploit the
superior physical. chemical and thermal properties of silcon nitride (Si3N4) and temary
SiCxNyceramic materials. The CUITent interest in new organosilicon precursors to Si3N4
results from the observed reactivity of ammonia in replacing organic substituents ori
silicon.1•2•3 Consequently, NH3 is being used in the reactive pyrolysis of
poly(carbosilane) (PCS),4,5.6 po1ysilane4•7, and polysilazane4,8.9 to form monolithic
SiCxNycomponents, and in the cbemical vapor deposition (CVD) procedures used for the
synthesis of thin films of these materials from monomeric silicon compounds.tO IntereSl
bas focussed mainly on ceramic yields and the composition of the ceramic produets. The
process ofcarbon removal from the organosilicon precursors in the presence of NH3 is not
weIl unde:tSlood despite the Caet that rigorous control over the composition of the SiCxNy
produets at different stages during the pyrolysis is important in achieving acontrol over the
1 Brown-WcnsIcy. KA; Sinclair. RA, U.5. Pat. No.4S37942, Aug. 27.19852 Ralle, J.; BujaJski, D~ EllI'op. Paz. Appt 200326, March 16. 19863 Okamwa. le.; Salo, M.; Hascgawa, Y. Ceam.ltIl~ 1!Jl7. 13.554 Burns. G.T.; CbaDdm, G. COmmlUl. Ame. Ce,am. Soc. 1989,72(2).333S TaIci, T.; Inui. M.; Okamwa. le.; Salo, M. J. Mazu. Sei. Leu. 1989, 8,11196 Corriu. RJ.P.; Leclerq. D.; Mutin, P.H.; ViOllX, A. C1IDn. Male'. 19!12. 4, 7117 Schmidt, W.R.; Marcheui, P.S.; Intemmte. L.V.; HurIey, WJ.; Lewis Jr~ RH.; Docanus. R.H.;Madel, G.E.. C1IDn. Mazu~ 1992, 4, 9378 Han, RN.; Lindquist, DA; Haggctty. J.5.; Scyfenb, D. C1IDn. Mazu~ 1!l92, 4, 70S9 MucaJo, M.R.; MiI_.N.B.; V"aclaidge. LC.; Swaïu. M.V. J. MateT. SeL 1994. 4, 44871Omrai. T.; Golo, T. J. Mazu. Sà.,198I, 16, 17

1
l-I11111111111111,1
105
tendency of Si3N4 to crystalliz.e. This control is also necessary for the synthesis of silicon
carbonittide materials. or for reducing the residual carbon content of the Si3N4 products.
Carbon/nitrogen exchange was observed by NMR spectroscopy during PCS pyrolysis
under NH3 al SOO-700"C.11 The possibility of thermal deeomposition of NH3 taking place
is not usual1] taken inta account, although there is evidence that the partial pressure of Hz
during pyrolysis cao dramatically affect the amount of residual carbon12 and the
crystallization range of Si3N4-13 On the other hand, the formation of unspecified active
species in the gas phase due ta the thermal decomposition of NH3 plays an important role
in the mineralization step ofpoly(ethylsilazane) precursors ta Si3N4 .14 The mineralization
step in this case is thought to take place via the nucleophilic cleavage of the ethyl groups
from silicon in the precursor induced by anack of these "active species", rather than the
homolytic cleavage observed during pyrolysis under NZ, a reaction Pat!tway also proposed
for polysilanes.1S
The behaviour of NH3 during preceramic polymer pyrolyses has been less
extensively studied. However, unlike the situation when inert atmospheres16 are used
(Si3N4 cao also be prepared by the pyrolysis of nitrogen-containing precursors such as
11Taki. T.; lDui. Mo: Okamura. K.; Salo. M. J. Mazer. Sei. Lert. 1989. S. 1119lZMach. R.; K1oez, B.-D.; Solute, K.-D.; Drost, H.; 0Ieszak. F.; Lacayo. G.; SzuIzewsIty. K.; Dorfel, I~Key Eng. Mazer. DM. 89-91, 4113NordcII. N.; N"JSbiDo, S.; Yang. J.-W; Jacob. c.; Pirouz. P. J. ElectroehDrt. Soc.. 1995. 142(2), S6S14Bac. Y.W.; Du. H.; Gallois. B.; Gonsalves, K.E.; WilkeDs. BJ. CMtrL Mazer. lm.4. 418ISSeyfenh, 0.; Wiseman, GA; Schwarlt. J.M.; Yu. Y.F.; Poumsse. CA in rllDrgonk aNlOrgDIIDmeraJlic Pol)7IIm; American Cbenùcal Society Symposiwn Series No. 360; ZeIdin. Mo Wyrme.lC.J. Allc:ock. H.R.. Cds.; American Oemical Society: Wasbington. DC.1!l88. 14316The terni -men" is onIy 1daIi~ as pyroIysis in an "'inenw NzllllllOSpbere was found ID CODtlIIlinalesrmic:oodllClOr SiC Iayels wiIh asigni6cantCOllC:CntIalioo oC IÙtrogeII-rda1ed dlargecaniezs.

1
~111111111111111
"1
106
silazanesI7.18.19 and siladiazanes20,2I,22 under N2), the pathways of the reactive pyrolyses
are highly dependent on the composition of the gas phase. Moreover. when :-ai3 is the
reactive annosphere in the pyrolysis, an additional complication arises due to the potential
thennal decomposition of NH3 at higher temperarures. For exarnple. a different pathway is
induced when NH3 is used instead of N2 during the synthesis of Si3N4 from Si02lC, and
faster reaction rates were observed in the l3SQ-1400°C range, due 10 the presence of"active
forms of hydrogen and nitrogen".23 Altematively, a catalytie effect was assumed in the
synthesis of Si3N4 films by double-source CVO using SiF424 or ethylsilazane2S and NH3.
This has been attributed to the lower activation energies of the process relative to the
thermal dissociation energies of the individual bonds in the precursors (> 340 kJ/mol26)
Despite its importance in the nitridation of polymerie precursors, especially due to
the involvement ofNH3 in dehydrocoupling and transamination reactions occurring 8t high
temperatures, the thermal decomposition of NH3 has not been considered in detail. No
attempts have been maèe 10 obtain quantitative data for the relationship between the thermal
decomposition of NH3 and the properties of the residual cerarnie produets. Decomposition
below 6S0°C was considered to be negligible during pyrolysis of a bulk polymerie
precursor, because of the absence of N2 in the gas phase.6 However, NH3 decomposition
8t SOOOC was observed during the synthesis of SiJN4 in a plasma reactor, proportional to
the NH3 fluxP Questions about the mechanism of the process, such as the involvement
17Laine,R.M.: Blum, Y.D.; Tse, D.; Glaser, R.llIOrgQlÙC œtd OrgtUlDlMlDllic Polymt:n; ZeIdiD. M:Wynne, ICJ.; ADcock, H.R., Eds; Ameri<:an Cbemical Society: Was!ùngton, D.C., 1988: 12418 Arides, B. J. Ekerrochem. Soc. 1986, 133, 23319Mazdyasni, K.$.; West, R.; David, LoD. J. Am. Qram. Soc~ 1978. 61(11-12). S042Oco1ombicz', C.; Cucr, J.P. Br. tlmItzIuk FR 2633301,1981121Colombiec, C. inProc.lst EIII'. Ceram. Soc. CDnf.Ed. G.de W"ldth, R. Terpsuaand R. MClSC1aar(Eds.).l98II (1), 14322He, J.; Uu, H.Q.; Hanod, J.F.; HyDCS, R. OrgQJIDmt:tDlllics,lIIII4, 13.33623Dwbam, B.G.: Munba, MJ.; Bumct. G. Adv. Ceram. Maler.ll188, 3(1),4524 Fuyuti, T.; Allain, B.; Perrin, J. J. Appl. Plrys.l9IIO, 68, 33222S Bac, Y.W.; Du, H.; Gallois, B.; Gonsalves. K.E.: Wilkens, BJ. Chem. MDler.l9II%,4, 47826 Walsb, R. Ace. Chem. Res.l9I1, 14, 246'J:l Davies, B.; Soucy. G. l'roc. 63·rd ACFAS Conf~ 22-26 May 19II5, Cbicoutimi, Qc

1
~1111111111111·11f1
107
of NH328 or NH2 radicals generated during decomposiùon of NH3 in the nitridation
process.29 or the relative dominance of high- and the low-temperawre reactions involved in
the carbon/nirrogen exchange,30,3t may only he addressed by an investigation of the
thermal decomposition of NH3. Two major reasons may be responsible for the carlier lack
of interest in studying the decomposition of NH3 under pyrolysis conditions:
(i) NH3 decomposition is considered to he negligible below 700°C. when the
carbon/nitrogen exchange process typical1y takes place in the precursor. In this case, two
questions arise. First, what is the role of NH3 at higher temperatures, and second, is this
assumption still valid at higher temperatures, since, as mentioned above, depending on the
pyrolysis conditions or the nature of the precursor (such as the structure of the backbone,
functional groups present or any previous curing), carbon displacement can be sigIÙficantly
retarded.
(ii) The possibility that factors other than the thermodynamic equilibrium are in
control. The low value for the free energy of formation of NH3, âC(j0298 =-3.94
kcal/mol32, makes NH3 thermodynamical1y unstable even at moderate œmperatures.
5.2. Experimental Section
Poly(methylsilane) (PMS) was prepared by DMZ-catalyzed dchydrocoupling of the
Wunz prepolymer, as described previously (Chapter 2). The substrates were [100]
oriented Czochra1ski-silicon single crystal wafers doped with boron and having resistivities
of 1-10n cm. These substrates were chemicaIly etehed 10 remove the native oxide layer
28 Peucten, Mo; Vaabs, T.; BIIck, M., Adv. MalU.,l99O, 2, 39829 Scyfenb, D. Siliœll-Based Polymer SciDfu. A Compre1rDJsi~ Reso",ce; Adv. Clrem. Ser•• 224;zeigIer, J.M.,Fearoo, F.W~ Eds.; American Oaemieal Society; Wasbingtoa, 1990. S6S30 Van DijeD, F.K.; P1uijmalœrs, J~J. Elu. COQ/IL Soc~ 1989, S.38S31 Van DijeD, F.K.; P1uflllllllœrs, J. J. Elu. Ceram. Soc. 198!1, S, 38S32Noyes, R.; Ammollia QIId S)'Itlhesis Gas, 1967, Noyes Dew!qlmertl Ca." -Hioa. New Je=y

1
~111111111111111l'1
lOS
and then passivaled thennally under UHP Ar. The layers were loaded inlo a sealed. fused
quanz, fumace tube (2.S cm diam, 60 cm long) under UHP Ar. Ali other manipulations
were also perfonned under UHP Ar in a dry box. The fumace tube was attached 10 inlel
and ouùet flowmelers adapted to provide independem flow rates and pressure adjustments
for NH3 and Ar flows. Prepurified NH3 (Matheson) was further dried in-tine in a 7S-mm
diam/l.S-m long drying column o.er predried, f1aked KOH followed by passage through a
mixed bed of 3 to 4 Amolecular sieves al a flow rate of S0-2OO ml/min.
Samples of 2Q-40 mg of the polymer precursor were deposited on the silicon
wafers. The flow rate for NH3 used during the experiments on the thermal decomposition
of NH3 ensured a molar excess of 10/1 of NH3 over the substrate, based on the
assumption that the kïneties of the reaction are diffusion-controlled in the PMS layer and
the diffusion coefficient of NH3 is similar to that obtained for the diffusion of oxygen in
PMS layers.33 For the "on-line" quantitative analysis of the NH3 content in the gas phase,
the exÏting gas was passed through a 1-M Ha solution using a carefully adjusted flow rate
and an analysis was performed with an automatic Metder DL-2l titrator. To ensure
reproducibility. after the flow rate and pressure of the system were adjusted, a series of
experiments was performed during which only the temperature in the fumaœ was changed.
The ratio of the decomposed NH3 to the total NH3 flow in the fumace was calcu1ated from
the volume required to neutralize a constant amount of the 1-M HO solution. The results
were calibrated agaittst an initial run with NH3 at 10SoC (chosen as the minimum
temperature in the fumace in arder to minimize moisture contamination), on the assumption
that the decomposition is negligib1e at this temperature.
The pyrolyses were condueted at 5-S torr over atmospheric pressure and were
undenaken in a Lindberg single-zone programmab'e fumaœ equipped with a Eurotherm
PID temperature controller with a maximum temperature of 11000C and providing an
accuracy of ±loC at lOOO"C. The elemental analyses were performed al Galbraith Lab.
33 5carleœ, Mo; ButJcr. LS.; Hamld, J.F. CIrDrL MQIQ. 1994. 6. m

1
~111111111111111,1
109(Knoxville) for the pure binary products and using a 2400 YA Control Equipment
elemental analyzer for the intermediate temary species. 29Si and !3C MAS NMR spectra
were recorded on a Chemagnetics CMX-300 solid-state NMR spectrometer. The Fr-IR
spectra were obtained in the 4000-620 cm'! region using the substrates as IR windows on a
Bruker IFS-48 spectrometer equipped with a microscope. a Mcr-B detector. and a Sony
Trinitron PYM 1340 color monitor for specttaJ display. The EPR spectra were measured
on solid products at room temperature on a Bruker ESP 300E instrument. The TGAJOTA
analyses were performed on a Seiko 220 instrument; the samples were heated in alumina
pans similar 10 those used during the pyrolyses.
5.3. Results and Discussion
5.3.1. Nitrogenation of PMS via Law-Temperature Reactions
Nirrogenation of PMS During Low·temperalUTe Reactions
The carbon. hydrogen. nitrogen and silicon contents of the samples produced by
heating in the 300-11OO°C range using a NH3 flow rate of 70 sccm (standard cubic
centimeters per minute) are given in Table S.l. Considering a generic formula orthe initial
polymer as (CH3SiH)o.6s(CH3Si)o.3s.34 complete consumption of the Si-H function via
Si-H/N-H dehydrocoupling [a reaction specttoscopically detected at 300"C(sec Chapter 4»
should result in 0.32-0.65 nitrogen atoms per silicon (0.65 if ooly an aminosilane is
fonned. and 0.32 if ooly a silazane species is produced). Preferential silazane over
aminosilane formation was observed, an effect that limits nitrogen incorporation. A similar
result has becn observed for other Si-N-H systems, e.g., for CVD-deposited,
hydrogenated silicon niaide produced from SïH4 and NH3 or N2. Prolonged reactiOD al
34Mu, Y.; Laine, R.M.; Harrod, J.F. Appl. Organotnet. CMm. 1994. 8. 9S
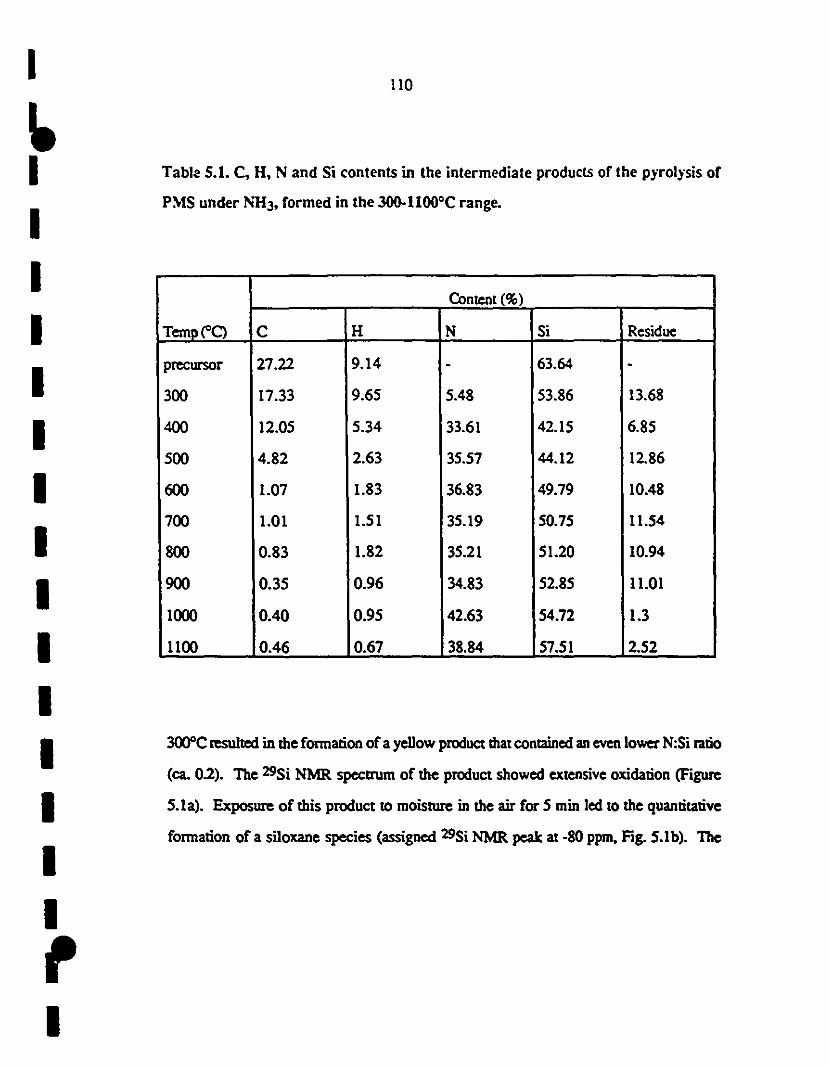
1
~111111111111111,1
110
Tabll! 5.1. C, H. N and Si contents in the intermediate products of the pyrolysis of
PMS under NH3. formed in the 300-1100°C range.
Content (%)
TemllCOC> C H N Si Residue
precursor 27.22 9.14 . 63.64 .
300 17.33 9.65 5.48 53.86 13.68
400 12.05 5.34 33.61 42.15 6.85
500 4.82 2.63 35.57 44.12 12.86
600 1.07 1.83 36.83 49.79 10.48
700 1.01 1.51 35.19 50.75 11.54
800 0.83 1.82 35.21 51.20 10.94
900 0.35 0.96 34.83 52.85 11.01
1000 0.40 0.95 42.63 54.72 1.3
1100 0.46 0.67 38.84 57.51 2.52
300"C resulted in the fonnation ofa yeUow produet that contained an even lower N:Si ratio
(ca. 0.2). The 29Si NMR specaum of the produet showed extensive oxidation (Figure
5.1a). Exposure of this produet ta moistIII'C in the air for 5 min led to the quantitative
fonnation of a siloxane species (assigned 29Si NMR peak at -80 ppm, Fig. 5.1b). The
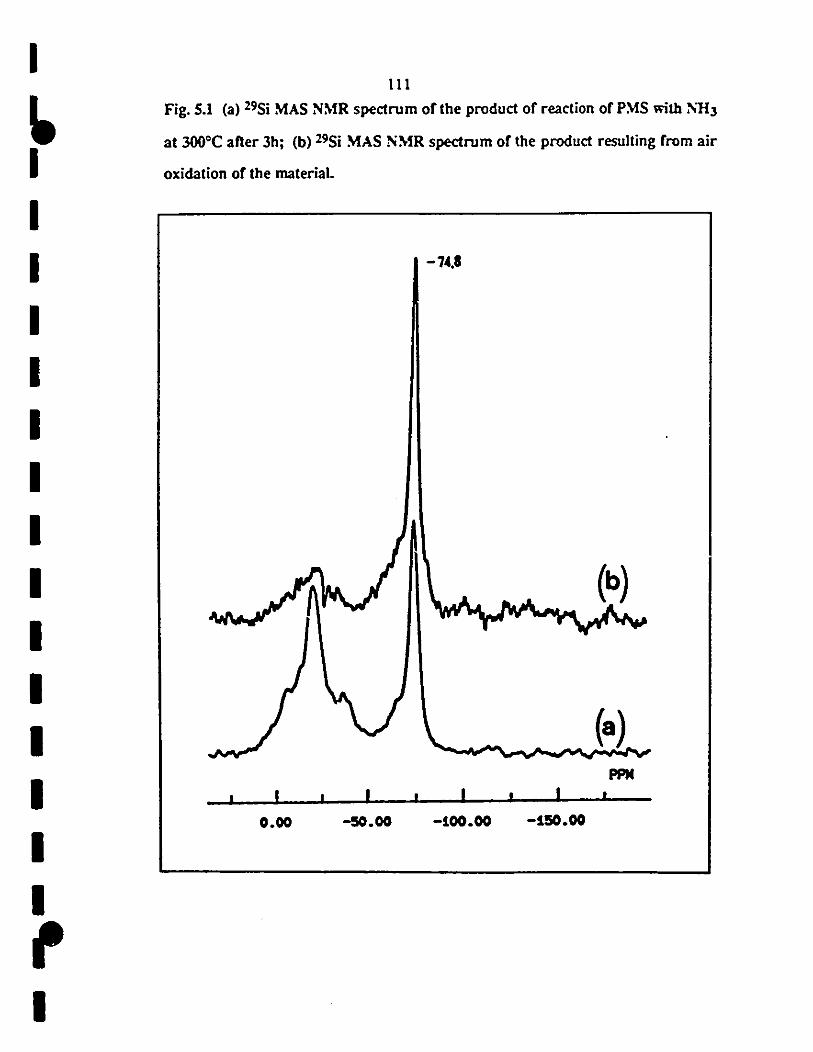
-74,8
III
Fig. 5.1 (a) 29Si MAS NMR spectrum of the product of reaction of PMS with :-'113
at 300°C aller 3h; (b) 29Si MAS N~fR spectrum of the product resulling from air
oxidation of the material.
, ,PPM
(a)
-lSO.00-100.00
,-50.000.00
1
~111111111111111,1
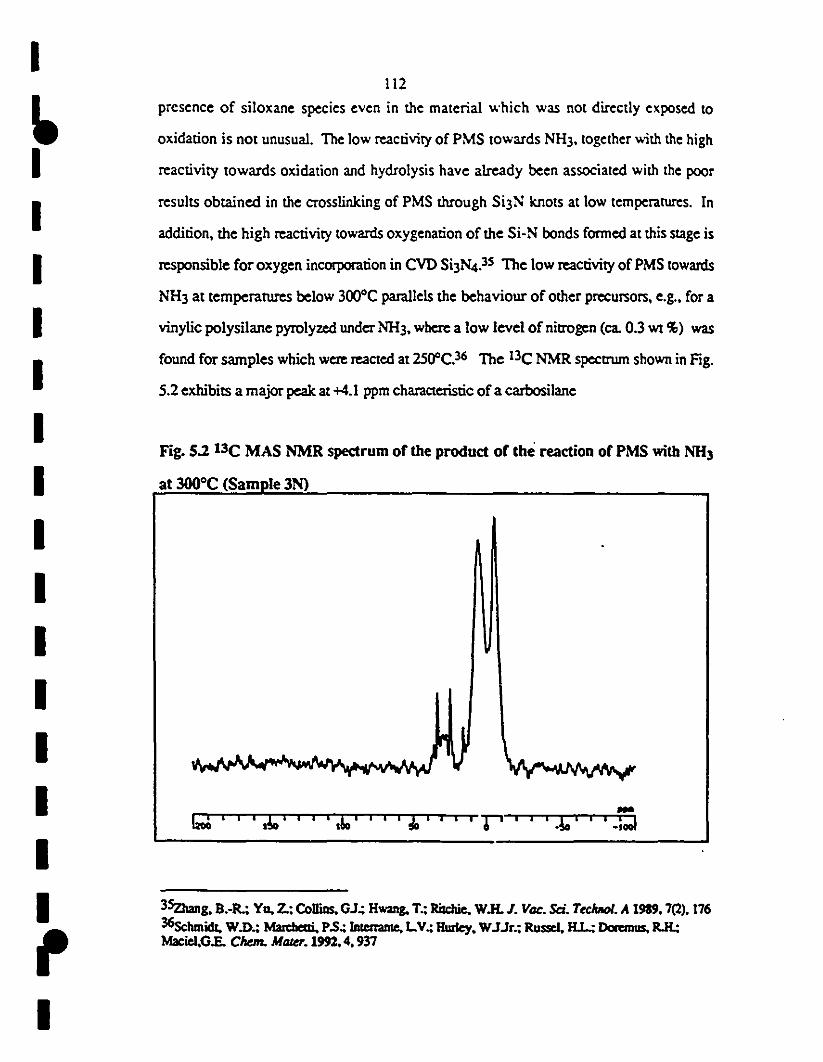
Fig. 5.2 13C MAS NMR spectrum of the product of the reaction of PMS with NH3
at 300°C (Sam le 3N)
3Szhang, B.-R.; Yu, z.; Collins, GJ.; Hwang, T.; RiIdüc, W.H. J. VIZC. Sei. Teclutol. A 1919.7(2), 17636sctunidt, W.D.; MaIdIeai, PS.; Ialernlllie, LV.; HurIey. W.J.Jr.; Russel, HL.; Dcxemus, R.H.:Macicl,G.E. Chem. MflIU. lm. 4, 937
112
presence of siloxane species even in the malerial .....hich was nOl directly exposed 10
oxidation is nOI unusual. The low reactivity of PMS lowards NH3. logelher with the high
reactivity lowards oxidation and hydrolysis have already been associaled with lhe poor
results obtained in the crosslinking of PMS through Si3N knOlS al low lemperatures. In
addition, me high reactivity 10wards oxygenation of the SioN bonds formed al this stage is
responsible for oxygen incorporation in evo Si3N4.3S The low reactivity of PMS lowards
NH3 al temperatures below 3000 e parallels the behaviour of other preeursors. e.g., for a
vinylic polysilane pyrolyzed under NH3. where a low level of nitrogen (ca. 0.3 Wl %) was
found for samples which were reacled al2S00C.36 The I3e NMR spectrum shown in Fig.
5.2 exhibits a major peak al +4.1 ppm charaCleristic of a carbosilane
-• 1001-.i :&; i'r'i JO 1i .1lO i• sL; i
1
~111111111111111,1

1
~111111111111111,1
113
SpeciCS.37 A new peak. not observed during pyrolysis under Ar. appeared at ·6.6 ppm; its
presence is associated with a species fonned when carbosilane precursors are pyrolyzed
under NH3.38
Massive incorporation of nitrogen was observed for the sample heated under NH3
at 400"C (sample 4N39). Elemental analysis of this brown produet indicated a Si:N ratio of
1:1.6. The three peaks in the 29Si NMR spectrum at -7.1, -23.0, and -42.2 ppm (Fig. 5.3)
are amibuted to SiN2X2 (X = H, C), SiN3X and SiN4 environments, respective1y. The
presence of the latter specics can only he'explained by assuming that there is cleavage ofSi
C bonds. The absence of the l3C peaks at 4 and -6 ppm in the produet at 300"C (Figure
5.4) indicatcs a complete change in the carbon environmenL No eenain assignment for the
peak at 51.2 ppm can he offered.40 The Si:N ratio al this stage of the pyrolysis shows an
excess of niaogen relative to Si3N4. This result may he compared to other systems, such
as PCS or polysi\azane precursors, as weIl as ~eposited silicon niaide,41,42 where a
silicon excess was observed al 400"c.
The Si:N ratio for sample 5N is still 1.6, but significant loss of carbon has
occurred. The 29Si NMR peak al -7 ppm (SiN2X2> bas disappeared and the intensity of the
37Tm T.; Inui, Mo; 01cam1llll, K.; SalO, Mo J. Mazu. Sei. Un. 1989,8, 111938 The small pcaks al 32. 28 and 23 ppm indicate bexaDe (soivent) stil1 present iD the sample39 The foUowiDg œmiDg scbcme was usee! throughout lhis wade e.g.. iD4N, "4" SlaIlds for4OO"C, wlùJc"N" means 1IIatNH3 was the atmospbc:ze duriDg pyrolysis.40 The peak al 112 ppm is aD anifact due 10 lbe probe.41A1CXl1l1drov, S.E.; Hi'cbman., M.L.: Sbalian, SA, J. Mazu. ChDn., 1994. 4(12), 184342Rocbdem', R.E.; ZbaDg, z.: Niles, D.W.; Mason, A.. J. Appl. PIrys.,1992, 72(1), 282
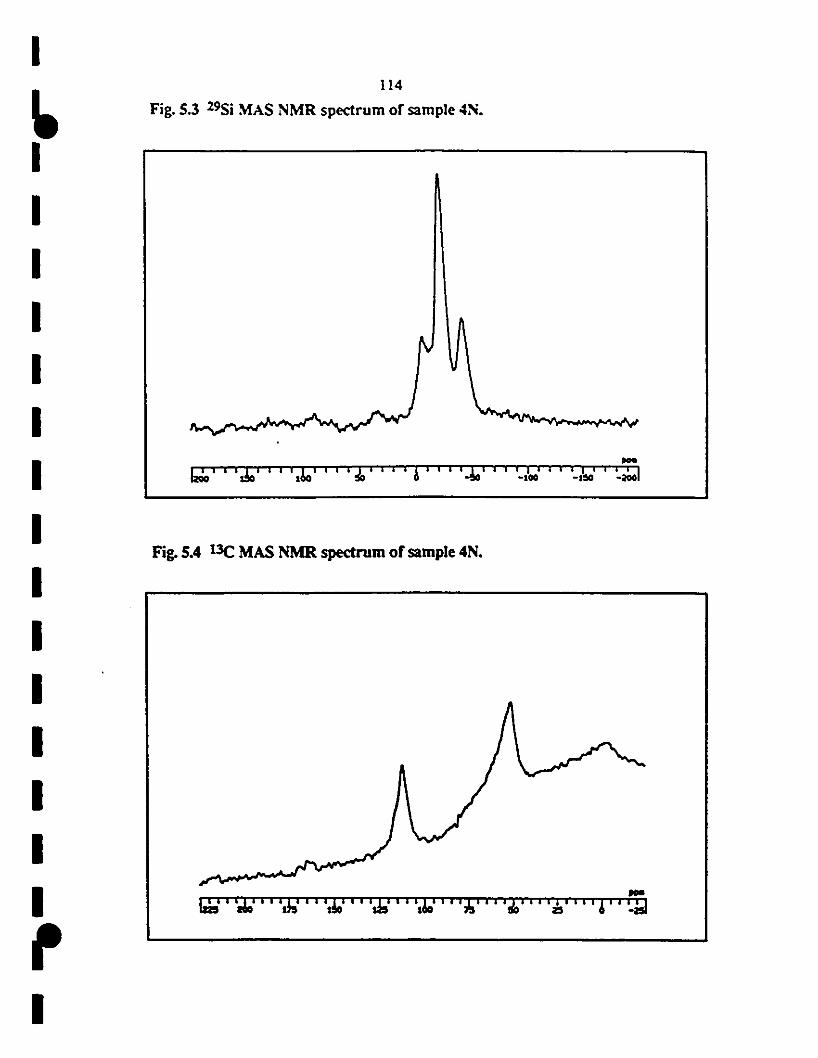
Gi!5' i 'l&i Il ilb' i i'SC i i 'l'A i. i,1i i •• ;&. i i 'JO' i i izb' i i Iii •~Q
114
Fig.5.3 29Si MAS NMR spectrum of sample 4N.
1
~111111111111111l'1
~ i i 'JO' i i i 1101 i i i JO i i 1 i i i i i ':t l i i i..1'oo' i i
Fig. 5.4 13(: MAS NMR spectrum of sample 4N.
i i i-SOO -i i 1-aoo
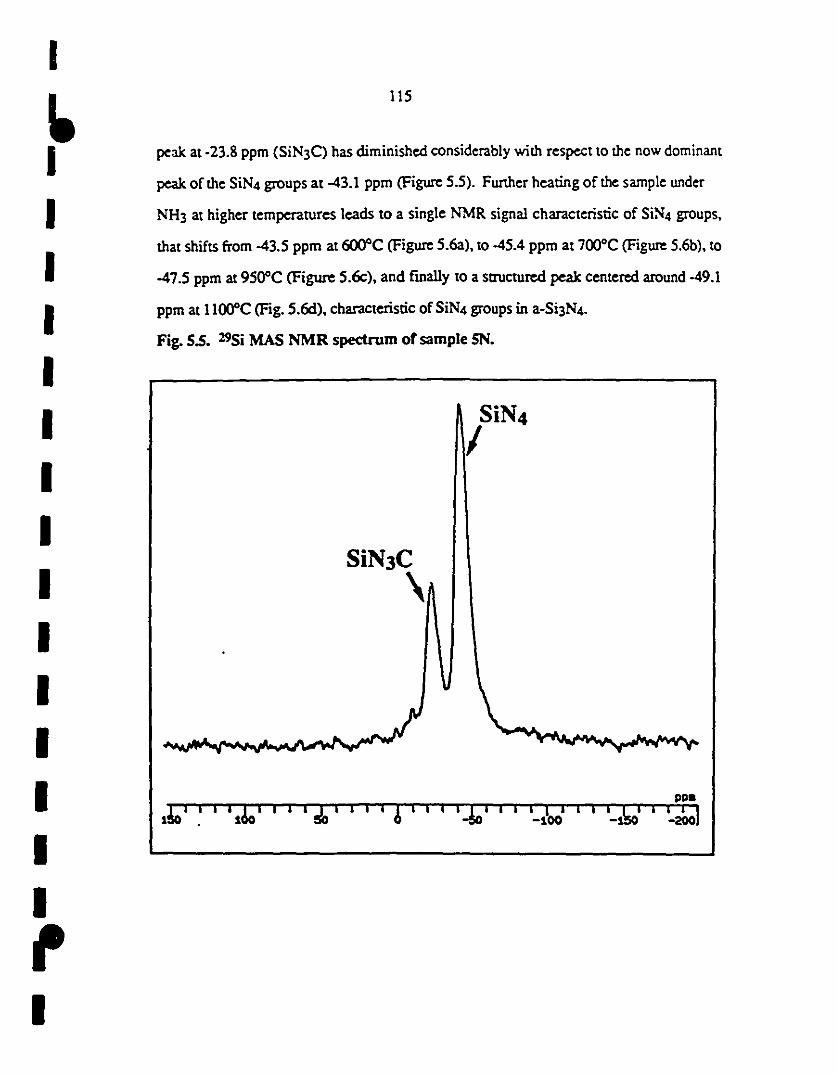
115
PP.i .-, ,i 1-1"50 -200
, 1 i-100'1)', Ai JO i
peak at -23.8 ppm (SiN3C) has diminished considerably with respect to the now dominant
peak of the SiN4 groups at -43.1 ppm (Figure 5.5). Further heating of the sample under
NH3 at higher temperatures leads to a single NMR signal characteristic of Sil'l'4 groups,
that shifts from -43.5 ppm at 600"C (Figure 5.00), to -45.4 ppm at 700°C (Figure 5.6b), to
-47.5 ppm at 950"C (Figure 5.6c), and fma1ly to a struetured peak centered around -49.1
ppm at ll000C (Fig. 5.6d), characteristic of SiN4 groups in a-Si3N4.
Fig. S.s. 29Si MAS NMR spectrum ofsample SN.
1
-111111111111111,1
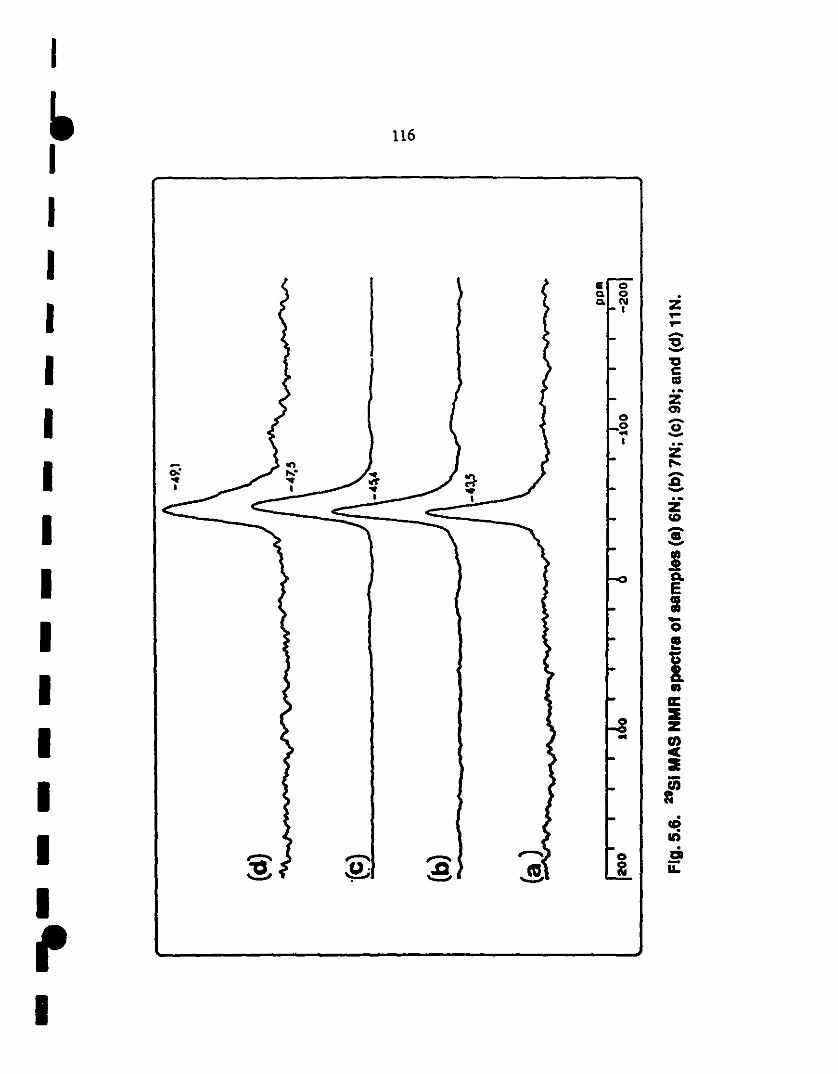
1
~111111111111111,1
116
• 0Q 0Q, N
1
oo..1
oli:
:a.E1'0l!
1.,a::Ez~:E
r
117
StaIting from 400°C. elementaI analysis indicates that the nitrogen content in the
sarnple (1.6) is greater than that in Si3N4 (1.33). The aClUal value may be compared ID the
case of complete amination of Si-H bonds resulting from the Kumada rearrangement.
where the maximum increase in the nitrogen content is 1 nitrogen atom per silicon if
Assuming both completion of the Kumada rearrangement at 400"C and complete
amination of the Si-H functionaliry. the maximum Si:N ratio that can be obtained from the
precursor with the formula (MeSiH)o.6s(MeSi)o.3S is 1:1.65. This is based on reaction
with gas phase NH3 only. excluding any involvement in the Si-H/N-H dehydrocoupling
reaetions of the NHx groups already present in the polymer. and assuming that DO Si-H
homodehydrocoupling occurs. This situation. however. is quite improbable. sinee
intramolecular attaek of NHx species already present in the polymer on the silicon center
should bc favored by decreased diffusivity of the reaetant species from the gas phase
through the reacting layer. A similar phenomenon was observee! in the case ofdiffusion
controlled oxidation of PMS. where the siloxane layer acts as a diffusion barrier for the
oxidation ofPMS.
Annealing sample 4N under Ar al 7000C resulted in a similar 29Si NMR panern ID
that of sample 5N (Figure 5.7). Therefore, annealing sample 4N aI SOOOC under NH3 has
the same effect as annealing under an inerl atmosphere aI 400-7000C. Sample SN was
subsequently heated under AraI 7000C and exhibited a 29Si NMR pattern similar ID that of
sample 6N (beated under NH3 up ID 600"C). as shown in Figure 5.8. Sample 6N. heated
1
~111111111111111
~1
aminosilane species form. as shown in eq. 5.1.
H NH NH21 3 1 AT.,Si··..·,,"ui ., ---Si·....," •
\ 1 "'CH3 CH
3
NH2.--Si...."" ,
\ (,)
Hi'
(5.1)
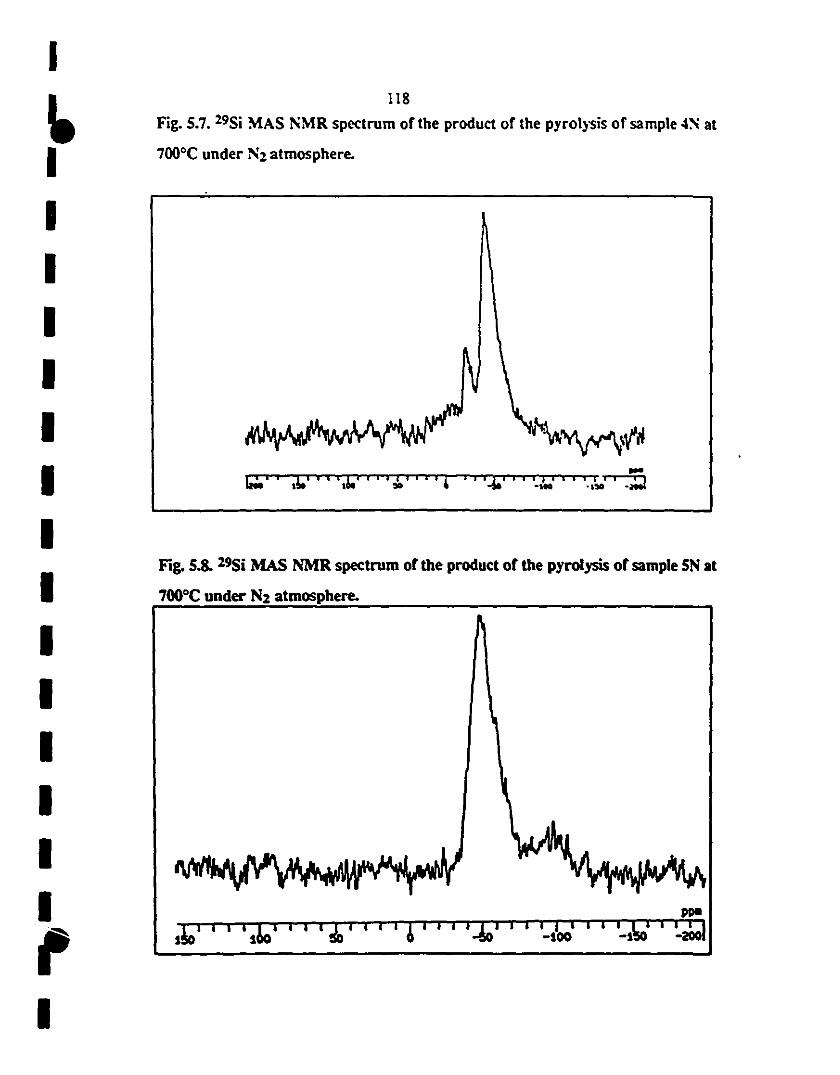
Fig. 5.8. 19Si MAS NMR spectrum of the product of tbe pyrolysis of sample SN at
700°C under Nl atmosphere.
II' III-s!50 -2001 J i-soo
Ji'"",' • i i J i i i 1 i i , ;;l-,eo .•,..
i 111 JO 1
lII:II" ,&;' i i i ,&;' i i i ~ i i i i 1
1SAOIJOI
IlsFig. 5.7. 19Si MAS NMR spectrum or the product or the pyrol)'sis or sample 4:'oi at
700°C under !\il atmosphere.
1
~111111111111111,1
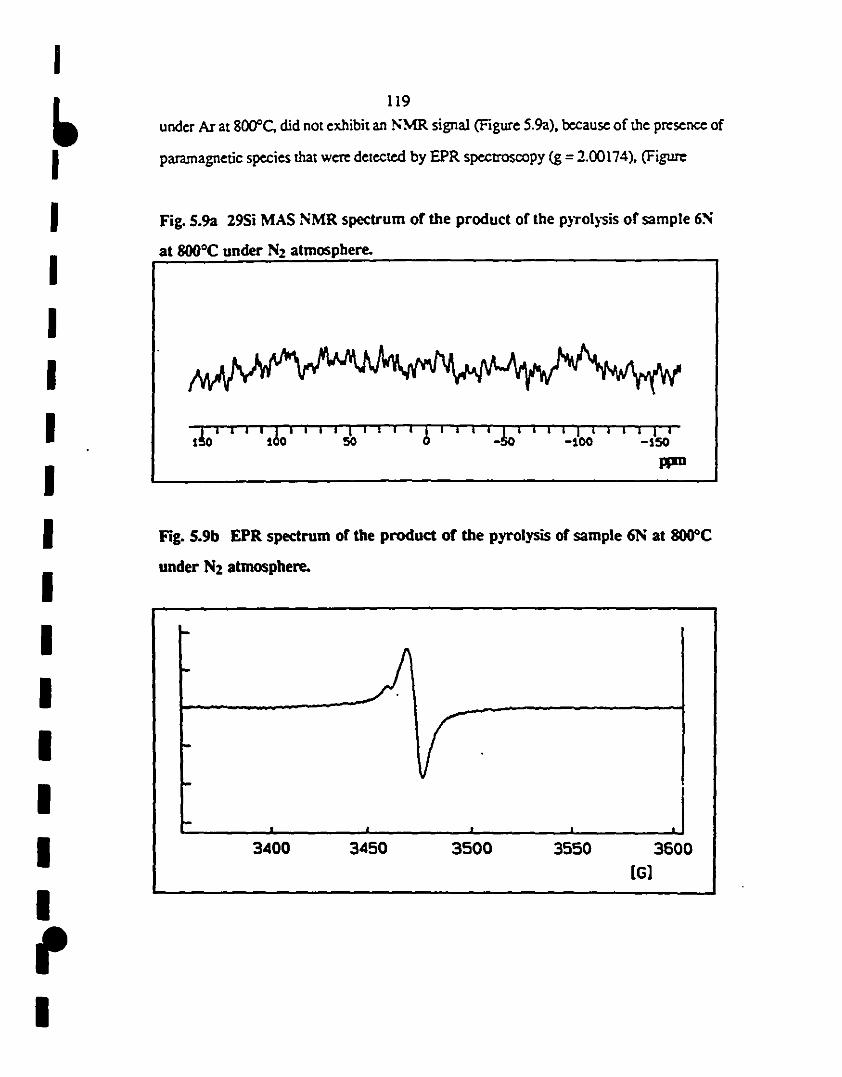
Fig. 5.9a 29Si MAS NMR spectrum of the product of the p)Tol)"Sis of sample 6~
at BOOoC under N2 atmosphere.
Fig. 5.9b EPR spectrum of the product of the pyrolysis of sample 6N at SOOOC
under N2 atmosphere.
119
under AI at SOO"C, did not exhibit an l'MR signal (Figure S.9a>, because of the presence of
paramagnetic species!bat were detected by EPR spectroscopy (g =2.00174), (Figure
i i i-150
f9II
Iii-100
l_lOi1 1 150i 1Ao 1
-- Ji- ("-
.... . .3400 3450 3500 3550 3600
[G]
1
l-I11111111111111,1

1
~1
11111111111111ft1
120
S.9b).
To explain these results. we suggest that at 4OQ-6OQ'C the Si-C bonds are c1caved
(producing numerous dangling bonds), allowing the silicon-centers to redisuibute towards
nitrogen-carrying captors present either in the sample43 or from the gas phase. The
remaining carbon-related centers undergo hydrogen exchange with ·NH. groups. The
laner process explains the general absence of gas-phase species bearing CoN groups in the
effluent stream. Such a process would be thermodynarnically driven. If active ·H and
·NH. (x = 1,2) species. resulting from NH3 thermal decomposition, are present in the gas
phase, fast radical recombination will occur. which consumes the EPR active species.
therefore permining the observation of NMR signals. This is the case for samples heated
only under NH3. In the absence of these radicals. a much slower or incomplete process
restricted to me condensed phase is involved in the consumption of the dangling bonds at
700·S00°C (which may involve the NH. groups already present in the polymer). ln this
case, the higher concenoation of paramagnetic species leads to the disappearance of the
NMR signals. This laner process. as observed in our case. is activated only al -SOO"C.
The 29Si NMR spectrUm of sarnple 7N annealed at SOO°C still shows interference from the
EPR active species (the low signal-to-noise ratio) and exhibits only the 29Si chemical shift
characteristic of SiN4 groups (Figure 5.10).
From these experimentS, two major conclusions may be reachcd: (a) the presence of
NH3 in the pyrolysis atmosphere al temper3ture5 higher than SOOOC is not neressa'Y for the
formation of SiN4 groups. and (b) in the S<Xl-SOOOC range. NH3 acts as a "sinlc" for the
43 A tbcnnodynamically driven redisUibulioo pocess diRcted tawanls caplIIring IlÏlIOgell CCIllI:IS issuggested by lhe sùnilarbcbaviour ordie silicon dangling bonds genesaœd ÙI SÙ/llC8ClÏllg wilb carlloocetlœrs, e.g•• in lhe rearrangement of poIysi1ane io PCS during Kumada-type œal3Ilgemt.1lt oCpolysi1anes.The use ofmixlllreS of"si1icon·ricb" and "carbon-ricb" <l'g3IIOSilic:o pol)'lllClS in appropriaœquamilies1eads ta Ibe sitllalioD wbere lhe==Si and C react and produce mosdy Si~ boods, e.g., die lIIÎXIIIn: oCpolY(melbylsi1alle)1N"lCll1oo poly(carbosiIane), sec Seyfenh. D.; Yu, Y.-F.; KoppelldI, G.E. Us. PtIIDIt4719272 (1988).
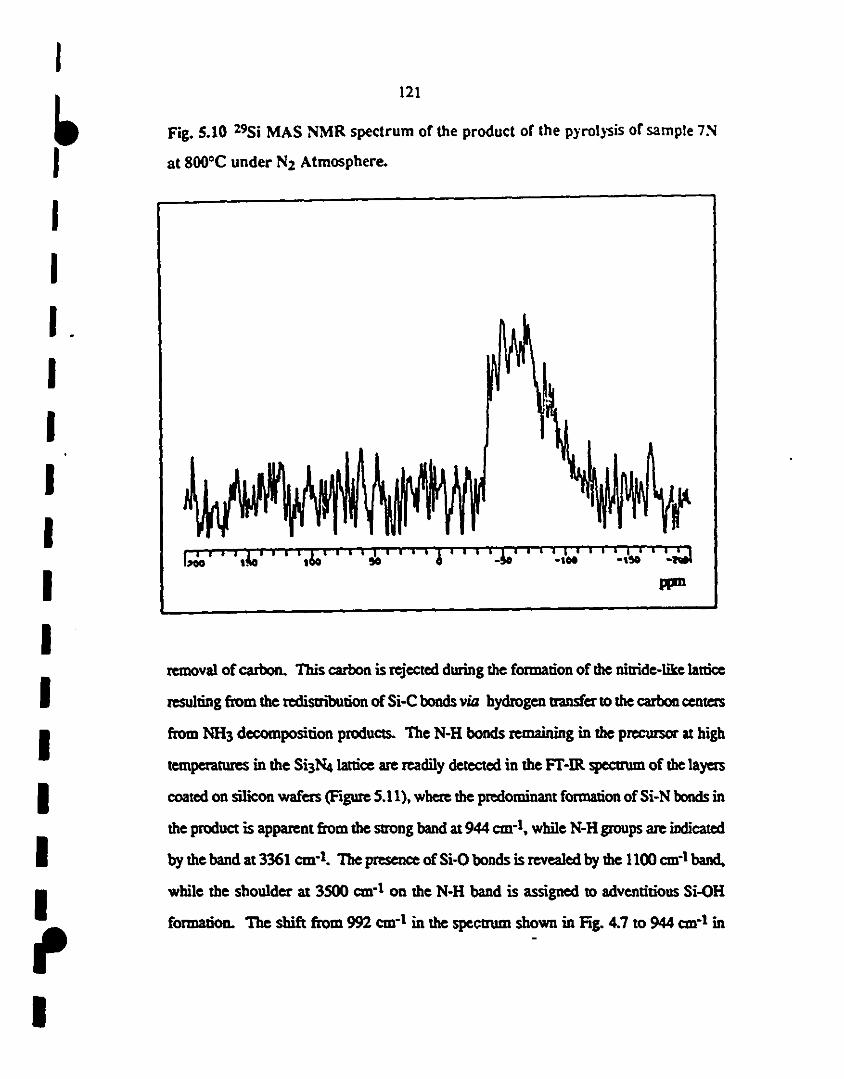
121
Fig. 5.10 29Si MAS NMR spectrum of the product of the p)"rol)"Sis of sarnple 7:"1
at 800°C under N2 Atmosphere.
liiiii.".,~ NIi A • i i i lOi , •• i i- -'01',10' i i 'Jo', i i , » " 1HO ,.
mnoval ofcarbon. This carbon is rejected dwing the formation of the niaide-like lanice
resu1ting ftom the redistribution of Si-C bonds via hycirogen tranSfer to the carbon centers
ftom NH3 decomposilÏon products. The N-H bonds remaining in the prccursor Il high
temperatureS in the Si3N4lattice are readily detected in the FT-IR specaum of the layers
coated on silicon wafers (Figure S.11), where the predominant formation ofSioN bonds in
the produe:t is apparent from the strong band at 944 cm-l, while N-Hgroups are indieated
by the band at 3361 cm-l• Thepresence of Si-O bonds is revealed by the 1100 cm,l band,
white the shoulder at 3500 cm-t on the N-H band is assigned to adventitious Si-QH
formation. The shift from 992 cm-l in the spee:trum shown in Fig. 4.7 to 944 cm-l in
1
~1111_11111111111,1
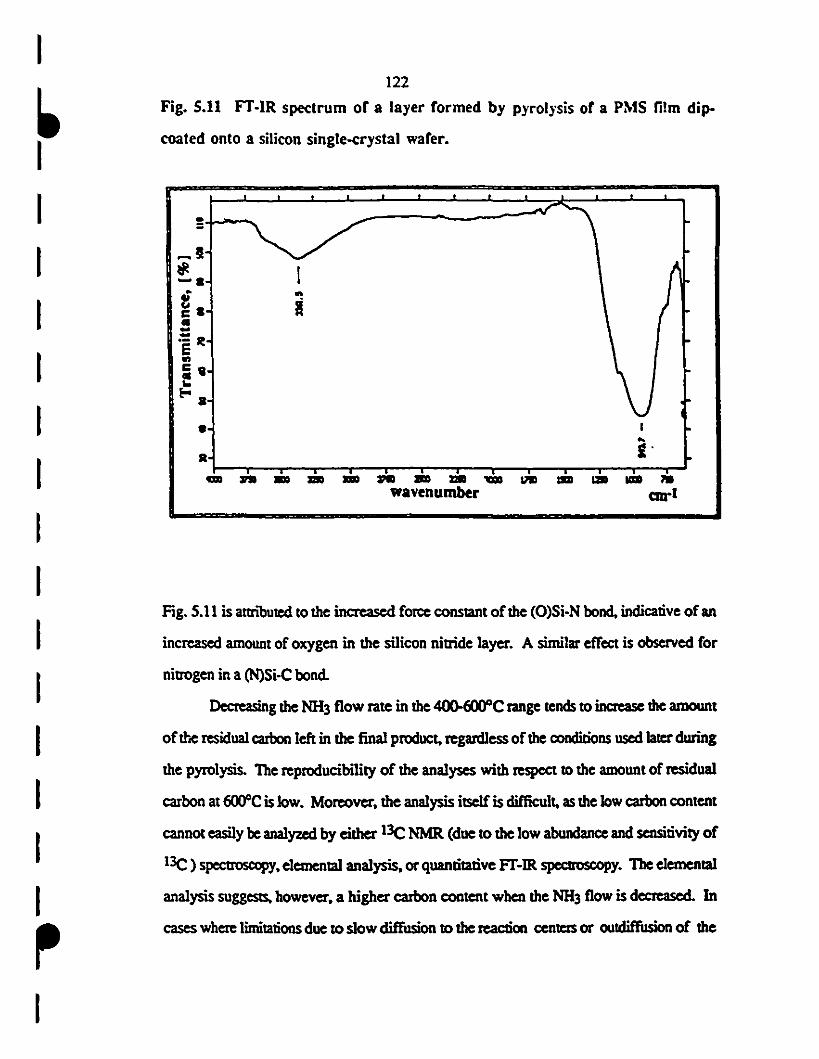
1
~1
1
1
1
1
1
1
1
1
1
1
1
1
1
1,.1
122
Fig. S.11 FT-IR spedrum of a layer formed by pyrol)'sis of a PMS film dip-
coated onto a silicon single-crystal wafer.
....r"\
~ ~
~!.
!1*' 1-a· ~• .. )~a li•..:~ ~E..
~1: e.f
E-o a.
• 1..• 2'
~- ft :BD - - - :BD - -- uil aD .. lIIlD 111wavenumber curl
Fig. 5.11 is 3nribulCd to the increased force constant of the (O)Si-N bond, indicative ofan
increased amount of oxygen in the silicon nitride layer. A similar effect is observed for
nitrogen in a (N)Si-C bond.
Decreasing the NB3 flow rate in the 400-6OO"C range tends ta increase the amount
of the residuaI carbon left in the final product, regardless of the conditions used larer during
the pyrolysis. The reproducibility of the analyses with respect to the amount of residual
carbon at 600"C is low. Moreover. the analysis itself is difficult, as the Iow carbon content
cannot easily lie analyzed by eïther 13C NMR (due ta the low abundance and sensitivity of
13C) spectroSCOpy. elementaI analysis. orquantitative Fr-IR speclluscopy. The elementaI
analysis suggests, however. a higher caIbon content when the NB3 flow is decreased. In
cases where linûlations due ta slow diffusion ID the reaction centelS or ourdiffusion of the

1
~111111111111111,1
123
prodUClS are not present (such as thin-film forming), this effect can also be associated with
significant NH3 decomposition (vide infra). Carbon removal at high temperatures may
occur either through the reaction C + NH3 --> HCN + H2, or by transamination.
Both of these reaction types requil'e a high partial pressure of NH3 in the gas phase, a
condition that might~ responsible for the lack of success in complete removal of carbon
from Si3N4 at temperatures above SOO°c, where .liffusion is fast, but NH3 is expected to
decompose. This carbonlnitrOgen exchange may also be viewed as involving two steps,
viz., fonnation ofdangling bonds followed by a thermodynamicaIly driven reorganization
to forro SioN bonds (as in the case of Kumada rearrangement where a similar
thermodynamic driving force is responsible for the reorganization of polysilanes to inctease
the concentration of Si-C bonds). These two processes may explain the formation of
temary phases in ammonia-poor pyrolyses. This suggestion is supported by a study of the
synthesis of nitrOgenated SiC layers, and the observed immiscibility ofC in Si3N4 obtained
from the pyrolysis oforganosilicon precursors in ammonia-rich systems (Chapter 3).
5.3.2. NH3 decomposition uoder thermodynamic equilibrium
NH3 Decomposition
The extent of NH3 deeomposition was investigated by titration of the NH3 stream
after it had left the reaction chamber. The experimental titration data indicate that
decomposition of NH3 is measurable, even at temperatureS as low as 400"c, and !bat the
fraction deeomposed during pyrolysis is a function of the flow rate and the Temperature
cycle. Data showing the volume parameter for titrating a constaI1t amount of lMHa

1
~1
1
1
1
11
111111111,.1
124
solution with the NH3 gas exiting from the fumace healed al different lemperarures for
different flow raIes are shown in Table 5.2.
Table 5.2. Volume parameter for the autotitration of lM HCI solution for a
given now rate of emuent NH3 from the pyrolysis furnace.
T,eC) 108 300 500 700 900 1100
V (sccm)
48 28 35 54 65 123 >500.57 24 28 41 50 68 130
70 20 23 33 36 45 118
Prior te studying the influence ofdifferent factors on the decomposition process.
a thermodynamic cquilibrium study was perfonned. The cquilibrium of the reaction
NH3 --> 1/2N2 + 3(2. H2 has been investigated experimentally and theoretically due to its
relevance to the Haber process. However, data for parameters different from the Haber
process conditions are not usually considered. Thermodynamic data have been used to
calcu1ate the cquilibriumconcentration of NH3 and the values oblained have been compared
to the experimental values associated with the pyrolysis. Using the Cp values Iisted in
Table 5.344 and the KiIchoff law, values were obtained for the fonctions of state ,1fHO-r,
,1fSO-r and ,1fGOor (Table 5.4). From ln Kp =,1f(;0/RT, the curve-fitted dependence of Kp
versus T is presented in Figure 5.12.4S
44 GmeÜIIS Handblldt der AnorgQlÙSt:he Chemïe,1975, 2, 68S4SKaleidagrapb, AbelbecJ: Software, Synergy Software (PCS 1Dc:.) Ed, 1994

(5.1)
1
~11111111111111.1t'1
125
Table 5.3 Cp(T) Data for NH3. N2 and H2232
Cn - a+bT+cT2 a l()3b 107c
N2 6.3 1.819 -3.45
H2 6.88 6.6 -2.79
NH. 5.92 8.963 -1.764
Table 5.4 Calculated functions of state for NH3 decomposition at various
.emperatures at nonnal pressure.
T("C) 300 400 500 600 700 800 900 1000
Mf CkI/mol) 46.42 48.58 50.67 52.13 53.32 54.35 55.14 55.75
Mi (J!KImol) 99.39 105.70 110.10 112.77 114.59 115.93 116.69 117.51
6G (kJ/mol) 16.57 4.70 -2.66 -7.87 -11.54 -14.41 -16.67 -18.54
Kft (atm) 0.0013 0.242 1.898 4.854 7.283 8.753 9.307 9.327
Within the same high 1evel of confidence (correlation coefficient =0.99984), the mole
fraction of NH3 in the equilibrium composition was calculated. The reaction coordinate cao
be kept as the independent variable if the amount of H2 produced by side reactions
involving the sample is negligible with respect to the H2 produced by thermal
decomposition. Since Kp = Kx*P, (Kx= equilibrium constant in terms of mole fractions)
the expression for Kp becomes
Kp =P [27112 (1-y)2]II6y
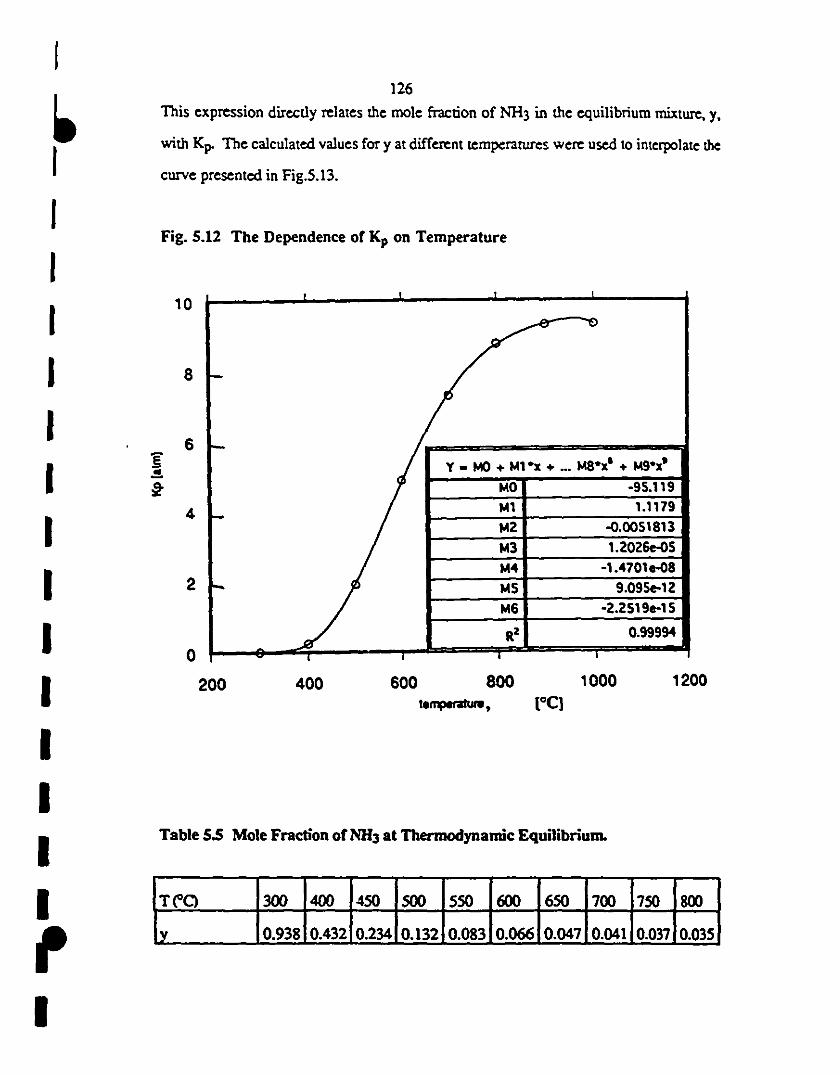
T("C) 300 400 450 SOO 550 600 650 700 750 800
Iv 0.938 0.432 0.234 0.132 0.083 0.066 0.047 0.041 0.037 0.035
126
This expression directly relaies the mole fraction of NH3 in the equiIibrium mixlure. y.
with Kp- The calculaled values for y al differenl lCmperalUre5 were used 10 inlerpolate the
curve presenled in Fig.5.13.
12001000800600400
. . • .
"V
~
~
y • MO + Ml *le + _. MS·x· + M9·x·
MO -95.119
~Ml 1.1179M2 -o.0051S13M3 t.2026e-05M4 -1.4701..oS
"- M5 9.095e-12M6 -2.2519e-15
RZ 0.99994
f 1 1
2
o200
4
6
8
10
Fig. 5.12 The Dependence or Kp on Temperature
Table 55 Mole Fraction or NH3 at Thermodynanûc Equilibrium.
1
~1
1
1
111111111111,1
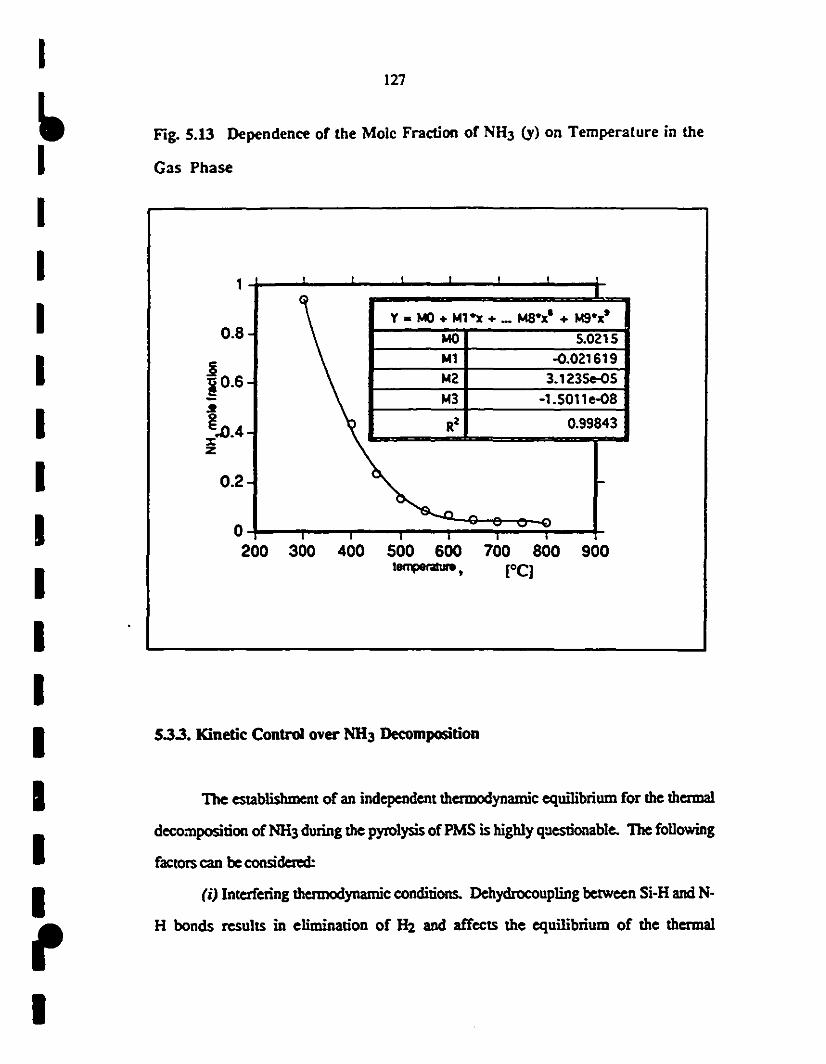
Fig. 5.13 Dependence or the Mole Fraction or NH3 (y) on Temperature in the
Gas Phase
The establishment of an independent thermodynamic equilibrium for the thermal
decomposition of NH3 during the pyrolysis of PMS is highly q:lestionable. The following
factors cao beconsidcred:
(i) Interfering thennodynamic conditions. Dehydrocoupling between Si-H and N
H bonds results in eliminatioD of H2 and affects the equilibrium of the thermal
127
900700 SOO[oC]
SOO 600lelTll8ra11n ,
400
• , , • , , ,•
y • MO + 141·x + _. Mll·x· + 149·x·- 1040 5.0215
141 -0.021619
- 142 3.123Se-OS
143 -1.S011e-08
- RZ 0.99843
- -JO> _
~
• 1 1 •o200 300
1
0.2
0.8
5.3.3. Kinetic Control over NB3 Decomposition
1
~111111111111111,.1

46Han. HoN.; Undquist, D.A.; Haggeny, J.s.; Seyferth, D. Chem. MQlQ. 1992,4, 70s
where D =diffusion coefficient. de = concentration gradient in the boundary layer, s = area
of the film. and li is the thickness of the boundary layer. Therefore, if a l-cm2 silicon
substrate is used, assu.-ning a diffusion coefficient of5 x 10.2cm2s·l and if the thiclcness
128
decomposition of NH3. Also. methane loss during thermal cracking of the polymer nuy
interfere with the equilibrium due to the added hydrogen pressure at high temperatures. In
both cases, irreproducible behavior in the solid ma; '·~d to irreproducible results in the gas
phase.
(ii) Kinetic control over decomposition. The flow rate, the pyrolysis cycle or the
residence time of NH3 in the pyrolysis fumace may dictate the steady-state conditions,
rather than thermodynamics. For example, during the transformation of PMS to Si3N4 \'ia
pyrolysis under NH3, the NH3 flow rate was a parameter affecting the amount of the
residual carbon, a process that could otherwise be interpreted as being related to the extent
of thermal decomposition of NH3.
(iii) Kinetic control associated with the solidlgas reaction. Diffusion-controlled
processes in the bulk materials (e.g., solvent evaporation,46 diffusion to the reaction center
in the solid or outdiffusion of the gaseous products formed during pyrolysis) may lead to
nonequilibrium conditions in the system.
For the decomposition process to be independent of the chemical reaction with the
precursor, the concentration of CF4 or H2 resulting from the polymer must be low so as
not to interfere with the decomposition equilibrium. A reasonable degree of independence
is achieved if the NH3 is present in a IQ-fold excess. Considering the reacti;>n of the
precursor with NH3 to be diffusion-controlled, the highest rate of consumption of NH3
occurs during the initial stages, due 10 the maximum value for de in the following relation
1
~111111111111111,.1
dm= -Ds de dta (5.2)
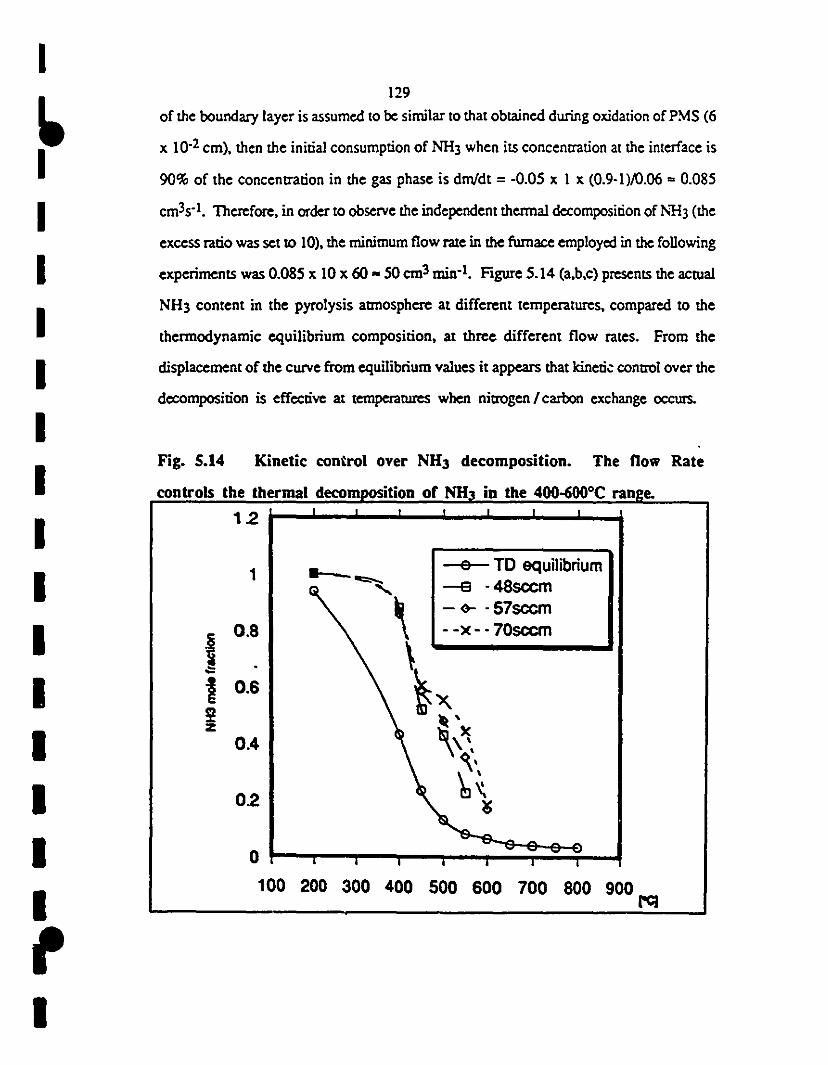
129
of the boundary layer is assumed to he similar to that obtained during oxidation of PMS (6
x 10-2 cm). then the initial consumption of NH3 when its concentration at the interface is
90% of the concentration in the gas phase is dm/dt = -0.05 x 1 x (0.9-1 )10.06 = 0.085
cm3s- l . Therefore. in orderto observe the independent thermal decomposition of/l;1i3 (the
excess ratio was set te 10). the minimum flow rate in the fumace employed in the following
experiments was 0.085 x 10 x 60 - 50 cm3 min-l. Figure 5.14 (a.b.c) presents the actual
NH3 content in the pyrolysis atmosphere at different temperatures. compared to the
thermodynamic equilibrium composition. al three different f10w rates. From the
displacement of the curve from equilibrium values it appears that kinetic control over the
decomposition is effective at temperatureS when nitrogen 1carbon exchange occurs.
r NH •
Kinetic control over NH3 decomposition. The now RateFig. 5.14
Is hcontro t e thermal decomoosltlon 0 h ID the range.
12 1 1 1 1 1 1 1
e TD equilibrium1 ----...." -€l -48sccm, - ~ -S7sccm
t 0.8 - -x _. 70sccm
. \-~~,1 0.6
!2z\\~\0.4
~'.02 '\
~
"""0 1 • 1 1 • 1 1
100 200 300 400 SOO 600 700 800 900 rel
1
l-I11111111111111l'1

1
~111111111111111,1
130However. changes in the flow rate do not appear to be effective up to 400"C. The thenno-
dynamic equilibrium is not reached. but other factors than the flow rate control the thenna1
decomposition of NH3. In the 4OQ-6QO°C range. an increase in the flow rate from 48 to 70
scern leads to an increase of -20% in the partia1 pressure of NH3 at SOO°C. Therefore. the
associated increased formation of radical species in the gas phase may be responsible for
the trend of increasing removal of the carbon from the precursor. At the S3I1l\. rime. the
increased decomposition of NH3 results in an increase by about 25% in the partial pressure
of H2 in the gas phase, retarding carbon removal due to the formation of CS4 unilS.4'
This combination of effects may he responsible for the variations observed in the amount of
residual carbon in the produet at 6QOOc.
5.4. Conclusions
The composition of the intermediate species produced during the reactive pyrolysis
of poly(methylsilane) under NH3 to form Si3N4 has been investigated by elemental
analysis and multinuclear (13C, 29Si) solid State NMR spectroscopy. The resullS can be
correlated with previously reported vibrational specttoscopic data. At 400"c, the nitrogen
content exceeds the stoichiometric ratio for N in Si3N4- Carbon displacement is a lWO-step
proccss !bat involves bomolytic cleavage of the Si-C bonds, followed by carbon removal
through chemical reaction with the prodUClS of the NH3 decomposition. Thermodynamic
control over the redistribution of Si-C to SioN bonds is effective in the 400-8OO"C range
and is responsible for the formation of SiN4 groups at 400°C. The resulting silicon
dangling bonds reaet with 'NHx species, either from the gas phase or already present in die
precursor, while hydrogen transfer from the ·NHxgroups to carbon forms C':H4 leading to
47 Mach, R.; K1ocz. IL-O.; SuIukc, K.·D.; Drosl. H.; Oleszak, F.; Lacayo. G.; SzuIzewsky. Je.; D<lrfel, LiD Key EIIg. Mazu. 1994,89-91.41

1
~111111111111111,1
131
elimination of residual carbon from the solid produet. The displacement of residual carbon
at temperatures >800°C is difficult, not only because of restricted diffusion of the gaseous
spccies in the extended crosslinked residues. but also due to insufficiem partial pressure of
NH3 in the gas phase to sustain HCN formaùon or any transaminaùon reacùons.
Experiments performed on milligram-scale samples have permi::ed the thermal
decomposition of NH3 tO be monitored by minimizing the extent of the chemical reactions
of NH3- Kinetic control over the thermal decomposition of NH3 was achieved by varying
the f10w rate in the 400-6OO"C temperature range.

6.1. Contributions to Knowledge
1
~111111111111111,1
1
2
3
4
132
CHAPTER6
Overall Conclusions
The main contributions to knowledge are seven listed below.
Fr-IR analysis of milligram-size samples of the polymeric precWSOlS, and
their chemical reactions during pyrolysis have been conducted under a
variety <;,f conditions on thin layers of the precursor deposited on silicon
single-crystal wafers.
Quantitative measurements of the oxygen content in poly(methysi1ane) in
the ppma range can be performed via a calibration procedure using the
interstitial oxygen content in the silicon single aystal wafers. ü these
wafers are used as substrates and IR windows.
The onset for the Kumada rearrangement of poly(methylsilane) to
poly(caIbosilane) has been studied by monitoring the Cll(Si-CH2-Si) mode,
at 1050 cm-1 in the IR spectra and bas been shown to occur al a lower
temperaturC titan previously reported.
Thin layers ofamorphous siliCOll-based.:eramic maœriaIs can be depositeci
on various substrates. such as alumina, silicon wafers. graphite. or
quartz, by c1assical methods (clip- or spin-coating) or by condensation of
the volatile species formed during the thermal cracking of the precursor
under pyrolysis conditions (CP-VO).

6.2. Suggestions for Further Work
1
~111111111111111,1
5
6
7
1
2
133
The amount of nitrogen incorporated in the ceramic residue can be
controlIed from very low levels up to complete CIN exchange, by varying
the NH3 partial pressure in the gas phase during pyrolysis of PMS.
A thermodynamica11y-driven process sünilar to the Kumada rearrangement
is responsible for the preferentiai •...distribution of Si· dangling bonds
formed by therma1 cracking ofthe organic groups bonded to SioN bonds.
Kinetic control over NH3 thermal decomposition has to be achieved in
order to control the CIN exchange process.
It wou1d be wonhwhile investigating whether ceramic materials with
precise elemental composition in the Si-o-C-N system can be obtained
using po1ymeric precursors, e.g., SiOC and SiON.
Investigation into the reactions producing volatile species by the thermal
cracking ofpoly(methylsilane) during pyro1ysis under anunonia may 1ead
to bener results in the synthesis of thin films of Si3N4. Increased
volatilization yields might be achieved in the CP-VD process under
conditions favouring transamination over cross\inlcing in the precursor.
ln $iru monitoring of the gas phase species simultaneously during the
deposition process by CP-VD may be performed by coupling a TG-MS to
a Fr-IR instrument equipped with silicon substates operating as IR
windows to determine the rate ofC-bearing species as well as H2.

1
'-111111111111111,1
134
6.3. Publications in Refereed Journals
1. M. Scarlete. S. Brienne. I.S. Butler and 1.F. Harrod. Infrared Spectroscopie Study
of Poly(methylsilane). Its Oxidaùon and Its Transfonnation into Poly(carbosilane) on the
Surfaces of Silicon single Crystal Wafers. Chem. Mazu.• 1994. 6. 977.
2. M. Scarlete; J.F. Harrod; I.S. Butler. Nitrogenation of Silicon Carbide Layers
Deposited on the Surface of Silicon Single Crystal Wafers. Chem. Mazu., 1995,7, 1214.
3. M. Scarlete; J. He; J.F. Harrod; I.S. Butler, Poly(methylsilane) and
Poly(methylsiladiazane) as Precursors for Silicon-Containing Ceramics, in Applicarions of
Organometallic Chemistry in the Synthesis and Processing of Advanced Materials
(&Is.: Laine. R.M.; Harrod, J.F.), Kluwer Academic Publishers, Dordrecht, 1995.
Proceedings of the NATO ARW, Sept. 4 - 9, 1994, Cap d'Agde, France.
4. M. Scarlete, I.S. Butler and J.F. Harrod, Investigation into the Formation of
Silicon Nitride and Oxynitride During the Reactive Pyrolysis of Poly(methylsilane) with
Ammonia, Chem.Mater., to be submined.
5. M. Scarlete, J. He, J.F. Harrod and I.S. Butler, Low-temperature Nitrogen
Carbon Exchange During Reactive Pyrolysis of Poly(methylsilane) under Ammonia,
Chem. Mater., to be submined.